 Dai Wang1†Kexin Li2,3,4†Lijuan Wang5†Zhongqiu Teng6Xia Luo6Hui Sun6Ying Huang6
Dai Wang1†Kexin Li2,3,4†Lijuan Wang5†Zhongqiu Teng6Xia Luo6Hui Sun6Ying Huang6 Songnian Hu4
Songnian Hu4 Xuefang Xu6*
Xuefang Xu6* Zilong He2,3*
Zilong He2,3*- 1Xiamen Key Laboratory of Perinatal-Neonatal Infection, Xiamen Women and Children's Hospital, Department of Pathology, State Key Laboratory of Molecular Vaccinology and Molecular Diagnostics, Department of Laboratory Medicine, School of Public Health, Xiamen University, Xiamen, Fujian, China
- 2School of Engineering Medicine, Beihang University, Beijing, China
- 3Beijing Advanced Innovation Center for Big Data-Based Precision Medicine, Interdisciplinary Innovation Institute of Medicine and Engineering, Beihang University, Beijing, China
- 4State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, Beijing, China
- 5Pediatric Intensive Care Unit, Beijing Children’s Hospital, National Center for Children's Health, Capital Medical University, Beijing, China
- 6National Key Laboratory of Intelligent Tracking and Forecasting for Infectious Diseases and National Institute for Communicable Diseases Control and Prevention, Chinese Center for Disease Control and Prevention, Beijing, China
Background: Infant botulism is caused by botulinum neurotoxin (BoNT), which is mainly produced by Clostridium botulinum. However, there is a lack of longitudinal cohort studies on infant botulism. Herein, we have constructed a cross-sectional and longitudinal cohort of infants infected with C. botulinum. Our goal was to reveal the differences in the intestinal microbiota of botulism-infected and healthy infants as well as the dynamic changes over time through multi-omics analysis.
Methods: We performed 16S rRNA sequencing of 20 infants’ stools over a period of 3 months and conducted whole genome sequencing of isolated C. botulinum strains from these laboratory-confirmed cases of infant botulism. Through bioinformatics analysis, we focused on the changes in the infants’ intestinal microbiota as well as function over time series.
Results: We found that Enterococcus was significantly enriched in the infected group and declined over time, whereas Bifidobacterium was significantly enriched in the healthy group and gradually increased over time. 18/20 isolates carried the type B 2 botulinum toxin gene with identical sequences. In silico Multilocus sequence typing found that 20\u00B0C. botulinum isolates from the patients were typed into ST31 and ST32.
Conclusion: Differences in intestinal microbiota and functions in infants were found with botulism through cross-sectional and longitudinal studies and Bifidobacterium may play a role in the recovery of infected infants.
Introduction
Infant botulism is one of the 6 forms of botulism (infant botulism, foodborne botulism, wound botulism, adult intestinal colonization, iatrogenic botulism, and inhalation botulism) (Shirey et al., 2015). Botulism is caused by botulinum neurotoxin (BoNT), which is mainly produced by Clostridium botulinum and rarely, by neurotoxigenic Clostridium baratii or Clostridium butyricum (Johnson et al., 1979). The illness is usually found in infants less than 1 year of age. C. botulinum, C. baratii, C. butyricum and C. sporogenes (BoNT-producing Clostridia) are responsible for infant and adult intestinal colonization botulism and can germinate, reproduce, and release BoNTs into the gut. The toxin is absorbed via lymphatic into the blood and transported to the neuromuscular junction, where it binds with soluble N-ethylmaleimide-sensitive factor attachment protein receptor, blocks acetylcholine release, and leads to flaccid paralysis (Arnon et al., 2006; Payne et al., 2018). Infant botulism is the major form of botulism in the United States (CDC, 2012) and has been found across the world (Drivenes et al., 2017; Dilena et al., 2021; Jeon et al., 2021; Avila et al., 2022; Douillard et al., 2022; Nga et al., 2022; Oliveira et al., 2022; Goldberg et al., 2023). Infant botulism was first reported in China in 1990 (Wu et al., 1990) and more cases have been reported in recent years with better diagnostic technologies (Xin et al., 2019; Ge et al., 2020; Zhang et al., 2022).
There are a few studies on the infection source and epidemiology of infant botulism (Koepke et al., 2008; Gladney et al., 2021). Some studies showed that BoNT-producing Clostridia spores are prevalent in soil which is the most likely source of infant botulism. A recent study in Colorado, United States found that the isolates from infant botulism cases were classified into a cluster from soil and dust, indicating a close relationship in genome sequences and a possible source from soil (Gladney et al., 2021). Infant botulism is known to affect the gut microbiota. Shirey TB et al. found significant differences in Proteobacteria, Firmicutes, and Enterobacteriaceae abundances in the fecal microbiota of infants with botulism compared to non-confirmed cases samples (Shirey et al., 2015). However, there is a lack of longitudinal cohort studies on gut microbiota variation in infant botulism in the short and long term, especially in gut microbiota following recovery.
In this study, we constructed a cross-sectional and longitudinal cohort of infants infected with C. botulinum. Stool samples for 16S rRNA sequencing were collected from 1 to 90 days of confirmed infected infants. Our goal was to reveal the differences in the intestinal microbiota and metabolism changes of botulism-infected infants compared with healthy infants as well as the dynamic changes over time through multi-omics analysis following treatment. The aim of the study was to gain a better understanding of the changes in intestinal microbiota caused by infant botulism and during recovery, to provide a better theoretical basis for the treatment of the disease.
Materials and methods
Sample collection
This study was conducted in accordance with the guidelines of the Helsinki Declaration and Rules of Good Clinical Practice. In compliance with human subjects’ exemption protocol (SHERLL 2019057) approved by the Ethics Committee of the Capital Institute of Pediatrics. Stool samples for 16S rRNA gene sequencing were collected from 20 infants who were all confirmed as BoNT gene positive by quantitative PCR from 2015 to 2021 (Huang et al., 2019). In addition, 10 stool samples were obtained from 10 healthy infants aged from 2 to 8 months for 16S rRNA gene sequencing analysis as BoNT gene negative by quantitative PCR. Besides, at least one botulism isolate from each patient was cultured and sequenced. Detailed meta information for 16S rRNA gene sequencing samples and isolated strains is listed in Table 1.
