 Haitham Hussain1†
Haitham Hussain1† Amer Nubgan2†§
Amer Nubgan2†§ César Rodríguez3
César Rodríguez3 Korakrit Imwattana4,5
Korakrit Imwattana4,5 Daniel R. Knight4,6
Daniel R. Knight4,6 Valerija Parthala2
Valerija Parthala2 Peter Mullany1‡
Peter Mullany1‡ Shan Goh2*‡
Shan Goh2*‡- 1Department of Microbial Diseases, Eastman Dental Institute, University College London, London, United Kingdom
- 2Department of Clinical, Pharmaceutical and Biological Sciences, University of Hertfordshire, Hatfield, United Kingdom
- 3Facultad de Microbiología and Centro de Investigación en Enfermedades Tropicales (CIET), Universidad de Costa Rica, San José, Costa Rica
- 4School of Biomedical Sciences, The University of Western Australia, Perth, WA, Australia
- 5Department of Microbiology, Faculty of Medicine Siriraj Hospital, Mahidol University, Salaya, Thailand
- 6Department of Microbiology, PathWest Laboratory Medicine WA, Queen Elizabeth II Medical Centre, Nedlands, WA, Australia
Clostridioides difficile is an emerging pathogen of One Health significance. Its highly variable genome contains mobile genetic elements (MGEs) such as transposons and prophages that influence its biology. Systematic deletion of each genetic element is required to determine their precise role in C. difficile biology and contribution to the wider mobilome. Here, Tn5397 (21 kb) and ϕ027 (56 kb) were deleted from C. difficile 630 and R20291, respectively, using allele replacement facilitated by CRISPR-Cas9. The 630 Tn5397 deletant transferred PaLoc at the same frequency (1 × 10−7) as 630 harboring Tn5397, indicating that Tn5397 alone did not mediate conjugative transfer of PaLoc. The R20291 ϕ027 deletant was sensitive to ϕ027 infection, and contained two unexpected features, a 2.7 kb remnant of the mutagenesis plasmid, and a putative catalase gene adjacent to the deleted prophage was also deleted. Growth kinetics of R20291 ϕ027 deletant was similar to wild type (WT) in rich medium but marginally reduced compared with WT in minimal medium. This work indicates the commonly used pMTL8000 plasmid series works well for CRISPR-Cas9-mediated gene deletion, resulting in the largest deleted locus (56.8 kb) described in C. difficile. Removal of MGEs was achieved by targeting conjugative/integrative regions to promote excision and permanent loss. The deletants created will be useful strains for investigating Tn5397 or ϕ027 prophage contribution to host virulence, fitness, and physiology, and a platform for other mutagenesis studies aimed at functional gene analysis without native transposon or phage interference in C. difficile 630 and R20291.
1 Introduction
Clostridioides difficile, also known as Clostridium difficile (Lawson et al., 2016), is a Gram-positive, anaerobic, endospore-forming bacterium that causes gastrointestinal illness. It is a leading cause of antibiotic-associated diarrhea (He et al., 2013; Smits et al., 2016), and recurrent C. difficile infection (CDI) is difficult to treat with antibiotics alone (van Prehn et al., 2021). It is an important nosocomial and community-acquired pathogen worldwide (Magill et al., 2014; Collins et al., 2020; Finn et al., 2021; Viprey et al., 2022). Sources of infection include community spaces, environmental water and soil, animals, and the food chain (Candel-Pérez et al., 2019; Knight and Riley, 2019; Jo et al., 2022). Genetically similar strains from pigs and humans have been reported, indicating possible zoonotic or anthropogenic transmission (Knetsch et al., 2014; Moloney et al., 2021).
Clostridioides difficile has a highly variable genome with up to 30% being made up of mobile genetic elements (MGEs) (Sebaihia et al., 2006). These are very common in bacteria and can loosely be defined as any genetic element that can mediate its own transfer from one part of the genome to another. C. difficile contains a range of MGEs from the very simple, such as insertion sequences (IS) to complex integrative conjugative elements (ICE, also sometimes referred to as conjugative transposons) and integrated phage genomes called prophage (reviewed in Roberts et al., 2014). These MGEs can have a profound effect on the biology of C. difficile. For example, ICE often encode resistance to antibiotics; e.g., Tn916, and Tn5397 (tetracycline resistance). A large ICE, 023_CTnT found in C. difficile clade 3 strains contains genes encoding a sortase, putative sortase substrates, lantibiotic ABC transporters and a putative siderophore biosynthetic cluster (Shaw et al., 2019). Similar genes are found throughout the gut microbiome indicating that ICE have a role in allowing organisms to adapt to their local environment and can transfer through the gut microbiome. C. difficile also contains integrative mobilizable elements such as Tn4453a/b (chloramphenicol resistance), and Tn5398 (erythromycin resistance), which spread via conjugation, between and beyond C. difficile (Roberts et al., 2014). Furthermore, bioinformatic analysis of C. difficile ICE show that they contain different sigma factors implying that they can have a global role in gene expression in the organism (Brouwer et al., 2011). Prophage and ICE are modular MGEs and both typically enter the host bacterial genome via the activity of site-specific recombinases (Johnson and Grossman, 2015). These belong to two different families namely serine or tyrosine. The amino acid named referring to that responsible for cutting DNA at the active site (Johnson and Grossman, 2015). Comparison of ICE and phage show further relationships indicating that they can exchange modules and have an intertwined evolutionary history (Johnson and Grossman, 2015).
Lysogeny is frequently observed in C. difficile (Sebaihia et al., 2006; Ramirez-Vargas et al., 2018), with prophages most commonly belonging to the order Siphoviridae and Myoviridae, and most commonly identified with ϕC2 (Goh et al., 2005), ϕMMP04 (Meessen-Pinard et al., 2012), ϕCD119 (Govind et al., 2006), ϕCDHM1 (Hargreaves et al., 2014), ϕCD38-2 (Fortier and Moineau, 2007), and ϕCD27 (Mayer et al., 2008), ranging in size from 31 to 56 kb with a GC content similar to that of the C. difficile genome (28–30%) (Knight et al., 2015). C. difficile prophages can influence host toxin regulation (Goh et al., 2005; Govind et al., 2009, 2011; Sekulovic et al., 2011; Riedel et al., 2017), quorum sensing (Hargreaves et al., 2014), biofilm formation (Slater et al., 2019) and fitness including transduction (Goh et al., 2013), phage immunity (Boudry et al., 2015; Sekulovic et al., 2015; Li et al., 2020), and plasmid/prophage maintenance (Peltier et al., 2020). Some of these studies were carried out by infecting C. difficile with a phage of interest and examining changes to the transcriptome or selected phenotype. However due to the presence of prophages in the studied strains, it can be difficult to attribute changes solely to the infecting phage.
To prove the role of ICE and prophage in C. difficile biology it is necessary to make clean scarless deletions of these large genetic elements. The best understood C. difficile ICE is Tn5397, which encodes resistance to tetracycline and is capable of broad host range transfer within several Gram-positive organisms (Wang et al., 2006). Tn5379 translocates between strains by excising and forming a circular molecule, which is then capable of conjugal transfer to a suitable recipient or reintegration into the host genome (Supplementary Figure S1). In C. difficile 630 this element integrates into the genome close to the region of the chromosome which encodes the major virulence factors of the organism toxins A and B, termed the PaLoc (Brouwer et al., 2013). The later element can transfer at low frequency to non-toxigenic C. difficile strains converting them to toxin producers. The PaLoc does not have any genes which are obviously involved in its own transfer, so it was proposed that one of the C. difficile ICE mediated its transfer via a mechanism like Hfr in E. coli (Brouwer et al., 2013).
Two studies have deleted a prophage from C. difficile 630, lysogenic for two inducible prophages, ϕCD630-1 and ϕCD630-2 (Sebaihia et al., 2006; Goh et al., 2007). Hong et al deleted ϕCD630-2 using CRISPR-Cpf1 (Hong et al., 2018), and Peltier et al deleted ϕCD630-2 using a toxin-antitoxin system to select for double crossover mutants (Peltier et al., 2020). R20291 is a hypervirulent epidemic C. difficile strain that is well-characterized, and its genome was predicted to contain a complete prophage genome (Stabler et al., 2009), ϕ027, shown to spontaneously excise from the bacterial chromosome and circularize to exist extrachromosomally (Sekulovic and Fortier, 2015). ϕ027 has not been characterized as a functional phage, perhaps due to the lack of a suitable indicator strain for phage infection.
C. difficile possesses the type I-B CRISPR-Cas system utilizing several Cas proteins (Boudry et al., 2015; Andersen et al., 2016), which is different to the commonly used type II system utilizing a Cas9 or Cas12a protein. The use of heterologously expressed type II systems in C. difficile could avoid native type I-B CRISPR-Cas systems interference, which was successfully re-programmed for gene deletion in both C. difficile 630Δerm and R20291 (Maikova et al., 2019). Indeed, native CRISPR-Cas systems of C. difficile 630 and R20291 (Boudry et al., 2015) did not interfere with several reports of successful gene deletants selected using Cas9 or Cas12a (McAllister et al., 2017; Hong et al., 2018; Wang et al., 2018; Ingle et al., 2019) expressed from pMTL8000 plasmids (Heap et al., 2009). In this study, we used a similar strategy of CRISPR-Cas9 to select deletants of Tn5397 from C. difficile 630 and ϕ027 prophage from R20291(Stabler et al., 2009), allowing their contribution to C. difficile biology to be determined.
2 Materials and methods
2.1 Bacterial strains and growth conditions
The C. difficile and Escherichia coli strains used in this study are listed in Table 1. All bacterial strains were stored at −80°C in their respective medium [brain heart infusion broth (BHIB, Neogen, UK) for C. difficile and Luria-Bertani (LB, Neogen, UK) for E. coli] with 20% (v/v) glycerol. C. difficile agar cultures were freshly prepared weekly from −80°C stocks on Brazier’s agar (Neogen, UK) supplemented with 1% defibrinated horse blood (Thermo Scientific, UK), 250 μg/mL cycloserine and 8 μg /mL cefoxitin (Merck, UK) incubated anaerobically (Don Whitley DG250: 10% H2, 5% CO2, 85% N2) at 37°C for 2–3 days. C. difficile broth cultures were prepared from agar cultures either in BHI, BHI supplemented with 5 g/L yeast extract (Oxoid, UK) and 0.1% L-cysteine (Merck, UK) (BHIS), or BHIS supplemented with the following antibiotics/inducer when appropriate: thiamphenicol (Tm, 15 μg/mL, Merck, UK), D-cycloserine (250 μg/mL) and kanamycin (50 μg/mL, Merck UK), and incubated 16–18 h or BHIS agar supplemented with the following antibiotics/inducer when appropriate: 7% defibrinated horse blood, Tm (15 μg/mL), D-cycloserine (250 μg/mL) and kanamycin (50 μg/mL), and incubated 2–3 days. Log phase cultures were prepared from 1 mL of 16–18 h cultures in 10 mL pre-reduce BHIB incubated anaerobically for 4 h at 37°C. E. coli NEB® 5 -alpha or NEB® 10-beta (New England Biolabs or NEB, UK) was used as the general host for plasmid construction and gene cloning. E. coli CA434 was used as the donor for conjugation with C. difficile. Transformation of E. coli was carried out by heat-shock at 42°C for either 45 s (E. coli CA434) or 30 s (E. coli NEB® 5 -alpha or NEB® 10-beta), and transformants were selected on LB agar plates (Difco, UK) supplemented with 25 μg/mL chloramphenicol (Biological Life Science USA), and grown in LB broth (Neogen, UK) with 12.5 μg/mL chloramphenicol.
