 Ajay Kumar Mishra
Ajay Kumar Mishra Poonam Yadav
Poonam Yadav Sheetal Sharma
Sheetal Sharma Piyush Maurya
Piyush Maurya
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 13 February 2025
Sec. Terrestrial Microbiology
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1444903
Soil microbial species diversity and distribution of microbial communities are vital for soil and crop health, nutrient cycling, availability, and subsequent plant growth. These soil dynamics are highly influenced and altered by various soil management practices, inputs, and agricultural techniques. In the present study, the effects of chemical and organic management practices on soil microbial diversity and community structure were examined and compared using amplicon sequencing of the 16S and ITS regions. Two contrasting soil samples were selected from each crop fields at the International Rice Research Institute-South Asia Regional Centre (IRRI-SARC) in Varanasi: one field followed conventional chemical fertilizer inputs, while the other implemented natural farming practices, including tillage, on-farm crop residue management, and water management. Soil samples from each field were analyzed for bacterial and fungal diversity. Our findings showed that the two differently managed soils exhibited distinct microbial community compositions, with the organically managed soil exhibiting a higher diversity of decomposer bacteria and fungi, showing 40 unique elements in organic soil samples and 19 in chemically managed soil. Natural farming practices also demonstrated a higher relative abundance of bacterial and fungal phyla. Our results emphasize the significance of sustainable soil management techniques, suggesting that organic inputs can increase soil microbial diversity and richness. The functional roles of these microbial communities in soil ecosystems and their potential impact on crop yield and nutrient cycling warrant further study.
Soil is a complex and dynamic system essential for sustaining life on Earth. A critical component of soil, and a key indicator of its sustainability, is soil microbial communities. These communities play a vital role in soil sustainability (Hartmann and Six, 2023) and crop productivity by providing crucial ecosystem services (Bardgett and van der Putten, 2014), such as nutrient recycling, nutrient availability to plants (Poonam Srivastava et al., 2017), carbon sequestration, and disease control through direct and indirect mechanisms (Van Der Heijden et al., 2008; Sun and Badgley, 2019; Krause et al., 2023). Further, soil micro-biota contributes to plant stress tolerance, systemic resistance and immunity build-up (Singh et al., 2023). However, the composition and function of these microbial communities are significantly influenced by soil management practices, especially the type and rate of fertilizers applied (Hartman et al., 2008; Enebe and Babalola, 2020).
To restore the soil health and sustainability, many practices have been employed for soil management to promote sustainable agricultural development and enhanced yield (Montanarella, 2015). Traditional tillage methods widely practised in conventional farming practices disrupt soil structure, causing increased soil erosion and the imbalance of the global carbon cycle (Van Oost et al., 2007; Bednář and Šarapatka, 2018). Conservation tillage and fallow rotation systems have been introduced to improve soil organic carbon storage, enhance nutrient uptake, and increase crop yields (Hussain et al., 2021; Krause et al., 2022; Li et al., 2023). These practices also reduce carbon dioxide emissions, maintain soil ecosystem health, and play a critical role in mitigating climate change’s impact on agricultural production (Sun and Badgley, 2019; Wagg et al., 2014; Yadav et al., 2021).
Two widely used soil management approaches, organic and chemical fertilization, have contrasting effects on soil microbial diversity and structure. Organic fertilization, involving plant-or animal-derived materials such as compost or manure, adds organic matter and improves soil health (Lupatini et al., 2013a,b; Krause et al., 2023; Xu et al., 2022). It has been shown to enhance microbial diversity, increase enzyme activity, and improve soil physicochemical properties. For instance, in one study (Xu et al., 2022), organic cultivation increased the abundance of dominant bacterial phyla, such as Proteobacteria and Acidobacteria, as well as fungal phyla like Ascomycota and Basidiomycota. In contrast, chemical fertilization provides concentrated nutrients that can boost crop yields, but long-term use may lead to reduced microbial diversity, degraded soil structure, and increased greenhouse gas emissions (Singh et al., 2011; Liang et al., 2019; Sun and Badgley, 2019). Therefore, organic fertilization has a more sustainable impact on soil health than chemical fertilizers (Rousk et al., 2009).
Metagenomics has emerged as a reliable and powerful biotechnological tool to comprehensively evaluate the effects of these practices on soil microbial communities. It enables the study of microbial diversity by analyzing the genetic composition of entire microbial populations in soil ecosystems (Suyal et al., 2019). Advances in next-generation sequencing technologies, such as amplicon sequencing of the 16S and ITS regions, allow for rapid and detailed profiling of bacterial and fungal communities (Saharan et al., 2023). Metagenomics technique has been widely accepted to investigate how environmental factors, including chemical fertilization and organic soil management practices affect microbial communities (Rousk et al., 2009; Saharan et al., 2023).
The theoretical framework for this study integrates the Biodiversity-Ecosystem Function Theory (Loreau et al., 2002) and the Soil Health Paradigm (Doran and Zeiss, 2000), which highlight the role of microbial diversity in maintaining soil functionality and resilience. Adverse impact of climate change and increased pesticide use can severely disrupt microbial communities, reduce biodiversity and crop yield, and weaken soil health. It is highly evident now that climate change can alter temperature and soil moisture levels (Yadav et al., 2021), directly impacting microbial activity and diversity (Compant et al., 2010), while excessive pesticide use can lead to soil degradation and the loss of beneficial microbes (Saharan et al., 2023). So, it is highly needed to understand how various farming practices and soil amendments are influencing soil microbial diversity and how we can develop a sustainable agricultural system with these findings that can also withstand environmental stresses, maintain productivity, and reverse the negative impacts of pesticides and climate change on soil ecosystems. Additionally, microbial diversity and composition in agricultural soils vary with crop/plant species, existing environmental conditions, irrigation and soil amendments/inputs practices (Majumdar et al., 2024). Microbes, especially in the rhizosphere and endosphere, influence plant growth and stress resilience (Upadhyay et al., 2018). Studies supported that organic farming promotes higher microbial diversity, thus enhancing crop/plant health yield (Bulgarelli et al., 2012; Edwards et al., 2015; Saharan et al., 2023).
This study aims to investigate the differences in microbial biodiversity and community structure between soils managed with organic and chemical fertilization strategies. The study’s novelty is based on the location, soil, and practice-specific study of the rice-wheat cropping system. There are few studies to justify the findings, so it is also very important to map the significance of such practices in agriculture. We hypothesize that organically managed fields with zero tillage will exhibit richer microbial diversity than chemically treated soils. Data were collected to assess differences in tillage frequency, microbial community composition, and photosynthetic bacterial inoculation in organic and conventionally managed plots over the rice-growing season.
Next-generation amplicon sequencing of 16S and ITS regions was used to identify and comparatively evaluate the variation in soil microorganisms’ community structure and diversity between these management systems. As this study integrates multiple organic management practices, including reduced and zero tillage, bio-input fertilization (Azolla, blue-green algae, vermicompost, and vermi-wash), diverse sowing methods, and reduced water use, in contrast to conventional practices makes it novel. This is the first study to examine the collective effects of these practices on soil microbes, offering valuable insights into sustainable soil management.
Our findings will have important implications for developing sustainable soil management practices that prioritize promoting healthy microbial communities (Yang et al., 2014). The integration of practices and technologies can result in more accessible and better soil and agricultural production system management that is more sustainable.
Two experimental fields were selected for soil sample collection: an organically managed paddy field and one conventionally managed paddy field. Organic practice has been done at this location for the last 3 years. The study was conducted during the year 2020–21 and 2021–22 at the agri-experimental farm of the International Rice Research Institute South Asia Regional Centre, Varanasi (25.302877–82.947973, 83 m above mean sea level), Uttar Pradesh, India.
The min-max temperatures during the experimental year range between 15.5°C and 36°C (mean values), respectively. Annual rainfall for the same period was between 900 and 1,113 mm range, and most of the rains (>70%) occurred during the monsoon months of July to September. Soil samples were randomly collected from (upper plow layer 0–15 cm) each replicated field trials and plots, and processed for 16S and ITS metagenomics study. Tillage, seed rate, cropping system and agronomic management practices followed in organic and conventional farming systems and the five practices, treatment and crop establishment table and initial soil properties are provided in Tables 1–3, respectively. Treatment under the natural farming were combined as various tillage and crop residue management practices along with alternated wetting-drying irrigation practices whereas in conventional practices traditional irrigational knowledge and use of chemical pesticides were applied on the Rice-Wheat-Mung cropping systems for the assessment.
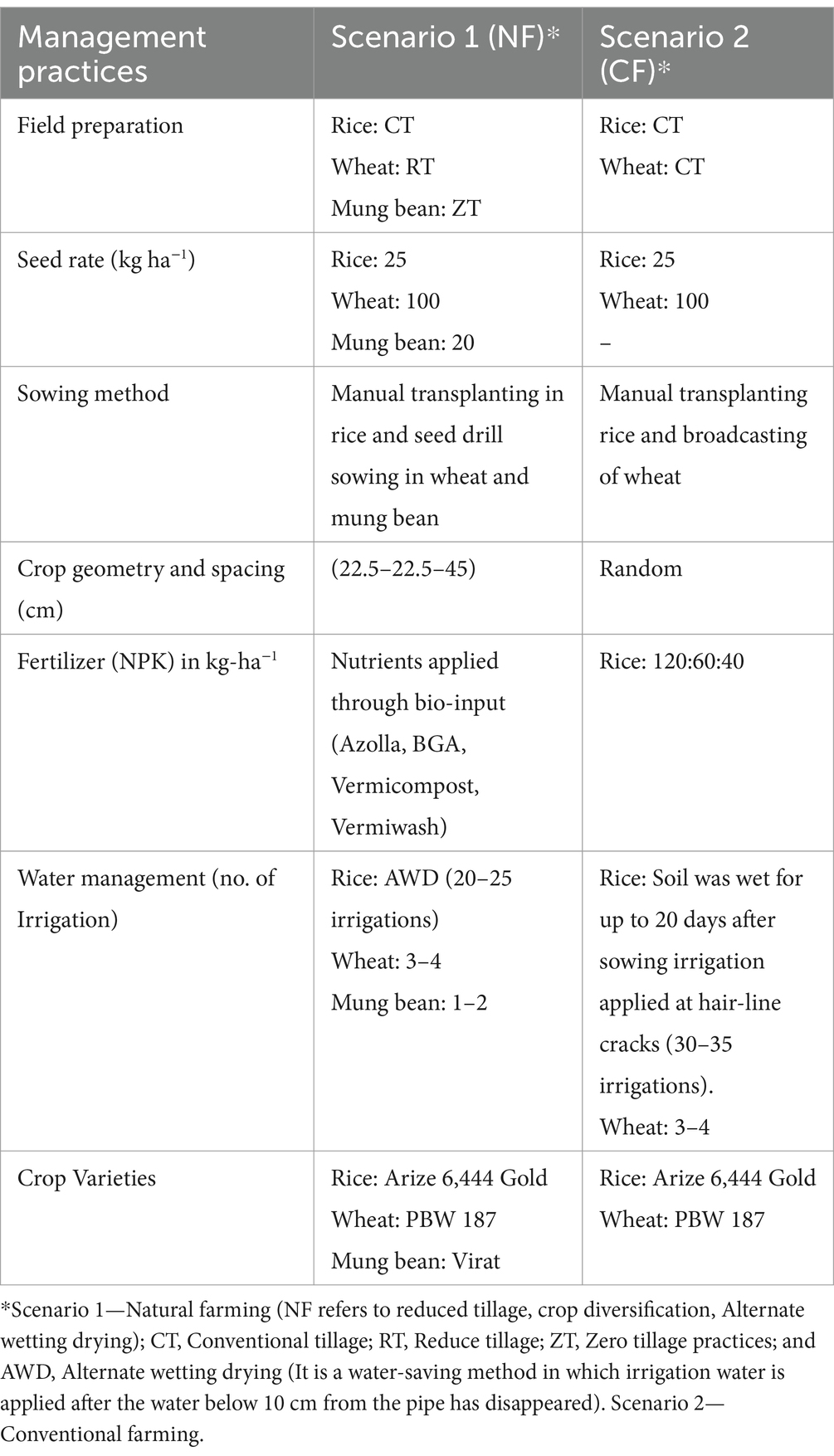
Table 1. Experimental design, and related agronomic management practices followed in organic and conventional farming systems.

