 Kuang-Hung Liu
Kuang-Hung Liu Yu-Xin Xiao
Yu-Xin Xiao Ruwen Jou
Ruwen Jou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 18 September 2024
Sec. Infectious Agents and Disease
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1439532
Introduction: Multidrug-resistant tuberculosis (MDR-TB) remains a challenge in the TB program of Taiwan, where 0.5% of new cases and 2.1% of previously treated cases were resistant to at least rifampin (RIF) and isoniazid (INH). Since >80% of our MDR-TB are new cases, genotyping of MDR Mycobacterium tuberculosis is implemented to facilitate contact investigation, cluster identification, and outbreak delineation.
Methods: This is a population-based retrospective cohort study analyzing MDR-TB cases from 2019 to 2022. Whole genome sequencing (WGS) was performed using the Illumina MiSeq and analyzed using the TB Profiler. A single nucleotide polymorphism (SNP) threshold of ≤ 12 and phylogenetic methods were used to identify putative transmission clusters. An outbreak was confirmed using genomic data and epidemiologic links.
Results: Of the 297 MDR-TB cases, 246 (82.8%), 45 (15.2%), and 6 (2.0%) were simple MDR, extensively drug-resistant tuberculosis (pre-XDR-TB) and extensively drug-resistant tuberculosis (XDR-TB), respectively. The sublineage 2.2 modern Beijing was the predominant (48.8%) MDR-TB strain in Taiwan. Phylogenetic analysis identified 25.3% isolates in 20 clusters, with cluster sizes ranging from 2 to 13 isolates. Nevertheless, only 2 clusters, one household and one community, were confirmed as outbreaks. In this study, we found that males had a higher risk of MDR-TB transmission compared to females, and those infected with the sublineage 2.1-proto-Beijing genotype isolates were at a higher risk of transmission. Furthermore, 161 (54.2%) isolates harbored compensatory mutations in the rpoC and non-rifampicin resistant determinant region (non-RRDR) of the rpoB gene. MDR-TB strains containing rpoB S450L and other compensatory mutations concurrently were significantly associated with clusters, especially the proto-Beijing genotype strains with the compensatory mutation rpoC E750D or the modern Beijing genotype strains with rpoC D485Y/rpoC E1140D.
Discussion: Routine and continuous surveillance using WGS-based analysis is recommended to warn of risks and delineate transmission clusters of MDR-TB. We proposed the use of compensatory mutations as epidemiological markers of M. tuberculosis to interrupt putative MDR-TB transmission.
The global emergence of multidrug-resistant tuberculosis (MDR-TB) poses a threat to achieving the End TB targets. MDR-TB can arise from delayed diagnosis, prolonged treatment, and the transmission of MDR Mycobacterium tuberculosis isolates. Although defining and tracking TB transmission has been particularly challenging due to the variability in disease progression after infection, genotyping of M. tuberculosis provides vital insight into investigating TB transmission patterns and aids in identifying associated risk factors (Gardy et al., 2011; Lalor et al., 2018).
Understanding these transmission patterns is crucial for guiding effective public health interventions aimed at preventing the spread of MDR-TB (Nathanson et al., 2010). Furthermore, identifying the risk factors associated with MDR-TB clustering is crucial for improving health outcomes through coordinated efforts to prevent transmission (Denholm et al., 2024; GBD 2021 Forecasting Collaborators, 2024). Compared to conventional genotyping methods (Barnes and Cave, 2003; Allix-Béguec et al., 2008), whole genome sequencing (WGS) provides a more accurate resolution of phylogenetic clusters, making it a powerful tool for TB surveillance and control (Luo et al., 2014; Yang et al., 2017; Guthrie et al., 2018, 2019).
Rifampicin-resistant (RR) isolates exhibit mutations within the RNA polymerase (rpoB) gene, with rpoB S450L being the most prevalent mutation (Comas et al., 2011; Brandis and Hughes, 2013). These RR isolates harbor compensatory mutations in RNA polymerase subunits, such as the rpoA and rpoC genes, or in regions outside the rpoB rifampin-resistance determining region (RRDR; non-RRDR). Specific mutations in the rpoA and rpoC genes can result in high-fitness MDR strains (Comas et al., 2011).
However, the relationship between drug resistance and compensatory mutations has been inconsistently reported (Cohen and Murray, 2004). Previous studies revealed that compensatory evolution can facilitate the spread of MDR M. tuberculosis isolates by mitigating the fitness costs associated with mutations, thereby increasing resistance rates (de Vos et al., 2013; Li et al., 2016; Merker et al., 2018). Conversely, other studies have indicated that compensatory mutations have a minimal impact on MDR-TB clustering in China (Liu et al., 2018; Chen et al., 2022).
The prevalence of MDR-TB in Taiwan is estimated to be over 80% (Chuang et al., 2016; Wu et al., 2023), yet the pattern of transmission remains unclear. To enhance the management of MDR-TB, we conducted a cohort study using WGS and epidemiological information to investigate putative MDR-TB transmission.
In our TB program, bacteriological examination is required for any presumptive TB cases, with universal drug susceptibility testing (DST) achieved through the TB laboratory network. This population-based retrospective study analyzed culture-confirmed MDR-TB cases reported to the Taiwan Centers for Disease Control (Taiwan CDC) between 2019 and 2022. For each case, one initial M. tuberculosis isolate was analyzed using WGS.
M. tuberculosis isolates were cultured and manipulated in a certified biosafety level 3 laboratory. Demographic information, bacteriological data, and epidemiological investigation information were obtained from the National TB Registry. A new case was defined as one that had never been previously reported or recorded in the TB Registry as an MDR-TB case. Previously treated MDR-TB cases included those with recurrent cases, those treated after loss to follow-up, those treated after failure, and other previously treated cases. We used a standardized questionnaire to investigate epidemiological links among MDR-TB cases.
Decontaminated specimens were inoculated on both solid and liquid media simultaneously. M. tuberculosis isolates were subjected to DST using the agar proportion method (APM) with 7H10 and 7H11 media (Coning Technology Limited Company, Taiwan). The APM procedure is described as follows: M. tuberculosis was grown in 7H9 complete media [0.2% glycerol, 0.1% Tween, 10% albumin, dextrose catalase (ADC) supplement] to log phase. Cultures were adjusted to 0.5–1 MacFarland standard using 7H9 complete media and serially diluted (10−2 and 10−4). A 0.1 ml aliquot of each culture mixture was inoculated onto 7H10 agar plates supplemented with 10% oleic acid, albumin, dextrose, and catalase (OADC). According to WHO recommendations, the critical concentrations of the tested drugs in 7H10 media were RIF, 1 mg/L; INH, 1.0 mg/L and 0.2 mg/L; ethambutol (EMB), 5 and 10 mg/L; streptomycin (STM), 2 mg/L and 10 mh/L; levofloxacin (LFX), 1 mg/L; and moxifloxacin (MXF), 0.5 mg/L. The critical concentrations of the tested drugs in 7H11 media were amikacin (AMK), 6 mg/L; kanamycin (KAN), 6 mg/L; capreomycin (CAP), 10 mg/L; ethionamide (ETO), 10 mg/L; and para-aminosalicylic acid (PAS), 8.0 mg/L. Resistance to pyrazinamide (PZA), 100 mg/L, was tested using the Bactec MGIT 960 system (Becton Dickinson Diagnostic Systems, Sparks, MD) as previously described (WHO, 2018). Growth on a control medium was compared to growth on the corresponding drug-containing medium to determine susceptibility. Cultures were incubated at 37°C for 3 weeks, and colony-forming units (CFUs) were counted. Resistance was defined as CFUs on the antibiotic quadrant over 1% of the CFUs on the antibiotic-free quadrant (Woods et al., 2011). The DST result was used to determine resistance or susceptibility. The tests were validated based on the susceptibility of M. tuberculosis H37Rv. Since STM was initially tested with RIF, INH, and EMB as first-line drugs, we categorized STM as a first-line drug. MDR is defined as an M. tuberculosis isolate that is resistant to at least INH and RIF. Pre-XDR is defined as an MDR isolate that is resistant to either fluoroquinolone (FQs; pre-XDR fluo) or at least one of the injectable drugs (pre-XDR inj; Banerjee et al., 2008). XDR was defined as MDR TB plus resistance to an FQ and at least one second-line injectable drug (SLID; WHO, 2007).
