 Gillian O. N. Ndhlovu1,2
Gillian O. N. Ndhlovu1,2 Kiran G. Javkar3,4
Kiran G. Javkar3,4 Takudzwa Matuvhunye1,2Froodia Ngondoh1,2
Takudzwa Matuvhunye1,2Froodia Ngondoh1,2 Dorota Jamrozy5
Dorota Jamrozy5 Stephen Bentley5
Stephen Bentley5 Adebayo O. Shittu6,7
Adebayo O. Shittu6,7 Felix S. Dube1,2*
Felix S. Dube1,2*- 1Department of Molecular and Cell Biology, Faculty of Science, University of Cape Town, Cape Town, South Africa
- 2Institute of Infectious Disease & Molecular Medicine, University of Cape Town, Cape Town, South Africa
- 3Department of Computer Science, University of Maryland, College Park, MD, United States
- 4Joint Institute for Food Safety and Applied Nutrition, University of Maryland, College Park, MD, United States
- 5Parasites and Microbes Programme, Wellcome Sanger Institute, Hinxton, United Kingdom
- 6Department of Microbiology, Obafemi Awolowo University, Ile-Ife, Osun, Nigeria
- 7Institute of Medical Microbiology, University Hospital Munster, Munster, Germany
Importance: Staphylococcus aureus frequently colonizes the skin and nose of patients with atopic dermatitis (AD), a disease associated with skin barrier dysfunction and chronic cutaneous inflammation. Published genomic studies on AD-associated S. aureus in pediatric populations in sub-Saharan Africa are limited.
Objectives: To investigate the phenotypic and genomic diversity of S. aureus in children with and without AD during early childhood.
Data, setting and participants: A cross-sectional study of 220 children (aged 9–38 months) with AD (cases) and without AD (controls) from Cape Town and Umtata, South Africa.
Main outcomes and measures: S. aureus phenotypic and genomic diversity were investigated using whole-genome sequencing, antibiotic susceptibility testing and biofilm microtiter assay.
Results: Of the 124 S. aureus isolates recovered from 220 children, 96 isolates (79 cases and 17 controls) with high-quality sequences were analyzed. Isolates from cases showed greater phenotypic resistance to gentamicin (10%), rifampicin (4%), chloramphenicol (4%), and exhibited multidrug resistance (9%) than in controls. Furthermore, the isolates from cases formed stronger biofilms than those from controls (76% vs. 35%, p = 0.001), but showed no dominance of any virulence factor gene or mobile genetic elements. There was no significant difference in the distribution of immune evasion cluster types between cases and controls. However, IEC type G was identified only among cases.
Conclusion and relevance: AD-associated S. aureus has phenotypic and genetic features that are important for successful pathogenic colonization and survival. Further studies are needed to assess the pathological implications of colonization of various S. aureus lineages in vivo to elucidate their pathological contribution to AD pathogenesis and pathophysiology.
Introduction
Pathological skin and nasal colonization with Staphylococcus aureus is common in patients with atopic dermatitis (AD)—a chronic or recurrent inflammatory skin disease which affects about 20% of children globally (Gilaberte et al., 2020). Due to the frequent association of S. aureus infections with AD, antibiotics are routinely used in the management of AD. However, the effectiveness of antibiotics in AD is limited by the rapid recolonisation with S. aureus following treatment cessation (di Domenico et al., 2019a). The excessive use of antibiotics may lead to significant changes in the skin microbial community and emergence of antibiotic-resistant isolates, which are associated with severe forms of AD (Jung et al., 2015; Shi et al., 2016). S. aureus contributes to AD pathology through virulence factors and biofilms, which are commonly detected in skin lesions of children with AD (Allen et al., 2014; Sonesson et al., 2017; di Domenico et al., 2018). Further, adhesins, toxins, proteases and significant changes in antigens enable the bacterium to adhere to host tissues, leading to impaired skin barrier function and inflammation (di Domenico et al., 2019b). Previous studies have shown that patients that are colonized with S. aureus strains which form strong biofilms, experience severe forms of the disease (Ogonowska et al., 2020; Gonzalez et al., 2021).
Some genes associated with antibiotic resistance, biofilm formation, and virulence in S. aureus are carried on mobile genetic elements (MGEs), such as plasmids, bacteriophages (prophages) and staphylococcal pathogenicity islands (SaPIs) (Malachowa and DeLeo, 2010). These are central to the adaptation of S. aureus in different environments (Malachowa and DeLeo, 2010). (Harkins et al., 2018) Moreover, lineage-specificity has been observed among MGEs (McCarthy et al., 2012; McCarthy and Lindsay, 2012). Recent data suggest that AD is associated with specific S. aureus lineages (Harkins et al., 2018), with clonal complex (CC) 1 being positively associated with AD in European children, while CC30 being negatively associated with AD (Rojo et al., 2014; Harkins et al., 2018). These differences in S. aureus lineages appear to be accompanied by differences in the carriage of virulence factors and adaptability to AD (Hwang et al., 2021).
It is therefore important to identify genomic markers of S. aureus that are associated with AD pathology in children with AD from their non-AD counterparts. Moreover, there is limited data on S. aureus strain biology from sub-Saharan Africa. We therefore investigated the phenotypic and genomic factors important for S. aureus adaptation on the skin and in the anterior nares of children with AD compared with healthy pediatric participants.
Materials and methods
Collection of Staphylococcus aureus isolates
We used the S. aureus isolates collected from our previously published work (Ndhlovu et al., 2021). Briefly, samples were collected using sterile nylon-tipped flocked swabs (Cat. no. 516C; Copan Italia, Brescia, Italy) from the lesional and non-lesional skin and anterior nares of children (n = 220) with and without AD from Umtata and Cape Town, South Africa (HREC/REF: 451/2014). The swabs were placed into skim milk-tryptone-glucose-glycerol (STGG) and transported to the laboratory within 2 h of collection at 4°C and stored in the freezers (−80°C) for further analysis. A total of 124 S. aureus isolates were recovered after the swabs were inoculated onto Mannitol Salt Agar (MSA) (National Health Laboratory Services [NHLS], Green Point Media Laboratory Cape Town, South Africa), and incubated at 37°C for 48 h in aerobic conditions. The S. aureus colonies were confirmed by testing for mannitol fermentation and DNase production (Kateete et al., 2010). Detailed information on the isolate collection can be found in Ndhlovu et al. (2021).
Assessment of biofilm formation of Staphylococcus aureus isolates
S. aureus isolates were assessed for biofilm formation in vitro using the crystal violet biofilm microtiter assay (MTA) as previously described (Stepanović et al., 2007; Gonzalez et al., 2021). Biofilm biomass OD readings were interpreted as follows: (OD ≤ OD negative control [ODc], non-producer; ODc < OD < 2·ODc, weak producer; 2·ODc < OD < 4·ODc, moderate producer; and OD > 4·ODc, strong producer) (Stepanović et al., 2007).
Antibiotic susceptibility testing of Staphylococcus aureus isolates
Antibiotic susceptibility testing (AST) was conducted using the Kirby-Bauer disk diffusion method as previously described and resistance to cefoxitin used as a surrogate for methicillin resistance (Wang et al., 2005). S. aureus ATCC 25923 was used as the quality control. The D-test was used to distinguish constitutive and inducible resistance to the macrolide-lincosamide-streptogramin B (MLSB) group of antibiotics (Yao et al., 2019). The AST was interpreted according to 2021 EUCAST guidelines and breakpoints. Isolates that were resistant to at least three classes of antibiotics were classified as multidrug-resistant (MDR) (Magiorakos et al., 2012).
DNA extraction, library preparation, and whole-genome sequencing
Total DNA extraction was performed using a heat lysis method as described previously in Ndhlovu et al (Ndhlovu et al., 2021), and quantified using the Biotium Accuclear Ultra-high sensitivity dsDNA Quantitative kit. Samples were cherry-picked to 200 ng/120 μL using the Tecan liquid handling platform and sheared to 450 bp using a Covaris LE220 instrument.
