
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 31 July 2024
Sec. Systems Microbiology
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1419914
This article is part of the Research TopicEmerging Trends and Advances in the Socioeconomic Applications of Beneficial MicrobesView all 11 articles
 Yunkun Teng1,2Shuai Feng1,2Zhuoxuan Gu1,2Chunqi Hou1,2Haoran Xu1,2
Yunkun Teng1,2Shuai Feng1,2Zhuoxuan Gu1,2Chunqi Hou1,2Haoran Xu1,2 Zhiqiang Li1,2Jing Zhao1,2Yi Fang1,2
Zhiqiang Li1,2Jing Zhao1,2Yi Fang1,2 Xin Ma1,2Hongyu Liu1,2Jing Guo1,2
Xin Ma1,2Hongyu Liu1,2Jing Guo1,2 Jun Wang1,2He Ding1,2*
Jun Wang1,2He Ding1,2* Wenfa Lu1,2*†
Wenfa Lu1,2*†Microbiota in the reproductive tract of cattle play a vital role in maintaining normal reproduction. However, the information on microbiota in different parts of reproductive tracts with different genetic background is few. The aim of the present study was to describe and compare the microbiota in vagina, cervix and uterus of Yanbian cattle and Yanhuang cattle. The results showed that microbial diversity increases from the vagina to the uterus. The top three bacterial phyla in bovine reproductive tract were Proteobacteria, Firmicutes and Bacteroidetes, accounting for more than 85%. From the vagina to the uterus, the relative abundance of Proteobacteria gradually decreased, while that of Firmicutes gradually increased. Phylum-level Firmicutes and genus-level UCG_010 were significantly enriched in the uterus of Yanbian cattle and Yanhuang cattle. Comparing the same parts of the two breeds, it was found that there was no significant difference in alpha diversity, but significant differences in beta diversity. In addition, microbiota with significant differences in the relative abundance of the reproductive tract were found. These findings lay a foundation for a comprehensive understanding of the structure of the genital tract microbiota of cows and its regulatory mechanisms.
The genital tract microbiota plays an important role in cattle reproductive health. The reproductive tract microbiota can inhibit the invasion and proliferation of pathogens by forming biofilms (Tang et al., 2008; Tachedjian et al., 2017). Certain lactobacilli can protect fetal development during pregnancy and promote healthy delivery (Romero et al., 2014). Recent research shows that reproductive tract microbiota can transmit chemical signals between species by producing pheromones (Srinivasan et al., 2021). The dysbiosis leads to changes in the microbiota, including a decrease in the abundance of lactobacilli and an increase in the population of facultative anaerobes, leading to a predisposition of the host to a variety of diseases (Valenti et al., 2018; Saraf et al., 2021). Exploring the reproductive tract microbiota structure of healthy cattle will provide a solid theoretical basis for studying the occurrence of reproductive diseases and reproductive obstacles.
However, the microbes in the reproductive tract are not fixed. Bacteria can enter the vagina from the outside, including skin and feces, and transmitted to sites such as the cervix and uterus (Sheldon and Dobson, 2004). Bacteria can also enter the reproductive tract through the bloodstream route (Jeon et al., 2017). The vagina is still considered the main source of microbiota in the uterus, cervix and other parts of the body (Galvão et al., 2019). Pathogenic bacteria that cause uterine infections such as Prevotella, Fusobacterium necrophorum, Escherichia coli, Arcanobacterium pyogenes, etc. are often proven to be associated with the vagina (Deng et al., 2019). Therefore, the microbiota composition of the vagina, cervix, and uterus is expected to be closely related. Although previous studies have separately reported the structure of vaginal microbiota and uterine microbiota in cows (Vitale et al., 2021), and there is a lack of systematic exploration of the complete reproductive tract microbiota.
For studying the composition of microbiota in various parts of the cow’s reproductive tract, in addition to studying the correlation between the microbiota in various parts, its changes under breed factors should also be considered. Studies on Gyr (Giannattasio-Ferraz et al., 2019) and Nellore (Laguardia-Nascimento et al., 2015) cattle found significant differences in vaginal microbiota. However, previous studies on the impact of genetic factors on reproductive tract microbiota may be more affected by sampling region, season and nutritional factors, and there is a lack of research on the cervix and uterus. For this reason, it is necessary to eliminate interfering factors such as region, feeding management and feed differences that affect the reproductive tract microbiota as much as possible, and systematically study the impact of breed factors on the entire reproductive tract microbiota.
Yanbian cattle, one of the five major local fine-bred cattle in China, originated from 1850 to 1870 and was formed by cross-breeding Korean cattle and Mongolian cattle (Shen et al., 2020). Yanhuang cattle are made from Limousin cattle as the male parent and Yanbian cattle as the female parent, through cross-breeding, cross-fixation and group selection, this breed contains 75% of Yanbian cattle genes and 25% of Limousin cattle genes. Compared with Yanbian cattle, the growth and development of Yanhuang cattle at various stages has been significantly improved than that of Yanbian cattle. Yanhuang cattle have obvious advantages in slaughter performance and feed conversion ratio, and the digestibility of dietary nutrients is also higher than that of Yanbian cattle (Ji et al., 2014).
The aim of the study is to investigate the commonality and uniqueness of microorganisms in different parts of the reproductive tract of Yanbian cattle and Yanhuang cattle, as well as the influence of breed factors on the composition of microorganisms in the bovine reproductive tract, which laid a foundation for a comprehensive understanding of the microecological composition and regulation of bovine genital tract.
This study selected Yanbian cattle and Yanhuang cattle from Benfu Ranch in Yanbian Korean Autonomous Prefecture, China. Multiparous cattle with healthy body condition and aged 3–5 years were selected. Samples were collected from November and December 2022. In order to avoid cows in different physiological cycles affecting the structure of the reproductive tract microbiota, we use estrus identification technology to select cows that were naturally in estrus (external observation combined with vaginal examination). Cotton swabs was used to collect samples on the day of estrus, and samples from the reproductive tract including the vagina, cervix and uterus were collected. A total of 98 samples from 21 Yanbian cattle (vagina: n = 14; cervix: n = 17; uterus: n = 17) and 19 Yanhuang cattle (vagina: n = 17; cervix: n = 17; uterus: n = 16) were collected for subsequent sequencing analysis. Samples were collected on sterile cotton swabs, placed in cryovials, group the samples (A.V: Yanbian cattle vagina; A.C: Yanbian cattle cervix; A.U: Yanbian cattle uterus; B.V: Yanhuang cattle vagina; B.C: Yanhuang cattle cervix; B.U: Yanhuang cattle uterus), and stored in liquid nitrogen at −196°C until used for microbiome analysis.
Genomic DNA was extracted by CTAB method, and DNA purity and concentration were detected using 1% agarose gel electrophoresis. Take an appropriate amount of sample DNA in a centrifuge tube and use sterile water to dilute the sample to 1 ng/μL. PCR amplification of the bacterial 16S rRNA gene V3–V4 region was performed using the forward primer 341F (5′-CCTAYGGGRBGCASCAG -3′) and the reverse primer 806R (5′-GGACTACNNGGGTATCTAAT-3′) (Table 1). Add 15 μL Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM primer and 10 ng genomic DNA template to the PCR mixture, perform the first denaturation at 98°C for 1 min, and then denature at 98°C (10 s) for 50°C (30 s) and 72°C (30 s) for 30 cycles, and it was maintained at 72°C for 5 min to obtain the PCR product. The expected amplified product fragment was 470 bp. PCR products are detected by electrophoresis using 2% concentration agarose gel; qualified PCR products are purified with magnetic beads and quantified using enzyme labeling. Equal amounts of samples are mixed according to the concentration of the PCR product. After mixing thoroughly, use 2% agarose gel electrophoresis to detect the PCR product, and use a universal DNA purification and recovery kit (TianGen) to recover the product of the target band. NEB Next® Ultra™ II FS DNA PCR-free Library Prep Kit (New England Biolabs) was used for library construction. The constructed library was quantified by Qubit and Q-PCR. After the library was qualified, NovaSeq 6000 (518 cycles) was used for PE 250 On-machine sequencing. All sequences used in this study are publicly available at the NCBI Sequence Read Archive under accession ID PRJNA1129596.
