 Bo Zhang1†
Bo Zhang1† Honglin Ren1†
Honglin Ren1† Xiaoxu Wang2†Cheng Han1Yuanyuan Jin1Xueyu Hu1Ruoran Shi1Chengwei Li1Yuzhu Wang1Yansong Li1Shiying Lu1Zengshan Liu1
Xiaoxu Wang2†Cheng Han1Yuanyuan Jin1Xueyu Hu1Ruoran Shi1Chengwei Li1Yuzhu Wang1Yansong Li1Shiying Lu1Zengshan Liu1 Pan Hu1*
Pan Hu1*- 1State Key Laboratory for Diagnosis and Treatment of Severe Zoonotic Infectious Diseases, Key Laboratory for Zoonosis Research of the Ministry of Education, Institute of Zoonosis, and College of Veterinary Medicine, Jilin University, Changchun, China
- 2Institute of Special Animal and Plant Sciences, Chinese Academy of Agricultural Sciences, Changchun, Jilin, China
As a common foodborne pathogen, infection with L. monocytogenes poses a significant threat to human life and health. The objective of this study was to employ comparative genomics to unveil the biodiversity and evolutionary characteristics of L. monocytogenes strains from different regions, screening for potential target genes and mining novel target genes, thus providing significant reference value for the specific molecular detection and therapeutic targets of L. monocytogenes strains. Pan-genomic analysis revealed that L. monocytogenes from different regions have open genomes, providing a solid genetic basis for adaptation to different environments. These strains contain numerous virulence genes that contribute to their high pathogenicity. They also exhibit relatively high resistance to phosphonic acid, glycopeptide, lincosamide, and peptide antibiotics. The results of mobile genetic elements indicate that, despite being located in different geographical locations, there is a certain degree of similarity in bacterial genome evolution and adaptation to specific environmental pressures. The potential target genes identified through pan-genomics are primarily associated with the fundamental life activities and infection invasion of L. monocytogenes, including known targets such as inlB, which can be utilized for molecular detection and therapeutic purposes. After screening a large number of potential target genes, we further screened them using hub gene selection methods to mining novel target genes. The present study employed eight different hub gene screening methods, ultimately identifying ten highly connected hub genes (bglF_1, davD, menE_1, tilS, dapX, iolC, gshAB, cysG, trpA, and hisC), which play crucial roles in the pathogenesis of L. monocytogenes. The results of pan-genomic analysis showed that L. monocytogenes from different regions exhibit high similarity in bacterial genome evolution. The PCR results demonstrated the excellent specificity of the bglF_1 and davD genes for L. monocytogenes. Therefore, the bglF_1 and davD genes hold promise as specific molecular detection and therapeutic targets for L. monocytogenes strains from different regions.
Introduction
Listeria monocytogenes, a foodborne pathogen, is a Gram-positive rod-shaped bacterium belonging to the genus Listeria within the phylum Firmicutes et Bacillota. It is a facultative anaerobe (Wang et al., 2021; Lourenco et al., 2022). Currently, there are 30 species of Listeria registered in the prokaryotic nomenclature database LPSN (last accessed on November 7, 2023) (Lu et al., 2022). Among the Listeria genus, L. monocytogenes is commonly considered a pathogenic strain and the most prevalent species (Sheng et al., 2017; Cain et al., 2023). It can cause listeriosis in humans, particularly in immunocompromised individuals such as neonates, elderly, pregnant women, and those with weakened immune systems, and can present with various symptoms including mild diarrhea, meningitis, and sepsis (Locatelli et al., 2017; Radoshevich and Cossart, 2018). The pathogenicity of bacteria and their unique ability to adapt to their habitats have a distinctive genetic basis. Numerous relationships between genes and pathogenic phenotypes have been identified and studied in L. monocytogenes (Fox et al., 2016; Disson et al., 2021). However, the genetic basis of L. monocytogenes pathogenicity and environmental adaptability is not fully understood and requires further elucidation.
In recent years, with the widespread application of next-generation sequencing and third-generation sequencing technologies, a large number of L. monocytogenes genomes have been sequenced and shared (Jordan and McAuliffe, 2018). Comparative genomics analysis of L. monocytogenes strains in different regions can deepen our understanding of their genetic mechanisms for adapting to different environments and their pathogenic lifestyles (Lomonaco et al., 2015). The objective of this study was to employ comparative genomics to unveil the biodiversity and evolutionary characteristics of L. monocytogenes strains from different regions, screening for potential target genes and mining novel target genes, thus providing significant reference value for the specific molecular detection and therapeutic targets of L. monocytogenes strains. Therefore, we conducted comparative genomics research on L. monocytogenes strains from different regions including America, Europe, and Asia. The pan-genomes, core genomes, and potential target genes of each L. monocytogenes strain were analyzed, and each strain was subjected to multilocus sequence typing (MLST). Functional analysis of the potential target genes in L. monocytogenes was conducted using GO and KEGG annotations. Furthermore, a protein-protein interaction (PPI) network was constructed for potential target genes of L. monocytogenes, and eight different hub gene analysis methods were utilized to screen novel target genes from the potential target genes. Finally, the virulence genes, antibiotic resistance genes, plasmids, prophages, and CRISPR-Cas systems of each L. monocytogenes strain were investigated.
Materials and methods
Data retrieval and management
In this study, a total of 355 genome sequences were retrieved and downloaded from the NCBI genome database (last accessed on November 7, 2023), including 343 L. monocytogenes strains from three different regions (223 from America, 91 from Europe, and 29 from Asia), as well as 12 other Listeria species and non-Listeria bacterial genomes. Detailed information of the studied L. monocytogenes genomes, such as GenBank accession numbers, strain names, genome size, GC content, number of contigs and N50, are summarized in Supplementary Tables S1, S2 (Palma et al., 2017; Chiaverini et al., 2021). To ensure greater representativeness, L. monocytogenes isolated from cerebrospinal fluid were prioritized, as these strains can induce severe clinical manifestations. In brief, this entails downloading all genomes of L. monocytogenes strains isolated from cerebrospinal fluid in NCBI databases pertaining to America, Europe, and Asia for subsequent analysis. To ensure the specificity of the target genes obtained, non-Listeria bacterial genomes were selected based on their high coverage and homology with Listeria sequences, using Gram-positive reference strains for analysis. Typically, bacteria belonging to the Gram-positive rods exhibit a genomic coverage and homology percentage exceeding 95% (Wang et al., 2022).
Pan-genomic analysis of Listeria monocytogenes and non-target bacterial strains from different regions
The analysis of pan-genomic comparison of L. monocytogenes and non-target strains can be used to screen potential target genes. The potential target genes refer to those genes that are unique to L. monocytogenes strains and are absent in non-target strains (Li et al., 2021a). In brief, all analyzed genome sequences were annotated using Prokka v1.14.6 (Seemann, 2014), and the output results of Prokka were used for pan-genomic analysis with Roary v3.11.2 (Page et al., 2015). A core genome was determined for each isolate using a 99% cutoff, with a BLASTP identity cutoff of 85% (Pang et al., 2019). Genes that matched with all L. monocytogenes strains genome sequences were considered highly conserved and used for subsequent comparisons with other Listeria species and non-Listeria bacterial genomes.
Pan-genome clusters were defined as core-genes: present in all isolates; soft-core genes: present in at least 95% of isolates; shell-genes (accessory genes): present between 15 and 95% of isolates; and cloud-genes (unique genes): present in less than 15% of isolates (Mafuna et al., 2022).
The potential target genes were screened according to the following criteria: 100% presence in L. monocytogenes strains and no presence in non-target bacterial strains. Then, these potential target genes were used screened against the nucleotide collection (nr/nt) databases using the online BLAST program to ensure specificity (Li et al., 2021b).
Multilocus sequence typing analysis
L. monocytogenes was subjected to MLST using 7 housekeeping genes (abcZ, bglA, cat, dapE, dat, ldh, and lhkA) as markers (Henri et al., 2016). MLST profiles were obtained from the Listeria database hosted by the Pasteur Institute, France. The MLST v.2.18.0 was used to align reads against these profiles to determine the sequence types (STs), Clonal Complex values (CC) and lineage for each genome (Mafuna et al., 2022).
Phylogenetic analysis
To investigate the phylogenetic relationships between the 343 L. monocytogenes from different regions, all the core single-copy genes were extracted and aligned using MAFFT v7.490 (Lu et al., 2022). Then, the aligned sequences were concatenated for each strain with a uniform gene order, and GBLOCKS 0.91b was utilized to remove the poorly aligned positions and divergent regions (Lu et al., 2022). MEGA 11 was used to compute the maximum likelihood (ML) phylogenetic tree (Lu et al., 2022). The online tool Interactive Tree of Life (iTOL) v6 was used to visualize the tree with midpoint rooting, and the geographic location, and the ST typing of each strain were annotated on the tree (Lu et al., 2022).
Functional characteristics of potential target genes
In order to investigate the functional characteristics of genes present exclusively in L. monocytogenes strains and absent in non-target bacterial strains (potential target genes), annotation analysis was performed using Gene Ontology enrichment analysis (GO analysis) and Kyoto Encyclopedia of Genes and Genomes enrichment analysis (KEGG analysis) (Gao et al., 2021), and the results were integrated.
Protein-protein interaction network analysis and identification of novel target genes
In this study, the STRING database was utilized to construct PPI networks, and these networks were visualized using Cytoscape v3.10.1 (Li et al., 2021c). The CytoHubba function in Cytoscape v3.10.1 was employed to identify hub genes (novel target genes) from the PPI. The CytoHubba function employs eight distinct algorithms to rank genes in the PPI network, which include Degree, Betweenness, BottleNeck, Closeness, Edge Percolated Component (EPC), Maximum Neighborhood Component (MNC), Radiality, and Stress. The top 10 genes with the highest scores are selected as hub genes (Zhang et al., 2019).
Prediction of virulence factors and antibiotic resistance genes of Listeria monocytogenes
The prediction of virulence factor-related genes and antibiotic resistance genes in the L. monocytogenes genome was conducted to determine their presence. The Virulence Factors of Pathogenic Bacteria (VFDB) database and The Comprehensive Antibiotic Resistance (CARD) database were employed to detect virulence genes and antibiotic resistance genes in the L. monocytogenes genome (Tan et al., 2015; Mafuna et al., 2021), and the results are summarized and presented in a heatmap.
Prediction of MGEs of Listeria monocytogenes
Mobile Genetic Elements (MGEs) refer to a class of genetic elements capable of spreading or transferring within a genome, such as plasmids and prophages, which can facilitate the evolution of microorganisms (Castro et al., 2021). The plasmid database PLSDB and the PHAge Search Tool-Enhanced Release (PHASTER) prophage database were utilized to detect MGEs in the L. monocytogenes genome (Bosi et al., 2017; Matle et al., 2019). The detection results were summarized and presented in a heatmap.
Prediction of CRISPR-Cas systems of Listeria monocytogenes
Predict the genome of L. monocytogenes to determine the presence of the CRISPR-Cas system. CRISPRCasFinder was used for the detection and typing of the Clustered Regularly Interspaced Short Palindromic Repeats and Cas genes (CRISPR-Cas) system of the L. monocytogenes (Parsons et al., 2021). To obtain the functional CRISPR-Cas system, the presence of both the CRISPR sequence and Cas genes was considered as evidence for an actual CRISPR-Cas system and used for the further analysis, and the detection results are summarized and displayed in a heatmap.
