 Wei-Chin Tsao
Wei-Chin Tsao Yi-Hsuan Li
Yi-Hsuan Li Yi-He Tu1†
Yi-He Tu1† Yu-Shin Nai
Yu-Shin Nai Chih-Li Wang
Chih-Li Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 24 September 2024
Sec. Microbe and Virus Interactions with Plants
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1431813
Moth orchids (Phalaenopsis spp.) are globally popular ornamental flowers. However, effective management strategies for Phalaenopsis leaf yellowing remain elusive, making the disease a challenging obstacle affecting moth orchids at various growth stages. This disease manifests as collar rot, leaf yellowing, leaf abscission, and eventually, plant death. The lack of effective management strategies is likely attributed to a limited understanding of the disease pathogenesis and pathogen dissemination pathways. Fusarium phalaenopsidis sp. nov. was established in this study to stabilize the classification status of Phalaenopsis leaf yellowing pathogens using molecular and morphological features. The genome of the holotype strain was sequenced and assembled, revealing its genome structures. Analyses of virulence-related elements, including transposon elements, secondary metabolite biosynthetic gene clusters, effectors, and secreted carbohydrate-active enzymes, shed light on the potential roles of three fast core chromosomes in virulence. Two species-specific primers were designed based on unique gene sequences of two virulence-related proteins through comparative genomics and BLAST screening. The specificity of these primers was validated using isolates of F. phalaenopsidis, non-target species in the Fusarium solani species complex, other Fusarium species complexes, and saprophytic fungi. These results are intended to accelerate the identification of the pathogens, facilitate the study of disease pathogenesis, and pave the way for elucidating pathogen dissemination pathways. Ultimately, they aim to contribute to the formulation of effective control strategies against Phalaenopsis leaf yellowing.
Moth orchids (Phalaenopsis spp.) are globally recognized as popular ornamental flowers and significant economy elements in Taiwan. The exported value from moth orchids accounts for approximately 70% of the total value of exported flowers. Leaf yellowing disease of Phalaenopsis is a significant challenge to the orchid industry, manifesting at various growth stages (Chung et al., 2011; Liao et al., 2012). Despite intensive fungicide treatments, the disease still emerges, especially under the stresses during ocean freight shipping, leading to considerable economic losses (Liao et al., 2012). The disease often occurs with incidence rates frequently ranging between 30 and 60% in susceptible orchid varieties (Su et al., 2010). Affected orchids exhibit necrotic rot at the leaf collar, resulting in leaf chlorosis, leaf abscission, and ultimately plant death.
Phalaenopsis leaf yellowing is attributed to Fusarium solani f. sp. phalaenopsis. Pathogenicity assays have shown that this pathogen can also affect Cymbidium sp. with mild symptoms but does not impact Oncidium sp., Dendrobium sp., Cattleya sp., Pisum sativum, Chrysanthemum indicum, and Cucumis melo (Chung et al., 2011). Fusarium solani, based on the morphological species concept, is now recognized as the Fusarium solani species complex (FSSC), encompassing multiple phylogenetic clades (Schroers et al., 2016). To enhance taxonomic clarity within the FSSC, Schroers et al. (2016) epitypified the type strain of F. solani sensu stricto within FSSC 5. Subsequently, several new species within the FSSC have been established, including F. stercicola, F. witzenhausenense (Šišić et al., 2018a), F. euwallaceae (Freeman et al., 2013), F. cucurbiticola, F. petroliphilum, and F. phaseoli (Geiser et al., 2021). Given that the phylogenetic relationships between these species and F. solani f. sp. phalaenopsis remain unexplored, it is essential to determine the phylogenetic position of F. solani f. sp. phalaenopsis within the FSSC and establish its taxonomic classification.
Although leaf yellowing disease poses a significant challenge to Phalaenopsis spp. production, effective disease management strategies remain elusive (Liao et al., 2012). This gap in disease management is due to the limited understanding of disease pathogenesis and pathogen dissemination. Notably, despite the economic importance of Fusarium species within the Fusarium solani species complex (FSSC) as agricultural pathogens, research on their virulence factors to plant hosts is sparse (Coleman, 2016). Advancements in whole-genome sequencing technology have uncovered genomes of key FSSC pathogens, including F. vanettenii (Coleman et al., 2009), F. virguliforme associated with soybean SDS (Srivastava et al., 2014), species in ambrosia Fusarium clade (AFC) (Short et al., 2017), and F. solani-melongenae (Xie et al., 2022). This genomic information paves the way for comprehensive molecular research on these pathogens. Therefore, establishing a genome database for the pathogen of Phalaenopsis leaf yellowing is imperative.
Comparative genomics has been utilized to construct genome structures and identify unique genes within a species (Rep and Kistler, 2010; Ma et al., 2013). Fusarium vanettenii 77-13-4 (formerly known as N. haematococca mpVI 77-13-4) was the first species within the Fusarium solani species complex (FSSC) to undergo chromosome-level genome assembly using optical mapping. This assembly revealed the presence of 17 chromosomes, with 3 supernumerary chromosomes (LSCs) identified through pulsed-field gel electrophoresis analysis of various isolates (Coleman et al., 2009). Hence, the well-defined F. vanettenii 77-13-4 genome was often employed to compare with novel genome of Fusarium spp. (Williams et al., 2016; Hoh et al., 2022). These studies revealed core chromosomes (CCs) and lineage-specific chromosomes (LSCs) in Fusarium genomes. The LSCs are often associated with adaptation to diverse environments and implicated in virulence and pathogenicity (Coleman et al., 2009; Ma et al., 2013).
The pathways of pathogen dissemination and the infection progression of Phalaenopsis leaf yellowing remain poorly understood, making it challenging to pinpoint the optimal timing for disease control. To address this knowledge gap, it is imperative to develop and utilize specific primers for pathogen detection, elucidating its dissemination pathways. Whole-genome comparison serves not only to assess genetic similarities among related organisms but also to identify candidate genes of a species for primer design. For instance, Dobbs et al. (2020) developed specific primers for detecting highly virulent strains of F. oxysporum f. sp. koae by comparing genomes of strains with varying virulence levels. In addition, Short et al. (2017) conducted genome comparisons of seven species within the ambrosia Fusarium clade (AFC) to pinpoint specific sequences for primer design, enabling the discrimination and monitoring of AFC symbionts in ambrosia beetles across the United States.
In this investigation, Fusarium phalaenopsidis sp. nov. was established as the pathogen of Phalaenopsis leaf yellowing, supported by pathogenicity assay, phylogenetic analysis, and distinctive morphological characteristics. Through comparative genome analysis, the genomic architecture of this economically significant pathogen was unveiled. By comparing the proteomes of various Fusarium spp., unique genes for the development of specific primers targeting F. phalaenopsidis were identified. The specific primers were subsequently validated with multiple species within the FSSC. The results of the study provide fundamental information and a research tool for future investigations of the significant pathogen. The intended application of these specific primers is to facilitate pathogen surveillance and reveal pathogen dissemination pathways in orchid greenhouses.
Pathogens were isolated from leaf collars and basal stems. Several pieces of tissue (5 × 5 mm) cut from diseased or healthy plants were surface-sterilized in 1% or 3% sodium hypochlorite (NaClO) solution for 1 min, respectively, and then rinsed three times in sterile distilled water for 1 min. After drying, the pieces of tissue were placed on 2% water agar medium (WA) to reduce bacterial growth and incubated at room temperature for 1 week. Subsequently, all isolates were sub-cultured on potato dextrose agar (PDA, Becton Dickinson and Company Difco™) and further purified through single-spore isolation.
The type strain FuZ10s was cultured on PDA and carnation leaf agar (CLA; 2% water agar sprinkled with 5 pieces of propylene oxide-fumigated carnation leaf) for morphological characterization (Leslie and Summerell, 2006). The micromorphology of the pathogen was examined and measured both on CLA and on artificially inoculated leaves of Phalaenopsis Sogo Yukidian “V3”. Colonies on PDA were cultured for 10 days at 25°C under 12/12 h light–dark cycles. Fungal materials were immersed in sterile distilled water and observed and measured using an upright microscope (Olympus BX53) equipped with differential interference contrast (DIC) for microscopic examination and a stereo microscope (Olympus SZX9) for macroscopic examination. The dimensions of each feature were recorded using a digital CMOS camera (E3ISPM Series) and calculated based on measurements from 30 randomly selected samples using Micrometrics SE Premium software (ACCU-SCOPE).
To harvest mycelia, each strain was cultured in 50 mL of yeast extract peptone dextrose broth (YEPD broth; 3 g yeast extract, 10 g peptone, 20 g dextrose/L) in an orbital shaker with 110 rpm at 25°C. After 3 days, the mycelia were collected with miracloth (Merck Millipore). The genomic DNA was extracted with the phenol-chloroform method (Green and Sambrook, 2012) and treated with RNase A (ProTECH, Taiwan) to remove RNA. Subsequently, the genomic DNA was precipitated with isopropanol, washed with 75% ethanol, dried in oven, and then dissolved in sterilized double distilled water.
The extracted DNA was subjected to PCR amplification. Four gene sequences of 19 studied isolates were amplified and used to construct phylogeny to infer organismal phylogeny, including translation elongation factor 1 alpha (TEF1α), internal transcribed spacer (ITS), large subunit of ribosomal ribonucleic acid (LSU), and the second largest subunit of the RNA polymerase II (RPB2). Gene sequences of the reference strains were retrieved from GenBank. To construct phylogeny, DNA sequences of each gene from all studied isolates and reference strains were aligned separately through MAFFT version 7 (Katoh et al., 2019) with default value parameter. Afterward, aligned gene sequences of each isolate were combined in the same order to generate a concatenated four-gene alignment dataset for phylogenetic analysis. Moreover, to enhance the phylogenetic resolution, sequences of three additional genes, calmodulin (CAM), ATP citrate lyase 1 (ACL1), and the largest subunit of the RNA polymerase II (RPB1), were incorporated with the four-gene alignment to generate a seven-gene alignment for phylogenetic analysis. Three isolates from the 19 isolates were selected for this further analysis. The primers used to amplify above genes are listed in Supplementary Table S1.
The phylogeny analyses were conducted through maximum likelihood (ML) (MEGA version 10) (Kumar et al., 2018) and Bayesian inference (BI) (Ronquist et al., 2012) method. The ML phylogeny was constructed with an appropriate model found by the Find Best DNA/Protein Models in MEGA version 10 and set 1,000 bootstraps for analysis. The BI phylogenies were constructed with MrBayes 3.2.7a and performed 1,000,000 times Markov chain Monte Carlo (MCMC). The trees were sampled every 1,000 generation (samplefreq = 1,000). The average standard deviation of slit frequencies of final analysis result was below 0.05, and the beginning 25% of sampled trees were discarded as burn-in.
Each isolate was cultured on PDA for 5 days at 25°C in 12/12 h light–dark cycles and prepared in spore suspension with a concentration of 105 conidia/mL for inoculation. The plant materials used for inoculation were Phalaenopsis Sogo Yukidian “V3”. For inoculating detached leaves, the second and third mature leaves of 3.5-inch pots of “V3” plants were used and cleaned with 0.1% sodium hypochlorite solution. Next, the inoculation sites of leaves were wounded with the sterilized toothpick and dropped with 100 μL of conidium suspension. Subsequently, a 8 × 0.7 mm (diameter x thickness) filter paper disk (ADVANTEC) was covered on each inoculation site for moisture. Sterile water treatment was used as a negative control. The inoculated plants were placed in an airtight plastic box moistened with sterile water in dark at 25°C for 5 days. Leaf collar inoculation was performed on living Phalaenopsis “V3” plants in 2.5-inch pots with the same inoculation procedures described above. Three plants were used for a biological replicate, and three replicates were done.
For whole-genome sequencing, the mycelia of FuZ10s were ground in liquid nitrogen to extract genomic DNA with the phenol-chloroform method (Green and Sambrook, 2012). After assessment of quality with 1.8–2.0 of O.D. 260/280 and ≥ 2.0 of O.D. 260/230, genomic DNA was used to generate sequencing library through NEBNext® Ultra™ DNA Library Prep Kit for Illumina sequencing (NEB, United States). The Illumina NovaSeq 6,000 platform performed the 151 bp paired-end sequencing. For Nanopore sequencing (Oxford Nanopore Technology), the long DNA fragments were selected by BluePippin system (Sage Science, United States). The ends of DNA fragments were added A-tail through NEB Next Ultra II End Repair/dA-tailing Kit (NEB, United States). Subsequently, Qubit® 3.0 Fluorometer (Invitrogen, United States) was employed to assess library fragment sizes, and Nanopore GridION X5 (Oxford Nanopore Technologies, United Kingdom) was applied to conduct sequencing.
The raw reads with mean qscore template <7 were removed, while high quality reads (Phred score > Q7) were selected by longQC v1.2.0 (Fukasawa et al., 2020) to assemble into the contigs performed by Flye v2.8.3 (Kolmogorov et al., 2019). Next, the contigs were polished by Racon v1.4.21 (Vaser et al., 2017) and to calibrate sequencing errors which were removed through Medaka v1.2.51 and Homopolish v0.0.2 (Huang et al., 2021). For optimization of genome completeness, the contigs were polished with Illumina reads through Pilon v1.23 (Walker et al., 2014).