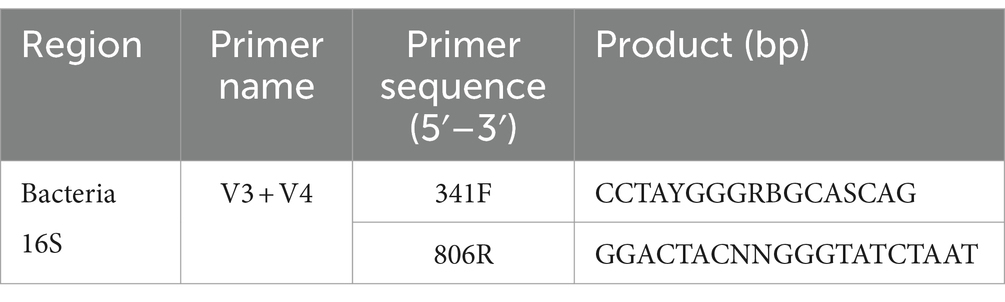
Table 1. Primers sequences for PCR.
Illumina NovaSeq sequencing platform was used for double-terminal sequencing of the library. Briefly, according to the Barcode sequence and PCR amplification primer sequence, each sample data is split from the offline data. Double-end data splicing uses FLASH (Version 1.2.11, http://ccb.jhu.edu/software/FLASH/) to truncate the Barcode and primer sequences to splice the reads of each sample, and the resulting spliced sequence is: Raw Tags data (Raw Tags). Use fastp software (Version 0.23.1) to perform strict filtering on the spliced Raw Tags to obtain high-quality Tags data (Clean Tags). The Tags obtained after the above processing need to be processed to remove chimera sequences. The Tags sequences are compared with the species annotation database (Silva database https://www.arb-silva.de/ for16S) to perform comparison and detection of chimeric sequences, and finally remove the chimeric sequences to obtain the final effective data (Effective Tags). For the obtained EffectiveTags, the DADA2 module or deblur in the QIIME2 (VersionQIIME2-202006) software was used for denoising (DADA2) to obtain the final ASVs (Amplicon Sequence Variants) and feature tables. Classify-sklearn algorithm of QIIME2 is adopted (Bokulich et al., 2018; Bolyen et al., 2019) annotated species for each ASV using a pre-trained Naive Bayes classifier.
Sequence data analysis was mainly performed using the QIIME2 and R packages. According to the results of ASVs annotation and the characteristic table of each sample, the top 10 dominant species were screened from the species abundance table at phylum and genus level, and the relative species abundance histogram was generated. A Venn diagram was generated to visualize the shared and unique OTUs among samples or groups using the R (Version 3.5.3) package “Venn Diagram.” Use QIIME2 software to calculate α diversity indices such as Chao1 index and Shannon index. β-diversity analysis was performed using the Bray–Curtis index to investigate structural changes in microbial communities in the sample and visualized by generating PCoA distribution maps. LDA effect size (LDA score >4) was performed to detect differentially abundant taxa across groups using the default parameters. In R (Version 3.5.3), a t-test was used to compare the abundance of taxa at the phyla and genus level within the group.
Through the species relative abundance column chart, we can visually see the species and their proportions with higher relative abundance in each group at the phylum and genus level. As shown in Figure 1A, the top three dominant bacterial phyla in the reproductive tract of Yanbian cattle are Proteobacteria (uterus: 38.63%, cervix: 47.65%, vagina 59.97%), Firmicutes (uterus: 41.19%, cervix: 34.56%, vagina: 21.27%), Bacteroidota (uterus: 12.90%, cervix: 10.11%, vagina: 9.97%). The proportion of the top three dominant bacterial phyla in various reproductive tract parts of Yanbian cattle is as high as 91.35–92.72%. The top three bacterial phyla in the reproductive tract of Yanhuang cattle are Proteobacteria (uterus: 21.37%, cervix: 33.60%, vagina: 30.30%), Firmicutes (uterus: 50.48%, cervix: 40.50%, vagina: 30.30%), Bacteroidota (uterus: 16.25%, cervix: 14.58%, vagina: 11.12%) (Figure 1D). The proportion of the top three dominant bacterial phyla in various reproductive tract parts of Yanhuang cattle is as high as 86.18–88.68%. The results showed that the top three dominant phyla of the two breeds of cattle were Proteobacteria, Firmicutes and Bacteroidetes, and these three phyla showed a consistent trend in the reproductive tract: the relative abundance of Proteobacteria showed a gradient decreasing trend from the vagina to the uterus, and the relative abundance of Firmicutes showed a gradient increasing trend from the vagina to the uterus, there was no obvious change in the Bacteroidetes phylum. From the genus level, Ralstonia occupies a high abundance in the reproductive tract of Yanbian cattle and Yanhuang cattle, and the relative abundance gradually decreases from the vagina to the uterus. As another dominant genus, UCG-010 has a relative abundance that gradually increases from the vagina to the uterus of Yanbian cattle and Yanhuang cattle. The Histophilus only occupies a greater advantage in the reproductive tract of Yanhuang cattle, and the relative abundance of Histophilus showed an increasing trend from the vagina to the uterus of Yanhuang cattle (Figures 1B,E).
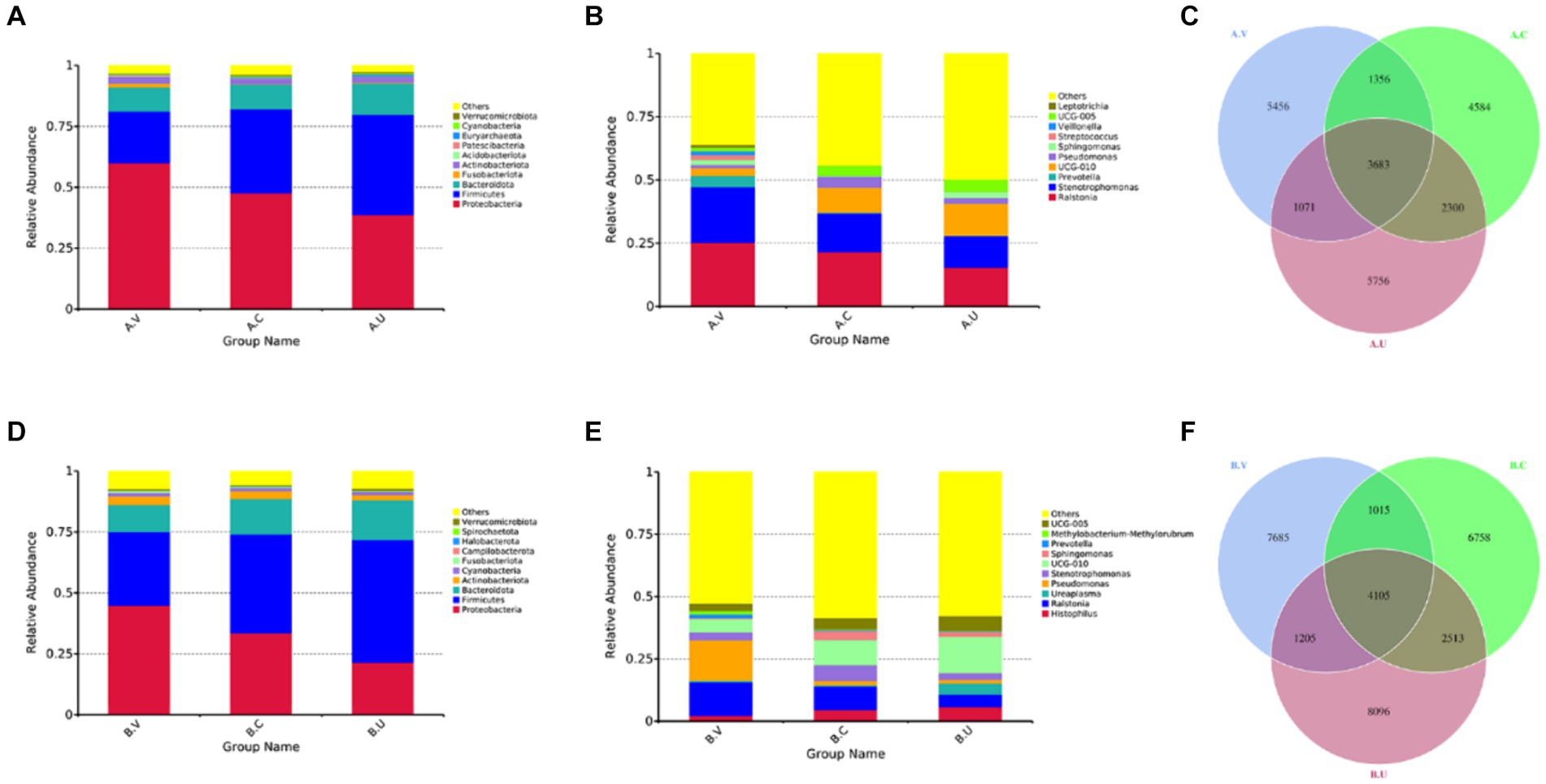
Figure 1. Composition and proportion of vaginal, uterine and cervical microbiota. (A,B,D,E) are the species annotations and abundance information at the phylum and genus level of the vagina, cervix, and uterus of Yanbian cattle and Yanhuang cattle. The top 10 species with the highest abundance were selected to generate a column accumulation chart of species relative abundance, the abscissa is the group name; the ordinate represents the relative abundance; others represents the sum of the relative abundance of all other phyla except these 10 phyla in the figure. Each circle in the figures C,F represents a group. The number in the overlapping part of the circle and the circle represents the number of OTUs shared between the groups. The number in the non-overlapping part represents the number of unique OTUs of the group.