Specific primer design and PCR detection conditions for Listeria monocytogenes
Primer design for the sequences of bglF_1 and davD genes was performed using Primer Premier 5 software (Table 1) (He et al., 2022). The primers were synthesized by Sangon Biotech Co., Ltd., Shanghai, China. Primer specificity was tested by PCR analysis of strains from the laboratory collection. Total reaction volume was 25 μL, including 12.5 μL of 2 × Es Taq MasterMix (CWBIO, Beijing, China), 1 μL each of forward and reverse primers (10 μM), 8.5 μL of sterile water, and 2 μL of the purified bacterial genomic DNA as a template. An equal volume of sterile distilled water was used instead of the template as a negative control. PCR thermal cycling involved an initial denaturation step at 95°C for 10 min, followed by 35 cycles of denaturation at 95°C for 30 s, annealing at 56°C for 30 s, and elongation at 72°C for 1 min, with a final elongation at 72°C for 10 min. PCR products were evaluated by 2% agarose electrophoresis.
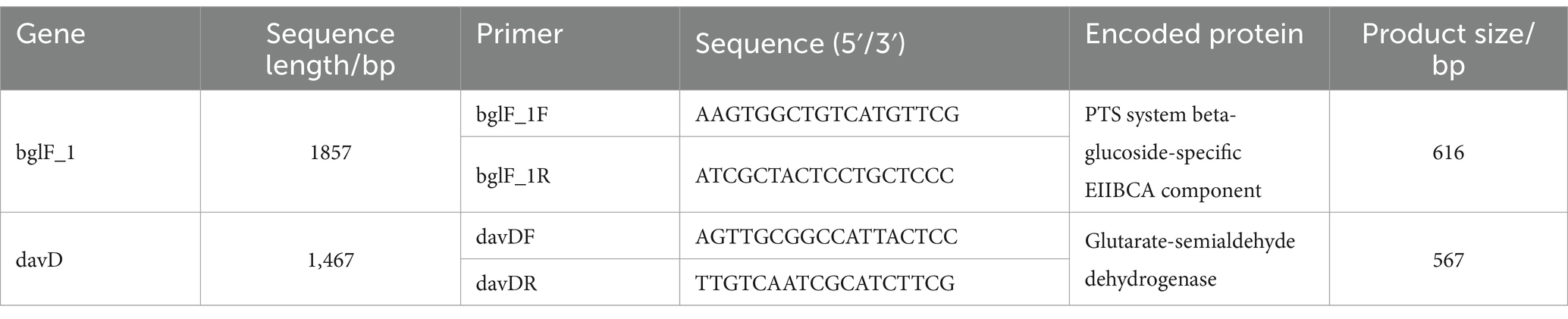
Table 1. Specific target genes and primers used for the detection of L. monocytogenes.
Results
Genome statistics and general features
By querying the NCBI genomic database, we identified 343 strains of L. monocytogenes. We downloaded and curated the whole-genome sequences of these L. monocytogenes strains from the NCBI genomic database, along with the corresponding information (Supplementary Tables S1, S2). Among them, there were 223 isolates from America, with an average genome size of 2.98 (2.8–3.2) Mbp, an average GC content of 37.98 (37.5–38.0)%, the number of contigs ≤214 and an average N50 of 334,248. In Europe, there were 91 L. monocytogenes isolates, with an average genome size of 2.96 (2.9–3.2) Mbp, an average GC content of 37.98 (37.5–38.0)%, the number of contigs ≤42 and an average N50 of 656,340. In Asia, there were 29 L. monocytogenes isolates, among which, one possesses a complete genome, with an average genome size of 2.96 (2.8–3.1) Mbp, an average GC content of 38%, the number of contigs ≤72 and an average N50 of 699,124.
Pan-genomic analysis of Listeria monocytogenes strains in different regions
Based on pan-genomic classification, the analysis of L. monocytogenes strains from different regions revealed the following gene distribution within the pan-genome: there were 1847 (15%) core genes, 314 (2.6%) soft-core genes, 1,237 (10.1%) shell genes, and 8,860 (72.3%) cloud genes (Figure 1A). The pan-genomic composition of L. monocytogenes varies across different regions. In America, L. monocytogenes strains possess 1866 core genes, 149 soft-core genes, 1,351 shell genes, and 7,714 cloud genes. In Europe, L. monocytogenes strains possess 2,133 core genes, 103 soft-core genes, 1,267 shell genes, and 3,782 cloud genes. In Asia, L. monocytogenes strains possess 2,178 core genes, 16 soft-core genes, 1,306 shell genes, and 1,649 cloud genes (Figures 1B,C). By analyzing and summarizing the genomic, pan-genomic, and core genomic features of L. monocytogenes strains from different regions, it was revealed that these strains possess open genomes, which provide a genetic basis for their adaptation to diverse environments.
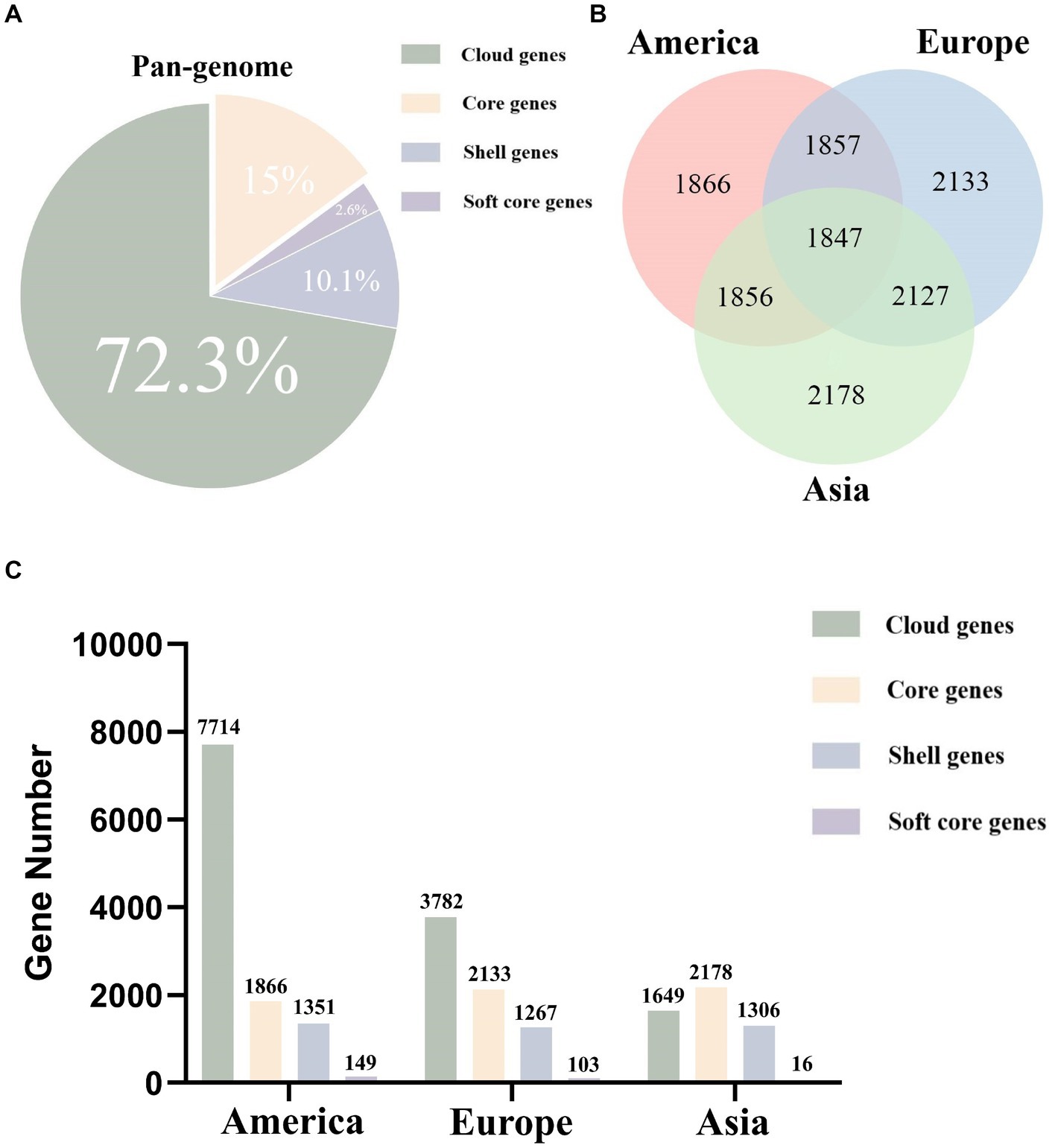
Figure 1. The proportion and quantity of various parts of the pan-genome in L. monocytogenes from different regions as analyzed through pan-genomics. (A) The pan-genome proportion of L. monocytogenes in three different regions. (B) The core genome Venn diagram of L. monocytogenes in three different regions. (C) The number of core genes, soft core genes, shell genes, and cloud genes in L. monocytogenes from three different regions.
Pan-genomic analysis of Listeria monocytogenes strains from different regions and non-target bacterial strains for the screening of potential target genes
To identify potential target genes in L. monocytogenes strains from different regions, we conducted pan-genomic analysis of these strains as well as non-target bacterial strains. Among them, due to the presence of non-target strains, the quantities of core genes are 0, the quantity of soft-core genes are 1919, the quantity of shell genes are 1,458, the quantity of cloud genes are 41,390, and the total quantity of genes are 44,767. A total of 357 potential target genes were detected in L. monocytogenes strains from different regions (Supplementary Table S3). These potential target genes were present in the L. monocytogenes strains included in this study, while being absent in non-target bacterial strains investigated. These potential target genes have the potential to serve as novel target genes for L. monocytogenes strains in different regions, but further screening of these potential target genes is still required.
MLST and phylogenetic analysis
To investigate the correlation among L. monocytogenes strains from different regions, MLST was employed to genotype the strains at the whole-genome level, aiming to determine the phylogenetic relationships among different sequence types (STs) and their associations with the disease. In America, the most common among L. monocytogenes strains was ST1 (n = 32, 14.3%), Clonal Complex 1 (CC1) (n = 34, 15.2%) and Lineage I (n = 161, 72.2%) (Figure 2A; Supplementary Table S4). In Europe, the most common was ST1 (n = 14, 15.4%), CC1 (n = 14, 15.4%) and Lineage I (n = 52, 57.1%) (Figure 2B; Supplementary Table S4). In Asia, the most common was ST8 (n = 7, 24.1%), CC8 (n = 8, 27.6%) and Lineage II (n = 18, 62.1%) (Figure 2C; Supplementary Table S4). In strains of L. monocytogenes in America and Europe, a higher proportion is observed for strains of ST1 and CC1 types. In strains of L. monocytogenes in Asia, a higher proportion is observed for strains of ST8 and CC8 types.