The sequence annotations of Fusarium vanettenii 77-13-4 from NCBI RefSeq and GenBank database were used as reference. Coding DNA sequences of genes were annotated against the reference by BRAKER2 (Brůna et al., 2021). Annotations of transfer RNA and ribosomal RNA were generated by tRNAscan-SE (Chan and Lowe, 2019) and Barrnap, respectively (Seemann, 2013). The functions of genes were searched using phmmer (Potter et al., 2018). The proteins not found in reference proteins were defined as hypothetical proteins. Gene ontology and Pfam domains of the coding DNA sequences were analyzed by InterProScan (Quevillon et al., 2005).
To better understand the genomic information of F. phalaenopsidis FuZ10s, several genomic features were analyzed, including GC content, repeat region density, TE region density, gene density, and locations of secreted proteins, carbohydrate-active enzymes (CAZys), secreted CAZys, and effectors. Fusarium phalaenopsidis FuZ10s (Accession number: PRJNA785279) was compared with five Fusarium spp., namely, Fusarium proliferatum (Accession number: PRJNA576857), Fusarium oxysporum f. sp. lycopersici (Accession number: PRJNA948560), Fusarium solani FSSC 5 MPI-SDFR-AT-0091 (Accession number: PRJNA801211), Fusarium vanettenii 77-13-4 (Accession number: PRJNA51499), and Fusarium ambrosium NRRL 20438 (Accession number: PRJNA389173). To identify the repeat region and transposable element (TE) regions, the tools of repeatModeler2 (Flynn et al., 2020) and RepeatMasker (Chen, 2004) were used. The MUMmer v4.0.0 (Delcher et al., 2003) was used to identify the lineage-specific region (LS region) of F. phalaenopsidis that was further compared the LS regions of the five Fusarium species. All analyzed information was visualized with Circos (Krzywinski et al., 2009). Moreover, a chromosome-level comparison between F. phalaenopsidis FuZ10s and F. vanettenii 77-13-4 was performed by using MUMmer (Delcher et al., 2003). The genome regional linkage among these two species was visualized with Circos.
Signal peptides of proteins were predicted with SignalP 5.0 (Almagro Armenteros et al., 2019), and transmembrane domains were predicted by TMHMM 2.0 (Krogh et al., 2001). Proteins possessing a signal peptide without transmembrane domains were considered as secreted proteins. The secreted proteins smaller than 350 amino acids were further predicted as effectors by EffectorP-fungi 3.0 (Sperschneider and Dodds, 2022). Pathogen–host interaction (PHI) database version 4.1 (Urban et al., 2022) was utilized to predict the putative PHI genes in F. phalaenopsidis FuZ10s genome. The prediction of secondary metabolites was analyzed via antiSMASH 6.0 (Blin et al., 2021).
To obtain specific gene sequences for primer design, the total proteins of F. phalaenopsidis FuZ10s were compared with those of five others Fusarium spp. utilizing OrthoVenn2 (Xu et al., 2019). The five species included Fusarium proliferatum (Accession number: PRJNA576857), Fusarium oxysporum f. sp. lycopersici (Accession number: PRJNA948560), Fusarium solani FSSC 5 MPI-SDFR-AT-0091 (Accession number: PRJNA801211), F. vanettenii 77-13-4 (Accession number: PRJNA51499), and F. ambrosium NRRL 20438 (Accession number: PRJNA389173). Next, these gene sequences of either unique proteins of FuZ10s or proteins which did not be clustered (singletons) by OrthoVenn2 were screened using megablast in NCBI to select gene sequences without hits. These no-hit candidates were further divided based on the presence or absence of signal peptides using SignalP 5.0. The proteins with signal peptides were further predicted to be effector proteins or not using EffectorP 3.0, resulting in two protein groups: effector and non-effector proteins. In addition, the proteins without any signal peptides were further compared to virulence-related proteins in the pathogen–host interactions database (PHI-base) with E-value <104. Afterward, these gene sequences of virulence-related proteins were screened for non-similarity using blastn in NCBI with E-value >10−20. Subsequently, proteins containing specific gene sequences were selected for specific primer design. Primer pairs were designed to amplify amplicons between 400 and 600 bp and were avoided with related sequences of organisms in Fusarium oxysporum species complex (FOSC). The specificity of primer pairs was evaluated by detecting 19 isolates of F. phalaenopsidis, 16 other members of FSSC (three isolates of F. petroliphilum, four isolates of F. solani-melongenae, five isolates of F. falciforme, two isolates of F. solani sensu stricto, and two isolates of F. keratoplasticum), six Fusarium species of non-FSSC (Supplementary Table S2), two orchid pathogens of Phytophthora (Phytophthora nicotiana and Phytophthora palmivora), and four species of saprophytic fungi (Penicillium sp., Aspergillus sp., Trichoderma sp., and Rhizopus sp.) commonly isolated from orchids.
For the PCR reactions, 12.5 μL of KAPA Taq ReadyMIX (KK1024, KAPA Biosystems), 9.5 μL of dH2O, 1 μL of the DNA sample (100 ng/μl), and 1 μL of each primer (10 μM) were mixed. The reaction conditions were as follows: initial denaturation at 95°C for 5 min, followed by 35 cycles of denaturation at 95°C for 30 s, and annealing and extension at 72°C for 30 s. The final extension step was performed at 72°C for 7 min. The annealing temperature was adjusted based on different primer pairs (Supplementary Table S1). The ITS sequences were amplified as DNA positive control. All primers used in this study are listed in Supplementary Table S1.
Sixteen Fusarium isolates obtained from symptomatic Phalaenopsis spp. displaying leaf yellows and three isolates collected from symptomless leaf blades in various locations across Taiwan were chosen for pathogenicity assays (Supplementary Table S2). Since the pathogen did not infect unwounded leaves, the inoculation was conducted on detached Phalaenopsis leaves. The results demonstrated that all 19 isolates induced necrotic symptoms. The leaf collar rot symptoms induced by isolate FuZ10s (= TNM F0036501) resembled those observed in the field affected by Phalaenopsis leaf yellowing (Supplementary Figure S1). To ascertain the taxonomic classification of these isolates within the FSSC, a phylogenetic analysis was performed using concatenated sequences from four genes: ITS (450 bp), TEF1α (458 bp), LSU (467 bp), and RPB2 (1,289 bp). The resulting phylogenetic tree revealed that the 19 isolates formed a distinct, well-supported monophyletic clade with a bootstrap value of 100% and a Bayesian posterior probability of 1 (Supplementary Figure S2). Notably, this clade did not contain isolates from any recognized species and was closely related to the clade representing F. solani sensu stricto (FSSC 5). Moreover, to enhance the phylogenetic resolution, sequences from three additional genes, namely, CAM (361 bp), ACL1 (367 bp), and RPB1 (737 bp), were incorporated for three selected isolates. The resulting analysis indicated that these three isolates formed a cohesive monophyletic group with a bootstrap value of 100% and a Bayesian posterior probability of 1, distinct from any known species, suggesting a novel species within FSSC (Figure 1). Hence, the name Fusarium phalaenopsidis was proposed for this newly identified clade within FSSC, which encompasses isolate cpy01a, previously classified as F. solani f. sp. phalaenopsis (Chung et al., 2011).
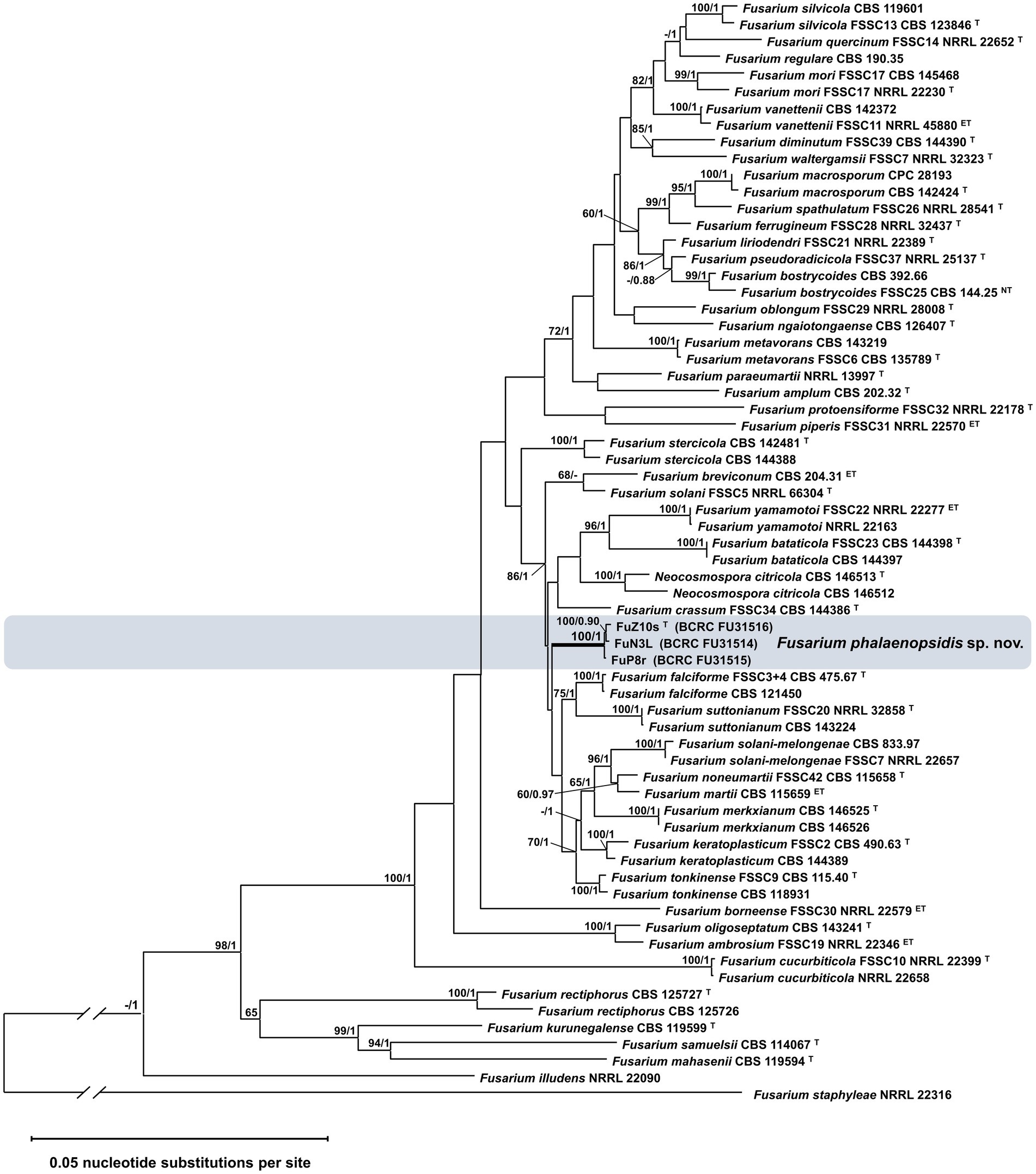
Figure 1. Phylogenetic analysis of Fusarium phalaenopsidis isolates. The phylogenetic tree inferred from concatenated sequences of ITS, TEF1, LSU, caM, acl1, RPB1, and RPB2 was generated by the maximum likelihood (ML) method with the general time reversible model. Values at nodes indicate bootstrap values >60% (1,000 replications) and the Bayesian posterior probability values >0.9. The tree is rooted to Fusarium staphyleae NRRL 22316. T, ex-type strain. ET, ex-epitype strain. The symbol “//” means abbreviation of 1 time of length based on the scale bar.
Fusarium phalaenopsidis C.L. Wang, & W.C. Tsao, sp. nov.
MycoBank: MB849930.
Etymology: Name refers to the former forma specialis.
Typification: Taiwan, Changhua County, from a necrotic leaf sheath with a yellowing leaf of Phalaenopsis sp., 14 July 2018, C. L. Wang, and W.C. Tsao. (holotype TNM F0036501 was deposited at the National Museum of Natural Science, Taiwan; ex-holotype strain BCRC FU31516 was preserved in the Bioresource Collection and Research Center, Taiwan).
Sexual and asexual morphs produced on artificially inoculated leaves of Phalaenopsis Sogo Yukidian “V3”. Perithecia solitary to gregarious, superficial, rarely immersed in the host leaves, sometimes on stroma of sporodochia, orange to red or dark red, globose to obpyriform with a papillate ostiole; wall is rough and warty and composed of textura globulosa to angularis. Asci unitunicate, clavate with truncate apex, 71.8–104.2 × 9.0–11.6 μm (n = 13). Ascospores hyaline to pale brown, fusiform to ellipsoid, 10.4–12.7 × 4.6–6.2 μm (n = 22), 1-septate, distinctly constricted at the septum, often with slightly unequal size of two cells, longitudinally striated surface, obliquely uniseriate or biseriate at the apical one-third to half of asci. Aerial conidiophores mostly simple or branched with terminal monophialides; Phialides long cylindrical, 24.2–53.8 × 2.8–3.8 μm (n = 7), conidiogenous loci with flared collarette and inconspicuous periclinal thickening. Aerial conidia 0- to 1-septate; 0-septate conidia hyaline, ellipsoidal to obovoidal, 8.8–17.3 × 3.3–5.7 μm (n = 46); 1-septate conidia hyaline, ellipsoidal to slightly falcate, 12.5–22.9 × 4.0–6.1 μm (n = 34). Sporodochia white or cream to pale orange, abundant on host leaves, often dry, with stromata-like structure. Sporodochial conidiophores densely penicillate with compact matula, often arise from the stromatic-like cell, terminally bear single or whorl of two to three monophialides. Sporodochial phialides subulate to subcylindrical, 12.0–26.3 × 2.5–4.1 μm (n = 17), conidiogenous loci collarette flared and inconspicuous periclinal thickening. Sporodochial conidia 2- to 5-septate, mostly 4- to 5-septate; 2-septate conidia hyaline, clavate with slightly curved, 21.1–27.4 × 4.8–6.2 μm (n = 8); 3-septate conidia hyaline, clavate and falcate, 22.7–45.4 × 4.1–5.8 μm (n = 39); 4- to 5-septate conidia hyaline, falcate, almost straight in ventral side, slightly curved in dorsiventral side, apex slightly curved to hooked, basal cell papillate to well-developed, 4-septate: 34.9–52.0 × 4.2–6.2 μm (n = 29); 5-septate: 43.0–57.4 × 4.6–6.7 μm (n = 41). Chlamydospores not observed (Figure 2).