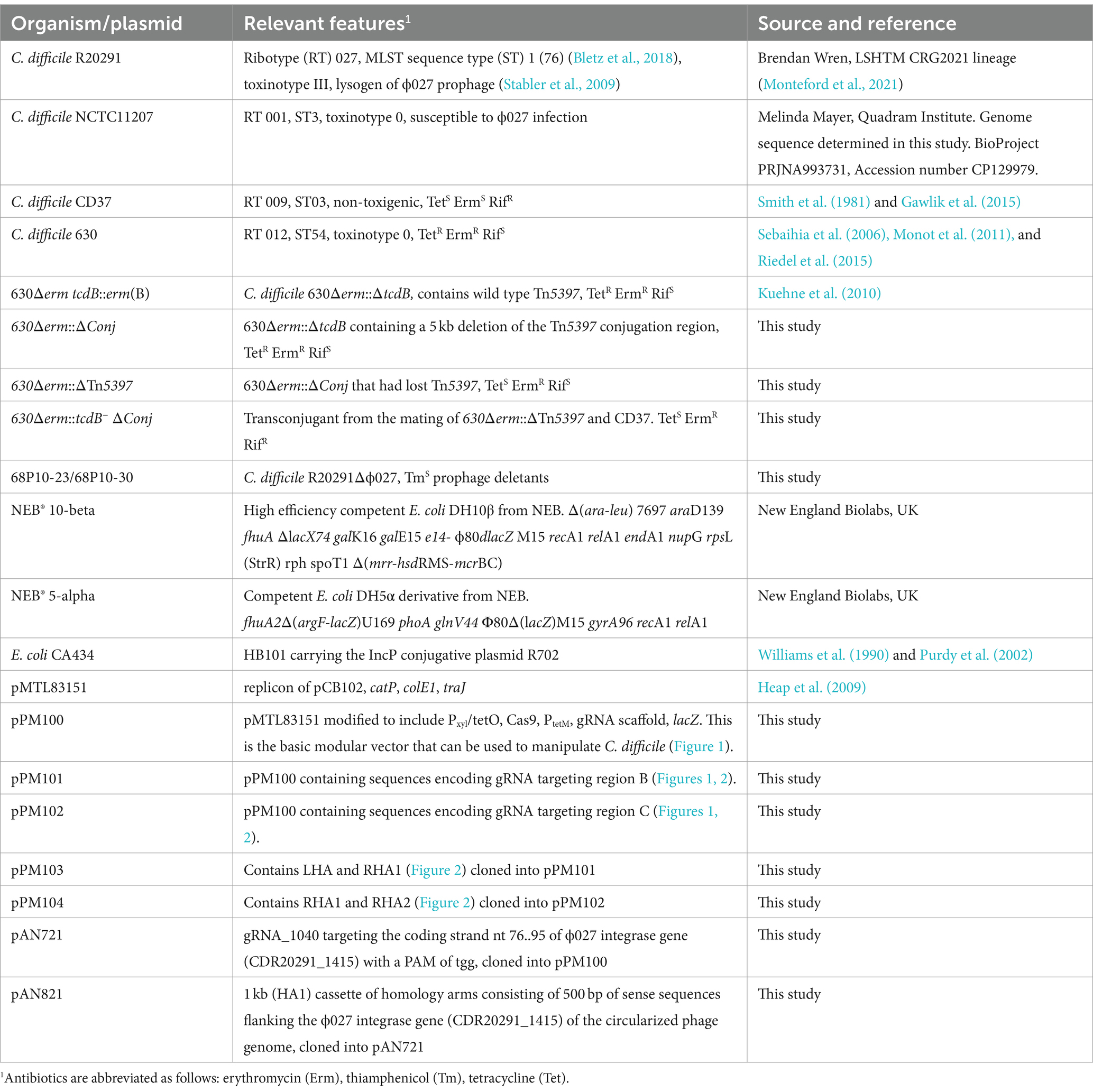
Table 1. Bacterial strains and plasmids used in this study.
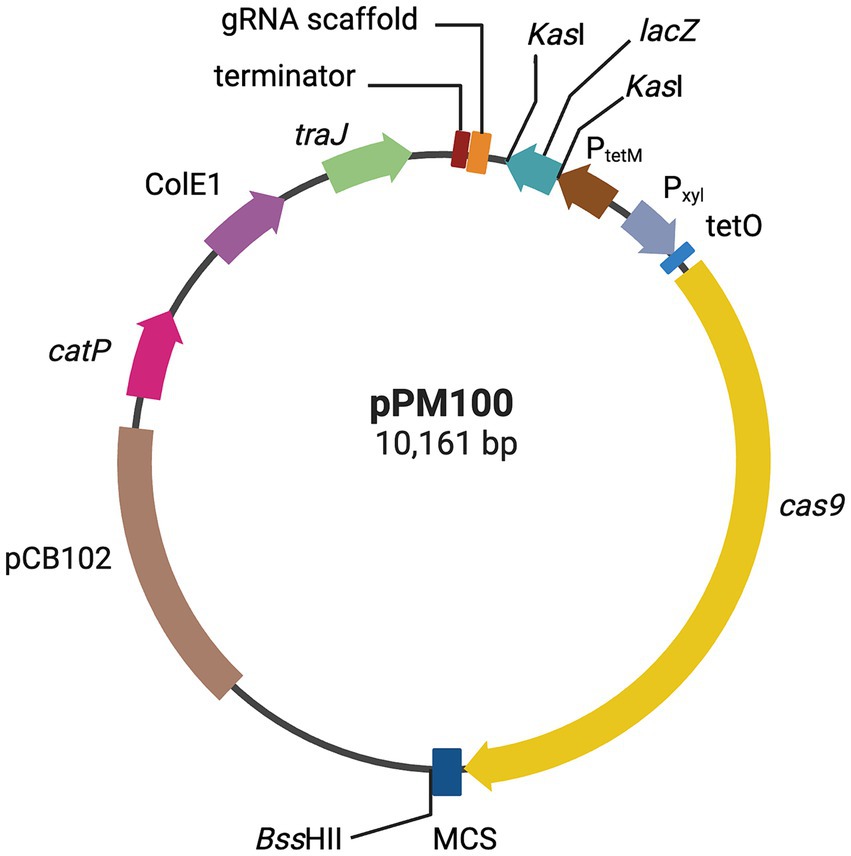
Figure 1. CRISPR-Cas9 genome editing vector. Plasmid map representation of the CRISPR-Cas9 vector developed in this study, pPM100. The S. pyogenes cas9, is under the control of the Pxyl-tetO promoter; a gRNA scaffold, consisting of a gRNA handle under the control of C. difficile PtetM promoter, and a multiple cloning site (MCS) for cloning of an editing template, containing upstream and downstream chromosomal regions flanking a deletion target site. The editing regions are cloned into the MCS (see text for more details). Created with biorender.com.
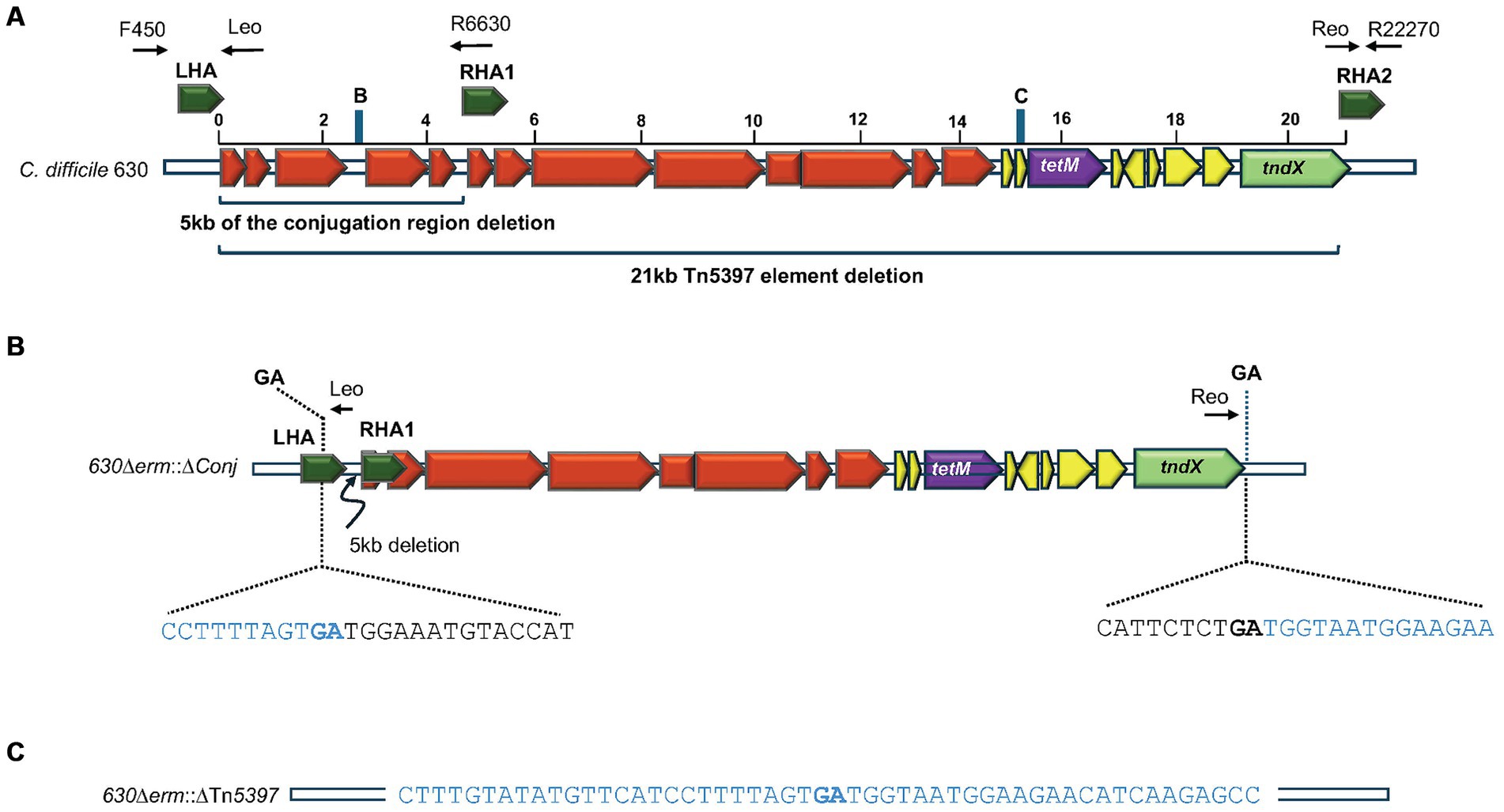
Figure 2. Deletion of Tn5397 from C. difficile 630. (A) Schematic representation of C. difficile Tn5397 conjugative transposon sequence. Black arrows above the figure represent binding sites of primers used in PCR analysis, the sequences of these are shown in Supplementary Table S1. The conjugation region genes are shown in red, the tetM gene is shown in purple, regulatory genes are shown in yellow, excision/insertion (tndX) shown in green. Brackets indicate the 5 kb conjugation region deleted from Tn5397, and the whole 21 kb Tn5397. Homology arms flanking regions for deletion are in deep green. Blue bars represent the positions of 20-nt sgRNA retargeting sequences within the region targeted for deletion from Tn5397 labelled B and C, corresponding to the sequences in Supplementary Table S2. (B) 630Δerm::ΔConj carrying the 5 kb deletion and sequence at the ends of the remaining element. Bacterial sequences are in blue, Tn5397 sequences are in black, GA in bold is the junction between the element. (C) Regenerated DNA sequences of 630Δerm::ΔTn5397 after deletion of whole element.
Growth curves of C. difficile R20291 and R20291Δϕ027 were generated from OD600 nm readings over 18 h in 96-well plates. Microtiter plates, sealing films, BHIB and C. difficile minimal medium (CDMM) (Cartman and Minton, 2010) were pre-reduced before use. Bacterial cultures in BHIB of 18 h were anaerobically diluted in growth media to OD600 nm of 0.1, and 200 μL volumes distributed into triplicate wells (3 technical repeats). Growth media alone were similarly distributed, serving as blanks and negative controls. The 96-well microtiter plate was sealed anaerobically, then transferred to a microtiter plate reader set at 37°C and kinetic measurements taken for 18 h every 15 min after 5 s of agitation. The experiment was repeated four times from which average OD600 nm values and standard deviation were calculated in Microsoft Excel and plotted in Prism 10 (GraphPad).
2.2 Phage induction, propagation, and purification
To induce ϕ027, known to exist within C. difficile R20291 (Stabler et al., 2009), a 16–18 h culture of C. difficile R20291 in 10 mL BHIB was treated with 3 μg/mL of mitomycin C (Merck, UK) for 6 h at 37°C. The induced culture was centrifuged at 4500 x g for 15 min and the supernatant was filtered through a sterile 0.45 μm membrane filter (Fisher Scientific, UK). Plaque assays were carried out in anaerobe basal agar (Oxoid, UK) with 600 μL of 4 h log phase cultures of NCTC11207 in BHIB and 100 μL of R20291 filtrate as previously described (Goh et al., 2005). A no-phage control was included with every plaque assay to ensure no spontaneously induced prophages from NCTC11207 were co-cultivated with ϕ027. Two rounds of single plaque propagation were carried out on NCTC11207, followed by whole plate assays for phage propagation to obtain crude phage suspensions as previously described (Goh et al., 2005). Crude phage suspensions were treated with DNase I (2 U/μL, Merck, UK) and RNase A (10 μg/mL, Merck, UK), precipitated by 1 M NaCl and 10% w/v PEG 8000, and recovered with chloroform to yield semi-pure suspensions. These were then either purified through a pre-formed CsCl density gradient of 1.3 g/mL, 1.5 g/mL, and 1.7 g/mL at 60,000 x g for 2 h at 4°C (Sorvall WX 80+ Ultracentrifuge, AH 650 swing out rotor) and dialyzed as previously described (Sambrook and Russell, 2001a) to yield purified suspensions, or concentrated by ultrafiltration through Amicon Ultra-15 3 kDa MWCO Centrifugal Filter Devices (Merck, Germany) spun at 4,000 × g for 60 min at room temperature.