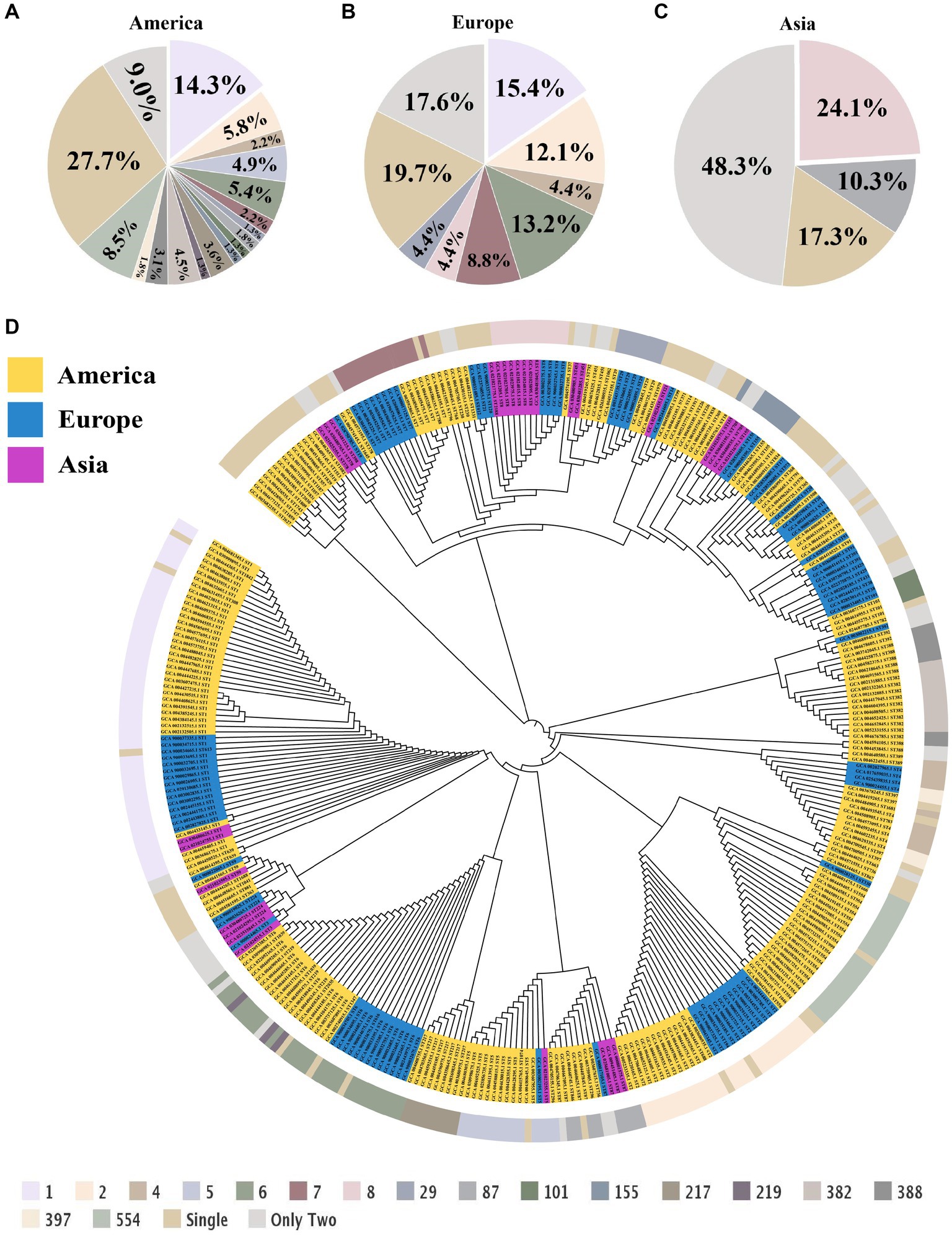
Figure 2. Multilocus sequence typing and phylogenetic tree of L. monocytogenes from different regions. (A) The proportion of STs in L. monocytogenes from the Americas region. (B) The proportion of STs in L. monocytogenes from the European region. (C) The proportion of STs in L. monocytogenes from the Asian region. (D) Phylogenetic trees of L. monocytogenes from different regions, with the outer circle indicating the corresponding ST for each strain.
Medically intriguingly, Listeria species inhabit diverse ecological niches, but only L. monocytogenes and L. ivanovii exhibit pathogenicity (Orsi and Wiedmann, 2016; Lu et al., 2022). To elucidate the evolutionary patterns of L. monocytogenes strains from different regions, we conducted a phylogenetic analysis of L. monocytogenes using conserved amino acid sequences of all single-copy genes (Figure 2D). Through the phylogenetic tree analysis, we can observe that L. monocytogenes strains in different regions share a common ancestor, all falling within this major root of the Listeria genus. Although the geographical locations of the L. monocytogenes strains we studied vary significantly, some strains from different locations still cluster within the same branch, indicating they share a relatively similar phylogenetic relationship. This indicates that despite being in different geographic locations and under varying environmental conditions, L. monocytogenes exhibits certain similarities in evolutionary mechanisms and genetic variations in response to environmental pressures, displaying a strong adaptability to diverse environments.
Enrichment analysis of the functional characteristics of potential target genes using GO and KEGG
To investigate the functional characteristics of 357 potential target genes in L. monocytogenes strains from different regions, we performed functional annotation and classification of these genes using GO and KEGG databases. The detailed information of the potential target genes is presented in Supplementary Table S3. The GO database categorizes gene functions into three main categories, namely Biological Processes (BP), Cellular Components (CC), and Molecular Functions (MF). In the BP category, the most enriched biological processes were cellular process (n = 124, 34.7%), nucleobase-containing compound metabolic process (n = 33, 9.2%), localization (n = 31, 8.7%), and transmembrane transport (n = 27, 7.6%). Within the CC category, the most abundant cellular components were cytoplasm (n = 50, 14%). In the MF category, the most enriched molecular functions were organic cyclic compound binding (n = 76, 21.3%), and purine nucleotide binding (n = 31, 8.7%) (Figure 3A). We integrated and ranked all the GO enrichment analysis results of potential target genes, and generated a bubble chart (Figure 3B) to display the top 20 functional features in GO enrichment analysis based on the number of genes and the significance of p values.
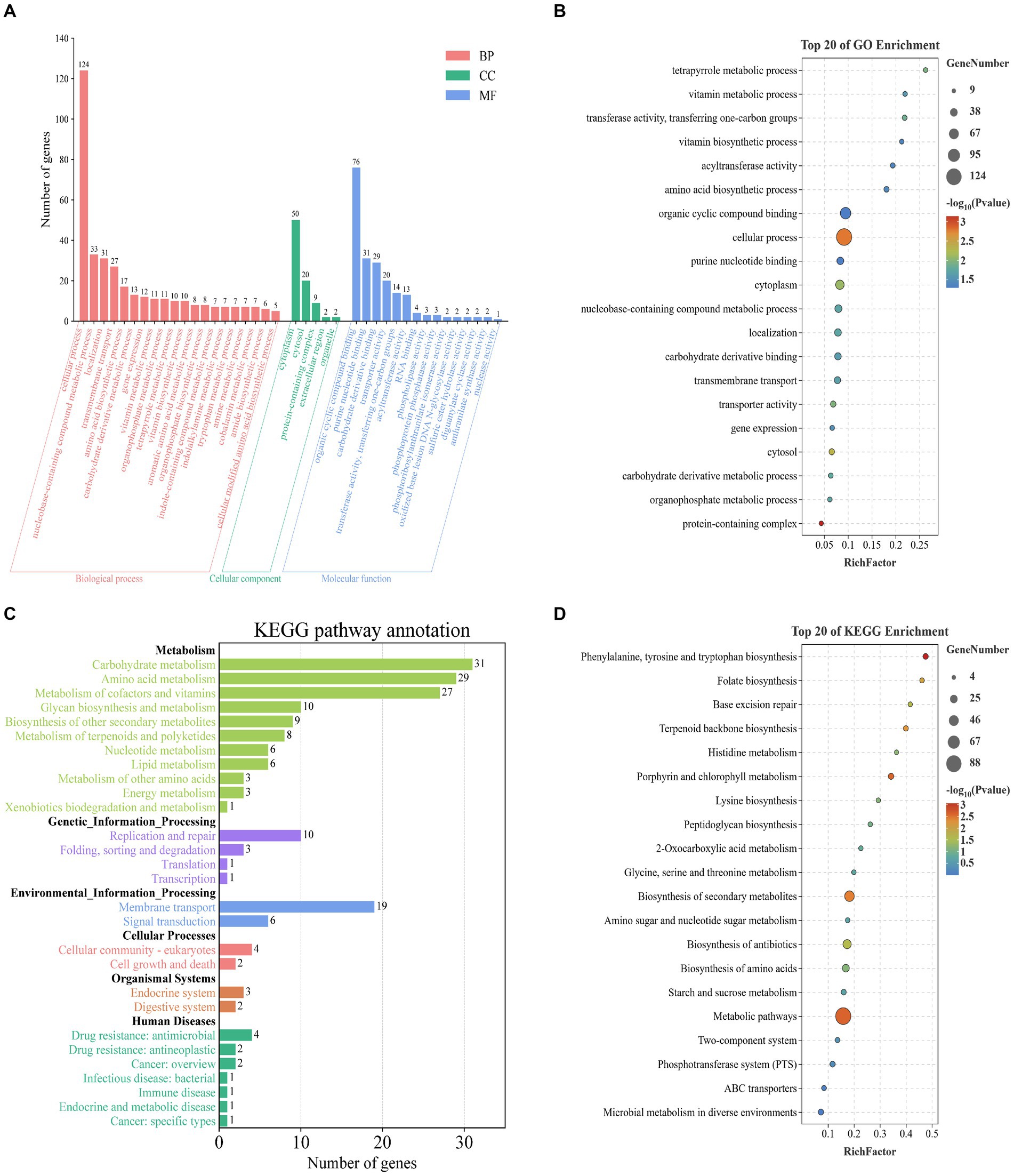
Figure 3. Enrichment analysis of potential target genes in L. monocytogenes from different regions based on the GO, and KEGG databases. (A) Enrichment analysis based on the GO database. (B) Enrichment analysis based on GO database with the top 20 enriched terms listed. (C) Enrichment analysis based on the KEGG database. (D) Enrichment analysis based on KEGG database with the top 20 enriched terms listed.
The pathway database of KEGG is the most widely used public database for metabolic pathways, which classifies biological metabolic pathways into six categories: Metabolism, Genetic Information Processing, Environmental Information Processing, Cellular Processes, Organismal Systems, and Human Diseases. The potential target genes were annotated using KEGG in six categories. Among these, the most enriched pathways in the Metabolism category were carbohydrate metabolism (n = 31, 8.7%), amino acid metabolism (n = 29, 8.1%), and metabolism of cofactors and vitamins (n = 27, 7.6%). In the Genetic Information Processing category, replication and repair (n = 10, 2.8%) were the most enriched pathways. In the Environmental Information Processing category, membrane transport (n = 19, 5.3%) and signal transduction (n = 6, 1.7%) were the most enriched pathways. In the Cellular Processes category, cellular community – prokaryotes (n = 4, 1.1%) and cell growth and death (n = 2, 0.6%) were the most enriched pathways. In the Organismal Systems category, endocrine system (n = 3, 0.8%) and digestive system (n = 2, 0.6%) were the most enriched pathways. In the Human Diseases category, drug resistance: antimicrobial (n = 4, 1.1%) and drug resistance: antineoplastic (n = 2, 0.6%) were the most enriched pathways (Figure 3C). We integrated and ranked all the KEGG enrichment analysis results of potential target genes, and generated a bubble chart (Figure 3D) to display the top 20 functional features in KEGG enrichment analysis based on the number of genes and the significance of p values.
In summary, the enrichment analysis of functional characteristics of 357 potential target genes using GO and KEGG databases indicates that these genes are primarily associated with metabolic processes, compound binding, protein localization, and transmembrane transport in L. monocytogenes. Examples include cellular metabolic process, carbohydrate metabolism and organic substance biosynthetic process. However, there were still some genes with unclear functional information, which warrants further investigation in future studies. The potential target genes are closely associated with fundamental biological processes and infection pathogenesis of L. monocytogenes, playing crucial roles in sustaining basic life activities, invading the host, and exerting pathogenic effects.