Table 2. Crop establishment method, cropping pattern and residue management under two different scenarios.
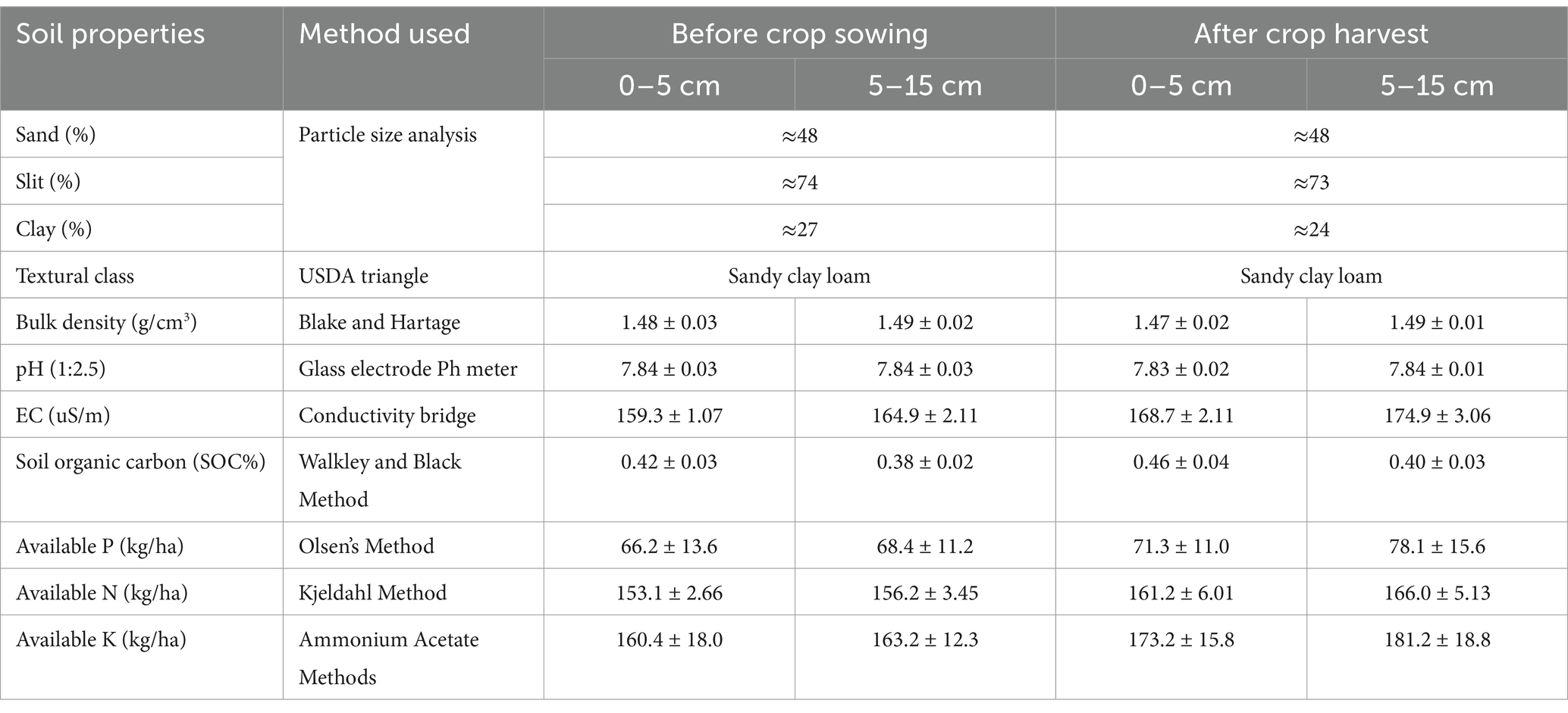
Table 3. Initial soil properties of the agriculture field before crop establishment.
The collected soil samples were homogenized and spread out in trays to remove any extraneous materials, such as small stems, root fragments, and leaves. Afterward, the samples were sieved and stored in plastic bags for further analysis. The soil collection process, and analysis methods were standardized to ensure consistency for both the treatments plots. Following sample collection, soil properties such as pH, organic matter content, available phosphorus (P), and available potassium (K) were analyzed (Li et al., 2023).
During sample collection, 3 samples/sites/replicates were collected and pooled from organically fields and same was done for conventional fields. Initial soil parameter of agricultural soil before sowing and harvest were done (Table 3).
Sampled soil was further assayed for microbial count using the standard serial dilution method (Saharan et al., 2023). A hundred milligrams of each soil sample were added to 900 mL of sterilized distilled water. After homogenization for 30 min, this solution was decimally diluted (10–1 to 10–9) and aliquots of the resulting solution, i.e., 10–9, were used for plating on appropriate culture media (nutrient agar) by pour plate method. After incubation at 30 ± 0.5°C, the colony-forming units (CFU) were counted.
DNA extraction, amplification and purification was conducted at Biokart India Pvt. Ltd. DNA was extracted from each treatments replicates using a commercial soil DNA extraction kit of Xploregen as per the manufacturer’s recommendations. To minimize DNA extraction bias, DNA samples from three replicates were pooled. Using Nano Drop and Gel electrophoresis, the DNA isolated from the samples was evaluated for quality. High-quality DNA was indicated by Nano Drop measurements (Absorbance at 260 nm/Absorbance at 280 nm) of about 1.8 to 2.0. The DNA samples were broken up into 600 bp length fragments using the KAPA fragmentation method (KAPA HyperPlus Kit including KAPA Frag Enzyme, KAPA Frag Buffer (10X) Conditioning Solution; a proprietary enzymatic systems) for library preparation by following the protocol mentioned in the Kit. Using a Hyper-Prep-plus ERAT (end-repair and A-tailing) enzyme combination, the fragmented materials were treated for end repair and A-tailing. For end-repair and A-tailing, 7 μL ERAT buffer and 3 μL ERAT enzyme mix was added to each sample (also previously fragmented gDNA samples) and incubated at 20°C for 30 min (end-repair) followed by 65°C for 30 min (A-tailing). Next, the adapters were introduced and ligated to the end repaired DNA segments using DNA ligase just after the end repair and A-tailing. Illumina primers were used to library amplify the adapter ligated samples. The V3V4 region of the 16S rRNA gene and the ITS region (In this study, we used the ITS region as a genetic marker for assessing fungal diversity in soil samples, following established methods for fungal community analysis) were amplified using universal primers (Caporaso et al., 2011; White et al., 1990). For bacterial diversity, the universal primers targeting the 16S rRNA gene (V3–V4 regions) were used: 341F (CCTACGGGNGGCWGCAG) and 805R (GACTACHVGGGTATCTAATCC). For fungal diversity, the Internal Transcribed Spacer (ITS) region was targeted using the universal primers: ITS1-F (CTTGGTCATTTAGAGGAAGTAA) and ITS2 (GCTGCGTTCTTCATCGATGC).
To perform the amplification, 40 ng of the isolated DNA and 10 pM of each primer were used. Denaturation was started at 98°C, followed by four cycles of amplification (denaturation at 98°C for 15 s, annealing at 60°C for 30 s, extension at 72°C for 30 s), and finally final extension at 72°C for 1 min. For the detailed protocol and steps, Saharan et al. (2023) can be followed.
Two types of amplicon sequencing were performed (a) 16S rRNA gene metagenomics sequencing and (b) ITS metagenomics sequencing to understand the diversity of bacterial and fungal community in both kind of samples (organic and chemical). The amplicons from each sample were purified with Ampure beads (Beckman Coulter, Brea, CA, United States) to remove unused primers. An additional 8 cycles of PCR was performed using Illumina barcoded adapters (using primers complementary to the Illumina flow cell adapters) to prepare the sequencing libraries. For this, PCR products were purified using Ampure XP beads. The beads were added to the PCR products at a 1.8:1 ratio, followed by incubation to bind the DNA. The beads were separated using a magnetic stand, and the supernatant was discarded. After two ethanol washes (70%) and brief air drying, the DNA was eluted with nuclease-free water or buffer and collected for further use as per the protocol of Beckman Coulter (Zhang et al., 2020). Libraries were purified using Ampure beads and quantitated using Qubit dsDNA High Sensitivity assay kit (Thermo Fisher Scientific Inc., Waltham, Massachusetts, United States). Sequencing was performed using Illumina Miseq with 2 × 300 PE v3-v4 and ITS sequencing kit. The PCR products were sequenced using Illumina MiSeq platform (Illumina, San Diego, CA, United States) by following the standard protocol,1 and the resulting raw sequence reads of Sample 1 and Sample 2 were quality checked using FastQC (Version 0.11.9) and MultiQC (Version 1.10.1) tools. Adaptors from the raw data were trimmed out with Trimgalore version 0.6.6 and further were processed using Biokart Metagenomics pipelines to get the final OTU (Operational Taxonomic Units) tables. Taxonomy was assigned using the NCBI and UNITE reference databases for 16S and ITS sequences with the confidence of 0.05, respectively (Nilsson et al., 2019; Schloss et al., 2009).
For Illumina sequencing, primers were designed with specific overhangs to allow for indexing and adapter ligation during the sequencing workflow. The primers were prepared by incorporating Illumina adapter sequences into the forward and reverse primers used for amplifying the target regions of bacterial 16S rRNA and fungal ITS genes.
The forward primer contains the i5 overhang sequence (5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG-3′) and is combined with the specific region targeting sequence for either the bacterial 16S or fungal ITS region. Similarly, the reverse primer includes the i7 overhang sequence (5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG-3′) followed by the target region-specific primer. These overhangs facilitate the addition of sequencing adapters during library preparation, which are essential for Illumina sequencing.
Rarefaction curves are plots of the number of individuals on the x-axis (sequence count) against the number of species on the y-axis (species richness). Sample sizes (N) may differ, but the relevant sections of the curves can still be visually compared. Alpha diversity metrics, such as richness and diversity indices (Chao1, Shannon, Simpson and Fisher), were calculated for each sample (Deluz et al., 2020). Past4.03 was used to generate the plots related to the alpha diversity indices and rarefaction curve with default parameters.
