
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 10 July 2024
Sec. Antimicrobials, Resistance and Chemotherapy
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1413618
 Yang Che1†
Yang Che1† Yewei Lu2†
Yewei Lu2† Yelei Zhu3
Yelei Zhu3 Tianfeng He1
Tianfeng He1 Xiangchen Li2Junli Gao2Junshun Gao2Xiaomeng Wang3
Xiangchen Li2Junli Gao2Junshun Gao2Xiaomeng Wang3 Zhengwei Liu3*
Zhengwei Liu3* Feng Tong1*
Feng Tong1*Background: Leveraging well-established DNA-level drug resistance mechanisms, whole-genome sequencing (WGS) has emerged as a valuable methodology for predicting drug resistance. As the most effective second-line anti-tuberculosis (anti-TB) drugs, fluoroquinoloness (FQs) are generally used to treat multidrug-resistant tuberculosis (MDR-TB, defined as being resistant to resistant to rifampicin and isoniazid) or rifampicin-resistant tuberculosis (RR-TB). However, FQs are also commonly used in the management of other bacterial infections. There are few published data on the rates of FQs resistance among rifampicin-susceptible TB. The prevalence of FQs resistance among TB patients who are rifampicin-susceptible has not been studied in Zhejiang Province, China. The goal of this study was to provide a baseline characterization of the prevalence of FQs resistance, particularly among rifampicin-susceptible TB in Zhejiang Province, China.
Methods: Based on WGS, we have investigated the prevalence of FQs resistance among rifampicin-susceptible TB in Zhejiang Province. All pulmonary TB patients with positive cultures who were identified in Zhejiang area during TB drug resistance surveillance from 2018 to 2019 have enrolled in this population-based retrospective study.
Results: The rate of FQs resistance was 4.6% (32/698) among TB, 4.0% (27/676) among rifampicin-susceptible TB, and 22.7% (5/22) among RR-TB. According to WGS, strains that differ within 12 single-nucleotide polymorphisms (SNPs) were considered to be transmission of FQ-resistant strains. Specifically, 3.7% (1/27) of FQs resistance was caused by the transmission of FQs-resistant strains among the rifampicin-susceptible TB and 40.7% (11/27) of FQs resistance was identified as hetero-resistance.
Conclusion: The prevalence of FQs resistance among TB patients who were rifampicin-susceptible was severe in Zhejiang. The emergence of FQs resistance in TB isolates that are rifampicin-susceptible was mainly caused by the selection of drug-resistant strains. In order to prevent the emergence of FQs resistance, the WGS-based surveillance system for TB should be urgently established, and clinical awareness of the responsible use of FQs for respiratory infections should be enhanced.
As the most critical second-line anti-tuberculosis drugs, fluoroquinolones (FQs) have been recommended as Group A agents for use in multidrug-resistant tuberculosis (MDR-TB, defined as being resistant to resistant to rifampicin and isoniazid) or rifampicin-resistant tuberculosis (RR-TB) regimens by the World Health Organization (WHO) (World Health Organization, 2020). WHO also recommended levofloxacin, a FQ antibiotic, in the treatment regimen of isoniazid-resistant tuberculosis (Hr-TB) (World Health Organization, 2022a). Moxifloxacin, a fourth-generation FQ antibiotic, is under consideration by WHO for inclusion in four-month regimens to treat drug-susceptible tuberculosis because of its pharmacokinetics and drug penetration into macrophages (Dorman et al., 2021; World Health Organization, 2022b). Thus, as the cornerstone of the regimens for MDR/RR/Hr-TB, FQs will be the essential drugs in the shorter regimen for drug-susceptible TB in the future. The prevalence of FQs resistance among RR-TB has been investigated in many studies because it can significantly reduce the risk of treatment failure or relapse and death in RR-TB patients (Miotto et al., 2017; Siddiqui et al., 2019). Nevertheless, few studies have investigated the prevalence of FQs resistance among rifampicin-susceptible TB, and the majority of these studies show that FQs resistance in rifampicin-susceptible TB is uncommon (Hu et al., 2011; Schwalb et al., 2021; Zhang et al., 2022).
As a group of broad-spectrum antibiotics, FQs are widely used to treat other bacterial infections, especially respiratory infections. Additionally, FQs have been widely utilized in healthcare institutions for the diagnostic treatment of patients with suspected TB and for the empirical treatment of TB patients without positive drug susceptibility testing (DST) results in several studies (Wang et al., 2006; Ho et al., 2014). We should pay more attention to the emergence of FQs resistance in rifampicin-susceptible Mycobacterium tuberculosis (Mtb) isolates in TB high-burden settings by the inappropriate use and the vital role of FQs in TB treatment.