2.3 Bacterial, phage, and plasmid DNA extraction
Five milliliters of a 16–18 h C. difficile broth culture was pelleted and frozen at −20°C before genomic DNA was extracted using the GenElute Bacterial Genomic DNA Kit (Sigma-Aldrich, UK). Phage DNA was extracted from semi-purified or purified dialyzed phage suspensions using either phenol:chloroform:isoamyl alcohol (Invitrogen, UK) (Sambrook and Russell, 2001b) or Phage DNA Isolation kit (Norgen Biotek, Canada). Plasmid DNA was extracted from 1 mL of 16–18 h E. coli broth cultures grown aerobically with agitation at 200 rpm using either the plasmid miniprep kit (Qiagen, UK) or Monarch Plasmid Miniprep kit (NEB UK).
2.4 PCR of phage, and phage and bacterial attachment sites in Clostridioides difficile R20291
Primer sets LCF889/LCF 890 and LCF 887/LCF 888 from Sekulovic and Fortier (2015) were used to confirm phage attachment site for ϕ027 in R20291 (Supplementary Table S1). Six primer sets specific for ϕ027 orf 1415, 1416, 1417, 1418, 1419, and 1464a were used to confirm the presence of the prophage in R20291 (Supplementary Table S1). OneTaq DNA polymerase and reaction buffer (NEB UK) were used for PCR reactions according to cycling conditions recommended by the manufacturer.
2.5 Construction of plasmids for ICE and prophage deletion
A modular vector pPM100, i.e., where desired DNA modules can be inserted in a single step, was constructed (Table 1 and Figure 1). The starting point was the E. coli–C. difficile shuttle vector pMTL83151, which contains the origin of replication from plasmid pCB102 (this replicon is unstable in C. difficile allowing it to be used as conditional lethal vector), along with the catP selective marker, the ColEI replicon and the mob region from RK2 (Heap et al., 2009). The Cas9 gene cassette of Streptococcus pyogenes from pCas9 (Addgene, UK) and the inducible promotor P xyl/tetO from pRPF185 (Fagan and Fairweather, 2011) were used as templates for PCR utilizing Pxyl/tetO-F with Pxyl/tetO-R and Cas9-F with Cas9-R primer pair, respectively. The two fragments were fused using splicing by overlap extension (SOE) PCR utilizing Pxyl/tetO-F and Cas9-R primer pair. The Pxyl/tetO-Cas9 fragment was cloned into pMTL83151 upstream of the Fdx terminator between XmaI and SalI restriction enzyme (Thermo Scientific, UK) sites resulting in plasmid pPM100. For deleting Tn5397 from C. difficile 630, the sgRNA encoding fragment was synthesized by Thermo Fisher Scientific and consisted of: the strong Tn916 derived promoter (P tetM ) (Su et al., 1992) and a 20 bp gRNA targeting sequence that was selected using an algorithm for scoring and ranking potential target sites with the Benchling CRISPR design tool1 (Supplementary Table S2). The sgRNA fragment was annealed by heating for 5 min then cloned into pMTL83151-CRISPR-Cas9 (pPM100) upstream of CD0164 terminator between XmaI and NotI sites. The editing regions were amplified by PCR using two pairs of primers (Supplementary Table S1) to produce fragments homologous to sequences targeted for recombination, and they were cloned next to the multiple cloning site (Figures 1, 2). Individual editing fragments were then fused together by SOE PCR at the BssHII site resulting in plasmid (pPM103 and pPM104) respectively (Table 1 and Figure 2).
For deleting ϕ027 from C. difficile R20291, a gRNA sequence targeting the region of interest identified by Benchling was chosen to which pairs of self-complementary oligos (Supplementary Tables S1, S2) were annealed at 50 pmol/μL in annealing buffer (10 mM Tris pH 8, 50 mM NaCl, 1 mM EDTA pH 8), phosphorylated with 15 U T4 polynucleotide kinase (NEB, UK), ligated to KasI-linearized (10 U of SspD1, Thermo Scientific, UK) and gel-extracted plasmid (Monarch kit, NEB, UK) with 5 U of T4 DNA ligase (Thermo Scientific, UK), then transformed by chemically competent NEB® 5-alpha cells. Cloned gRNA was confirmed by PCR using OneTaq DNA polymerase (NEB, UK) and Sanger sequencing (Source Biosciences Ltd., UK) with primers pMTL83151bb_99 and PtetM_191 (Supplementary Table S1). NEBuilder® HiFi DNA Assembly Tool designed primers for amplification and assembly of homology arms (Supplementary Table S1). Phusion High-Fidelity DNA Polymerase (Thermo Scientific, UK) amplified 0.5 kb sequences flanking the ϕ027 integrase gene with primer pairs int_RLA_rev/phi027_1415_LHA_R and int_RLA_fwd/ phi027_1415_RHA_F. Amplicons were cloned with NEBuilder® HiFi DNA Assembly Master Mix (NEB) into BssHII linearized and gel-extracted pAN721. Assembled plasmids were transformed by heat-shock to NEB® 10-beta to generate pAN821. Cloned inserts were Sanger sequenced (Source Biosciences Ltd., UK) with primer pairs pHHCas9_HACS_F/R, and 14152HA_pwalk1/2 (Supplementary Table S1).
2.6 Mating experiments
2.6.1 Filter-mating experiments between Clostridium difficile 630 and CD37
To test for PaLoc and Tn5397 transfer, methods described in Brouwer et al. (2013) were used with the following modifications. C. difficile was grown in BHIB for 18 h anaerobically, then subcultured to fresh broth at 37°C until mid-exponential phase (OD600 nm of 0.45). Cultures of C. difficile 630 (TetR ErmR RifS) or 630Δerm tcdB::erm(B) (TetR ErmR RifS) donors and CD37 (TetS ErmS RifR) recipient were mixed, and 200 μL was spread on nitrocellulose 0.45 μm pore-size filters on BHI agar and incubated for 18 h at 37°C in an anaerobic environment. The filters were removed from the agar plates and placed in 20 mL bottles and vigorously washed with 1 mL BHIB. Aliquots (100 μL) were spread on BHI agar supplemented with either 10 μg/mL tetracycline, 100 μg/mL erythromycin, 15 μg/mL thiamphenicol, or (25 μg/mL) rifampicin and incubated anaerobically for 48 h. Putative transconjugants were subcultured on fresh selective plates and incubated for a further 48 h. Selection of transfer of Tn5397 from 630Δerm tcdB::erm(B) to CD37 was made on plates containing rifampicin and tetracycline. Transfer of the PaLoc from 630Δerm tcdB::erm(B) and strains that had lost Tn5397, or contained a deletion of part of the conjugation region, was made on plates containing erythromycin and rifampicin.
2.6.2 Transfer of plasmids from Escherichia coli to Clostridium difficile
Single colonies of E. coli CA434 containing mutagenic plasmids were grown anaerobically overnight at 37°C in pre-reduced BHIS with 12.5 μg/mL chloramphenicol, 1 mL was pelleted and washed in pre-reduced BHIS. Two hundred microliters of overnight C. difficile culture was heated to 52°C for 5 min, cooled for 2 min, (the heating step was only required when R20291 was the recipient) then mixed with E. coli donor cell pellets and incubated for 8 h as described previously (Kirk and Fagan, 2016). The mating mixture was spotted onto BHIS agar, grown anaerobically for 8 h, harvested in 1 mL pre-reduced phosphate-buffered saline (PBS) and plated onto BHIS with 250 μg/mL cycloserine, 50 μg/mL kanamycin, and 15 μg/mL Tm (CKTm). After 24–48 h of growth, colonies were picked and transfer of mutagenic plasmid pPM103, pPM104 or pAN821 was confirmed by PCR (see Supplementary Table S1 and the results section for more details). C. difficile transconjugants were re-streaked onto BHIS agar with appropriate antibiotics. After 2 days, a single colony was inoculated into pre-reduced 10 mL BHIS supplemented with appropriate antibiotics and grown overnight for gDNA extraction for further PCR confirmation. Conjugation frequency was calculated against either donor or recipient in mating mixtures. Donor and recipient cultures were serially diluted 10-fold in pre-reduced PBS and plated onto LB and BHI plates, respectively. Colonies were counted after 24 h anaerobic incubation for E. coli and 48 h incubation for C. difficile. The conjugation frequency was calculated as colony forming units (CFU) of transconjugants/CFU of donor or recipients.
2.7 Induction of CRISPR-Cas9 system and MGE deletion
Clostridioides difficile containing mutagenic plasmid on BHIS CKTm and 7% defibrinated horse blood plates were grown in 10 mL BHIB with Tm (15 μg/mL) for 16–18 h, serially diluted 10-fold in pre-reduced sterile 1 x PBS, spread-plated onto dried and pre-reduced BHIS + anhydrotetracycline (aTC, Merck, UK, 30 ng/mL) + Tm (15 μg/mL) plates and grown for 2 days. Five to 10 colonies were screened by colony PCR for ϕ027 prophage or Tn5397 deletion. For prophage deletion primer pairs phiR2_1415_F/phiR2_1,415_R, phi027_1464a_F/phi027_1464a_R, LCF887/LCF889, LCF888/LCF890 and catP_2/3 were used (Supplementary Table S1). To confirm Tn5397 conjugation region deletion, primers Tn5397(F450) and Tn5397(R6630), and Tn5397(R450) and Tn5397(R22270) were used to determine if the whole element was absent (Supplementary Table S1).
2.8 Plasmid curing
To cure the plasmid from strains with mutant or deleted target MGE (ϕ027 or Tn5397), a single colony of the mutant with the desired deletion (i.e., deletant) was subcultured in BHIB with no antibiotics. After 18 h, 100 μL of the culture was used to inoculate 10 mL of BHIB. This subculture was repeated daily for up to 10 days. Ten-fold serial dilutions of deletant culture in pre-reduced 1 x PBS were made after each subculture for Tn5397 deletants, but only at the end of 10 days for ϕ027 deletants, and 100 μL of the 10−5 dilution was spread onto BHI agar plates without antibiotic. Replica plating was performed on agar supplemented with 15 μg/mL Tm, tetracycline (Tet) or erythromycin (Erm) to identify colonies that have lost the mutagenesis plasmid (i.e., either pPM103, pPM104 or pAN821). Colonies that had lost antibiotic resistance at the least number of subcultures were isolated for further study to avoid excessive subculture. Loss of the plasmid was confirmed by PCR using plasmid-specific primers (Cas9-F and Cas9-R, or catP_2/3) (Supplementary Table S1). For Tm sensitive (TmS) prophage deletants putatively cured of plasmid, gDNA was extracted and checked with 1464a_F/R, NF1643/44, and catP_2/3 primers, then sequenced by Illumina sequencing. To screen for further loss of a truncated Cas9 plasmid remnant in sequenced TmS prophage deletant strains, single colonies were picked for colony PCR using primers 68P1023_LF/LR, specific for the left junction of the integrated plasmid remnant (Supplementary Table S1). Putatively negative colonies were grown in BHIB for gDNA extraction and confirmation by PCR with the same primers in addition to 68P1023_RF/RR, and pHHCas9_3F/end.
2.9 Genomic DNA library preparation for Nanopore sequencing
For long-read Oxford Nanopore Technology (ONT, UK) sequencing of NCTC11207, 5 mL of 18 h culture was pelleted and frozen at −20°C before DNA extraction using the Qiagen MagAttract High Molecular Weight DNA Kit (Qiagen, UK). DNA quality and quantity were assessed using NanoDrop, Qubit (Thermo Fisher Scientific, UK), and Agilent TapeStation instruments (Agilent, UK). ONT sequencing libraries were prepared by multiplexing DNA from C. difficile isolates per flow cell using a Nanopore protocol for native barcoding of genomic DNA (version NBE_9065_v109_revAC_14Aug2019). This firstly involved DNA repair and end-prep carried out with NEBNext FFPE DNA Repair Mix (M6630, NEB, UK), NEBNext Ultra II End repair/dA-tailing module (E7546, NEB, UK), and AMPure XP beads (Beckman Coulter, UK). Secondly, native barcode ligation was carried out using the Native Barcoding Expansion kit (EXP-NBD104; ONT, UK) and NEB Blunt/TA Ligase Master mix (M0367, NEB, UK). Thirdly, adapter ligation using Ligation Sequencing Kit (SQK-LSK109, ONT, UK), NEBNext Quick T4 DNA Ligase (E6057, NEB UK), NEBNext Quick Ligation Reaction Buffer (B6058, NEB, UK). Sequencing libraries were loaded onto a R9 generation flow cell (FLO-MIN106) and sequenced in MinION Mk1C (ONT, UK), stopping after 25 h.