Alpha diversity indices were visualized using box plots to illustrate the distribution of diversity across both farming practices. Statistical significance was determined with p-values, considering p < 0.05 as significant. Heatmap was generated to analyse the pattern of higher and lower abundance spread between the samples using Euclidean distance method with top 50 organisms at genus level of taxonomy with ClusVis.2 Comparative analysis was done to find the unique and common taxon between the samples in terms of 16S and ITS metagenomics. The analysis entirely depends on the category of organisms at genus level. The comparative Venn diagram was built using the tool Venny 2.1.3
Soil sample were collected from organically and conventionally farmed fields and subjected to soil nutrients analysis before crop sowing and after crop harvesting. On comparing the soil data from both fields, there were no any significant changes observed in sand, slit, clay, texture class, bulk density and pH before sowing and after harvesting and along the soil depth profile. Whereas, EC was increased at initial and final sampling along the soil depth. SOC was found slightly increased at two time points. Similar trend as SOC was followed by N-P-K concentration along with the soil depth profile (Table 3). Soil properties take a significantly longer time to change their natural characteristics, that is why in our case no considerable changes in soils were noted.
A total of 200,171 bacterial sequences and 17,058 fungal sequences were obtained for each sample after quality filtering. The taxonomic profiling reveals a total of 724 OTUs bacteria and 230 OTUs of fungus were observed in amplicon based metagenomics study of soil sample from scenario 1 and 2. At the phylum level, both the organic sample (Sc 1) and the conventional sample (Sc 2) had a higher relative abundance of Firmicutes (49.66; 51.12%), Actinobacteria (21.79; 1,611%) and Proteobacteria (20.64; 25.41%) bacteria (Figure 1) whereas in fungus category Ascomycota (85.15; 78.91%) and Basidiomycota (14.85; 15.42%) showed the highest relative abundance. If analysing the data at the genus level, both the organic sample (Sc 2) and the conventional sample (Sc 1) had a higher relative abundance of Lactobacillus (24.17; 20.45%) and Pseudomonas (14.72; 11.31%) (Figure 2). The ITS analysis showed that both the organic sample (Sc 2) and the conventional sample (Sc 2) had a higher relative abundance of Ascomycota and Basidiomycota (Figure 1). At the genus level, both the organic sample and the conventional sample had a higher relative abundance of Unclassified Trichocomaceae and Aspergillus (Figure 2).
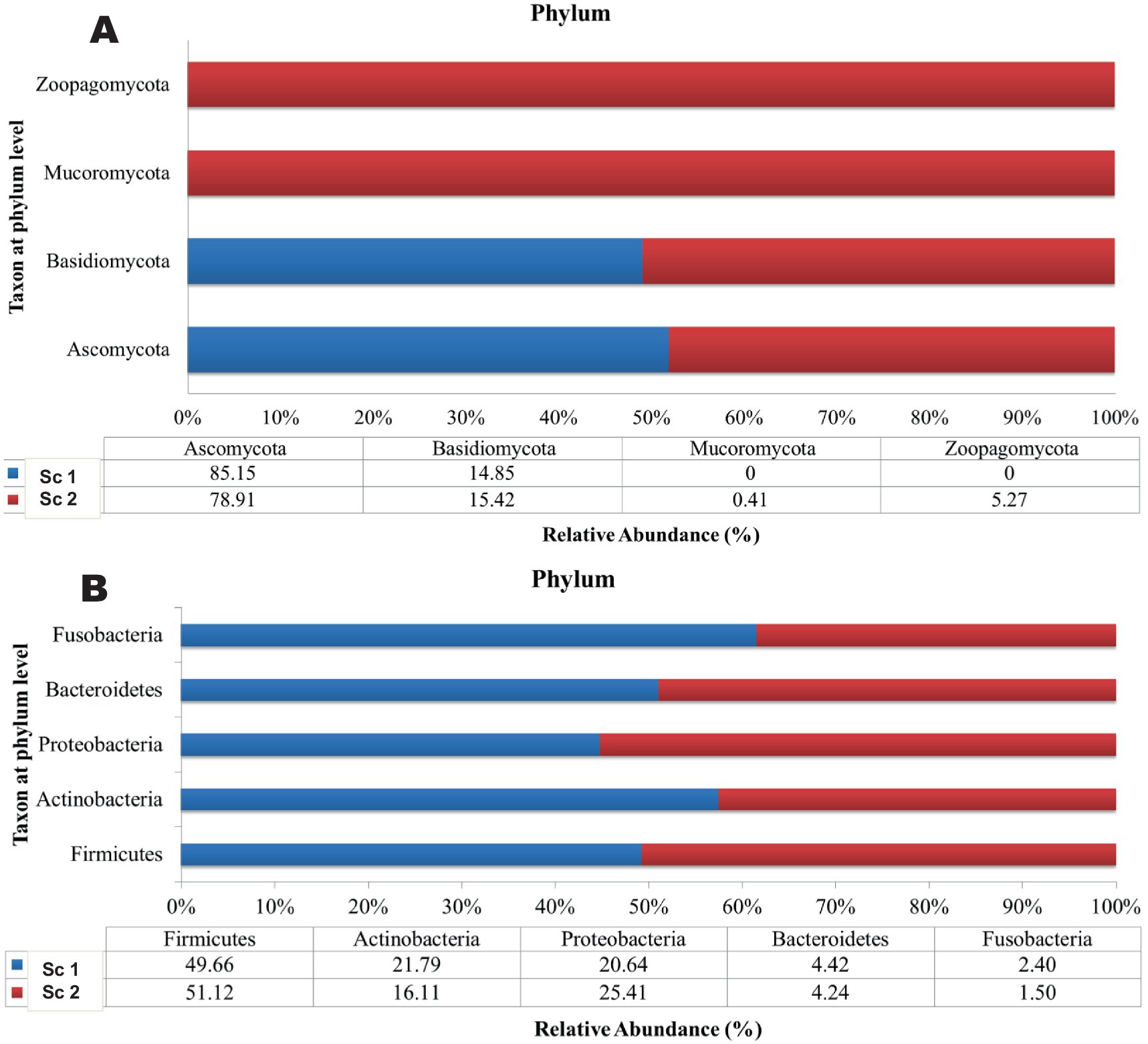
Figure 1. The plots reveals that the most abundant phylum of bacteria (A) is Firmicutes [Sc 1(49.7%) and Sc 2 (51.12%)]. The most abundant phylum of fungi (B) is Ascomycota [Sc 1(85.15%) and Sc 2 (78.9%)].
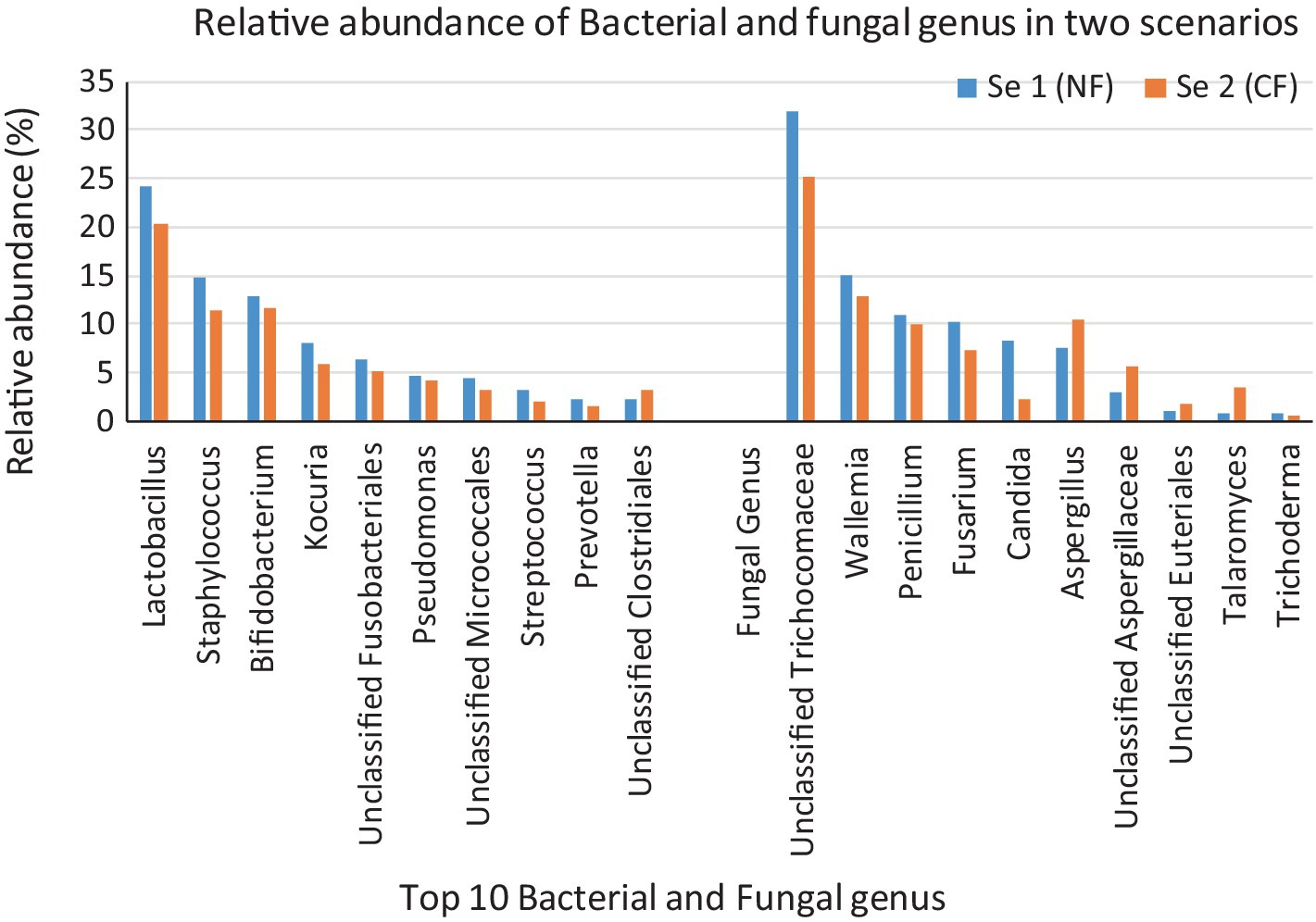
Figure 2. The above 100% stacked bar plots gives us the idea of the top 10 most abundant bacteria and fungus present in the Sc 1 and Sc 2. 16S study revealed that Lactobacillus is the most abundant bacteria observed in Sc 1(25.17%) and Sc 2 (20.45%). ITS study says that most abundant fungus observed in Sc 1 (31.92%) and Sc 2 (25.17%) is Unclassified Trichocomaceae.
The number of individuals on the x-axis (sequence count) was plotted against the number of species on the y-axis (species richness) to visualize the rarefaction curve of the organic and conventional samples. Sample sizes (N) may differ, but the relevant sections of the curves can still be visually compared. In terms of 16S study the Sc 1 has higher sequence count and higher species richness compared to Sc 2 in terms of 16S metagenomics study whereas in ITS study the Sc 1 has higher species richness and lower sequence count while Sc 2 has higher sequence count and lower species richness (Figure 3).