Based on DNA sequencing platforms, whole-genome sequencing, which reconstructed the complete genome’s DNA sequence, can provide high-resolution genotyping and identification (Witney et al., 2016; Walker et al., 2017; Cohen et al., 2019). FQs resistance in Mtb has been mainly attributed to mutations in the gyrA and gyrB genes, which code for two subunits of DNA gyrase (Ruiz, 2003; Andriole, 2005). Hetero-resistance, which is defined as the coexistence of drug-susceptible and drug-resistant isolates in clinical samples and may exhibit varying levels of drug susceptibility, appears to be more frequent in FQs resistance (Eilertson et al., 2014). The accuracy of WGS in predicting phenotypic resistance to rifampicin, isoniazid, and FQs is high in extensive comparative studies (Farhat et al., 2017; Meehan et al., 2019).
Understanding the evolution of FQs resistance in rifampicin-susceptible TB may help assess the effectiveness of TB programs and interventions and provide guidance for TB prevention and care in the future. However, little is known about FQs resistance in rifampicin-susceptible TB that emerged in China (Zhang et al., 2022). Zhejiang Province, as the first province in China to launch sentinel surveillance, has conducted drug resistance surveys following the initiation of the WHO/International Union Against Tuberculosis and Lung Disease global anti-TB drug resistance surveillance project in 1994. Zhejiang Province was also the first province in China to conduct periodic surveys for drug-resistant tuberculosis (DR-TB) since WHO integrated the country into its surveillance network for DR-TB in 1999.
To the best of our knowledge, our study is the first to address the varying prevalence patterns of FQs resistance among rifampicin-susceptible TB in a well-developed province based on the TB drug-resistance survey project. We provide the findings of a retrospective WGS analysis of all Mtb isolates collected from pulmonary TB cases in the Zhejiang area during TB drug resistance surveillance from 2018 to 2019. We aimed to investigate the prevalence of FQs resistance, especially among rifampicin-susceptible TB. Furthermore, we identified the instances of hetero-resistance in FQs-resistant strains and quantified the FQs resistance due to the transmission of FQs-resistant strains.
The study was a retrospective study that included all the culture-positive patients diagnosed with TB at local TB dispensaries in Zhejiang Province during TB drug-resistance surveillance from Jan 1, 2018, to Dec 31, 2019. The Zhejiang ProvincialCenter for Disease Control and Prevention’s TB reference laboratory collected all clinical isolates from pulmonary TB patients for further species identification, strain preservation and WGS before each patient started anti-TB or other relevant clinical treatments. Records related to demographics, clinical and microbiology were retrieved from the national TB information management system.
WGS was performed for each clinical isolate after it was collected. Mtb culture products were inactivated, and genomic DNA was extracted using a bacterial DNA extraction kit (QIAGEN Inc., Dusseldorf, Germany), following the manufacturer’s instructions. The purified genomic DNA was quantified using a TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA, United States) to ensure compliance with quality requirements for library preparation, sequencing, and detection. Genomic DNA samples were treated and fragmented to approximately 400 bp. Sequencing libraries were generated using the NEXTflex™ Rapid DNA-Seq Kit, followed by multiplexing and loading onto the Illumina NovaSeq 6000 PE150 system (San Diego, CA92122, United States). Sequencing employed a 2 × 150 paired-end configuration. Raw sequencing data underwent processing with fastp v0.20.1 to remove adapter sequences and filter out low-quality bases (Chen et al., 2018). Subsequently, high-quality sequence data were then input into Kraken v1.1.1 for species identification. Samples identified as other species or with an Mtb proportion below 90% were rejected as contaminated (Wood and Salzberg, 2014). The remaining samples’ sequencing data were aligned to the H37Rv reference genome (NC_000962.3) using BWA v0.7.17 (Li, 2013). Samples meeting criteria with an average sequencing depth ≥ 20× and average genome coverage ≥95% were selected for subsequent data analysis. The SAMtools/BCFtools suite v1.13 facilitated the calling of fixed single-nucleotide polymorphisms (SNPs) (frequency ≥ 90%) at loci where the alternate alleles were supported by at least five reads (combining both forward and reverse reads) (Danecek et al., 2021).