Post sheared samples (in 96 well plates) were purified using SPRISelect beads and rearrayed into a in 384 plate on the Hamilton STAR. Library construction (ER, A-tailing, and ligation) was performed using the ‘NEB Ultra II custom kit’ on an Agilent Bravo WS automation system. PCR setup was performed using KapaHiFi Hot start mix and 384 well UDI tag barcodes on the Agilent Bravo WS automation system. PCR conditions were as follows: initial incubation at 95°C for 5 min followed by five cycles of 98°C for 30 s, 65°C for 30 s and 72°C for 1 min, and a final incubation step at 72°C for 10 min. Post PCR plates were pooled using equal volumes and purified using Agencourt AMPure XP SPRI beads on the Hamilton STAR. Libraries were quantified using an Agilent Bioanalyser, normalized to 2.8 nM, and prepared for sequencing on a NovaSeq (Page et al., 2016).
Genome assembly, MLST analysis, gene prediction and gene clustering
All bioinformatics tools listed herewith were used with their default parameter settings unless any custom parameter was explicitly stated. Pre-processed reads were de novo assembled using Velvet v1.2.10 (Zerbino, 2010). The species identity of each genome was confirmed using KmerFinder v3.2 (Hasman et al., 2014; Larsen et al., 2014). High-quality genomes with <150 contigs and N50 > 50,000 (Manara et al., 2018) were included in the subsequent analyses. In silico multi-locus sequence typing (MLST) and spa typing were conducted using the MLST tool v2.0 and spaTyper v1.0, respectively. MLST clonal complexes (CCs) were inferred from PubMLST.1 Virulence factors were detected using the VirulenceFinder v2.0 tool (Kleinheinz et al., 2014) and the Virulence Factors of Pathogenic Bacteria (VFPB 2022) database (Liu et al., 2019). Antibiotic resistance determinants were identified using the Resistance Gene Identifier v5.2.0; accessed via the Comprehensive Antibiotic Resistance Database (CARD) v4.0.2 (Alcock et al., 2020). and ResFinder v4.4.3 (Florensa et al., 2022). The staphylococcal cassette chromosome mec (SCCmec) types of mecA-positive MRSA isolates were determined by the SCCmecFinder v1.2 (Kaya et al., 2018). Identification of putative plasmids was performed by using the PlasmidFinder v2.1.1 (Carattoli and Hasman, 2020). The size and associated genes of plasmids identified by PlasmidFinder were obtained from NCBI (Boratyn et al., 2013). The PHAge Search Tool Enhanced Release (PHASTER) algorithm (Arndt et al., 2016) was used to identify prophage sequences from the S. aureus genomes. Insertion sequence (IS) elements were identified from Prokka v1.14.6 (Seemann, 2014) and annotated genomes using the ISfinder v2.0 (Siguier et al., 2006). Analyses were limited to replicon, prophage, and IS elements identified in 10 or more genomes.
Phylogenetic tree construction and annotation
Single nucleotide polymorphism based phylogenetic analysis was conducted using the Reference sequence Alignment based Phylogeny builder (REALPHY) v1.13 (Bertels et al., 2014) with default settings. Staphylococcus aureus NCTC 8325 was used as a reference. The generated phylogenetic trees were visualised and annotated using the iTOL (Interactive Tree of Life) tool v6 (Letunic and Bork, 2021).
Statistical analyses
Unless stated otherwise, all statistical analyses were performed in STATA SE 15.1 (StataCorp LP, Texas, USA). The two-sample z-test was applied to compare the proportions. Cohen’s kappa statistic was performed to assess the agreement between phenotypic and genotypic susceptibility and resistance to antibiotics. The κ coefficient was interpreted as no agreement (κ < 0–0.2), minimal agreement (κ = 0.21–0.39), weak agreement (κ = 0.4–0.59), moderate agreement (κ = 0.6–0.79), strong agreement (κ = 0.8–0.9), and almost perfect agreement (κ >0.9) (McHugh, 2012).
Results
Participant characteristics and Staphylococcus aureus population structure
Supplementary Table S1 describes the participant’s characteristics. Of the 124 S. aureus isolates, high-quality genomes were obtained from 96 isolates (79 cases and 17 controls) (see Materials and Methods section for the high-quality genome selection criteria). The average size of the assembled genomes was 2.80 Mb (2.71–2.98 Mb), with an average of 25 (10–79) contigs per genome. Each genome had approximately 2,665 (2538–2,936) predicted protein sequences and a GC content of 32.7% (32.6–32.9%). The S. aureus isolates were grouped into 20 STs, which further clustered into nine CCs and three singletons (Figure 1). Of these CC5 (ST5, ST650, and ST4005) and CC8 (ST8, ST72, and ST612) were predominant, accounting for 24% (23/96) and 25% (24/96) of the isolates, respectively (Supplementary Table S2).
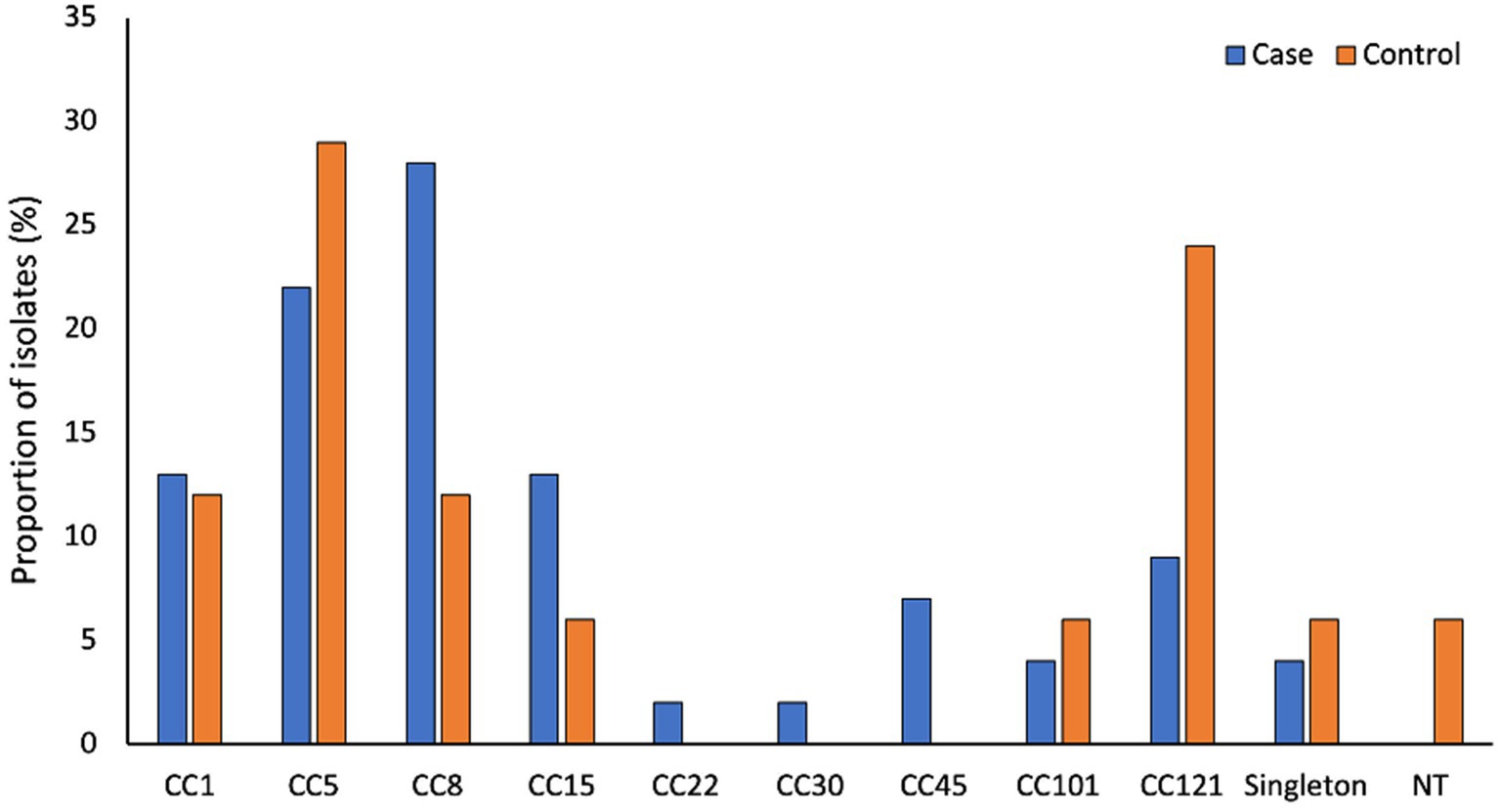
Figure 1. Genetic diversity of S. aureus associated with AD. CC1 (ST1), CC5 (ST5, ST4005, ST650), CC8 (ST8, ST72 and ST612), CC15 (ST15, ST199 and ST2126), CC22 (ST22), CC30 (ST31), CC45 (ST45 and SST508), CC101 (ST101) and CC121 (ST121). Singletons include ST12 (n = 2), ST20 (n = 1) and ST88 (n = 1).