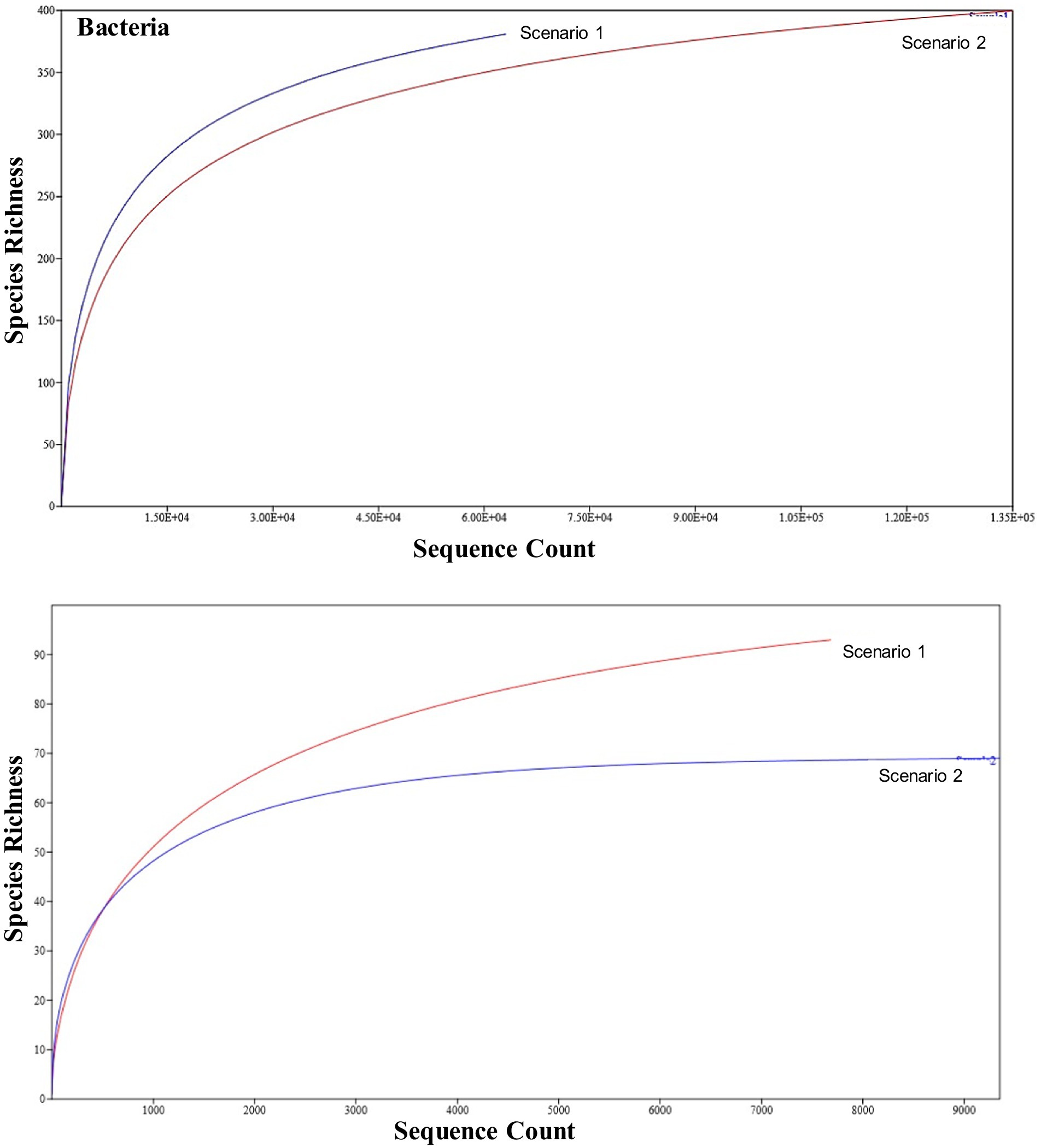
Figure 3. Representation of rarefaction curve for the NF (Sc 1) and conventional sample (Sc 2) on the basis of bacterial and fungal results using Past4.03.
The analysis revealed that the conventional sample (Sc 2) had higher microbial diversity and richness than the organic sample (Sc 1) (Table 4), as indicated by the Simpson, Shannon, Fisher and Chao1 indices (Chao and Lee, 1992; Fierer et al., 2005). Alpha diversity is used to measure the diversity present within a sample or community. Alpha diversity can be characterized via the total number of species (richness), the abundances of the species (evenness) or measures that considered both richness and evenness. How these measures estimate the diversity is need to be considered when performing alpha-diversity analysis. Chao1 estimate the richness by inferring out the number of rare organisms that may have lost due to under sampling. The indices such as Shannon, Simpson and Fisher infer the number (richness) along with the abundance of organisms (evenness) and measured to describe the actual diversity of a community (Pielou, 1966).
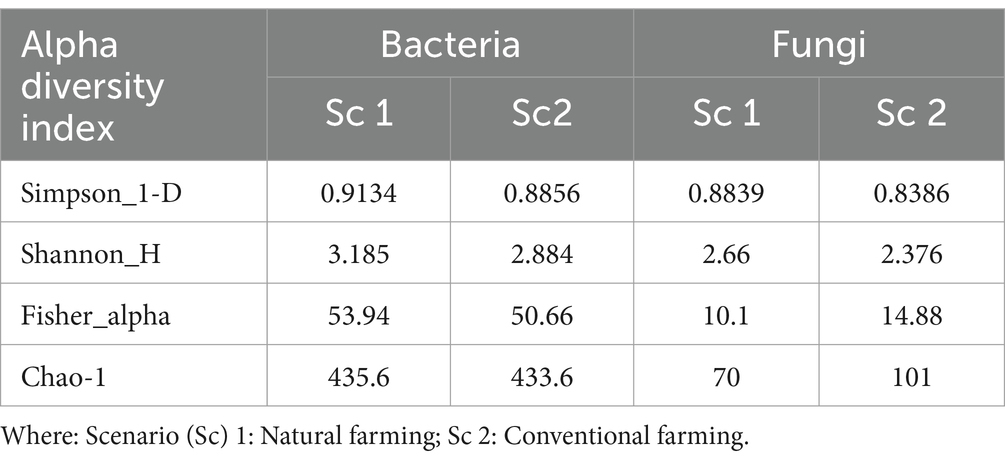
Table 4. Representing the alpha diversity measure of organic and conventional soil samples for both bacterial and fungal communities.
In terms of 16S metagenomics, even though the Sc 1 has higher sequence count and high species richness, but from the alpha diversity we can observe that Sc 2 has higher diversity. While in terms of ITS metagenomics Sc 1 has higher diversity compared to Sc 2 and, Sc 2 has higher abundance (Figures 4–6).
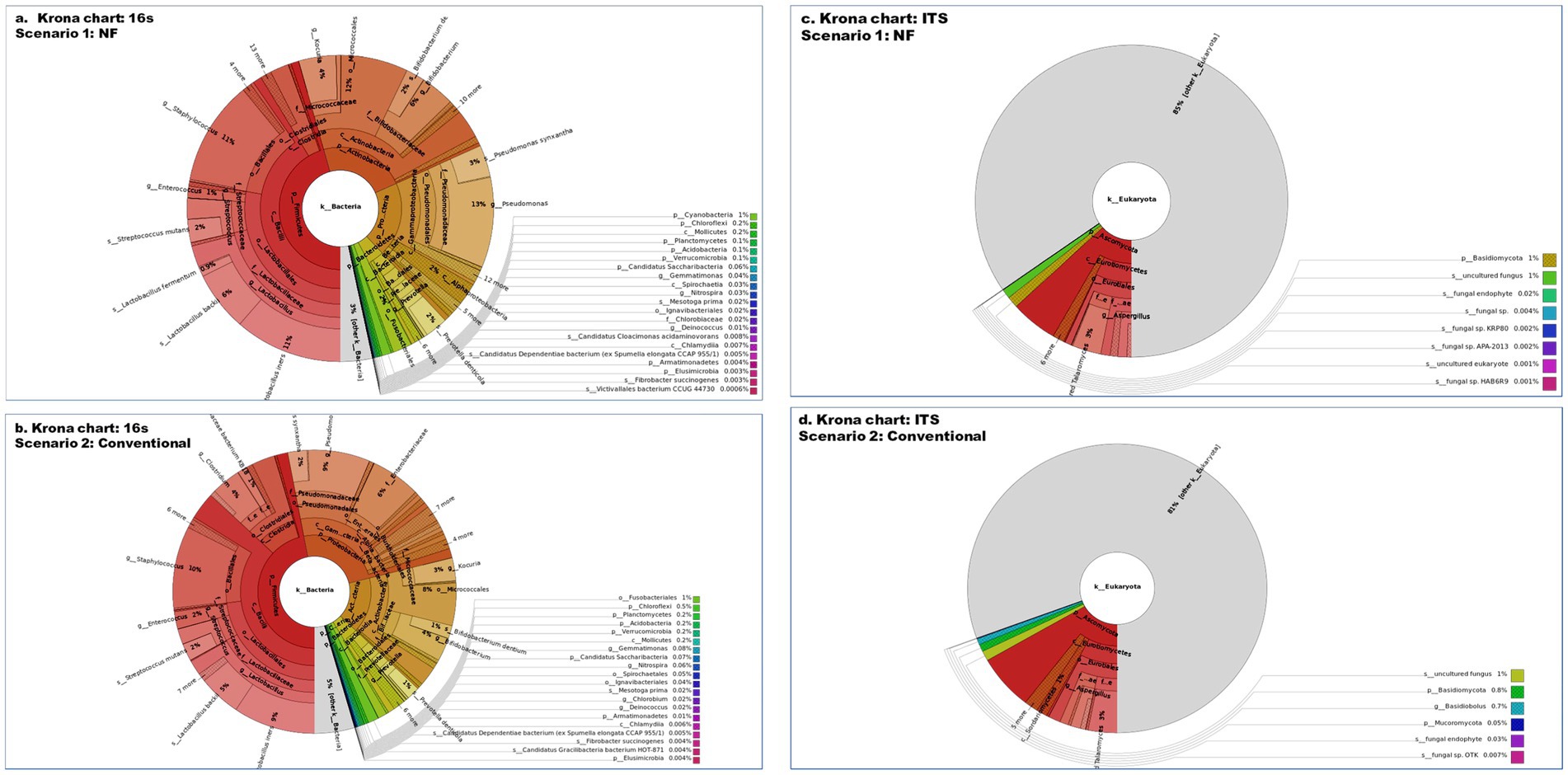
Figure 4. The phylogenetic tree diagram of soil bacterial (A,B) and fungal (C,D) communities in organic farming (NF, Sc 1) and conventional farming (CF, Sc 2) as identified in 16S and ITS. The phylogenetic tree shows the overall sample, from the phylum to the genus (from the inner to the outer ring arranged in sequence). The size of the point represents the average relative abundance of the taxon, and the relative abundance of the first 20 taxa is also identified with letters.