Clean sequencing data were input into the local version of TB-Profiler v4.4.2 and aligned to the reference genome of H37Rv to identify the genotype of resistance-associated mutations and detect the resistance profile of 9 anti-TB drugs. These drugs encompassed isoniazid, rifampicin, streptomycin, ethambutol, fluoroquinolones (levofloxacin and moxifloxacin), amikacin, kanamycin, and capreomycin (Phelan et al., 2019). Mutations with a frequency of less than 10% were excluded from the analysis. WGS-based DST results were deduced by assessing the presence or absence of mutations (Tier 1 and Tier 2 mutations) in a comprehensive database containing drug resistance-associated mutations, adhering to evidence levels recommended by WHO (Walker et al., 2022). In this study, hetero-resistance was defined based on the observation that the frequency of resistant alleles in the sequence reads was below 99%.
The fixed SNPs, excluding those located in proline-glutamic acid (PE)/proline-proline-glutamic acid (PPE) genes, insertion elements, repetitive regions, and genes associated with drug resistance, were combined into a concatenated alignment (Meehan et al., 2019). Maximum-likelihood (ML) phylogenetic trees were inferred from the concatenated alignment using IQ-Tree v2.2.5 (Nguyen et al., 2015). The best-scoring ML trees were rooted using M. canettii (RefSeq: NC_015848.1) as the outgroup and were visually represented with the Interactive Tree of Life (iTOL) (Letunic and Bork, 2021). A genomic threshold (≤12 SNPs) was applied to identify clusters of isolates potentially consistent with recent transmission referred to as genomic clusters (Yang et al., 2017). Primary FQs resistance (transmitted FQs resistance) was defined as the FQs resistance-conferring mutation shared by two or more strains within a genomic cluster. Any other FQs resistance-conferring mutations were categorized as acquired FQs resistance.