PPI network analysis of potential target genes and identification of novel target genes
The PPI network plays a crucial role in various biological processes within organisms. To further assess the interconnections among potential target genes of L. monocytogenes strains in different regions, PPI analysis was carried out using the STRING database. The PPI network of potential target genes comprised 357 genes, and visualization of the PPI network was performed using Cytoscap_v3.10.1 software. They were clustered together, indicating strong physical interaction or functional association.
To further analyze potential target genes of L. monocytogenes for the selection of novel target genes, we employed the CytoHubba function of Cytoscape v3.10.1 software to identify hub genes. Hub genes are key factors in protein-protein interaction networks that exhibit high connectivity in gene expression networks, indicating their ability to regulate multiple genes. The genes in the PPI network were screened using eight different algorithms available in the CytoHubba function. The top 10 genes with the highest scores were selected as hub genes, and their ranking is presented in Table 2. A comprehensive analysis of the results obtained from the eight algorithms was performed, and raincloud plots illustrating the scores of the hub genes for each algorithm was generated (Figure 4B). The top 10 genes with the highest scores identified by the Degree algorithm were ultimately determined as novel target genes, and a PPI network was constructed based on their scores (Figure 4A). Among them, bglF_1 and davD genes had the highest score of 54, followed by menE_1 gene with a score of 52, tilS gene with a score of 50, dapX gene with a score of 48, iolC gene with a score of 46, gshAB gene with a score of 42, cysG gene with a score of 42, trpA gene with a score of 40, and hisC gene with a score of 38. The detailed information regarding these 10 genes, including their functional roles, gene lengths, etc., is provided in Table 3. These 10 hub genes play crucial roles in sustaining basic life activities and infection invasion of L. monocytogenes strains, with the potential to become novel target genes for L. monocytogenes strains, particularly the top-scoring genes, bglF_1 and davD.
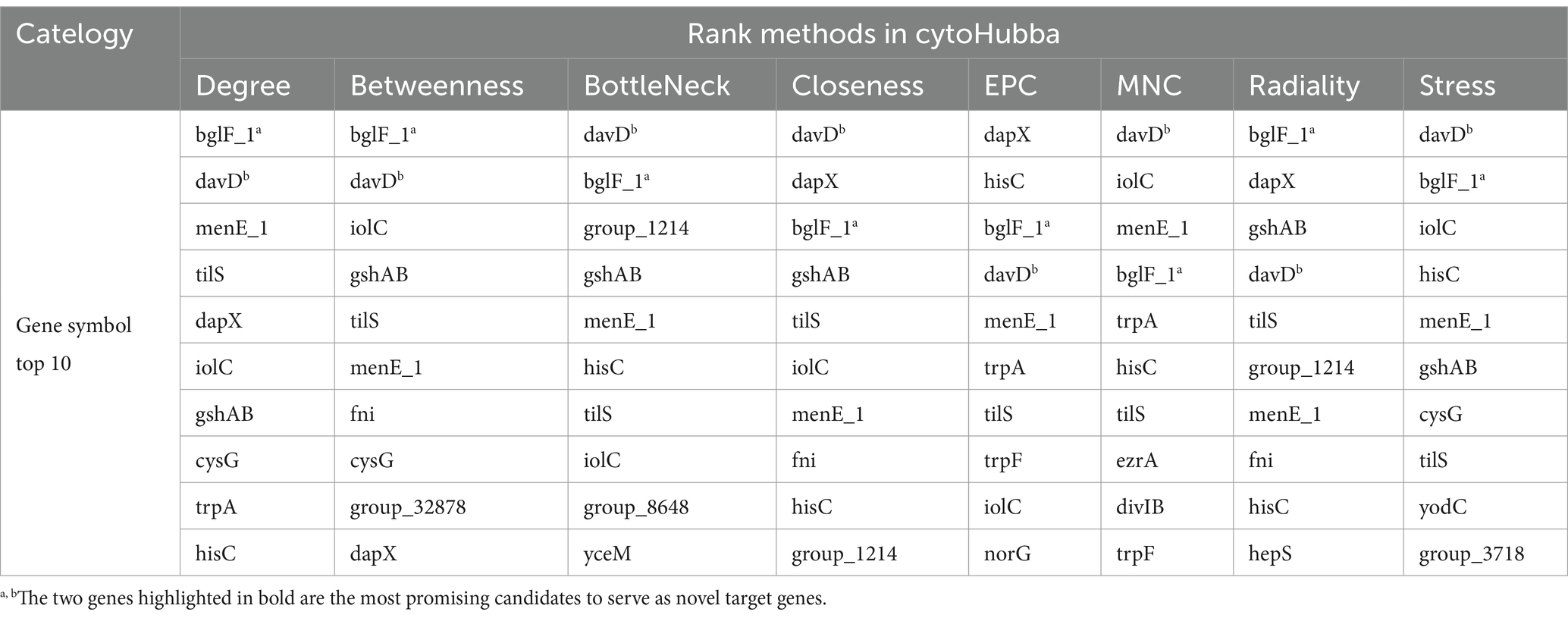
Table 2. Top 10 hub genes ranked by scoring in eight different algorithms.
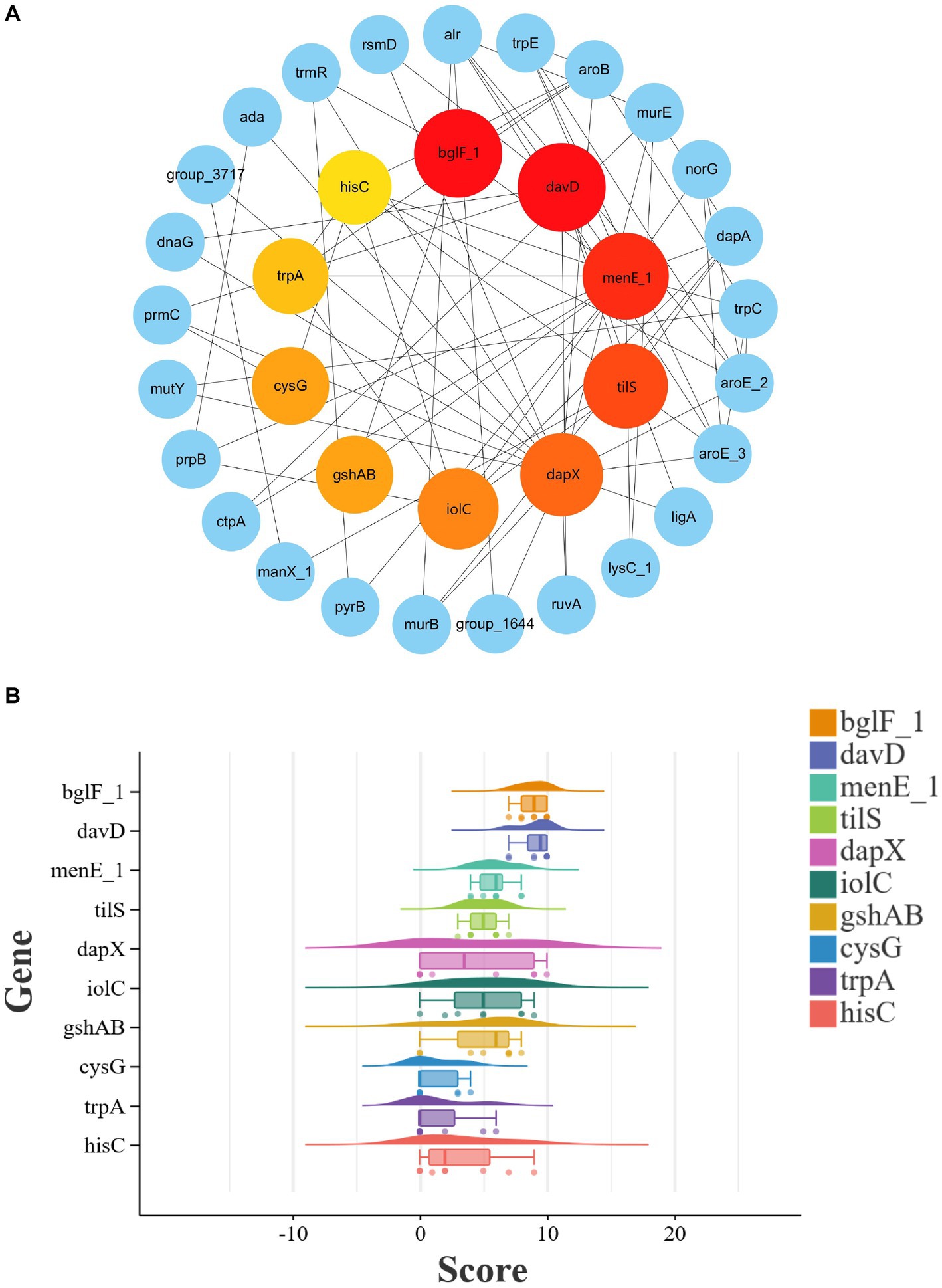
Figure 4. PPI network analysis and identification of novel target genes among potential target genes in L. monocytogenes from different regions. (A) Visualization of PPI network among the top 10 hub genes ranked by Degree algorithm. The larger the circle and the deeper the red color, the higher the score of the gene. (B) Scoring of hub genes across eight different algorithms.
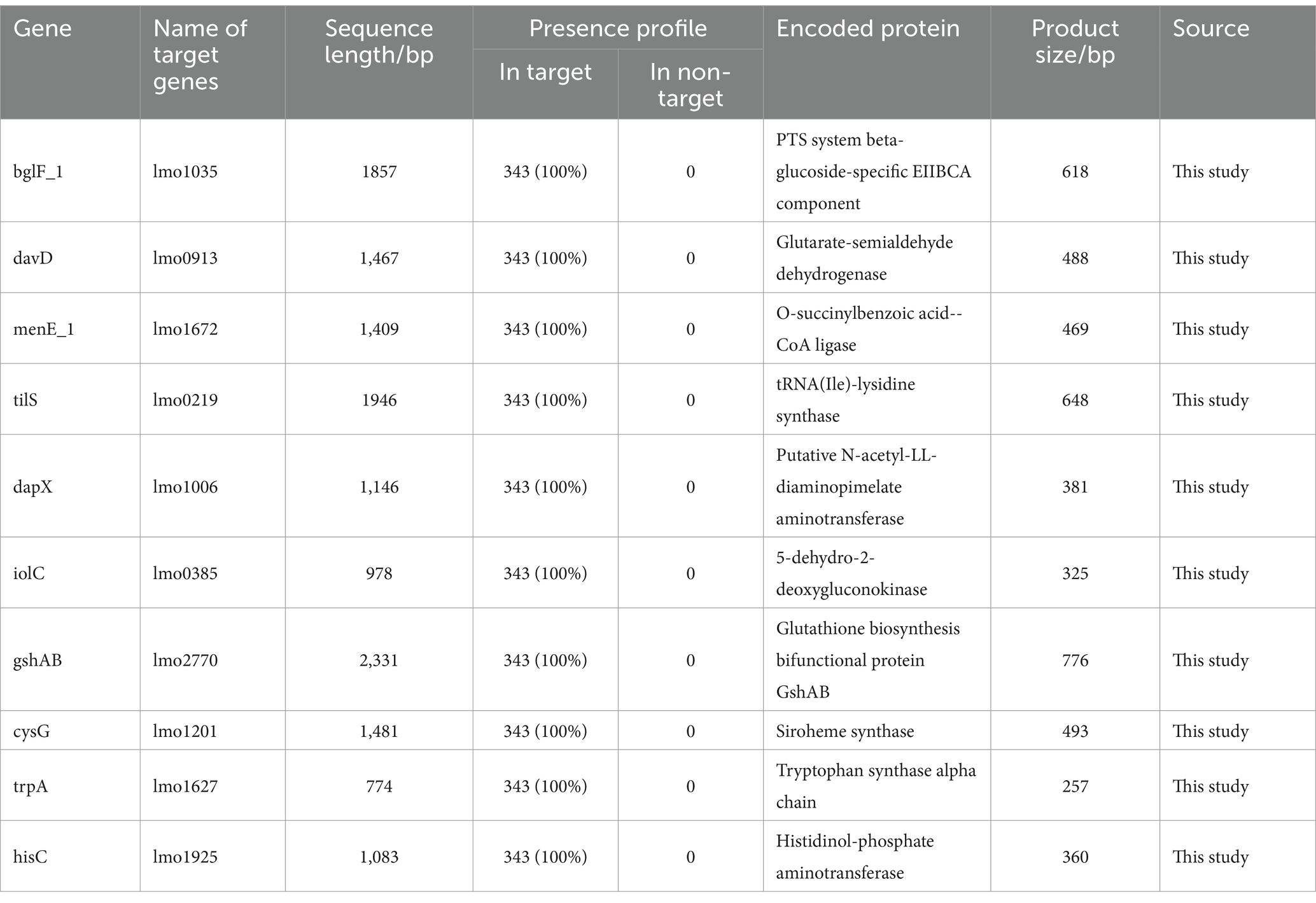
Table 3. Detailed information of top 10 hub genes ranked by scoring in Degree algorithm.
