 Qian Li1,2†
Qian Li1,2† Yulei Chen1†
Yulei Chen1† Hao Tang3,4Baichao Shu1Zhengyue Zhang1Jiaye Tang1Dang Li1Linjia Jiang1Jiwei Shen1Yaojun Yang2Hanyu Wang2*
Hao Tang3,4Baichao Shu1Zhengyue Zhang1Jiaye Tang1Dang Li1Linjia Jiang1Jiwei Shen1Yaojun Yang2Hanyu Wang2* Menggen Ma1*
Menggen Ma1*- 1College of Resources, Sichuan Agricultural University, Chengdu, China
- 2Bamboo Diseases and Pests Control and Resources Development Key Laboratory of Sichuan Province, College of Life Science, Leshan Normal University, Leshan, China
- 3Key Laboratory of Land Resources Evaluation and Monitoring in Southwest (Sichuan Normal University), Ministry of Education, Chengdu, China
- 4Institute of Landscape Ecology, University of Münster, Münster, Germany
This study examined the phenol degradation capabilities and oxidative stress responses of Candida tropicalis SHC-03, demonstrating its metabolic superiority and resilience compared to Saccharomyces cerevisiae BY4742 in a culture medium with phenol as the sole carbon source. Through comparative growth, transcriptomic, and metabolomic analyses under different phenol concentrations, this study revealed C. tropicalis SHC-03’s specialized adaptations for thriving in phenol as the sole carbon source environments. These include a strategic shift from carbohydrate metabolism to enhanced phenol degradation pathways, highlighted by the significant upregulation of genes for Phenol 2-monoxygenase and Catechol 1,2-dioxygenase. Despite phenol levels reaching 1.8 g/L, C. tropicalis exhibits a robust oxidative stress response, efficiently managing ROS through antioxidative pathways and the upregulation of genes for peroxisomal proteins like PEX2, PEX13, and PMP34. Concurrently, there was significant upregulation of genes associated with membrane components and transmembrane transporters, enhancing the cell’s capacity for substance exchange and signal transduction. Especially, when the phenol concentration was 1.6 g/L and 1.8 g/L, the degradation rates of C. tropicalis towards it were 99.47 and 95.91%, respectively. Conversely, S. cerevisiae BY4742 shows limited metabolic response, with pronounced growth inhibition and lack of phenol degradation. Therefore, our study not only sheds light on the molecular mechanisms underpinning phenol tolerance and degradation in C. tropicalis but also positions this yeast as a promising candidate for environmental and industrial processes aimed at mitigating phenol pollution.
1 Introduction
Phenol, a widely used industrial chemical in the pharmaceutical, resin synthesis, and plastics manufacturing sectors, is a prevalent contaminant in industrial effluents (Schmidt, 2005; Motamedi et al., 2022). Its high stability and toxicity necessitate effective and eco-friendly methods for its removal, essential for environmental protection (Guerra, 2001; Kujawski et al., 2004; Pinto et al., 2005). The adverse effects of phenol on aquatic life and human health, ranging from headaches and dizziness to severe respiratory, neurological, and cardiovascular issues, underscore the urgency for efficient remediation techniques (Panigrahy et al., 2022; Grace Pavithra et al., 2023). Biodegradation stands out as an eco-friendly and efficient approach to addressing phenol pollution. Microorganisms play a crucial role in this process, converting phenol into less toxic substances (Agarry et al., 2008; Gu et al., 2016). Although bacteria are commonly utilized for their rapid growth and adaptability, their efficiency and stability often diminish at high phenol concentrations or in complex environmental conditions (Gu et al., 2016; Li et al., 2019; Mahgoub et al., 2023).
Yeast, particularly Saccharomyces cerevisiae and Candida tropicalis, have long been subjects of intense study due to the distinct physiological and metabolic characteristics (Phalgune et al., 2013; Hegazy et al., 2024). Recent advances have highlighted their differential adaptation strategies to environmental stresses, including phenol and other phenolic compounds (Adav et al., 2007; Yang et al., 2012; He et al., 2022). S. cerevisiae has been studied for its resilience against phenolic compound concentrations (Tekarslan et al., 2015). These studies have demonstrated that S. cerevisiae combats phenolic stress by detoxifying harmful compounds through oxidation–reduction processes, enhancing cellular defenses against oxidative stress, maintaining membrane integrity, and ensuring energy production and cellular function through mitochondrial activities (Ding et al., 2012; Gu et al., 2019).
Additionally, C. tropicalis has been recognized for its remarkable phenol biodegradation capabilities, employing specific enzymatic pathways for converting phenol to carbon dioxide and water, even under high concentrations that typically exert inhibitory effects (Varma and Gaikwad, 2009; Phalgune et al., 2013). This efficiency is due to a unique genetic and metabolic framework, including the activation of genes encoding for phenol hydroxylase and other key enzymes involved in the catabolism of phenolic compounds, enabling C. tropicalis to excel in phenol biodegradation tasks (Adav et al., 2007; Phalgune et al., 2013; He et al., 2022). In our previous studies, we discovered that C. tropicalis exhibits high tolerance to phenol (Wang et al., 2020). Using a complete Yeast extract peptone dextrose (YPD) supplemented with phenol, we observed that C. tropicalis does not activate its phenol degradation mechanisms when other more readily degradable carbon sources (such as glucose) are present. This finding suggests that C. tropicalis possesses a regulatory mechanism that prioritizes the utilization of less toxic carbon sources over phenol under nutrient-rich conditions (Wang et al., 2020). Building on this foundation and guided by prior research, this study employs a medium where phenol is the sole carbon source. This experimental shift is crucial for unveiling the comprehensive adaptability of C. tropicalis to metabolize phenol in the absence of alternative carbon sources, offering insights into the yeast’s specific metabolic pathways and regulatory networks involved in phenol tolerance and degradation.
Therefore, this study seeks to investigate the specific pathways and regulatory mechanisms that enable C. tropicalis to utilize phenol as a sole carbon source, contrasting with the phenol response strategies of S. cerevisiae. This exploration aims to provide a comprehensive understanding of yeast adaptability and potential biotechnological applications in environmental remediation, highlighting the sophisticated metabolic engineering that underpins phenol degradation in yeasts.
2 Materials and methods
2.1 Strain and culture conditions
C. tropicalis SHC-03 was isolated from the distillery lees of Shehong Brewery in Sichuan Province, while S. cerevisiae BY4742 from Open Biosystems, Inc. (United States).
Yeast extract Peptone Dextrose (YPD) (Wang et al., 2020) Medium Preparation: YPD medium was prepared with the following composition per liter: 20 g domestic peptone, 10 g yeast extract powder, and 20 g glucose. The pH of the medium was adjusted to 6.0. For solid media, 20 g of agar per liter was added. The medium was sterilized by autoclaving at 121°C for 20 min. Glucose was autoclaved separately at 115°C for 20 min to avoid Maillard reactions that could occur at higher temperatures. All reagents used for the YPD medium were sourced from Chengdu Haoboyou Company.
Phenol-Only Carbon Source Medium Preparation (Yoneda et al., 2016): The medium for phenol utilization studies was composed of the following per liter: 0.4 g K2HPO4, 0.2 g KH2PO4, 0.1 g NaCl, 0.1 g MgSO4, 0.01 g MnSO4·H2O, 0.01 g Fe2(SO4)3·H2O, 0.01 g Na2MoO4·2H2O, and 0.4 g (NH4)2SO4. This medium was autoclaved at 121°C for 20 min. Phenol was added post-sterilization to achieve the desired concentration (If glucose was used in place of phenol, it served as the sole carbon source). Reagents for medium were obtained from Chengdu Haoboyou Company.
Other Reagents: 2′,7′-Dichlorofluorescein diacetate was procured from Sigma-Aldrich, and other reagents, including ammonia, NH4Cl, 4-aminoantipyrine, and K3(FeCN)6, were purchased from Chengdu Haoboyou Company.
2.2 Incubated strains and measured growth curves
C. tropicalis SHC-03 and S. cerevisiae BY4742 were inoculated into YPD liquid medium and incubated on a constant temperature shaker set at 30°C and 200 rpm overnight to cultivate the cells. Following incubation, the cell suspension was washed three times with sterile water to remove residual medium components. Subsequently, the cells were transferred into a medium containing phenol as the sole carbon source, prepared at varying concentrations, and incubated under identical conditions (30°C and 200 rpm). Growth was monitored by measuring the optical density at 600 nm (OD600) using an ultraviolet spectrophotometer at various time intervals. The OD600 readings were used to determine cell concentration and construct growth curves for each strain across the different phenol concentrations.