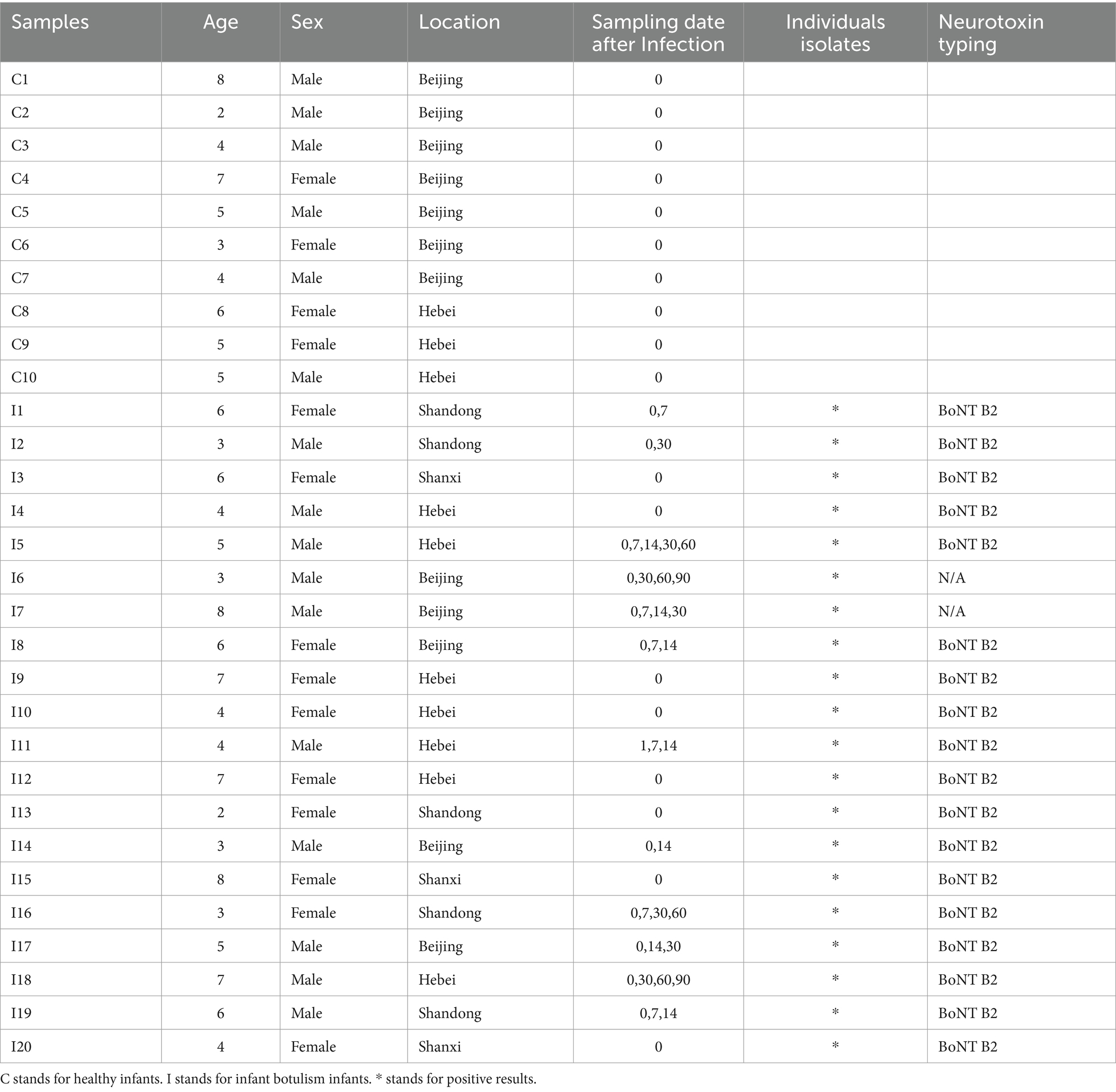
Table 1. Metadata of infant cohort.
DNA extraction and sequencing
All the stool samples for 16S rRNA sequencing were collected and frozen at −80°C after sampling. DNA was extracted using the E.Z.N.A stool DNA kit as the manufacturer’s instructions for 16S rRNA sequencing. The 16S rRNA gene were amplified for PCR amplification using universal primers 341F (5’-CCTAYGGGRBGCASCAG-3′) and 806R (3’-GGACTACNNGGGTATCTAAT-5′). According to the manufacturer’s instructions, the library was constructed using TruSeq DNA PCR-free sample preparation kit. Each PCR reaction was comprised of 5 μL of 10 × PCR buffer, 0.5 μL of dNTP (10 mM), 0.5 μL of PCR forward primer (50 μM), 0.5 μL of revese primer (50 μM), 0.5 μL of Platinum Taq (5 U/μL) (Thermo Fisher Scientific, USA) and 20 ng DNA in a total volume of 50 μL. PCR conditions comprised of initial denaturation at 95°C for 5 min, followed by 29 cycles of denaturation at 94°C for 30 s, annealing at 50°C for 60 s and elongation at 72°C for 60 s, with the final extension at 72°C for 7 min. Each sample was indexed and sequenced using the Illumina MiSeq PE-300. Pure strains of C. botulinum isolated from laboratory-confirmed cases of infant botulism were submitted to DNA extraction, and the total DNA of these isolated strains was purified from overnight culture using Qiagen Genomic-tip 100/G columns (Qiagen, Germantown, MD, United States) under the manufacturer’s instructions. The purity and integrity of the DNA were tested by agarose gel electrophoresis and quantified by Qubit. Qualified DNA samples detected by electrophoresis were randomly broken into fragments. Libraries were prepared using a Nextera XT DNA library preparation kit (Illumina, Inc., Cambridge, UK). Whole genome sequencing (WGS) was performed using Illumina NovaSeq 6,000 with PE-150 model.