2.10 Nanopore sequence analysis of NCTC11207
Before assembly, long read sequences were filtered using Filtlong v0.2.0, keeping the minimum length of 1,000 bp and 90% of best quality sequences2. Genome assembly was performed using Flye assembler v2.9 (Kolmogorov et al., 2019) and Trycycler v0.5.0 (Wick et al., 2021), with a standard protocol recommended by the developers (Wick et al., 2021). The final assembly graph was visualized and polished with Bandage v0.8.1 (Wick et al., 2015). Multi-Locus Sequence Typing was performed in silico using BIGSdb v1.32.0 hosted at PubMLST3 and the scheme of Griffiths et al. (2010). Prophage screening of the NCTC11207 chromosome was performed using PHASTER (Arndt et al., 2016). The final circular genome was annotated using the NCBI Prokaryotic Genomes Annotation Pipeline (PGAP) (Tatusova et al., 2016) and is now available in GenBank under BioProject PRJNA993731 (accession number CP129979). Similarity of the ϕ027 genome to genomes of NCTC11207 prophages 1 and 2 was determined using VIRIDIC (Moraru et al., 2020).
2.11 Illumina sequencing of ϕ027 deletants and deletion/insertion confirmations
Clostridioides difficile gDNA was extracted using either GenElute Bacterial kit (Merck, UK) or Qiagen MagAttract High Molecular Weight DNA Kit (Qiagen, UK), checked for quality, and paired-end sequenced at Microbes NG (UK, 2 × 250 bp, 30 x coverage) or SeqCenter (USA, 2 × 151 bp, 30 x coverage). Trimmed Illumina reads facilitated by these vendors were mapped against the genome of C. difficile R20291 (accession number NZ_CP029423.1). Unmapped reads were afterwards de novo assembled using Unicycler v0.4.8 and mapped against pAN821 using the BWA-MEM algorithm (arXiv:1303.3997v2). To detect ORF 1465 in deletant and WT by PCR, primers LF1 and RR1 (Supplementary Table S1) were used on two batches of genomic DNA prepared from WT and deletant cultures as described in 2.3. OneTaq DNA polymerase and reaction buffer (NEB UK) were used for PCR reactions according to cycling conditions recommended by the manufacturer.
3 Results
3.1 Construction of a modular vector for gene knock out in C. difficile
A simple modular vector, pPM100, was constructed for generating CRISPR-directed mutations in C. difficile (see Materials and Methods and Figure 1). This plasmid is unstable in C. difficile and therefore an ideal delivery vector. Furthermore, all the modules on this vector can be easily replaced or modified making it a useful and efficient tool for relatively easy manipulation of C. difficile.
3.2 Editing regions designed to delete the conjugation region of Tn5397 resulted in a mixture of clones some of which had lost just the conjugation region and some the whole of Tn5397
To investigate the role of Tn5397 in genome mobility and to generate a Tet sensitive (TetS) derivative of 630Δerm tcdB::erm(B) [this has a Clostron insertion conferring Erm resistance (ErmR) in the tcdB gene to allow for the selection of PaLoc transfer (Kuehne et al., 2011; Brouwer et al., 2013)], we initially wanted to precisely delete the whole of Tn5397. This was attempted by generating a CRISPR-Cas9 vector (pPM103) with gRNA encoding sequences targeting region C of Tn5397 (Figure 2A and Supplementary Table S2) and editing regions flanking the insertion site of Tn5397 in 630Δerm tcdB::erm(B) (LHA and RHA2, Figures 1, 2A). This plasmid was conjugated into 630Δerm tcdB::erm(B) and the resulting four Tm-resistant (TmR) transconjugants were subject to Cas9 induction and then screened by PCR for loss of Tn5397 using primers flanking Tn5397. All four transconjugants still had Tn5397. It was assumed that the reason for this failure was that the region we were trying to delete is too large. Therefore, it was decided to delete part of the conjugation region. To do this, the CRISPR-Cas9 vector (pPM104) containing a gRNA encoding region targeting region B at 2500 bp on Tn5397 and an editing region consisting of LHA and RHA1 was used (Figure 2A and Supplementary Table S2). Plasmids were transferred by conjugation to 630Δerm tcdB::erm(B), and five TmR colonies arose. These were subject to PCR with primers F450 and R6630 and these yielded a product of 1.5 kb (no product was obtained with strains carrying wild-type Tn5397 presumably because the product was too large) (Figure 2A). DNA sequence analysis of this product confirmed that a precise 5 kb deletion of part of the conjugation region had occurred (Figure 2B and Supplementary Figure S2).
One of the strains, 630Δerm::ΔConj, carrying the 5 kb deletion was selected for further study. It was grown for 18 h in drug free broth then plated onto drug free media, 600 colonies were screened for loss of resistance to Tet, and 2 of 600 were sensitive, hence the mutation efficiency was 0.3%.
PCR analysis of the two tetracycline-sensitive mutants using primers flanking Tn5397 (F450 and R22270 in Figure 2A) gave a product of 1.2 kb (no product was obtained in strains carrying wild-type Tn5397 or those carrying the 5 kb deletion). One of these strains was selected for further study and designated 630Δerm::ΔTn5397. DNA sequencing of the PCR product showed that the target site of Tn5397 had been regenerated (Figure 2C) and that the whole of the transposon had been lost. This implies that deletion of part of the conjugation region destabilizes Tn5397 so that it can still excise from the host chromosome and circularize but presumably due to the large deletion some circular molecules are lost. This idea was supported by the fact that we could detect the presence of a circular form of the element using primers Tn5397 (Leo) and Tn5397 (Reo) in the 630Δerm::ΔConj mutants that contained the 5 kb deletion. These primers read out from the ends of Tn5397 and will only form a product when the ends are ligated together in a circular form of the element (Supplementary Figure S1 and Figure 2A). Diagrams showing these events in wild-type Tn5397 have been previously published and are summarized in Supplementary Figure S1 (Wang et al., 2000; Wang and Mullany, 2000; Brouwer et al., 2011). No product was obtained from the tetracycline sensitive strains.
3.3 Strains that have lost Tn5397 can still transfer the PaLoc at the same frequency as WT
Tn5397 is the nearest MGE to the PaLoc in the 630Δerm tcdB::erm(B) chromosome (Brouwer et al., 2013) and it was proposed that this element might be responsible for its mobilization. However, mutants that contain a deletion of the conjugation region (630Δerm, ΔConj) and those that have lost Tn5397 completely (630Δerm, ΔTn5397) both transfer the PaLoc at the same frequency of around 1 × 10−7 of ermR transconjugants per donor (encoded by the Clostron inserted in the tcdB gene) as the WT contains an intact Tn5397. This shows that a genetic element other than Tn5397 is responsible for PaLoc transfer, although we cannot completely rule out a role for this element.
3.4 Ф027 is a functional phage integrated in R20291
ϕ027 was first identified as a putative prophage integrated into the chromosome of C. difficile R20291 (GenBank accession numbers FN545816.1 and CP029423.1) (Stabler et al., 2009). This phage and its bacterial attachment sites, attP and attB, were later identified by PCR (Sekulovic and Fortier, 2015), as there was a population of spontaneously excised and re-circularized ϕ027 genomes in DNA preparations of the lysogen. In this study, we firstly confirmed R20291 was PCR positive for 6 predicted genes of ϕ027 (Supplementary Figure S3). Then we found that ϕ027 in C. difficile R20291 of CRG2021 lineage (i.e., closest to the original R20291 clinical isolate and less amenable to conjugation) (Monteford et al., 2021) is a functional and inducible phage that can be propagated on C. difficile NCTC11207 (GenBank accession CP129979), obtaining yields of 108–109 plaque forming units (pfu)/mL. NCTC11207 is a ribotype (RT) 001 strain (Table 1), which was sequenced here and predicted to contain two intact prophages whose features are summarized in Supplementary Table S3. To ensure that NCTC11207 prophages were not co-propagated with ϕ027, a phage buffer (i.e., no phage) control was included with every batch of propagated phage to ensure no plaques were derived from spontaneously induced NCTC11207 prophages. ϕ027 virion DNA was extracted and used for PCR to confirm the attP sequence 5′ tattacaacttaagtaaata 3′, is as previously found in circularized ϕ027 prophage DNA within R20291 (Sekulovic and Fortier, 2015). The linear phage DNA annotation is re-arranged to convention in Supplementary Figure S4 and Supplementary Table S4. We also obtained similar PCR results when using R20291 bacterial genomic DNA, confirming previous observations that ϕ027 spontaneously excises, and exists extra-chromosomally and as an integrated prophage located at nt. 1670843…1,726,837 encompassing CDS CDR20291_1415 to CDR20291_1464a (Genbank accession number FN545816.1 and Supplementary Figure S4; Sekulovic and Fortier, 2015).
As ϕ027 prophage spontaneously excises and circularizes within host cells, we hypothesized that removal of the integrase gene (CDR20291_1415) from the circular form would lead to loss of the phage as the circular form would not be able to reintegrate (Figure 3).
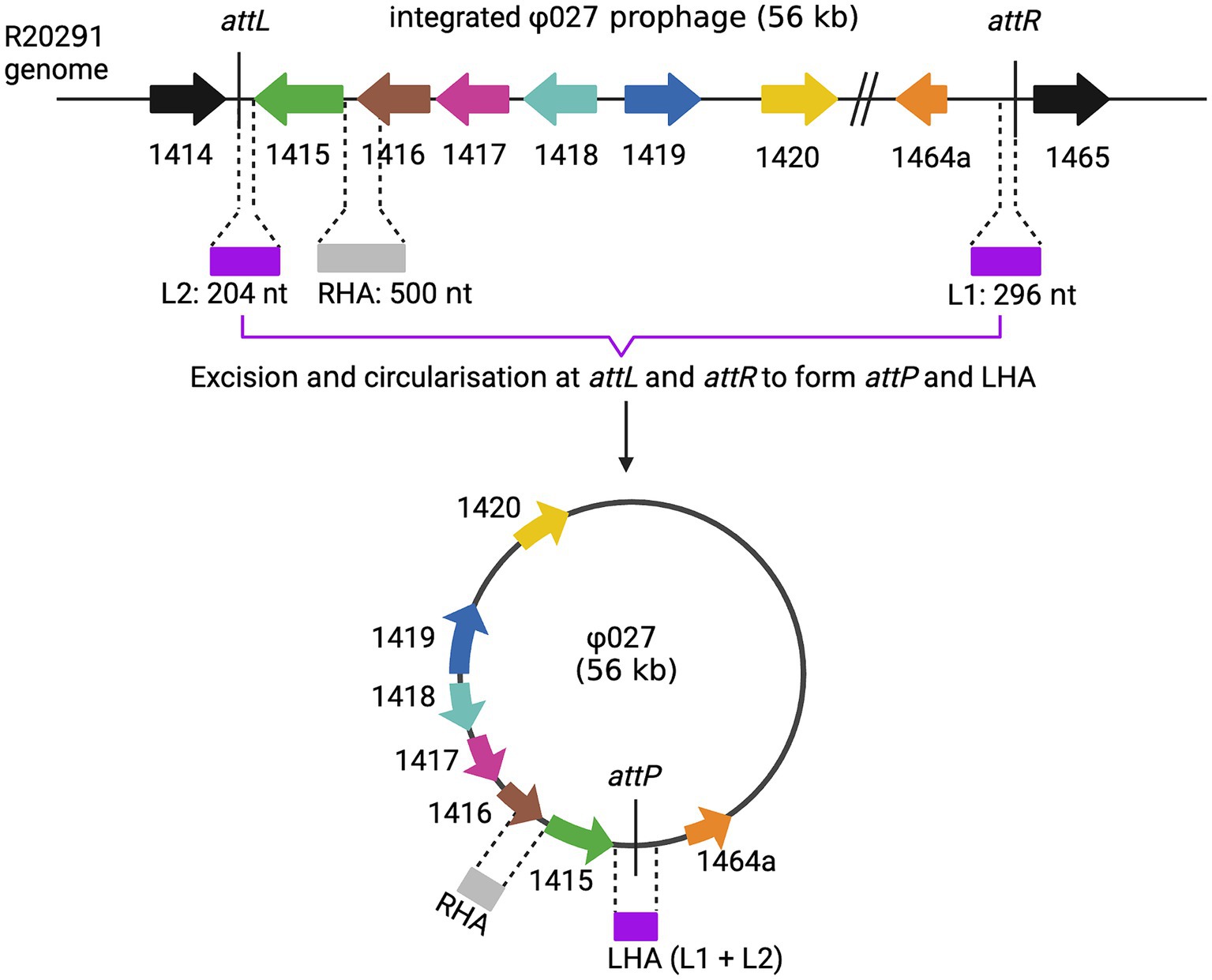
Figure 3. Homology arms for prophage deletion. The top line shows the integrated form of ϕ027, the junction between phage and bacterial DNA is shown at attL and attR. The phage can excise and circularize by site-specific recombination between attL and attR to generate a circular molecule with attP at the joint of the circular form. The location of homology arms that were used in mutagenesis experiments are shown. Note that LHA is only present at the joint of the circular form as it is composed of attP. Created with biorender.com.