Statistical analyses were conducted using the R package gtsummary (Sjoberg et al., 2021). The Pearson’s chi-squared or Fisher’s exact test was used for comparison of categorical variables, such as demographic, bacteriological, and clinical characteristics.
A total of 837 culture-positive patients diagnosed with TB in Zhejiang area during TB drug-resistance surveillance from January 1, 2018, to December 31, 2019 were enrolled in this study. Out of these patients, 139 (16.6%) were excluded from analysis due to strain contamination, recovery failure, WGS failure or loss of epidemiological data (Figure 1). The remaining 698 patients had an average age of 51 years (ranging from 13 to 91 years). 493 (70.6%) of them were male and 645 (92.4%) were new TB cases. Based on the results of WGS-based DST, 22 (3.2%) were diagnosed with RR-TB. Notably, RR-TB patients were relatively younger, exhibiting a statistically significant difference compared to rifampicin-susceptible patients (median age 62 years vs. 67.5 years, p = 0.009). Further analysis revealed RR-TB patients were more likely to be migrant (59% vs. 38%, p = 0.049) and have been previously treated for TB (22.7% vs. 7.1%; p = 0.020) than rifampicin-susceptible TB patients (Table 1).
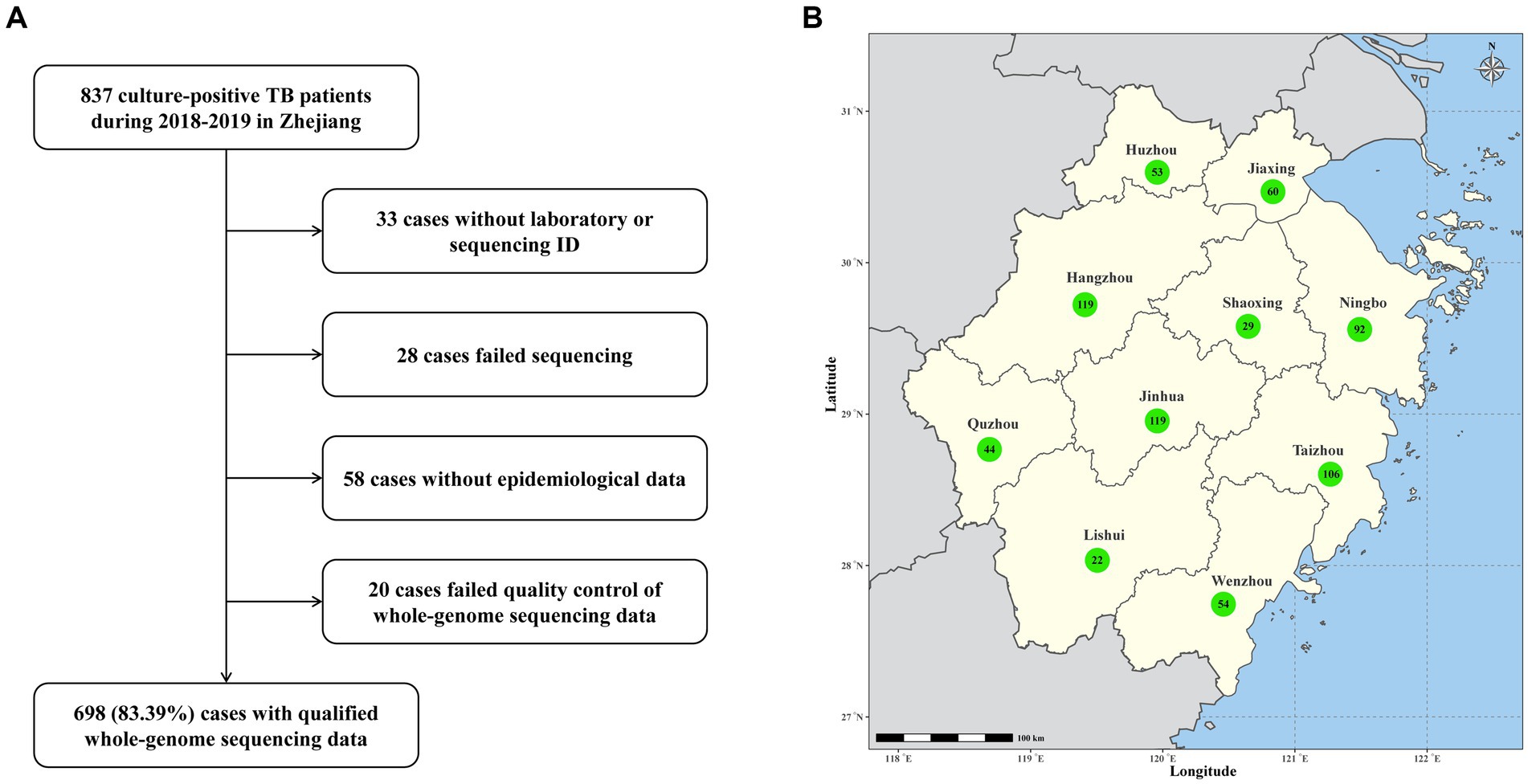
Figure 1. (A) Sample enrollment and study flowchart. (B) Map of Zhejiang Province with the number of TB cases from each prefecture-level city.
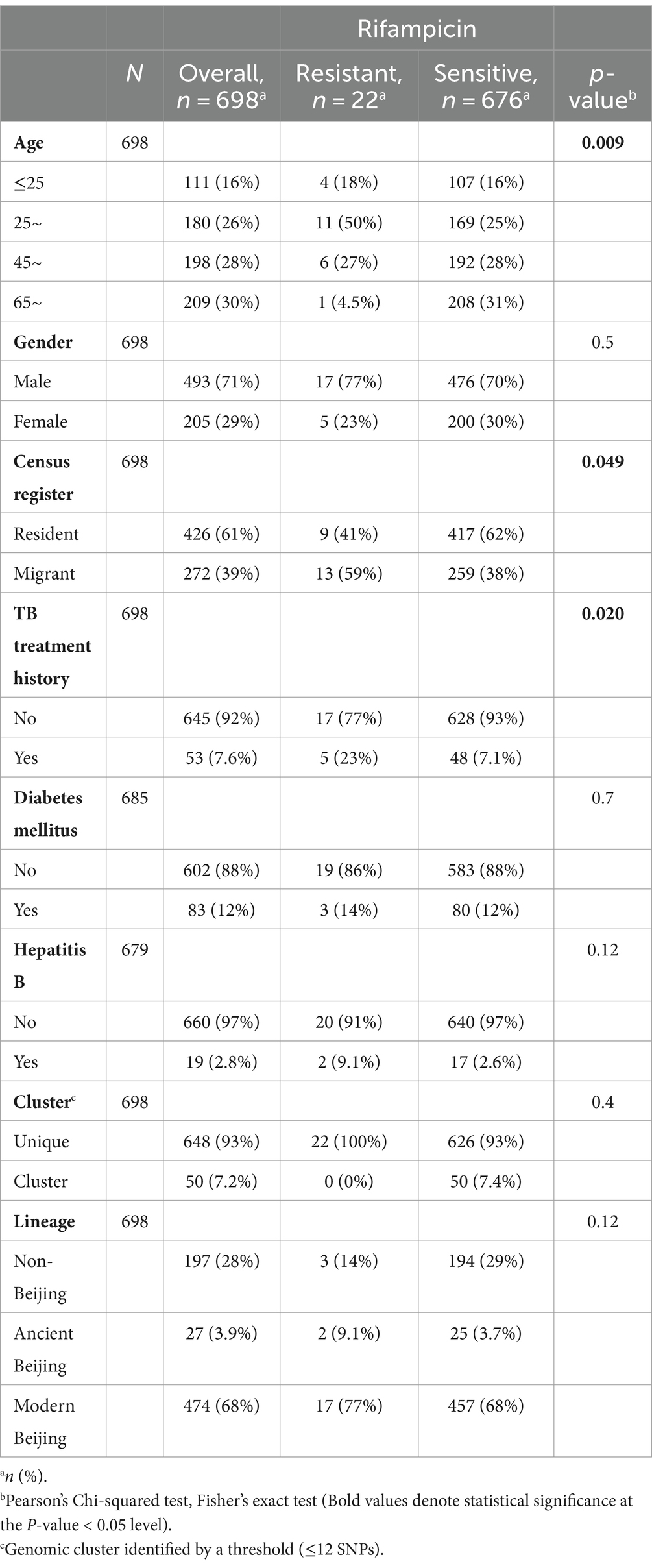
Table 1. Demographic, clinical, and bacteriological characteristics of RR-TB and rifampicin-susceptible TB cases in Zhejiang Province.