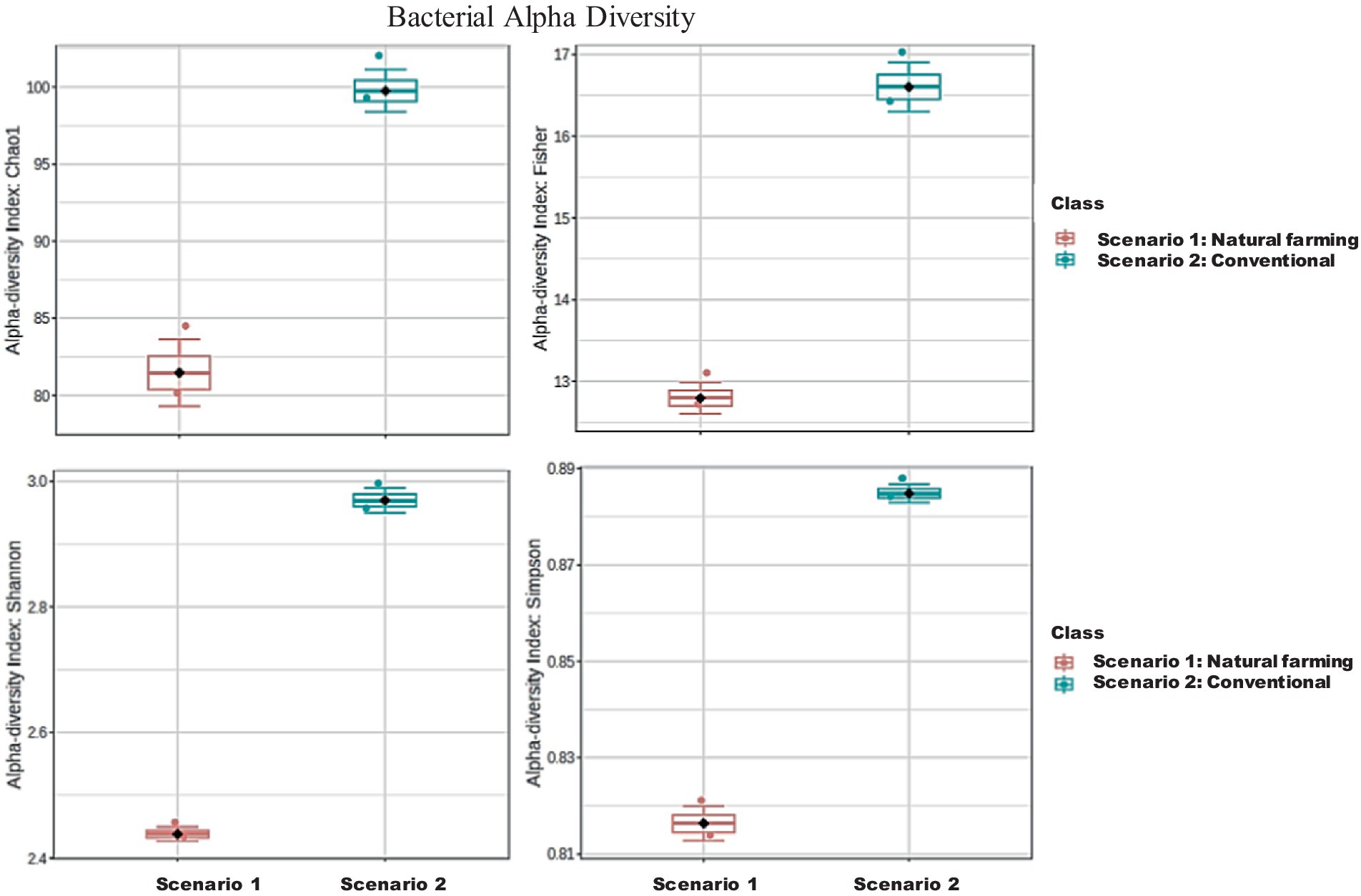
Figure 5. Alpha diversity of bacterial community in between NF (Sc 1) and conventional (Sc 2) soil samples.
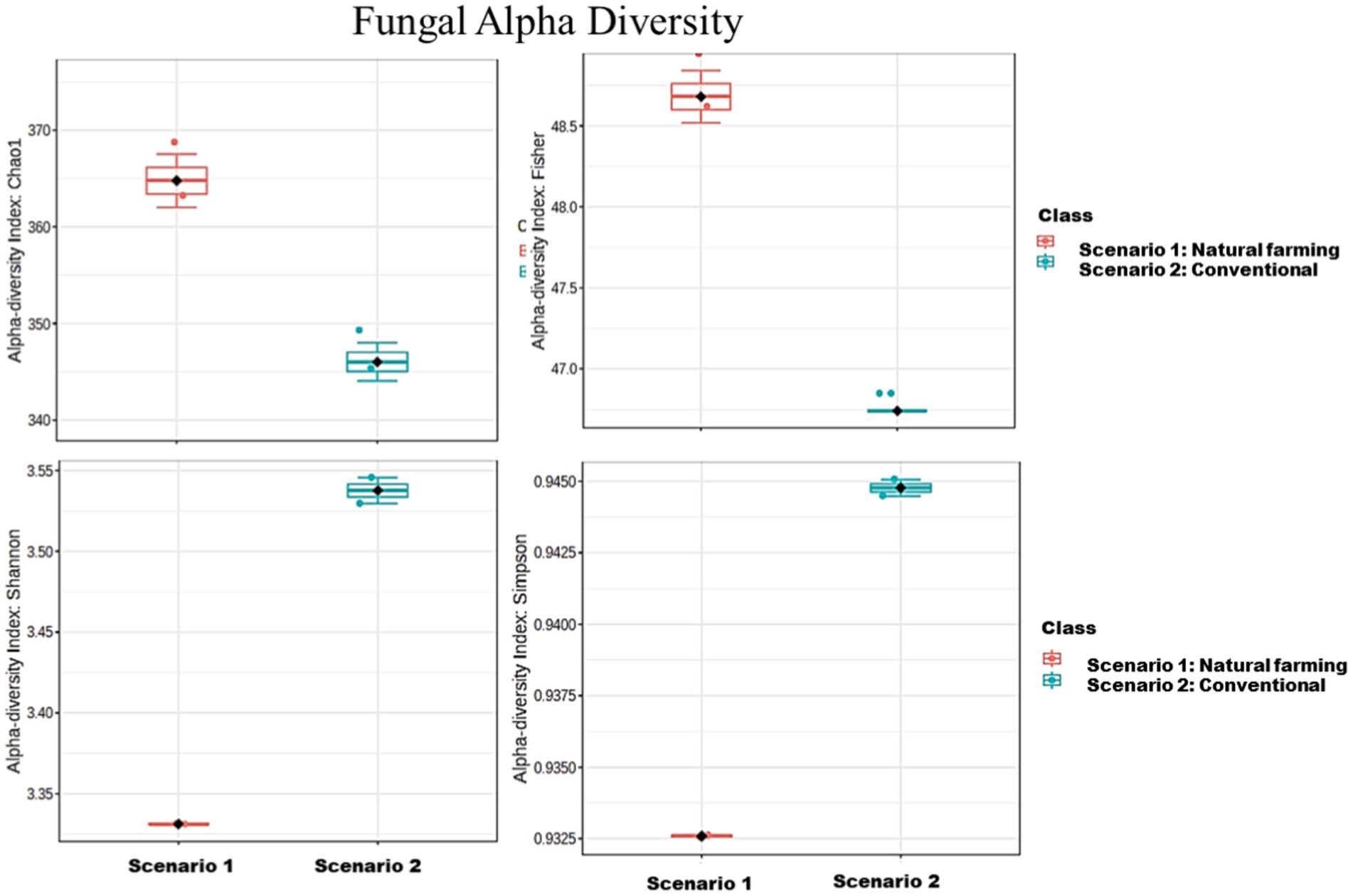
Figure 6. Comparison of Alpha diversity of fungal community between natural farming (NF) (Sc 1) and conventional (Sc 2) soil samples.
Top 50 soil micro-organisms were clustered using correlation distance and average linkage with unit variance scaling applied on to the sequence abundance to construct the heatmaps. The heat map infers,
1. Differences in bacterial and fungal community composition: The heatmap reveals the differences in the abundance of different taxa between the organic and conventional soil samples. Among the top 50 organisms there are 44 taxa of bacteria which are more abundant in organic soil sample and there are 34 taxa of fungi which are more abundant in organic sample compare to the conventional soil sample (Sc 2), this could indicate that the farming practices used in conventional agriculture are affecting the composition of the soil microbiome.
2. Relationships between different taxa: This heatmap also reveals the patterns of co-occurrence or exclusion between different bacterial and fungal taxa. There are 44 bacterial taxa and 34 fungal taxa that are consistently found to be abundant in Sc 01, this could suggest that they have a mutualistic relationship and the same applies for Sc 02
Samples were then compared on the basis of higher abundance. Taxon with 0 abundance are eliminated to find which among the organism are unique to each sample of study and observation (see Figure 7).
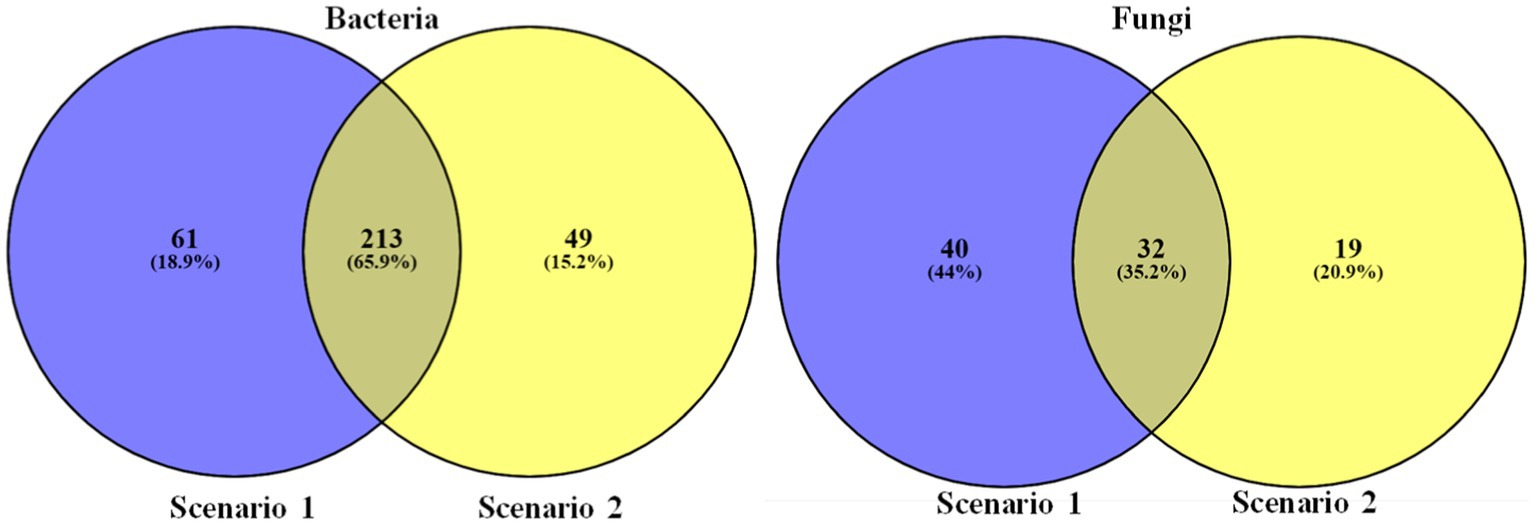
Figure 7. Venn diagrams representing the comparison of unique and common taxa between the two farming scenarios.
On comparing the two contarsting crop productions systems and practices, it was observed that chemical input based agriculture practises in the CF system showed more yield in comparison to the nature based inputs and management practices in NF scenarios. Our data observation and analysis for the year 2020–21 and 2021–22 shows that organic amendments increased the crop yields for the 2021–22 as compared to 2020–21, specially for rice crop approx (Table 5). 17% increase was noted. Wheat (3.12 to 3.45 t/ha) and Mungbean (0.30 to 1.71 t/ha) were also observed with an increase in crop yield though increase in yield of wheat was not significant. Rice equivalent was also increased in NF from 2020–21 to 2021–22 (7.33 to 8.64 t/ha). Whereas, there was a significant difference in rice equivalent system yield under the CF and NF for both the experimental years. It is a well-known fact that crop is drop in crop yield under the organic practices but here as mentioned in the treatment table, multiple approaches were combined (Tables 1, 2) and tested that gave enhanced yield in next year. This may be because of the established soil sustainability and health under the NF practices. Modern agricultural is going through lots of challenges and transforming our food system for the sustainable production is crucial. Also, the food production systems should produce the appropriate quality and quantity without challenging the environmental resources. Additionally, focus of sustainable agriculture practices is to have low Global Warming Potential (GWP) and GHG emission factors (Sun and Badgley, 2019). In this regard, many organic/natural and circular bio-economy based agriculture are proved to be climate friendly. A reduced yield is compensated by the low input and management cost. To this, an increased crop yield can be recorded after 3–4 years of natural and organic practices.