Processing of 16S rRNA sequencing data
All 16S rRNA sequencing data were processed with the Quantitative Insights into Microbial Ecology version 2 (QIIME2) software (Bolyen et al., 2019). The DADA2 plugin was used for quality control, filtering chimeric sequences, and assembling reads (Callahan et al., 2016). Q2-feature-classifier plugin with the Greengene database was used to do taxonomic annotation (DeSantis et al., 2006). Alpha diversity and Principal Coordinates Analysis (PCoA) based on Bray-Curtis diversity were performed by vegan and ggplot2 package in R software (version 3.6.1). The LEfSe was applied to determine the microbial taxa with significantly differential abundance between groups (LDA > 3.0) (Segata et al., 2011). Metabolic pathways were predicted by PICRUSt2 using the KEGG database (Douglas et al., 2020). The co-abundance networks of microbial taxa were visualized by Cytoscape version 3.72.
Processing of whole genome sequencing data
Low quality and adapter reads were removed by trimmomatic v0.39. The reads were assembled using SPAdes v3.14.0 with the “- careful” option (Prjibelski et al., 2020). QUAST v4.6 and BUSCO v5 were used to conduct a quality check of the genomes and infer the annotation completeness (Gurevich et al., 2013; Manni et al., 2021). Prokka v1.13 was used to predict and annotate the genome assemblies (Seemann, 2014). The pan genome analysis was performed using Roary v3.13 with default parameters (Page et al., 2015). Multilocus sequence typing (MLST) schemes were determined using the PubMLST C. botulinum database.1 The multi-sequence alignment was performed by mafft and visualized by MEGA X (Katoh et al., 2002; Kumar et al., 2018).
Statistical analysis
R scripts with packages ggplot2 and vegan in R v3.6.1 were used for statistical analysis and visualization. The heatmap was drawn by the pheatmap package.
Result
The overview of infant cohorts and sequencing data
Stool samples were collected from a total of 30 infants for the study (10 healthy and 20 infected infants). The 30 infants came from northern cities in China and ranged in age from 2 months to 8 months, with a sex ratio of nearly 1:1 (Table 1). To investigate the ongoing effects of C. botulinum on infant gut microbiota, we collected the fecal samples from 12 of 20 infected infants at days 1, 7, 14, 30, 60 and 90. A total of 47 longitudinal cohort samples were collected for 16S rRNA analysis. On average, each 16S sample contained 71,427 ± 11,050 reads across all samples. In addition, 20 strains of C. botulinum were isolated from infected infant fecal samples for whole genome sequencing. The total size of whole genomes sequencing for strains was 26.46 G, with an average sequencing coverage of 339 ± 122.36 × per strain. Following pre-processing, all samples were saturated by genome completion assessment and ready for subsequent analysis. The specific analysis flow is shown in Supplementary Figure S1.
The composition of intestinal microbiota in healthy and Clostridium botulinum-infected infants
Based on the standard 16S data analysis protocol, 16 phyla, 24 orders, 34 orders, 54 families, 138 genera and 195 species were identified in healthy and C. botulinum -infected infants. In terms of taxonomic composition, the top 3 phyla were Actinobacteriota (37.46% mean abundance), Firmicutes (29.65%) and Proteobacteria (26.20%). At the genus level (Figure 1A), the most abundant genera were Bifidobacterium (31.58%), Escherichia (15.87%) and Enterococcus (10.24%). In terms of diversity, differences in the overall distribution between the healthy and infected groups were found using PCoA analysis based on the Bray–Curtis dissimilarity method (PERMANOVA, p value <0.05) (Figure 1B). The Shannon index and the Simpson index of the healthy group were higher than those of the infected group, but the difference was not statistically significant (Figure 1C).
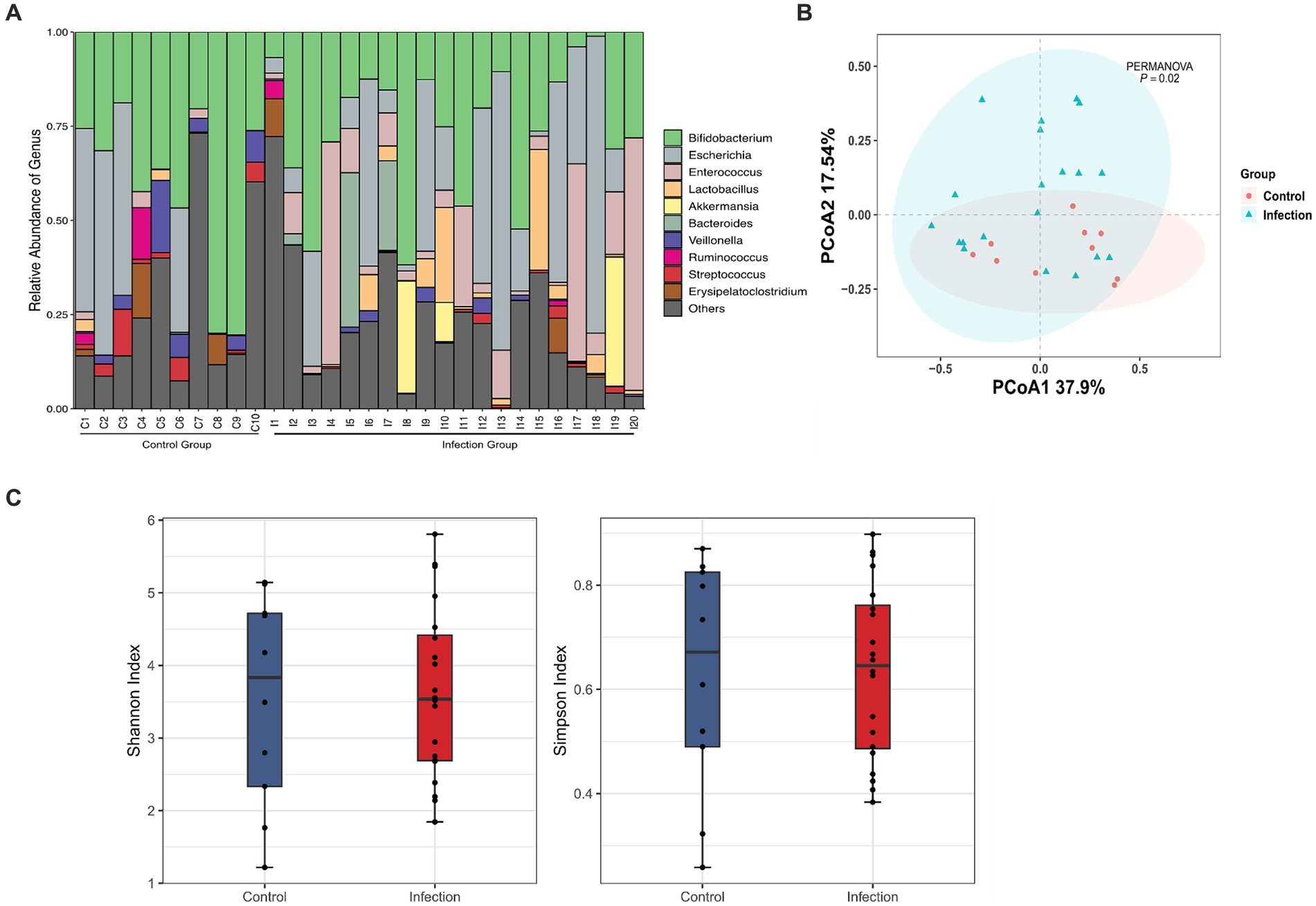
Figure 1. Diversity analysis based on 16S rRNA analysis. (A) Taxonomic classification at the genus level between control and infection groups (day1). (B) Principal coordinate analysis plot between control and infection groups based on Bray Curtis of bacterial communities (PERMANOVA, FDR, p < 0.05). (C) Shannon and Simpson indexes between Control (red) and Infection (blue) samples.