M. tuberculosis isolates were subcultured on 7H11 medium, and genomic DNA was extracted using the cetyl-trimethyl-ammonium-bromide method (van Soolingen et al., 1991). The TruSeq DNA PCR-Free LT Sample Preparation Kit (Illumina, Inc., San Diego, CA, USA) was used to prepare paired-end libraries following the manufacturer's instructions. The average fragment size (500–600 bp) of the DNA libraries was checked by the Agilent 2100 Bioanalyzer in combination with the High Sensitivity DNA Kit. The concentration of the DNA libraries was measured by quantitative PCR with the KAPA Library Quantification Kit (Roche Sequencing Solutions, Inc., Pleasanton, CA, USA). Using the MiSeq Reagent Kit ver. 3 and an Illumina MiSeq system (Illumina, Inc., San Diego, CA, USA), the 24 pure DNA libraries were pooled (11 pM) and sequenced (600 cycles) with an expected coverage of 100X. We analyzed compensatory mutations in the rpoA, rpoC, and non-RRDR of the rpoB genes. Putative compensatory mutations were identified based on the following criteria: (1) the presence of non-synonymous mutations in the rpoA, rpoB, or rpoC genes in RIF-resistant isolates; (2) each putative compensatory mutation has been independently observed in at least two isolates (Liu et al., 2018).
We conducted a dry laboratory analysis of WGS data (Behzadi and Ranjbar, 2019). Paired-end Illumina reads were checked using FastQC (www.bioinformatics.babraham.ac.uk/projects/fastqc/) for primary assessment of data quality, and any adapter fragment and low-quality reads were removed using Trimmomatic. Bowtie2 was used to map paired-end reads to the reference genome H37Rv (GenBank AL123456). The strict SNP filtering (closed SNP set) template within the BioNumerics 7.6.3 software was used to generate a list of high-quality, informative SNPs for each cluster. The SNP filtration was based on the following criteria: have total coverage of 5 reads, not contain ambiguous bases, not contain gaps, and not be within 12 base pairs of adjoining SNPs. Non-informative SNPs (identical in all samples) were also excluded. No genomic regions were specifically excluded from the analysis. The TB Profiler v.4.1.1 was used to assign lineages and predict gene mutations associated with drug resistance (Phelan et al., 2019). A phylogenetic tree was built from 3,453 identified SNP positions, excluding repetitive genomic regions (PE/PEE), using the maximum likelihood method with the Tamura-Nei model and 1,000 bootstrap replicates in MEGA 7.0. iTOL v6 (https://itol.embl.de) was used to annotate and visualize the phylogenetic tree. Isolates were defined as clustered or unique based on a genetic distance of ≤ 12 or > 12 SNPs, respectively. The sequence data were deposited in the National Center for Biotechnology Information Sequence Read Archive under BioProject ID PRJNA1141184. A minimum spanning tree was constructed using BioNumerics 7.6.3 software.
Descriptive statistics were performed on patients' demographics, including lineage, sex, age, previous treatment history, and clustering of MDR isolates. Logistic regression analysis was used to calculate statistical significance. Odd ratios with 95% confidence intervals (CI) were calculated, and variables with P-values < 0.05 were considered potential new risk factors. The odds ratio calculator from MedCalc Software Ltd. (version 20.013) was used for the statistical analysis.
The Taiwan CDC Institutional Review Board approved this study (TwCDC IRB No. 106211). All procedures were conducted in accordance with applicable guidelines and regulations. As the study only involved archived isolates, written informed consent from participants was not required.
In this population-based study, 297 cases of MDR-TB accounted for 1.0% of the 30,193 TB cases from 2019 to 2022 (Figure 1). Of these 297 MDR-TB cases, 219 (73.7%) were male, and 78 (26.3%) cases were female, with a median age of 63 years (ranging from 50 to 76 years). Among the cases, 242 (81.5%) were new cases, 52 (17.5%) were previously treated cases, and 3 (1.0%) had an unknown treatment history. A maximum-likelihood phylogenetic tree was constructed based on 3,453 SNPs in non-repetitive regions of the studied isolates, which included 45 (15.2%) pre-XDR and 6 (2.0%) XDR-TB cases (Figure 2). Lineage 2 isolates were predominant, including 43 (14.5%) sublineage 2.1 (proto-Beijing) and 145 (48.8%) sublineage 2.2 (modern Beijing) isolates (Table 1). Data for all 297 MDR-TB cases are shown in Supplementary Table 3.
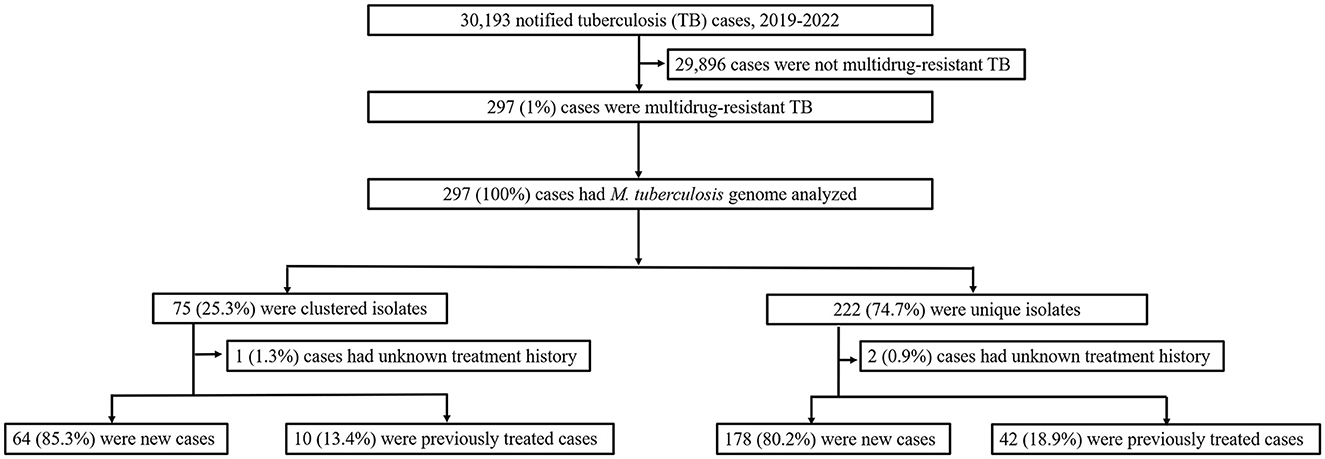
Figure 1. Classification of multidrug-resistant tuberculosis based on treatment history and genomic analysis. PTB, pulmonary tuberculosis; DST, drug susceptibility testing; MDR-TB, multidrug-resistant tuberculosis.