As shown in Figures 1C,F, the number of OTUs that coexisted in the vagina, cervix and uterus of Yanbian cattle reached 3,683, 5,039 OTUs coexisted in the vagina and cervix, 5,983 OTUs coexisted in the cervix and uterus, and 4,754 OTUs coexisted in the vagina and uterus. The number of OTUs that coexisted in the vagina, cervix and uterus of Yanhuang cattle reached 4,105, 5,120 OTUs coexisted in the vagina and cervix, 6,618 OTUs coexisted in the cervix and uterus, and 5,310 OTUs coexisted in the vagina and uterus. In addition, there are still relatively high amounts of OTU in the vagina, cervix, and uterus of the two breeds of cattle, which are unique to each part.
By comparing the differences in Shannon index and Chao 1 index of the vagina, cervix and uterus of Yanbian cattle and Yanhuang cattle (Figures 2A,B,E,F), it was found that the alpha diversity changes of the reproductive tract microbiota of Yanbian cattle and Yanhuang cattle showed consistent changes. There was a significant difference in the alpha diversity of the vagina and uterus between the two breeds (p < 0.05). However, there was no significant difference in alpha diversity between vagina-cervix and cervix-uterus (p > 0.05). Based on the PCoA distribution of vaginal, cervical and uterine samples from the two breeds of cattle, it was found that the distribution of vaginal samples was significantly different from the distribution of uterine and cervical samples, while the distribution of cervical and uterine samples was more similar and the community structure was more similar (Figures 2C,G). In order to further determine the significance of distribution differences between sample groups, using the Wilcox rank sum test method based on Bray–Curtis, there were significant differences in vaginal-cervical and vaginal-uterine beta diversity (p < 0.05), and there was no significant difference in cervical-uterine beta diversity (p > 0.05) (Figures 2D,H).
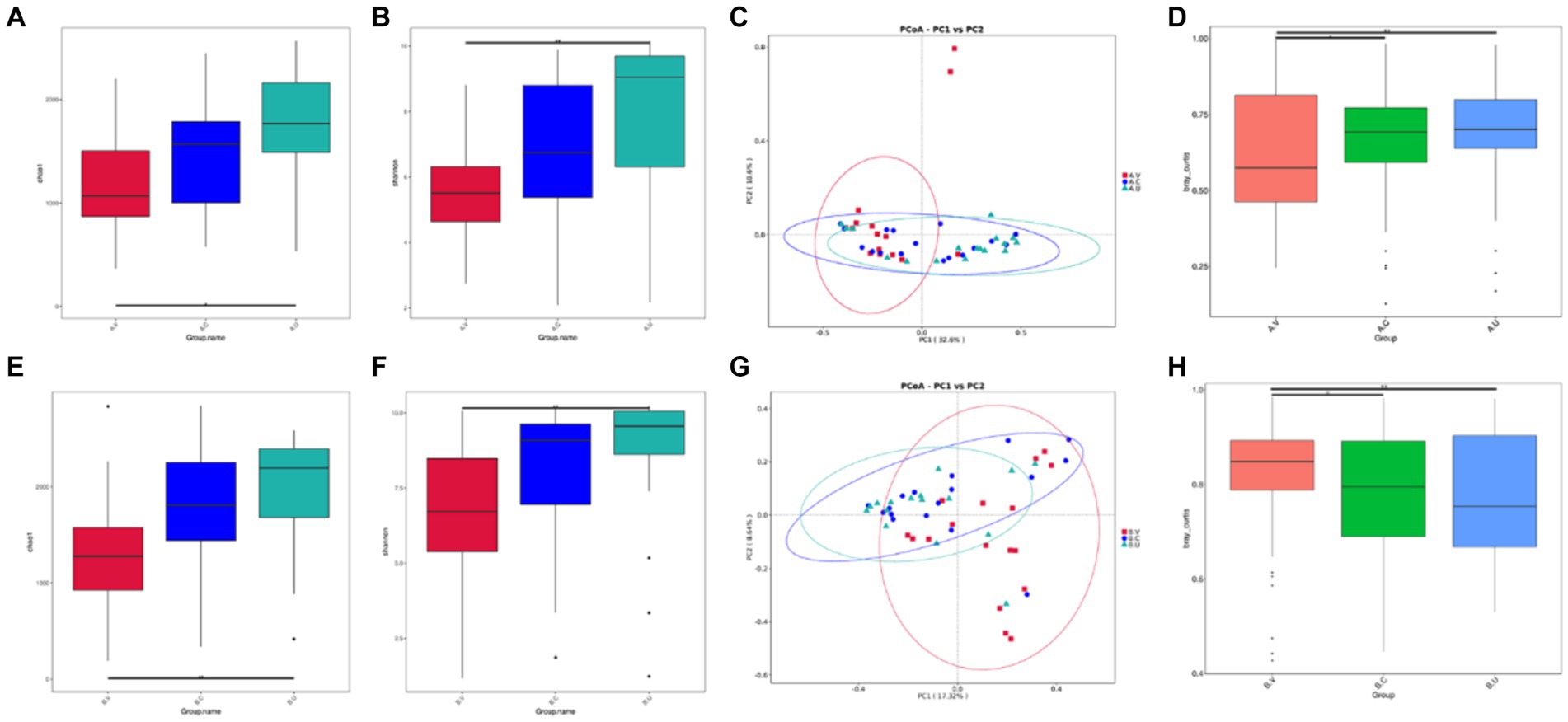
Figure 2. Comparison of the alpha and beta diversity of the vaginal, cervical and intrauterine microbiota of Yanbian cattle and Yanhuang cattle. Alpha diversity, including Chao 1 and Shannon index (A,B,E,F) of the two groups of samples, C,G based on the beta diversity of the Bray–Curtis metric of Yanbian cattle and Yanhuang cattle. The abscissa represents one principal component, the ordinate represents another principal component, and the percentage represents the contribution of the principal component to the sample difference; each point in the figure represents a sample, and samples in the same group are represented by the same color. (D,H) The Wilcox rank sum test was used to analyze the Bray–Curtis differences among each group. *Means significant difference (p < 0.05), **means extremely significant difference (p < 0.01).
In order to find out the species with significant differences in different genital tract parts between Yanbian cattle and Yanhuang cattle, statistics of significantly enriched bacterial groups in each group were carried out through LEfSe analysis. As shown in Figure 3. o_Veillonellales_Selenomonadales was significantly enriched in the vagina of Yanbian cattle, and c_Gammaproteobacteria, o_Pseudomonadales, and f_Pseudomonadaceae were significantly enriched in the vagina of Yanhuang cattle. In the uterus of Yanbian cattle and Yanhuang cattle, f_Rikenellaceae, g_UCG_010, f_UCG_010, p_Firmicutes, and c_Clostridia were significantly enriched. g_UCG-005, f_Oscillospiraceae, and o_Oscillospirales were only enriched in the uterus of Yanbian cattle. g_Rikenellaceae_RC9_gut_group was only significantly enriched in the uterus of Yanhuang cattle. Thus, phylum-level Firmicutes and genus-level UCG_010 were significantly enriched in the uterus of two breeds of cattle, UCG-005 was significantly enriched only in the uterus of Yanbian cattle, and Rikenellaceae_RC9_gut_group was significantly enriched only in the uterus of Yanhuang cattle (Figures 3A,C).