Distribution of virulence genes and antibiotic resistance genes in Listeria monocytogenes strains in different regions
To investigate the relationship between L. monocytogenes strains in different regions and their pathogenic mechanisms, we predicted virulence factor-encoding genes of the entire genome of L. monocytogenes. Based on VFDB prediction and annotation, virulence factors of L. monocytogenes were classified into 12 categories including Adherence, Bile resistance, Enzyme, Immune modulator, Intracellular survival, Invasion, Iron uptake, Nucleation-promoting factor, Peptidoglycan modification, Regulation, Surface protein anchoring, and Toxin.
In this study, the virulence genes dltA, fbpA, lap, plcB, stp, inlK, oppA, prsA2, inlB, lpeA, hbp2, pdgA, agrC, cheA, lisK, lisR, prfA, virR, and virS were found to be present in 100% of L. monocytogenes strains from different regions (Figure 5). Our predictive findings indicate the presence of numerous virulence genes, including inlB, and plcB, which play pivotal roles in L. monocytogenes infection and host invasion, across L. monocytogenes strains from various regions. It is well known that L. monocytogenes is pathogenic within the Listeria genus, owing to its abundance of virulence genes, which contribute to its pathogenicity. Through the prediction of virulence genes, L. monocytogenes strains from different regions exhibit high pathogenicity.
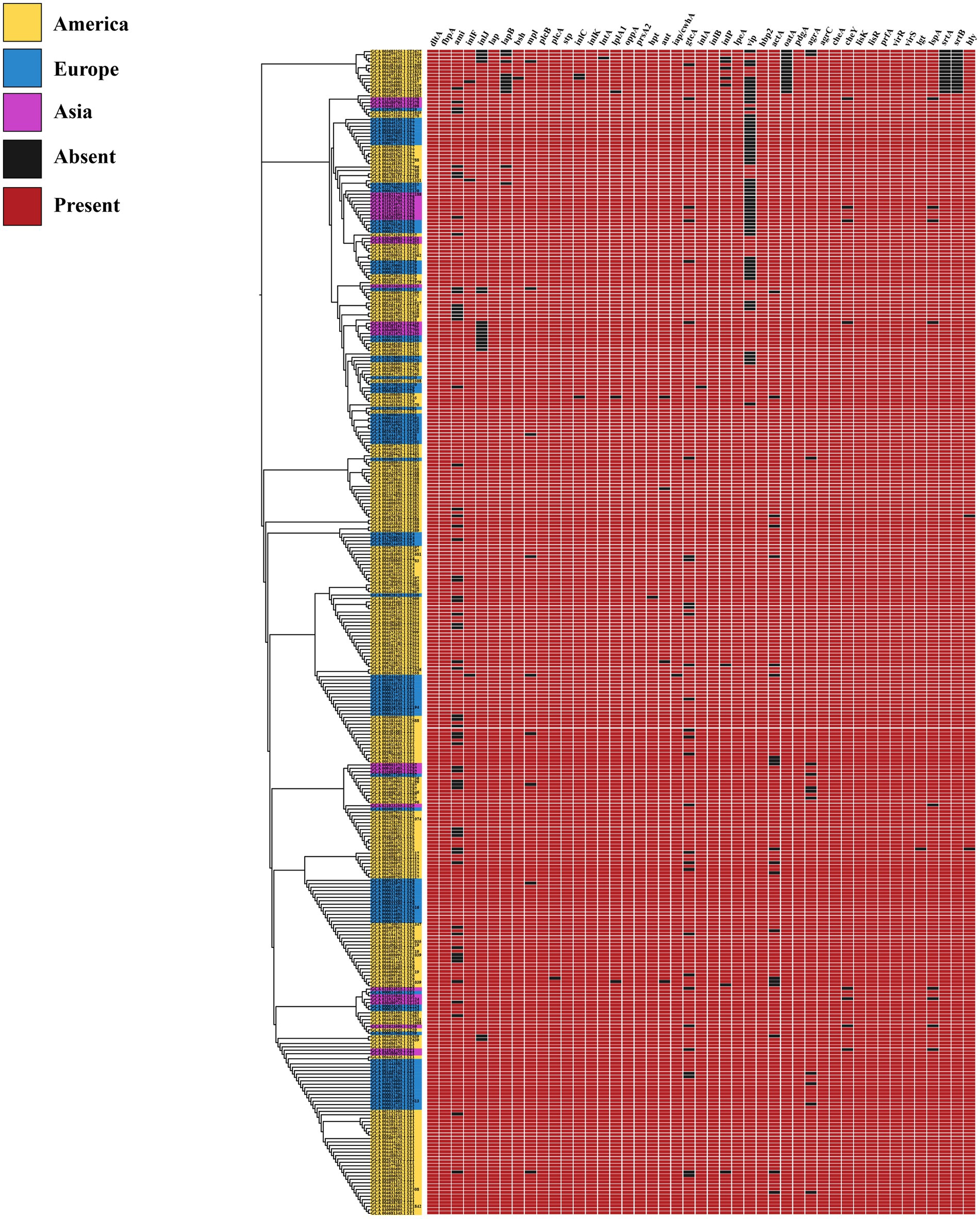
Figure 5. The distribution of virulence genes in L. monocytogenes from different regions.
With the widespread use of antibiotics, the antimicrobial resistance of foodborne strains has increased in many countries. The distribution of antimicrobial resistance genes in L. monocytogenes was investigated using the CARD database. In this study, a total of 10 antimicrobial resistance genes belonging to 9 drug classes and exhibiting 5 resistance mechanisms were identified among the 343 L. monocytogenes genomes analyzed. Among the L. monocytogenes strains from different regions, 100% were found to harbor four types of antibiotic resistance genes, including phosphonic acid antibiotic gene (FosX), glycopeptide antibiotic genes (vanTG, vanYM), lincosamide antibiotic gene (lin), and peptide antibiotic gene (mprF) (Figure 6).
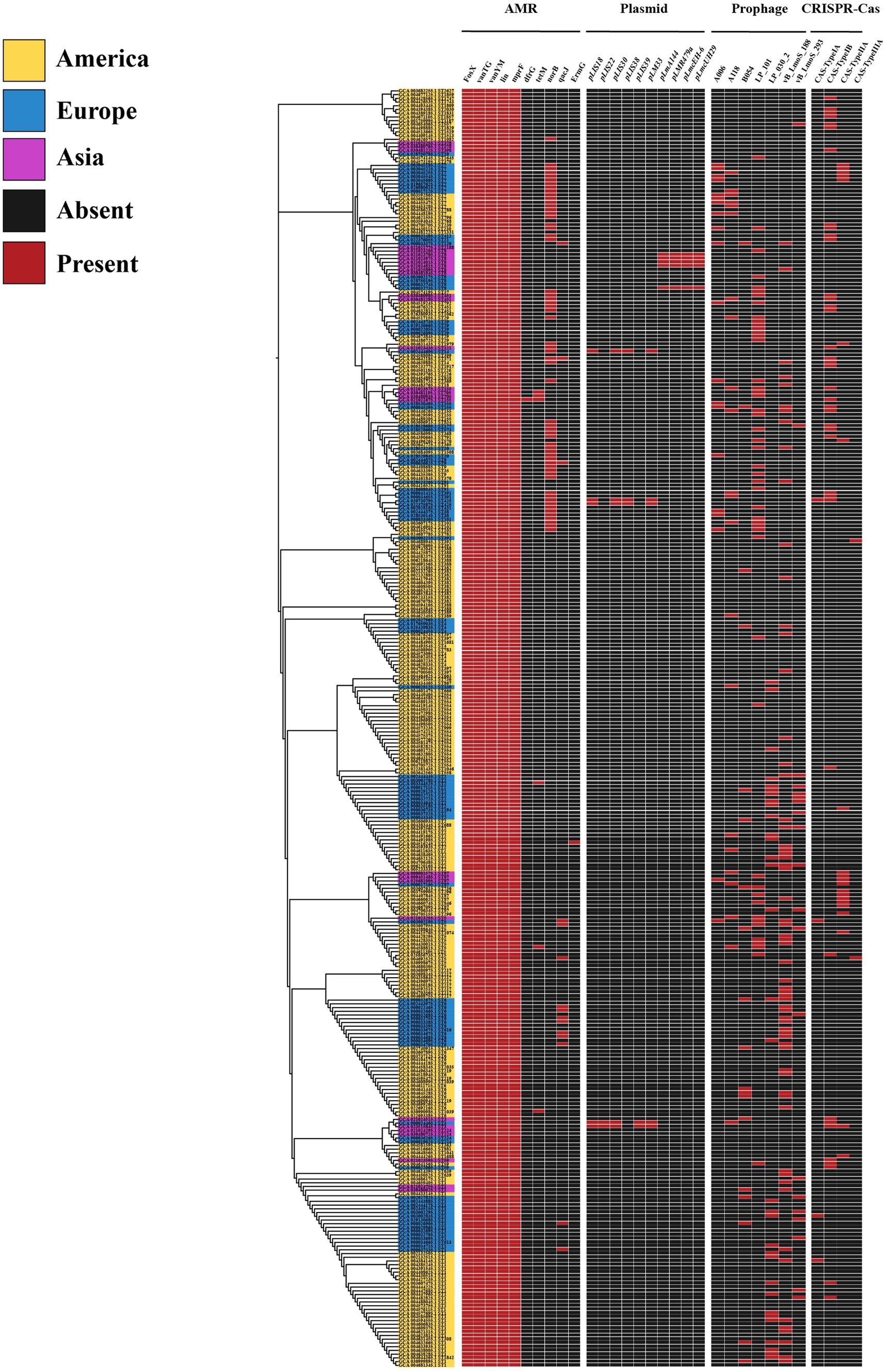
Figure 6. The distribution of antibiotic resistance genes, plasmids, prophages, and CRISPR-Cas systems in L. monocytogenes from different regions.