The classification and functional differences in intestinal microorganisms between healthy and Clostridium botulinum infected infant cohorts
To systematically assess the differences in intestinal microbiota between healthy and infected infants, we analyzed the microbial communities with different abundance and the function of differential microorganisms. We detected 15 differentially abundant microbiotas at the genus level (LDA > 2), with Bifidobacterium showing the greatest degree of difference among those with higher abundance in the healthy group (LDA > 4). Gemella, Lachnoanaerobaculum, and Veillonella were also different in the two groups. Besides, differential abundant microbiota with higher abundance in the infected group were Enterococcus (LDA > 4) and Lactobacillus (LDA > 3) (Figure 2A).
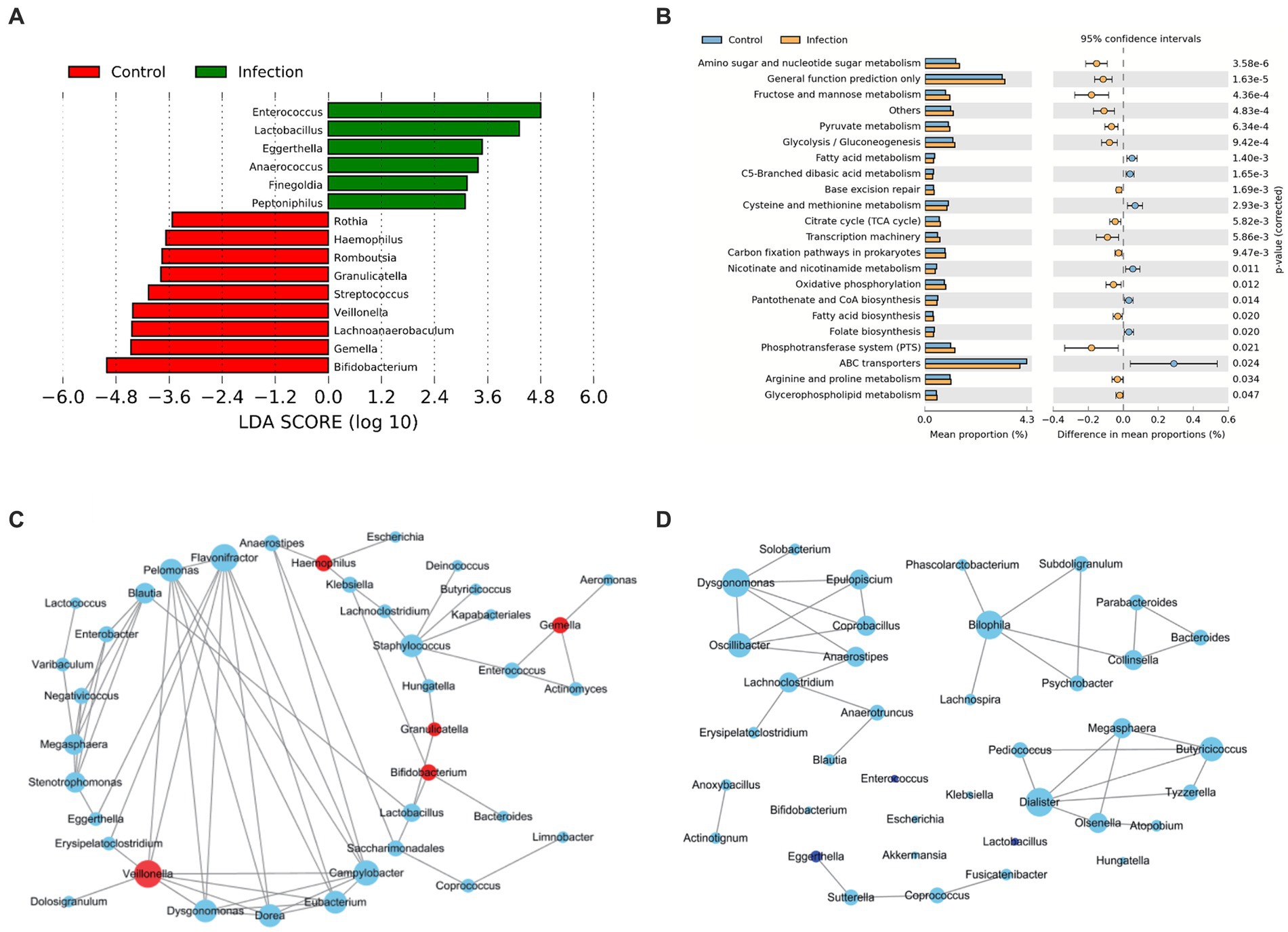
Figure 2. Bacteria analysis and functional categories of the bacterial communities predicted by PICRUSt2 analysis. (A) Lefse analysis at the genus level between control and infection groups (Wilcox test, FDR, p < 0.05). (B) Functional categories of the differentiated pathways at KEGG level 3 between control and infection groups (Welch’s t-test, p < 0.05). The bars in the graph represent the mean proportion (%) of the functional categories. The 95% confidence intervals reflect the difference in mean proportions (%), and corrected p values are displayed. (C,D) Co-abundance networks of genera between control (C) and infection (D) groups. The red dots represent the differentially abundant genera for each group. The blue dots represent intestinal genera that were not differentially abundant genera. The size of the dots represents the degree. The links represent the interactions of genera. The correlations with r > 0.75 and p < 0.05 are shown.