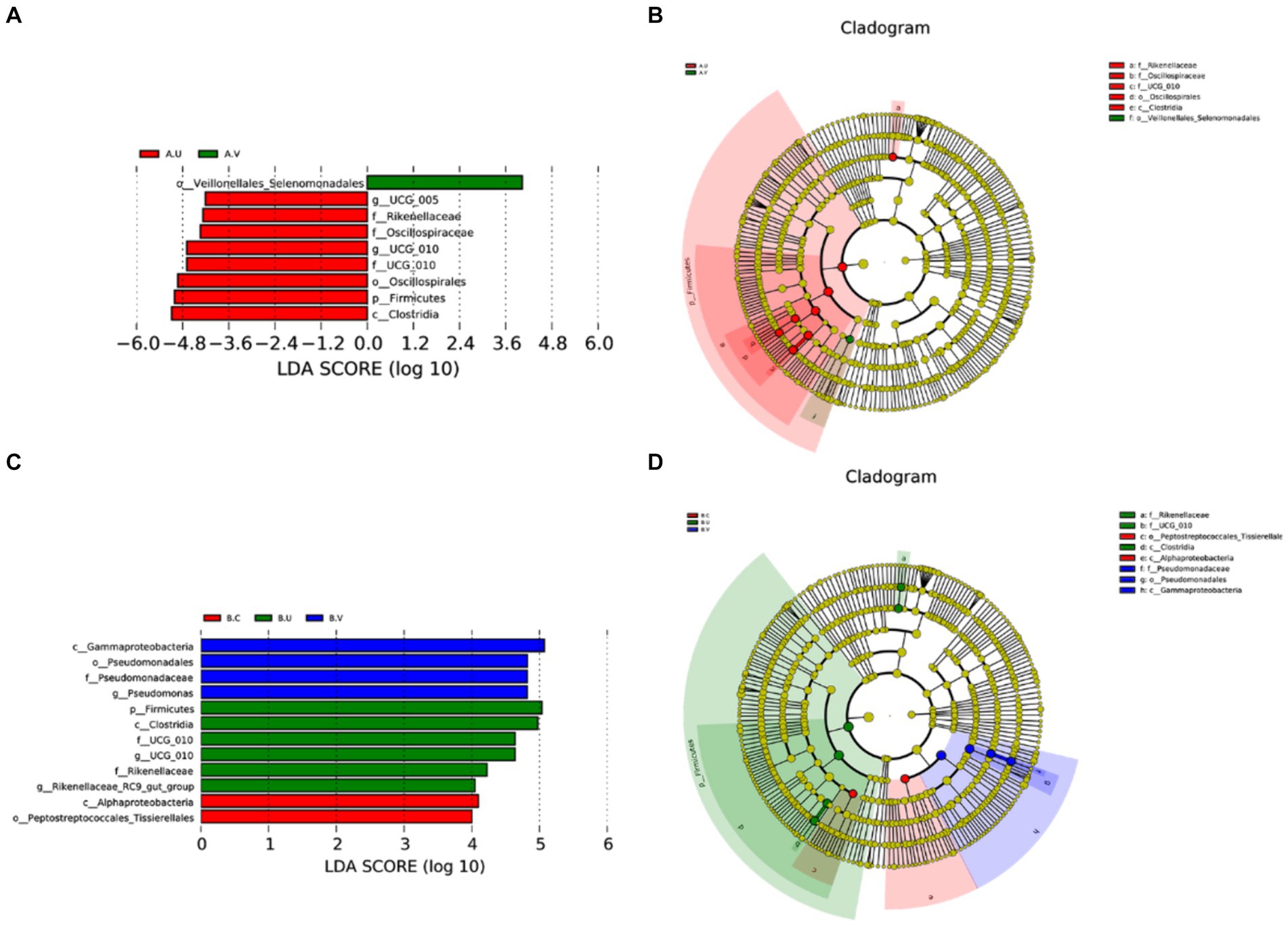
Figure 3. Comparison of the microbiota in the vagina, cervix and uterus of Yanbian cattle and Yanhuang cattle. The LDA value distribution histogram shows species whose LDA score is greater than the set value of 4, that is, species that are significantly enriched in each group. The length of the bar graph represents the effect size (LDA score) of differential species (A,C). In a cladogram, the circles radiating from the inside to the outside represent the classification levels from phylum to genus (or species). Each small circle at a different classification level represents a classification at that level, and the diameter of the small circle is proportional to the relative abundance (B,D).
In order to study the main composition and proportion of microbiota in the same reproductive tract of Yanbian cattle and Yanhuang cattle, the top 10 species with the highest abundance were selected to generate a column accumulation chart of species relative abundance. In order to visually observe the distribution of dominant species among groups. The results showed that at the phylum level, the top three bacterial phyla were Proteobacteria, Firmicutes, and Bacteroidetes. Among them, the relative abundance of Proteobacteria in the vagina, cervix and uterus of Yanbian cattle was higher than that of Yanhuang cattle, while the relative abundance of Firmicutes and Bacteroidetes was lower than that of Yanhuang cattle (Figures 4A,D,G). In addition, at the genus level, Ralstonia and Stenotrophomonas were both higher in the vagina, cervix and uterus of Yanbian cattle to a certain extent than in Yanhuang cattle (Figures 4B,E,H). Interestingly, the cervix and uterus of Yanhuang cattle also contain a relatively high abundance of Histophilus. The OTU of the vagina, cervix and uterus of the two breeds of cattle showed that the number of OTU from the vagina to the uterus of the two breeds of cattle increased (vagina: 3039 OTUs, cervix: 4918 OTUs, uterus: 5488 OTUs) (Figures 4C,F,I).
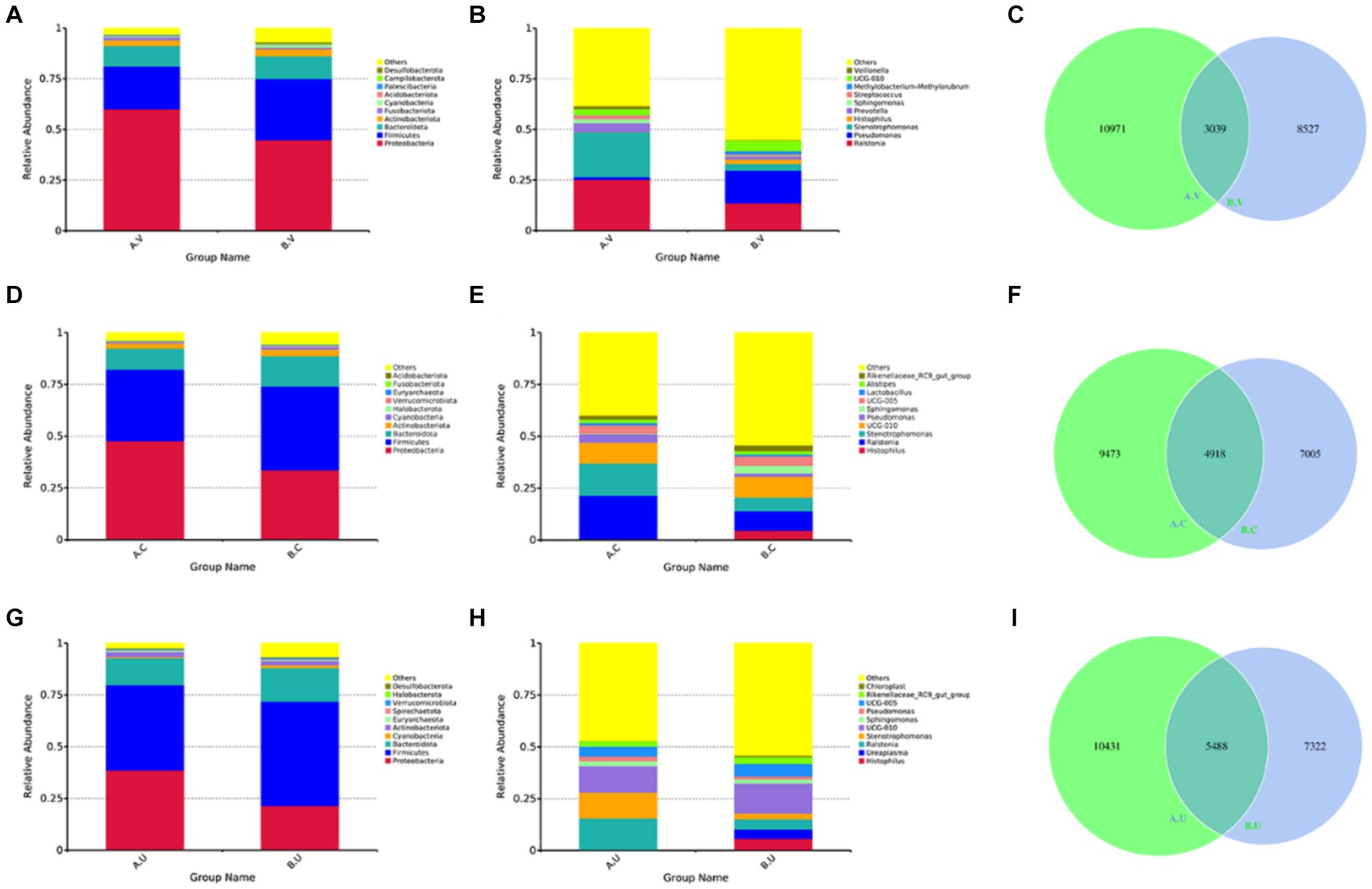
Figure 4. The composition and proportion of the microbiota in the same reproductive tract of Yanbian cattle and Yanhuang cattle. (A,B,D,E,G,H) Are the species annotations and abundance information at the phylum and genus level in the same reproductive tract of Yanbian cattle and Yanhuang cattle. Select the top 10 species with the highest abundance to generate a species relative abundance column. It is a cumulative graph. The abscissa (group name) is the group name; the ordinate (relative abundance) represents the relative abundance; others represents the sum of the relative abundance of all other phyla except the 10 phyla in the graph. Each circle in the figures C,F,I represent a group. The number in the overlapping part of the circle and the circle represents the number of OTUs shared between the groups. The number in the non-overlapping part represents the number of unique OTUs of the group.