2.3 Determination of phenol degradation rate
The residual concentration of phenol in the medium was quantified using the 4-aminoantipyrine spectrophotometric method. This method is based on the reaction between phenol, 4-aminoantipyrine, and potassium ferricyanide in an alkaline environment, resulting in the formation of a red complex. The intensity of the color, indicative of the phenol concentration, was measured by recording the absorbance at 510 nm (OD510) (Mosae Selvakumar, 2018).
2.4 Intracellular reactive oxygen species detection
Cells incubated overnight were washed thrice and incubated in a glucose-only carbon source medium for 3 h (cells collected at this point were labeled as 0 h). After another wash, cells were transferred to phenol-only carbon source medium which with 1.8 g/L phenol and cultured for 6 h at 30°C and 200 rpm, and cells collected at 6 h were labeled accordingly. Cells from both time points were adjusted to OD600 = 1.0 in 1.5 mL Eppendorf tubes. The cell pellet obtained after centrifugation was stained with 2′,7′-DCFdiacetate (pre-thawed), and fluorescence microscopy (equipped with a GFP filter) was used for visualization. Statistical analysis was performed to evaluate ROS accumulation in cells.
2.5 Sample preparation for transcriptome and metabolome analyses
Primary cultures of strains SHC-03 and BY4742 were washed thrice and incubated in glucose-only carbon source medium for 3 h (samples collected at this stage were designated as 0 h for both transcriptome and metabolome analyses). The remaining cells were washed thrice with sterile water and cultured in phenol-only carbon source medium for 6 h according to the initial concentration OD600 = 0.3. Then the cells were collected (labeled 6 h for transcriptome and metabolome analysis). For the reliability of experimental data, three biological replicates were prepared for transcriptomics, and six for metabolomics. The remaining culture was monitored every 6 h to assess growth and ensure accurate sampling.
2.6 Transcriptome analysis
Total RNA was extracted using the TRIzol reagent (Thermo Fisher, Catalog No. 15596018). The quantity and integrity of the RNA were evaluated using the RNA Nano 6,000 Assay Kit on the Bioanalyzer 2,100 system (Agilent Technologies, CA, United States). Gene expression levels were estimated using the FPKM (Fragments Per Kilobase of transcript per Million mapped reads) method. For differential expression analysis, the DESeq2 package in R was utilized. Significantly differentially expressed genes (DEGs) were identified with a threshold of |log2(foldchange)| ≥ 1 and an adjusted p-value (padj) ≤ 0.05. Enrichment analyses for KEGG pathways and GO terms of the DEGs were conducted using the DAVID tool.1
2.7 Metabolome analysis
Untargeted metabolomic analysis was performed using a Vanquish UHPLC system (Thermo Fisher, Germany) coupled to an Orbitrap Q Exactive™ HF mass spectrometer (Thermo Fisher, Germany). Raw data files underwent processing with Compound Discoverer 3.1 (CD3.1, Thermo Fisher) for peak alignment, picking, and quantitation of each metabolite. Statistical analyses of the metabolomic data were conducted using R and Python programming languages. Metabolites were annotated against the KEGG database and conducting enrichment analysis using the MetaboAnalyst tool2 to identify their metabolic pathways.
2.8 Integrated analysis of transcriptome and metabolome
The interaction between metabolites and genes was illustrated through network mapping, selecting the top 10 differentially expressed genes and top 5 differentially expressed metabolites for this purpose. Both sets of data were mapped against the KEGG pathway database to identify common pathways, focusing on major metabolic and signal transduction pathways. The iPath tool3 was used to visualize the enrichment pathways common to both differential metabolites and genes, providing a comprehensive overview of metabolic pathways enriched by both metabolomic and transcriptomic analyses.
3 Results and analysis
3.1 Effects of different phenol concentrations on cell growth and phenol degradation
This study investigated the impact of various phenol concentrations on the growth of C. tropicalis SHC-03, using two sets of culture conditions. One set used media supplemented with 20.0 g/L glucose as the sole carbon source to serve as a control group without inhibitors. The other set employed media where phenol was the sole carbon source, with phenol added at concentrations of 1.6 g/L, 1.8 g/L, 2.0 g/L, and 2.2 g/L, with no glucose included. C. tropicalis SHC-03 demonstrated normal growth in media with 20.0 g/L glucose (Figure 1A). However, growth was notably inhibited at phenol concentrations of 1.6 g/L and 1.8 g/L, with cellular growth resuming only after 24 h of incubation (Figure 1A). Notably, after 96 h of cultivation, phenol degradation rates were observed to reach 99.47 and 95.91%, respectively, for 1.6 g/L and 1.8 g/L concentrations (Figure 1C). Conversely, in media with 2.0 g/L and 2.2 g/L phenol, growth was entirely inhibited, with the phenol concentration in the culture medium remaining largely unchanged, suggesting a complete halt in cellular activity under these conditions (Figure 1C). So, a phenol concentration of 1.8 g/L was selected for subsequent experiments. This concentration effectively maintains the cells in a recoverable state of growth inhibition, allowing for further investigation into the degradation capabilities and adaptability of C. tropicalis SHC-03 under phenolic stress.
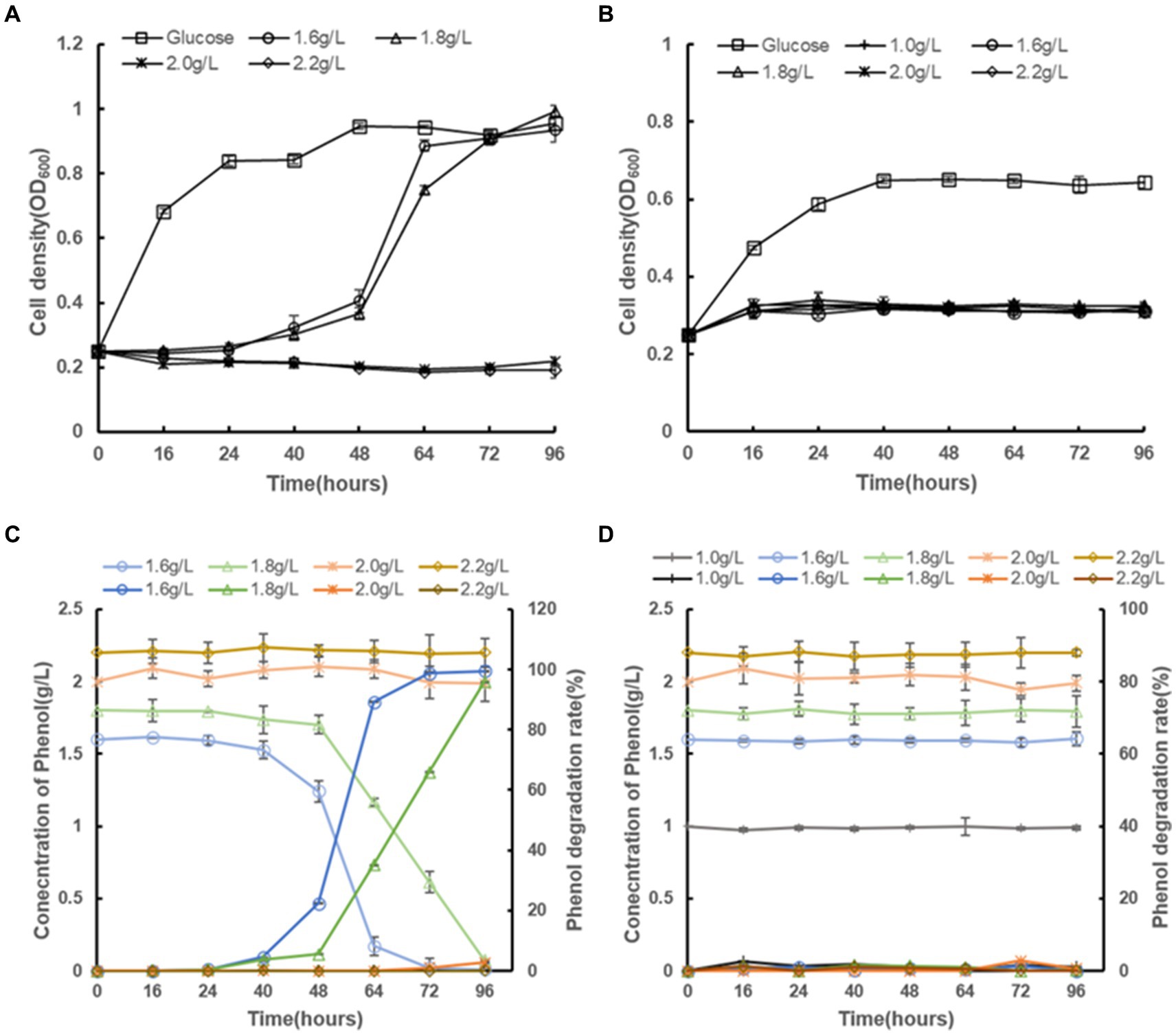
Figure 1. The growth profiles of C. tropicalis SHC-03 and S. cerevisiae BY4742 in different culture media, as well as the residual phenol concentrations and phenol degradation rates in the culture media. (A) Growth curve of C. tropicalis SHC-03. (B) Growth curve of S. cerevisiae BY4742. (C) Phenol concentration and phenol degradation rate in the culture medium (C. tropicalis SHC-03). (D) Phenol concentration and phenol degradation rate in the culture medium (S. cerevisiae BY4742).