Distribution of mobile genetic elements (MGEs) among cases and controls
Plasmid analysis revealed 27 unique plasmids based on rep genes. Plasmid replicons were not identified in four isolates (4.2% [4/96]), all from cases. Lineage-association was limited to three plasmids, namely pBORa53 (CC8), SAP074A (CC5), and SAP057A (CC121) (Supplementary Table S3). Plasmids pN315, pSJH101 and pMSSA476, carrying penicillin (blaZ) and cadmium (cadD) resistance genes, were the most frequently identified in both cases (47% [37/79], 19% [17/79], and 41% [32/79], respectively) and controls (41% [7/17], 35% [6/17], and 24% [4/17], respectively) (Table 1). Plasmid pE-1(EDINA), which carries the enterotoxin E (see), edinA, as well as cadmium and arsenic resistance genes, was more common among controls than cases (29% [5/17] vs. 6% [5/79], p = 0.005) (Table 1).
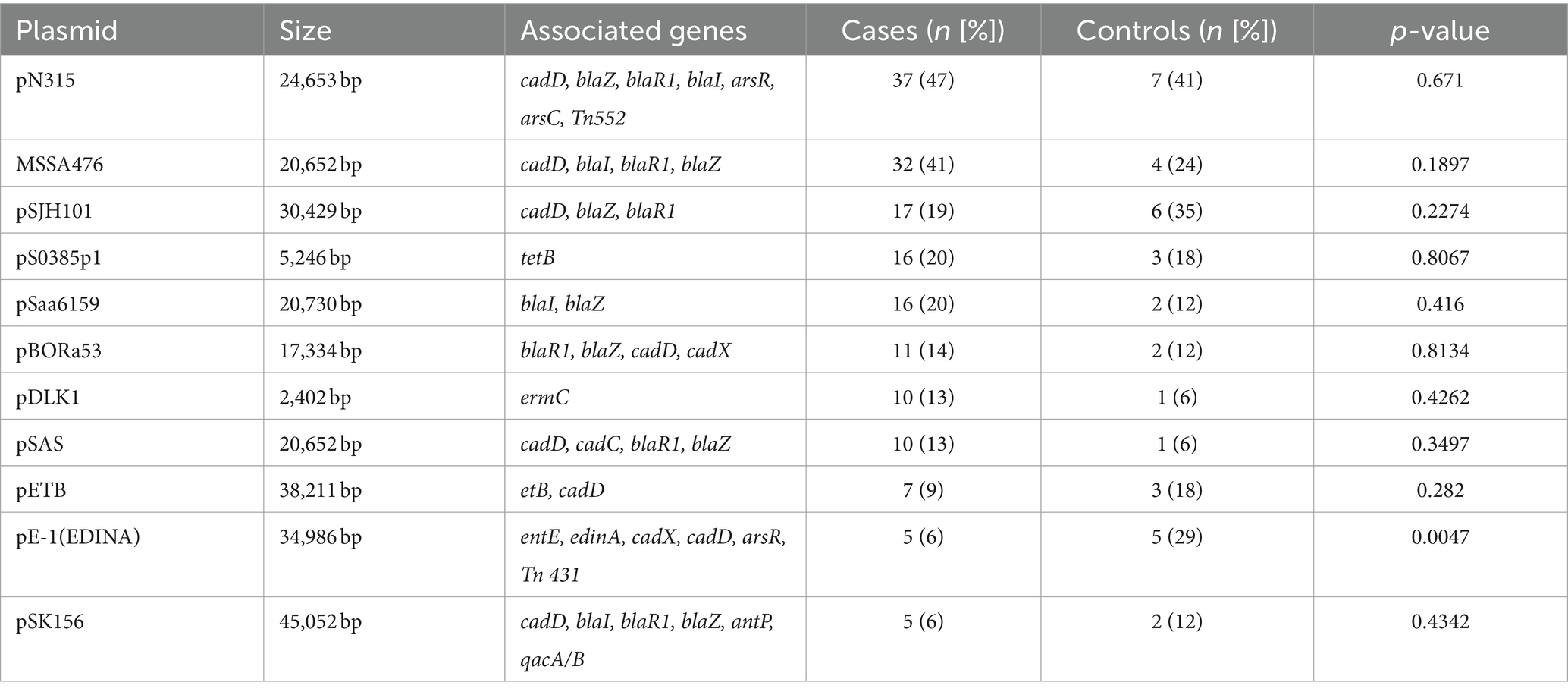
Table 1. The distribution of plasmids, present in at least 10 genomes, in cases and controls.
We predicted 392 prophage genomes from 96 S. aureus isolates, of which, 150 (38%) were intact/complete. The 150 intact prophages were identified in 95% (91/96) of the isolates: 96% (76/79) of cases and 88% (15/17) of controls. All intact prophages belonged to the family Siphoviridae and various genera including Biseptimavirus, Dubowvirus, Peeveelvirus, Phietavirus, and Triavirus (Supplementary Table S4). Phage integrase genes were found in 68% (102/150) of the intact prophages. The predicted intact prophages were grouped into 31 distinct prophages. No significant association was observed between the lineage and the identified intact prophages (Supplementary Table S4). However, some prophages, such as Staphylococcus phages phiETA (CC121), SA97, and phiN315 (CC5), and phage 71 (CC45) only had one lineage (Supplementary Table S4). We limited our analysis to prophages that were identified in at least 10 genomes (Table 2). Of these, Staphylococcus prophage phi2958PVL was the most common (34% [33/96]) and was more frequently identified among cases than controls (41% [32/79] vs. 6% [1/17], p = 0.0064) (Table 2).
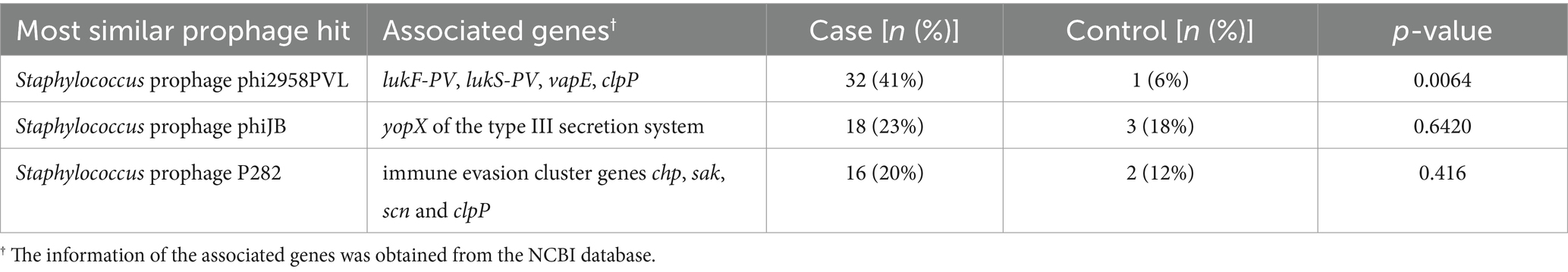
Table 2. The distribution of prophages, present in at least 10 genomes, in cases and controls.