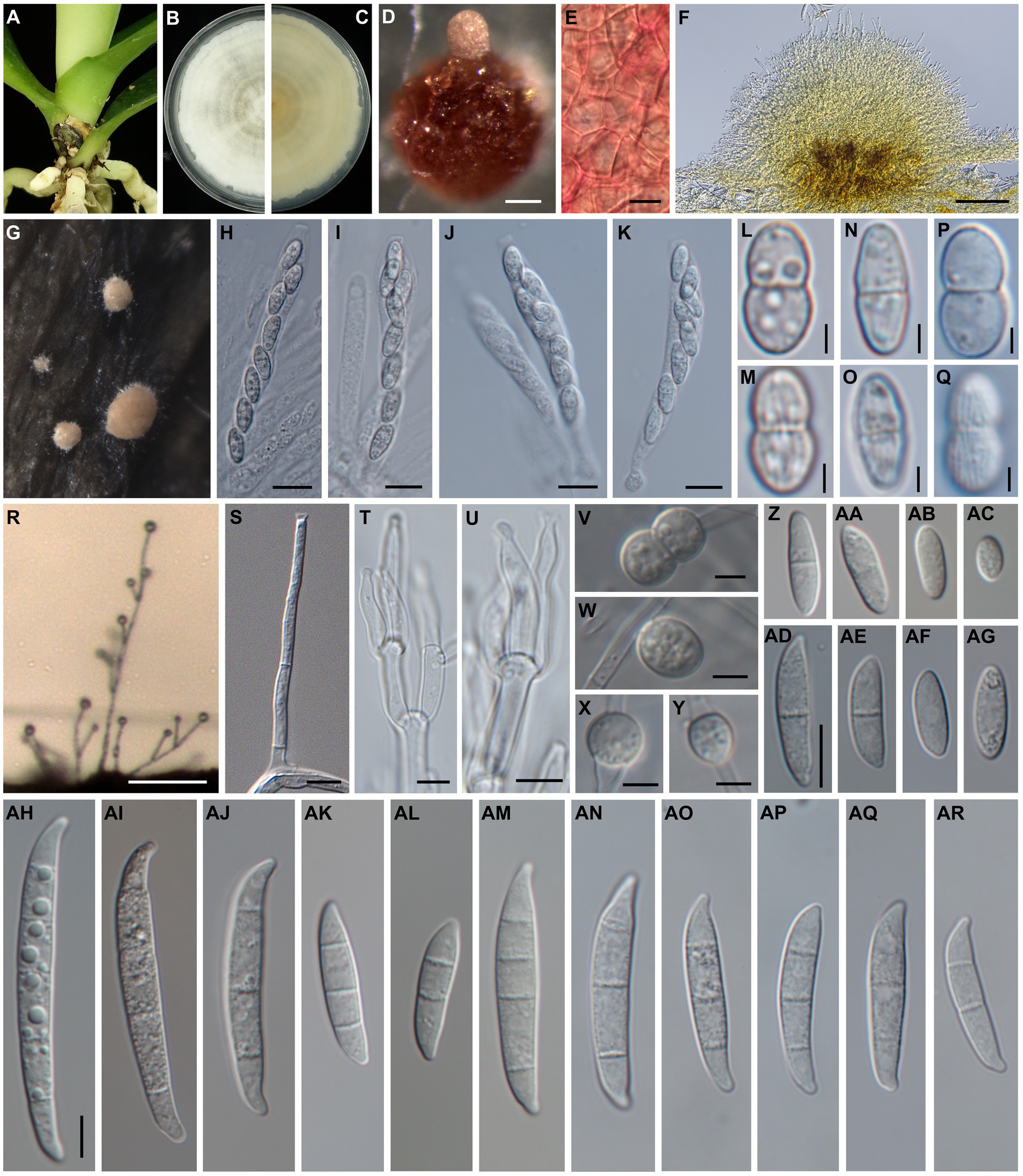
Figure 2. Morphology of Fusarium phalaenopsidis sp. nov. (A) The natural occurring symptoms on Phalaenopsis spp. caused by F. phalaenopsidis. (B,C) The upper and reverse sides of a colony on PDA. (D) A perithecium on CLA. (E) Perithecial wall cells. (F) A sectional view of a sporodochium. (G) Sporodochia on inoculated plants. (H,I) Asci from an inoculated plant. (J,K) Asci from CLA. (L–O) Ascospores from an inoculated plant. (P,Q) Ascospores from CLA. (R) Aerial conidiophores on an inoculated plant. (S) Aerial conidiophores on CLA. (T,U) Sporodochial conidiophores and phialides. (V–Y) Chlamydospores. (Z–AC) Microconidia from an inoculated plant. (AD–AG) Microconidia from CLA. (AH–AL) Sporodochial macroconidia from an inoculated plants. (AM–AR) Aerial macroconidia from CLA. Scale bar: 2.5 μm (L–Q); 5 μm (S–Y); 10 μm (E,H–K,Z–AR); 100 μm (D,F,R). (Z–AG) use same scale bar on (AD); (AH–AR) use same scale bar on (AH).
Sexual and asexual morphs developed on carnation leaf agar. Perithecia solitary to gregarious, superficial, orange to red or dark red, globose to obpyriform with a papillate ostiole, wall is rough and warty and comprise textura globulosa to angularis. Asci unitunicate, clavate, apex flat, 56.7–98.1 × 8.1–11.6 μm (n = 30). Ascospores are hyaline to pale brown, fusiform to ellipsoid, 9.9–13.3 × 4.5–6.7 μm (n = 36), 1-septate, obviously constricted at the septum with slightly unequal sizes of two cells, longitudinally striated surface, obliquely uniseriate or biseriate at apical one-third to half of asci. Aerial conidiophores mostly simple or branched with monophialides; phialides mostly cylindrical or slightly subulate, flared collarette and inconspicuous periclinal thickening, 38.6–79.6 × 2.6–4.2 μm (n = 35). Aerial microconidia 0- to 1- septate, 0-septate hyaline, often ellipsoidal to obovoidal, 6.0–17.7 × 2.5–4.7 μm (n = 53); 1-septate conidia hyaline, ellipsoidal straight curved, 13.8–24.4 × 3.6–5.4 μm (n = 39); macroconidia 2- to 5-septate, mostly 3-septate, hyaline, almost straight and slightly curved at the ends, apex blunt to slightly beaked, basal cell papillate; 2-septate macroconidia: 20.5–31.3 × 4.4–6.2 μm (n = 10), 3-septate macroconidia: 23.8–47.7 × 4.3–6.6 μm (n = 37), 4-septate macroconidia: 34.7–49.0 × 5.2–7.2 μm (n = 17), 5-septate macroconidia: 46.9–52.7 × 6.1–6.9 μm (n = 3). Sporodochia are not observed on CLA. Chlamydospores are hyaline or pale brown, thick-walled, globose to subglobose, single or paired, terminal or intercalary, abundantly produced, 6.3–9.8 × 5.0–9.2 μm (n = 11) (Figure 2).
Colonies achieve diameter 61.8 to 63.1 mm at 7 day on PDA with an average growth rate of 4.4 to 4.5 mm/day at 25°C, 12 h light photoperiods, white to cream, upper side margin entire with circular concentric rings or not, cream to buff in underside.
Although F. phalaenopsidis was placed in a clade of 86% bootstrap value and 1 Bayesian posterior probability that encompasses 14 known species in the FSSC phylogenetic tree (Figure 1), there were some morphological characters distinguishing F. phalaenopsidis from others in this clade. For instance, most of species in this clade did not record sexual stage or maybe heterothallic, except for F. yamamotoi, F. solani-melongenae, F. keratoplasticum (Sandoval-Denis and Crous, 2018; Sandoval-Denis et al., 2019; Crous et al., 2021; Guarnaccia et al., 2021), and F. phalaenopsidis recorded in this study. Among the four species, the sporodochial conidia of F. yamamotoi (6-septate of largest, overall: 34.5–78.5 × 4–6.5 μm) and F. solani-melongenae (9-septate of largest, overall: 28.0–95.5 × 4.5–7.5 μm) revealed longer and more septa than F. phalaenopsidis (5-septate of largest, overall: 21.1–57.4 × 4.8–6.7 μm). Although F. keratoplasticum (overall: 13.2–60.1 × 2.8–8.2 μm) was closed to F. phalaenopsidis in sporodochial conidia, its ecologic niches of animal substrates (Short et al., 2013) were distinct from F. phalaenopsidis. In addition, the two species were phylogenetically distant from each other in this clade. The TEF1α gene sequence of F. phalaenopsidis ex-type strain (BCRC FU31516) was only 93.7% identical to that of F. keratoplasticum (CBS 490.63) with 10 bp polymorphisms, 13 bp insertions, and 4 bp deletions. Notably, F. phalaenopsidis did not generate sporodochia on CLA. Stroma-like structures in the described sporodochia developing on the inoculated Phalaenopsis leaves were a distinct feature as well.
To facilitate future molecular investigations of F. phalaenopsidis, the holotype strain FuZ10s was subjected to whole-genome sequencing. The de novo assembly of the FuZ10s strain genome combined the sequence databases from Nanopore (long-read data) and Illumina (short-read data) sequencing and produced a total length of 52,271,178 bp (52.2 Mb) distributed across 57 contigs, with an N50 length of 3,659,495 bp (3.65 Mb) (Table 1). The BUSCO assessment yielded a score of 98.80%, indicating high quality and completeness of the sequenced FuZ10s genome. In the synteny analysis, the genome of F. phalaenopsidis FuZ10s was compared with the genome of F. vanettenii 77-13-4, which has been well assembled at the chromosome level. This comparison revealed 12 mapped regions that exhibited significant synteny with the 12 CCs of F. vanettenii 77-13-4 (Figure 3). Consequently, these 12 mapped regions were designated as CCs, while the contigs that did not align with the CCs of F. vanettenii 77-13-4 were assigned as a lineage-specific (LS) region of F. phalaenopsidis.
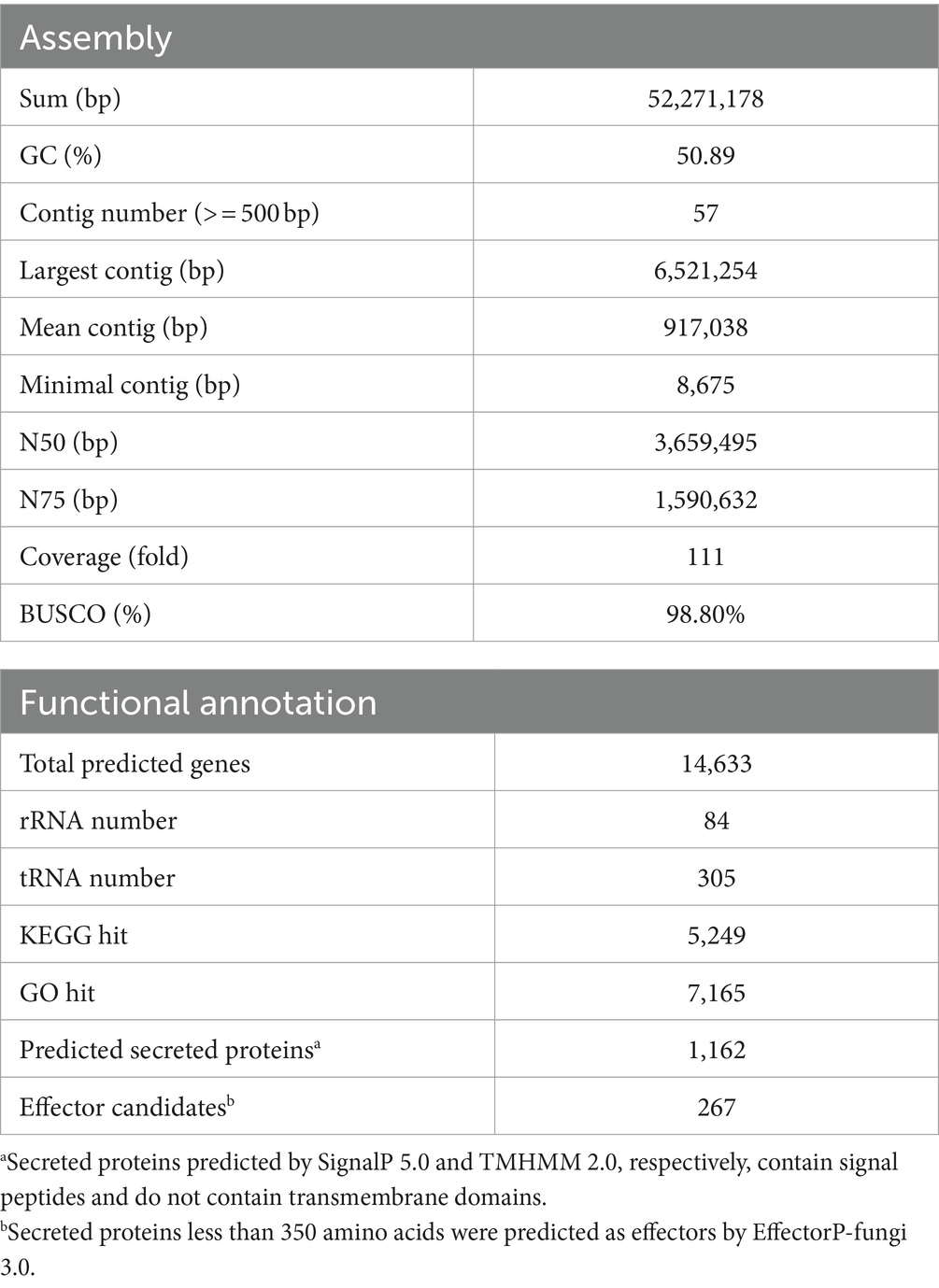
Table 1. Genome de novo assembly and gene functional annotation statistics for Fusarium phalaenopsidis FuZ10s generated with Nanopore sequencing polished with Illumina reads.