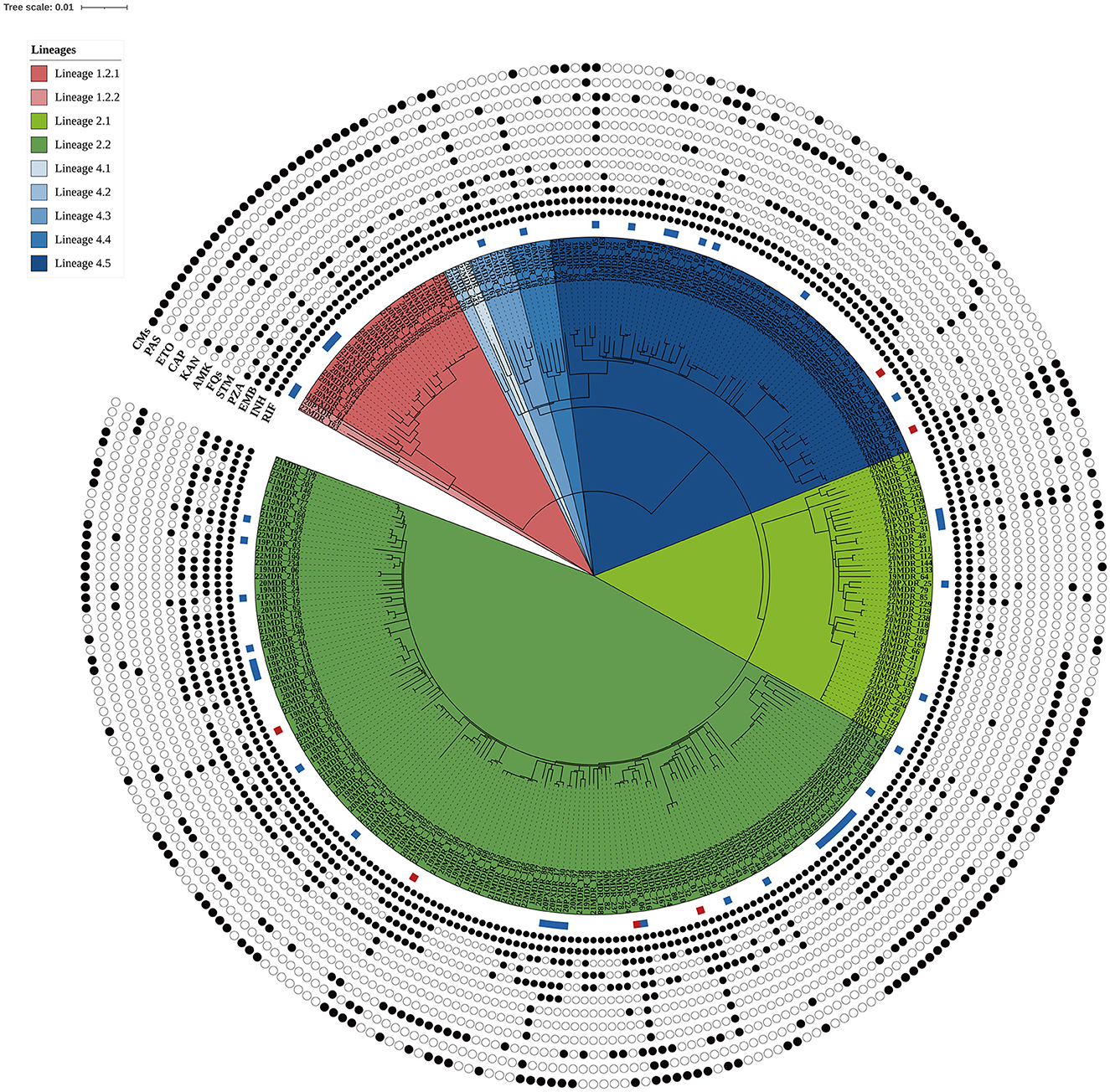
Figure 2. Maximum likelihood phylogenetic tree of the 297 MDR-TB isolates from Taiwan. The tree was constructed based on 3,453 genome-wide SNPs. Lineages are represented by different colored blocks. Mutations encoded resistance were represented by filled circles (presence of mutation) or empty circles (absence of mutations) icons. The square beside the nodes shows the presence of pre-extensively drug-resistant (pre-XDR, blue) and extensively drug-resistant (XDR, red) phenotypes among the multidrug-resistant tuberculosis strains. The figure was generated using iTOL v6 (https://itol.embl.de). RIF, rifampicin; INH, isoniazid; EMB, ethambutol; PZA, pyrazinamide; STM, streptomycin; FQs, fluoroquinolones; AMK, amikacin; KAM, kanamycin; CAP, capreomycin; ETO, ethionamide; PAS, para-aminosalicylic acid; CMs, compensatory mutations.
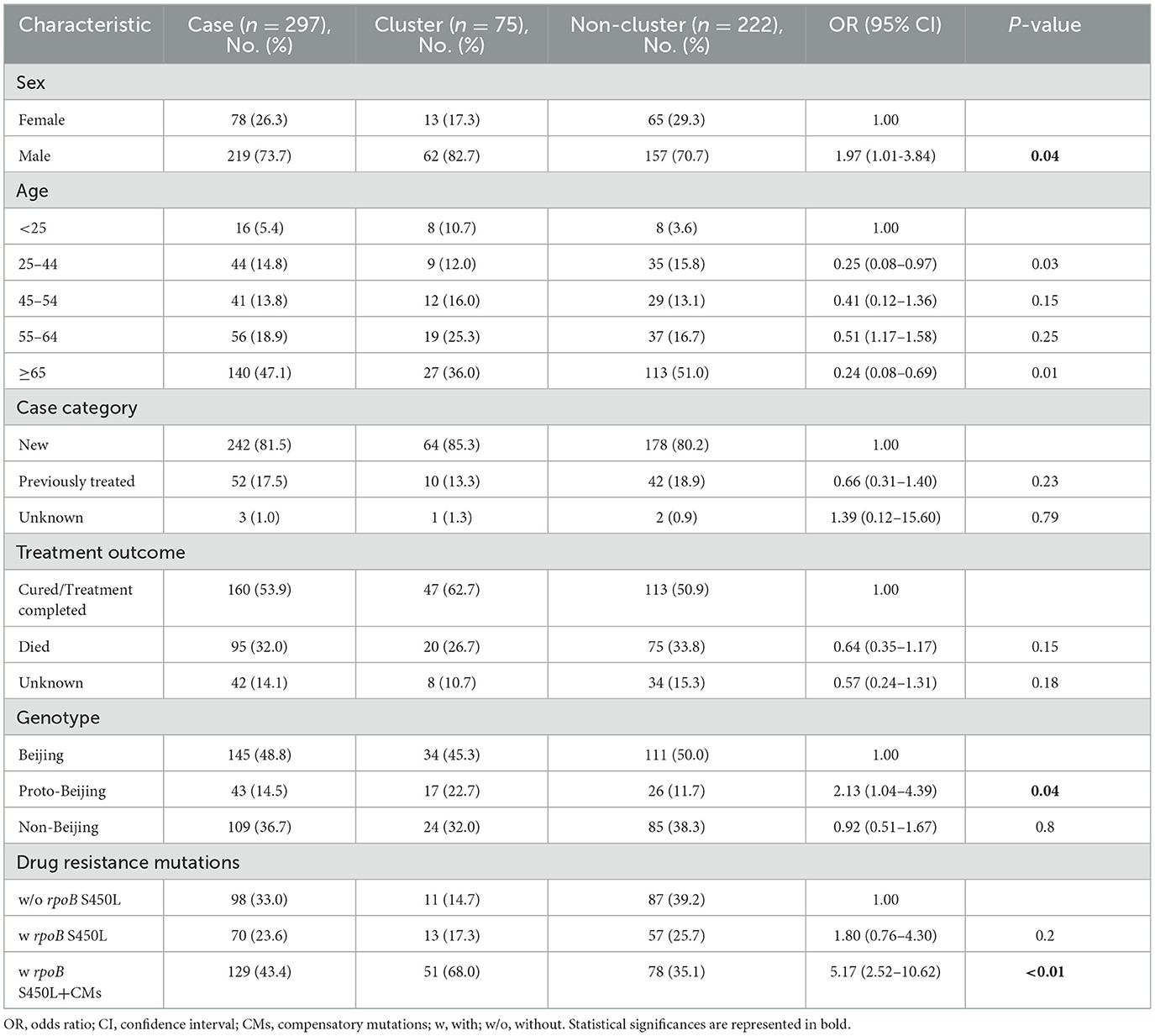
Table 1. Demographic and other factors associated with TB clustering (≤12 SNPs difference).