Table 3 shows the distribution of IS elements in cases and controls. ISSau3 was detected in all case and control isolates. ISSau5 was only identified in cases (14% [11/79]) and ISSep3 was more prevalent in cases than controls (54% [43/79] vs. 29% [5/17], p = 0.061). Also, ISCsp1 and ISSau6 were more common among controls than cases (71% [12/17] vs. 37% [29/79], p = 0.0104 and 53% [9/17] vs. 23% [18/79], p = 0.0121, respectively).
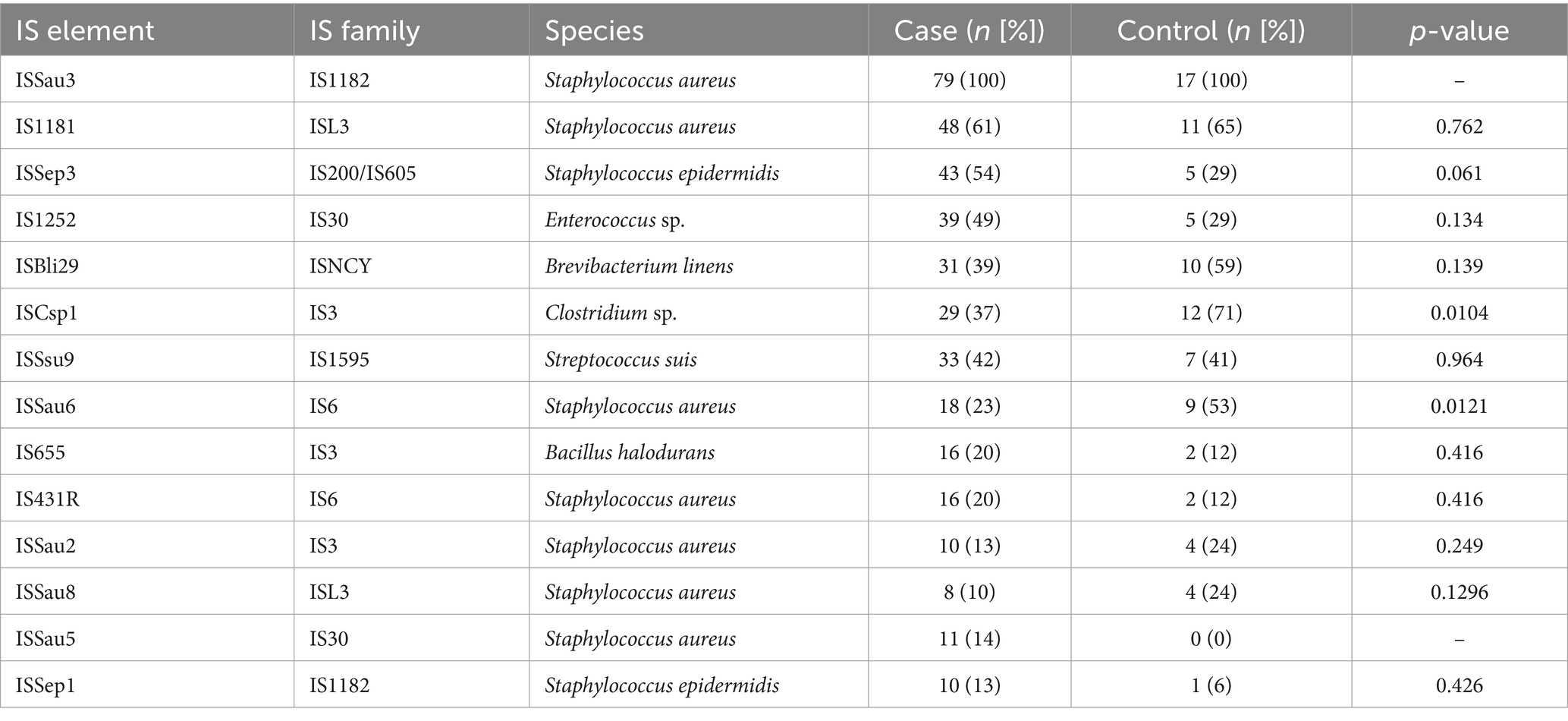
Table 3. The distribution of IS elements, present in at least 10 genomes, in cases and controls.
Prevalence of phenotypic and genotypic antibiotic non-susceptibility
Phenotypic susceptibility to all antibiotics was similar in cases (61%; 48/79) and controls (59%; 10/17, p = 0.882) (Supplementary Figure S2). Phenotypic resistance to chloramphenicol (4% [3/77]), gentamicin (9% [7/77]) and rifampicin (4% [3/78]) was low, being detected exclusively among cases. The prevalence of MRSA was also low in both cases (8% [6/78]) and controls (6% [1/17]). Phenotypic MDR isolates were only detected in cases (9% [7/79]) (Supplementary Figure S1). In silico determination of antibiotic resistance revealed ARGs for methicillin (mecA), tetracycline (tet45, tetM, and tetK), gentamicin (acc(6′)-le-aph(2″)-la), rifampicin (double mutation in RpoB; H481N and I527M), chloramphenicol (cat(pC221)), and MLSB (ermC and msrA) (Supplementary Figure S3). There was moderate to almost perfect concordance between phenotypic and genotypic resistance to all antibiotics tested, except for trimethoprim-sulfamethoxazole (Supplementary Table S6). MRSA was identified in cases comprising the multidrug-resistant CC5-ST5-SCCmecIV and CC8-ST612-SCCmecIV (Supplementary Figures S5, S6).
Phenotypic biofilm formation and biofilm-associated genes
Overall, most isolates were strong biofilm producers (72% [69/96]), followed by moderate (17% [16/96]) and weak (11% [11/96]). Strong biofilm producers were more common in cases (76% [60/79]) than in controls (35% [6/17], p = 0.001). The prevalence of isolates with moderate (14% [11/79] cases vs. 29% [5/17] controls, p = 0.120) and weak biofilm formation (10% [8/79] cases vs. 18% [3/17] controls, p = 0.377) was similar in the two groups (Figure S4).
Prevalence of virulence genes
There was a low prevalence of staphylococcal superantigen genes, except for the enterotoxin gene cluster (EGC; comprises of seg, sei, sem, sen, seo, and seu) (48% [46/96]) (Figure 2). About 20% (20/96) of the isolates analysed were positive for the cytolysin genes lukF-PV and lukS-PV. More than 90% (90/96) of all isolates were lukED-positive. The toxin genes edinC (5% [4/79]), etB (4% [3/79]) and sed (9% [7/79]) were detected only in cases. Furthermore, edinA and seb were more frequently identified among controls than cases (24% [4/17] vs. 6% [5/79], p = 0.049 and 29% [5/17] vs. 1% [1/79], p = 0.001, respectively) (Figure 2).