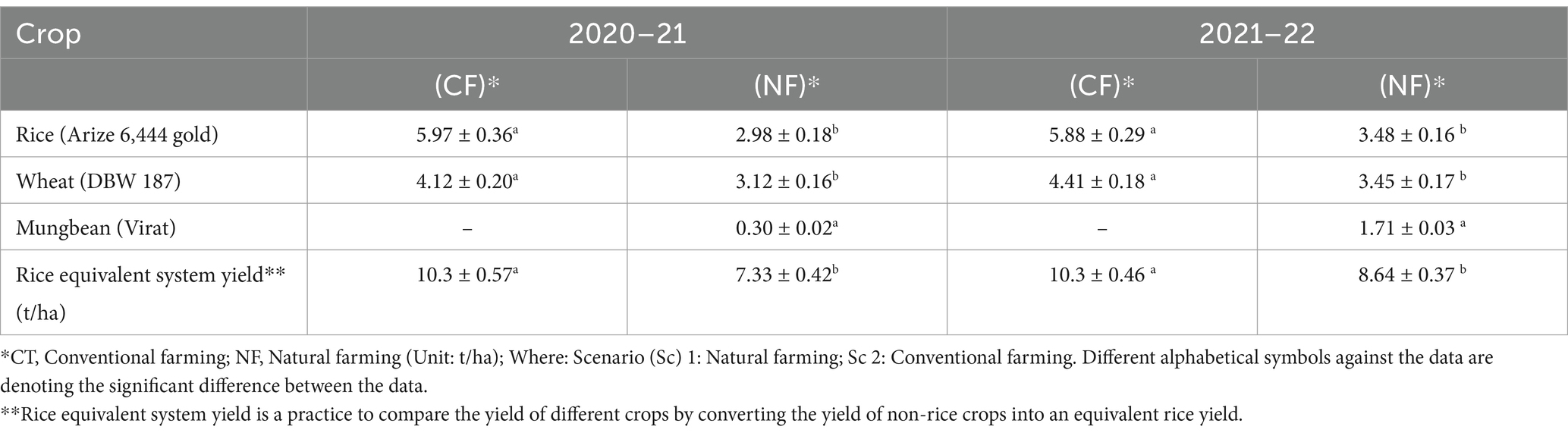
Table 5. Crop yield under different scenarios in the year 2020–22 (Unit: t/ha).
Soil properties related to the microbial action has also been reported to improve under the Sc 1 as compared to the Sc 2 (Table 6). Soil microbial biomass carbon and nitrogen (SMBC and SMBN) along with dehydrogenase, alkaline phosphatase, urease and TGRSP were reported in increased amount (73.51; 7,431; 36.81; 13.43 ± 4.54; 185.55 ± 1.5; and 2.23 ± 0.16, respectively) in the naturally managed soil as compared to the conventional soil (40.85 ± 5.14; 34.18 ± 16.11; 25.85 ± 0.88; 7.96 ± 0.62; 180.55 ± 2.58 and 1.78 ± 0.09, respectively). Soil enzymes are the indicators of the soil sustainability, fertility and health and organically soil shows higher activities of the microbial community under the degradable materials. While chemically hazardous inputs and intensively managed farming systems can greatly reduce the microbial population and biomass, application of organic amendments (crop-residue) and manure is a potential strategy to strengthen the microbial biomass and improve soil health and resilience (see Table 7).
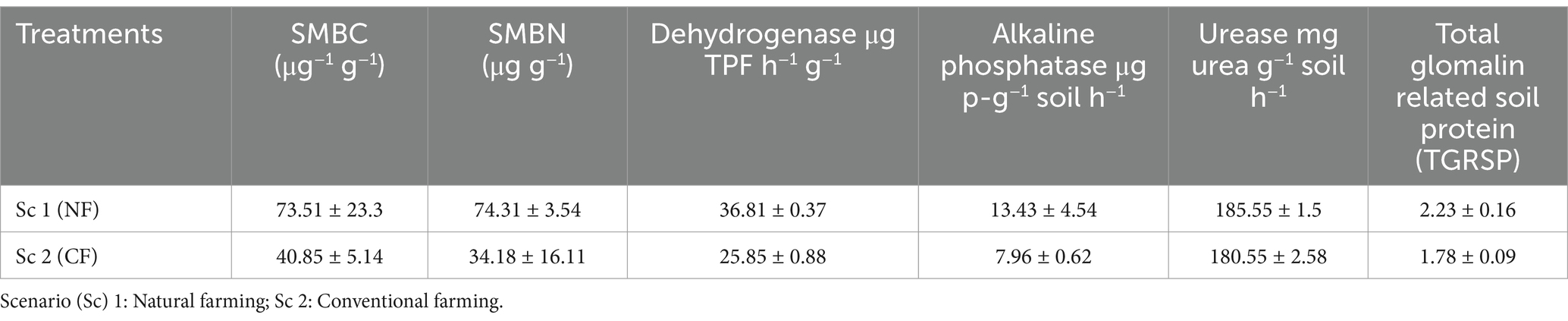
Table 6. Effect of cropping system and different agriculture practices on soil microbial properties after 1 year of experiment.
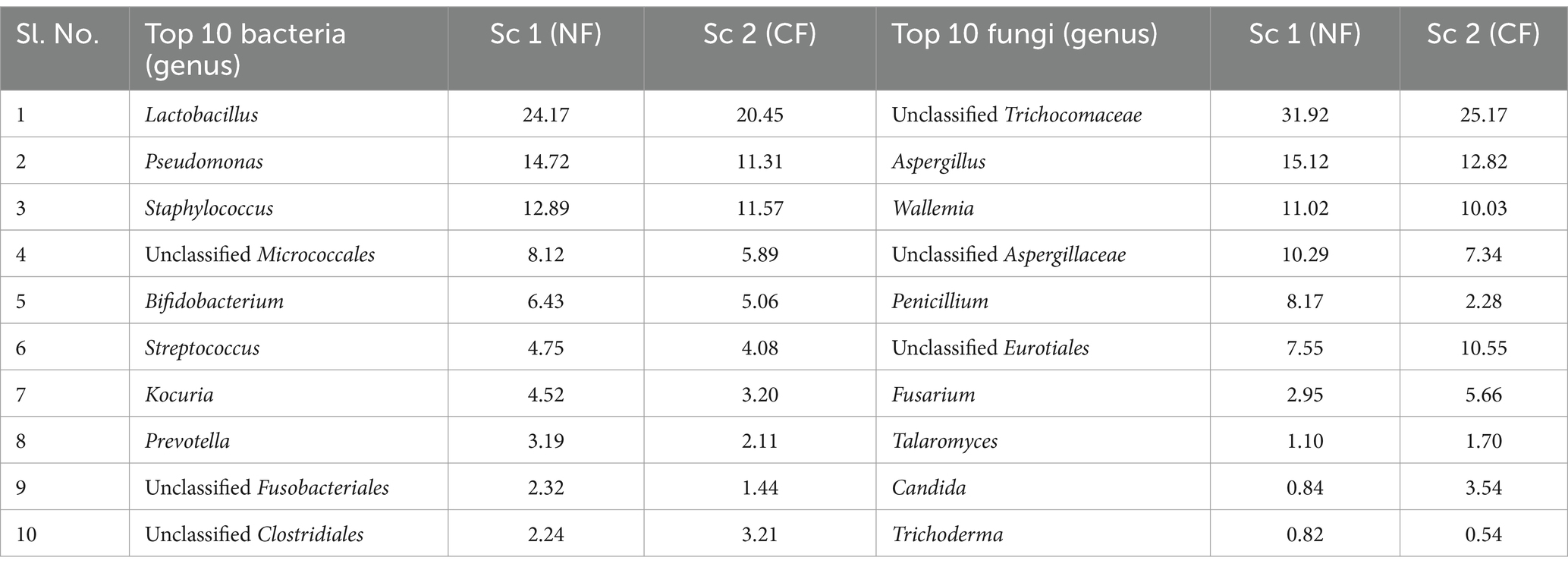
Table 7. List of top 10 genus of bacteria and fungi with their relative abundance (%).
Various activities, such as agricultural practices, significantly alter soil microbial function and communities, causing heavy use of toxic chemicals in crop fields and environmental pollution (Teng and Chen, 2019). The difference in the taxonomic profiles between the organic and conventional soil samples, as determined by 16S and ITS metagenomics, may be attributed to differences in the management practices between the two soil types. Firmicutes and Actinobacteria are commonly associated with soils that are high in organic matter, and the increased levels of these phyla in the organic soil sample may be indicative of the presence of more decomposing organic material, which could contribute to soil fertility (Fierer and Jackson, 2006). On the other hand, Ascomycota and Basidiomycota are fungal phyla commonly found in soils and are involved in the decomposition of organic matter (Xu et al., 2022). These phyla may also contribute to the nutrient cycling processes in the soil, and their increased abundance in the ITS metagenomics data from the organic soil sample may indicate a higher level of organic matter decomposition in this soil type (Rousk et al., 2010).
The conventional soil sample, on the other hand, may have lower levels of organic matter and thus lower levels of Firmicutes, Actinobacteria, Ascomycota, and Basidiomycota (Xu et al., 2022). It is also possible that the conventional soil sample has been subjected to higher levels of disturbance or chemical inputs, which may have impacted the microbial community composition and diversity (Zhou et al., 2012). A larger number of microorganisms in organically treated soils (fungi and bacteria) and associated biomass carbon have been reported (Saharan et al., 2023). Meanwhile, soil data from chemical fertilizers have shown more effects on microbial composition and diversity.
In a contrasting farming practice experimental study, Enebe and Babalola (2020) reported the response of various maize-related microflora (bacterial, fungi and archaeal communities) to compost and inorganic fertilization as we analyzed. The study also reports that both fertilizer practices influenced the maize rhizosphere microbial community, but the organic amendments provided the most stable microbial community. Similar results were documented by Zhan et al. (2018), stating that a higher level of NPK input in the soil negatively affects the microbial community’s abundance and structure. Moisture and mineral content also play a significant role in decomposing bacteria diversity.
Overall, the differences in microbial community composition between the organic and conventional soil samples suggest that management practices can significantly impact soil microbiota. The higher abundance of Firmicutes and Actinobacteria in the organic soil sample and the higher abundance of Ascomycota and Basidiomycota in the ITS metagenomics data may be indicative of a more diverse and active microbial community in the organic soil, which could contribute to improved soil health and fertility. Other studies have also reported a taxonomically diverse group of decomposer soil microbes that are related to the Firmicutes, Actinobacteria, Proteobacteria and Bacteroidetes phyla (Zhan et al., 2018; Bernard et al., 2007; España et al., 2011; Semenov et al., 2012). Growth of Proteobacteria and Acidobacteriota was associated with the amount of/and increasing soil organic carbon content and limitation of nutrients in the system. Plant species-dependent effects were also noted to impact the soil fungal community significantly (Hannula et al., 2019; He et al., 2022). On the other hand, soil bacteria are found to show higher species richness than fungal communities in the same soil-quality habitat. It can be concluded that the bacteria are better adapted to non-extreme soil disturbances due to greater diversity (He et al., 2017).