Using a 12-SNP cut-off, 25.3% (75/297) MDR-TB cases were grouped into 20 clusters, with cluster sizes ranging from 2 to 13 cases (Tables 1, 2). Cluster 04 was the largest cluster, with 84.6% (11/13) of cases originating from northern Taiwan, all of which had identical drug-resistance gene mutations, including fabG1 t-8c and rpoB S450L, along with a compensatory mutation, rpoC E750D (Table 2 and Supplementary Table 2). The univariate analysis revealed a correlation between MDR-TB clusters and male sex. Individuals carrying the sublineage 2.1-proto-Beijing genotype had a higher risk of transmitting the infection (Table 1). Of the 20 clustered MDR-TB cases, 8 (40%) had definite epidemiological links. Minimum Spanning Tree (MST) analysis showed household and community links in clusters 10 and 11 (Figures 3, 4). In cluster 10, the SNP differences among the 7 cases ranged from 0 to 1 (Figure 3A). Cases 19MDR_01, 19MDR_09, 19MDR_10, and 19MDR_39 lived in the same household, while cases 19MDR_01 and 19MDR_44 attend the same school (Figure 3B). However, 19MDR_36 and 20MDR_103 had no known epidemiological links with other cases in the cluster. In cluster 11, cases 21PXDR_32, 21PXDR_47, and 19PXDR_09 were pre-XDR TB cases with SNP differences ranging from 0 to 8 (Figure 4A) and were from a high-burden village with presumptive infection from the community (Figure 4B). Cases 21PXDR_32 and 21PXDR_41 were household members (Figure 4B). Although the SNP difference between cases 20MDR_100 and 21PXDR_32 was 5, no epidemiological links were recalled.
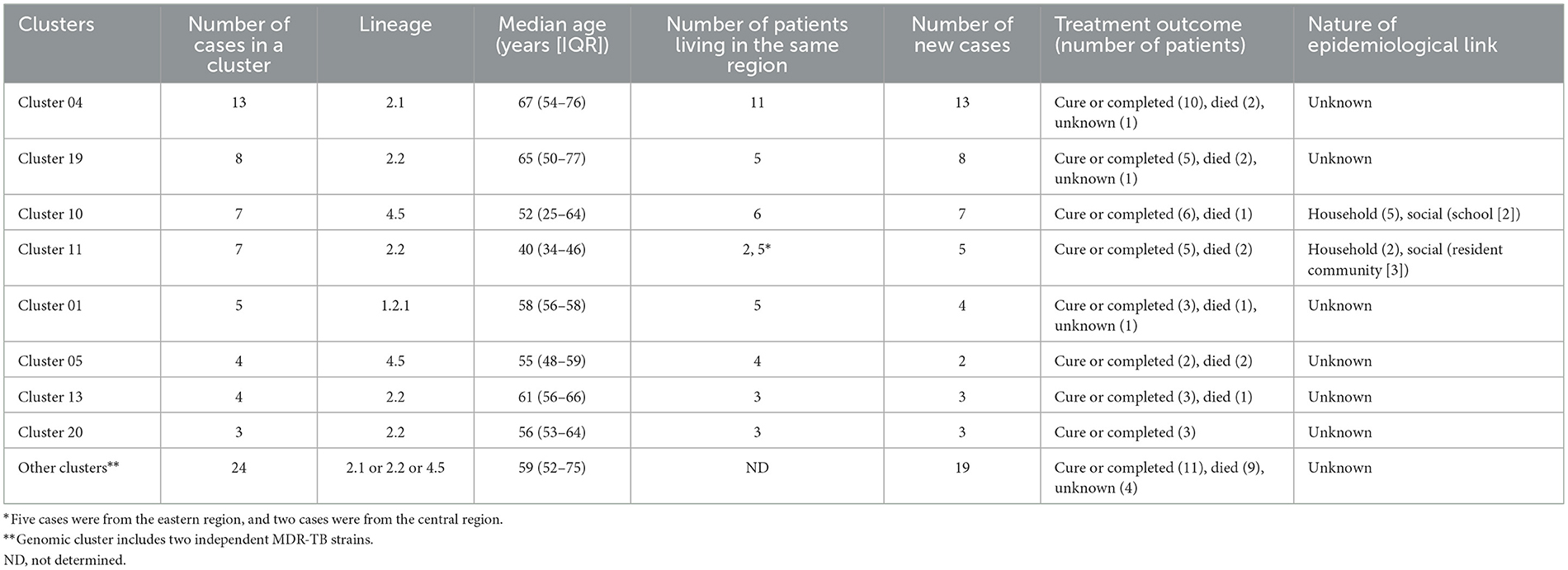
Table 2. Characteristics of MDR-TB clusters based on whole genome sequencing analysis.
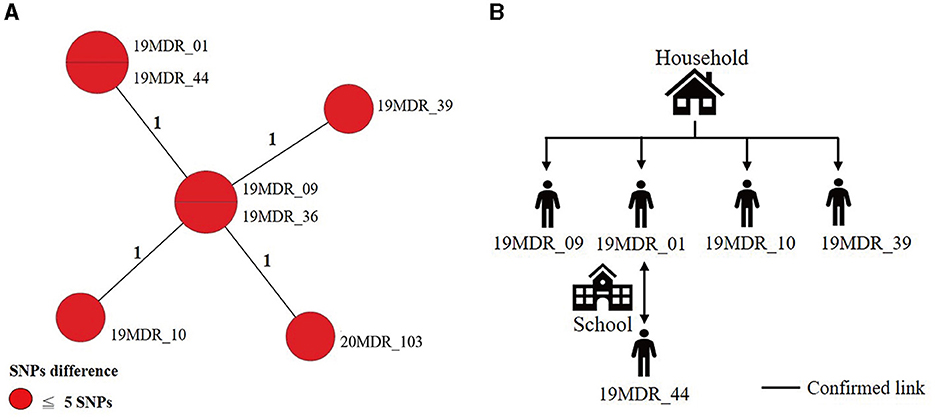
Figure 3. The minimum spanning tree of genomic cluster 10. (A) Minimum spanning tree of genomic cluster 10. (B) Putative transmission network based on epidemiological links. The numbers on the black lines are the difference in the number of SNPs between isolates. The first two digits of the case's identification indicated the year MDR-TB was notified.