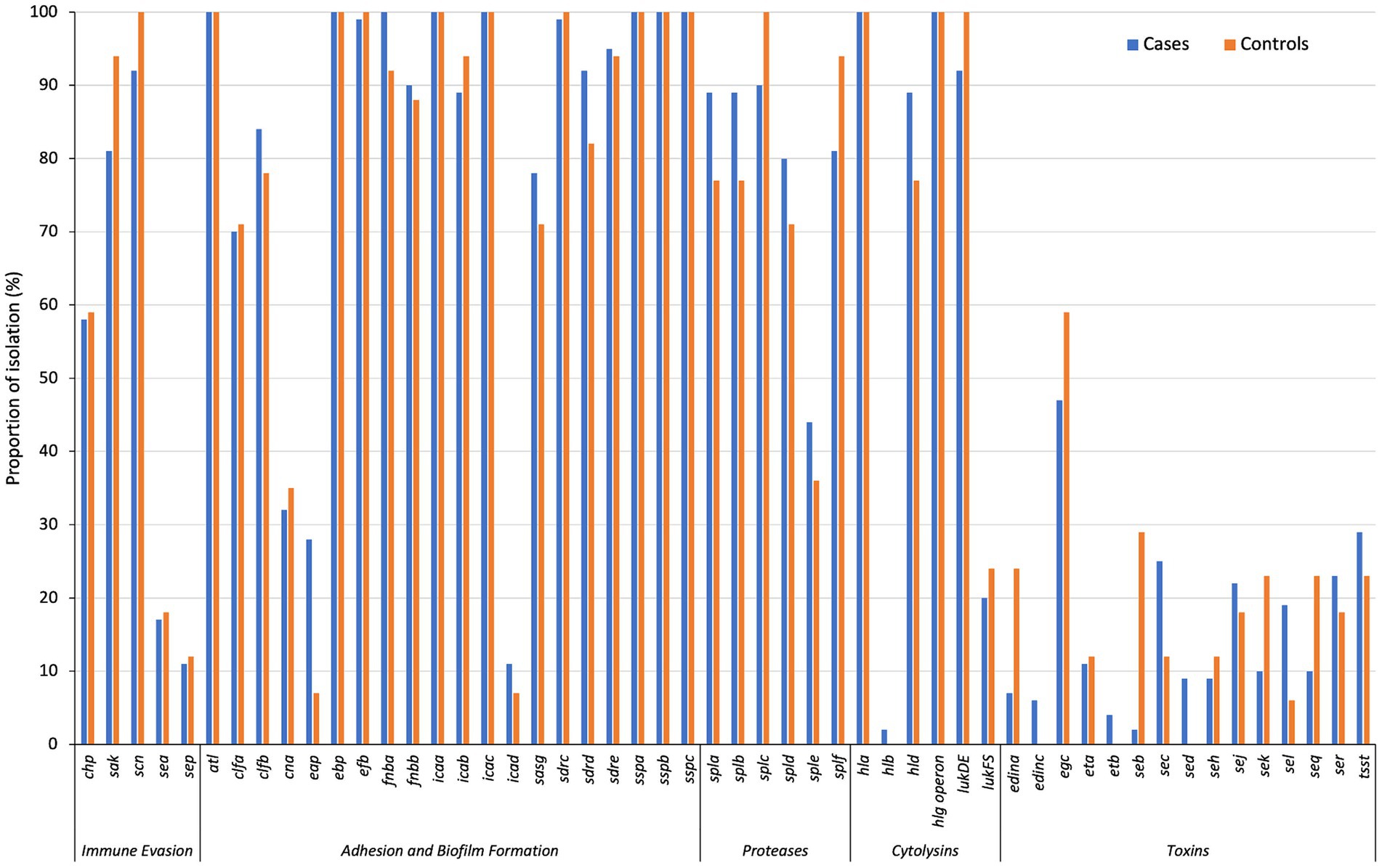
Figure 2. The distribution of virulence-associated genes in cases and controls, grouped by functional class. *p < 0.05; ***p < 0.001.
Clustering of CCs with distinct antibiotic and virulence gene profiles Figure S6. CC1 isolates were seh-positive, while 58% (7/12) and 75% (9/12) were positive for the lukFS-PV and sea/sek/seq genes, respectively. The egc cluster was present in all CC5-ST5, CC8-ST72, CC121 and CC45-ST508 isolates. Most CC8-ST8 isolates carried tsst (78% [14/18]) and sej (72% [13/18]). The exfoliative toxin encoding genes (eta and etb) were detected only in CC15-ST15 (n = 4; eta only) and CC121 (n = 8; eta and or etb). The epidermal cell differentiation inhibitor (EDIN) genes were identified only in CC5-ST5 (n = 9, edinA) and CC121 (n = 4; edinC).
Distribution of the immune evasion cluster genes
Regarding the immune evasion cluster (IEC) genes, most isolates carried scn (94%, 90/96), followed by sak (83%, 80/96), chp (57%, 55/96), sea (17%, 16/96) and sep (11% [11/96]); with frequencies similar between cases and controls (Figure S5). Six isolates, all from cases, did not carry any of the IEC genes. Of the various IEC groups, type B (sak-chp-scn) was predominant (37% [33/90]), followed by type E (sak-scn; 22% [20/90]), type D (sea-sak-scn; 13% [12/90]), type C (chp-scn; 11% [10/90]), type F (sep-sak-chp-scn; 8% [7/90]), and type A (sea-sak-chp-scn) and type G (sep-sak-scn); each with four isolates (4%).
Discussion
This study shows that S. aureus isolates associated with AD tended to form stronger biofilms and carried genetic features that are linked to antibiotic resistance and virulence with potential clinical relevance to AD pathology.
The low prevalence of antibiotic resistance in our study is consistent with previous reports from Irish (Harkins et al., 2018), Polish (Wrobel et al., 2018), and Italian (di Domenico et al., 2018) pediatric patients with AD. The prevalence of MRSA strains was also low in both cases (8%) and controls (6%), as previously demonstrated (Harkins et al., 2018). Moreover, MDR S. aureus isolates were only detected among cases, suggesting frequent use of antibiotics or exposure to resistant strains, possibly from a hospital environment. Considering the low prevalence of AD in our setting, the proportion of MDR strains among children with AD may not reflect the true burden. Larger studies may be needed to further explore the impact of S. aureus antibiotic resistance on AD pathology.
S. aureus produces a variety of toxins, cytotoxins, proteases, antigens and adhesins that are key to its successful colonization, pathogenicity, and survival (di Domenico et al., 2019b). Epidemiological studies have shown that AD-associated S. aureus carry more virulence genes than controls (Na et al., 2012; Rojo et al., 2014; Cavalcante et al., 2015; Wang et al., 2016), which is also linked to increased disease severity (Rojo et al., 2014). Presently, the cases did not exhibit a predominance of virulence factor genes. Although no significant virulence factor genes were observed, it is likely that the disease environment could trigger more expression of the virulence factors resulting in disease pathology (Poh et al., 2022).
Biofilm formation is associated with increased disease severity (Pascolini et al., 2011; Allen et al., 2014; di Domenico et al., 2018). Particularly strong biofilms, protect S. aureus from (i) environmental factors (including antibiotics), (ii) the bactericidal effects of host antimicrobial peptides (AMPs) and, (iii) host immune responses (di Domenico et al., 2019a). In this regard, strong biofilm producers were more common among cases similar to previous studies (Allen et al., 2014; Sonesson et al., 2017; di Domenico et al., 2018). This suggests that AD-associated S. aureus isolates were better able to form biofilms and adapted for persistent colonization. These findings support the importance of strong biofilm formation in driving persistent colonization in children with AD. Also, persistent colonization with S. aureus in AD may be detrimental to the child as this provides a continuous source of virulence factors that will further perpetuate the clinical manifestations of AD and contribution to disease flares. As such, treatment strategies in AD need to have anti-biofilm capabilities to effectively reducing S. aureus colonization and mitigating its harmful effects.