The conjugation frequencies of pPM100 (the plasmid backbone), pAN721 (targeting integrase and not containing homology arms), and pAN821 (targeting integrase and containing the 1 kb homology arms for integrase deletion) are shown in Supplementary Table S5. Conjugation frequencies of cells harboring pPM100 and pAN721 were comparable to those of cells harboring pAN821, indicating cas9 expression is likely to be repressed in the absence of the inducer and did not affect cell viability.
3.5 Generation of Clostridioides difficile R20291 ΔФ027
Four of 8 transconjugants containing pAN821 with intact sgRNA, cas9 and deletion cassette (Supplementary Figure S5) were devoid of prophage after aTC induction of Cas9 (Figure 4), could not be induced by mitomycin C to form plaques, and were susceptible to reinfection by ϕ027. The mutation efficiency was 50%. Curing of pAN821 from prophage deletants was attempted by passaging in non-selective BHIB for 10 days, then screening colonies for loss of susceptibility to Tm. Cultures containing control plasmids pPM100 (the plasmid backbone) and pAN721 (targeting integrase and not containing homology arms) were also screened for plasmid curing in the same way (Table 2). Just 1.5% (2/137) of colonies from the culture containing pAN821 had lost plasmid-encoded resistance after 10 passages. In contrast, plasmids lacking homology arms were all rapidly cured in this time from R20291 (Table 2). It is possible the homology arms allowed pAN821 to survive by recombining with the host genome. The actual mechanism for this requires further investigation. Susceptibility of two cured prophage deletants to ϕ027 infection was confirmed (Figure 5).
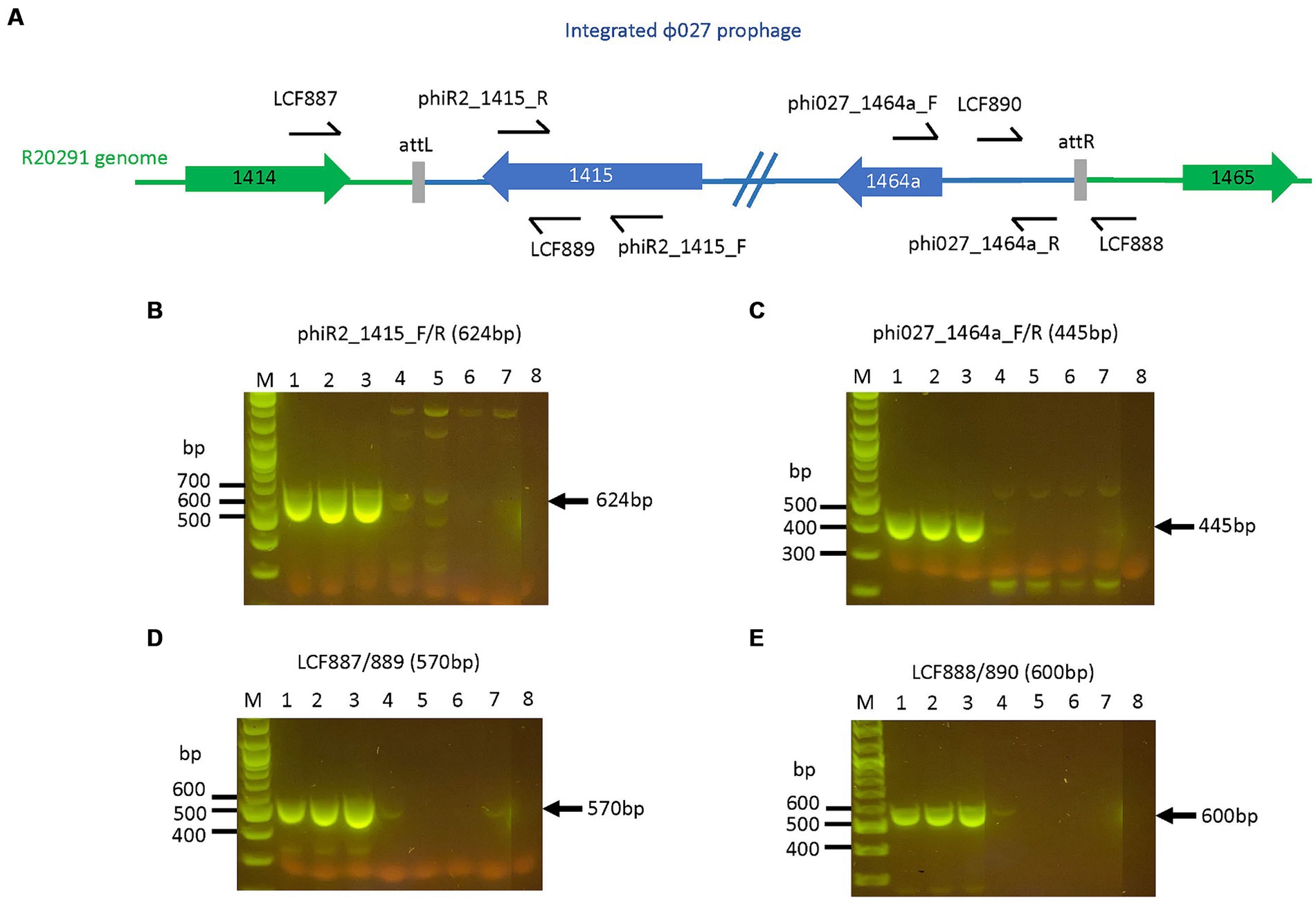
Figure 4. R20291 prophage deletants devoid of prophage features. Primers targeting selected features of the prophage are shown in (A). The loss of (B) prophage orf 1415 (integrase), (C) orf 1464a, (D) attL, and (E) attR were observed in R20291 deletants. Lane 1: WT R20291, 2: transconjugant of pPM100, 3: transconjugant of pAN721, 4–7: transconjugants of pAN821, 8: No template control. M: 1 kb Plus DNA ladder (NEB, UK).
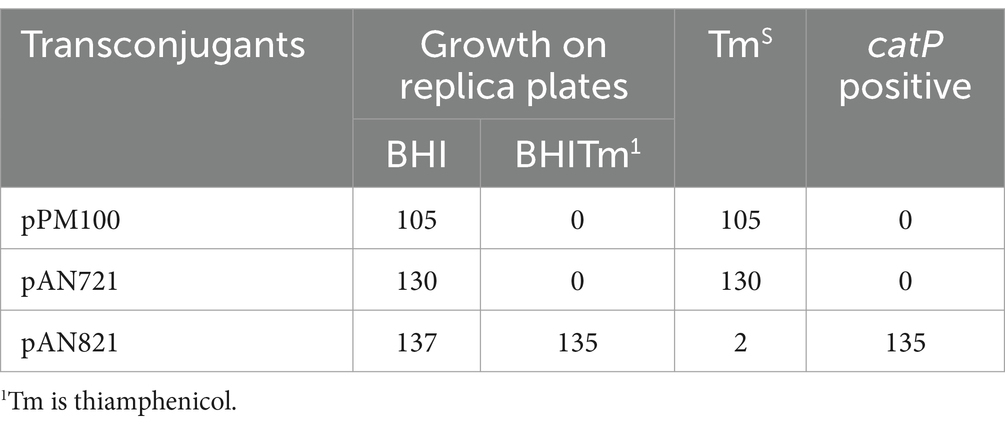
Table 2. Colonies screened for loss of mutagenesis plasmid.

Figure 5. Phage susceptibility of R20291 prophage deletants. Susceptibility to ϕ027 infection was determined by plaque assays of TmS prophage deletants (68P10-23 and 68P10-30) that had lost plasmid-encoded resistance, the indicator strain NCTC11207 which is susceptible to ϕ027, and the lysogen WT R20291, which is resistant to ϕ027. White arrowheads indicate plaques, which vary in size and clarity.
3.6 Whole-genome sequencing confirmed Ф027 deletion
Two TmS prophage deletants, 68P10-23 and 68P10-30, were subject to Illumina sequencing and had identical sequences, having the phage attachment site but neither the ϕ027 prophage genome nor the entire pAN821. Also ORF 1465 (678 bp), which was downstream of the attR site, hence predicted to be a bacterial gene (Figure 3), was deleted. Essentially a 56.8 kb locus containing the prophage and ORF 1465, was removed at the expected locations of nt. 1670843..1726837 (Figures 6A,B). However, 48–52% of the sequence reads contained a 2.7 kb remnant of pAN821 where the prophage was previously integrated (Figure 6C). The 2.7 kb remnant of pAN821 aligned with 1767 of 4,107 bases of the 3′ end of cas9, a 262 nt downstream intergenic region, 500 nt of the RHA, and 204 nt of the LHA (Figure 6D). Its presence was confirmed by PCR in all of 254 single colonies of 68P10-23 tested, and all of 160 single colonies of 68P10-30 tested (results not shown). To see if ORF 1465 could be absent from the WT genome naturally due to prophage excision, PCR and Sanger sequencing was carried out to show that deletion of ORF 1465 was likely a consequence of the mutagenic plasmid (Supplementary Figure S6). There was no other genomic difference between the prophage deletants and the WT. Batch culture growth curves of R20291Δϕ027 (68P10-23) was similar to WT in rich medium but marginally reduced compared with WT in minimal medium (Figure 7).
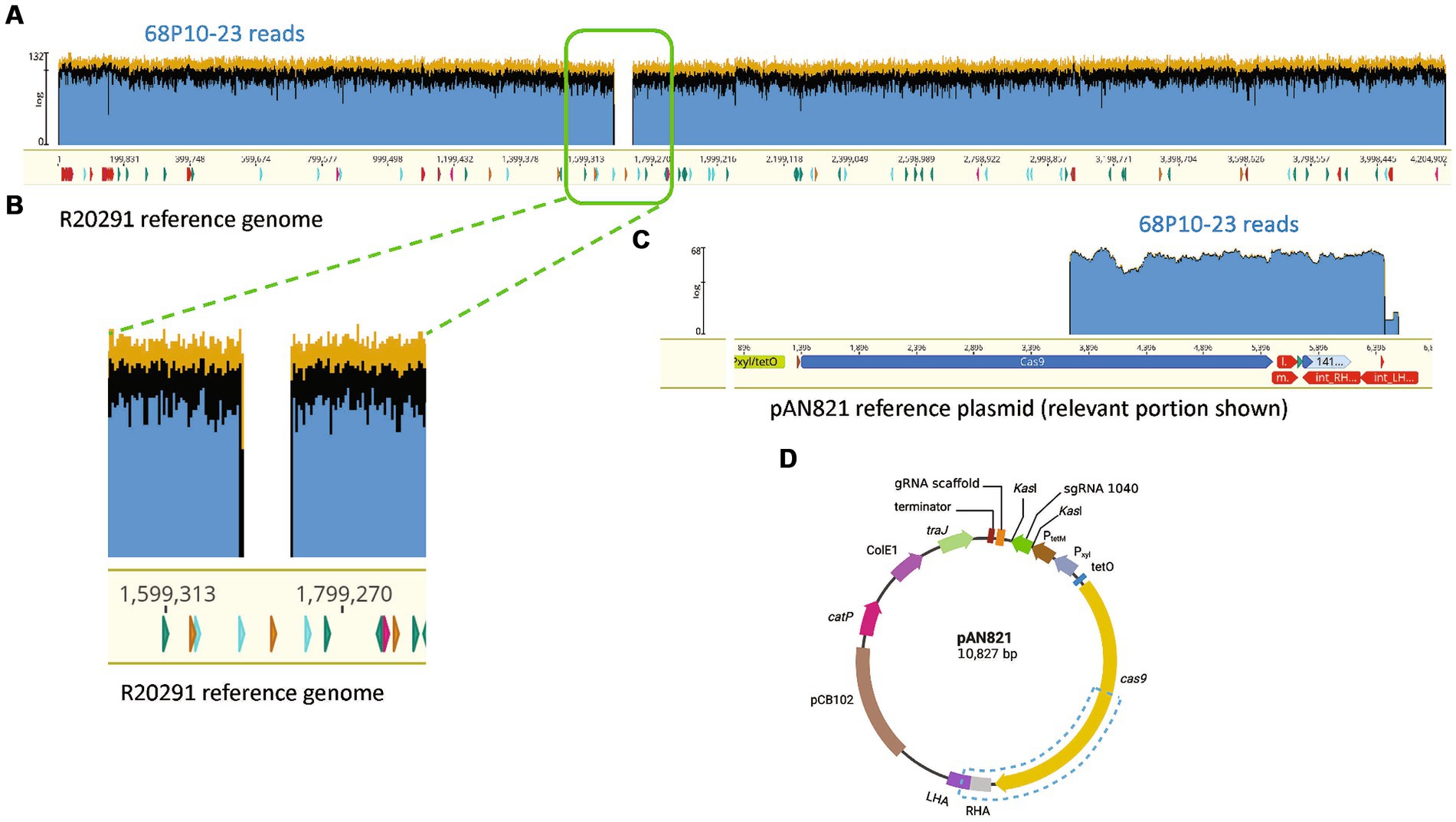
Figure 6. R20291Δϕ027 (68P10-23) contained a fragment of the mutagenesis plasmid. (A) Sequence reads of 68P10-23 (top blue bar) mapped to WT R20291 genome (bottom yellow bar with nucleotide numbers) showed missing reads as a gap, which belonged to the prophage, indicating its deletion. (B) Magnified section of where prophage sequence reads were missing from the WT genome. (C) A population of reads not mapping to R20291 was aligned to a 2.7 kb fragment of the pAN821 plasmid (bottom yellow bar) encompassing part of cas9 and downstream sequences. (D) Blue dashed box shows the location of the 2.7 kb plasmid portion remaining in the prophage deletant. Created with biorender.com.