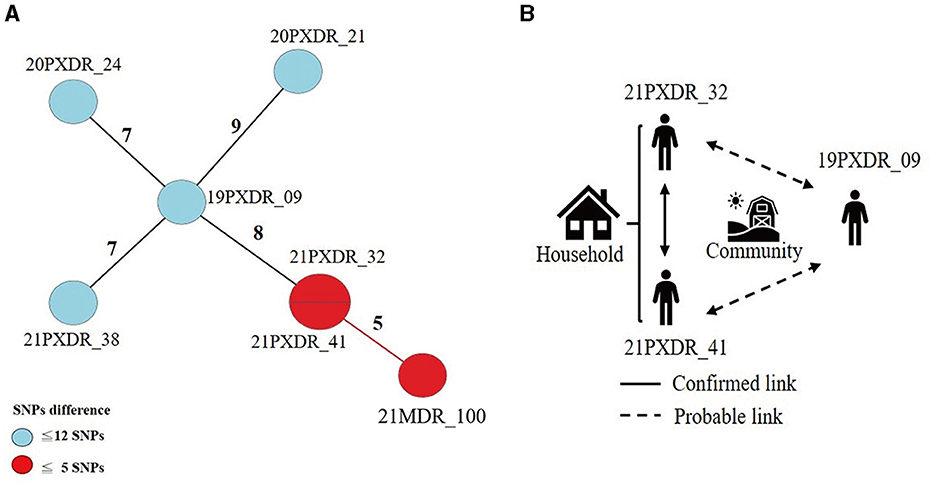
Figure 4. The results of the epidemiology investigation and genomic analysis of cluster 11. (A) Minimum spanning tree of genomic cluster 11 (B) Putative transmission network based on epidemiological links. The numbers on the black and red lines are the difference in the number of SNPs between isolates. The first two digits of the case's identification indicated the year MDR-TB was notified.
We identified putative compensatory mutations in the rpoA, rpoC, or non-RRDR of the rpoB genes in 161 (54.2%) MDR isolates. The most predominant mutation was rpoC V483G (17/161, 10.6%), one of the high-probability compensatory mutations (HCMs) identified by Comas et al. (2011), followed by the rpoC A172V (14/161, 8.7%) and rpoC E750D (14/161, 8.7%) mutations (Supplementary Table 1). The putative compensatory mutation rpoC E750D was not previously reported and was found only among sublineage 2.1 isolates (Supplementary Table 1). We observed that 43.43% (129/297) of M. tuberculosis isolates harbored concurrent compensatory mutations with rpoB S450L mutation. Notably, these mutations were significantly associated with clustering (Table 1). In addition, the rpoC E750D in sublineage 2.1 and the rpoC D485Y/rpoC E1140D in sublineage 2.2 were associated with MDR-TB transmission (Table 3).
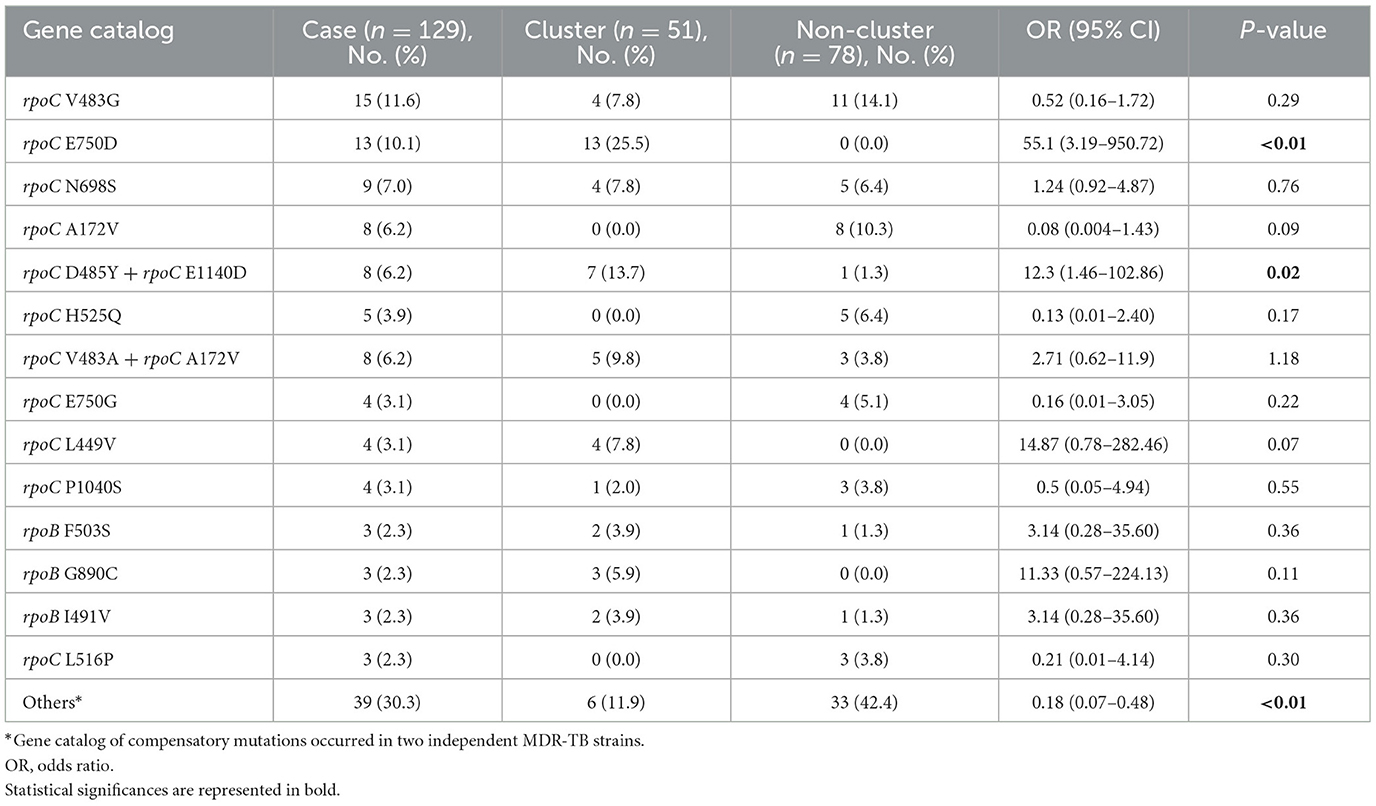
Table 3. Univariable analysis of MDR-TB strains harbored rpoB S450L with compensatory mutations associated with clustering.
To contain and mitigate ~80% of new MDR-TB cases confirmed annually in Taiwan, we applied WGS analysis alongside epidemiological investigation to delineate the putative transmission network and guide public health responses. We found that the MDR-TB clustering rate was 25.3%, consisting of 20 clusters with two outbreaks. In comparison, the clustering rates were 20 and 15% in the US and UK, respectively (Moonan et al., 2013; Anderson et al., 2014). The Beijing genotype was predominant, with 39.5% of the proto-Beijing genotype isolates found in clusters (Table 1). Our results recapitulated that MDR isolates harboring the low fitness cost rpoB S450L mutation and compensatory mutations in the rpoC gene have an impact on MDR-TB transmission (de Vos et al., 2013). Particularly, sublineage 2.1 MDR isolates harboring the rpoB S450L and a compensatory mutation in rpoC E750D had a heightened risk of clustering.
Our results supported previous findings indicating that lineage 2 isolates were the most common in MDR-TB clusters (Yang et al., 2017; Yin et al., 2022; Xiao et al., 2023). Previous studies revealed that lineage 2 isolates displayed greater virulence than other lineages in East Asia. Specifically, lineage 2 isolates induce reduced lower levels of pro-inflammatory cytokines TNF and IL-12p40 and proliferate rapidly within monocyte-derived macrophages (Sarkar et al., 2012; Smith et al., 2022). These characteristics facilitate immune evasion, contributing to the widespread transmission of lineage 2 isolates in densely populated East Asian regions (Borrell and Gagneux, 2009; Chai et al., 2020; Allué-Guardia et al., 2021; Chandra et al., 2022). Notably, we identified 14.5% of isolates as belonging to sublineage 2.1, a prevalence comparable to that in southern China, Guangxi (9.3%), and Hainan Island (27.2%; Liang et al., 2023; Wang et al., 2023), but not in central China, Ningbo (0.9%), or Japan (2.35%; Che et al., 2024; Yokoyama et al., 2010). This suggests that sublineage 2.1 exhibits considerable geographic variability, potentially influenced by local environmental conditions and specific genetic adaptations that may confine its epidemic presence to certain areas. Additionally, research has shown that sublineage 2.1 isolates in MDR or XDR-TB cases have been linked to transmission in Thailand, although the reasons remain unclear (Srilohasin et al., 2020). Investigating host immune responses to sublineage 2.1 strains might further clarify the relationship between geographic distribution and epidemiological patterns.