Immune evasion by S. aureus is mediated by the immune evasion cluster (IEC), which modulates innate immune responses in humans (Verkaik et al., 2011). IEC includes SCIN, and a varying combination of SAK, CHIPS and staphylococcal enterotoxins, SEA or SEP, across different IEC types (van Wamel et al., 2006). We detected IEC genes in the S. aureus control group and most (92%) of the cases. Further, cases and controls did not differ substantially in the distribution of IEC types. Similar to previous reports (van Wamel et al., 2006; Verkaik et al., 2011; Silva et al., 2020), IEC type B, were independent of disease status, whereas the type G isolates were only identified among cases (Benito et al., 2016). Although the specific functions of the IEC genes and their contribution to AD are addressed in the literature (Rojo et al., 2014; Jian et al., 2021), the clinical relevance of different IEC types in AD is lacking and warrants further study.
Insertion sequences (IS) are small MGEs and are generally between 700–2,500 bp, affecting the genomic plasticity, diversity, and pathogenic potential of S. aureus (Kuroda et al., 2001). We found that ISCsp1 and ISSau6 were more common among controls. While there was no IS element more common in cases than controls, we observed that ISSau3, IS1181, ISSep3 were the three most frequently detected IS elements in cases. ISSau3 has been shown to increase resistance to beta-lactam antibiotics (Wang et al., 2021), the roles of IS1181 and ISSep3 are poorly studied in literature, especially in disease context.
There is increasing evidence that specific S. aureus lineages are adapted for induction of disease activity and survival in AD patients (Yeung et al., 2011; Byrd et al., 2017; Fleury et al., 2017; Harkins et al., 2018). In this study, we noted no significant difference in the clonality of S. aureus isolates from cases and controls. However, CC8 isolates predominated among cases, while CC121 isolates were the most common among controls. The specific features or mechanisms that favor the predominance of certain lineages in AD remain unknown (Geoghegan et al., 2018). However, it has been suggested that in AD, differences in the virulence potential across lineages are associated with the proliferation of some lineages and other lineages are associated with asymptomatic colonization in controls (Harkins et al., 2018).
Limitations to this study include the small sample size and our investigation focused on the recovery of a single colony which does not provide a comprehensive understanding of the clonality of S. aureus colonization in AD (Byrd et al., 2017). Lastly, we noted a higher prevalence of incomplete and questionable prophages compared to intact prophages. This is likely because intact prophages are usually under strong selection or genetic degradation for rapid deletion from bacterial genomes (Lawrence et al., 2001). However, since genomes are divided into contigs, some prophage sequences may have been split into different contigs and therefore misidentified as incomplete or questionable prophages by PHASTER (Marques et al., 2021). Moreover, the PHASTER tool also relies on previously annotated prophage sequences and, therefore, cannot identify novel prophage sequences. Subsequent studies will be focused on exploring the overall genomic factors beyond a specific set of genes, using tools like PRAWNS (Javkar et al., 2023).
Conclusion
This study provides an understanding of the genomic differences of S. aureus in early childhood AD, in a previously unstudied population in South Africa. Although we did not observe a clear phylogenetic and clonal differentiation of S. aureus based on AD disease, specific phenotypic and genetic signatures distinguished AD isolates from controls. Specifically, AD-associated isolates exhibited phenotypic features related to biofilm formation, and genomic features associated with DNA damage repair and toxins which are important for the persistent colonization and propagation of AD disease.
Data availability statement
Supplementary results on S. aureus colonization based on AD and health, S. aureus colonization based on disease severity, study spa types and MLST sequence types are available in the in the Supplementary Data Sheet S3. The sequence data presented in this study are available on online repositories, European Nucleotide Archive (accession numbers are in Supplementary Table S2).
Ethics statement
The studies involving humans were approved by Human Research Ethics Committee of the Faculty of Health Sciences, University of Cape Town and the Western Cape Provincial Child Health Research Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
GN: Formal analysis, Writing – review & editing, Data curation, Writing – original draft, Methodology, Investigation, Validation. KJ: Data curation, Formal analysis, Methodology, Software, Validation, Writing – review & editing, Writing – original draft. TM: Investigation, Methodology, Writing – review & editing, Writing – original draft. FN: Investigation, Writing – original draft, Formal analysis, Writing – review & editing, Methodology. DJ: Software, Writing – review & editing, Formal analysis, Data curation, Writing – original draft, Methodology, Investigation. SB: Supervision, Conceptualization, Writing – original draft, Writing – review & editing. AS: Conceptualization, Writing – review & editing, Supervision, Writing – original draft. FD: Resources, Investigation, Writing – original draft, Funding acquisition, Visualization, Project administration, Validation, Formal analysis, Conceptualization, Data curation, Writing – review & editing, Methodology, Supervision.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. We acknowledge the Medical Research Council of South Africa, Nestlé Foundation, Mylan, Thermo Fisher Scientific funding of the parent study, and the Allergy Society of South Africa (ALLSA) for funding of this study. GN acknowledges the National Research Foundation and the University of Cape Town Vice Chancellor’s Research Scholarship for their financial assistance towards her PhD degree. AS is currently an awardee of the Georg Forster Research Fellowship (for Experienced Researchers) of the Alexander von Humboldt Foundation. FD is supported by the National Research Foundation of South Africa (112160), Future Leaders – African Independent Research (FLAIR) Fellowship, NIHR-MPRU, the University of Cape Town and the Allergy Society of South Africa (ALLSA). FD was supported by the National Research Foundation of South Africa, The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgments
We thank Susannah Salter, University of Cambridge, Anne von Gottberg, Linda de Gouveia and the staff of the Center for Respiratory Diseases and Meningitis (CRDM), National Institute for Communicable Diseases (NICD) of the National Health Laboratory Service for training, sharing of standard operating procedures, supplying control isolates.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1422902/full#supplementary-material
Footnotes
References
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Allen, H. B., Vaze, N. D., Choi, C., Hailu, T., Tulbert, B. H., Cusack, C. A., et al. (2014). The presence and impact of biofilm-producing staphylococci in atopic dermatitis. JAMA Dermatol. 150, 260–265. doi: 10.1001/jamadermatol.2013.8627