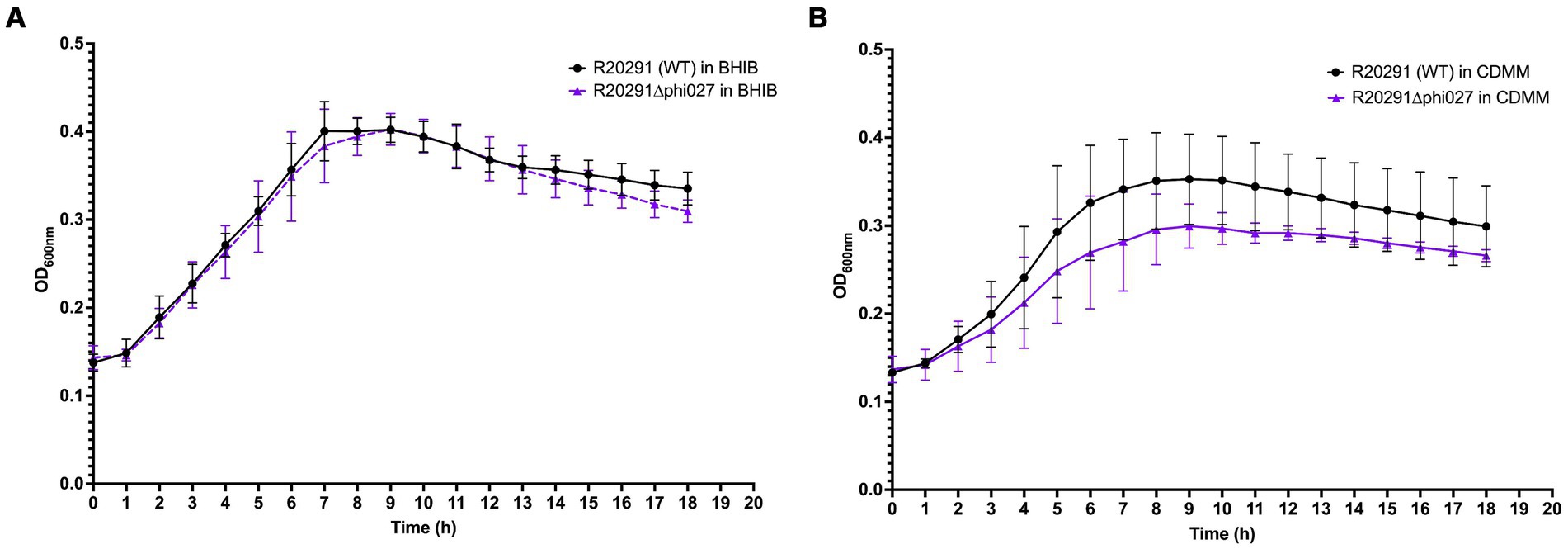
Figure 7. Growth of R20291 WT and Δϕ027 in (A) BHIB and (B) CDMM (n = 4).
4 Discussion
Large MGEs such as prophages and ICE use integrase enzymes to facilitate their entry and exit from the host chromosome. There are two general families, i.e., the tyrosine and the serine recombinases. The former requires an accessory protein, Xis to excise the MGE while the serine recombinases can mediate both integration and excision (Stark, 2014). In this work, we showed that a prophage could be deleted from the C. difficile genome by targeting the integrase and that the ICE Tn5397 can be cleanly removed by targeting the conjugation region. These observations show that it should be possible to specifically remove any of the large integrated MGEs from the C. difficile genome and determine their contribution to the organism’s biology. In this work, we ruled out a direct role for Tn5397 in the transfer of the PaLoc. However, C. difficile does contain many different ICE and phages which have the potential to mediate chromosomal transfer, and systematic deletion of each of these is required to determine their precise role in the organism’s biology and their contribution to the wider mobilome.
Tn5397 and the closely related genetic element Tn916 are both very stable in bacterial genomes. The rate of loss of Tn5397 being much less than the 2 in 600 observed in this work; we have tested 3,000 colonies containing wild-type Tn5397 with no loss of the element (unpublished data). Therefore, it is likely that the deletion of part of the conjugation region destablises the element. There have only been a small number of studies examining gene regulation in the Tn916/5397 family in genetic elements and these have all been done in Tn916 (Scornec et al., 2017). This work has shown that transcription is tightly regulated and expression of the conjugation region requires transcription initiating at the strong tet(m) promoter progressing over the joint of the circular form into the conjugation region. Our mutant that lacks part of the conjugation region still contains the ends of the element on which TndX can act (explaining why circular forms of the element are still detected). It is possible that the deletion of part of the conjugation region results in premature transcriptional termination and that not enough TndX is produced to allow reintegration, hence the element is lost. However more work is required to determine the exact mechanism for loss of Tn5397.
Our hypothesis for transfer of the PaLoc is that integrated origins of transfers (oriT) result in the mobilization of the chromosome from donor to recipient (Brouwer et al., 2013). As Tn5397 was the nearest oriT to the PaLoc this seemed like a good candidate for mobilizing the PaLoc. However, as the PaLoc still transferred from strains lacking Tn5397, this element is obviously not required for PaLoc transfer. The C. difficile genome does contain a number of integrated oriTs that could transfer the PaLoc, or it may transfer by a completely different mechanism, for example cell fusion to form a zygote. Further work is required to determine the exact mechanism of PaLoc transfer.
Clean deletion of large DNA fragments (up to 8,000 bp) in Clostridia using CRISPR-Cas9 is challenging (Wang et al., 2016, 2018). The potential causes include: (i) the Cas9 protein is toxic to the host; (ii) the Cas9-carrying plasmid is often large and therefore potentially unstable; (iii) homologous arms present on the mutagenesis plasmid enable repeated re-integration after double crossover events. Here, we demonstrate the successful deletion of the ~56 kb ϕ027 prophage in R20291 using the pMTL83151 backbone modified with CRISPR-Cas9. Four previous studies which described gene deletions in C. difficile 630 or R20291 by CRISPR-Cas 9 or Cas12a were built on pMTL84151 (McAllister et al., 2017), pMTL82151 (Hong et al., 2018; Wang et al., 2018), and pMTL83151 (Ingle et al., 2019). The main difference between these plasmids is the replicon, with pMTL82151 having a replicon from pBP1, pMTL83151 a replicon from pCB102, and pMTL84151 a replicon from pCD6 (Heap et al., 2009). Compared to other reports, our mutation efficiency of 0.3% for deleting Tn5397 (21 kb) in C. difficile 630 was very low. This could be because of the size of the deletion and the limits of the Cas9 nuclease in C. difficile 630, since other studies which used Cas9 for selecting deletions at a high efficiency targeted sequences up to 3.6 kb. In C. difficile 630, Cas9 on pMTL82151 selected spo0A deletants (825 bp) at 100% efficiency (Wang et al., 2018), and Cas9 on pMTL83151 allowed selection of pyrE (234 bp), and ermB1 and ermB2 (3.6 kb) deletants at 89 and 96% efficiency, respectively (Ingle et al., 2019). Interestingly, Hong et al. was unable to obtain transconjugants when they attempted to use Cas9 on pMTL82151 for deletion of ϕCD630-2 (49 kb) in C. difficile 630. However, they succeeded using Cas12a (Cpf1) nuclease to select for deleted prophage ϕCD630-2 (49.2 kb) at mutation efficiencies of 37.5–58.3% (Hong et al., 2018). They also deleted fur (390 bp), cwp66 (1.8 kb), tetM (1.9 kb), ermB1 and ermB2 (3.2 kb), and tcdA (8.1 kb) at mutation efficiencies of 25–100% (Hong et al., 2018). Our 50% mutation efficiency of deleting ϕ027 (56 kb) prior to plasmid curing in R20291 was comparable to McAllister et al (McAllister et al., 2017). They deleted pyrE (585 bp) and selD (951 bp) at 50 and 20%, respectively (McAllister et al., 2017). However, we were unable to completely cure the mutagenesis plasmid. It is worth noting that (Maikova et al., 2019) re-programmed the endogenous Cas I-B system in R20291 and achieved 90% mutation efficiency for deleting a 261 bp gene. This strategy could be useful to avoid toxic effects of Cas9.
In this work, the pAN821 mutagenesis plasmid deleted the ϕ027 prophage and a downstream predicted bacterial gene (CDR20291_1,465) from R20291. However, the plasmid was not completely cured; a truncated cas9 and the RHA from the plasmid remained stably integrated in a population of bacterial cells. This likely occurred from an imprecise double crossover event and was detected by whole genome sequencing, although previous studies in C. difficile did not report this phenomenon, perhaps because it is undetectable by standard PCR assays for loss of gene targets. Primers flanking the attL and attR sites (LCF887/888, Figure 4) were not used to check for prophage deletion to avoid false positive results of prophage deletion, since the prophage could spontaneously exist extrachromosomally. A study in C. beijerinckii reported difficulties in plasmid curing, which was overcome with the inclusion of CRISPR-Cas9 self-targeting of the mutagenesis plasmid (Wang et al., 2016). This could be explored in future. It was not possible to quantify the subset of cells based on the number of sequence reads due to amplification bias in sequencing. However, cells with the plasmid remnant appear to be the dominant cell type based on PCR screening of single colonies. The truncated cas9 translates to amino acid (aa) residues 781 to 1,368, which consists of the HNH, RuvCIII, Topo-homology, and PI domains that function in nuclease and PAM recognition, i. e. the “nuclease lobe” (Jinek et al., 2014; Nishimasu et al., 2014). However, without the other Cas9 protein domains that form the “recognition lobe” for facilitating guide RNA binding to DNA, this truncated cas9 will likely be inactive if translated in the prophage deletant (Jinek et al., 2014; Nishimasu et al., 2014). Interestingly, Ingle et al. used the same backbone pMTL83151 for CRISPR-Cas9 deletion in C. difficile 630 and found truncation of Cas9 (Ingle et al., 2019). However, their truncated Cas9 was missing 87 aa from the N-terminus (Ingle et al., 2019) while the cas9 remaining in our ϕ027 deletant would be missing approximately 780 aa (or 2,340 bases) from the N-terminus (or 5′ end of the gene) as mentioned above, if it was translated.
The C. difficile gene deleted adjacent to the prophage CDR20291_1465 is homologous to a putative manganese-containing catalase found in the Bacillus subtilis spore coat protein CotJC. In R20291, two other genes encode CotJC1 (CotCB) and CotJC2 (CotD), which have 70 and 50% amino acid sequence similarity, respectively, to CotJC (Permpoonpattana et al., 2011). Insertional mutations of cotCB and cotD in C. difficile 630 resulted in a reduction of catalase activity, but otherwise no significant defect to spore coat formation (Permpoonpattana et al., 2013). This suggests functional redundancy of CDR20291_1465 in R20291, the deletion of which would be unlikely to affect spore coat formation.
We were able to assay for ϕ027 plaque formation and hence propagate the phage using NCTC11207, although it was of a different ribotype to R20291 and contained two predicted prophage genomes. This indicates the potential of ϕ027 to lysogenize isolates other than RT 027, in which it is commonly found (He et al., 2013). Sensitivity of R20291 to ϕ027 was restored after prophage deletion, indicating in R20291 superinfection exclusion provided by the lysogenic prophage was the main mechanism of superinfection immunity. For instance, ϕ027 prophage encoded an Abi-like protein with similarity to abiD from Lactococcus lactis. Abi proteins protect uninfected bacterial cells from phage infection by infected cell suicide, hence aborting further phage infection (Lopatina et al., 2020). CRISPR-Cas is another system which provides immunity against phage. CRISPR arrays found in R20291 did not target ϕ027, though it is noteworthy that ϕ027 carried two CRISPR arrays which very likely conferred immunity to 12 phages (Boudry et al., 2015). Toxin-antitoxin systems, which is another possible phage defense system, has not been predicted in R20291. Since ϕ027 harbored two phage-defense systems, its deletion from R20291 may increase bacterial susceptibility to phage infection. The prophage deletant did not differ significantly in growth compared to WT in rich or minimal medium, although its growth in minimal medium was consistently lower than WT under nutrient limiting conditions (i.e., in the late stationary and death phase of growth curves) and could indicate the prophage was involved in regulation of genes for survival under those conditions. Our prophage deletion approach resulted in an unexpected genetic feature in the prophage deletant that could affect bacterial behavior. This could be determined by re-lysogenizing the prophage deletant with ϕ027 and comparing it to WT. Nevertheless, we anticipate the R20291 prophage deletant to be a useful strain for investigating prophage contribution to host virulence, fitness, and physiology, and a platform for other mutagenesis studies aimed at functional gene analysis without native phage interference.