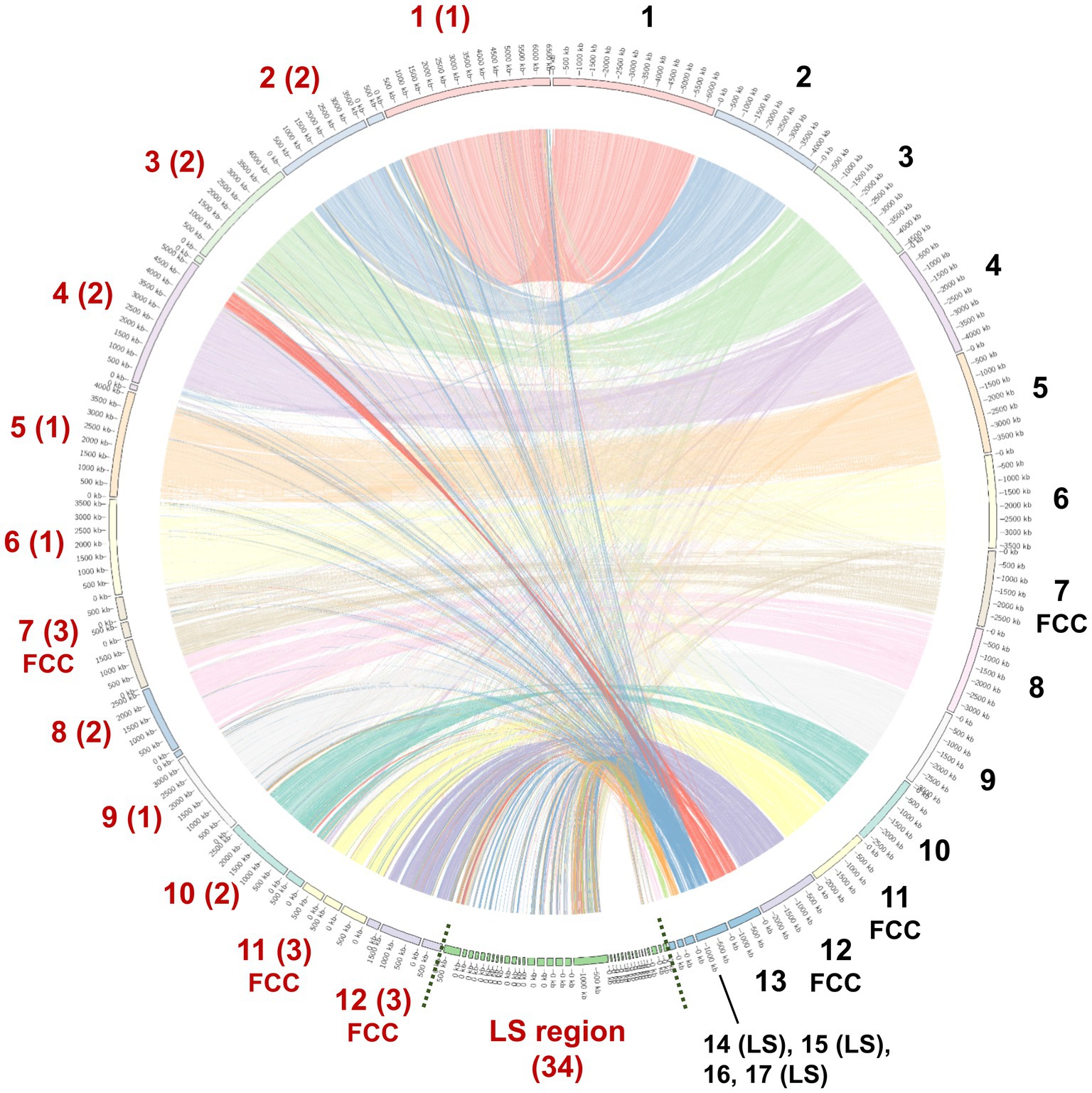
Figure 3. Genome regional linkage map between Fusarium phalaenopsidis FuZ10s (Fp) and Fusarium vanettenii 77-13-4 (Fv). The map is a comparing synteny between Fp and Fv genomes. The red numbers in addition to the map indicated inferred Fp chromosomes and lineage-specific (LS) region, whereas the black numbers indicate Fv chromosomes. Fast core chromosomes (FCCs) and LS region of two genomes are indicated. The numbers in parentheses indicate the contig number comprising each chromosome of F. phalaenopsidis.
Gene prediction identified a total of 14,633 genes in the assembled genome of strain FuZ10s. To delve deeper into genes associated with pathogenesis, analysis was conducted on the 12 CCs and the LS region of F. phalaenopsidis FuZ10s, focusing on secreted proteins, transposon elements, and secondary metabolite biosynthetic gene clusters (Figures 4, 5; Supplementary data sheets S1–S4). Among these, 1,162 genes were predicted to encode secreted proteins based on the presence of signal peptides and the absence of transmembrane domains (Figure 4; Supplementary data sheets S2–S4). In addition, analysis of secreted proteins smaller than 350 amino acids revealed 267 effector candidates of F. phalaenopsidis (Supplementary data sheets S2–S4). Notably, the LS region exhibited a significantly higher abundance of transposon elements compared to other CCs (Figure 5). Chromosomes 6 and 11, along with the LS region, displayed an enrichment of predicted secondary metabolite biosynthetic gene clusters relative to other chromosomes, with the LS region containing all types of predicted secondary metabolite biosynthetic gene clusters. In addition, 12 out of 43 predicted secondary metabolite biosynthetic gene clusters in the genome were predicted to generate known compounds, while the remaining ones were predicted to produce unknown compounds (Supplementary Table S3). Gene ontology analysis of the LS region indicated enrichment for transmembrane transporter activity, DNA and ATP/ADP binding activity, as well as oxidoreductase, monooxygenase, and hydrolase activity among 307 out of 900 total proteins (Supplementary data sheet S5). Moreover, the LS region contained only 16 effector genes (Supplementary data sheet S4), of which 4 effectors with predicted functional domains in Pfam (Sonnhammer et al., 1997) were a PAN domain-containing protein (g6841), a hydrophobin (g13479), a glycosyl hydrolase family 61 (g6084), and a peptidase A4 family (g6210). Furthermore, virulence-related genes, including effectors and secreted carbohydrate-active enzymes (CAZys), were found to be enriched in chromosomes 7, 11, and 12 (Figure 5), being consistent with the characteristics of the “fast core chromosomes” observed in FSSC (Hoh et al., 2022). These secreted proteins encompassed glycosyl hydrolases (GH), necrosis-inducing proteins (NPP), cerato-platanin, and tuberculosis necrotizing toxin (TNT)-like proteins, which could potentially contribute to the necrotic symptoms associated with Phalaenopsis leaf yellowing (Table 2).
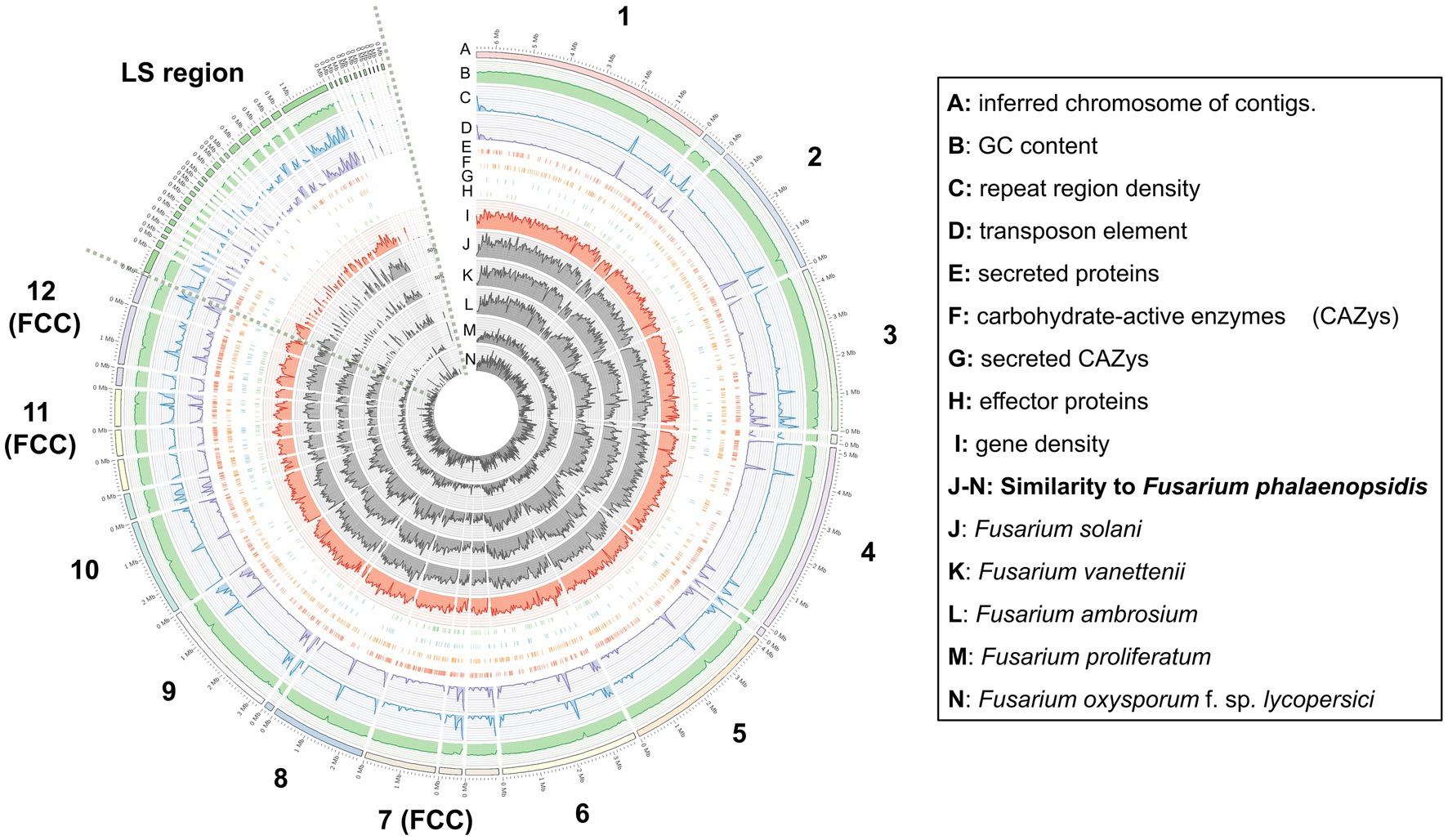
Figure 4. Distribution and abundance of featured elements on each chromosome and lineage-specific region of Fusarium phalaenopsidis. FCC, fast core chromosome; LS region, lineage-specific region.