In terms of tertiary metabolic functions, the two groups differed in 21 pathways (p < 0.05) (Figure 2B). The metabolic pathways with the greatest differences were mainly amino sugar and nucleotide sugar metabolism, fructose and mannose metabolism, and pyruvate metabolism, which were significantly enriched in the infected group. Compared with the control group, fatty acid metabolism, cysteine and methionine metabolism, and nicotinate and nicotinamide metabolism decreased in the infection group.
In addition, we constructed microbial co-abundance networks for the healthy group and the infected group of infants, respectively, (Figures 2C,D). We found that the network in the healthy group was more tightly linked than the network in the infected group, with more connections between nodes. In the healthy group, the degree of all the differential abundant microbiota was less than or equal to 3, except for Veillonella, which had a linkage greater than 5. Flavonifractor (degree = 8) was the most linked taxon in the healthy group network (Figure 2C). The overall degree of the infected group network was not high. The most linked taxa in the infected group network were Dysgonomonas, Bilophila, and Dialister, all of which had a linkage of 5 (Figure 2D).
Longitudinal analysis reveals the dynamics of key microbiota from Clostridium botulinum infected infants
In the differential abundant microbiota analysis described above, we found that Bifidobacterium and Enterococcus differed most in the two cohorts. To examine the temporal dynamics of these two groups, we followed up on some of the infected infants (12/20) and collected stool samples for the longitudinal study. The cross-sectional 16S data analysis also showed significant differences in Enterococcus (p < 0.05) (Figures 3A,B). The overall abundance of Bifidobacterium increased significantly after 30 days and the dispersion throughout decreased, whereas Enterococcus decreased rapidly after 14 days and was almost undetectable after 30 (Figures 3C,D). We also compared the samples of patients at the initial stage of infection (Infection group) with the samples of patients at 60 days and 90 days after disease diagnosis (Recovery group). We found that the differential abundant bacteria between the two groups were also different between the infected group and the healthy group, except for Bifidobacterium (Supplementary Figure S2). Secondary metabolic pathway analysis revealed that the microbial-related metabolic pathways like Glycan biosynthesis and metabolism, amino acid metabolism, metabolism of cofactors and vitamins, and biosynthesis of other secondary metabolites in the infected group gradually returned to the levels of the control group over time (Figure 3E).
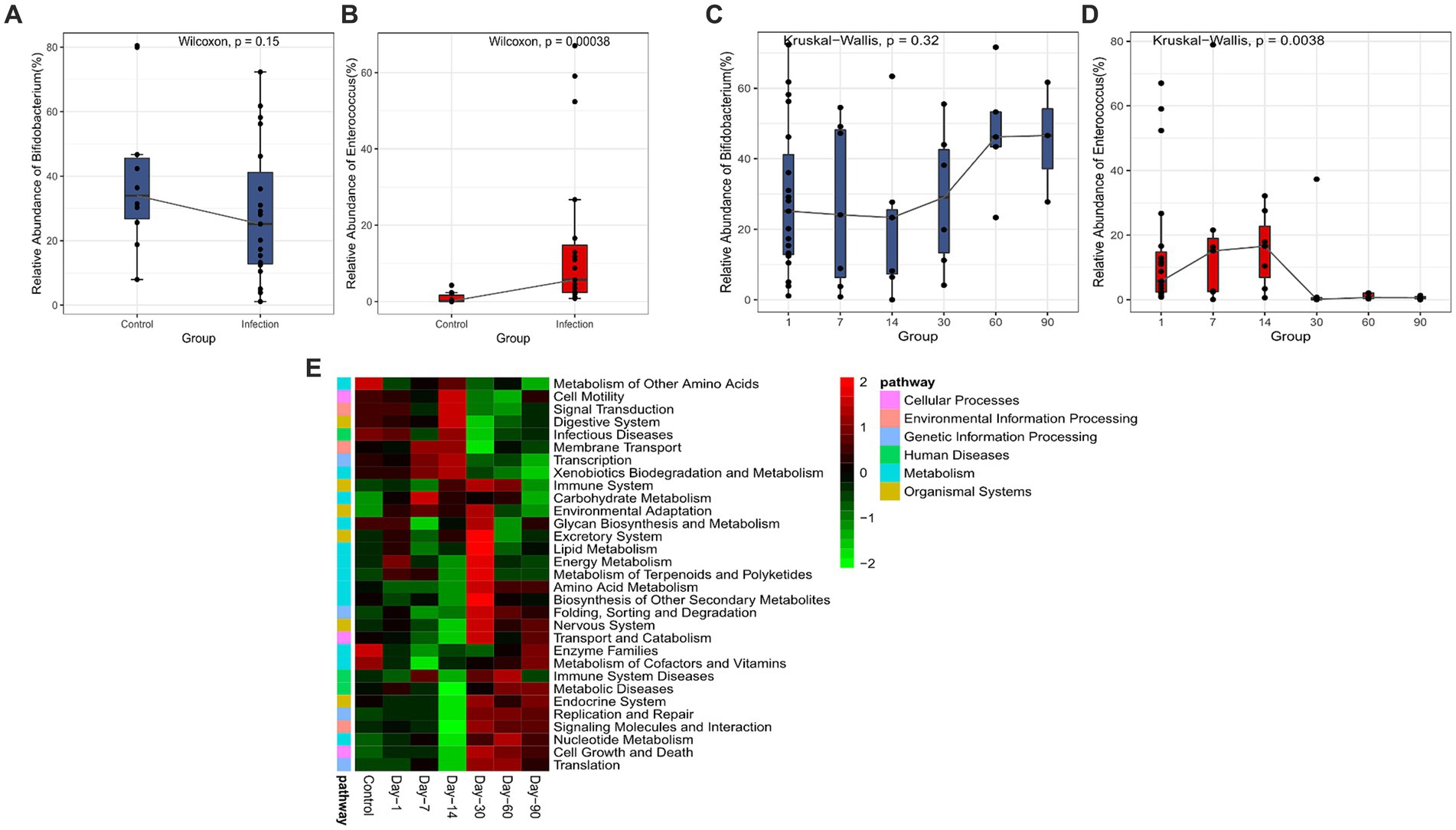
Figure 3. Analysis of significant bacteria and functional categories in different groups. (A) Bifidobacterium in Control and Infection groups. (B) Enterococcus in Control and Infection groups. (C) Changing of Bifidobacterium in days 1, 7, 14, 30, 60, 90. (D) Changing of Enterococcus in day 1, 7, 14, 30, 60, 90. (E) Heatmap of the relative abundances of pathways in different groups predicted by PICRUSt2 analysis at KEGG level 3 based on z-scores.