Our study demonstrated a significant association between rpoC compensatory mutations and MDR-TB clustering. Previous studies have shown that RIF-resistant M. tuberculosis isolates with concurrent compensatory mutations significantly impact increased transmission rates (de Vos et al., 2013; Casali et al., 2014; Li et al., 2016; Merker et al., 2018; Gygli et al., 2021; Goig et al., 2023). A study in South Africa showed that clustered M. tuberculosis isolates had a significantly higher prevalence of rpoC mutants than non-clustered isolates (30.8 vs. 9.4%). Although the association between rpoC gene mutations and MDR-TB dissemination has been hypothesized, the transmission mechanism remains unclear (de Vos et al., 2013). We observed a high-probability compensatory mutation, rpoC V483G, in 10.6% of MDR isolates, which was prevalent in high-burden MDR isolates (Comas et al., 2011). The compensatory mutation in rpoC V483G could restore the significant fitness cost caused by the low fitness rpoB S450L under stringent growth conditions (Song et al., 2014).
When exposed to RIF, MDR isolates carrying the rpoB S450L and F503S mutations demonstrated increased in vitro growth (Zhong et al., 2010). Furthermore, studies have indicated that the presence of the rpoB S450L mutation, along with additional compensatory mutations, is associated with higher transmission rates (Conkle-Gutierrez et al., 2024). A study from Russia further demonstrated that MDR isolates with the rpoB S450L mutation and the compensatory mutation, rpoB E761D, could enhance transmission (Casali et al., 2014).
In addition, Brunner et al. showed that isolates harboring the rpoB S450L mutation along with compensatory mutations promoted lineage and cluster formation during in vitro growth (Brunner et al., 2024). These compensatory mutations likely facilitate the clustering of MDR isolates with rpoB S450L, possibly due to their effects on protein structure. The rpoB mutation might disrupt the structural confirmation and interactions among the β′, β, and α subunits of RNA polymerase, negatively impacting growth and RNA transcription. However, compensatory mutations could reverse these impacts (Gagneux et al., 2006; Brandis et al., 2012; Li et al., 2016; Brunner et al., 2024).
Previous studies suggest that the survival and evolution of MDR isolates may involve epistatic interactions between various drug-resistant and compensatory mutations (Phillips, 2008; Borrell and Gagneux, 2011). The epistatic interactions between specific mutations, such as rpoB S450L and rpoC compensatory mutations, particularly novel and high-confidence mutations, warrant further investigation.
Compensatory mutations could be lineage-specific. The lineage markers, tlyA N236K and rpoC G594E, were identified in lineage 4.6.2 (Cameroon genotype) and lineage 4.1.2 (Haarlem lineage), respectively (Comas et al., 2011; Walker et al., 2018). In our study, all lineage 1.2.1 and 1.2.2 isolates carried the compensatory mutation rpoC A172V, consistent with findings in Indo-Oceanic genotype EAI isolates in India (Comas et al., 2011; Advani et al., 2019). A cluster-specific gene marker has been used in Shigella and enteroinvasive Escherichia coli (EIEC) for epidemiological and diagnostic inquiries (Zhang et al., 2021). Our research showed that compensatory mutations in the rpoC gene could prompt clustering and transmission (Table 3 and Supplementary Table 2), indicating their potential use as epidemiological gene markers or lineage-specific markers to predict putative clusters. Compensatory mutations may serve as prognostic indicators, facilitating epidemic alerts and responses to MDR-TB clustering and outbreaks.
The strength of this study lies in its population-based cohort design, which effectively demonstrates MDR-TB clustering. However, there are some limitations to be considered. First, the lack of detailed epidemiological data hinders the ability to definitively identify true outbreaks among cases within the same cluster. Additionally, the lack of spatial transmission analysis limits our comprehensive understanding of the transmission dynamics. To address this, we plan to employ Geographic Information Systems (GIS) to conduct spatial analysis to enhance molecular epidemiological investigations.
In conclusion, this study provides the first report on the population-based genomic epidemiological analysis of MDR-TB in Taiwan, revealing that 25.3% of cases were clustered. The sublineage 2.1-proto-Beijing genotype MDR isolates were identified as having a high risk of transmission. We found that 54.2% of isolates harbored compensatory mutations in the rpoC gene and non-RRDR regions of the rpoB gene.
Notably, proto-Beijing genotype isolates with concurrent rpoB S450L/rpoC E750D mutations and modern Beijing genotype isolates harboring rpoC D485Y/rpoC E1140D mutations were significantly associated with MDR clusters.
We propose that specific compensatory mutations could serve as epidemiological markers to detect clusters and putative outbreaks. Overall, we modernized and strengthened laboratory services and surveillance by systemically integrating WGS-based analysis with public health investigations to elucidate the genetic basis of MDR-TB clusters.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
K-HL: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft. Y-XX: Data curation, Formal analysis, Software, Writing – review & editing. RJ: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by grants MOHW111-CDC-C-315-133113, MOHW112-CDC-C-315-000105, and MOHW113-CDC-C-315-114112 from the Taiwan Centers for Disease Control, Ministry of Health and Welfare, Taiwan.
The authors would like to thank Hseuh-Chien Hsiao and Hsin-Hua Chan for their technical support.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1439532/full#supplementary-material
Advani, J., Verma, R., Chatterjee, O., Pachouri, P. K., Upadhyay, P., Singh, R., et al. (2019). Whole genome sequencing of Mycobacterium tuberculosis clinical isolates from India reveals genetic heterogeneity and region-specific variations that might affect drug susceptibility. Front. Microbiol. 10:309. doi: 10.3389/fmicb.2019.00309
Allix-Béguec, C., Fauville-Dufaux, M., and Supply, P. (2008). Three-year population-based evaluation of standardized mycobacterial interspersed repetitive-unit-variable-number tandem-repeat typing of Mycobacterium tuberculosis. J. Clin. Microbiol. 46, 1398–1406. doi: 10.1128/JCM.02089-07
Allué-Guardia, A., García, J. I., and Torrelles, J. B. (2021). Evolution of drug-resistant Mycobacterium tuberculosis strains and their adaptation to the human lung environment. Front. Microbiol. 12:612675. doi: 10.3389/fmicb.2021.612675
Anderson, L. F., Tamne, S., Brown, T., Watson, J. P., Mullarkey, C., Zenner, D., et al. (2014). Transmission of multidrug-resistant tuberculosis in the UK: a cross-sectional molecular and epidemiological study of clustering and contact tracing. Lancet Infect. Dis. 14, 406–415. doi: 10.1016/S1473-3099(14)70022-2
Banerjee, R., Allen, J., Westenhouse, J., Oh, P., Elms, W., Desmond, E., et al. (2008). Extensively drug-resistant tuberculosis in California, 1993-2006. Clin. Infect. Dis. 47, 450–457. doi: 10.1086/590009
Barnes, P. F., and Cave, M. D. (2003). Molecular epidemiology of tuberculosis. N. Engl. J. Med. 349, 1149–1156. doi: 10.1056/NEJMra021964
Behzadi, P., and Ranjbar, R. (2019). DNA microarray technology and bioinformatic web services. Acta Microbiol. Immunol. Hung. 66, 19–30. doi: 10.1556/030.65.2018.028
Borrell, S., and Gagneux, S. (2009). Infectiousness, reproductive fitness and evolution of drug-resistant Mycobacterium tuberculosis. Int. J. Tuberc. Lung Dis. 13, 1456–1466.