According to the results of WGS-based DST, 32 (4.6%) exhibited resistance to FQs, including 27 rifampicin-susceptible isolates and 5 rifampicin-resistant isolates. The rate of FQs resistance was 4.0% (27/676) among rifampicin-susceptible TB and 22.7% (5/22) among RR-TB. The drug resistance profile and epidemiological information of 32 FQs -resistant TB patients were shown in Supplementary Table S1.
Five rifampicin-resistant isolates harbored the FQs resistance-conferring mutations in the gyrA and gyrB genes, including two gyrA D94A, two gyrA A90V and one gyrB E501D. Among 676 rifampicin-susceptible isolates, 27 harbored diverse FQs resistance-conferring mutations, including ten gyrA D94A, seven gyrA A90V, six gyrA D94N, one gyrA D94Y, one gyrB A504V, and one gyrB D461N. Additionally, one rifampicin-susceptible isolates harbored more than one FQs resistance-conferring mutation, gyrA A90V + gyrA S91P.
Among 27 rifampicin-susceptible isolates with FQs resistance-conferring mutations, 11 (40.7%) were identified as FQ hetero-resistance. The allele frequencies of FQ hetero-resistance ranged from 10.1 to 98.7%. Regarding FQs hetero-resistant isolates, 26 had a single unfixed mutation and one had multiple unfixed mutations in gyrA. FQs hetero-resistance was only observed in one isolate among 5 rifampicin-resistant isolates (20%, 1/5). The allele frequencies of FQs resistance-conferring mutations in 32 FQs-resistant isolates were shown in Figure 2.
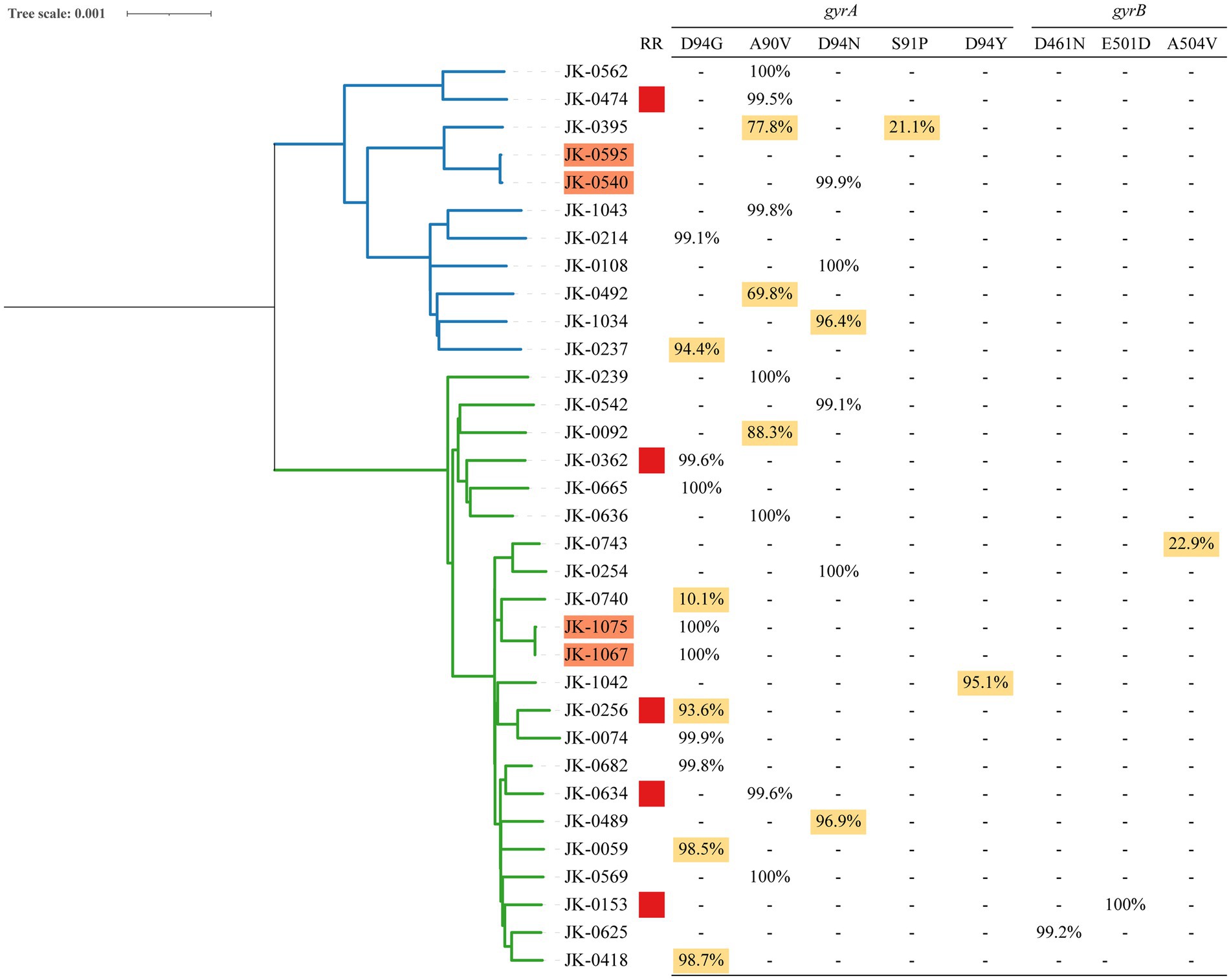
Figure 2. Phylogeny, clustering and FQs hetero-resistance profile of 32 FQs-resistant Mtb isolates. (1) Blue and green branches indicate non-Beijing and Beijing strains, respectively. (2) Genomic-clustered strains differing by ≤12 SNPs are highlighted in orange. (3) Red square indicates the Mtb isolate resistant to rifampicin. (4) Allele frequency of FQs resistance-conferring loci in gyrA and gyrB is shown in the right side. Hetero-resistance mutations are highlighted in yellow.