In order to further explore the influence of genetic factors on the diversity of reproductive tract microbiota, we compared the diversity of the same genital tract microbiota between Yanbian cattle and Yanhuang cattle. Regarding alpha diversity, as shown by the Chao 1 index (Figures 5A,E,I) and Shannon index (Figures 5B,F,J), the richness and evenness of the reproductive tract microbiota of Yanhuang cattle are slightly higher than those of Yanbian cattle, but there is no significant difference (p > 0.05). We analyzed beta diversity using the Bray–Curtis based Wilcox rank sum test method to examine differences in microbial communities between groups, there is a large dispersion in the distribution of vaginal, cervix, and uterine samples (Figures 5C,G,K), and the differences in the microbial communities in the same parts were statistically analyzed. As shown in Figures 5D,H,L, there were significant differences in beta diversity between the vagina, cervix, and uterus of Yanbian cattle and Yanhuang cattle.
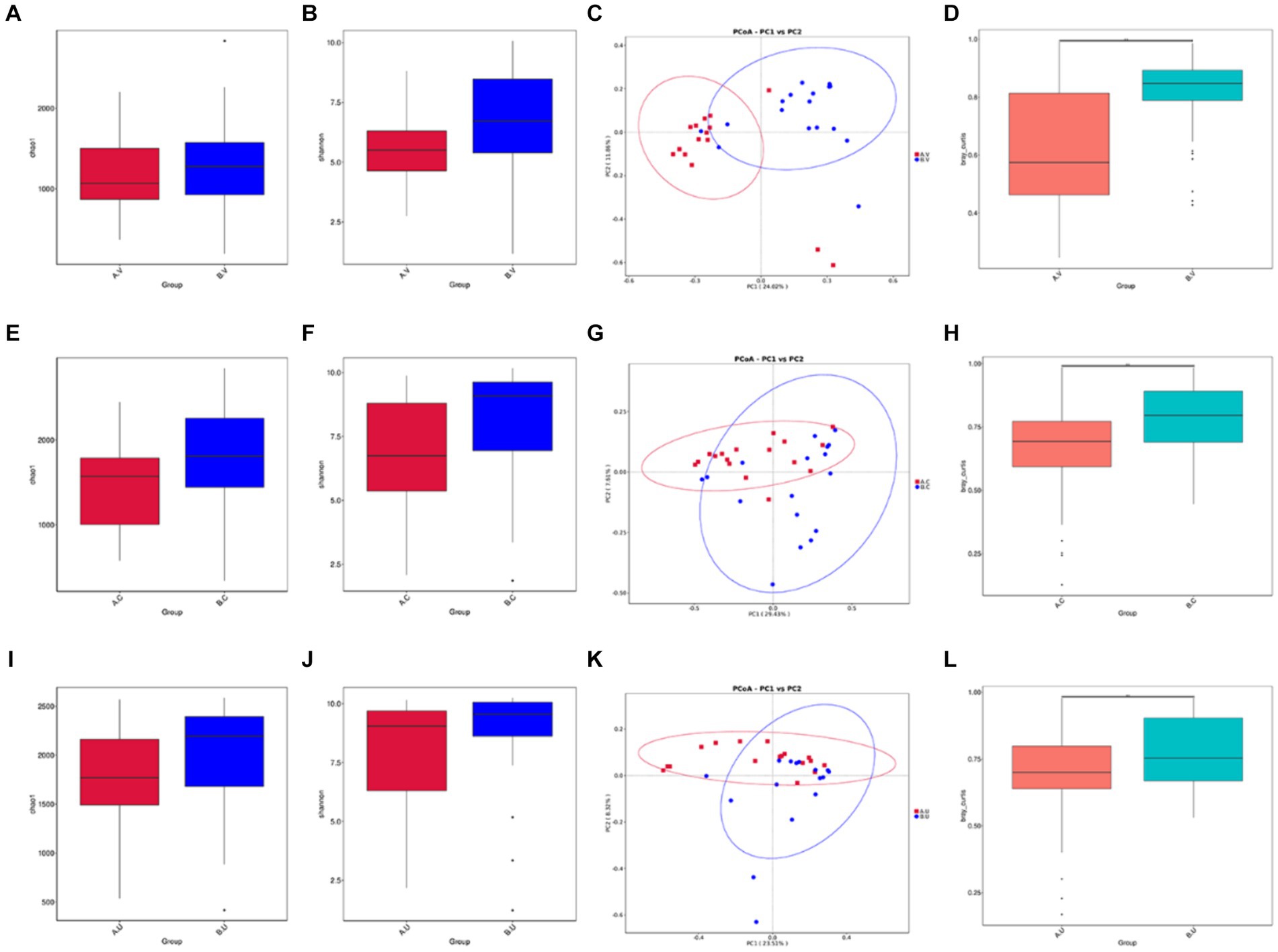
Figure 5. Comparison of the alpha and beta diversity of the microbiota in the same reproductive tract site of Yanbian cattle and Yanhuang cattle. Alpha diversity, including Chao 1 and Shannon index (A,B,E,F,I,J) of the two groups of samples, C,G,K based on the beta diversity of the Bray–Curtis metric of Yanbian cattle and Yanhuang cattle. The abscissa represents one principal component, the ordinate represents another principal component, and the percentage represents the contribution of the principal component to the sample difference; each point in the figure represents a sample, and samples in the same group are represented by the same color. (D,H,L) The Wilcox rank sum test was used to analyze the Bray–Curtis differences among each group. *Means significant difference (p < 0.05).
In order to further identify the differential species that affect the bacterial community structure, t-test was used to identify species with significant differences at the phylum and genus levels, and then clarify the next research direction. At the phylum level, the number of Cyanobacteria in the vagina and cervix of Yanbian cattle was significantly lower than that of Yanhuang cattle (p < 0.05), and the number of Desulfobacterota in the vagina of Yanbian cattle was significantly lower than that of Yanhuang cattle (Figures 6A,B). No phylum-level species differences were observed in the uterus of Yanbian cattle and Yanhuang cattle. At the genus level, Delftia was significantly higher in the vagina, cervix, and uterus of Yanbian cattle than in Yanhuang cattle; Stenobacteria was significantly higher in the vagina, uterus of Yanbian cattle. In Yanbian cattle. In addition, Bacteroides was significantly lower in the vagina, cervix, and uterus of Yanbian cattle than in Yanhuang cattle, and Mitochondria was significantly lower in the vagina and cervix of Yanbian cattle than in Yanhuang cattle (Figures 6C–E).