In parallel, S. cerevisiae BY4742 was subjected to the same conditions. While the strain BY4742 displayed normal growth in media where glucose served as the sole carbon source, it exhibited no growth in media supplemented with phenol, regardless of the concentration. This was accompanied by an unchanged phenol concentration within the culture medium, reinforcing the observation that S. cerevisiae lacks the capacity to utilize phenol as a carbon source under the tested conditions (Figures 1B,D).
3.2 Transcriptomic and metabolomic analysis under phenol stress conditions
Sequencing analysis revealed that both C. tropicalis SHC-03 and S. cerevisiae BY4742 undergo significant changes in gene expression upon phenol exposure. The sequencing quality metrics, with clean bases predominantly exceeding 6.0 G and both Q20 and Q30 above 90%, underscore the high fidelity of the data, setting a robust foundation for subsequent analyses (Supplementary Table S1). Correlation analysis between samples showed high consistency among biological replicates, further validating the reliability of the data for subsequent research analysis (Supplementary Figures S1, S2).
Upon analyzing the sequencing data of C. tropicalis SHC-03 and S. cerevisiae BY4742, a notable change in gene expression was observed following phenol exposure. The comparison between pre-treatment (0 h) and post-treatment (6 h) revealed differential expression in both strains. Specifically, the strain SHC-03 displayed 1,345 differentially expressed genes, consisting of 765 upregulated and 580 downregulated genes. In contrast, the strain BY4742 exhibited a total of 2,247 differentially expressed genes, with 1,188 being upregulated and 1,059 downregulated. Volcano plots (Figures 2A,B) visually summarize these differential expressions, with further analyses for these genes.
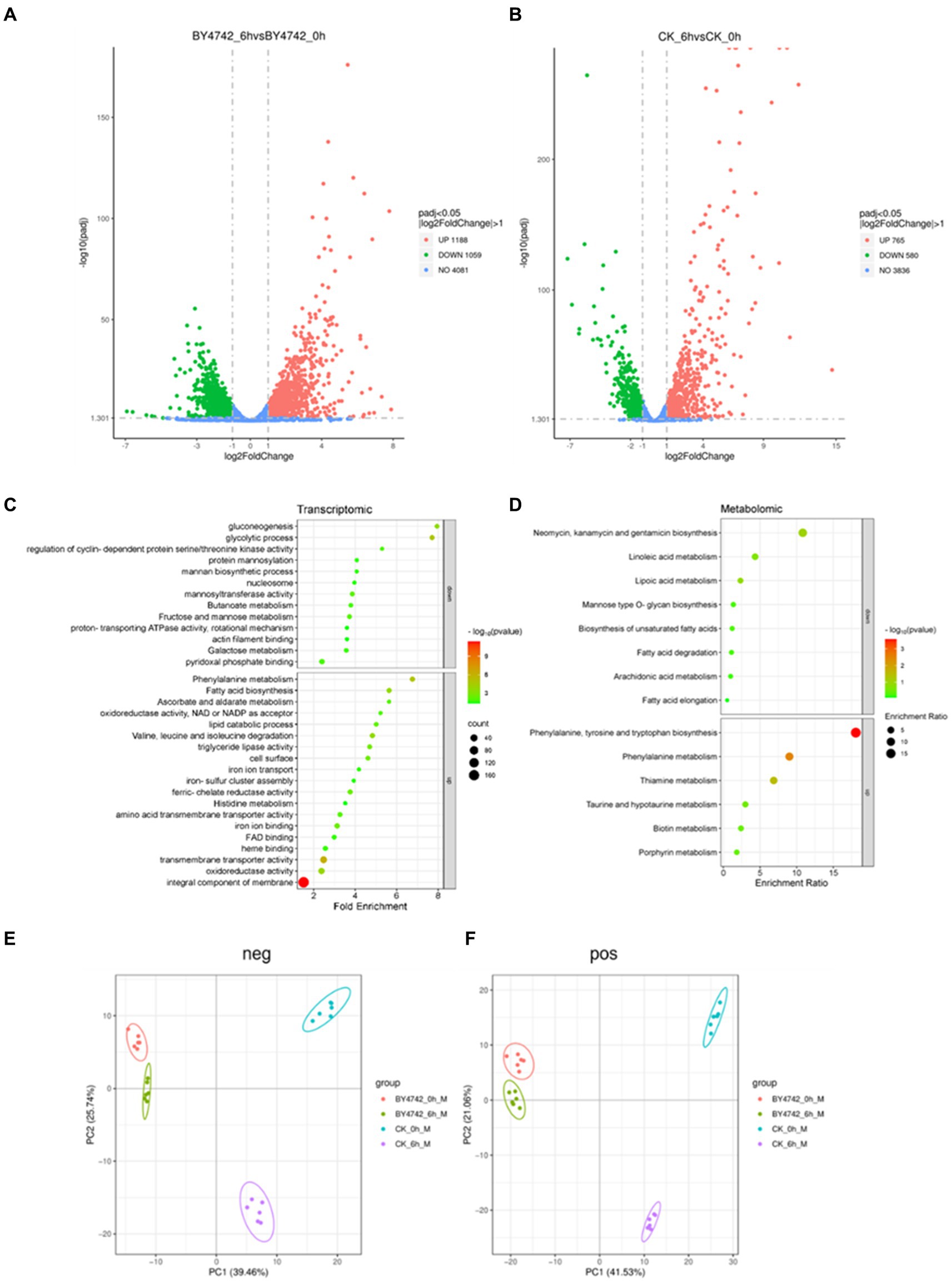
Figure 2. Transcriptome and metabolome analysis of C. tropicalis SHC-03 and S. cerevisiae BY4742. (A) Volcano plot of differentially expressed genes in Saccharomyces cerevisiae BY4742. (B) Volcano plot of differentially expressed genes in C. tropicalis SHC-03. (C) Transcriptionally specific upregulation and downregulation pathways in C. tropicalis SHC-03. (D) Metabolic pathways with specific upregulation and downregulation in C. tropicalis SHC-03. (E) Principal component analysis (PCA) plot of positive metabolites between different samples. (F) Principal component analysis (PCA) plot of negative metabolites between different samples (CK_0h: C. tropicalis SHC-03 0 h; CK_6h: C. tropicalis SHC-03 6 h).
Gene enrichment analyses, conducted via the DAVID tool. Upregulated genes of SHC-03 predominantly enriched pathways such as the “integral component of membrane” and “extracellular region,” whereas downregulated genes were mainly associated with pathways like “mitochondrion” and “plasma membrane” (Supplementary Tables S2, S3). For the strain BY4742, upregulated genes were enriched in pathways including “cellular response to heat” and “ribosome,” with downregulated genes enriched in “translation” and “cytoplasmic translation (Supplementary Tables S4, S5).” Specific upregulated pathways in the strain SHC-03 include “integral component of membrane,” “transmembrane transporter activity,” and “oxidoreductase activity,” among others, while specific downregulated pathways feature “nucleosome,” “glycolytic process,” and “gluconeogenesis,” to name a few (Figure 2C).
Pearson correlation analysis of metabolomic data showed a high correlation among replicate samples, validating the reliability of the dataset (Supplementary Figures S3, S4). Principal component analysis (PCA) highlighted distinct metabolomic profiles between strains SHC-03 and BY4742 post-phenol treatment, underscoring significant metabolic differences (Figures 2E,F). the strain SHC-03 experienced changes in 272 upregulated and 458 downregulated metabolites, while the strain BY4742 expressed 138 upregulated and 220 downregulated metabolites. MetaboAnalyst tool was utilized for enrichment analysis (Supplementary Figures S5–S8), with the strain SHC-03’s specific upregulated metabolites showed enrichment in “Phenylalanine, tyrosine and tryptophan biosynthesis,” and “Phenylalanine metabolism,” among others. Specific downregulated metabolites in the strain SHC-03 were enriched in pathways like “Fatty acid degradation (Figure 2D).”
In summary, C. tropicalis SHC-03 and S. cerevisiae BY4742 exhibit distinct transcriptomic and metabolomic responses under phenol stress, with the strain SHC-03 showed adaptations that may facilitate phenol degradation through specific metabolic pathways.