In conclusion, we have shown that it is feasible to make a clean deletion of the ICE Tn5397. A phage genome could also be precisely deleted from the host chromosome. However, it was also observed that a fragment of the vector used for generating phage deletion could not be completely removed from the host cells. This is probably due to continual recombination between the host genome and the vector DNA. We have also observed this type of interaction with other host vector systems (Hussain et al. unpublished). Our previous work has also shown that some vectors can transfer between E. coli and C. difficile without the requirement for an obvious oriT and that transfer is sensitive to DNase (Khodadoost et al., 2017). Therefore, it is recommended that researchers undertake whole genome sequence analysis after mutant construction to determine the exact genotype of their mutant as this could impact how downstream physiological experiments are interpreted. Furthermore, it is important that further work is done to get a deeper understanding of the mechanism of transfer of MGEs between C. difficile strains.
Data availability statement
The datasets generated for this study can be found in BioProject, accession number: PRJNA993731 and GenBank, accession number: CP129979.1. Materials generated in this study are available upon request from the corresponding author.
Author contributions
HH: Data curation, Investigation, Methodology, Visualization, Writing – review & editing, Writing – original draft. AN: Writing – original draft, Writing – review & editing, Data curation, Investigation, Methodology, Visualization. CR: Methodology, Resources, Visualization, Writing – review & editing, Data curation, Formal analysis. KI: Formal Analysis, Methodology, Writing – review & editing. DK: Data curation, Resources, Writing – review & editing. VP: Investigation, Visualization, Writing – review & editing. PM: Conceptualization, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation, Writing – original draft, Writing – review & editing. SG: Conceptualization, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. KI was supported by the Mahidol Scholarship. SG is supported by an MRC Equipment award (MC_PC_ MR/X012190/1). This work was supported by an Emerging Leaders Grant from WA Department of Health (WANMA2021) awarded to DK.
Acknowledgments
This research used the facilities and services of the Pawsey Supercomputing Centre, Perth, Western Australia.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1416665/full#supplementary-material
Footnotes
References
Andersen, J. M., Shoup, M., Robinson, C., Britton, R., Olsen, K. E. P., and Barrangou, R. (2016). CRISPR diversity and microevolution in Clostridium difficile. Genome Biol. Evol. 8, 2841–2855. doi: 10.1093/gbe/evw203
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bletz, S., Janezic, S., Harmsen, D., Rupnik, M., and Mellmann, A. (2018). Defining and evaluating a Core genome multilocus sequence typing scheme for genome-wide typing of Clostridium difficile. J. Clin. Microbiol. 56, e01987–17. doi: 10.1128/JCM.01987-17
Boudry, P., Semenova, E., Monot, M., Datsenko, K. A., Lopatina, A., Sekulovic, O., et al. (2015). Function of the CRISPR-Cas system of the human pathogen Clostridium difficile. MBio 6, e01112–e01115. doi: 10.1128/mBio.01112-15
Brouwer, M. S., Roberts, A. P., Hussain, H., Williams, R. J., Allan, E., and Mullany, P. (2013). Horizontal gene transfer converts non-toxigenic Clostridium difficile strains into toxin producers. Nat. Commun. 4:2601. doi: 10.1038/ncomms3601
Brouwer, M. S., Warburton, P. J., Roberts, A. P., Mullany, P., and Allan, E. (2011). Genetic organisation, mobility and predicted functions of genes on integrated, mobile genetic elements in sequenced strains of Clostridium difficile. PLoS One 6:e23014. doi: 10.1371/journal.pone.0023014
Candel-Pérez, C., Ros-Berruezo, G., and Martínez-Graciá, C. (2019). A review of Clostridioides [Clostridium] difficile occurrence through the food chain. Food Microbiol. 77, 118–129. doi: 10.1016/j.fm.2018.08.012
Cartman, S. T., and Minton, N. P. (2010). A mariner-based transposon system for in vivo random mutagenesis of Clostridium difficile. Appl. Environ. Microbiol. 76, 1103–1109. doi: 10.1128/AEM.02525-09
Collins, D. A., Sohn, K. M., Wu, Y., Ouchi, K., Ishii, Y., Elliott, B., et al. (2020). Clostridioides difficile infection in the Asia-Pacific region. Emerg. Microbes Infect. 9, 42–52. doi: 10.1080/22221751.2019.1702480
Fagan, R. P., and Fairweather, N. F. (2011). Clostridium difficile has two parallel and essential sec secretion systems. J. Biol. Chem. 286, 27483–27493. doi: 10.1074/jbc.M111.263889
Finn, E., Andersson, F. L., and Madin-Warburton, M. (2021). Burden of Clostridioides difficile infection (CDI) - a systematic review of the epidemiology of primary and recurrent CDI. BMC Infect. Dis. 21:456. doi: 10.1186/s12879-021-06147-y
Fortier, L. C., and Moineau, S. (2007). Morphological and genetic diversity of temperate phages in Clostridium difficile. Appl. Environ. Microbiol. 73, 7358–7366. doi: 10.1128/AEM.00582-07
Gawlik, D., Slickers, P., Engelmann, I., Müller, E., Lück, C., Friedrichs, A., et al. (2015). DNA-microarray-based genotyping of Clostridium difficile. BMC Microbiol. 15:158. doi: 10.1186/s12866-015-0489-2
Goh, S., Chang, B., and Riley, T. (2005). Effect of phage infection on toxin production by Clostridium difficile. J. Med. Microbiol. 54, 129–135. doi: 10.1099/jmm.0.45821-0
Goh, S., Hussain, H., Chang, B. J., Emmett, W., Riley, T. V., and Mullany, P. (2013). Phage phiC2 mediates transduction of Tn6215, encoding erythromycin resistance, between Clostridium difficile strains. MBio 4, e00840–e00813. doi: 10.1128/mBio.00840-13
Goh, S., Ong, P. F., Song, K. P., Riley, T. V., and Chang, B. J. (2007). The complete genome sequence of Clostridium difficile phage phiC2 and comparisons to phiCD119 and inducible prophages of CD630. Microbiology 153, 676–685. doi: 10.1099/mic.0.2006/002436-0
Goh, S., Riley, T. V., and Chang, B. J. (2005). Isolation and characterization of temperate bacteriophages of Clostridium difficile. Appl. Environ. Microbiol. 71, 1079–1083. doi: 10.1128/AEM.71.2.1079-1083.2005
Govind, R., Fralick, J. A., and Rolfe, R. D. (2006). Genomic organization and molecular characterization of Clostridium difficile bacteriophage PhiCD119. J. Bacteriol. 188, 2568–2577. doi: 10.1128/JB.188.7.2568-2577.2006
Govind, R., Fralick, J. A., and Rolfe, R. D. (2011). In vivo lysogenization of a Clostridium difficile bacteriophage ФCD119. Anaerobe 17, 125–129. doi: 10.1016/j.anaerobe.2011.05.012
Govind, R., Vediyappan, G., Rolfe, R. D., Dupuy, B., and Fralick, J. A. (2009). Bacteriophage-mediated toxin gene regulation in Clostridium difficile. J. Virol. 83, 12037–12045. doi: 10.1128/JVI.01256-09
Griffiths, D., Fawley, W., Kachrimanidou, M., Bowden, R., Crook, D. W., Fung, R., et al. (2010). Multilocus sequence typing of Clostridium difficile. J. Clin. Microbiol. 48, 770–778. doi: 10.1128/JCM.01796-09
Hargreaves, K. R., Kropinski, A. M., and Clokie, M. R. J. (2014). What does the talking?: quorum sensing Signalling genes discovered in a bacteriophage genome. PLoS One 9:e85131. doi: 10.1371/journal.pone.0085131
He, M., Miyajima, F., Roberts, P., Ellison, L., Pickard, D. J., Martin, M. J., et al. (2013). Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat. Genet. 45, 109–113. doi: 10.1038/ng.2478
Heap, J. T., Pennington, O. J., Cartman, S. T., and Minton, N. P. (2009). A modular system for Clostridium shuttle plasmids. J. Microbiol. Methods 78, 79–85. doi: 10.1016/j.mimet.2009.05.004
Hong, W., Zhang, J., Cui, G., Wang, L., and Wang, Y. (2018). Multiplexed CRISPR-Cpf1-mediated genome editing in Clostridium difficile toward the understanding of pathogenesis of C. difficile infection. ACS Synth. Biol. 7, 1588–1600. doi: 10.1021/acssynbio.8b00087
Ingle, P., Groothuis, D., Rowe, P., Huang, H., Cockayne, A., Kuehne, S. A., et al. (2019). Generation of a fully erythromycin-sensitive strain of Clostridioides difficile using a novel CRISPR-Cas9 genome editing system. Sci. Rep. 9:8123. doi: 10.1038/s41598-019-44458-y
Jinek, M., Jiang, F., Taylor, D. W., Sternberg, S. H., Kaya, E., Ma, E., et al. (2014). Structures of Cas9 endonucleases reveal RNA-mediated conformational activation. Science 343:1247997. doi: 10.1126/science.1247997
Jo, J., Gonzales-Luna, A. J., Lancaster, C. K., McPherson, J. K., Begum, K., Jahangir Alam, M., et al. (2022). Multi-country surveillance of Clostridioides difficile demonstrates high prevalence of spores in non-healthcare environmental settings. Anaerobe 75:102543. doi: 10.1016/j.anaerobe.2022.102543
Johnson, C. M., and Grossman, A. D. (2015). Integrative and conjugative elements (ICEs): what they do and how they work. Annu. Rev. Genet. 49, 577–601. doi: 10.1146/annurev-genet-112414-055018
Khodadoost, L., Hussain, H., and Mullany, P. (2017). Plasmids can transfer to Clostridium difficile CD37 and 630Δerm both by a DNase resistant conjugation-like mechanism and a DNase sensitive mechanism. FEMS Microbiol. Lett. 364. doi: 10.1093/femsle/fnx208
Kirk, J. A., and Fagan, R. P. (2016). Heat shock increases conjugation efficiency in Clostridium difficile. Anaerobe 42, 1–5. doi: 10.1016/j.anaerobe.2016.06.009
Knetsch, C. W., Connor, T. R., Mutreja, A., van Dorp, S. M., Sanders, I. M., Browne, H. P., et al. (2014). Whole genome sequencing reveals potential spread of Clostridium difficile between humans and farm animals in the Netherlands, 2002 to 2011. Euro Surv. 19:20954. doi: 10.2807/1560-7917.ES2014.19.45.20954
Knight, D. R., Elliott, B., Chang, B. J., Perkins, T. T., and Riley, T. V. (2015). Diversity and evolution in the genome of Clostridium difficile. Clin. Microbiol. Rev. 28, 721–741. doi: 10.1128/CMR.00127-14
Knight, D. R., and Riley, T. V. (2019). Genomic delineation of zoonotic origins of Clostridium difficile. Front. Public Health 7:164. doi: 10.3389/fpubh.2019.00164
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. A. (2019). Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 37, 540–546. doi: 10.1038/s41587-019-0072-8
Kuehne, S. A., Cartman, S. T., Heap, J. T., Kelly, M. L., Cockayne, A., and Minton, N. P. (2010). The role of toxin a and toxin B in Clostridium difficile infection. Nature 467, 711–713. doi: 10.1038/nature09397
Kuehne, S. A., Cartman, S. T., and Minton, N. P. (2011). Both, toxin a and toxin B, are important in Clostridium difficile infection. Gut Microbes 2, 252–255. doi: 10.4161/gmic.2.4.16109
Lawson, P. A., Citron, D. M., Tyrrell, K. L., and Finegold, S. M. (2016). Reclassification of Clostridium difficile as Clostridioides difficile (hall and O'Toole 1935) Prévot 1938. Anaerobe 40, 95–99. doi: 10.1016/j.anaerobe.2016.06.008
Li, T., Zhang, Y., Dong, K., Kuo, C. J., Li, C., Zhu, Y. Q., et al. (2020). Isolation and characterization of the novel phage JD032 and global transcriptomic response during JD032 infection of Clostridioides difficile Ribotype 078. mSystems 5, e00017–20. doi: 10.1128/mSystems.00017-20
Lopatina, A., Tal, N., and Sorek, R. (2020). Abortive infection: bacterial suicide as an antiviral immune strategy. Ann. Rev. Virol. 7, 371–384. doi: 10.1146/annurev-virology-011620-040628
Magill, S. S., Edwards, J. R., Bamberg, W., Beldavs, Z. G., Dumyati, G., Kainer, M. A., et al. (2014). Multistate point-prevalence survey of health care-associated infections. N. Engl. J. Med. 370, 1198–1208. doi: 10.1056/NEJMoa1306801
Maikova, A., Kreis, V., Boutserin, A., Severinov, K., and Soutourina, O. (2019). Using an endogenous CRISPR-Cas system for genome editing in the human pathogen Clostridium difficile. Appl. Environ. Microbiol. 85, e01416–19. doi: 10.1128/AEM.01416-19
Mayer, M. J., Narbad, A., and Gasson, M. J. (2008). Molecular characterization of a Clostridium difficile bacteriophage and its cloned biologically active endolysin. J. Bacteriol. 190, 6734–6740. doi: 10.1128/JB.00686-08
McAllister, K. N., Bouillaut, L., Kahn, J. N., Self, W. T., and Sorg, J. A. (2017). Using CRISPR-Cas9-mediated genome editing to generate C. difficile mutants defective in selenoproteins synthesis. Sci. Rep. 7. doi: 10.1038/s41598-017-15236-5
Meessen-Pinard, M., Sekulovic, O., and Fortier, L. C. (2012). Evidence of in vivo prophage induction during Clostridium difficile infection. Appl. Environ. Microbiol. 78, 7662–7670. doi: 10.1128/AEM.02275-12
Moloney, G., Eyre, D. W., Mac Aogáin, M., McElroy, M. C., Vaughan, A., Peto, T. E. A., et al. (2021). Human and porcine transmission of Clostridioides difficile Ribotype 078. Europe Emerg. Infect. Diseases 27, 2294–2300. doi: 10.3201/eid2709.203468
Monot, M., Boursaux-Eude, C., Thibonnier, M., Vallenet, D., Moszer, I., Medigue, C., et al. (2011). Reannotation of the genome sequence of Clostridium difficile strain 630. J. Med. Microbiol. 60, 1193–1199. doi: 10.1099/jmm.0.030452-0
Monteford, J., Bilverstone, T. W., Ingle, P., Philip, S., Kuehne, S. A., Minton, N. P., et al. (2021). What's a SNP between friends: the lineage of Clostridioides difficile R20291 can effect research outcomes. Anaerobe 71:102422.