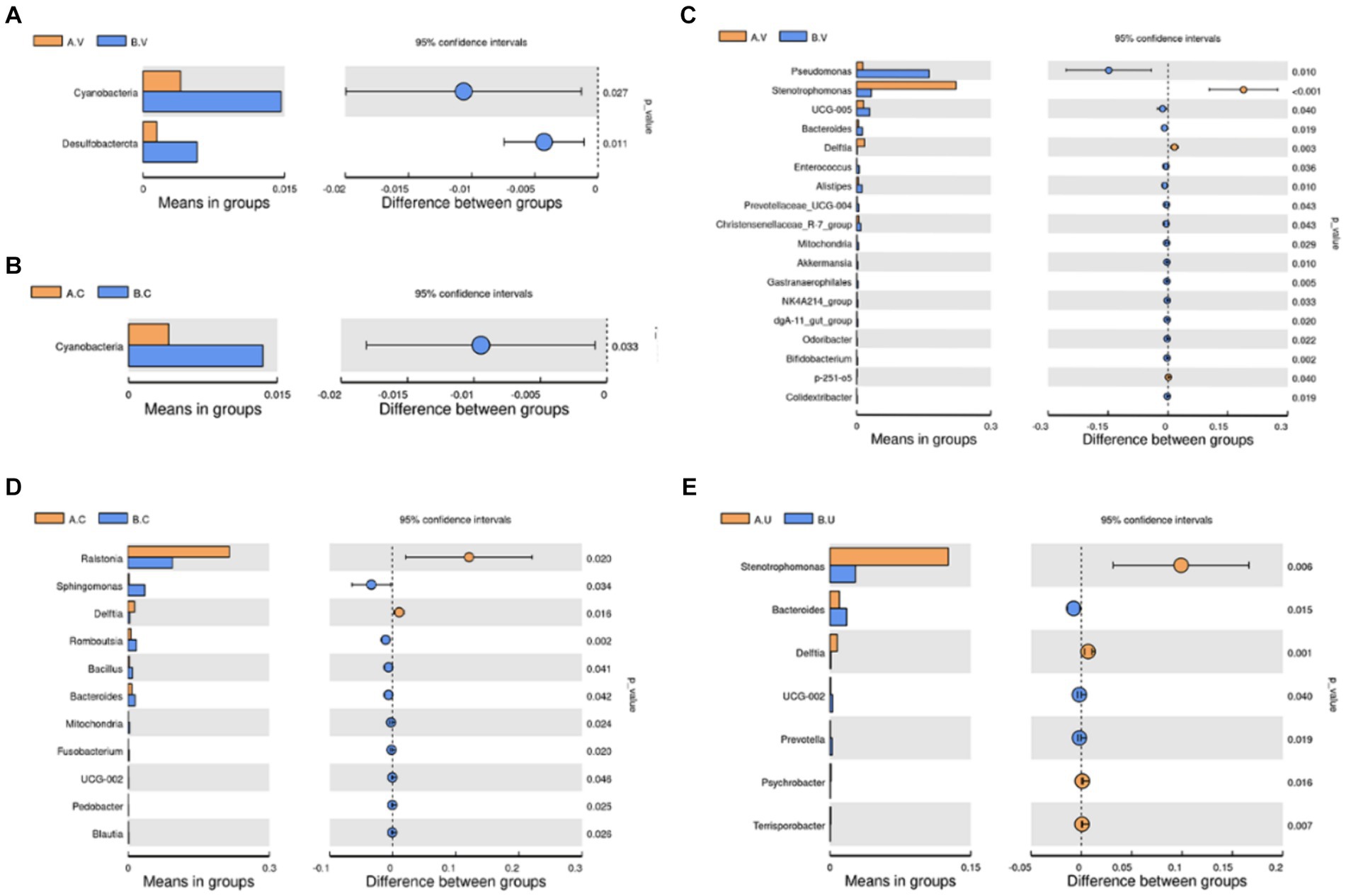
Figure 6. Comparison of the microbiota in the same reproductive tract of Yanbian cattle and Yanhuang cattle. Use t-test to identify species with significant differences between groups at the phylum (A,B) and genus (C–E) levels (p < 0.05). The picture on the left shows the difference in species abundance between groups. Each bar in the picture represents the mean value in each group of species with significant differences in abundance between groups. The picture on the right shows the confidence level of the difference between groups. The leftmost endpoint of each circle in the picture represents the lower limit of the 95% confidence interval of the mean difference, and the rightmost endpoint of the circle represents the upper limit of the 95% confidence interval of the mean difference. The center of the circle represents the difference between the means, and the color of the circle represents the p-value of the significant difference test between groups for the corresponding species.
The results of this study show that the abundance and composition of bacteria in the vagina, cervix and uterus are common and different. In addition, there were differences in the composition of microbiota in the same part between the two breeds. In this study, the top three bacterial phyla in the vagina-cervix-uterus are Proteobacteria, Firmicutes, and Bacteroidetes. From the vagina to the uterus, the relative abundance of Proteobacteria shows a gradient decreasing trend. From the vagina to the uterus, the relative abundance of Muricobacteria showed a gradient increasing trend, and Yanbian cattle and Yanhuang cattle showed a consistent pattern. These three bacterial phyla dominate the digestive tract and may be related to the main source of reproductive tract microbiota (Zhu et al., 2023). Previous studies on the reproductive tract microbiota of humans (Liu et al., 2022) and cattle (Clemmons et al., 2017) have proven the dominance of these three bacterial phyla, but the proportions of each bacterial phylum are different, which may be related to environmental, nutritional and other factors. In the first three bacterial phyla, Proteobacteria include most of the well-known pathogenic bacteria and are potential diagnostic features of dysbiosis or disease risk (Chen et al., 2021). Studies have shown that Firmicutes in the digestive tract are mostly obligate anaerobic bacteria (Wozniak et al., 2022), this may be the reason for the presence of high-abundance Firmicutes in the anaerobic environment of the uterus. Studies have shown that the ratio of Firmicutes to Bacteroidetes is related to the occurrence of inflammation (Wu et al., 2021). Artificial control of the ratio of the bovine reproductive tract microbiota, such as the infusion of probiotics, has important potential in preventing inflammatory diseases in the bovine reproductive tract. Interestingly, we found that Histophilus was enriched in the reproductive tract of Yanhuang cattle, and the relative abundance increased from the vagina to the uterus. Histophilus is associated with several disease syndromes in cattle (Shirbroun, 2020). More recent studies indicate that Histophilus stimulates endothelial cell tissue factor activity and disrupts intercellular junctions (Behling-Kelly et al., 2016).
The results of the study showed an increasing trend in α diversity from the vagina to the uterus, and there are significant differences in alpha diversity between vagina and uterus, which is consistent with previous studies on human reproductive tract microbiota (Chen et al., 2017; Liu et al., 2022). However, previous studies of lactating Angus cattle have found that the alpha diversity of the vaginal flora is significantly higher than that of the uterine flora (Clemmons et al., 2017). Compared with our study, there are differences in season, region, breed, sampling method and animal feeding management mode, etc. These factors may lead to differences in the results of the two studies. From the perspective of physiological structure, the environment of tight cervix may also be an important reason for the difference in diversity of vaginal and uterine microbiota. As one of the important barriers to protect the uterine body from environmental pathogens, the cervix maintains a more stable environment of the uterus (Sheldon and Dobson, 2004; Azawi, 2008). The anaerobic environment in utero has a low biomass of bacteria, but corresponds to a high bacterial diversity (Chen et al., 2017). Besides, there are significant differences in beta diversity between vagina-cervix and vagina-uterus. This highlights the fact that there are differences in vaginal-cervical and vaginal-uterine microbiota profiles. Firmicutes were significantly enriched in the uterus of two breeds of cattle, although previous studies revealed that Firmicutes occupies a dominant position in the uterus (Machado et al., 2012; Santos and Bicalho, 2012; Clemmons et al., 2017), previous studies have also revealed the dominant position of Firmicutes in uterus. In addition, UCG-010 was significantly enriched in the uterus of two breeds of cattle. Previous studies have demonstrated the enrichment of UCG-010 in the intestine of cattle (Couch et al., 2021). The presence of this bacteria in the uterus may originate from the digestive tract. At the same time, the uterine environment may be more conducive to the colonization of UCG-010, but its role in the uterus has not yet been reported. However, UCG-005 was only significantly enriched in the uterus of Yanbian cattle, and Rikenellaceae_RC9_gut_group was only significantly enriched in the uterus of Yanhuang cattle. Studies have shown that members of Rikenellaceae_RC9_gut_group can neutralize cytotoxic reactive oxygen species and protect cells from oxidative stress, thereby reducing the likelihood of inflammation (Gryaznova et al., 2022). We found that Yanhuang cattle only had common marker species in the uterus, and the number of marker species showed an increasing trend from vagina to uterus. This finding suggests that the uterus may have a more stable environment, making the microbiota more similar in the uterus of different species. In addition to being affected by genetic factors between breeds, the vagina and cervix are more affected by opportunistic infections in the external environment.