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Benito, D., Aspiroz, C., Gilaberte, Y., Sanmartín, R., Hernández-Martin, Á., Alonso, M., et al. (2016). Genetic lineages and antimicrobial resistance genotypes in Staphylococcus aureus from children with atopic dermatitis: detection of clonal complexes CC1, CC97 and CC398. J. Chemother. 28, 359–366. doi: 10.1179/1973947815Y.0000000044
Bertels, F., Silander, O. K., Pachkov, M., Rainey, P. B., and van Nimwegen, E. (2014). Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 31, 1077–1088. doi: 10.1093/molbev/msu088
Boratyn, G. M., Camacho, C., Cooper, P. S., Coulouris, G., Fong, A., Ma, N., et al. (2013). BLAST: a more efficient report with usability improvements. Nucleic Acids Res. 41, W29–W33. doi: 10.1093/nar/gkt282
Byrd, A. L., Deming, C., Cassidy, S. K. B., Harrison, O. J., Ng, W. I., Conlan, S., et al. (2017). Staphylococcus aureus and Staphylococcus epidermidis strain diversity underlying pediatric atopic dermatitis. Sci. Transl. Med. 9:397. doi: 10.1126/scitranslmed.aal4651
Carattoli, A., and Hasman, H. (2020). PlasmidFinder and in silico pMLST: identification and typing of plasmid replicons in whole-genome sequencing (WGS). Methods Mol. Biol. 2075, 285–294. doi: 10.1007/978-1-4939-9877-7_20
Cavalcante, F. S., Abad, E. D., Lyra, Y. C., Saintive, S. B., Ribeiro, M., Ferreira, D. C., et al. (2015). High prevalence of methicillin resistance and PVL genes among Staphylococcus aureus isolates from the nares and skin lesions of pediatric patients with atopic dermatitis. Braz. J. Med. Biol. Res. 48, 588–594. doi: 10.1590/1414-431X20154221
di Domenico, E. G., Cavallo, I., Bordignon, V., Prignano, G., Sperduti, I., Gurtner, A., et al. (2018). Inflammatory cytokines and biofilm production sustain Staphylococcus aureus outgrowth and persistence: a pivotal interplay in the pathogenesis of atopic dermatitis. Sci. Rep. 8:9573. doi: 10.1038/s41598-018-27421-1
di Domenico, E. G., Cavallo, I., Capitanio, B., Ascenzioni, F., Pimpinelli, F., Morrone, A., et al. (2019a). Staphylococcus aureus and the cutaneous microbiota biofilms in the pathogenesis of atopic dermatitis. Microorganisms 7:301. doi: 10.3390/microorganisms7090301
di Domenico, E. G., Cavallo, I., Capitanio, B., Ascenzioni, F., Pimpinelli, F., Morrone, A., et al. (2019b). Exploring the role of Staphylococcus aureus toxins in atopic dermatitis. Toxins (Basel) 11:321. doi: 10.3390/toxins11060321
Fleury, O. M., McAleer, M. A., Feuillie, C., Formosa-Dague, C., Sansevere, E., Bennett, D. E., et al. (2017). Clumping factor B promotes adherence of Staphylococcus aureus to Corneocytes in atopic dermatitis. Infect. Immun. 85:1128. doi: 10.1128/IAI.00994-16
Florensa, A. F., Kaas, R. S., Clausen, P., Aytan-Aktug, D., and Aarestrup, F. M. (2022). ResFinder - an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom. 8:000748. doi: 10.1099/mgen.0.000748
Geoghegan, J. A., Irvine, A. D., and Foster, T. J. (2018). Staphylococcus aureus and atopic dermatitis: a complex and evolving relationship. Trends Microbiol. 26, 484–497. doi: 10.1016/j.tim.2017.11.008
Gilaberte, Y., Pérez-Gilaberte, J. B., Poblador-Plou, B., Bliek-Bueno, K., Gimeno-Miguel, A., and Prados-Torres, A. (2020). Prevalence and comorbidity of atopic dermatitis in children: a large-scale population study based on real-world data. J. Clin. Med. 9:1632. doi: 10.3390/jcm9061632
Gonzalez, T., Stevens, M. L., Baatyrbek kyzy, A., Alarcon, R., He, H., Kroner, J. W., et al. (2021). Biofilm propensity of Staphylococcus aureus skin isolates is associated with increased atopic dermatitis severity and barrier dysfunction in the MPAACH pediatric cohort. Allergy 76, 302–313. doi: 10.1111/all.14489
Harkins, C. P., McAleer, M. A., Bennett, D., McHugh, M., Fleury, O. M., Pettigrew, K. A., et al. (2018). The widespread use of topical antimicrobials enriches for resistance in Staphylococcus aureus isolated from patients with atopic dermatitis. Br. J. Dermatol. 179, 951–958. doi: 10.1111/bjd.16722
Harkins, C. P., Pettigrew, K. A., Oravcová, K., Gardner, J., Hearn, R. M. R., Rice, D., et al. (2018). The microevolution and epidemiology of Staphylococcus aureus colonization during atopic eczema disease flare. J. Invest. Dermatol. 138, 336–343. doi: 10.1016/j.jid.2017.09.023
Hasman, H., Saputra, D., Sicheritz-Ponten, T., Lund, O., Svendsen, C. A., Frimodt-Møller, N., et al. (2014). Rapid whole-genome sequencing for detection and characterization of microorganisms directly from clinical samples. J. Clin. Microbiol. 52, 139–146. doi: 10.1128/JCM.02452-13
Hwang, J., Thompson, A., Jaros, J., Blackcloud, P., Hsiao, J., and Shi, V. Y. (2021). Updated understanding of Staphylococcus aureus in atopic dermatitis: from virulence factors to commensals and clonal complexes. Exp. Dermatol. 30, 1532–1545. doi: 10.1111/exd.14435
Javkar, K., Rand, H., Strain, E., and Pop, M. (2023). PRAWNS: compact pan-genomic features for whole-genome population genomics. Bioinformatics 39:btac844. doi: 10.1093/bioinformatics/btac844
Jian, Y., Zhao, L., Zhao, N., Lv, H. Y., Liu, Y., He, L., et al. (2021). Increasing prevalence of hypervirulent ST5 methicillin susceptible Staphylococcus aureus subtype poses a serious clinical threat. Emerg. Microbes Infect. 10, 109–122. doi: 10.1080/22221751.2020.1868950
Jung, M. Y., Chung, J. Y., Lee, H. Y., Park, J., Lee, D. Y., and Yang, J. M. (2015). Antibiotic susceptibility of Staphylococcus aureus in atopic dermatitis: current prevalence of methicillin-resistant Staphylococcus aureus in Korea and treatment strategies. Ann. Dermatol. 27, 398–403. doi: 10.5021/ad.2015.27.4.398
Kateete, D. P., Kimani, C. N., Katabazi, F. A., Okeng, A., Okee, M. S., Nanteza, A., et al. (2010). Identification of Staphylococcus aureus: DNase and mannitol salt agar improve the efficiency of the tube coagulase test. Ann. Clin. Microbiol. Antimicrob. 9, 1–7. doi: 10.1186/1476-0711-9-23
Kaya, H., Hasman, H., Larsen, J., Stegger, M., Johannesen, T. B., Allesøe, R. L., et al. (2018). SCCmecFinder, a web-based tool for typing of staphylococcal cassette chromosome mec in Staphylococcus aureus using whole-genome sequence data. Clin. Vaccine Immunol. 3:e00612-17. doi: 10.1128/mSphere.00612-17
Kleinheinz, K. A., Joensen, K. G., and Larsen, M. V. (2014). Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and E. coli virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage 4:e27943. doi: 10.4161/bact.27943
Kuroda, M., Ohta, T., Uchiyama, I., Baba, T., Yuzawa, H., Kobayashi, I., et al. (2001). Whole genome sequencing of meticillin-resistant Staphylococcus aureus. Lancet 357, 1225–1240. doi: 10.1016/s0140-6736(00)04403-2
Larsen, M. V., Cosentino, S., Lukjancenko, O., Saputra, D., Rasmussen, S., Hasman, H., et al. (2014). Benchmarking of methods for genomic taxonomy. J. Clin. Microbiol. 52, 1529–1539. doi: 10.1128/JCM.02981-13
Lawrence, J. G., Hendrix, R. W., and Casjens, S. (2001). Where are the pseudogenes in bacterial genomes? Trends Microbiol. 9, 535–540. doi: 10.1016/s0966-842x(01)02198-9
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Magiorakos, A. P., Srinivasan, A., Carey, R. B., Carmeli, Y., Falagas, M. E., Giske, C. G., et al. (2012). Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 18, 268–281. doi: 10.1111/j.1469-0691.2011.03570.x
Malachowa, N., and DeLeo, F. R. (2010). Mobile genetic elements of Staphylococcus aureus. Cell. Mol. Life Sci. 67, 3057–3071. doi: 10.1007/s00018-010-0389-4