Genomic analysis of Clostridium botulinum isolates from infected infants
The 20 C. botulinum isolates from the stool samples of the 20 infected infants with one isolate per case were sequenced using Illumina sequencing. The average genome length ranged from 3.8 to 4.3 M, with an average N50 of around 846 kb (Supplementary Table S1). 18/20 isolates carried the botulinum toxin gene with identical nucleotide sequence encoding the type B 2 botulinum toxin (Supplementary Figure S3). In silico MLST typing found that the 20 C. botulinum isolates from the patients were typed into two sequence types (ST), ST31 and ST32 with 10 isolates each. KEGG enrichment analysis based on the core genome of 20 strains of C. botulinum showed that ABC transporters and two-component system pathways had the highest gene enrichment degree (Supplementary Figure S4).
Discussion
The initial sterile gut in the infant is an oxidized environment favorable to be colonized by facultative aerobes such as Lactobacillus, Prevotella, and Sneathia sp. With less oxygen in the gut, it becomes more suitable for anaerobic bacteria to grow (Penders et al., 2006). Therefore, BoNT-producing Clostridia infection mostly occurs in infants aged 0 to 8 months. Because the intestinal microbiota of infants is not stable, it is likely to carry spores into the gut through the mouth, thus causing poisoning. At present, there are few studies on the intestinal microbiota of infants with botulism. Only one study in the US reported intestinal microbial profiling of 14 infected infants (Shirey et al., 2015). Most botulinum toxin studies are associated with strains isolated from humans or the environment, but there is a lack of cohort comparison in cross-sectional and longitudinal analysis of intestinal microbiota in infant botulism. In this study, we first collected the fecal samples of infants infected with C. botulinum from China. In addition, some of the isolates were isolated from infant feces and the surrounding environment. To study the dynamic changes of intestinal microbiota in infants infected with C. botulinum on a time scale, we tracked and collected fecal samples from some infants at multiple time points. The strategy of cross-sectional and longitudinal study based on metagenome and whole genome sequencing of bacteria may help us better understand the infection process and mechanism of botulism.
Shirey et al. examined 14 infant fecal samples including 8 botulism samples and 6 non-confirmed which were used as control (Shirey et al., 2015). Due to the lack of metadata on the samples, the non-confirmed samples might not be strictly healthy controls. In this study, we investigated 20 botulism samples which were not only confirmed by PCR but also by strain isolation. 10 healthy infant fecal samples were collected as control covering all the age spectrum of the confirmed samples. Shirey et al. illustrated that the abundance of Enterobacteriaceae in infant intestinal microbiota of botulism is significantly increased, which is consistent with our findings (Shirey et al., 2015). We found that the abundance of Escherichia and Enterococcus were all increased in the infected group compared with the healthy group. In the time scale analysis, we also found that the abundance of Enterococcus in the infected group gradually decreased after 14 days until it could not be detected, indicating that Enterococcus may be involved in the occurrence of the infection. Interestingly, it was also reported an increase in Enterococcus in children infected with Clostridioides difficile (Ling et al., 2014). In addition, the LDA score of Bifidobacterium was very high in the healthy group compared with the infected group. It is well known that Bifidobacterium, as a probiotic, plays an important role in the regulation of intestinal microbiome (Stuivenberg et al., 2022). Many studies have reported that Bifidobacterium plays a positive role in promoting intestinal health (O'Callaghan and van Sinderen, 2016; Turroni et al., 2019; Derrien et al., 2022). In our study, the abundance of Bifidobacterium in the healthy group was higher than that in the infected group (LDA > 4). The time scale analysis showed that the abundance of Bifidobacterium increased significantly after 30 days in the infection group, indicating that the abundance of Bifidobacterium decreased after the initial infection, and increased during the recovery of infection. This phenomenon is consistent with previous reports indicating that infected children progressively return to a “healthy microbiota status” (Becker-Dreps et al., 2015; Singh et al., 2015). Hence, it may be indicated that the supplement of Bifidobacterium during the infection process may help in the treatment of the infection. Furthermore, metabolic changes caused by microflora changes were also studied here. Amino acid metabolism including arginine, proline, cysteine, and methionine were altered. We know that high protein concentration was needed in the growth of C. botulinum. Therefore, we infer that these amino acid changes are related to C. botulinum growth.
In this study, genomic analysis of C. botulinum isolates identified two STs, neither of which has been reported in previous studies. Previous studies have reported genotyping of C. botulinum from infants to be predominantly ST2, with fewer strains of other ST typing (Brunt et al., 2020). In terms of botulinum toxin identification, 18/20 strains were found to carry the botulinum toxin gene. Interestingly, all the botulinum toxin genes identified were type B 2 and the protein sequences were highly consistent, suggesting that the botulinum toxins prevalent in northern China are likely to be closely related as previously reported (Vanhomwegen et al., 2013; Ma et al., 2022). What’s more, we downloaded 68 complete-level genome sequences of C. botulinum in NCBI database. We found B5 was the most toxin subtype (n = 23), followed by A1 (n = 22), A2 (n = 12), and, as reported here, B2 (n = 10) (Supplementary Figure S5).