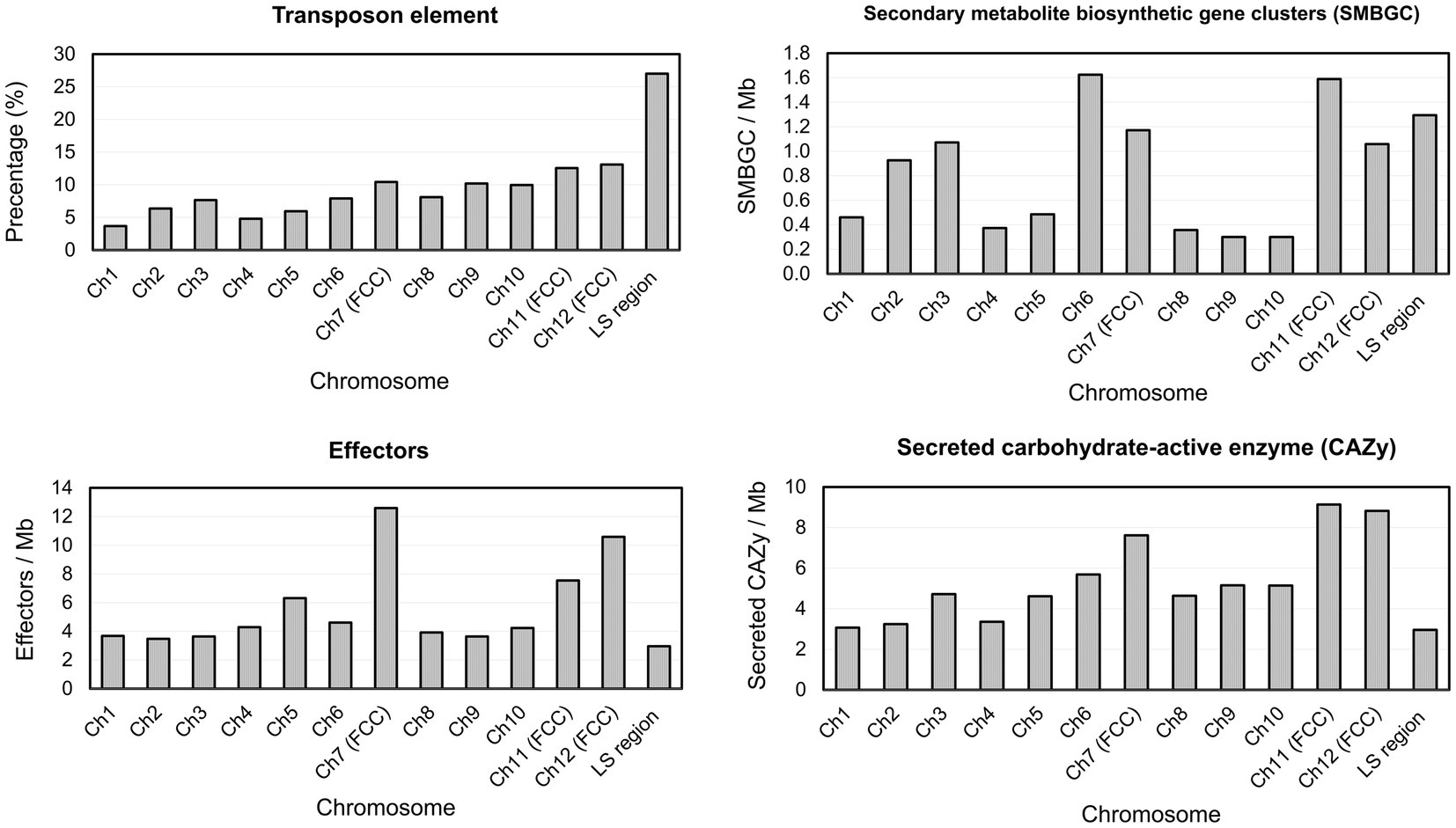
Figure 5. Percentage of transposon element and density of virulence-related genes on each Fusarium phalaenopsidis chromosome. FCC, fast core chromosome; LS region, lineage-specific region.
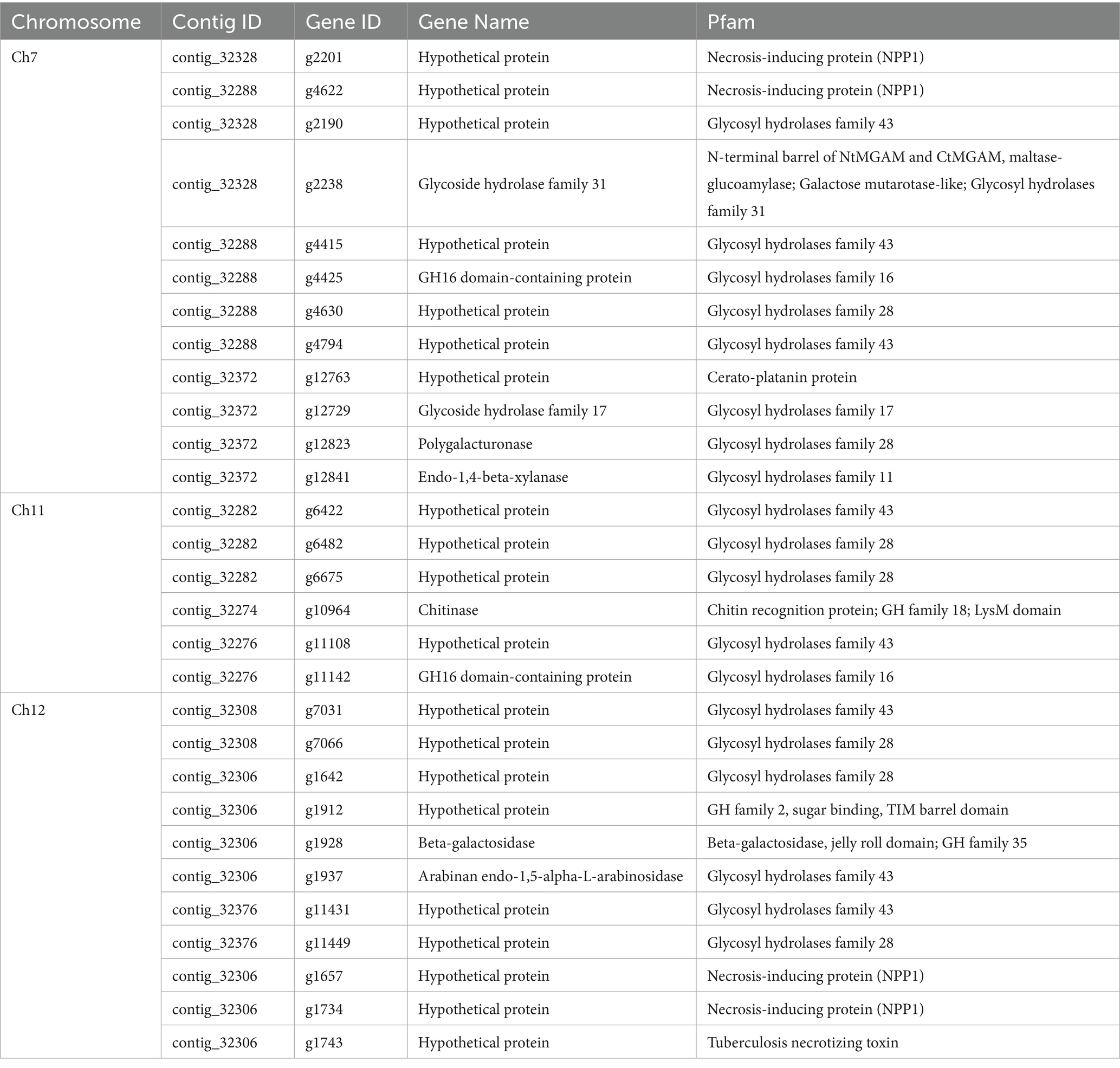
Table 2. Necrosis-related proteins in fast core chromosome (FCC).
To identify candidate proteins for developing specific primers of F. phalaenopsidis, OrthoVenn2 was employed to compare the total proteins of F. phalaenopsidis FuZ10s with those of five Fusarium species, including three species of FSSC (F. ambrosium NRRL 20438, F. vanettenii 77-13-4, and F. solani MPI-SDFR-AT-0091), one species of F. oxysporum species complex (F. oxysporum f. sp. lycopersici 4,287), and one species of F. fujikuroi species complex (F. proliferatum ET1) (Figure 6). This analysis identified 57 proteins in 22 clusters that were unique to FuZ10s among the six species. In addition, 505 singleton proteins were not clustered in OrthoVenn2. Subsequently, a total of the 562 gene sequences of proteins were screened by comparing them with the GenBank database through NCBI megablast (with the setting of “Highly similar sequences” at NCBI), revealing that 150 gene sequences had no matches (no hits) for any similar genes.
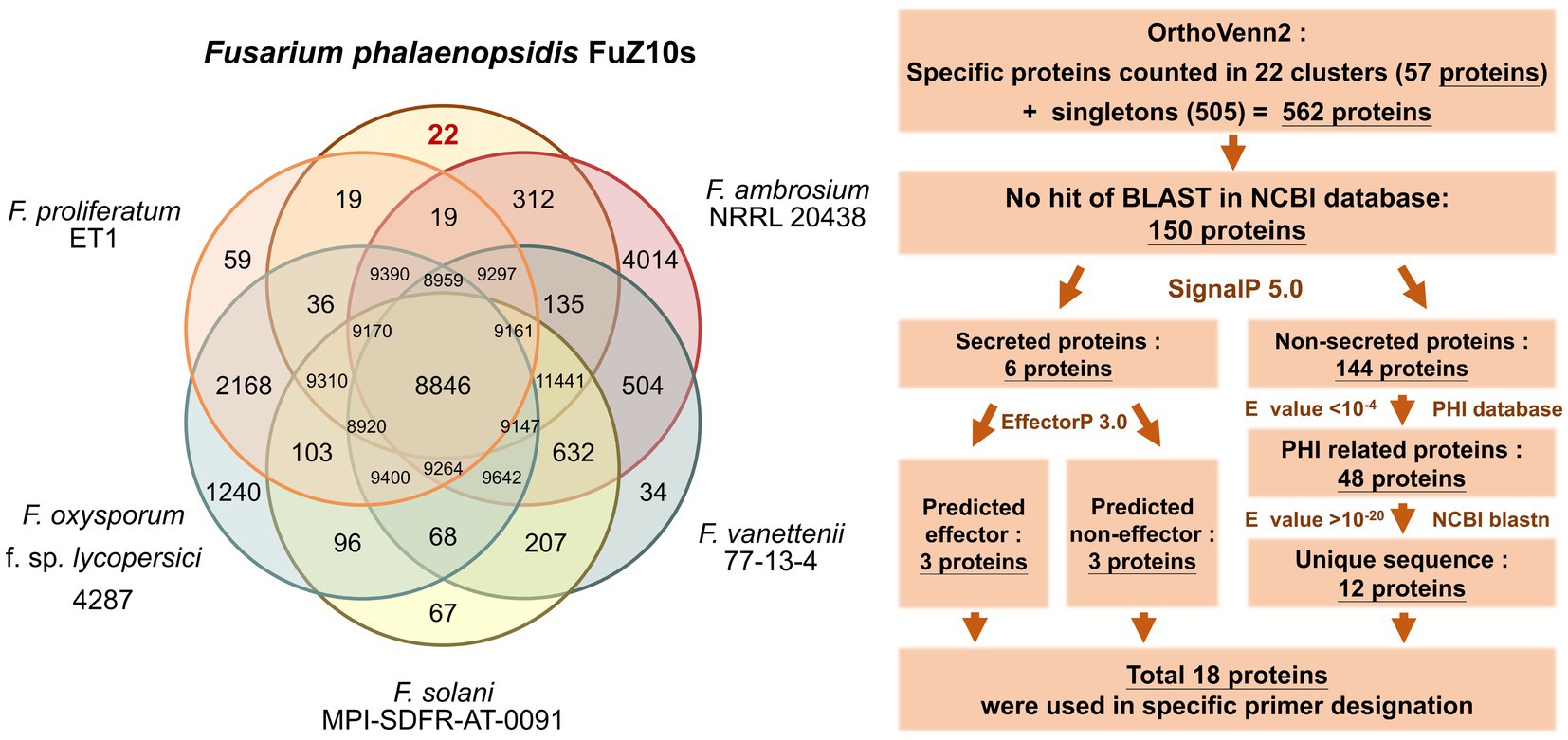
Figure 6. Comparison of orthologues protein clusters and the flowchart of process to screen proteins for specific primer designation.
These 150 proteins were categorized based on the presence or absence of signal peptides. Among them, six proteins, including three predicted effectors and three non-effectors, were predicted to be secreted, while the remaining 144 proteins were not. The 144 proteins were then compared with the PHI-base, revealing 48 virulence-related proteins with E-value <10−4. Further comparison of the gene sequences of these 48 proteins with the GenBank database through the BLASTN program (with the setting of “Somewhat similar sequences” at NCBI) unveiled 12 unique genes of virulence-related proteins with E-value >10−20. Finally, a total of 18 gene sequences of proteins, including the 12 virulence-related proteins and the six secreted proteins, were selected for designing specific primers (Table 3; Supplementary Table S1 and screening process in Supplementary Table S4).