Distribution of MGEs in Listeria monocytogenes strains in different regions
Currently, many scientists have begun to pay extensive attention to the significant role of horizontal transfer of MGEs in bacterial genome evolution and adaptation to specific environmental pressures. We employed PLSDB and PHASTER databases for the detection of MGEs, and the results only documented plasmids with an identity score of 1 and intact prophage regions. In this study, the L. monocytogenes genome contained a total of 10 plasmids and 7 intact prophage regions. Among them, plasmids pLmA144, pLMR479a, pLmcEH-6, and pLmcUH29 were exclusively present in ST8, while plasmids pLIS22 and pLIS39 were only found in ST3 (Figure 6). The most prevalent prophage was PHAGE_Lister_vB_LmoS_188 [NC_028871] (n = 64, 18.7%), followed by PHAGE_Lister_LP_101 [NC_024387] (n = 48, 14%) (Figure 6). Intriguingly, despite the geographical disparity of L. monocytogenes isolates, identical phage genomes have been detected, suggesting a certain degree of similarity in MGEs across strains from different regions.
Distribution of CRISPR-Cas system types in Listeria monocytogenes strains in different regions
The CRISPR-Cas system is a bacterial adaptive immune system that protects bacteria from viral infections, which is also associated with the virulence and pathogenicity of pathogens. In this study, we characterized the CRISPR-Cas systems in 343 L. monocytogenes genomes and identified four types of CRISPR-Cas systems. Each CRISPR-Cas system type exhibited distinct cas genes, with a total of 14 cas genes detected (Table 4). The CRISPR-Cas system types detected in L. monocytogenes included CAS-TypeIA (4/343), CAS-TypeIB (39/343), CAS-TypeIIA (21/343), and CAS-TypeIIIA (2/343). Approximately one-fifth of the L. monocytogenes genomes (66/343) harbored at least one CRISPR-Cas system, with CAS-TypeIB (11.4%) and CAS-TypeIIA (6%) being the most prevalent (Figure 6). CAS-TypeIA was only detected in ST1, ST5, and ST425, while CAS-TypeIIIA was found exclusively in ST5 and ST392. The CAS-type IA system detected in the L. monocytogenes isolates in this study was composed of csa5_TypeIA and casRa_TypeIA. The CAS-TypeIB system was composed of cas5b_TypeIB, cas6_TypeI-III, cas8a1b_TypeIB, cas7b_TypeIB, cas3_TypeI, cas2_TypeI-II-III, cas4_TypeI-II, and cas1_TypeIB. The CAS-TypeIIA system was composed of csn2_TypeIIA, cas2_TypeI-II-III, cas1_TypeII, and cas9_TypeII. The CAS-type IIIA system was composed of csm2_TypeIIIA.
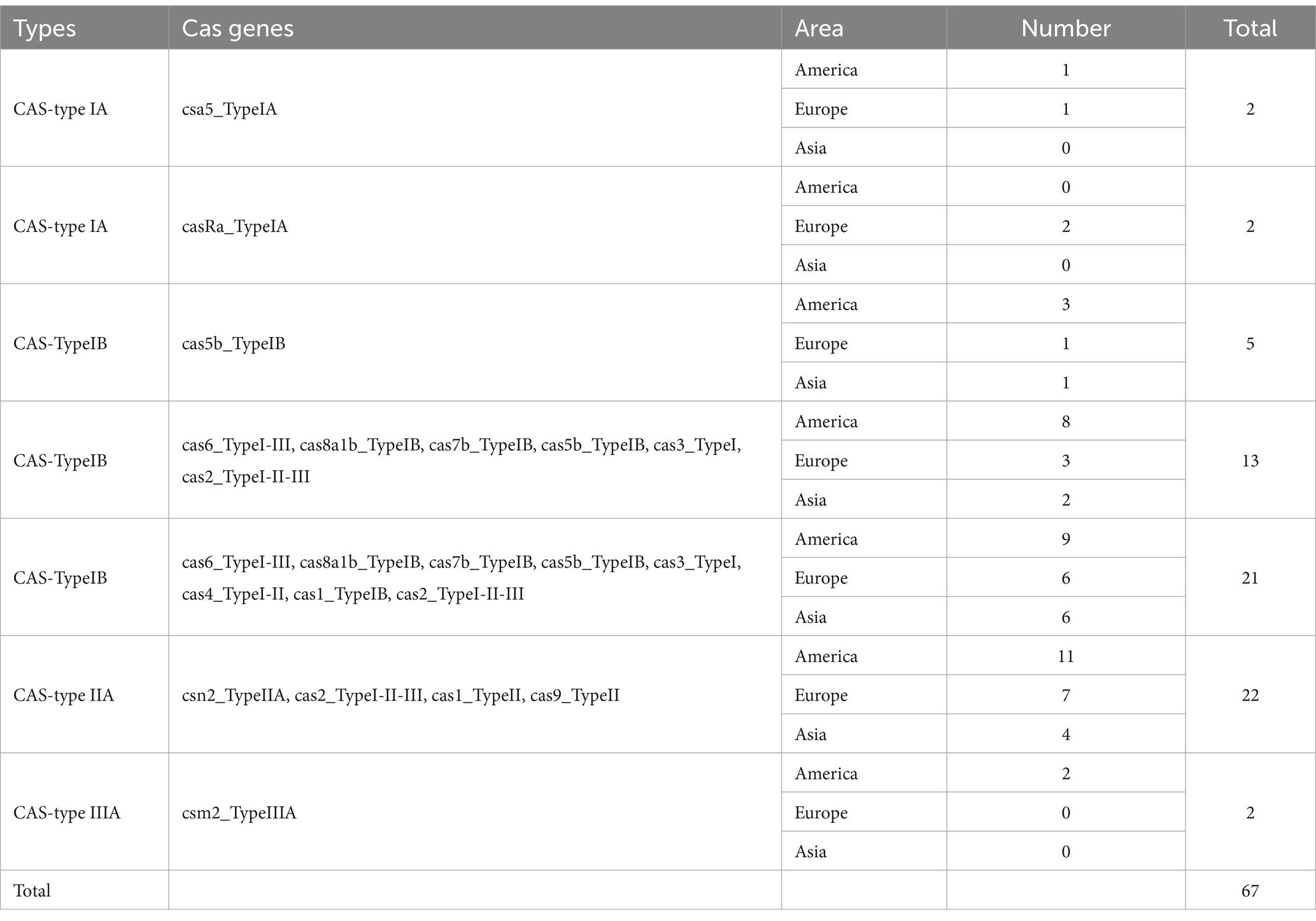
Table 4. Identification of CRISPR-Cas system types and the corresponding cas genes detected in L. monocytogenes strains from different regions.
Detection of Listeria monocytogenes using specific primers by PCR
To validate the potential of bglF_1 and davD genes as specific molecular detection and therapeutic targets in L. monocytogenes strains from different regions, primers were designed for bglF_1 and davD genes, followed by PCR experiments to assess their specificity. The PCR results revealed a distinct band at 616 bp for L. monocytogenes in the bglF_1 gene primer system, while non-L. monocytogenes samples showed no band (Figure 7A). Similarly, a clear band at 567 bp was observed for L. monocytogenes in the davD gene primer system, with no band detected in non-L. monocytogenes samples (Figure 7B). The results demonstrated the excellent specificity of the bglF_1 and davD genes for L. monocytogenes. Therefore, the bglF_1 and davD genes hold promise as specific molecular detection and therapeutic targets for L. monocytogenes strains from different regions.
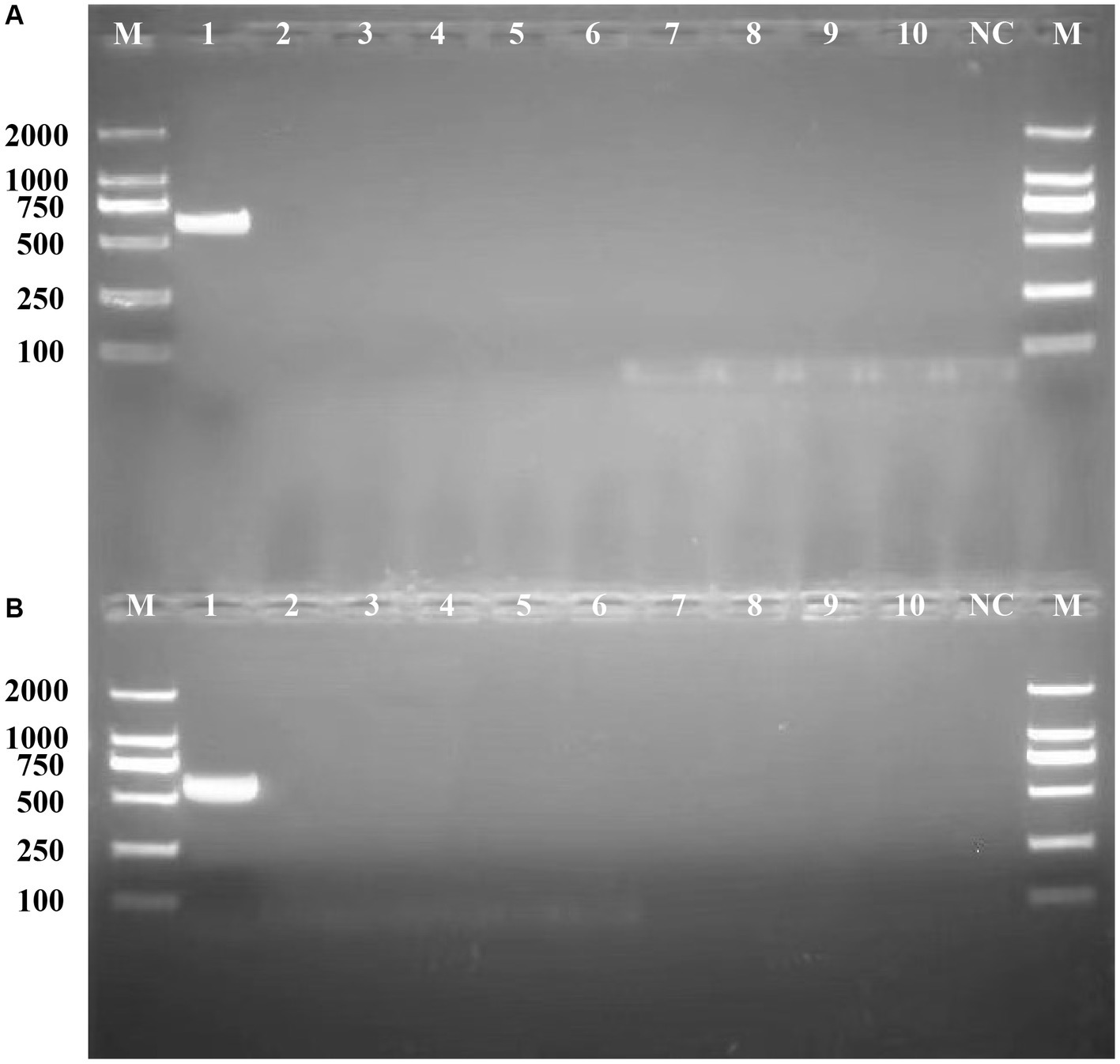
Figure 7. Validation of the specificity of the primers for the bglF_1 and davD genes in L. monocytogenes. (A) The PCR results of the primer system targeting the bglF_1 gene. (B) The PCR results of the primer system targeting the davD gene. Lane M: DL DNA 2000 marker, lane NC: negative control, and lanes 1–10: represent 10 different strains, including lane 1: Listeria monocytogenes, lane 2: Listeria innocua, lane 3: Listeria ivanovii, lane 4: Listeria welshimeri, lane 5: Escherichia coli, lane 6: Salmonella, lane 7: Klebsiella Pneumoniae, lane 8: Acinetobacter baumannii, lane 9: Pseudomonas aeruginosa, lane 10: Pneumocystosis jirovecii.