In addition to fungi like Aspergillus, Penicillium, and Fusarium, the study by Saharan et al. (2023) also highlights beneficial bacteria such as Bacillus and Pseudomonas as critical contributors to soil health in zero-budget natural farming. This microbial diversity enhances nutrient cycling and plant resilience to stress. Previous studies by Singh et al. (2011) and Edwards et al. (2015) also support the beneficial roles of these bacteria in crop health and soil fertility, as they are also reported in our study (Figure 8).
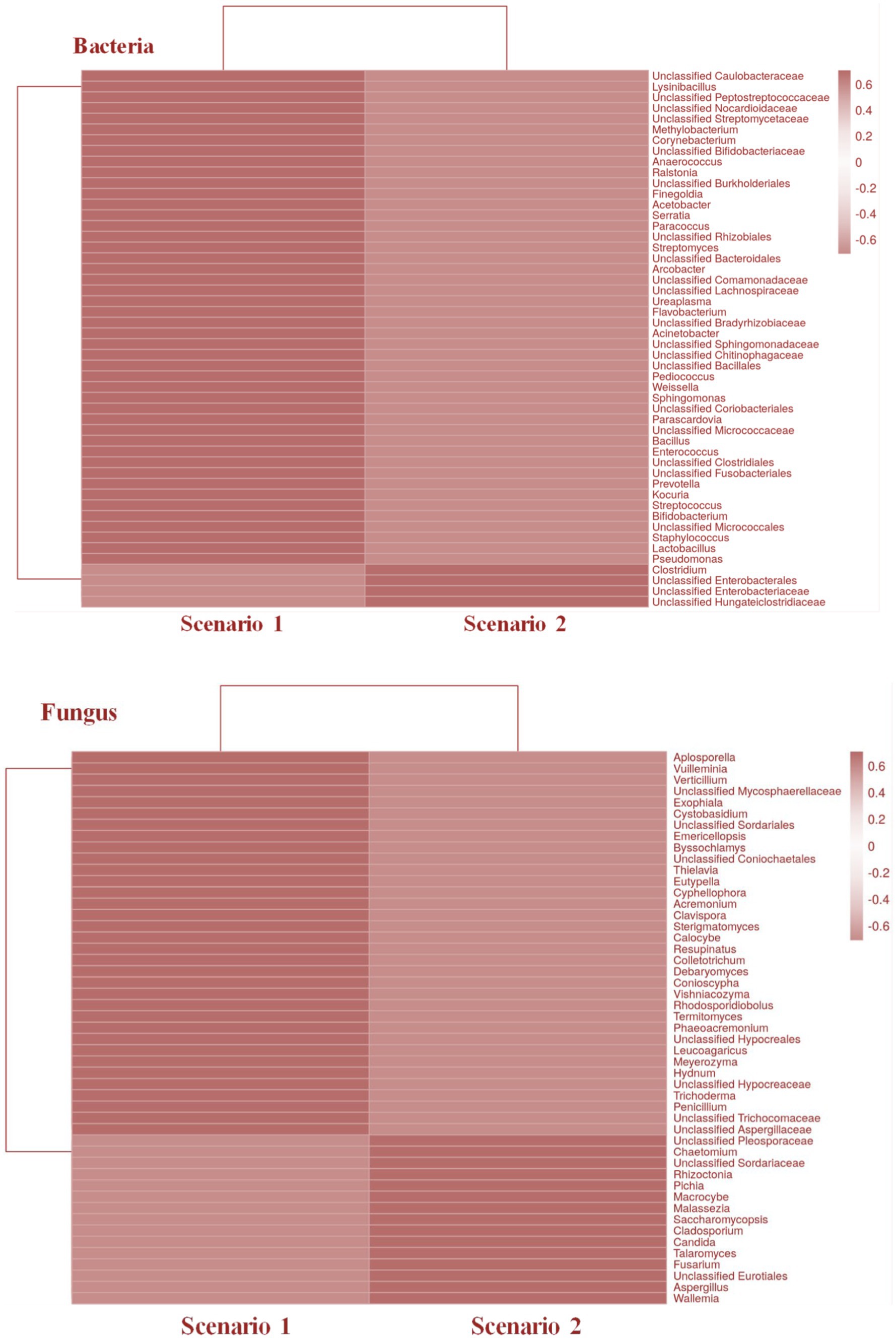
Figure 8. Heatmaps representing the taxonomical relationship between the two farming scenarios in both bacterial and fungal community.
Additionally, crop yield has been found to reduce, but on comparing the economics, natural practices are cost-effective. The findings demonstrate that soil microbial activity, represented by SMBC, SMBN, and enzyme activities (e.g., dehydrogenase, alkaline phosphatase, and urease), was significantly higher in naturally managed soils (Scenario 1) compared to conventional systems (Scenario 2). This suggests enhanced soil health, fertility, and sustainability under organic and water-saving (AWD) management practices (Majumdar et al., 2021, 2024; Das et al., 2024; Gunapala and Scow, 1998). Organic amendments like crop residues and manure promote microbial biomass, while intensive chemical inputs reduce microbial diversity and soil quality (Mandal et al., 2023).
Many people agree that organic farming is a successful approach to sustainably managing agriculture. Organic farming is typically thought to enhance crop/grain and soil quality. However, it is observed that farmers (special small land holding farmers) are hesitated to promote organic farming at large-scale because of the lower crop yields compared to conventional farming (based on the personal observations and interaction with the farmers). Though, yield cannot be the only deciding parameter to promote any practice, overall economic analysis and environmental sustainability along with farmers’ benefit should also be taken care in promoting them at larger level. The global adoption of environmentally friendly techniques will help preserve ecosystems and reduce the negative impacts of agrochemicals. However, further research must provide substantial evidence supporting these natural approaches for broader acceptance and implementation.
The qualitative and quantitative nature of the metagenomic DNA representing different environmental samples decides the success of metagenomic approaches. From the study, it can be concluded that use of different fertilizer use strategies in soil management systems has a significant role on the diversity and distribution of microbial communities. It can be a potential application-based practice in agriculture for soil health management under climate change impact management. The analysis of both 16S and internal transcribed spacer (ITS) amplicon sequencing data revealed distinct patterns of microbial community composition between the organic and chemical-based fertilizer treatments, with a greater diversity of bacteria and fungi observed in the soil treated with organic fertilizer. Results of the study provides valuable insight into the organically amended soil, and impact of different chemical based fertilization strategies on soil microbial communities and underscores the importance of sustainable soil management practices that prioritize organic fertilization. Further, more studies are needed to explore and support the functional implications of microbial communities in soil ecosystems and their potential for influencing crop productivity, nutrient cycling and fighting the climate change impacts.
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The manuscript presents research on animals that do not require ethical approval for their study.
AM: Project administration, Supervision, Validation, Writing – review & editing, Conceptualization, Data curation, Investigation. PY: Data curation, Validation, Writing – original draft, Writing – review & editing. SS: Validation, Writing – review & editing, Project administration, Supervision. PM: Formal analysis, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors are thankful to the International Rice Research Institute (IRRI) and also thanks to ISARC, Varanasi, UP, India for providing the research and development facility and support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Bardgett, R. D., and van der Putten, W. H. (2014). Belowground biodiversity and ecosystem functioning. Nature 515, 505–511. doi: 10.1038/nature13855
Bednář, M., and Šarapatka, B. (2018). Relationships between physical–geographical factors and soil degradation on agricultural land. Environ. Res. 164, 660–668. doi: 10.1016/j.envres.2018.03.042
Bernard, L., Mougel, C., Maron, P. A., Nowak, V., Lévêque, J., Henault, C., et al. (2007). Dynamics and identification of soil microbial populations actively assimilating carbon from 13C-labelled wheat residue as estimated by DNA-and RNA-SIP techniques. Environ. Microbiol. 9, 752–764. doi: 10.1111/j.1462-2920.2006.01197.x
Bulgarelli, D., Rott, M., Schlaeppi, K., Loren, V., van Themaat, E., Ahmadinejad, N., et al. (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 488, 91–95. doi: 10.1038/nature11336
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-Lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl Acad. Sci. USA 108, 4516–4522. doi: 10.1073/pnas.1000080107
Chao, A., and Lee, S. M. (1992). Estimating the number of classes via sample coverage. J. Am. Stat. Assoc. 87, 210–217. doi: 10.1080/01621459.1992.10475194
Compant, S., Van Der Heijden, M. G., and Sessitsch, A. (2010). Climate change effects on beneficial plant–microorganism interactions. FEMS Microbiol. Ecol. 73, 197–214. doi: 10.1111/j.1574-6941.2010.00900.x
Das, S., Biswas, S., Ramakrishnan, B., Das, T. K., Purakayastha, T. J., Gawade, B. H., et al. (2024). Biological soil health with conventional and qPCR based indicators under conservation agriculture based rice-wheat cropping system in Indo-Gangetic Plain. Appl. Soil Ecol. 193:105128. doi: 10.1016/j.apsoil.2023.105128
Deluz, C., Nussbaum, M., Sauzet, O., Gondret, K., and Boivin, P. (2020). Evaluation of the potential for soil organic carbon content monitoring with farmers. Front. Environ. Sci. 8:113. doi: 10.3389/fenvs.2020.00113
Doran, J. W., and Zeiss, M. R. (2000). Soil health and sustainability: managing the biotic component of soil quality. Appl. Soil Ecol. 15, 3–11. doi: 10.1016/S0929-1393(00)00067-6
Edwards, J., Johnson, C., Santos-Medellín, C., Lurie, E., Podishetty, N. K., Bhatnagar, S., et al. (2015). Structure, variation, and assembly of the root-associated microbiomes of rice. Proc. Natl. Acad. Sci. 112, E911–E920. doi: 10.1073/pnas.1414592112
Enebe, M. C., and Babalola, O. O. (2020). Effects of inorganic and organic treatments on the microbial community of maize rhizosphere by a shotgun metagenomics approach. Ann. Microbiol. 70:49. doi: 10.1186/s13213-020-01591-8
España, M., Rasche, F., Kandeler, E., Brune, T., Rodriguez, B., Bending, G. D., et al. (2011). Identification of active bacteria involved in decomposition of complex maize and soybean residues in a tropical Vertisol using 15N-DNA stable isotope probing. Pedobiologia 54, 187–193. doi: 10.1016/j.pedobi.2011.03.001
Fierer, N., and Jackson, R. B. (2006). The diversity and biogeography of soil bacterial communities. Proc. Natl Acad. Sci. USA 103, 626–631. doi: 10.1073/pnas.0507535103
Fierer, N., Jackson, J. A., Vilgalys, R., and Jackson, R. B. (2005). Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl. Environ. Microbiol. 71, 4117–4120. doi: 10.1128/AEM.71.7.4117-4120.2005
Gunapala, N., and Scow, K. M. (1998). Dynamics of soil microbial biomass and activity in conventional and organic farming systems. Soil Biol. Biochem. 30, 805–816. doi: 10.1016/S0038-0717(97)00162-4
Hannula, S. E., Kielak, A. M., Steinauer, K., Huberty, M., Jongen, R., De Long, J. R., et al. (2019). Time after time: Temporal variation in the effects of grass and forb species on soil bacterial and fungal communities. MBio 10, e02635–e02619. doi: 10.1128/mBio.02635-19
Hartman, W. H., Richardson, C. J., Vilgalys, R., and Bruland, G. L. (2008). Environmental and anthropogenic controls over bacterial communities in wetland soils. Proc. Natl Acad. Sci. USA 105, 17842–17847. doi: 10.1073/pnas.0808254105
Hartmann, M., and Six, J. (2023). Soil structure and microbiome functions in agroecosystems. Nat. Earth Environ. 4, 4–18. doi: 10.1038/s43017-022-00366-w
He, G., Peng, T., Guo, Y., Wen, S., Ji, L., and Luo, Z. (2022). Forest succession improves the complexity of soil microbial interaction and ecological stochasticity of community assembly: Evidence from Phoebe bournei-dominated forests in subtropical regions. Front. Microbiol. 13:1021258. doi: 10.3389/fmicb.2022.1021258
He, D., Shen, W., Eberwein, J., Zhao, Q., Ren, L., and Wu, Q. L. (2017). Diversity and co-occurrence network of soil fungi are more responsive than those of bacteria to shifts in precipitation seasonality in a subtropical forest. Soil Biol. Biochem. 115, 499–510. doi: 10.1016/j.soilbio.2017.09.023
Hussain, S., Hussain, S., Guo, R., Sarwar, M., Ren, X., Krstic, D., et al. (2021). Carbon sequestration to avoid soil degradation: A review on the role of conservation tillage. Plan. Theory 10:2001. doi: 10.3390/plants10102001
Krause, H. M., Ono-Raphel, J. G., Karanja, E., Matheri, F., Lori, M., Cifuentes, Y., et al. (2023). Organic and conventional farming systems shape soil bacterial community composition in tropical arable farming. Appl. Soil Ecol. 191:105054. doi: 10.1016/j.apsoil.2023.105054
Krause, H. M., Stehle, B., Mayer, J., Mayer, M., Steffens, P., Mäder, P., et al. (2022). Biological soil quality and soil organic carbon change in biodynamic, organic, and conventional farming systems after 42 years. Agron. Sustain. Dev. 42:117. doi: 10.1007/s13593-022-00843-y
Li, Y., Wang, C., Chang, H., Zhang, Y., Liu, S., and He, W. (2023). Metagenomics reveals the effect of long-term fertilization on carbon cycle in the maize rhizosphere. Front. Microbiol. 14:1170214. doi: 10.3389/fmicb.2023.1170214
Liang, Y., Li, G., van der Werf, W., Zhang, R., Li, H., Li, X., et al. (2019). Long-term fertilization practices alter aluminum fractions and bacterial diversity of a moderately acid soil. Soil Biol. Biochem. 129, 136–143. doi: 10.1016/j.soilbio.2018.11.016
Loreau, M., Naeem, S., and Inchausti, P. (Eds.) (2002). Biodiversity and ecosystem functioning: synthesis and perspectives. Oxford: OUP.