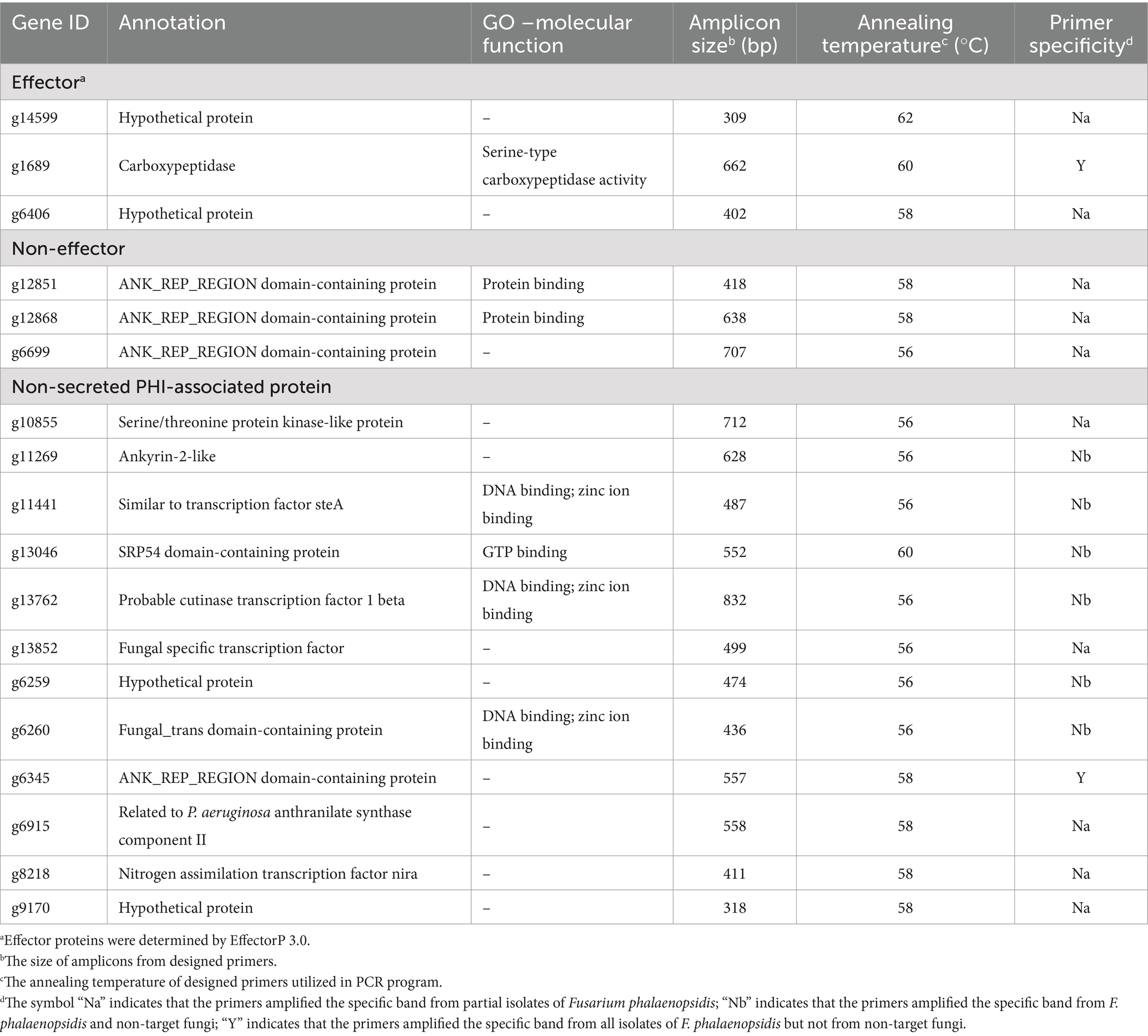
Table 3. Genes of Fusarium phalaenopsidis used in primer design and primer specificity.
Eighteen primer pairs were designed based on the gene sequences of these proteins, with 35 primers targeting the coding region of these gene sequences, while the forward primer of gene g1689 was designed in the non-coding region. An applicability assessment indicated that primer pairs of 10 genes only amplified target bands from partial tested F. phalaenopsidis isolates (Table 3). The remaining eight primer pairs that passed the applicability assessment were further used to assess specificity with a total of 28 non-target species of fungi or fungus-like organisms, including 16 other members of FSSC, six Fusarium species of non-FSSC (Supplementary Table S2; Supplementary Figure S3), two Phytophthora spp., and four species of fungi commonly isolated from Phalaenopsis spp. The results indicated that only two primer pairs designed from genes g1689 and g6345 could amplify specific bands from all F. phalaenopsidis but not from non-target fungi (Table 3; Figure 7; Supplementary Figure S4). These primers were named FphSP1F and FphSP1R for gene g1689 and FphSP6F and FphSP6R for gene g6345. To further assess sensitivity, the two pairs of specific primers were used to amplify F. phalaenopsidis FuZ10s DNA at different concentrations. The results suggested that these primer pairs could amplify specific bands at least at a DNA concentration of 100 pg./μL (Supplementary Figure S5).
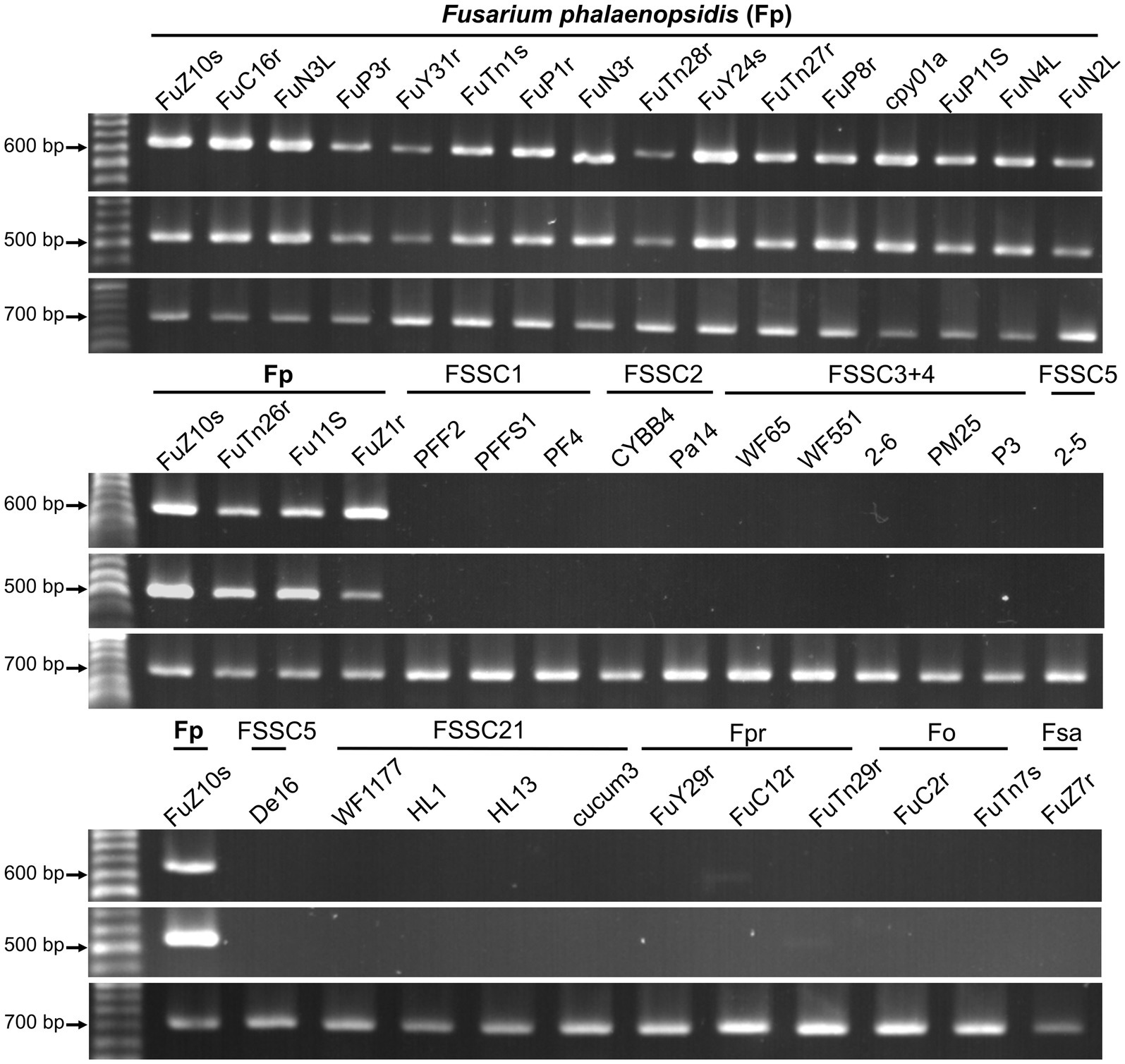
Figure 7. Primer applicability and specificity assays. The primer sets used in assays are FphSP1F/FphSP1R (upper panel) and FphSP6F/FphSP6R (middle panel) for detecting Fusarium phalaenopsidis (Fp) and V9G/ITS4 (lower panel) for verifying quality of DNA from all tested isolates. FSSC1, Fusarium petroliphilum; FSSC2, Fusarium keratoplasticum; FSSC3 + 4, Fusarium falciforme; FSSC5, Fusarium solani; FSSC21, Fusarium solani-melongenae; Fpr, Fusarium proliferatum; Fo, Fusarium oxysporum; Fsa, Fusarium sacchari.
Phalaenopsis leaf yellowing was first reported in Taiwan in 2010 and attributed to F. solani (Su et al., 2010). Later, the pathogen was further recognized as F. solani f. sp. phalaenopsis based on its host specificity to Phalaenopsis (Chung et al., 2011). In 2016, the presence of F. solani f. sp. phalaenopsis in Australia was verified and associated with orchid species of Cymbidum, Cattleya, and Laelia by sequencing the cultures collected in 1996, suggesting a wider host range of the pathogen (Laurence et al., 2016). On the other hand, F. solani isolates identified prior to the study of Schroers et al. (2016) or without comparison with FSSC 5 should be considered as members of F. solani sensus lato, a species complex encompassing isolates in polyphyletic clades. Thus, although F. solani has been reviewed to cause various symptoms on orchids, including Phalaenopsis, Cymbidium, Dendrobium, and Miltonia, in several countries, such as Korea, Japan, Malaysia, and USA (Hawaii) (Srivastava et al., 2018), it is still obscure if those pathogens are F. solani f. sp. phalaenopsis. Moreover, there May be contradictions in the use of “forma specialis,” leading to misunderstandings about the host pathogenicity (Šišić et al., 2018a). For instance, Fusarium solani f. sp. pisi (current name F. vanettenii) is named based on its pathogenicity on Pisum sativum. However, the virulence test on 62 species of legumes indicated that F. vanettenii is capable of infecting 33 species of legumes, implicating that forma specialis system cannot well define the pathogen (Šišić et al., 2018b). Our phylogenetic analysis indicates that all Phalaenopsis leaf yellowing pathogens, including F. solani f. sp. phalaenopsis cpy01a (Chung et al., 2011) (Supplementary Figure S2), were clustered in a monophyletic clade with strongly supported and did not include any known species, indicating that phalaenopsis leaf yellow pathogen is a novel phylogenetic species. To stabilize the nomenclature status of the economic important pathogen, F. phalaenopsidis was introduced as a new species based on the molecular information and morphological features.
Hoh et al. (2022) sequenced six genomes of five FSSC species in chromosome level to illustrate chromosome structures of species in FSSC. Among them, Fusarium sp. Ph1 isolate obtained from tissues of Phalaenopsis leaf yellowing is likely a member of F. phalaenopsidis. The research also explored transcriptomes of F. falciforme and F. keratoplasticum infecting sea turtles, without delving into the F. phalaenopsidis aspect. In the current study, the genome of F. phalaenopsidis holotype strain FuZ10s (BCRC FU31516) was sequenced and further compared with F. vanettenii 77-13-4. The results suggested that F. phalaenopsidis possess 12 core chromosomes and the LS region. Three FCCs 7, 11, and 12 of FSSC were delineated which harbor higher FSSC-specific orthologues when compared with non-FSSC Fusarium species including F. oxysporum, F. graminearum, and F. fujikuroi (Hoh et al., 2022). These FCCs exhibit an enrichment in the number of effectors, CAZys, and SMBGCs (Hoh et al., 2022), corresponding to features observed in F. phalaenopsidis FuZ10s FCCs (Figure 5). Furthermore, some effectors within F. phalaenopsidis FCCs are potential in contributing to the necrotic symptoms of Phalaenopsis leaf yellowing, such as necrosis-inducing proteins (NPP) (g2201, g4622, g1657, and g1734) (Gijzen and Nürnberger, 2006), cerato-platanin proteins (g12763) (Pazzagli et al., 2006), and tuberculosis necrotizing toxin (TNT)-like proteins (g1743) (Sun et al., 2015) (Table 2). Many glyscosyl hydrolases (GHs) in FCCs belong to enzyme families of GH11 (g12841), GH17 (g12729), and GH28 (g4630, g12823, g6482, g6675, g7066, g1642, and g11449) which are likely associated with plant cell wall degradation and virulence (Brito et al., 2006; Have et al., 1998; Liu et al., 2023; Kars et al., 2005). For instance, Magnaporthe oryzae Ebg1, an exo-β-1,3-glucanase belonging to the GH17 family, is crucial for pathogenicity through suppressing plant immunity during the invasive hyphal growth stage within rice cells (Liu et al., 2023). In addition, Botrytis cinerea endopolygalacturonase BcPG1 and BcPG2 belonging to GH28 are required for virulence on tomato (Have et al., 1998; Kars et al., 2005). Botrytis cinerea xylanase BcXyl1 belonging to GH11 is a virulence factor on tomato leaves and grape fruit (Brito et al., 2006). In addition, gene g12763 located on FCC 7 was predicted to encode a cerato-platanin protein that has been implicated as an elicitor, inducing cell death in hosts (Pazzagli et al., 2006) and contributing to virulence in Sclerotinia sclerotiorum (SsCP1), B. cinerea (BcSpl1), and F. oxysporum f. sp. cubense Tropical Race 4 (Frías et al., 2011; Yang et al., 2018; Liu et al., 2019). Moreover, four genes resided on FCC 7 (g2201 and g4622) and FCC 12 (g1657 and g1734) were predicted to encode the NPP1 domain and were considered members of a large protein family capable of inducing cell death in host. They also serve as virulence factors, accelerating disease progression (Gijzen and Nürnberger, 2006). Interestingly, the gene g1743 located on FCC 12 was predicted to encode a tuberculosis necrotizing toxin (TNT) domain-containing protein which acts as necrosis factor of human bacteria pathogen Mycobacterium tuberculosis (Sun et al., 2015). However, the function of TNT in plant fungal pathogens remains largely unknown. Further investigation into the roles of these potential NPPs, GHs, cerato-platanin proteins, and cerato-platanin proteins in the pathogenicity of F. phalaenopsidis is warranted.