Discussion
L. monocytogenes, as a significant foodborne pathogen, is widely prevalent worldwide, posing a serious threat to human life and health. Therefore, we conducted comparative genomic analysis of L. monocytogenes strains from different regions to explore their biodiversity and evolutionary characteristics, identify potential target genes, and further mining novel target genes, aiming to provide novel specific molecular detection and therapeutic strategies for L. monocytogenes strains.
In this study, we conducted a pan-genomic comparative analysis of 343 L. monocytogenes strains from different regions to investigate the biodiversity and evolutionary characteristics of strains. To assess the genomic biodiversity of L. monocytogenes strains in different regions, we conducted core/pan-genome analysis. Core and accessory genomes were analyzed based on the whole genomes of L. monocytogenes. The core genome represents the essential portion necessary for the presence and shared phenotypic features of specific strains, while the accessory genome provides unique characteristics for a species or strain that are not essential for their basic survival, but offer selective advantages for ecological adaptation and antibiotic resistance (Deng et al., 2010; Zhang et al., 2017; Lu et al., 2022). Although the studied strains of L. monocytogenes are geographically diverse, there still exist 1847 core genes that constitute the fundamental components of L. monocytogenes survival and development. In America, the number of core genes in L. monocytogenes strains is 1866, in Europe it is 2,133, and in Asia it is 2,178. It can be observed that the number of core genes in L. monocytogenes strains varies across different regions. Apart from the core genes they share, L. monocytogenes strains in different regions also possess unique core genes that are present only in one region and absent in others. These unique core genes may be the primary reason for the distinctiveness of L. monocytogenes strains in one region compared to those in other regions. The primary reason for this phenomenon may be attributed to the different environments in which the strains reside. Hence, in order to adapt to these unique environments, the strains have evolved genes that are specific to these environments to counteract environmental pressures (Liao et al., 2023). This is also very intriguing, as it allows for the exploration of the differences among L. monocytogenes strains in different regions, analyzing the unique characteristics of strains in different areas, thereby studying the evolutionary patterns of strains in that region, and subsequently devising targeted prevention and control measures for that region. As the number of genomes increases, the pan-genome size continues to rise while the core genome decreases and tends to plateau. This indicates that the studied L. monocytogenes possesses an open pan-genome, which provides a genetic basis for the adaptation of L. monocytogenes to different environments. The potential target genes are exclusively present in L. monocytogenes strains in different regions, while they are absent in non-target strains. This indicates that the potential target genes play a crucial role in the pathogenicity of L. monocytogenes strains in different regions. These genes are indispensable for the survival, virulence, and invasion of L. monocytogenes, making them essential for maintaining life activities and infection. Therefore, investigating potential target genes can facilitate the analysis of the biodiversity and evolutionary characteristics of L. monocytogenes, aiding in the selection of novel specific molecular detection and therapeutic target genes.
To investigate the biodiversity and evolutionary characteristics of L. monocytogenes strains in different regions, we conducted MLST typing analysis. The results revealed that L. monocytogenes strains from America and Europe were predominantly characterized by ST1 and CC1 types, whereas those from Asia were predominantly characterized by ST8 and CC8 types. Wang et al. (2012) identified the three most common L. monocytogenes types in China as ST8, ST9, and ST87. Amarasekara et al. (2024) identified a significant presence of ST1 and CC1 types among L. monocytogenes isolates from agricultural markets in the United States. Additionally, Toledo et al. (2018) found that the most prevalent type of L. monocytogenes in samples from Chile was ST1. Our results are consistent with the findings reported in the above-mentioned literature. Our findings indicate that the L. monocytogenes strains isolated from America, Europe, and Asia exhibit different types. The underlying reasons for this phenomenon could be attributed to variations in the transmission routes and environmental conditions of L. monocytogenes, as well as genetic variability among strains. Based on our analysis, although L. monocytogenes strains originate from diverse geographical regions, they exhibit relatively similar phylogenetic relationships in the constructed phylogenetic tree. This indicates that while the L. monocytogenes strains are present in different environments, they exhibit a certain degree of genetic similarity in terms of bacterial variability. Under various environmental pressures, L. monocytogenes gradually evolves into life forms adapted to these specific environments, undergoing extensive genetic variations. Bacterial genetic variations result in distinct predominant types of L. monocytogenes strains in America, Europe, and Asia. Interestingly, the predominant sequence type of L. monocytogenes in both America and Europe is ST1, which may be attributable to the relatively close geographical proximity of these regions resulting in fewer environmental disparities. Additionally, trade between these regions may contribute to the mutual dissemination of L. monocytogenes strains. This discovery provides valuable insights into the reasons for the differences in the predominant ST and CC types of L. monocytogenes strains in America, Europe, and Asia.
It is well known that within the genus Listeria, only L. monocytogenes and L. ivanovii are considered pathogenic, with L. monocytogenes exhibiting higher pathogenicity. Moreover, the high pathogenicity of L. monocytogenes typically relies on a plethora of virulence genes as its foundation. Our analysis findings align with this observation, as strains of L. monocytogenes in different regions harbor a significant abundance of virulence genes. During invasion of the host by L. monocytogenes, the bacterium first utilizes the inlA and inlB genes to bind with the E-Cadherin and Met receptors of the host’s eukaryotic cell membrane, respectively, thereby inducing bacterial uptake through receptor-mediated endocytosis. After internalization, the bacterium is encapsulated within a vacuole, and releases the hly, plcA, and plcB genes to mediate vacuole escape. Subsequently, the actA gene is utilized to induce actin polymerization and generate sufficient force for the bacterium to spread from one cell to another. During the invasion process of L. monocytogenes, LIPI-1 (prfA, plcA, hly, mpl, actA, and plcB) and LIPI-2 (inlA, inlB, inlC, inlE, inlF, inlG, inlH, inlJ, and inlK) play pivotal roles (Mejía et al., 2023). Interestingly, in this study, the genes prfA, plcB, inlK, and inlB were found to be present in 100% of the selected L. monocytogenes strains, whereas hly, actA, and inlA were not always present at 100%, but their presence probability exceeded 99%. This phenomenon could be attributed to prediction errors in the database or possibly due to genetic variations occurring in individual strains under specific environmental conditions (Li et al., 2021d). These virulence genes are essential for infecting and invading hosts, highlighting the high pathogenicity of L. monocytogenes. Furthermore, the potential target genes we screened also include these virulence genes. The potential target genes play crucial roles in the fundamental life activities and infective invasion of L. monocytogenes. Selecting these virulence genes as molecular detection and therapeutic targets may be a viable option, however, it may lack novelty, as previous studies have validated genes such as inlA, inlB, and hly as targets for the detection and treatment of L. monocytogenes. Therefore, although these virulence genes were also selected as potential target genes in this study, they were not directly chosen as targets for detection and treatment. This study employed hub gene screening methods to further select hub genes from numerous potential target genes. As is well known, hub genes, also known as key genes, refer to genes that play a crucial role in a particular disease or biological process (Li et al., 2022). Therefore, we selected highly scoring hub genes from potential target genes as novel target genes.
In this study, L. monocytogenes strains demonstrated relatively high resistance to phosphonic antibiotics, glycopeptide antibiotics, lincosamide antibiotics, and peptide antibiotics. Therefore, it is recommended to avoid selecting these four classes of antibiotics when undergoing treatment. Other types of antibiotics may yield better therapeutic effects, such as ampicillin, gentamicin, and penicillin. Analyzing MGEs can provide insights into the evolution of bacterial genomes. In this study, despite the different geographical locations of the L. monocytogenes strains, they exhibited certain similarities at the MGEs level. This suggests that although L. monocytogenes is exposed to diverse external environments, there still exists a degree of similarity in terms of bacterial genome evolution.
By performing PPI network analysis and conducting GO and KEGG enrichment analyses on potential target genes, we aimed to understand the role of these genes in L. monocytogenes strains in different regions (Adnan et al., 2022). Functional annotation results revealed that the potential target genes encompassed a significant number of transport and metabolism genes, as well as virulence-associated genes, which play crucial roles in the fundamental life activities and pathogenicity of L. monocytogenes. However, some genes still lack clear functional information, necessitating further investigation in future studies. Hub genes, which are the most crucial genes in PPI networks, were selected to mining novel target genes (Li et al., 2022). Ten highly connected hub genes (bglF_1, davD, menE_1, tilS, dapX, iolC, gshAB, cysG, trpA, hisC) were identified from the pool of potential target genes. These ten hub genes play crucial roles in the fundamental life activities and infective invasion of L. monocytogenes. Among them, bglF_1 and davD genes scored the highest and showed closer connections with other proteins, indicating their potential to serve as specific molecular detection and therapeutic targets for L. monocytogenes strains. The inhibitors or antagonists of these genes hold promise as novel therapeutic agents. The PCR results demonstrated the excellent specificity of the bglF_1 and davD genes for L. monocytogenes. Therefore, the bglF_1 and davD genes hold promise as specific molecular detection and therapeutic targets for L. monocytogenes strains from different regions.
Conclusion
In summary, we employed comparative genomic analysis to investigate the biodiversity and evolutionary characteristics of L. monocytogenes strains from different regions. Although L. monocytogenes strains originate from different regions, they exhibit a high degree of similarity in bacterial genome evolution, harboring numerous potential target genes that sustain the essential life activities and infection invasion of L. monocytogenes. Through further exploration of potential target genes and validation of PCR results, the bglF_1 and davD genes emerged as promising candidates for specific molecular detection and therapeutic targets in L. monocytogenes strains. This study provides significant reference value for the specific molecular detection and therapeutic targets of L. monocytogenes strains.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
BZ: Data curation, Project administration, Validation, Visualization, Writing – original draft, Writing – review & editing. HR: Supervision, Writing – review & editing. XW: Supervision, Writing – review & editing. CH: Validation, Writing – original draft. YJ: Validation, Writing – original draft. XH: Validation, Writing – original draft. RS: Validation, Writing – original draft. CL: Validation, Writing – original draft. YW: Validation, Writing – original draft. YL: Supervision, Writing – review & editing. SL: Supervision, Writing – review & editing. ZL: Supervision, Writing – review & editing. PH: Supervision, Writing – review & editing.
Funding
The work was supported by the National Key Research and Development Program of China (2023YFD1801000).