The research on the factor of variety shows that the relative abundance of Proteobacteria in the vagina, cervix and uterus of Yanbian cattle is higher than that of Yanhuang cattle, and the relative abundance of Firmicutes and Bacteroidetes is lower than that of Yanhuang cattle. The number of OTU in the uterus of Yanbian cattle and Yanhuang cattle was the highest. In previous studies, a total of 2075 OTUs were found in the vaginas of Holstein and Fleckvieh cattle (Nesengani et al., 2017), close to the results of this study. This further proves that there is a more stable environment in the uterus, making the uterus of different breeds of cattle have more similar microbiota. Previous studies have also revealed high similarities in the uterine microbiota of dairy cows in the same state (Santos et al., 2011). There is no significant difference in the alpha diversity of the vagina, cervix and uterus of the two breeds of cattle, but the alpha diversity of the microbiota in various parts of the reproductive tract of Yanhuang cattle is slightly higher than that of Yanbian cattle. The differences in species composition between different habitats were analyzed through beta diversity. There are significant differences in the characteristics of the microbiota in the same reproductive tract of cattle breeds. This diversity difference can be explained by different breeds (Appiah et al., 2020). To this end, we further identified the species responsible for this difference at the phylum and genus level. We found that the number of Cyanobacteria in the vagina and cervix of Yanbian cattle was significantly lower than that of Yanhuang cattle, Cyanobacteria are also highly enriched in the intestines of other animals, such as yaks (Wang et al., 2021), but there is currently no scientific evidence that Cyanobacteria play a role in the reproductive tract. The number of Desulfobacteria in the vagina of Yanbian cattle was significantly lower than that of Yanhuang cattle. Although Desulfobacteria can participate in catabolic reactions in the intestine by reducing sulfur compounds and degrading butyrate, etc. (Bai et al., 2022). However, the specific roles played by these two bacterial phyla in the bovine reproductive tract remain to be explored. Affected by the genetic factors of the two breeds, we found differences in bacterial genera in the reproductive tracts of the two breeds of cattle. Delftia was significantly higher in the vagina, cervix, and uterus of Yanbian cattle than in Yanhuang cattle; Bacteroides is lower than that of Yanhuang cattle. Delftia may serve as predictor of HPV lesion evolution (Gardella et al., 2022); Bacteroides metabolize polysaccharides and oligosaccharides to provide nutrients, vitamins, and other functions to the host and other intestinal microbial residents (Zafar and Saier, 2021). These results indicate that the key microbiota is significantly different in the reproductive tract of Yanbian cattle and Yanhuang cattle. The role of these species with significant differences in abundance in the reproductive tract of the two breeds remains to be explored. The relationship between these differences and the reproductive traits of Yanbian cattle and Yanhuang cattle is worth further exploration.
In conclusion, our study found the commonalities and differences in the structure of the microbiota in different parts of the bovine genital tract, as well as the influence of breed factors on the composition of the bovine genital tract. These findings provide a solid theoretical basis for us to understand the reproductive health status of cattle, reveal the microecological balance of bovine reproductive tract, and guide the prevention and treatment of bovine reproductive diseases. The difference of reproductive tract microflora between Yanbian cattle and Yanhuang cattle reveals the possibility of microflora playing a role in the reproductive traits of the two breeds, which is worthy of further study.
The data that supports the findings of this study are available from the corresponding author upon reasonable request. All sequences used in this study are publicly available at the NCBI Sequence Read Archive under accession ID PRJNA1129596.
The animal studies were approved by the Ethical Committee of Jilin Agricultural University Approval Code: 20230824001. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
YT: Formal analysis, Investigation, Software, Validation, Writing – original draft, Writing – review & editing. SF: Writing – review & editing. ZG: Formal analysis, Resources, Writing – review & editing. CH: Formal analysis, Validation, Writing – review & editing, Writing – original draft. HX: Investigation, Supervision, Writing – review & editing. ZL: Formal analysis, Writing – review & editing. JZ: Software, Supervision, Writing – review & editing. YF: Supervision, Validation, Writing – review & editing. XM: Supervision, Validation, Writing – review & editing. HL: Software, Supervision, Writing – review & editing. JG: Methodology, Supervision, Writing – review & editing. JW: Data curation, Formal analysis, Supervision, Writing – review & editing. HD: Conceptualization, Supervision, Validation, Writing – review & editing. WL: Data curation, Formal analysis, Supervision, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The authors thank the funds supported by the National Natural Science Foundation of China International Cooperative Research and Exchange Program (31861143014), National Natural Science Foundation of China (Grant No. U20A2053) and Jilin Province Beef Cattle Industrialization Development Major Science and Technology Project: High-quality Beef Cattle Large-scale Breeding Technology Research and Demonstration (YDZJ202203CGZH041).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Appiah, M. O., Wang, J., and Lu, W. (2020). Microflora in the reproductive tract of cattle: a review. Agriculture 10:232. doi: 10.3390/agriculture10060232
Azawi, O. I. (2008). Postpartum uterine infection in cattle. Anim. Reprod. Sci. 105, 187–208. doi: 10.1016/j.anireprosci.2008.01.010
Bai, J., Wan, Z., Zhang, Y., Wang, T., Xue, Y., and Peng, Q. (2022). Composition and diversity of gut microbiota in diabetic retinopathy. Front. Microbiol. 13:926926. doi: 10.3389/fmicb.2022.926926
Behling-Kelly, E., Rivera-Rivas, J., and Czuprynski, C. J. (2016). Interactions of Histophilus somni with host cells. Curr. Top. Microbiol. Immunol. 396, 71–87. doi: 10.1007/82_2015_5010
Bokulich, N. A., Kaehler, B. D., Rideout, J. R., Dillon, M., Bolyen, E., Knight, R., et al. (2018). Optimizing taxonomic classification of marker-gene amplicon sequences with QIIME 2’s q2-feature-classifier plugin. Microbiome 6:90. doi: 10.1186/s40168-018-0470-z
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Chen, C., Song, X., Wei, W., Zhong, H., Dai, J., Lan, Z., et al. (2017). The microbiota continuum along the female reproductive tract and its relation to uterine-related diseases. Nat. Commun. 8:875. doi: 10.1038/s41467-017-00901-0
Chen, L., Wang, Z., Wang, P., Yu, X., Ding, H., Wang, Z., et al. (2021). Effect of long-term and short-term imbalanced Zn manipulation on gut microbiota and screening for microbial markers sensitive to zinc status. Microbiol. Spectr. 9:e0048321. doi: 10.1128/Spectrum.00483-21
Clemmons, B. A., Reese, S. T., Dantas, F. G., Franco, G. A., Smith, T. P. L., Adeyosoye, O. I., et al. (2017). Vaginal and uterine bacterial communities in postpartum lactating cows. Front. Microbiol. 8:1047. doi: 10.3389/fmicb.2017.01047