All 698 isolates were divided into 24 clusters defined as strains that differed by 12 or fewer SNPs, among which two clusters included FQs-resistant isolates. There are two basic ways that drug resistance emerges: acquired drug resistance arising from insufficient therapy or primary drug resistance as a result of the transmission of drug-resistant strains. Notably, we were able to observe primary and acquired drug resistance in each of the clusters with FQs-resistant isolates, separately, as depicted in Figure 2. Within one cluster (JK-1075 and JK-1067), we identified a uniform fixed mutation conferring FQsresistance (gyrA D94G) present in both isolates, with a 100% allele frequency. This observation strongly suggests primary drug resistance resulting from transmission. In another cluster (JK-0595 and JK-0540), acquired drug resistance was evident, where only one of the two isolates exhibited FQs resistance, featuring a fixed mutation conferring resistance (gyrA D94N, 99.88%).
Rapid, reliable, and increasingly affordable WGS technology can help with TB prevention and care, including diagnosis, treatment, and surveillance (Satta et al., 2018; van der Werf and Ködmön, 2019). It has demonstrated efficacy in discerning genetic drug resistance profiles (Jabbar et al., 2019; Wu et al., 2022). In this study, we have investigated the prevalence of FQs resistance and its transmission in all Mtb isolates from pulmonary TB patients during TB drug-resistance surveillance in Zhejiang Province. Our findings reveal that the majority (84.4%, 27/32) of FQs resistant TB were susceptible to rifampicin, with a notable proportion (40.7%, 11/27) identified as hetero-resistance. WGS data indicate that the transmission of FQs-resistant strains contributed to 3.7% (1/27) of FQs resistance in rifampicin-susceptible TB.