Acknowledgments
The authors would like to thank all the reviewers who participated in the review, as well as other members in the team of PH.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1424868/full#supplementary-material
References
Adnan, M., Siddiqui, A. J., Noumi, E., Hannachi, S., Ashraf, S. A., Awadelkareem, A. M., et al. (2022). Integrating network pharmacology approaches to decipher the multi-target pharmacological mechanism of microbial biosurfactants as novel green antimicrobials against Listeriosis. Antibiotics (Basel) 12:5. doi: 10.3390/antibiotics12010005
Amarasekara, N. R., Swamy, A. S., Paudel, S. K., Jiang, W., Li, K., Shen, C., et al. (2024). Hypervirulent clonal complex (CC) of Listeria monocytogenes in fresh produce from urban communities. Front. Microbiol. 15:1307610. doi: 10.3389/fmicb.2024.1307610
Bosi, E., Fondi, M., Orlandini, V., Perrin, E., Maida, I., Pascale, D., et al. (2017). The pangenome of (Antarctic) Pseudoalteromonas bacteria: evolutionary and functional insights. BMC Genomics 18:93. doi: 10.1186/s12864-016-3382-y
Cain, R. J., Scortti, M., Monzó, H. J., and Vázquez, J. A. (2023). Listeria InlB expedites vacuole escape and intracellular proliferation by promoting Rab7 recruitment via Vps34. MBio 14:e0322122. doi: 10.1128/mbio.03221-22
Castro, H., Douillard, F. P., Korkeala, H., and Lindström, M. (2021). Mobile elements harboring heavy metal and bacitracin resistance genes are common among Listeria monocytogenes strains persisting on dairy farms. mSphere 6:e0038321. doi: 10.1128/mSphere.00383-21
Chiaverini, A., Guidi, F., Torresi, M., Acciari, V. A., Centorotola, G., Cornacchia, A., et al. (2021). Phylogenetic analysis and genome-wide association study applied to an Italian Listeria monocytogenes outbreak. Front. Microbiol. 12:750065. doi: 10.3389/fmicb.2021.750065
Deng, X., Phillippy, A. M., Li, Z., Salzberg, S. L., and Zhang, W. (2010). Probing the pan-genome of Listeria monocytogenes: new insights into intraspecific niche expansion and genomic diversification. BMC Genomics 11:500. doi: 10.1186/1471-2164-11-500
Disson, O., Moura, A., and Lecuit, M. (2021). Making sense of the biodiversity and virulence of Listeria monocytogenes. Trends Microbiol. 29, 811–822. doi: 10.1016/j.tim.2021.01.008
Fox, E. M., Allnutt, T., Bradbury, M. I., Fanning, S., and Chandry, P. S. (2016). Comparative genomics of the Listeria monocytogenes ST204 subgroup. Front. Microbiol. 7:2057. doi: 10.3389/fmicb.2016.02057
Gao, Z., Zhong, W., Liu, T., Zhao, T., and Guo, J. (2021). Global proteomic analysis of Listeria monocytogenes response to linalool. Food Secur. 10:2449. doi: 10.3390/foods10102449
He, P., Wang, H., Yan, Y., Zhu, G., and Chen, Z. (2022). Development and application of a multiplex fluorescent PCR for Shigella detection and species identification. J. Fluoresc. 32, 707–713. doi: 10.1007/s10895-021-02876-0
Henri, C., Félix, B., Guillier, L., Leekitcharoenphon, P., Michelon, D., Mariet, J. F., et al. (2016). Population genetic structure of Listeria monocytogenes strains as determined by pulsed-field gel electrophoresis and multilocus sequence typing. Appl. Environ. Microbiol. 82, 5720–5728. doi: 10.1128/AEM.00583-16
Jordan, K., and McAuliffe, O. (2018). Listeria monocytogenes in foods. Adv. Food Nutr. Res. 86, 181–213. doi: 10.1016/bs.afnr.2018.02.006
Li, J. X., Huang, Y. Y., Huang, Z. M., Cao, X. J., Xie, L. M., and Guo, X. G. (2022). Screening of potential hub genes involved in cutaneous Leishmaniasis infection via bioinformatics analysis. Acta Trop. 236:106645. doi: 10.1016/j.actatropica.2022.106645
Li, C., Li, J., and Lu, P. (2021a). Identification of key genes involved in Brg1 mutation-induced cataract using bioinformatics analyses with publicly available microarray data. Acta Biochim. Pol. 68, 733–737. doi: 10.18388/abp.2020_5632
Li, F., Ye, Q., Chen, M., Shang, Y., Zhang, J., Ding, Y., et al. (2021b). Real-time PCR identification of Listeria monocytogenes serotype 4c using primers for novel target genes obtained by comparative genomic analysis. LWT 138:110774. doi: 10.1016/j.lwt.2020.110774
Li, F., Ye, Q., Chen, M., Zhang, J., Xue, L., Wang, J., et al. (2021c). Multiplex PCR for the identification of pathogenic Listeria in Flammulina velutipes plant based on novel specific targets revealed by Pan-genome analysis. Front. Microbiol. 11:634255. doi: 10.3389/fmicb.2020.634255
Li, F., Ye, Q., Chen, M., Zhou, B., Xiang, X., Wang, C., et al. (2021d). Mining of novel target genes through pan-genome analysis for multiplex PCR differentiation of the major Listeria monocytogenes serotypes. Int. J. Food Microbiol. 339:109026. doi: 10.1016/j.ijfoodmicro.2020.109026
Liao, J., Guo, X., Li, S., Anupoju, S. M. B., Cheng, R. A., Weller, D. L., et al. (2023). Comparative genomics unveils extensive genomic variation between populations of Listeria species in natural and food-associated environments. ISME Commun. 3:85. doi: 10.1038/s43705-023-00293-x
Locatelli, A., Lewis, M. A., and Rothrock, M. J. Jr. (2017). The distribution of Listeria in pasture-raised broiler farm soils is potentially related to University of Vermont Medium Enrichment Bias toward Listeria innocua over Listeria monocytogenes. Front. Vet. Sci. 4:227. doi: 10.3389/fvets.2017.00227
Lomonaco, S., Nucera, D., and Filipello, V. (2015). The evolution and epidemiology of Listeria monocytogenes in Europe and the United States. Infect. Genet. Evol. 35, 172–183. doi: 10.1016/j.meegid.2015.08.008
Lourenco, A., Linke, K., Wagner, M., and Stessl, B. (2022). The saprophytic lifestyle of Listeria monocytogenes and entry into the food-processing environment. Front. Microbiol. 13:789801. doi: 10.3389/fmicb.2022.789801
Lu, Q., Zhu, X., Long, Q., Yi, X., Yang, A., Long, X., et al. (2022). Comparative genomics reveal the utilization ability of variable carbohydrates as key genetic features of Listeria pathogens in their pathogenic lifestyles. Pathogens 11:1430. doi: 10.3390/pathogens11121430
Mafuna, T., Matle, I., Magwedere, K., Pierneef, R. E., and Reva, O. N. (2021). Whole genome-based characterization of Listeria monocytogenes isolates recovered from the food chain in South Africa. Front. Microbiol. 12:669287. doi: 10.3389/fmicb.2021.669287
Mafuna, T., Matle, I., Magwedere, K., Pierneef, R. E., and Reva, O. N. (2022). Comparative genomics of Listeria species recovered from meat and food processing facilities. Microbiol. Spectr. 10:e0118922. doi: 10.1128/spectrum.01189-22
Matle, I., Pierneef, R., Mbatha, K. R., Magwedere, K., and Madoroba, E. (2019). Genomic diversity of common sequence types of Listeria monocytogenes isolated from ready-to-eat products of animal origin in South Africa. Genes (Basel) 10:1007. doi: 10.3390/genes10121007
Mejía, L., Espinosa-Mata, E., Freire, A. L., Zapata, S., and González-Candelas, F. (2023). Listeria monocytogenes, a silent foodborne pathogen in Ecuador. Front. Microbiol. 14:1278860. doi: 10.3389/fmicb.2023.1278860
Orsi, R. H., and Wiedmann, M. (2016). Characteristics and distribution of Listeria spp., including Listeria species newly described since 2009. Appl. Microbiol. Biotechnol. 100, 5273–5287. doi: 10.1007/s00253-016-7552-2
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Palma, F., Pasquali, F., Lucchi, A., De, C. A., and Manfreda, G. (2017). Whole genome sequencing for typing and characterisation of Listeria monocytogenes isolated in a rabbit meat processing plant. Ital. J. Food Saf. 6:6879. doi: 10.4081/ijfs.2017.6879
Pang, R., Xie, T., Wu, Q., Li, Y., Lei, T., Zhang, J., et al. (2019). Comparative genomic analysis reveals the potential risk of Vibrio parahaemolyticus isolated from ready-to-eat foods in China. Front. Microbiol. 10:186. doi: 10.3389/fmicb.2019.00186
Parsons, C., Brown, P., and Kathariou, S. (2021). Use of bacteriophage amended with CRISPR-Cas systems to combat antimicrobial resistance in the bacterial foodborne pathogen Listeria monocytogenes. Antibiotics (Basel) 10:308. doi: 10.3390/antibiotics10030308
Radoshevich, L., and Cossart, P. (2018). Listeria monocytogenes: towards a complete picture of its physiology and pathogenesis. Nat. Rev. Microbiol. 16, 32–46. doi: 10.1038/nrmicro.2017.126
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sheng, L., Edwards, K., Tsai, H. C., Hanrahan, I., and Zhu, M. J. (2017). Fate of Listeria monocytogenes on fresh apples under different storage temperatures. Front. Microbiol. 8:1396. doi: 10.3389/fmicb.2017.01396
Tan, M. F., Siow, C. C., Dutta, A., Mutha, N. V., Wee, W. Y., Heydari, H., et al. (2015). Development of ListeriaBase and comparative analysis of Listeria monocytogenes. BMC Genomics 16:755. doi: 10.1186/s12864-015-1959-5
Toledo, V., Bakker, H. C., Hormazábal, J. C., González-Rocha, G., Bello-Toledo, H., Toro, M., et al. (2018). Genomic diversity of Listeria monocytogenes isolated from clinical and non-clinical samples in Chile. Genes (Basel) 9:396. doi: 10.3390/genes9080396
Wang, Y., Ji, Q., Li, S., and Liu, M. (2021). Prevalence and genetic diversity of Listeria monocytogenes isolated from retail pork in Wuhan, China. Front. Microbiol. 12:620482. doi: 10.3389/fmicb.2021.620482
Wang, C., Ye, Q., Ding, Y., Zhang, J., Gu, Q., Pang, R., et al. (2022). Detection of Pseudomonas aeruginosa serogroup G using real-time PCR for novel target genes identified through comparative genomics. Front. Microbiol. 13:928154. doi: 10.3389/fmicb.2022.928154
Wang, Y., Zhao, A., Zhu, R., Lan, R., Jin, D., Cui, Z., et al. (2012). Genetic diversity and molecular typing of Listeria monocytogenes in China. BMC Microbiol. 12:119. doi: 10.1186/1471-2180-12-119
Zhang, Y., Zheng, Y., Fu, Y., and Wang, C. (2019). Identification of biomarkers, pathways and potential therapeutic agents for white adipocyte insulin resistance using bioinformatics analysis. Adipocytes 8, 318–329. doi: 10.1080/21623945.2019.1649578
Keywords: Listeria monocytogenes, comparative genomics, pan-genomics, biodiversity, target genes
Citation: Zhang B, Ren H, Wang X, Han C, Jin Y, Hu X, Shi R, Li C, Wang Y, Li Y, Lu S, Liu Z and Hu P (2024) Comparative genomics analysis to explore the biodiversity and mining novel target genes of Listeria monocytogenes strains from different regions. Front. Microbiol. 15:1424868. doi: 10.3389/fmicb.2024.1424868
Edited by:
Xiangyu Fan, University of Jinan, ChinaReviewed by:
Sankarasubramanian Jagadesan, University of Nebraska Medical Center, United StatesLongxiang Xie, Henan University, China
Copyright © 2024 Zhang, Ren, Wang, Han, Jin, Hu, Shi, Li, Wang, Li, Lu, Liu and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pan Hu, aHVwYW44NEAxNjMuY29t
†These authors have contributed equally to this work and share first authorship