3.3 Activation of the oxidative stress response
This investigation-stained cells of C. tropicalis SHC-03 and S. cerevisiae BY4742 to quantify the accumulation of reactive oxygen species (ROS). Under fluorescence microscopy, ROS-accumulating cells were distinguished by green fluorescence (Allen et al., 2010; Wang et al., 2023; Liao et al., 2024), while cells without ROS accumulation did not fluoresce (Figure 3A). Statistical analysis at 0 h revealed that 25.82% of C. tropicalis SHC-03 cells and 54.85% of S. cerevisiae BY4742 cells exhibited ROS accumulation. By 6 h, these figures rose to 35.65 and 73.87%, respectively, highlighting a significantly higher ROS accumulation in S. cerevisiae at both time points, and the proportion of cells accumulating ROS in the strain BY4742 was significantly higher than that in the strain SHC-03 (Figure 3B).
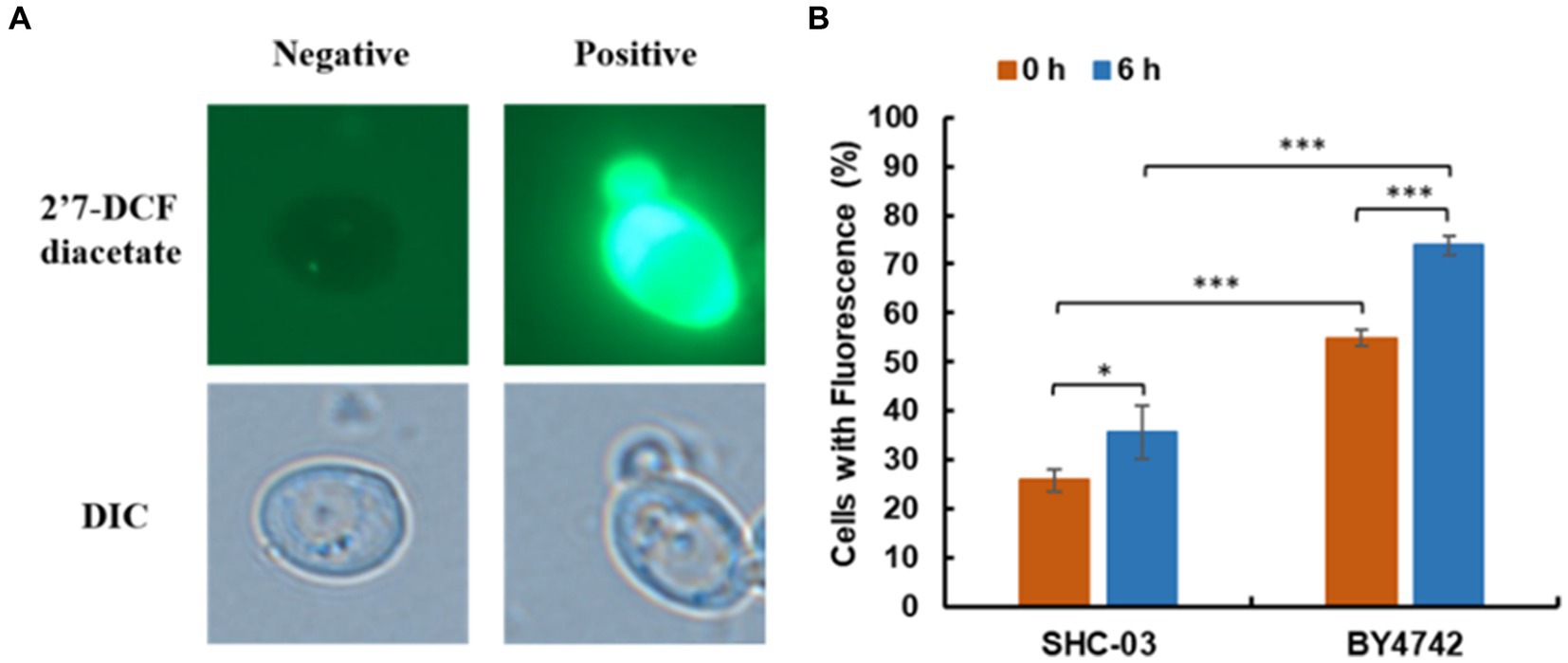
Figure 3. Reactive oxygen species accumulation in C. tropicalis SHC-03 and S. cerevisiae BY4742. (A) Accumulation of reactive oxygen species in cells. (B) The proportion of cells at 1.8 g/L phenol that contained reactive oxygen species after treatment for 0 and 6 h. 2′7′-DCF diacetate (top column): reactive oxygen species indicator dye. DIC (down column): differential interference microscope. Negative: no signal; Positive: there is a signal. * p < 0.05, *** p < 0.001 indicates significant differences. The data represent aver-ages of three experiments. At least 100 cells were examined on each bright-field image.
Further analysis of transcriptomic and metabolomic data to understand the distinct responses of the strains revealed significant upregulation of genes encoding Peroxisome, specifically PEX2, PEX13, and PMP34, in the peroxisome pathway of C. tropicalis SHC-03 (Table 1 and Figure 4). These proteins play crucial roles in the detoxification of reactive oxygen species, enhancing the cell’s ability to manage oxidative stress. In contrast, S. cerevisiae BY4742 displayed elevated gene expression levels exclusively for PEX3 and PEX19 within the same pathway, with fold changes of 1.49 and 1.33, respectively, (Figure 4). Notably, the expression of PEX1 decreased by 1.05-fold compared to baseline (0 h), suggesting a more limited peroxisomal response under phenol stress (Figure 4). Additionally, C. tropicalis SHC-03 showed increased expression in pathways associated with the oxidative stress response, including Oxidoreductase activity, Iron ion binding, FAD binding, and Ascorbate and aldarate metabolism (Figure 2C), with metabolites in Ascorbate and aldarate also exhibited upregulation (Supplementary Figure S5). This suggests a more robust and nuanced response to phenol-induced stress in the strain SHC-03 compared to the strain BY4742, with the former displaying a superior ability to counteract oxidative stress from phenol.
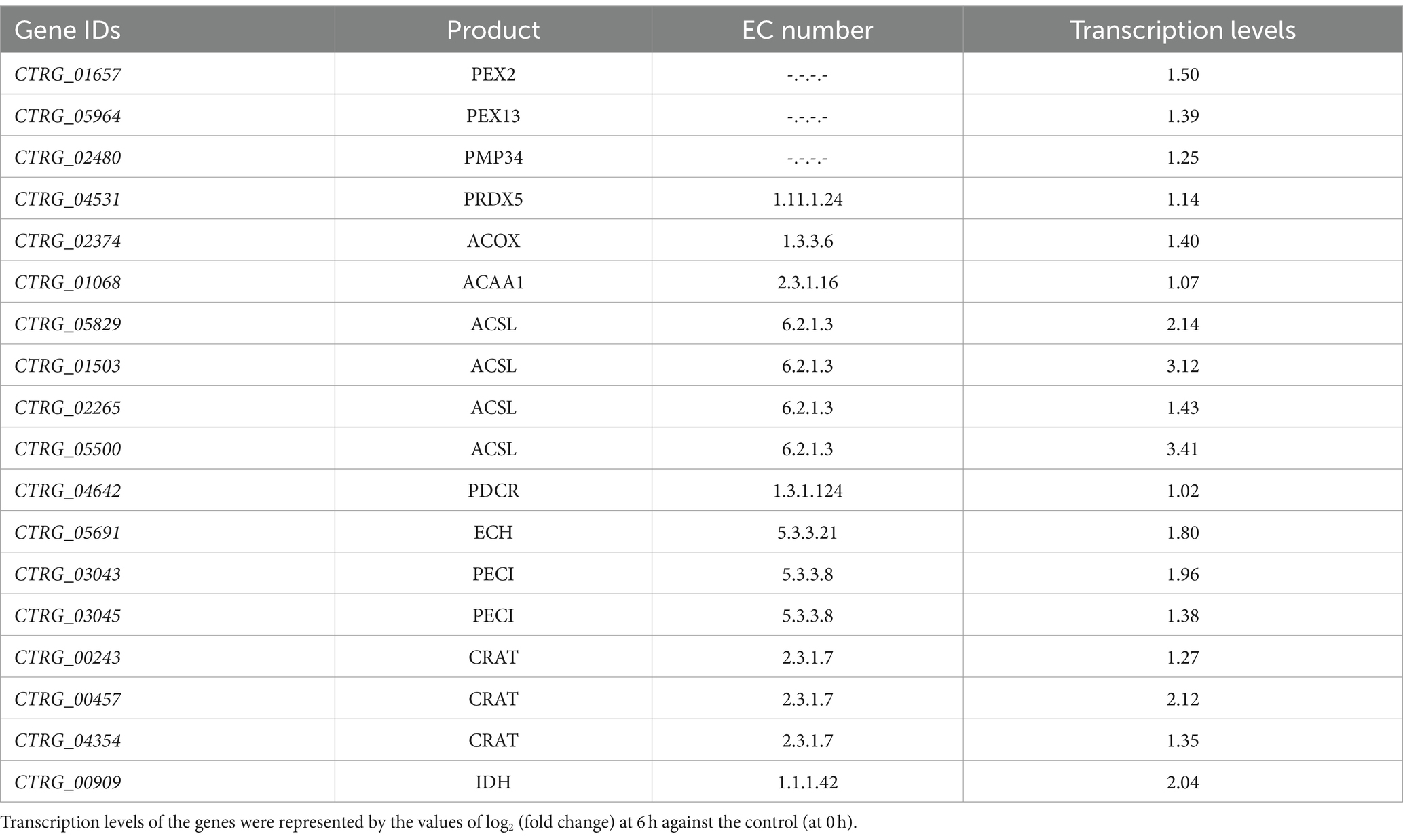
Table 1. Transcription levels of differentially expressed genes on the peroxisome pathway in Candida tropicalis SHC-03.