Borrell, S., and Gagneux, S. (2011). Strain diversity, epistasis and the evolution of drug resistance in Mycobacterium tuberculosis. Clin. Microbiol. Infect. 17, 815–820. doi: 10.1111/j.1469-0691.2011.03556.x
Brandis, G., and Hughes, D. (2013). Genetic characterization of compensatory evolution in strains carrying rpoB Ser531Leu, the rifampicin resistance mutation most frequently found in clinical isolates. J. Antimicrob. Chemother. 68, 2493–2497. doi: 10.1093/jac/dkt224
Brandis, G., Wrande, M., Liljas, L., and Hughes, D. (2012). Fitness-compensatory mutations in rifampicin-resistant RNA polymerase. Mol. Microbiol. 85, 142–151. doi: 10.1111/j.1365-2958.2012.08099.x
Brunner, V. M, and Fowler, P. W. (2024). Compensatory mutations are associated with increased in vitro growth in resistant clinical samples of Mycobacterium tuberculosis. Microb. Genom. 10:001187. doi: 10.1099/mgen.0.001187
Casali, N., Nikolayevskyy, V., Balabanova, Y., Harris, S. R., Ignatyeva, O., Kontsevaya, I., et al. (2014). Evolution and transmission of drug-resistant tuberculosis in a Russian population. Nat. Genet. 46, 279–286. doi: 10.1038/ng.2878
Chai, Q., Wang, L., Liu, C. H., and Ge, B. (2020). New insights into the evasion of host innate immunity by Mycobacterium tuberculosis. Cell Mol. Immunol. 9, 901–913. doi: 10.1038/s41423-020-0502-z
Chandra, P., Grigsby, S. J., and Philips, J. A. (2022). Immune evasion and provocation by Mycobacterium tuberculosis. Nat. Rev. Microbiol. 20, 750–766. doi: 10.1038/s41579-022-00763-4
Che, Y., Li, X., Chen, T., Lu, Y., Sang, G., Gao, J., et al. (2024). Transmission dynamics of drug-resistant tuberculosis in Ningbo, China: an epidemiological and genomic analysis. Front. Cell Infect. Microbiol. 14:1327477. doi: 10.3389/fcimb.2024.1327477
Chen, Y., Liu, Q., Takiff, H. E., and Gao, Q. (2022). Comprehensive genomic analysis of Mycobacterium tuberculosis reveals limited impact of high-fitness genotypes on MDR-TB transmission. J. Infect. 85, 49–56. doi: 10.1016/j.jinf.2022.05.012
Chuang, P. H., Wu, M. H., Fan, S. Y., Lin, K. Y., and Jou, R. (2016). Population-based drug resistance surveillance of multidrug-resistant tuberculosis in Taiwan, 2007-2014. PLoS ONE 11:e0165222. doi: 10.1371/journal.pone.0165222
Cohen, T., and Murray, M. (2004). Modeling epidemics of multidrug-resistant M. tuberculosis of heterogeneous fitness. Nat. Med. 10, 1117–1121. doi: 10.1038/nm1110
Comas, I., Borrell, S., Roetzer, A., Rose, G., Malla, B., Kato-Maeda, M., et al. (2011). Whole-genome sequencing of rifampicin-resistant Mycobacterium tuberculosis strains identifies compensatory mutations in RNA polymerase genes. Nat. Genet. 44, 106–110. doi: 10.1038/ng.1038
Conkle-Gutierrez, D., Ramirez-Busby, S. M., Gorman, B. M., Elghraoui, A., Hoffner, S., Elmaraachli, W., et al. (2024). Novel and reported compensatory mutations in rpoABC genes found in drug resistant tuberculosis outbreaks. Front. Microbiol. 14:1265390. doi: 10.3389/fmicb.2023.1265390
de Vos, M., Muller, B., Borrell, S., Black, P. A., van Helden, P. D., Warren, R. M., et al. (2013). Putative compensatory mutations in the rpoC gene of rifampin-resistant Mycobacterium tuberculosis are associated with ongoing transmission. Antimicrob. Agents Chemother. 57, 827–832. doi: 10.1128/AAC.01541-12
Denholm, J. T., Behr, M. A., de Vries, G., Anthony, R., Robinson, E., Backx, M., et al. (2024). Developing best practice public health standards for whole genome sequencing of Mycobacterium tuberculosis. Lancet Reg. Health West Pac. 46:101014. doi: 10.1016/j.lanwpc.2024.101014
Gagneux, S., Long, C. D., Small, P. M., Van, T., Schoolnik, G. K., and Bohannan, B. J. (2006). The competitive cost of antibiotic resistance in Mycobacterium tuberculosis. Science 312, 1944–1946. doi: 10.1126/science.1124410
Gardy, J. L., Johnston, J. C., Ho Sui, S. J., Cook, V. J., Shah, L., Brodkin, E., et al. (2011). Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N. Engl. J. Med. 364, 730–739. doi: 10.1056/NEJMoa1003176
GBD 2021 Forecasting Collaborators (2024). Burden of disease scenarios for 204 countries and territories, 2022–2050: a forecasting analysis for the Global Burden of Disease Study 2021. Lancet 403, 2204–2256. doi: 10.1016/S0140-6736(24)00685-8
Goig, G. A., Menardo, F., Salaam-Dreyer, Z., Dippenaar, A., Streicher, E. M., Daniels, J., et al. (2023). Effect of compensatory evolution in the emergence and transmission of rifampicin-resistant Mycobacterium tuberculosis in Cape Town, South Africa: a genomic epidemiology study. Lancet Microbe. 4, e506–e515. doi: 10.1016/S2666-5247(23)00110-6
Guthrie, J. L., Delli Pizzi, A., Roth, D., Kong, C., Jorgensen, D., Rodrigues, M., et al. (2018). Genotyping and whole-genome sequencing to identify tuberculosis transmission to pediatric patients in British Columbia, Canada, 2005–2014. J. Infect. Dis. 218, 1155–1163. doi: 10.1093/infdis/jiy278
Guthrie, J. L., Strudwick, L., Roberts, B., Allen, M., McFadzen, J., Roth, D., et al. (2019). Whole genome sequencing for improved understanding of Mycobacterium tuberculosis transmission in a remote circumpolar region. Epidemiol. Infect. 147:e188. doi: 10.1017/S0950268819000670
Gygli, S. M., Loiseau, C., Jugheli, L., Adamia, N., Trauner, A., Reinhard, M., et al. (2021). Prisons as ecological drivers of fitness-compensated multidrug-resistant Mycobacterium tuberculosis. Nat. Med. 27, 1171–1177. doi: 10.1038/s41591-021-01358-x
Lalor, M. K., Casali, N., Walker, T. M., Anderson, L. F., Davidson, J. A., Ratna, N., et al. (2018). The use of whole-genome sequencing in cluster investigation of a multidrug-resistant tuberculosis outbreak. Eur. Respir. J. 51:2017. doi: 10.1183/13993003.02313-2017
Li, Q. J., Jiao, W. W., Yin, Q. Q., Xu, F., Li, J. Q., Sun, L., et al. (2016). Compensatory mutations of rifampin resistance are associated with transmission of multidrug-resistant Mycobacterium tuberculosis Beijing genotype strains in China. Antimicrob. Agents Chemother. 60, 2807–2812. doi: 10.1128/AAC.02358-15
Liang, D., Song, Z., Liang, X., Qin, H., Huang, L., Ye, J., et al. (2023). Whole genomic analysis revealed high genetic diversity and drug-resistant characteristics of Mycobacterium tuberculosis in Guangxi, China. Infect. Drug Resist. 16, 5021–5031. doi: 10.2147/IDR.S410828