Moraru, C., Varsani, A., and Kropinski, A. M. (2020). VIRIDIC—A novel tool to calculate the Intergenomic similarities of prokaryote-infecting viruses. Viruses 12:1268. doi: 10.3390/v12111268
Nishimasu, H., Ran, F. A., Hsu, P. D., Konermann, S., Shehata, S. I., Dohmae, N., et al. (2014). Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell 156, 935–949. doi: 10.1016/j.cell.2014.02.001
Peltier, J., Hamiot, A., Garneau, J. R., Boudry, P., Maikova, A., Hajnsdorf, E., et al. (2020). Type I toxin-antitoxin systems contribute to the maintenance of mobile genetic elements in Clostridioides difficile. Commun. Biol. 3:718. doi: 10.1038/s42003-020-01448-5
Permpoonpattana, P., Phetcharaburanin, J., Mikelsone, A., Dembek, M., Tan, S., Brisson, M. C., et al. (2013). Functional characterization of Clostridium difficile spore coat proteins. J. Bacteriol. 195, 1492–1503. doi: 10.1128/JB.02104-12
Permpoonpattana, P., Tolls, E. H., Nadem, R., Tan, S., Brisson, A., and Cutting, S. M. (2011). Surface layers of Clostridium difficile endospores. J. Bacteriol. 193, 6461–6470. doi: 10.1128/JB.05182-11
Purdy, D., O'Keeffe, T. A., Elmore, M., Herbert, M., McLeod, A., Bokori-Brown, M., et al. (2002). Conjugative transfer of clostridial shuttle vectors from Escherichia coli to Clostridium difficile through circumvention of the restriction barrier. Mol. Microbiol. 46, e00276–15. doi: 10.1046/j.1365-2958.2002.03134.x
Ramirez-Vargas, G., Goh, S., and Rodriguez, C. (2018). The novel phages phiCD5763 and phiCD2955 represent two groups of big Plasmidial Siphoviridae phages of Clostridium difficile. Front. Microbiol. 9:26. doi: 10.3389/fmicb.2018.00026
Riedel, T., Bunk, B., Thürmer, A., Spröer, C., Brzuszkiewicz, E., Abt, B., et al. (2015). Genome resequencing of the virulent and multidrug-resistant reference strain Clostridium difficile 630. Genome Announc. 3, e00276–15. doi: 10.1128/genomeA.00276-15
Riedel, T., Wittmann, J., Bunk, B., Schober, I., Sproer, C., Gronow, S., et al. (2017). A Clostridioides difficile bacteriophage genome encodes functional binary toxin-associated genes. J. Biotechnol. 250, 23–28. doi: 10.1016/j.jbiotec.2017.02.017
Roberts, A. P., Allan, E., and Mullany, P. (2014). The impact of horizontal gene transfer on the biology of Clostridium difficile. Adv. Microb. Physiol. 65, 63–82. doi: 10.1016/bs.ampbs.2014.08.002
Sambrook, J., and Russell, D. W. (2001a). Protocol 8. Purification of bacteriophage lambda particles by isopycnic centrifugation through CsCl gradients. Molecular cloning: A laboratory manual. 3rd Edn. New Yor: Cold Spring Harbor Laboratory Press.
Sambrook, J., and Russell, D. W. (2001b). Protocol 11. Extraction of bacteriophage lambda DNA from large-scale cultures using proteinase K and SDS. Molecular cloning: A laboratory manual. 3rd Edn. New York: Cold Spring Harbor Laboratory Press.
Scornec, H., Bellanger, X., Guilloteau, H., Groshenry, G., and Merlin, C. (2017). Inducibility of Tn916 conjugative transfer in Enterococcus faecalis by subinhibitory concentrations of ribosome-targeting antibiotics. J. Antimicrob. Chemother. 72, 2722–2728. doi: 10.1093/jac/dkx202
Sebaihia, M., Wren, B. W., Mullany, P., Fairweather, N. F., Minton, N., Stabler, R., et al. (2006). The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat. Genet. 38, 779–786. doi: 10.1038/ng1830
Sekulovic, O., and Fortier, L. C. (2015). Global transcriptional response of Clostridium difficile carrying the phiCD38-2 prophage. Appl. Environ. Microbiol. 81, 1364–1374. doi: 10.1128/AEM.03656-14
Sekulovic, O., Meessen-Pinard, M., and Fortier, L. C. (2011). Prophage-stimulated toxin production in Clostridium difficile NAP1/027 lysogens. J. Bacteriol. 193, 2726–2734. doi: 10.1128/JB.00787-10
Sekulovic, O., Ospina Bedoya, M., Fivian-Hughes, A. S., Fairweather, N. F., and Fortier, L. C. (2015). The Clostridium difficile cell wall protein CwpV confers phase-variable phage resistance. Mol. Microbiol. 98, 329–342. doi: 10.1111/mmi.13121
Shaw, H. A., Khodadoost, L., Preston, M. D., Corver, J., Mullany, P., and Wren, B. W. (2019). Clostridium difficile clade 3 (RT023) have a modified cell surface and contain a large transposable island with novel cargo. Sci. Rep. 9:15330. doi: 10.1038/s41598-019-51628-5
Slater, R. T., Frost, L. R., Jossi, S. E., Millard, A. D., and Unnikrishnan, M. (2019). Clostridioides difficile LuxS mediates inter-bacterial interactions within biofilms. Sci. Rep. 9:9903. doi: 10.1038/s41598-019-46143-6
Smith, C. J., Markowitz, S. M., and Macrina, F. L. (1981). Transferable tetracycline resistance in Clostridium difficile. Antimicrob. Agents Chemother. 19, 997–1003. doi: 10.1128/AAC.19.6.997
Smits, W. K., Lyras, D., Lacy, D. B., Wilcox, M. H., and Kuijper, E. J. (2016). Clostridium difficile infection. Nat. Rev. Dis. Primers 2:16020. doi: 10.1038/nrdp.2016.20
Stabler, R. A., He, M., Dawson, L., Martin, M., Valiente, E., Corton, C., et al. (2009). Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 10:R102. doi: 10.1186/gb-2009-10-9-r102
Stark, W. M. (2014). The serine recombinases. Microbiol Spectr 2:6. doi: 10.1128/microbiolspec.MDNA3-0046-2014
Su, Y. A., He, P., and Clewell, D. B. (1992). Characterization of the tet(M) determinant of Tn916: evidence for regulation by transcription attenuation. Antimicrob. Agents Chemother. 36, 769–778. doi: 10.1128/AAC.36.4.769
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
van Prehn, J., Reigadas, E., Vogelzang, E. H., Bouza, E., Hristea, A., Guery, B., et al. (2021). European Society of Clinical Microbiology and Infectious Diseases: 2021 update on the treatment guidance document for Clostridioides difficile infection in adults. Clin. Microbiol. Infect. 27, S1–s21. doi: 10.1016/j.cmi.2021.09.038
Viprey, V. F., Davis, G. L., Benson, A. D., Ewin, D., Spittal, W., Vernon, J. J., et al. (2022). A point-prevalence study on community and inpatient Clostridioides difficile infections (CDI): results from combatting bacterial resistance in Europe CDI (COMBACTE-CDI), July to November 2018. Euro Surv. 27, pii=2100704. doi: 10.2807/1560-7917.ES.2022.27.26.2100704
Wang, S., Hong, W., Dong, S., Zhang, Z. T., Zhang, J., Wang, L., et al. (2018). Genome engineering of Clostridium difficile using the CRISPR-Cas9 system. Clin. Microbiol. Infect. 24, 1095–1099. doi: 10.1016/j.cmi.2018.03.026
Wang, H., and Mullany, P. (2000). The large resolvase TndX is required and sufficient for integration and excision of derivatives of the novel conjugative transposon Tn5397. J. Bacteriol. 182, 6577–6583. doi: 10.1128/JB.182.23.6577-6583.2000
Wang, H., Roberts, A. P., Lyras, D., Rood, J. I., Wilks, M., and Mullany, P. (2000). Characterization of the ends and target sites of the novel conjugative transposon Tn5397 from Clostridium difficile: excision and circularization is mediated by the large resolvase. TndX. J Bacteriol. 182, 3775–3783. doi: 10.1128/JB.182.13.3775-3783.2000
Wang, H., Smith, M. C., and Mullany, P. (2006). The conjugative transposon Tn5397 has a strong preference for integration into its Clostridium difficile target site. J. Bacteriol. 188, 4871–4878. doi: 10.1128/JB.00210-06
Wang, Y., Zhang, Z.-T., Seo, S.-O., Lynn, P., Lu, T., Jin, Y.-S., et al. (2016). Bacterial genome editing with CRISPR-Cas9: deletion, integration, single nucleotide modification, and desirable “clean” mutant selection in Clostridium beijerinckii as an example. ACS Synth. Biol. 5, 721–732. doi: 10.1021/acssynbio.6b00060
Wick, R. R., Judd, L. M., Cerdeira, L. T., Hawkey, J., Méric, G., Vezina, B., et al. (2021). Trycycler: consensus long-read assemblies for bacterial genomes. Genome Biol. 22:266. doi: 10.1186/s13059-021-02483-z
Wick, R. R., Schultz, M. B., Zobel, J., and Holt, K. E. (2015). Bandage: interactive visualization of de novo genome assemblies. Bioinformatics 31, 3350–3352. doi: 10.1093/bioinformatics/btv383
Keywords: C. difficile , prophage deletion, transposon deletion, CRISPR-Cas9, site-specific recombinase
Citation: Hussain H, Nubgan A, Rodríguez C, Imwattana K, Knight DR, Parthala V, Mullany P and Goh S (2024) Removal of mobile genetic elements from the genome of Clostridioides difficile and the implications for the organism’s biology. Front. Microbiol. 15:1416665. doi: 10.3389/fmicb.2024.1416665
Edited by:
Axel Cloeckaert, Institut National de Recherche pour l’Agriculture, l’Alimentation et l’Environnement (INRAE), FranceReviewed by:
Khald Blau, University of Applied Sciences Emden/Leer, GermanyNozomu Obana, University of Tsukuba, Japan
§PRESENT ADDRESS:
Amer Nubgan, Department of Biology, College of Science, University of Baghdad, Baghdad, IraqCopyright © 2024 Hussain, Nubgan, Rodríguez, Imwattana, Knight, Parthala, Mullany and Goh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shan Goh, cy5nb2g1QGhlcnRzLmFjLnVr
†These authors share first authorship
‡These authors have contributed equally to this work and share last authorship