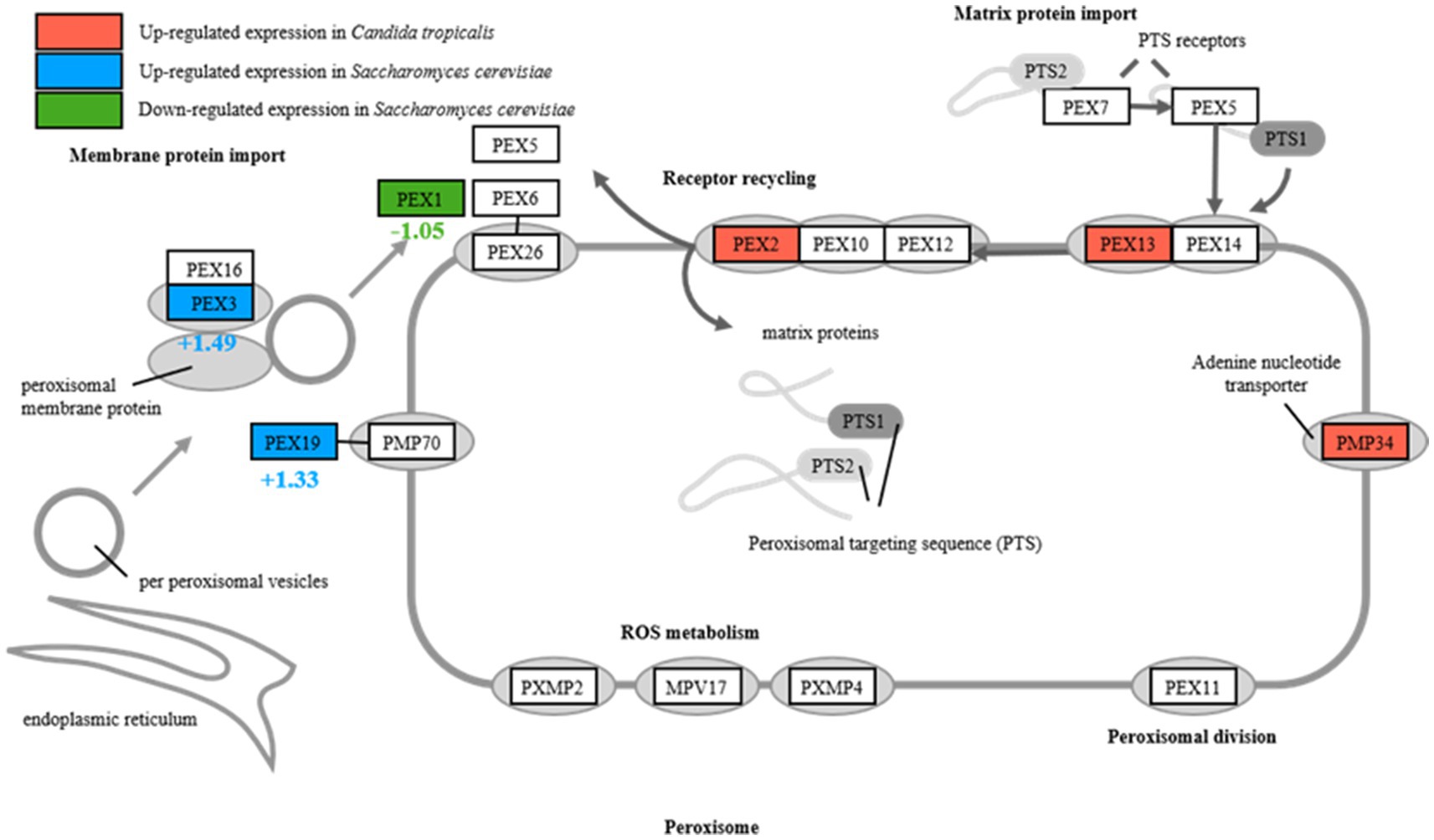
Figure 4. Differential expression of peroxisome in Candida tropicalis and Saccharomyces cerevisiae.
The comparative analysis illustrated the disparate oxidative stress responses and phenol resilience between C. tropicalis SHC-03 and S. cerevisiae BY4742. Specifically, the strain SHC-03 showed a markedly enhanced capacity for mitigating oxidative stress triggered by phenol, underscoring the significant differences in stress adaptation mechanisms between the two yeast strains.
3.4 Metabolic adaptation to phenol stress in C. tropicalis
Despite the non-detection of catechol and cis, cis-muconate and minimal changes in the phenol concentration after 6 h, these findings do not negate the onset of phenol metabolism by C. tropicalis. The upregulation of phenylalanine metabolism and associated pathways suggests that the yeast is undergoing preliminary metabolic adjustments in preparation for phenol degradation (Figures 2C,D).
Unlike S. cerevisiae, which cannot metabolize phenol as a sole carbon source, C. tropicalis undergoes significant metabolic reprogramming under phenolic stress. Within 6 h of exposure to a phenol-rich medium, a notable suppression was observed in carbohydrate metabolism pathways such as glycolysis, gluconeogenesis, and the metabolism of fructose and mannose (Figure 2C and Supplementary Table S3). This suppression signifies a strategic shift from carbohydrate utilization, redirecting the yeast’s metabolic capacity to process phenol as an alternative carbon source.
Concurrently, there was a marked increase in the expression of genes related to membrane components and transmembrane transporters, which play a crucial role in enhancing the cell’s ability for substance exchange and signal transduction (Figure 2C). This upregulation improves the cellular interface for environmental interactions, crucial for adapting to phenolic stress. Specifically, genes encoding Major Facilitator Superfamily (MFS) transporters, including CTRG_06081, CTRG_06100, CTRG_05562, CTRG_03960 and CTRG_03958, were significantly upregulated (Table 2). MFS transporters are integral membrane proteins that facilitate the passive transport of substrates such as sugars, polyols, drugs, and various organic acids across cellular membranes (dos Santos et al., 2014; Wang et al., 2018). In the context of phenol stress, these transporters likely play pivotal roles in modulating the cellular uptake of phenol and the efflux of its toxic metabolites, thereby enhancing cell survival and metabolic efficiency under stress conditions. Furthermore, our study observed a reduction in secondary metabolite synthesis, particularly in pathways for neomycin, kanamycin, and gentamicin biosynthesis (Figure 2D). The reduction suggests a strategic reallocation of cellular resources towards essential pathways for phenol detoxification and assimilation into the central metabolism.