Liu, Q., Zuo, T., Xu, P., Jiang, Q., Wu, J., Gan, M., et al. (2018). Have compensatory mutations facilitated the current epidemic of multidrug-resistant tuberculosis? Emerg. Microbes. Infect. 7:98. doi: 10.1038/s41426-018-0101-6
Luo, T., Yang, C., Peng, Y., Lu, L., Sun, G., Wu, J., et al. (2014). Whole-genome sequencing to detect recent transmission of Mycobacterium tuberculosis in settings with a high burden of tuberculosis. Tuberculosis 94, 434–440. doi: 10.1016/j.tube.2014.04.005
Merker, M., Barbier, M., Cox, H., Rasigade, J. P., Feuerriegel, S., Kohl, T. A., et al. (2018). Compensatory evolution drives multidrug-resistant tuberculosis in Central Asia. Elife 7:29. doi: 10.7554/eLife.38200.029
Moonan, P. K., Teeter, L. D., Salcedo, K., Ghosh, S., Ahuja, S. D., Flood, J., et al. (2013). Transmission of multidrug-resistant tuberculosis in the USA: a cross-sectional study. Lancet Infect. Dis. 13, 777–784. doi: 10.1016/S1473-3099(13)70128-2
Nathanson, E., Nunn, P., Uplekar, M., Floyd, K., Jaramillo, E., Lonnroth, K., et al. (2010). MDR tuberculosis–critical steps for prevention and control. N. Engl. J. Med. 363, 1050–1058. doi: 10.1056/NEJMra0908076
Phelan, J. E., O'Sullivan, D. M., Machado, D., Ramos, J., Oppong, Y. E. A., Campino, S., et al. (2019). Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 11:41. doi: 10.1186/s13073-019-0650-x
Phillips, P. C. (2008). Epistasis–the essential role of gene interactions in the structure and evolution of genetic systems. Nat. Rev. Genet. 9, 855–867. doi: 10.1038/nrg2452
Sarkar, R., Lenders, L., Wilkinson, K. A., Wilkinson, R. J., and Nicol, M. P. (2012). Modern lineages of Mycobacterium tuberculosis exhibit lineage-specific patterns of growth and cytokine induction in human monocyte-derived macrophages. PLoS ONE 7:e43170. doi: 10.1371/journal.pone.0043170
Smith, C. M., Baker, R. E., Proulx, M. K., Mishra, B. B., Long, J. E., Park, S. W., et al. (2022). Host-pathogen genetic interactions underlie tuberculosis susceptibility in genetically diverse mice. Elife 11:e74419. doi: 10.7554/eLife.74419
Song, T., Park, Y., Shamputa, I. C., Seo, S., Lee, S. Y., Jeon, H. S., et al. (2014). Fitness costs of rifampicin resistance in Mycobacterium tuberculosis are amplified under conditions of nutrient starvation and compensated by mutation in the β' subunit of RNA polymerase. Mol. Microbiol. 91, 1106–1119. doi: 10.1111/mmi.12520
Srilohasin, P., Prammananan, T., Faksri, K., Phelan, J. E., Suriyaphol, P., Kamolwat, P., et al. (2020). Genomic evidence supporting the clonal expansion of extensively drug-resistant tuberculosis bacteria belonging to a rare proto-Beijing genotype. Emerg. Microbes Infect. 9, 2632–2641. doi: 10.1080/22221751.2020.1852891
van Soolingen, D., Hermans, P. W., de Haas, P. E., Soll, D. R., and van Embden, J. D. (1991). Occurrence and stability of insertion sequences in Mycobacterium tuberculosis complex strains: evaluation of an insertion sequence-dependent DNA polymorphism as a tool in the epidemiology of tuberculosis. J. Clin. Microbiol. 29, 2578–2586. doi: 10.1128/jcm.29.11.2578-2586.1991
Walker, T. M., Merker, M., Knoblauch, A. M., Helbling, P., Schoch, O. D., van der Werf, M. J., et al. (2018). A cluster of multidrug-resistant Mycobacterium tuberculosis among patients arriving in Europe from the Horn of Africa: a molecular epidemiological study. Lancet Infect. Dis. 18, 431–440. doi: 10.1016/S1473-3099(18)30004-5
Wang, J., Yu, C., Xu, Y., Chen, Z., Qiu, W., Chen, S., et al. (2023). Analysis of drug-resistance characteristics and genetic diversity of multidrug-resistant tuberculosis based on whole-genome sequencing on the Hainan Island, China. Infect. Drug Resist. 16, 5783–5798. doi: 10.2147/IDR.S423955
WHO (2018). Technical Manual for Drug Susceptibility Testing of Medicines Used in the Treatment of Tuberculosis. Geneva: WHO.
Woods, G. L., Brown-Elliott, B. A., Conville, P. S., Desmond, E. P., Hall, G. S., Lin, G., et al. (2011). Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes, 2nd Edn. Wayne, PA: Clinical and Laboratory Standards Institute.
Wu, M. H., Hsiao, H. C., Chu, P. W., Chan, H. H., Lo, H. Y., and Jou, R. (2023). Surveillance of multidrug-resistant tuberculosis in Taiwan, 2008–2019. J. Microbiol. Immunol. Infect. 56, 120–129. doi: 10.1016/j.jmii.2022.08.004
Xiao, Y. X., Liu, K. H., Lin, W. H., Chan, T. H., and Jou, R. (2023). Whole-genome sequencing-based analyses of drug-resistant Mycobacterium tuberculosis from Taiwan. Sci. Rep. 13, 2540. doi: 10.1038/s41598-023-29652-3
Yang, C., Luo, T., Shen, X., Wu, J., Gan, M., Xu, P., et al. (2017). Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect. Dis. 17, 275–284. doi: 10.1016/S1473-3099(16)30418-2
Yin, J., Zhang, H., Gao, Z., Jiang, H., Qin, L., Zhu, C., et al. (2022). Transmission of multidrug-resistant tuberculosis in Beijing, China: an epidemiological and genomic analysis. Front. Public Health 10:1019198. doi: 10.3389/fpubh.2022.1019198
Yokoyama, E., Hachisu, Y., Hashimoto, R., and Kishida, K. (2010). Concordance of variable-number tandem repeat (VNTR) and large sequence polymorphism (LSP) analyses of Mycobacterium tuberculosis strains. Infect. Genet. Evol. 10, 913–918. doi: 10.1016/j.meegid.2010.05.013
Zhang, X., Payne, M., Nguyen, T., Kaur, S., and Lan, R. (2021). Cluster-specific gene markers enhance Shigella and enteroinvasive Escherichia coli in silico serotyping. Microb. Genom. 7:704. doi: 10.1099/mgen.0.000704
Keywords: Mycobacterium tuberculosis, tuberculosis, whole genome sequencing, multidrug-resistance, transmission, mutation
Citation: Liu K-H, Xiao Y-X and Jou R (2024) Multidrug-resistant tuberculosis clusters and transmission in Taiwan: a population-based cohort study. Front. Microbiol. 15:1439532. doi: 10.3389/fmicb.2024.1439532
Received: 28 May 2024; Accepted: 27 August 2024;
Published: 18 September 2024.
Edited by:
Abdelazeem Algammal, Suez Canal University, EgyptReviewed by:
Norhan Khairy Abd El-Aziz, Zagazig University, EgyptCopyright © 2024 Liu, Xiao and Jou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ruwen Jou, cndqQGNkYy5nb3YudHc=; cndqMjAwN0BnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.