Manara, S., Pasolli, E., Dolce, D., Ravenni, N., Campana, S., Armanini, F., et al. (2018). Whole-genome epidemiology, characterisation, and phylogenetic reconstruction of Staphylococcus aureus strains in a paediatric hospital. Genome Med. 10:82. doi: 10.1186/s13073-018-0593-7
Marques, A. T., Tanoeiro, L., Duarte, A., Gonçalves, L., Vítor, J. M. B., and Vale, F. F. (2021). Genomic analysis of prophages from Klebsiella pneumoniae clinical isolates. Microorganisms 9:2252. doi: 10.3390/microorganisms9112252
McCarthy, A. J., and Lindsay, J. A. (2012). The distribution of plasmids that carry virulence and resistance genes in Staphylococcus aureus is lineage associated. BMC Microbiol. 12:104. doi: 10.1186/1471-2180-12-104
McCarthy, A. J., Witney, A. A., and Lindsay, J. A. (2012). Staphylococcus aureus temperate bacteriophage: carriage and horizontal gene transfer is lineage associated. Front. Cell. Infect. Microbiol. 2:6. doi: 10.3389/fcimb.2012.00006
McHugh, M. L. (2012). Interrater reliability: the kappa statistic. Biochem. Med. 22, 276–282. doi: 10.11613/BM.2012.031
Na, S. Y., Roh, J. Y., Kim, J. M., Tamang, M. D., and Lee, J. R. (2012). Analysis of colonization and genotyping of the exotoxins of Staphylococcus aureus in patients with atopic dermatitis. Ann. Dermatol. 24, 413–419. doi: 10.5021/ad.2012.24.4.413
Ndhlovu, G. O. N., Abotsi, R. E., Shittu, A. O., Abdulgader, S. M., Jamrozy, D., Dupont, C. L., et al. (2021). Molecular epidemiology of Staphylococcus aureus in African children from rural and urban communities with atopic dermatitis. BMC Infect. Dis. 21:348. doi: 10.1186/s12879-021-06044-4
Ogonowska, P., Gilaberte, Y., Baranska-Rybak, W., and Nakonieczna, J. (2020). Colonization with Staphylococcus aureus in atopic dermatitis patients: attempts to reveal the unknown. Front. Microbiol. 11:567090. doi: 10.3389/fmicb.2020.567090
Page, A. J., de Silva, N., Hunt, M., Quail, M. A., Parkhill, J., Harris, S. R., et al. (2016). Robust high-throughput prokaryote de novo assembly and improvement pipeline for Illumina data. Microb. Genom. 2:e000083. doi: 10.1099/mgen.0.000083
Pascolini, C., Sinagra, J., Pecetta, S., Bordignon, V., de Santis, A., Cilli, L., et al. (2011). Molecular and immunological characterization of Staphylococcus aureus in pediatric atopic dermatitis: implications for prophylaxis and clinical management. Clin. Dev. Immunol. 2011:718708. doi: 10.1155/2011/718708
Poh, S. E., Koh, W. L. C., Lim, S. Y. D., Wang, E. C. E., Yew, Y. W., Common, J. E. A., et al. (2022). Expression of Staphylococcus aureus virulence factors in atopic dermatitis. JID Innov. 2:100130. doi: 10.1016/j.xjidi.2022.100130
Rojo, A., Aguinaga, A., Monecke, S., Yuste, J. R., Gastaminza, G., and España, A. (2014). Staphylococcus aureus genomic pattern and atopic dermatitis: may factors other than superantigens be involved? Eur. J. Clin. Microbiol. Infect. Dis. 33, 651–658. doi: 10.1007/s10096-013-2000-z
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shi, B., Bangayan, N. J., Curd, E., Taylor, P. A., Gallo, R. L., Leung, D. Y. M., et al. (2016). The skin microbiome is different in pediatric versus adult atopic dermatitis. J. Allergy Clin. Immunol. 138, 1233–1236. doi: 10.1016/j.jaci.2016.04.053
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference Centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Silva, V., Almeida, F., Carvalho, J. A., Castro, A. P., Ferreira, E., Manageiro, V., et al. (2020). Emergence of community-acquired methicillin-resistant Staphylococcus aureus EMRSA-15 clone as the predominant cause of diabetic foot ulcer infections in Portugal. Eur. J. Clin. Microbiol. Infect. Dis. 39, 179–186. doi: 10.1007/s10096-019-03709-6
Sonesson, A., Przybyszewska, K., Eriksson, S., Mörgelin, M., Kjellström, S., Davies, J., et al. (2017). Identification of bacterial biofilm and the Staphylococcus aureus derived protease, staphopain, on the skin surface of patients with atopic dermatitis. Sci. Rep. 7:8689. doi: 10.1038/s41598-017-08046-2
Stepanović, S., Vuković, D., Hola, V., Bonaventura, G. D., Djukić, S., Ćirković, I., et al. (2007). Quantification of biofilm in microtiter plates: overview of testing conditions and practical recommendations for assessment of biofilm production by staphylococci. APMIS 115, 891–899. doi: 10.1111/j.1600-0463.2007.apm_630.x
van Wamel, W. J., Rooijakkers, S. H., Ruyken, M., van Kessel, K. P., and van Strijp, J. A. (2006). The innate immune modulators staphylococcal complement inhibitor and chemotaxis inhibitory protein of Staphylococcus aureus are located on beta-hemolysin-converting bacteriophages. J. Bacteriol. 188, 1310–1315. doi: 10.1128/JB.188.4.1310-1315.2006
Verkaik, N. J., Benard, M., Boelens, H. A., de Vogel, C. P., Nouwen, J. L., Verbrugh, H. A., et al. (2011). Immune evasion cluster-positive bacteriophages are highly prevalent among human Staphylococcus aureus strains, but they are not essential in the first stages of nasal colonization. Clin. Microbiol. Infect. 17, 343–348. doi: 10.1111/j.1469-0691.2010.03227.x
Wang, W., Baker, M., Hu, Y., Xu, J., Yang, D., Maciel-Guerra, A., et al. (2005). Cefoxitin resistance as a surrogate marker for the detection of methicillin-resistant Staphylococcus aureus. J. Antimicrob. Chemother. 55, 506–510. doi: 10.1093/jac/dki052
Wang, W., Baker, M., Hu, Y., Xu, J., Yang, D., Maciel-Guerra, A., et al. (2016). Prevalence and odds of Staphylococcus aureus carriage in atopic dermatitis: a systematic review and meta-analysis. Br. J. Dermatol. 175, 687–695. doi: 10.1111/bjd.14566
Wang, W., Baker, M., Hu, Y., Xu, J., Yang, D., Maciel-Guerra, A., et al. (2021). Whole-genome sequencing and machine learning analysis of Staphylococcus aureus from multiple heterogeneous sources in China reveals Common genetic traits of antimicrobial resistance. mSystems 6:e0118520. doi: 10.1128/mSystems.01185-20
Wrobel, J., Tomczak, H., Jenerowicz, D., and Czarnecka-Operacz, M. (2018). Skin and nasal vestibule colonisation by Staphylococcus aureus and its susceptibility to drugs in atopic dermatitis patients. Ann. Agric. Environ. Med. 25, 334–337. doi: 10.26444/aaem/85589
Yao, W., Xu, G., Li, D., Bai, B., Wang, H., Cheng, H., et al. (2019). Staphylococcus aureus with an erm-mediated constitutive macrolide-lincosamide-streptogramin B resistance phenotype has reduced susceptibility to the new ketolide, solithromycin. BMC Infect. Dis. 19:175. doi: 10.1186/s12879-019-3779-8
Yeung, M., Balma-Mena, A., Shear, N., Simor, A., Pope, E., Walsh, S., et al. (2011). Identification of major clonal complexes and toxin producing strains among Staphylococcus aureus associated with atopic dermatitis. Microbes Infect. 13, 189–197. doi: 10.1016/j.micinf.2010.10.023
Keywords: genomic diversity 5, Staphylococcus aureus, pediatric, atopic dermatitis, South Africa
Citation: Ndhlovu GON, Javkar KG, Matuvhunye T, Ngondoh F, Jamrozy D, Bentley S, Shittu AO and Dube FS (2024) Investigating genomic diversity of Staphylococcus aureus associated with pediatric atopic dermatitis in South Africa. Front. Microbiol. 15:1422902. doi: 10.3389/fmicb.2024.1422902
Edited by:
Jens Andre Hammerl, Bundesinstitut für Risikobewertung, GermanyReviewed by:
Bernhard Krismer, University of Tübingen, GermanyMarco Ferrari, University of Sassari, Italy
Copyright © 2024 Ndhlovu, Javkar, Matuvhunye, Ngondoh, Jamrozy, Bentley, Shittu and Dube. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felix S. Dube, c2l6d2UuZHViZUB1Y3QuYWMuemE=