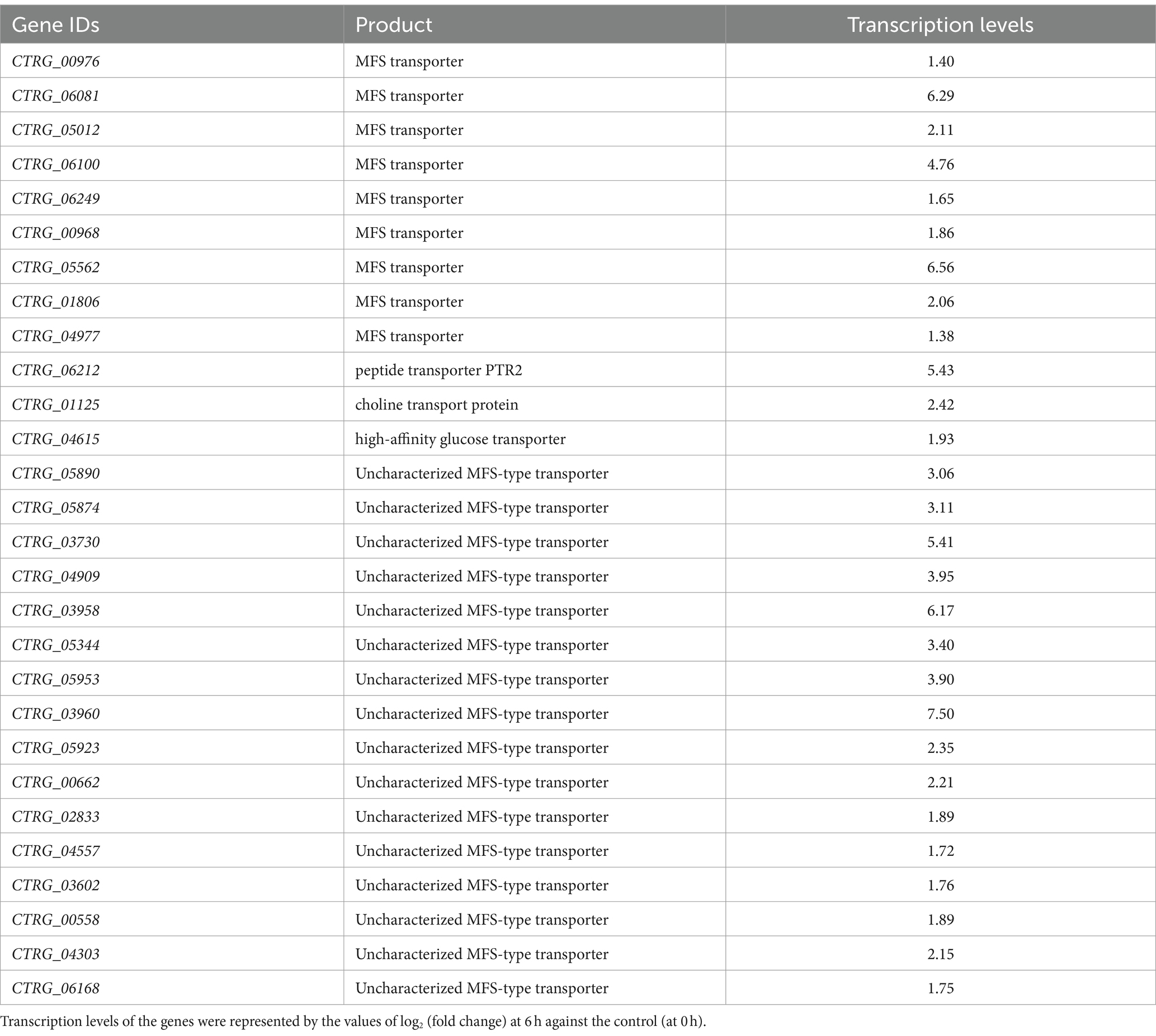
Table 2. Transcription levels of differentially expressed genes on transmembrane transporter activity in Candida tropicalis SHC-03.
Furthermore, significant upregulation was noted in genes encoding Phenol 2-monoxygenase (CTRG_00423 and CTRG_03102) were upregulated by 3.77-fold and 11.90-fold (Figure 5), initiating the degradation of phenol to catechol, a step that relies on NADPH. The increased expression of enzymes requiring NAD(P)H as a cofactor, especially those involved in oxidoreductive reactions (Supplementary Table S2), highlights a heightened demand for cofactors necessary for phenol metabolism. And the genes for Catechol 1,2-dioxygenase (CTRG_01732 and CTRG_00171) were upregulated by 7.80-fold and 7.01-fold (Figure 5), respectively, indicating the activation of the β-ketoadipate pathway for catechol conversion to cis, cis-muconate (Figure 5) (Yan et al., 2005; Long et al., 2014; Wang et al., 2020; He et al., 2022).
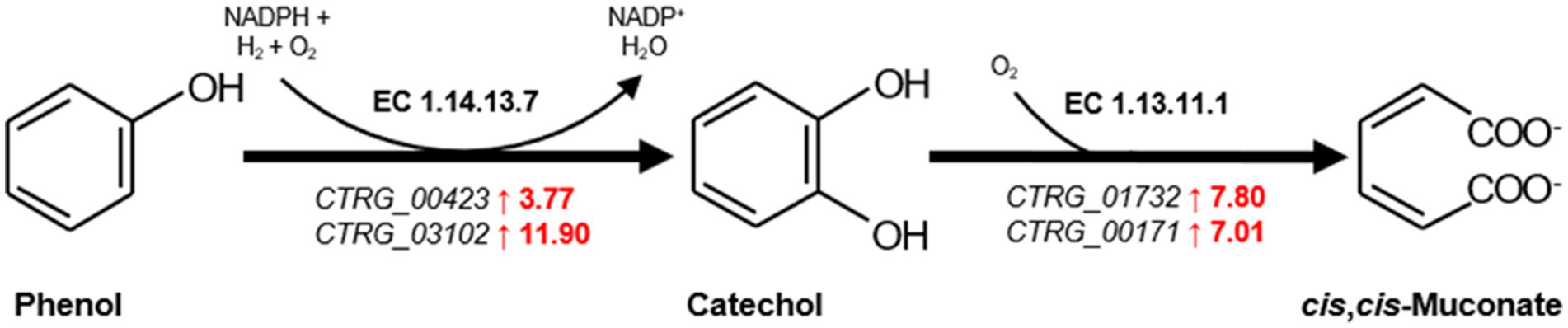
Figure 5. The first and second step of β-ketoadipate pathway in the biodegradation phenol. Genes significantly upregulated are highlighted in red. The numbers represent transcription levels, quantified as log2 (fold change) of gene expression at 6 h relative to the control at 0 h, indicating the magnitude of upregulation.
4 Discussion
Our results demonstrating C. tropicalis SHC-03’s superior ability to degrade phenol, showing degradation rates of 99.47 and 95.91% at phenol concentrations of 1.6 g/L and 1.8 g/L respectively, highlight the presence of a robust enzymatic system in C. tropicalis. Meanwhile, Phenol 2-monoxygenase and Catechol 1,2-dioxygenase, are pivotal for initiating the breakdown of phenol into less toxic intermediates (Adav et al., 2007; Long et al., 2014; Wang et al., 2020; He et al., 2022). The observed growth inhibition in C. tropicalis SHC-03 at higher phenol concentrations of 2.0 g/L and 2.2 g/L, aligns with findings by He et al., who reported a threshold phenol concentration beyond which cellular activity is significantly hampered (He et al., 2022). This suggests that C. tropicalis SHC-03 possesses a threshold for phenol tolerance and degradation, beyond which the toxic effects of phenol outweigh the yeast’s enzymatic capacity for degradation. In contrast, the absence of phenol degradation in S. cerevisiae underlines a lack of such enzymatic pathways or an effective phenolic stress response. This aligns with observations that S. cerevisiae may require genetic modification to efficiently metabolize phenolic compounds (Adeboye, 2016), further underscoring the metabolic versatility and resilience of C. tropicalis SHC-03 in phenol degradation and stress response.
In this study, significant metabolic reprogramming of C. tropicalis was observed, characterized by the suppression of primary carbohydrate metabolism and the concurrent activation of phenol degradation pathways, indicates a sophisticated prioritization of metabolic resources under phenolic stress. This strategic shift, underscored by the suppression of glycolysis, gluconeogenesis, and fructose and mannose metabolism pathways, demonstrates the yeast’s capability to dynamically reroute its metabolic machinery towards the utilization of phenol as an alternative carbon source (He et al., 2022). Furthermore, the upregulation of Major Facilitator Superfamily (MFS) transporters, such as CTRG_06081, CTRG_06100, CTRG_05562, CTRG_03960, and CTRG_03958, underscores a vital cellular mechanism for managing phenolic compounds. MFS transporters are not merely passive conduits for molecular traffic but active participants in the cell’s response to environmental changes. They facilitate the uptake and secretion of a wide range of substrates, including sugars, ions, and various xenobiotics, thus playing a pivotal role in cellular adaptability to environmental stresses. The increased expression of these transporters in C. tropicalis SHC-03 not only facilitates more efficient phenol uptake and detoxification but also highlights a sophisticated regulatory network that enhances resilience and adaptability to environmental stresses (dos Santos et al., 2014; Wang et al., 2018). This finding is in line with Wang et al. (2018), who emphasized the crucial role of MFS transporters in microbial responses to xenobiotic stress, aiding in the efflux of toxic substances and thus safeguarding cellular integrity. The downregulation of secondary metabolite synthesis pathways, particularly those associated with antibiotic biosynthesis, suggests a strategic reallocation of cellular resources. This adaptation, aimed at ensuring survival under phenolic stress by diverting energy and molecular precursors towards phenol detoxification pathways, mirrors observations by Montibus et al. (2015). They demonstrated that environmental stressors prompt a significant redirection of metabolic fluxes, optimizing microorganisms’ survival strategies in adverse conditions. The collective insights from these studies reinforce the concept that C. tropicalis’ metabolic reprogramming in response to phenolic stress is not an isolated phenomenon but part of a broader, highly conserved strategy among microorganisms to cope with toxic environments.