Couch, C. E., Stagaman, K., Spaan, R. S., Combrink, H. J., Sharpton, T. J., Beechler, B. R., et al. (2021). Diet and gut microbiome enterotype are associated at the population level in African buffalo. Nat. Commun. 12:2267. doi: 10.1038/s41467-021-22510-8
Deng, F., McClure, M., Rorie, R., Wang, X., Chai, J., Wei, X., et al. (2019). The vaginal and fecal microbiomes are related to pregnancy status in beef heifers. J. Anim. Sci. Biotechnol. 10:92. doi: 10.1186/s40104-019-0401-2
Galvão, K. N., Bicalho, R. C., and Jeon, S. J. (2019). Symposium review: the uterine microbiome associated with the development of uterine disease in dairy cows. J. Dairy Sci. 102, 11786–11797. doi: 10.3168/jds.2019-17106
Gardella, B., Pasquali, M. F., La Verde, M., Cianci, S., Torella, M., and Dominoni, M. (2022). The complex interplay between vaginal microbiota, HPV infection, and immunological microenvironment in cervical intraepithelial neoplasia: a literature review. Int. J. Mol. Sci. 23:7174. doi: 10.3390/ijms23137174
Giannattasio-Ferraz, S., Laguardia-Nascimento, M., Gasparini, M. R., Leite, L. R., Araujo, F. M. G., de Matos Salim, A. C., et al. (2019). A common vaginal microbiota composition among breeds of Bos taurus indicus (Gyr and Nellore). Braz. J. Microbiol. 50, 1115–1124. doi: 10.1007/s42770-019-00120-3
Gryaznova, M., Dvoretskaya, Y., Burakova, I., Syromyatnikov, M., Popov, E., Kokina, A., et al. (2022). Dynamics of changes in the gut microbiota of healthy mice fed with lactic acid bacteria and bifidobacteria. Microorganisms 10:1020. doi: 10.3390/microorganisms10051020
Jeon, S. J., Cunha, F., Vieira-Neto, A., Bicalho, R. C., Lima, S., Bicalho, M. L., et al. (2017). Blood as a route of transmission of uterine pathogens from the gut to the uterus in cows. Microbiome 5:109. doi: 10.1186/s40168-017-0328-9
Ji, S., Yang, R., Lu, C., Qiu, Z., Yan, C., and Zhao, Z. (2014). Differential expression of PPARγ, FASN, and ACADM genes in various adipose tissues and longissimus dorsi muscle from Yanbian yellow cattle and Yan yellow cattle. Asian Australas. J. Anim. Sci. 27, 10–18. doi: 10.5713/ajas.2013.13422
Laguardia-Nascimento, M., Branco, K. M., Gasparini, M. R., Giannattasio-Ferraz, S., Leite, L. R., Araujo, F. M., et al. (2015). Vaginal microbiome characterization of Nellore cattle using metagenomic analysis. PLoS One 10:e0143294. doi: 10.1371/journal.pone.0143294
Liu, F. T., Yang, S., Yang, Z., Zhou, P., Peng, T., Yin, J., et al. (2022). An altered microbiota in the lower and upper female reproductive tract of women with recurrent spontaneous abortion. Microbiol. Spectr. 10:e0046222. doi: 10.1128/spectrum.00462-22
Machado, V. S., Oikonomou, G., Bicalho, M. L., Knauer, W. A., Gilbert, R., and Bicalho, R. C. (2012). Investigation of postpartum dairy cows’ uterine microbial diversity using metagenomic pyrosequencing of the 16S rRNA gene. Vet. Microbiol. 159, 460–469. doi: 10.1016/j.vetmic.2012.04.033
Nesengani, L., Wang, J., Yang, Y., Yang, L., and Lu, W. (2017). Unravelling vaginal microbial genetic diversity and abundance between Holstein and Fleckvieh cattle. RSC Adv. 7, 56137–56143. doi: 10.1039/C7RA10553C
Romero, R., Hassan, S. S., Gajer, P., Tarca, A. L., Fadrosh, D. W., Nikita, L., et al. (2014). The composition and stability of the vaginal microbiota of normal pregnant women is different from that of non-pregnant women. Microbiome 2:4. doi: 10.1186/2049-2618-2-4
Santos, T. M., and Bicalho, R. C. (2012). Diversity and succession of bacterial communities in the uterine fluid of postpartum metritic, endometritic and healthy dairy cows. PLoS One 7:e53048. doi: 10.1371/journal.pone.0053048
Santos, T. M., Gilbert, R. O., and Bicalho, R. C. (2011). Metagenomic analysis of the uterine bacterial microbiota in healthy and metritic postpartum dairy cows. J. Dairy Sci. 94, 291–302. doi: 10.3168/jds.2010-3668
Saraf, V. S., Sheikh, S. A., Ahmad, A., Gillevet, P. M., Bokhari, H., and Javed, S. (2021). Vaginal microbiome: normalcy vs. dysbiosis. Arch. Microbiol. 203, 3793–3802. doi: 10.1007/s00203-021-02414-3
Sheldon, I. M., and Dobson, H. (2004). Postpartum uterine health in cattle. Anim. Reprod. Sci. 82-83, 295–306. doi: 10.1016/j.anireprosci.2004.04.006
Shen, J., Hanif, Q., Cao, Y., Yu, Y., Lei, C., Zhang, G., et al. (2020). Whole genome scan and selection signatures for climate adaption in Yanbian cattle. Front. Genet. 11:94. doi: 10.3389/fgene.2020.00094
Shirbroun, R. M. (2020). Histophilus somni: antigenic and genomic changes relevant to bovine respiratory disease. Vet. Clin. North Am. Food Anim. Pract. 36, 279–295. doi: 10.1016/j.cvfa.2020.02.003
Srinivasan, M., Adnane, M., and Archunan, G. (2021). Significance of cervico-vaginal microbes in bovine reproduction and pheromone production—a hypothetical review. Res. Vet. Sci. 135, 66–71. doi: 10.1016/j.rvsc.2021.01.003
Tachedjian, G., Aldunate, M., Bradshaw, C. S., and Cone, R. A. (2017). The role of lactic acid production by probiotic Lactobacillus species in vaginal health. Res. Microbiol. 168, 782–792. doi: 10.1016/j.resmic.2017.04.001
Tang, G., Kitten, T., Munro, C. L., Wellman, G. C., and Mintz, K. P. (2008). EmaA, a potential virulence determinant of Aggregatibacter actinomycetemcomitans in infective endocarditis. Infect. Immun. 76, 2316–2324. doi: 10.1128/IAI.00021-08
Valenti, P., Rosa, L., Capobianco, D., Lepanto, M. S., Schiavi, E., Cutone, A., et al. (2018). Role of lactobacilli and lactoferrin in the mucosal cervicovaginal defense. Front. Immunol. 9:376. doi: 10.3389/fimmu.2018.00376
Vitale, S. G., Ferrari, F., Ciebiera, M., Zgliczyńska, M., Rapisarda, A. M. C., Vecchio, G. M., et al. (2021). The role of genital tract microbiome in fertility: a systematic review. Int. J. Mol. Sci. 23:180. doi: 10.3390/ijms23010180
Wang, Y., Fu, Y., He, Y., Kulyar, M. F., Iqbal, M., Li, K., et al. (2021). Longitudinal characterization of the gut bacterial and fungal communities in yaks. J. Fungi 7:559. doi: 10.3390/jof7070559
Wozniak, H., Beckmann, T. S., Fröhlich, L., Soccorsi, T., Le Terrier, C., de Watteville, A., et al. (2022). The central and biodynamic role of gut microbiota in critically ill patients. Crit. Care 26:250. doi: 10.1186/s13054-022-04127-5
Wu, Y. F., Lee, W. F., Salamanca, E., Yao, W. L., Su, J. N., Wang, S. Y., et al. (2021). Oral microbiota changes in elderly patients, an indicator of Alzheimer’s disease. Int. J. Environ. Res. Public Health 18:4211. doi: 10.3390/ijerph18084211
Zafar, H., and Saier, M. H. Jr. (2021). Gut Bacteroides species in health and disease. Gut Microbes 13, 1–20. doi: 10.1080/19490976.2020.1848158
Zhu, S. L., Gu, F. F., Tang, Y. F., Liu, X. H., Jia, M. H., Valencak, T. G., et al. (2023). Dynamic fecal microenvironment properties enable predictions and understanding of peripartum blood oxidative status and non-esterified fatty acids in dairy cows. J. Dairy Sci. 107, 573–592. doi: 10.3168/jds.2022-23066
Keywords: Yanbian cattle, Yanhuang cattle, microbiota, reproductive tract, 16S rRNA gene
Citation: Teng Y, Feng S, Gu Z, Hou C, Xu H, Li Z, Zhao J, Fang Y, Ma X, Liu H, Guo J, Wang J, Ding H and Lu W (2024) Comparison of microbiota structure in reproductive tract of Yanbian cattle and Yanhuang cattle. Front. Microbiol. 15:1419914. doi: 10.3389/fmicb.2024.1419914
Edited by:
Pragya Tiwari, Yeungnam University, Republic of KoreaReviewed by:
Laith Khalil Tawfeeq Al-Ani, Universiti Sains Malaysia, MalaysiaCopyright © 2024 Teng, Feng, Gu, Hou, Xu, Li, Zhao, Fang, Ma, Liu, Guo, Wang, Ding and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: He Ding, ZGluZ2hlMTEzMEAxMjYuY29t; Wenfa Lu, d2VuZmEyMDA0QDE2My5jb20=
†ORCID Wenfa Lu, http://orcid.org/0000-0002-2417-1096
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.