In this project, we used a strategy of 16S sequencing and whole genome sequencing of single bacteria isolates to study C. botulinum infection. Unfortunately, 16S rRNA sequencing failed to obtain OTUs of C. botulinum, which may be due to the resolution of the 16S segments and thus the inability to accurately quantify the abundance of C. botulinum. Given the low microbial content of infant feces, the full-length 16S rRNA sequencing based on third-generation sequencing may be more suitable. This technology can pinpoint the species-level abundance of most microorganisms without requiring much raw DNA volume compared to normal 16S rRNA sequencing, which may be a better choice for the study of intestinal microbes in infants in the future. In addition, a combined strategy of full-length 16S sequencing and whole genome sequencing is also recommended.
Conclusion
In conclusion, we have systematically analyzed the differences in gut microbes and functions in botulism-affected infants through cross-sectional and longitudinal studies. We found that Enterococcus was significantly enriched in the infected group and declined over time, whereas Bifidobacterium was significantly enriched in the healthy group and gradually increased over time following infection in infected infants. Pathway analysis revealed that the metabolism of cofactors and vitamins, and amino acid metabolism gradually returned to normal levels over time. The findings provided a better understanding of the changes in intestinal microbiota caused by infant botulism and during recovery and a better theoretical basis for the management of the disease.
Data availability statement
Sequencing data were deposited in NCBI with accession numbers PRJNA908234 and PRJNA931660.
Ethics statement
The studies involving humans were approved by this study was conducted in accordance with the guidelines of the Helsinki Declaration and Rules of Good Clinical Practice. In compliance with human subjects’ exemption protocol (SHERLL 2019057) approved by Ethics Committee of the Capital Institute of Pediatrics. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants' legal guardians/next of kin.
Author contributions
DW: Writing – review & editing, Writing – original draft, Supervision, Software, Investigation, Funding acquisition, Data curation, Conceptualization. KL: Writing – review & editing, Writing – original draft, Software, Methodology, Investigation, Formal analysis, Data curation. LW: Writing – original draft, Resources. ZT: Writing – original draft, Methodology, Formal analysis, Data curation. XL: Writing – original draft, Methodology, Investigation. HS: Writing – original draft, Validation. YH: Writing – original draft, Validation. SH: Writing – review & editing, Resources. XX: Writing – review & editing, Writing – original draft, Validation, Supervision, Project administration, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. ZH: Writing – review & editing, Writing – original draft, Validation, Supervision, Software, Methodology, Investigation, Formal analysis, Data curation, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the NSFC grants (82272349/81971905), Shenzhen Science and Technology Program (JCYJ20220530143409021) to DW.
Acknowledgments
We thank Xiushan Ge for providing the infant stool samples.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1416879/full#supplementary-material
Footnotes
References
Arnon, S. S., Schechter, R., Maslanka, S. E., Jewell, N. P., and Hatheway, C. L. (2006). Human botulism immune globulin for the treatment of infant botulism. N. Engl. J. Med. 354, 462–471. doi: 10.1056/NEJMoa051926
Avila, C. E., Cardenas, M. M., Kaltenbach, G. H., Lazzarini, L., and Pierangeli, N. (2022). Infant botulism: a descriptive study in a pediatric intensive care unit. Arch. Argent. Pediatr. 121:e202202656. doi: 10.5546/aap.2022-02656.eng
Becker-Dreps, S., Allali, I., Monteagudo, A., Vilchez, S., Hudgens, M. G., Rogawski, E. T., et al. (2015). Gut microbiome composition in young Nicaraguan children during diarrhea episodes and recovery. Am. J. Trop. Med. Hyg. 93, 1187–1193. doi: 10.4269/ajtmh.15-0322
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Author correction: reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37:1091. doi: 10.1038/s41587-019-0252-6
Brunt, J., van Vliet, A. H. M., Carter, A. T., Stringer, S. C., Amar, C., Grant, K. A., et al. (2020). Diversity of the genomes and neurotoxins of strains of Clostridium botulinum group I and Clostridium sporogenes associated with foodborne, infant and wound botulism. Toxins 12:586. doi: 10.3390/toxins12090586
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
CDC. (2012). National botulism surveillance system. Available at: http://www.cdc.gov/nationalsurveillance/botulism-surveillance.html
Derrien, M., Turroni, F., Ventura, M., and van Sinderen, D. (2022). Insights into endogenous Bifidobacterium species in the human gut microbiota during adulthood. Trends Microbiol. 30, 940–947. doi: 10.1016/j.tim.2022.04.004
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dilena, R., Pozzato, M., Baselli, L., Chidini, G., Barbieri, S., Scalfaro, C., et al. (2021). Infant botulism: checklist for timely clinical diagnosis and new possible risk factors originated from a case report and literature review. Toxins 13:860. doi: 10.3390/toxins13120860
Douglas, G. M., Maffei, V. J., Zaneveld, J. R., Yurgel, S. N., Brown, J. R., Taylor, C. M., et al. (2020). PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 38, 685–688. doi: 10.1038/s41587-020-0548-6
Douillard, F. P., Derman, Y., Woudstra, C., Selby, K., Maklin, T., Dorner, M. B., et al. (2022). Genomic and phenotypic characterization of Clostridium botulinum isolates from an infant botulism case suggests adaptation signatures to the gut. MBio 13:e0238421. doi: 10.1128/mbio.02384-21
Drivenes, B., Krause, T. G., Andersson, M., Muller, L., Fuursted, K., Pedersen, T., et al. (2017). Infant botulism in Denmark from 1995 to 2015. Dan. Med. J. 64:A5404.