PEX2 functions as part of the RING complex, a ubiquitin ligase critical for the import of enzymes into peroxisomes, affecting the turnover and proliferation of these organelles and PEX13 is a component of the docking complex required for the import of matrix proteins into peroxisomes, facilitating the efficient sequestration and breakdown of ROS, while PEX34 is not a universally recognized PEX protein, assuming a similar role would suggest involvement in peroxisomal membrane assembly or function, potentially enhancing the compartmentalization and efficiency of peroxisomal processes (Jansen et al., 2021). The upregulation of these PEX genes in C. tropicalis suggests an enhanced capacity for peroxisome proliferation, import of antioxidative enzymes, and overall improved management of oxidative stress. This is particularly important under phenolic stress conditions, where elevated ROS levels can cause significant cellular damage. Conversely, S. cerevisiae exhibits upregulated expression of PEX3 and PEX19, indicating active peroxisomal maintenance but possibly a less adaptable response to oxidative stresses. The specific patterns of PEX gene expression in C. tropicalis underscore its superior ability to manage oxidative stress, likely due to more dynamic peroxisomal adaptations. At the same time, the genes related to oxidoreductase activities, iron ion and FAD binding, ascorbic acid and uronate metabolism pathway in C. tropicalis were also up-regulated. And it underscored a comprehensive strategy not merely aimed at detoxifying ROS but also at maintaining cellular redox equilibrium and protecting cellular components from oxidative damage (Dias-Gunasekara et al., 2006; Herrero et al., 2008; Xiao et al., 2021). Enhanced oxidoreductase activities, pivotal in converting ROS into less reactive molecules, underscore a critical adaptive response to the oxidative stress resultant from phenol metabolism (Herrero et al., 2008). The roles of iron and FAD are particularly noteworthy, given their involvement in redox reactions and oxidative stress management, suggesting that C. tropicalis may optimize its redox balance as a countermeasure to phenolic stress (Dias-Gunasekara et al., 2006). Additionally, the upregulation of the ascorbate and aldarate metabolism pathways, accompanied by increased expression of related metabolites, signifies a strategic enhancement of antioxidant defenses, which further highlights C. tropicalis’s adaptive mechanisms to mitigate and thrive under phenolic-induced oxidative stress conditions (Xiao et al., 2021).
The upregulation of genes encoding Phenol 2-monoxygenase and Catechol 1,2-dioxygenase, pivotal for the initial steps in phenol catabolism, does not merely signify an enhanced capacity for phenol degradation. It also reflects an intricate regulatory adaptation that enables C. tropicalis to efficiently convert phenol into catechol and subsequently to cis, cis-muconate via the β-ketoadipate pathway, a mechanism that is central to the degradation of aromatic compounds (Yan et al., 2005; Long et al., 2014; Wang et al., 2020; He et al., 2022). The reliance on NAD(P)H in these reactions further illuminates the critical role of redox balance in the yeast’s response to phenolic stress, suggesting an integrated response that encompasses both the efficient degradation of phenol and the maintenance of cellular redox homeostasis (Long et al., 2014).
In summary, the metabolic adaptation of C. tropicalis to phenolic stress is emblematic of a highly evolved response that integrates phenol degradation with broader cellular objectives, including the maintenance of redox balance and cellular integrity. This orchestrated response not only underscores the yeast’s resilience to environmental stressors but also highlights its potential as a biotechnological tool for the bioremediation of phenolic pollutants.
5 Conclusion
This study illuminates the sophisticated metabolic and oxidative stress response mechanisms of C. tropicalis under phenolic stress. Unlike S. cerevisiae, which lacks the metabolic flexibility to degrade phenol, C. tropicalis demonstrates an exceptional ability to not only metabolize phenol efficiently but also to manage the resultant oxidative stress effectively. The research highlights the critical roles of Phenol 2-monoxygenase and Catechol 1,2-dioxygenase in phenol degradation, alongside a strategic metabolic shift that prioritizes the utilization of phenol as an alternative carbon source. Furthermore, the enhanced expression of PEX genes and pathways involved in the oxidative stress response underscores C. tropicalis’s comprehensive strategy for maintaining cellular homeostasis and integrity in the face of phenolic-induced stress. Our results contribute to the field of microbial bioremediation, providing valuable insights into the adaptive mechanisms that enable C. tropicalis to thrive in phenol-contaminated environments. In conclusion this research not only enhances our understanding of yeast biology and environmental adaptation but also underscores the potential of C. tropicalis as a biotechnological tool for the remediation of phenolic pollutants, offering a promising avenue for addressing environmental contamination challenges.
Data availability statement
The raw data supporting the findings of this study have been deposited in publicly accessible repositories. The transcriptome sequencing data have been uploaded to the NCBI Sequence Read Archive (SRA) under the accession number PRJNA1102611. The metabolomics data are available in the MetaboLights repository and can be accessed via the following link: https://www.ebi.ac.uk/metabolights with the accession number MTBLS10757.
Author contributions
QL: Conceptualization, Data curation, Formal analysis, Investigation, Project administration, Writing – original draft, Writing – review & editing. YC: Formal analysis, Project administration, Writing –original draft. HT: Formal analysis, Investigation, Writing – original draft. BS: Formal analysis, Writing – review & editing. ZZ: Investigation, Writing – review & editing. JT: Data curation, Writing – review & editing. DL: Investigation, Writing – review & editing. LJ: Data curation, Writing – review & editing. JS: Data curation, Writing – review & editing. YY: Writing – review & editing. HW: Conceptualization, Writing – review & editing. MM: Conceptualization, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by the Chengdu Technology Innovation Research and Development Project (No. 2022-YF05-00671-SN), the Sichuan Science and Technology Program (No. 2020YFH0142), the National Natural Science Foundation of China (No. 32200044), the Key Project of Science and Technology Bureau of LeShan Town (No. 22ZDYJ0089), the Talent Research Startup Projects of Leshan Normal University (No. RC2022003), and the Scientific Research and Cultivation Plan of Leshan Normal University (No. 2022SSDJS010).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1442235/full#supplementary-material
Footnotes
References
Adav, S. S., Chen, M. Y., Lee, D. J., and Ren, N. Q. (2007). Degradation of phenol by aerobic granules and isolated yeast Candida tropicalis. Biotechnol. Bioeng. 96, 844–852. doi: 10.1002/bit.21148
Adeboye, T. P. (2016). Mapping phenolics metabolism and metabolic engineering of Saccharomyces cerevisiae for increased endogenous catabolism of phenolic compounds. Gothenburg: Chalmers University of Technology.