In addition to high percentage of the transposon elements, the LS region of F. phalaenopsidis encompasses protein enrichment of monooxygenase activity (25 proteins), oxidoreductase activity (49 proteins), and transmembrane transporter activity (58 proteins) (Supplementary data sheet S5). Research studies revealed that monooxygenases, dehydrogenases, and transmembrane transporters serve as virulence factors in plant fungal pathogen (Aliyu et al., 2019; Coleman, 2016; Enkerli et al., 1998; Klose and Kronstad, 2006; Liu et al., 2017; Roohparvar et al., 2007). For instance, the maackiain detoxification gene 1 (MAK1), which encodes a flavin adenine dinucleotide (FAD)-containing monooxygenase, is located on the LS chromosome 14 of F. vanettenii. The enzyme metabolizes maackiain to a less toxic form, suggesting that detoxification via this monooxygenase is a partial determinant of the pathogen’s ability to infect chickpea (Coleman, 2016; Enkerli et al., 1998). Dehydrogenases constitute a group of oxidoreductase. The short-chain acyl-CoA dehydrogenases MoSCAD1 and MoSCAD2 of Magnaporthe grisea were shown to play roles in stress tolerance, growth, conidiation, and pathogenicity (Aliyu et al., 2019). Ustilago Maydis MFE2 encoding a dehydrogenase protein contributed virulence in corn (Klose and Kronstad, 2006). In addition, major facilitator superfamily (MFS) domain-containing proteins, a group of transmembrane transporters, were largely present in F. phalaenopsidis LS region. The MFS transporters were considered to involve in multidrug resistance. For instance, Mycosphaerella graminicola MgMfs1 attributes tolerance to strobilurin fungicides and cercosporin (Roohparvar et al., 2007). Colletotrichum higginsianum ChMfs1 is involved in the development of primary and secondary hyphae, conidiation, and pathogenicity on Arabidopsis thaliana (Liu et al., 2017). Furthermore, the predicted effectors resided on the LS region of F. phalaenopsidis may play important roles in virulence. For example, the gene g13479 was predicted to encode hydrophobin. A class I hydrophobin encoded by Magnaporthe grisea MPG1 is essential in appressorium formation, conidiation, and is required for pathogenicity on rice (Talbot et al., 1996). In addition, the gene g6210 encodes a protein belonging in peptidase A4 family. Fusarium oxysporum f. sp. lycopersici peptidase FoAPY1 and aspartic protease FolAsp both contribute to virulence on tomato (Qian et al., 2022; Wang et al., 2023). This information suggests that the LS region of F. phalaenopsidis harbors many genes involved in the pathogenesis of Phalaenopsis leaf yellowing.
Previous studies have generated numerous specific primers for identification and detection of species within the FSSC. The majority of these primers are based on conserved housekeeping genes, such as TEF1α (Arif et al., 2012; Costa et al., 2017; Li and Hartman, 2003; Mehl and Epstein, 2007; Villarino et al., 2021) and RPB2 (Agee and Barthet, 2021). Here, whole-genome comparison approach was employed to uncover unique gene sequences of virulence-related proteins in F. phalaenopsidis for specific primer design. This strategy offers several advantages. First, whole-genome information is allowed to discover unique gene sequence and non-coding sequence differentiation among intra-genus species for species-specific primer design. For instance, Vázquez-Rosas-Landa et al. (2021) compared 63 species within FSSC associated with ambrosia beetles to discover genes of unique proteins for the designation of three sets of F. kuroshium-specific primers. In addition, a non-conserved intergenic region obtained via comparing genome of 29 Colletotrichum spp. was used to design a specific primer for C. siamense and C. fructicola on strawberry (Chung et al., 2022). Second, virulence factors and effectors are often targeted for primer design due to their potential in differentiation of host range among pathogens (Lievens et al., 2009). For instance, two secreted in xylem (SIX) effector genes, namely, SIX14 and SIX9, were applied to develop specific primers for identification of F. oxysporum f. sp. rapae and F. oxysporum f. sp. matthiolae, respectively (Chu et al., 2024). Finally, pathogenicity and virulence-related genes involved in prolonged pathogen–host interactions were considered to be absent in saprophytic fungi, making them suitable candidates for the detection of pathogenic fungi.
Currently, disease pathogenesis and pathogen dissemination of Phalaenopsis leaf yellowing is largely unknown, leading to a great limitation in developing disease control strategies. The genome analysis holds promise in characterization of gene functions and paves a way for elucidating potential virulence factors, thereby enhancing our knowledge of the pathogenesis. In comparative genomics, the genome structure of F. phalaenopsidis has been determined, and potential virulence factors were uncovered in three FCCs and the LS region. Moreover, by employing genomic comparisons, we have designed two sets of specific primers for F. phalaenopsidis to accelerate the identification procedures for investigating pathogen dissemination. Coupling with the stabilization of the taxonomic status of the pathogen of Phalaenopsis leaf yellowing, the pathogen can be identified by phylogenetic analysis or specific primers without pathogenicity assay in future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
W-CT: Data curation, Investigation, Writing – original draft, Writing – review & editing, Formal analysis, Software. Y-HL: Data curation, Investigation, Software, Writing – review & editing. Y-HT: Data curation, Formal analysis, Investigation, Writing – review & editing. Y-SN: Data curation, Methodology, Supervision, Writing – review & editing. T-CL: Funding acquisition, Resources, Writing – review & editing. C-LW: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Ministry of Science and Technology, Taiwan (MOST 110-2313-B-005-014-MY3), the Animal and Plant Health Inspection Agency, Ministry of Agriculture, Taiwan (112AS-5.3.4-BQ-B1, 113AS-5.3.4-VP-01), and the Smart Sustainable New Agriculture Research Center (SMARTer), Taiwan (NSTC 112-2634-F-005-002).
Partial data for this publication were provided from the master’s theses of WCT and YHT. The authors also appreciate Dr. Jyh-Nong Tsai in Taiwan Agricultural Research Institute with providing two isolates of Phytophthora nicotiana and P. palmivora. In addition, ChatGPT 3.5 was utilized to improve the manuscript by correcting grammatical errors and editing the draft. Additional edit was made and confirmed by the corresponding author.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1431813/full#supplementary-material
Agee, A., and Barthet, M. (2021). Development of species-specific primers for rapid identification by PCR of the ecologically important pathogen Fusarium keratoplasticum from isolated and environmental samples. Lett. Appl. Microbiol. 73, 607–615. doi: 10.1111/lam.13545
Aliyu, S. R., Lin, L., Chen, X., Abdul, W., Lin, Y., Otieno, F. J., et al. (2019). Disruption of putative short-chain acyl-CoA dehydrogenases compromised free radical scavenging, conidiogenesis, and pathogenesis of Magnaporthe oryzae. Fungal Genet. Biol. 127, 23–34. doi: 10.1016/j.fgb.2019.02.010
Almagro Armenteros, J. J., Tsirigos, K. D., Sønderby, C. K., Petersen, T. N., Winther, O., Brunak, S., et al. (2019). SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat. Biotechnol. 37, 420–423. doi: 10.1038/s41587-019-0036-z
Arif, M., Chawla, S., Zaidi, M., Rayar, J., Variar, M., and Singh, U. (2012). Development of specific primers for genus Fusarium and F. solani using rDNA sub-unit and transcription elongation factor (TEF-1α) gene. Afr. J. Biotechnol. 11, 444–447. doi: 10.5897/AJB10.489
Blin, K., Shaw, S., Kautsar, S. A., Medema, M. H., and Weber, T. (2021). The antiSMASH database version 3: increased taxonomic coverage and new query features for modular enzymes. Nucleic Acids Res. 49, D639–D643. doi: 10.1093/nar/gkaa978
Brito, N., Espino, J. J., and González, C. (2006). The endo-β-1, 4-xylanase Xyn11A is required for virulence in Botrytis cinerea. Mol. Plant-Microbe Interact. 19, 25–32. doi: 10.1094/MPMI-19-0025
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M., and Borodovsky, M. (2021). BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3:lqaa108. doi: 10.1093/nargab/lqaa108
Chan, P. P., and Lowe, T. M. (2019). tRNAscan-SE: searching for tRNA genes in genomic sequences: Methods Mol. Biol. 19, 1–14.
Chen, N. (2004). Using repeat masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics 5, 4–10. doi: 10.1002/0471250953.bi0410s05
Chu, H. H., Tsao, W. C., Huang, J. W., Chang, P. F. L., and Wang, C. L. (2024). Development of specific primers for Fusarium oxysporum formae speciales rapae and matthiolae with an integrated multiplex PCR for distinguishing four formae speciales on Brassicaceae. Plant Dis. 108, 1632–1644. doi: 10.1094/PDIS-08-23-1656-RE
Chung, W. C., Chen, L. W., Huang, J. H., Huang, H. C., and Chung, W. H. (2011). A new “forma specialis” of Fusarium solani causing leaf yellowing of Phalaenopsis. Plant Pathol. 60, 244–252. doi: 10.1111/j.1365-3059.2010.02376.x
Chung, P. C., Wu, H. Y., Chen, Y. C., Hung, T. H., and Chung, C. L. (2022). Development of a nested PCR assay for detecting Colletotrichum siamense and Colletotrichum fructicola on symptomless strawberry plants. PLoS One 17:e0270687. doi: 10.1371/journal.pone.0270687
Coleman, J. J. (2016). The Fusarium solani species complex: ubiquitous pathogens of agricultural importance. Mol. Plant Pathol. 17, 146–158. doi: 10.1111/mpp.12289
Coleman, J. J., Rounsley, S. D., Rodriguez-Carres, M., Kuo, A., Wasmann, C. C., Grimwood, J., et al. (2009). The genome of Nectria haematococca: contribution of supernumerary chromosomes to gene expansion. PLoS Genet. 5:e1000618. doi: 10.1371/journal.pgen.1000618
Costa, S. S., Moreira, G. M., and Pfenning, L. H. (2017). Development of a PCR protocol for the identification and detection of Fusarium solani f. sp. piperis from soil and roots of black pepper (Piper nigrum). Trop. Plant Pathol. 42, 55–59. doi: 10.1007/s40858-016-0124-2
Crous, P. W., Lombard, L., Sandoval-Denis, M., Seifert, K. A., Schroers, H.-J., Chaverri, P., et al. (2021). Fusarium: more than a node or a foot-shaped basal cell. Stud. Mycol. 98:100116. doi: 10.1016/j.simyco.2021.100116
Delcher, A. L., Salzberg, S. L., and Phillippy, A. M. (2003). Using MUMmer to identify similar regions in large sequence sets. Curr. Protoc. Bioinformatics Chapter 10:Unit 10.3. doi: 10.1002/0471250953.bi1003s00
Dobbs, J. T., Kim, M.-S., Dudley, N. S., Klopfenstein, N. B., Yeh, A., Hauff, R. D., et al. (2020). Whole genome analysis of the koa wilt pathogen (Fusarium oxysporum f. sp. koae) and the development of molecular tools for early detection and monitoring. BMC Genomics 21, 764–715. doi: 10.1186/s12864-020-07156-y
Enkerli, J., Bhatt, G., and Covert, S. F. (1998). Maackiain detoxification contributes to the virulence of Nectria haematococca MP VI on chickpea. Mol. Plant-Microbe Interact. 11, 317–326. doi: 10.1094/MPMI.1998.11.4.317
Flynn, J. M., Hubley, R., Goubert, C., Rosen, J., Clark, A. G., Feschotte, C., et al. (2020). RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. USA 117, 9451–9457. doi: 10.1073/pnas.1921046117
Freeman, S., Sharon, M., Maymon, M., Mendel, Z., Protasov, A., Aoki, T., et al. (2013). Fusarium euwallaceae sp. nov.—a symbiotic fungus of Euwallacea sp., an invasive ambrosia beetle in Israel and California. Mycologia 105, 1595–1606. doi: 10.3852/13-066
Frías, M., González, C., and Brito, N. (2011). BcSpl1, a cerato-platanin family protein, contributes to Botrytis cinerea virulence and elicits the hypersensitive response in the host. New Phytol. 192, 483–495. doi: 10.1111/j.1469-8137.2011.03802.x
Fukasawa, Y., Ermini, L., Wang, H., Carty, K., and Cheung, M. S. (2020). LongQC: a quality control tool for third generation sequencing long read data. G3 (Bethesda) 10, 1193–1196. doi: 10.1534/g3.119.400864
Geiser, D. M., Al-Hatmi, A. M., Aoki, T., Arie, T., Balmas, V., Barnes, I., et al. (2021). Phylogenomic analysis of a 55.1-kb 19-gene dataset resolves a monophyletic Fusarium that includes the Fusarium solani species complex. Phytopathology 111, 1064–1079. doi: 10.1094/PHYTO-08-20-0330-LE
Gijzen, M., and Nürnberger, T. (2006). Nep1-like proteins from plant pathogens: recruitment and diversification of the NPP1 domain across taxa. Phytochemistry 67, 1800–1807. doi: 10.1016/j.phytochem.2005.12.008
Green, M. R., and Sambrook, J. (2012). Molecular cloning: a laboratory manual 4th, 448. New York, USA: Cold spring Harbor Laboratory Press.