We observed that more than two-thirds of the FQs-resistant cases were identified in rifampicin-susceptible TB patients. Regard as the core agents for RR-TB treatment, moxifloxacin and levofloxacin are usually prescribed to RR-TB patients (Kayalı et al., 2021; World Health Organization, 2022a). This prescription practice aligns with the technical specifications on TB prevention and care. Surveillance data on the prevalence of FQs resistance in rifampicin-susceptible TB are scarce, and the DST of FQs is not routinely conducted for rifampicin-susceptible TB patients (Heyckendorf et al., 2018; Tagliani et al., 2021). Previous review studies showed that prevalence of FQs resistance in Mtb clinical isolates in Shanghai was higher (6.1%) than reported in other countries, where the range is typically 0 to 4.4% (Ginsburg et al., 2003; Ho et al., 2014; Kayalı et al., 2021; Schwalb et al., 2021; Zhang et al., 2022). FQs are also the most often recommended antibiotics in China for respiratory infections. Therefore, it is imperative to conduct surveillance on FQs resistance and monitor fluoroquinolones exposure in newly diagnosed TB patients in countries with a high TB burden.
Previous studies have indicated that mutations in the quinolones resistance determining region of gyrA or gyrB constitute the main mechanism for Mtb resistance to fluoroquinolones (Gröschel et al., 2021; Maruri et al., 2021). Within the scope of this study, the majority of FQs resistance-conferring mutations occurring in rifampicin-resistant Mtb were D94G (2/5) or A90V (2/5). Our findings suggest that Mtb strains harboring multiple other drug resistance-conferring mutations might be more likely to acquire FQs resistance-conferring mutations with a lower fitness cost. The profile of FQs resistance-conferring mutations occurring in rifampicin-susceptible Mtb exhibited greater diversity. Every instance of FQs hetero-resistance was observed in rifampicin-susceptible Mtb, with the exception of a single rifampicin-resistant Mtb isolate. Hetero-resistance represents a critical stage in the development of an initially drug-susceptible Mtb population toward complete resistance to a specific drug over the course of an infection (Dheda et al., 2018). Non-lethal drug concentrations facilitate the emergence and selection of drug mutations with a low fitness cost (Castro et al., 2020). According to the hypotheses, the characteristics of FQs resistance-conferring mutations in rifampicin-susceptible Mtb imply that the Mtb FQs resistance may have been induced by ineffective FQs treatment. Due to easy accessibility and improper use of FQs, several studies have indicated a high proportion of TB patients being exposed to FQs before their TB diagnosis (Yang et al., 2015; Yuen et al., 2015). Moreover, hetero-resistance hampers the effectiveness of rapid molecular assays in detecting drug resistance (O’Donnell et al., 2019). Utilizing WGS techniques, variant allele frequencies could be employed to identify hetero-resistance for drug resistance prediction (Chaidir et al., 2019). In our study, we identified FQs hetero-resistance in rifampicin-susceptible Mtb isolates, with hetero-resistance frequencies ranging from 10.1 to 98.7%. Our findings revealed that WGS is sensitive in detecting FQs resistance in hetero-resistant strains and the real burden of FQs resistance in rifampicin-susceptible TB might be underestimated.
Previous studies have demonstrated that the transmission of rifampicin-resistant strains facilitated the spread of FQs resistance (Takiff and Feo, 2015; Nikolayevskyy et al., 2019). In the present study, FQs resistance caused by transmission was also observed in rifampicin-susceptible Mtb isolates within the Zhejiang region. Comparative analysis of transmission and resistance-conferring mutations in FQs-resistant isolates revealed a minimal incidence of FQs resistance in rifampicin-susceptible isolates (3.7%, 1/27) and no occurrences in rifampicin-resistant isolates (0/5) resulting from the transmission of FQs-resistant strains. The higher prevalence of FQs resistance in rifampicin-susceptible TB cases, as compared to rifampin-resistant TB cases, is likely due to the wide use of FQs for antibiotic therapy throughout Zhejiang. Moreover, the empirical prescription of FQs for respiratory infections has been linked to delays in the detection and treatment of pulmonary tuberculosis. These delays may, in turn, contribute to the increased transmission of TB (Lin et al., 2016).