Agarry, S. E., Durojaiye, A. O., and Solomon, B. O. (2008). Microbial degradation of phenols: a review. Int. J. Environ. Pollut. 32, 12–28. doi: 10.1504/IJEP.2008.016895
Allen, S. A., Clark, W., McCaffery, J. M., Cai, Z., Lanctot, A., Slininger, P. J., et al. (2010). Furfural induces reactive oxygen species accumulation and cellular damage in Saccharomyces cerevisiae. Biotechnol. Biofuels 3:2. doi: 10.1186/1754-6834-3-2
Dias-Gunasekara, S., Van Lith, M., Gareth Williams, J. A., Kataky, R., and Benham, A. M. (2006). Mutations in the FAD binding domain cause stress-induced misoxidation of the endoplasmic reticulum oxidoreductase Ero1β. J. Biol. Chem. 281, 25018–25025. doi: 10.1074/jbc.M602354200
Ding, M. Z., Wang, X., Liu, W., Cheng, J. S., Yang, Y., and Yuan, Y. J. (2012). Proteomic research reveals the stress response and detoxification of yeast to combined inhibitors. PLoS One 7:e43474. doi: 10.1371/journal.pone.0043474
dos Santos, S. C., Teixeira, M. C., Dias, P. J., and Sá-Correia, I. (2014). MFS transporters required for multidrug/multixenobiotic (MD/MX) resistance in the model yeast: understanding their physiological function through post-genomic approaches. Front. Physiol. 5:180. doi: 10.3389/fphys.2014.00180
Grace Pavithra, K., Sundar Rajan, P., Arun, J., Brindhadevi, K., Hoang Le, Q., and Pugazhendhi, A. (2023). A review on recent advancements in extraction, removal and recovery of phenols from phenolic wastewater: challenges and future outlook. Environ. Res. 237:117005. doi: 10.1016/j.envres.2023.117005
Gu, Q., Wu, Q., Zhang, J., Guo, W., Wu, H., and Sun, M. (2016). Community analysis and recovery of phenol-degrading bacteria from drinking water biofilters. Front. Microbiol. 7:495. doi: 10.3389/fmicb.2016.00495
Gu, H., Zhu, Y., Peng, Y., Liang, X., Liu, X., Shao, L., et al. (2019). Physiological mechanism of improved tolerance of Saccharomyces cerevisiae to lignin-derived phenolic acids in lignocellulosic ethanol fermentation by short-term adaptation. Biotechnol. Biofuels 12:268. doi: 10.1186/s13068-019-1610-9
Guerra, R. (2001). Ecotoxicological and chemical evaluation of phenolic compounds in industrial effluents. Chemosphere 44, 1737–1747. doi: 10.1016/S0045-6535(00)00562-2
He, Y., Wang, Z., Li, T., Peng, X., Tang, Y., and Jia, X. (2022). Biodegradation of phenol by Candida tropicalis sp.: kinetics, identification of putative genes and reconstruction of catabolic pathways by genomic and transcriptomic characteristics. Chemosphere 308:136443. doi: 10.1016/j.chemosphere.2022.136443
Hegazy, G. E., Soliman, N. A., Farag, S., El-Helow, E. R., Hassan, H. Y., and Abdel-Fattah, Y. R. (2024). Isolation and characterization of Candida tropicalis B: a promising yeast strain for biodegradation of petroleum oil in marine environments. Microb. Cell Factories 23:20. doi: 10.1186/s12934-023-02292-y
Herrero, E., Ros, J., Bellí, G., and Cabiscol, E. (2008). Redox control and oxidative stress in yeast cells. Biochim. Biophys. Acta-Gen. Subj. 1780, 1217–1235. doi: 10.1016/j.bbagen.2007.12.004
Jansen, R. L. M., Santana-Molina, C., van den Noort, M., Devos, D. P., and van der Klei, I. J. (2021). Comparative genomics of peroxisome biogenesis proteins: making sense of the PEX proteins. Front. Cell Dev. Biol. 9:654163. doi: 10.3389/fcell.2021.654163
Kujawski, W., Warszawski, A., Ratajczak, W., Porȩbski, T., Capała, W., and Ostrowska, I. (2004). Removal of phenol from wastewater by different separation techniques. Desalination 163, 287–296. doi: 10.1016/S0011-9164(04)90202-0
Li, H., Meng, F., Duan, W., Lin, Y., and Zheng, Y. (2019). Biodegradation of phenol in saline or hypersaline environments by bacteria: a review. Ecotoxicol. Environ. Saf. 184:109658. doi: 10.1016/j.ecoenv.2019.109658
Liao, H., Li, Q., Chen, Y., Tang, J., Mou, B., Lu, F., et al. (2024). Genome-wide identification of resistance genes and response mechanism analysis of key gene knockout strain to catechol in Saccharomyces cerevisiae. Front. Microbiol. 15:1364425. doi: 10.3389/fmicb.2024.1364425
Long, Y., Yang, S., Xie, Z., and Cheng, L. (2014). Identification and characterization of phenol hydroxylase from phenol-degrading Candida tropicalis strain JH8. Can. J. Microbiol. 60, 585–591. doi: 10.1139/cjm-2014-0417
Mahgoub, S. A., Qattan, S. Y. A., Salem, S. S., Abdelbasit, H. M., Raafat, M., Ashkan, M. F., et al. (2023). Characterization and biodegradation of phenol by Pseudomonas aeruginosa and Klebsiella variicola strains isolated from sewage sludge and their effect on soybean seeds germination. Molecules 28:1203. doi: 10.3390/molecules28031203
Montibus, M., Pinson-Gadais, L., Richard-Forget, F., Barreau, C., and Ponts, N. (2015). Coupling of transcriptional response to oxidative stress and secondary metabolism regulation in filamentous fungi. Crit. Rev. Microbiol. 41, 295–308. doi: 10.3109/1040841X.2013.829416
Mosae Selvakumar, P. (2018). Phenol sensing studies by 4-Aminoantipyrine method-a review. Org. Med. Chem. Int. J. 5:555657. doi: 10.19080/omcij.2018.05.555657
Motamedi, M., Yerushalmi, L., Haghighat, F., and Chen, Z. (2022). Recent developments in photocatalysis of industrial effluents: a review and example of phenolic compounds degradation. Chemosphere 296:133688. doi: 10.1016/j.chemosphere.2022.133688
Panigrahy, N., Priyadarshini, A., Sahoo, M. M., Verma, A. K., Daverey, A., and Sahoo, N. K. (2022). A comprehensive review on eco-toxicity and biodegradation of phenolics: recent progress and future outlook. Environ. Technol. Innov. 27:102423. doi: 10.1016/j.eti.2022.102423
Phalgune, U. D., Rajamohanan, P. R., Gaikwad, B. G., Varma, R. J., and George, S. (2013). Biodegradation of phenol by the yeast candida tropicalis: an investigation by NMR spectroscopy. Appl. Biochem. Biotechnol. 169, 2029–2037. doi: 10.1007/s12010-013-0119-0
Pinto, R. T. P., Lintomen, L., Luz, L. F. L., and Wolf-Maciel, M. R. (2005). Strategies for recovering phenol from wastewater: thermodynamic evaluation and environmental concerns. Fluid Phase Equilib. 228–229, 447–457. doi: 10.1016/j.fluid.2004.09.005
Schmidt, R. J. (2005). Industrial catalytic processes - phenol production. Appl. Catal. A Gen. 280, 89–103. doi: 10.1016/j.apcata.2004.08.030
Tekarslan, H., Alkim, C., Hunte, C., and Çakar, Z. P. (2015). Physiological and genetic analysis of cellular sodium and lithium response/resistance behavior using the yeast Saccharomyces cerevisiae as a model organism. J. Pharm. Istanbul Univ. 45, 165–179.
Varma, R. J., and Gaikwad, B. G. (2009). Biodegradation and phenol tolerance by recycled cells of Candida tropicalis NCIM 3556. Int. Biodeterior. Biodegrad. 63, 539–542. doi: 10.1016/j.ibiod.2009.01.001
Wang, Q., Chen, D., Wu, M., Zhu, J., Jiang, C., Xu, J. R., et al. (2018). MFS transporters and GABA metabolism are involved in the self-defense against DON in fusarium graminearum. Front. Plant Sci. 9:438. doi: 10.3389/fpls.2018.00438
Wang, H., Li, Q., Peng, Y., Zhang, Z., Kuang, X., Hu, X., et al. (2020). Cellular analysis and comparative Transcriptomics reveal the tolerance mechanisms of Candida tropicalis toward phenol. Front. Microbiol. 11:544. doi: 10.3389/fmicb.2020.00544
Wang, H., Li, Q., Zhang, Z., Ayepa, E., Xiang, Q., Yu, X., et al. (2023). Discovery of new strains for furfural degradation using adaptive laboratory evolution in Saccharomyces cerevisiae. J. Hazard. Mater. 459:132090. doi: 10.1016/j.jhazmat.2023.132090
Xiao, M., Li, Z., Zhu, L., Wang, J., Zhang, B., Zheng, F., et al. (2021). The multiple roles of ascorbate in the abiotic stress response of plants: antioxidant, cofactor, and regulator. Front. Plant Sci. 12:598173. doi: 10.3389/fpls.2021.598173
Yan, J., Jianping, W., Hongmei, L., Suliang, Y., and Zongding, H. (2005). The biodegradation of phenol at high initial concentration by the yeast Candida tropicalis. Biochem. Eng. J. 24, 243–247. doi: 10.1016/j.bej.2005.02.016
Yang, J., Ding, M. Z., Li, B. Z., Liu, Z. L., Wang, X., and Yuan, Y. J. (2012). Integrated phospholipidomics and transcriptomics analysis of saccharomyces cerevisiae with enhanced tolerance to a mixture of acetic acid, furfural, and phenol. Omi. A J. Integr. Biol. 16, 374–386. doi: 10.1089/omi.2011.0127
Keywords: Candida tropicalis, Saccharomyces cerevisiae, phenol, degradation, reactive oxygen species
Citation: Li Q, Chen Y, Tang H, Shu B, Zhang Z, Tang J, Li D, Jiang L, Shen J, Yang Y, Wang H and Ma M (2024) Unveiling superior phenol detoxification and degradation ability in Candida tropicalis SHC-03: a comparative study with Saccharomyces cerevisiae BY4742. Front. Microbiol. 15:1442235. doi: 10.3389/fmicb.2024.1442235
Edited by:
Jianjun Chen, University of Florida, United StatesReviewed by:
Shraddha Maitra, University of Illinois at Urbana-Champaign, United StatesVarinia López, Instituto Tecnológico Superior de Irapuato, Mexico
Copyright © 2024 Li, Chen, Tang, Shu, Zhang, Tang, Li, Jiang, Shen, Yang, Wang and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hanyu Wang, SGFueXUud2FuZzYxNDNAb3V0bG9vay5jb20=; Menggen Ma, bWdlbkBzaWNhdS5lZHUuY24=
†These authors have contributed equally to this work