Ge, X. S., Xu, Q., Liu, X., Huang, S., Gao, Y., Liu, P., et al. (2020). Clinical analysis and laboratory diagnosis of three cases with infantile botulism caused by Clostridium botulinum type B. Chin. J. Pediatr. 58, 499–502. doi: 10.3760/cma.j.cn112140-20191101-00691C
Gladney, L., Halpin, J. L., and Luquez, C. (2021). Genomic characterization of strains from a cluster of infant botulism type a in a small town in Colorado, United States. Front. Microbiol. 12:688240. doi: 10.3389/fmicb.2021.688240
Goldberg, B., Danino, D., Levinsky, Y., Levy, I., Straussberg, R., Dabaja-Younis, H., et al. (2023). Infant Botulism, Israel, 2007-2021. Emerg. Infect. Dis. 29, 235–241. doi: 10.3201/eid2902.220991
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Huang, Y., Shi, Y., Ye, C., and Xu, X. (2019). Establishment of real-time PCR assays for rapid detection of Clostridium botulinum type a and B. Dis. Surveill. 34, 844–848. doi: 10.3784/j.issn.1003-9961.2019.09.015
Jeon, J. H., Choi, C. H., Kim, J. H., Hyun, J., Choi, E. S., Choi, S. Y., et al. (2021). Genetic characterization of Clostridium botulinum isolated from the first case of infant botulism in Korea. Ann. Lab. Med. 41, 489–492. doi: 10.3343/alm.2021.41.5.489
Johnson, R. O., Clay, S. A., and Arnon, S. S. (1979). Diagnosis and management of infant botulism. Am. J. Dis. Child. 133, 586–593
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Koepke, R., Sobel, J., and Arnon, S. S. (2008). Global occurrence of infant botulism, 1976-2006. Pediatrics 122, e73–e82. doi: 10.1542/peds.2007-1827
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Ling, Z., Liu, X., Jia, X., Cheng, Y., Luo, Y., Yuan, L., et al. (2014). Impacts of infection with different toxigenic Clostridium difficile strains on faecal microbiota in children. Sci. Rep. 4:7485. doi: 10.1038/srep07485
Ma, X., Li, K., Li, F., Su, J., Meng, W., Sun, Y., et al. (2022). Tracing foodborne botulism events caused by Clostridium botulinum in Xinjiang Province, China, using a Core genome sequence typing scheme. Microbiol. Spectr. 10:e0116422. doi: 10.1128/spectrum.01164-22
Manni, M., Berkeley, M. R., Seppey, M., and Zdobnov, E. M. (2021). BUSCO: assessing genomic data quality and beyond. Curr. Protoc. 1:e323. doi: 10.1002/cpz1.323
Nga, T. T., Hoang, L. H., Trang, L. T., Tram, N. T., Yen, P. B., Trung, N. T., et al. (2022). First confirmed case of infant botulism caused by Clostridium botulinum type a(B) in a 10-month-old infant in Hanoi, Vietnam. IJID Reg. 5, 18–20. doi: 10.1016/j.ijregi.2022.08.003
O'Callaghan, A., and van Sinderen, D. (2016). Bifidobacteria and their role as members of the human gut microbiota. Front. Microbiol. 7:925. doi: 10.3389/fmicb.2016.00925
Oliveira, L. M., Goncalves, D. B., Cabral, L. C. R., Bernardino, M. R. A., and Feitoza, P. V. S. (2022). Botulism in the Brazilian Amazon: a life-threatening disease in a neglected population. Arq. Neuropsiquiatr. 80, 1227–1232. doi: 10.1055/s-0042-1758651
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Payne, J. R., Khouri, J. M., Jewell, N. P., and Arnon, S. S. (2018). Efficacy of human botulism immune globulin for the treatment of infant botulism: the first 12 years post licensure. J. Pediatr. 193, 172–177. doi: 10.1016/j.jpeds.2017.10.035
Penders, J., Thijs, C., Vink, C., Stelma, F. F., Snijders, B., Kummeling, I., et al. (2006). Factors influencing the composition of the intestinal microbiota in early infancy. Pediatrics 118, 511–521. doi: 10.1542/peds.2005-2824
Prjibelski, A., Antipov, D., Meleshko, D., Lapidus, A., and Korobeynikov, A. (2020). Using SPAdes De novo assembler. Curr. Protoc. Bioinformatics 70:e102. doi: 10.1002/cpbi.102
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Shirey, T. B., Dykes, J. K., Luquez, C., Maslanka, S. E., and Raphael, B. H. (2015). Characterizing the fecal microbiota of infants with botulism. Microbiome 3:54. doi: 10.1186/s40168-015-0119-0
Singh, P., Teal, T. K., Marsh, T. L., Tiedje, J. M., Mosci, R., Jernigan, K., et al. (2015). Intestinal microbial communities associated with acute enteric infections and disease recovery. Microbiome 3:45. doi: 10.1186/s40168-015-0109-2
Stuivenberg, G. A., Burton, J. P., Bron, P. A., and Reid, G. (2022). Why are Bifidobacteria important for infants? Microorganisms 10:278. doi: 10.3390/microorganisms10020278
Turroni, F., Duranti, S., Milani, C., Lugli, G. A., van Sinderen, D., and Ventura, M. (2019). Bifidobacterium bifidum: a key member of the early human gut microbiota. Microorganisms 7:544. doi: 10.3390/microorganisms7110544
Vanhomwegen, J., Berthet, N., Mazuet, C., Guigon, G., Vallaeys, T., Stamboliyska, R., et al. (2013). Application of high-density DNA resequencing microarray for detection and characterization of botulinum neurotoxin-producing clostridia. PLoS One 8:e67510. doi: 10.1371/journal.pone.0067510
Wu, J. W., Cheng, S., and Zheng, X. (1990). A report of an infant botulism case. Chin. J. Pediatr. 28:1.
Xin, W., Huang, Y., Ji, B., Li, P., Wu, Y., Liu, J., et al. (2019). Identification and characterization of Clostridium botulinum strains associated with an infant botulism case in China. Anaerobe 55, 1–7. doi: 10.1016/j.anaerobe.2018.06.015
Keywords: infant botulism, 16S rRNA sequencing, whole genome sequencing, Bifidobacterium, longitudinal study
Citation: Wang D, Li K, Wang L, Teng Z, Luo X, Sun H, Huang Y, Hu S, Xu X and He Z (2024) Dissecting and tracing the gut microbiota of infants with botulism: a cross sectional and longitudinal study. Front. Microbiol. 15:1416879. doi: 10.3389/fmicb.2024.1416879
Edited by:
François J. M. A. Meurens, Montreal University, CanadaReviewed by:
Fabrizio Anniballi, National Institute of Health (ISS), ItalyChristelle Mazuet, Institut Pasteur, France
Copyright © 2024 Wang, Li, Wang, Teng, Luo, Sun, Huang, Hu, Xu and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuefang Xu, eHV4dWVmYW5nQGljZGMuY24=; Zilong He, aGV6aWxvbmdAYnVhYS5lZHUuY24=
†These authors have contributed equally to this work