Lupatini, M., Korthals, G. W., de Hollander, M., Janssens, S. B., Kuramae, E. E., and de Boer, W. (2013a). Soil microbiome is more heterogeneous in organic than in conventional farming systems. Front. Microbiol. 4:12. doi: 10.3389/fmicb.2016.02064
Lupatini, M., Suleiman, A. K. A., Jacques, R. J. S., Antoniolli, Z. I., Kuramae, E. E., de Oliveira Camargo, F. A., et al. (2013b). Soil-borne bacterial structure and diversity does not reflect community activity in Pampa biome. PLoS One 8:e76465. doi: 10.1371/journal.pone.0076465
Majumdar, A., Upadhyay, M. K., Giri, B., Srivastava, S., Srivastava, A. K., Jaiswal, M. K., et al. (2021). Arsenic dynamics and flux assessment under drying-wetting irrigation and enhanced microbial diversity in paddy soils: A four year study in Bengal delta plain. J. Hazard. Mater. 409:124443. doi: 10.1016/j.jhazmat.2020.124443
Majumdar, A., Upadhyay, M. K., Giri, B., Yadav, P., Moulick, D., Sarkar, S., et al. (2024). Sustainable water management in rice cultivation reduces arsenic contamination, increases productivity, microbial molecular response, and profitability. J. Hazard. Mater. 466:133610. doi: 10.1016/j.jhazmat.2024.133610
Mandal, M., Das, S., Roy, A., Rakwal, R., Jones, O. A., Popek, R., et al. (2023). Interactive relations between plants, the phyllosphere microbial community, and particulate matter pollution. Sci. Total Environ. 890:164352. doi: 10.1016/j.scitotenv.2023.164352
Montanarella, L. (2015). Agricultural policy: Govern our soils. Nature 528, 32–33. doi: 10.1038/528032a
Nilsson, R. H., Larsson, K. H., Taylor, A. F. S., Bengtsson-Palme, J., Jeppesen, T. S., Schigel, D., et al. (2019). The UNITE database for molecular identification of fungi: handling dark taxa and parallel taxonomic classifications. Nucleic Acids Res. 47, D259–D264. doi: 10.1093/nar/gky1022
Pielou, E. C. (1966). The measurement of diversity in different types of biological collections. J. Theor. Biol. 13, 131–144. doi: 10.1016/0022-5193(66)90013-0
Poonam Srivastava, S., Pathare, V., and Suprasanna, P. (2017). Physiological and molecular insights into rice-arbuscular mycorrhizal interactions under arsenic stress. Plant Gene 11, 232–237. doi: 10.1016/j.plgene.2017.03.004
Rousk, J., Baath, E., Brookes, P. C., Lauber, C. L., Lozupone, C., Caporaso, J. G., et al. (2010). Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 4, 1340–1351. doi: 10.1038/ismej.2010.58
Rousk, J., Brookes, P. C., and Baath, E. (2009). Contrasting soil pH effects on fungal and bacterial growth suggests functional redundancy in carbon mineralization. Appl. Environ. Microbiol. 75, 1589–1596. doi: 10.1128/AEM.02775-08
Saharan, B. S., Tyagi, S., Kumar, R., Vijay, O. H., Mandal, B. S., and Duhan, J. S. (2023). Application of Jeevamrit improves soil properties in zero budget natural farming fields. Agriculture 13:196. doi: 10.3390/agriculture13010196
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/AEM.01541-09
Semenov, A. V., Pereira e Silva, M. C., Szturc-Koestsier, A. E., Schmitt, H., Falcão Salles, J., and van Elsas, J. D. (2012). Impact of incorporated fresh 13C-potato tissues on the bacterial and fungal community composition of soil. Soil Biol. Biochem. 49, 88–95. doi: 10.1016/j.soilbio.2012.02.016
Singh, A., Mazahar, S., Chapadgaonkar, S. S., Giri, P., and Shourie, A. (2023). Phyto-microbiome to mitigate abiotic stress in crop plants. Front. Microbiol. 14:1210890. doi: 10.3389/fmicb.2023.1210890
Singh, J. S., Pandey, V. C., and Singh, D. P. (2011). Efficient soil microorganisms: a new dimension for sustainable agriculture and environmental development. Agric. Ecosyst. Environ. 140, 339–353. doi: 10.1016/j.agee.2011.01.017
Sun, S., and Badgley, B. D. (2019). Changes in microbial functional genes within the soil metagenome during forest ecosystem restoration. Soil Biol. Biochem. 135, 163–172. doi: 10.1016/j.soilbio.2019.05.004
Suyal, D. C., Joshi, D., Debbarma, P., Soni, R., Das, B., and Goel, R. (2019). “Soil metagenomics: Unculturable microbial diversity and its function” in Mycorrhizosphere and Pedogenesis. eds. A. Varma and D. Choudhary (Singapore: Springer), 355–362.
Teng, Y., and Chen, W. (2019). Soil microbiomes – a promising strategy for contaminated soil remediation: a review. Pedosphere 29, 283–297. doi: 10.1016/S1002-0160(18)60061-X
Upadhyay, M. K., Yadav, P., Shukla, A., and Srivastava, S. (2018). Utilizing the potential of microorganisms for managing arsenic contamination: a feasible and sustainable approach. Front. Environ. Sci. 6:24. doi: 10.3389/fenvs.2018.00024
Van Der Heijden, M. G., Bardgett, R. D., and Van Straalen, N. M. (2008). The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 11, 296–310. doi: 10.1111/j.1461-0248.2007.01139.x
Van Oost, K., Quine, T. A., Govers, G., De Gryze, S., Six, J., Harden, J. W., et al. (2007). The impact of agricultural soil erosion on the global carbon cycle. Science 318, 626–629. doi: 10.1126/science.1145724
Wagg, C., Bender, S. F., Widmer, F., and Van Der Heijden, M. G. (2014). Soil biodiversity and soil community composition determine ecosystem multifunctionality. Proc. Natl. Acad. Sci. 111, 5266–5270. doi: 10.1073/pnas.1320054111
White, T. J., Bruns, T. J., Lee, S. B., and Taylor, J. W. (1990). “Amplification and sequencing of fungal ribosomal RNA genes for phylogenetics” in PCR protocols: a guide to methods and applications. eds. M. A. Innis, D. H. Gelfand, J. J. Sninsky, and T. J. White (New York: Academic Press), 315–322.
Xu, C., Li, Y., Hu, X., Zang, Q., Zhuang, H., and Huang, L. (2022). The influence of organic and conventional cultivation patterns on physicochemical property, enzyme activity and microbial community characteristics of paddy soil. Agriculture 12:121. doi: 10.3390/agriculture12010121
Yadav, P., Jaiswal, D. K., and Sinha, R. K. (2021). “Climate change: Impact on agricultural production and sustainable mitigation” in Global climate change. eds. S. Singh, P. Singh, S. Rangabhashiyam, and K. K. Srivastava (Amsterdam: Elsevier), 151–174.
Yang, Y., Gao, Y., Wang, S., Xu, D., Yu, H., Wu, L., et al. (2014). The microbial gene diversity along an elevation gradient of the Tibetan grassland. ISME J. 8, 430–440. doi: 10.1038/ismej.2013.146
Zhan, Y., Liu, W., Bao, Y., Zhang, J., Petropoulos, E., Li, Z., et al. (2018). Fertilization shapes a well-organized community of bacterial decomposers for accelerated paddy straw degradation. Sci. Rep. 8:7981. doi: 10.1038/s41598-018-26375-8
Zhang, R., Liu, L., Yoder, A., Palys, J., Daniels, B., Gautreau, I., et al. (2020). PCR reaction setup and AMPure XP application: reproducible and efficient automation of PCR reaction setup and purification using the Biomek 4000 Workstation and AMPure XP. Available at: https://ls.beckmancoulter.co.jp/files/products/laboratory-automation/AppNote/AMPureXPOnB4K.pdf (accessed September 27, 2024)
Keywords: metagenomics, soil sustainability, organic farming, amplicon sequencing, 16S-ITS
Citation: Mishra AK, Yadav P, Sharma S and Maurya P (2025) Comparison of microbial diversity and community structure in soils managed with organic and chemical fertilization strategies using amplicon sequencing of 16 s and ITS regions. Front. Microbiol. 15:1444903. doi: 10.3389/fmicb.2024.1444903
Edited by:
Ranjith Kumavath, Pondicherry University, IndiaReviewed by:
Jamseel Moopantakath, Central University of Kerala, IndiaCopyright © 2025 Mishra, Yadav, Sharma and Maurya. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ajay Kumar Mishra, YS5rLm1pc2hyYUBpcnJpLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.