Guarnaccia, V., Van Niekerk, J., Crous, P., and Sandoval-Denis, M. (2021). Neocosmospora spp. associated with dry root rot of citrus in South Africa. Phytopathol. Mediterr. 60, 79–100. doi: 10.36253/phyto-12183
Have, A. T., Mulder, W., Visser, J., and van Kan, J. A. (1998). The endopolygalacturonase gene Bcpg1 is required for full virulence of Botrytis cinerea. Mol. Plant-Microbe Interact. 11, 1009–1016. doi: 10.1094/MPMI.1998.11.10.1009
Hoh, D. Z., Lee, H. H., Wada, N., Liu, W. A., Lu, M. R., Lai, C. K., et al. (2022). Comparative genomic and transcriptomic analyses of trans-kingdom pathogen Fusarium solani species complex reveal degrees of compartmentalization. BMC Biol. 20:236. doi: 10.1186/s12915-022-01436-7
Huang, Y. T., Liu, P. Y., and Shih, P. W. (2021). Homopolish: a method for the removal of systematic errors in nanopore sequencing by homologous polishing. Genome Biol. 22, 95–17. doi: 10.1186/s13059-021-02282-6
Kars, I., Krooshof, G. H., Wagemakers, L., Joosten, R., Benen, J. A., and Van Kan, J. A. (2005). Necrotizing activity of five Botrytis cinerea endopolygalacturonases produced in Pichia pastoris. Plant J. 43, 213–225. doi: 10.1111/j.1365-313X.2005.02436.x
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief. Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Klose, J., and Kronstad, J. W. (2006). The multifunctional β-oxidation enzyme is required for full symptom development by the biotrophic maize pathogen Ustilago maydis. Eukaryot. Cell 5, 2047–2061. doi: 10.1128/ec.00231-06
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Laurence, M., Howard, C., Summerell, B., and Liew, E. (2016). Identification of Fusarium solani f. sp. phalaenopsis in Australia. Aust Plant Dis Notes 11, 1–3. doi: 10.1007/s13314-016-0200-y
Leslie, J. F., and Summerell, B. A. (2006). The Fusarium laboratory manual. Professional, Iowa, USA: Blackwell Publishing.
Li, S., and Hartman, G. (2003). Molecular detection of Fusarium solani f. sp. glycines in soybean roots and soil. Plant Pathol. 52, 74–83. doi: 10.1046/j.1365-3059.2003.00797.x
Liao, G., Hsieh, T., and Chen, H. (2012). Effect of chemical treatment on prevention of Fusarium yellow of Phalaenopsis seedlings under simulated conditions of shipment. J. Taiwan Agric. Res. 61, 124–131. doi: 10.6156/JTAR/2012.06102.05
Lievens, B., Houterman, P. M., and Rep, M. (2009). Effector gene screening allows unambiguous identification of Fusarium oxysporum f. sp. lycopersici races and discrimination from other formae speciales. FEMS Microbiol. Lett. 300, 201–215. doi: 10.1111/j.1574-6968.2009.01783.x
Liu, H., Lu, X., Li, M., Lun, Z., Yan, X., Yin, C., et al. (2023). Plant immunity suppression by an exo-β-1, 3-glucanase and an elongation factor 1α of the rice blast fungus. Nat. Commun. 14:5491. doi: 10.1038/s41467-023-41175-z
Liu, S., Wu, B., Yang, J., Bi, F., Dong, T., Yang, Q., et al. (2019). A cerato-platanin family protein FocCP1 is essential for the penetration and virulence of Fusarium oxysporum f. sp. cubense tropical race 4. Int. J. Mol. Sci. 20:3785. doi: 10.3390/ijms20153785
Liu, L., Yan, Y., Huang, J., Hsiang, T., Wei, Y., Li, Y., et al. (2017). A novel MFS transporter gene ChMfs1 is important for hyphal morphology, conidiation, and pathogenicity in Colletotrichum higginsianum. Front. Microbiol. 8:1953. doi: 10.3389/fmicb.2017.01953
Ma, L. J., Geiser, D. M., Proctor, R. H., Rooney, A. P., O'Donnell, K., Trail, F., et al. (2013). Fusarium pathogenomics. Ann. Rev. Microbiol. 67, 399–416. doi: 10.1146/annurev-micro-092412-155650
Mehl, H., and Epstein, L. (2007). Identification of Fusarium solani f. sp. cucurbitae race 1 and race 2 with PCR and production of disease-free pumpkin seeds. Plant Dis. 91, 1288–1292. doi: 10.1094/PDIS-91-10-1288
Pazzagli, L., Pantera, B., Carresi, L., Zoppi, C., Pertinhez, T. A., Spisni, A., et al. (2006). Cerato-platanin, the first member of a new fungal protein family: cloning, expression, and characterization. Cell Biochem. Biophys. 44, 512–521. doi: 10.1385/CBB:44:3:512
Potter, S. C., Luciani, A., Eddy, S. R., Park, Y., Lopez, R., and Finn, R. D. (2018). HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204. doi: 10.1093/nar/gky448
Qian, H., Song, L., Wang, L., Wang, B., and Liang, W. (2022). The secreted FoAPY1 peptidase promotes Fusarium oxysporum invasion. Front. Microbiol. 13:1040302. doi: 10.3389/fmicb.2022.1040302
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Rep, M., and Kistler, H. C. (2010). The genomic organization of plant pathogenicity in Fusarium species. Curr. Opin. Plant Biol. 13, 420–426. doi: 10.1016/j.pbi.2010.04.004
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Roohparvar, R., De Waard, M. A., Kema, G. H., and Zwiers, L. H. (2007). MgMfs1, a major facilitator superfamily transporter from the fungal wheat pathogen Mycosphaerella graminicola, is a strong protectant against natural toxic compounds and fungicides. Fungal Genet. Biol. 44, 378–388. doi: 10.1016/j.fgb.2006.09.007
Sandoval-Denis, M., and Crous, P. W. (2018). Removing chaos from confusion: assigning names to common human and animal pathogens in Neocosmospora. Persoonia 41, 109–129. doi: 10.3767/persoonia.2018.41.06
Sandoval-Denis, M., Lombard, L., and Crous, P. W. (2019). Back to the roots: a reappraisal of Neocosmospora. Persoonia 43, 90–185. doi: 10.3767/persoonia.2019.43.04
Schroers, H. J., Samuels, G. J., Zhang, N., Short, D. P., Juba, J., and Geiser, D. M. (2016). Epitypification of Fusisporium (Fusarium) solani and its assignment to a common phylogenetic species in the Fusarium solani species complex. Mycologia 108, 806–819. doi: 10.3852/15-255
Seemann, T. (2013). Barrnap 0.9: rapid ribosomal RNA prediction. Available at: https://github.com/tseemann/barrnap (Accessed September, 2021).
Short, D. P., O’Donnell, K., Stajich, J. E., Hulcr, J., Kijimoto, T., Berger, M. C., et al. (2017). PCR multiplexes discriminate fusarium symbionts of invasive Euwallacea ambrosia beetles that inflict damage on numerous tree species throughout the United States. Plant Dis. 101, 233–240. doi: 10.1094/PDIS-07-16-1046-RE
Short, D. P., O’Donnell, K., Thrane, U., Nielsen, K. F., Zhang, N., Juba, J. H., et al. (2013). Phylogenetic relationships among members of the Fusarium solani species complex in human infections and the descriptions of F. keratoplasticum sp. nov. and F. petroliphilum stat. nov. Fungal Genet. Biol. 53, 59–70. doi: 10.1016/j.fgb.2013.01.004
Šišić, A., Al-Hatmi, A. M., Baćanović-Šišić, J., Ahmed, S. A., Dennenmoser, D., de Hoog, G. S., et al. (2018a). Two new species of the Fusarium solani species complex isolated from compost and hibiscus (Hibiscus sp.). Antonie Van Leeuwenhoek 111, 1785–1805. doi: 10.1007/s10482-018-1068-y
Šišić, A., Baćanović-Šišić, J., Al-Hatmi, A. M., Karlovsky, P., Ahmed, S. A., Maier, W., et al. (2018b). The “forma specialis” issue in Fusarium: A case study in Fusarium solani f. sp. pisi. Sci. Rep. 8:1252. doi: 10.1038/s41598-018-19779-z
Sonnhammer, E. L., Eddy, S. R., and Durbin, R. (1997). Pfam: a comprehensive database of protein domain families based on seed alignments. Proteins 28, 405–420. doi: 10.1002/(sici)1097-0134(199707)28:3<405::aid-prot10>3.0.co;2-l
Sperschneider, J., and Dodds, P. N. (2022). EffectorP 3.0: prediction of apoplastic and cytoplasmic effectors in fungi and oomycetes. Mol. Plant-Microbe Interact. 35, 146–156. doi: 10.1094/MPMI-08-21-0201-R
Srivastava, S. K., Huang, X., Brar, H. K., Fakhoury, A. M., Bluhm, B. H., and Bhattacharyya, M. K. (2014). The genome sequence of the fungal pathogen Fusarium virguliforme that causes sudden death syndrome in soybean. PLoS One 9:e81832. doi: 10.1371/journal.pone.0081832
Srivastava, S., Kadooka, C., and Uchida, J. Y. (2018). Fusarium species as pathogen on orchids. Microbiol. Res. 207, 188–195. doi: 10.1016/j.micres.2017.12.002
Su, J., Lee, Y., Chen, C., Hsieh, T. F., and Huang, J. (2010). Sheath and root rot of Phalaenopsis caused by Fusarium solani. Acta Hortic. 878, 389–394. doi: 10.17660/ActaHortic.2010.878.49
Sun, J., Siroy, A., Lokareddy, R. K., Speer, A., Doornbos, K. S., Cingolani, G., et al. (2015). The tuberculosis necrotizing toxin kills macrophages by hydrolyzing NAD. Nat. Struct. Mol. Biol. 22, 672–678. doi: 10.1038/nsmb.3064
Talbot, N. J., Kershaw, M. J., Wakley, G. E., De Vries, O. M., Wessels, J. G., and Hamer, J. E. (1996). MPG1 encodes a fungal hydrophobin involved in surface interactions during infection-related development of Magnaporthe grisea. Plant Cell 8, 985–999. doi: 10.2307/3870210
Urban, M., Cuzick, A., Seager, J., Wood, V., Rutherford, K., Venkatesh, S., et al. (2022). PHI-base in 2022: A multi-species phenotype database for pathogen-host interactions. Nucleic Acids Res. 50, D837–D847. doi: 10.1093/nar/gkab1037
Vaser, R., Sović, I., Nagarajan, N., and Šikić, M. (2017). Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746. doi: 10.1101/gr.214270.116
Vázquez-Rosas-Landa, M., Sánchez-Rangel, D., Hernández-Domínguez, E. E., Pérez-Torres, C. A., López-Buenfil, A., de Jesús García-Ávila, C., et al. (2021). Design of a diagnostic system based on molecular markers derived from the ascomycetes pan-genome analysis: the case of Fusarium dieback disease. PLoS One 16:e0246079. doi: 10.1371/journal.pone.0246079
Villarino, M., De Cal, A., Melgarejo, P., and Larena, I. (2021). Development of a multiplex PCR for the identification of Fusarium solani and F. oxysporum in a single step. J. Plant Dis. Prot. 128, 1275–1290. doi: 10.1007/s41348-021-00475-6
Walker, B. J., Abeel, T., Shea, T., Priest, M., Abouelliel, A., Sakthikumar, S., et al. (2014). Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. doi: 10.1371/journal.pone.0112963
Wang, C., Zheng, Y., Liu, Z., Qian, Y., Li, Y., Yang, L., et al. (2023). The secreted FolAsp aspartic protease facilitates the virulence of Fusarium oxysporum f. sp. lycopersici. Front. Microbiol. 14:1103418. doi: 10.3389/fmicb.2023.1103418
Williams, A. H., Sharma, M., Thatcher, L. F., Azam, S., Hane, J. K., Sperschneider, J., et al. (2016). Comparative genomics and prediction of conditionally dispensable sequences in legume–infecting Fusarium oxysporum formae speciales facilitates identification of candidate effectors. BMC Genomics 17, 191–124. doi: 10.1186/s12864-016-2486-8
Xie, S. Y., Ma, T., Zhao, N., Zhang, X., Fang, B., and Huang, L. (2022). Whole-genome sequencing and comparative genome analysis of Fusarium solani-melongenae causing fusarium root and stem rot in sweetpotatoes. Microbiol. Spectr. 10, e00683–e00622. doi: 10.1128/spectrum.00683-22
Xu, L., Dong, Z., Fang, L., Luo, Y., Wei, Z., Guo, H., et al. (2019). OrthoVenn2: a web server for whole-genome comparison and annotation of orthologous clusters across multiple species. Nucleic Acids Res. 47, W52–W58. doi: 10.1093/nar/gkz333
Keywords: moth orchid, Fusarium phalaenopsidis, Fusarium solani f. sp. phalaenopsis, phylogeny, comparative genomics, virulence gene, primer
Citation: Tsao W-C, Li Y-H, Tu Y-H, Nai Y-S, Lin T-C and Wang C-L (2024) Identification and molecular detection of the pathogen of Phalaenopsis leaf yellowing through genome analysis. Front. Microbiol. 15:1431813. doi: 10.3389/fmicb.2024.1431813
Edited by:
Ravinder Kumar, Indian Agricultural Research Institute (ICAR), IndiaReviewed by:
Xiao-Ren Chen, Yangzhou University, ChinaCopyright © 2024 Tsao, Li, Tu, Nai, Lin and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chih-Li Wang, Y2x3YW5nQG5jaHUuZWR1LnR3
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.