The retrospective study design of our study limited the access to information regarding FQs prescriptions for TB patients prior to diagnosis, including details such as prescription date, dosage, specific type of FQs, and duration of supply. However, to the best of our knowledge, this study represents the first investigation into the prevalence of FQs resistance among rifampicin-susceptible TB cases in Zhejiang Province (Zhang et al., 2022). Analysis of WGS data revealed that 50% of FQs resistance in rifampicin-susceptible TB cases manifested as hetero-resistance, and the transmission of FQs -resistant strains contributed to the development of FQ-resistant TB. In order to mitigate the emergence of FQ resistance, it is imperative to promptly establish a WGS-based surveillance system for TB. Additionally, raising clinical awareness regarding the judicious use of FQs for respiratory infections is essential.
The prevalence of FQs resistance among TB patients who were rifampicin-susceptible was severein Zhejiang. The emergence of FQs resistance in TB isolates susceptible to rifampicin mainly stems from the selection of drug-resistant strains. In order to prevent the development of FQs resistance, the WGS-based surveillance system for TB should be urgently established, and clinical awareness of the responsible use of FQs for respiratory infections should be enhanced.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://ngdc.cncb.ac.cn/bioproject/browse/PRJCA018552.
This study was approved by the Ethics Committee of the Zhejiang Provincial Center for Disease Control and Prevention. All eligible participants who agreed to participate in the program and signed an informed consent form were required to complete a questionnaire and provide at least one sputum specimen for subsequent studies.
YC: Writing – review & editing, Writing – original draft, Methodology, Funding acquisition, Formal analysis, Conceptualization. YL: Writing – review & editing, Writing – original draft, Formal analysis, Data curation, Conceptualization. YZ: Writing – review & editing, Formal analysis, Data curation. TH: Writing – review & editing, Formal analysis, Data curation. XL: Writing – review & editing, Formal analysis, Data curation. JunlG: Writing – review & editing, Data curation. JunsG: Writing – review & editing, Data curation. XW: Writing – review & editing, Formal analysis, Data curation. ZL: Writing – review & editing, Funding acquisition, Data curation. FT: Writing – review & editing, Data curation.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was supported by National Natural Science Foundation of China No. 82373649, Zhejiang Provincial Natural Science Foundation of China under Grant No. LTGY23H190001 and No. LTGY23H190002, Zhejiang Provincial Medical Research Project No. 2022KY1189, Ningbo Top Medical and Health Research Program No. 2023020713, the Project of Ningbo Key R&D Plan and “Unveiling and Leading” No. 2023Z174. The funders had no role in study design, data collection, analysis, interpretation of data and writing the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1413618/full#supplementary-material
Andriole, V. T. (2005). The quinolones: past, present, and future. Clin. Infect. Dis. 41, S113–S119. doi: 10.1086/428051
Castro, R. A., Ross, A., Kamwela, L., Reinhard, M., Loiseau, C., Feldmann, J., et al. (2020). The genetic background modulates the evolution of fluoroquinolones-resistance in Mycobacterium tuberculosis. Mol. Biol. Evol. 37, 195–207. doi: 10.1093/molbev/msz214
Chaidir, L., Ruesen, C., Dutilh, B. E., Ganiem, A. R., Andryani, A., Apriani, L., et al. (2019). Use of whole-genome sequencing to predict Mycobacterium tuberculosis drug resistance in Indonesia. J. Glob. Antimicrob. Resist. 16, 170–177. doi: 10.1016/j.jgar.2018.08.018
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Cohen, K. A., Manson, A. L., Desjardins, C. A., Abeel, T., and Earl, A. M. (2019). Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: progress, promise, and challenges. Genome Med. 11:45. doi: 10.1186/s13073-019-0660-8
Danecek, P., Bonfield, J. K., Liddle, J., Marshall, J., Ohan, V., Pollard, M. O., et al. (2021). Twelve years of SAMtools and BCFtools. Gigascience 10:giab008. doi: 10.1093/gigascience/giab008
Dheda, K., Lenders, L., Magombedze, G., Srivastava, S., Raj, P., Arning, E., et al. (2018). Drug-penetration gradients associated with acquired drug resistance in patients with tuberculosis. Am. J. Respir. Crit. Care Med. 198, 1208–1219. doi: 10.1164/rccm.201711-2333OC
Dorman, S. E., Nahid, P., Kurbatova, E. V., Phillips, P. P. J., Bryant, K., Dooley, K. E., et al. (2021). Four-month Rifapentine regimens with or without moxifloxacin for tuberculosis. N. Engl. J. Med. 384, 1705–1718. doi: 10.1056/NEJMoa2033400
Eilertson, B., Maruri, F., Blackman, A., Herrera, M., Samuels, D. C., and Sterling, T. R. (2014). High proportion of Heteroresistance in gyrA and gyrB in fluoroquinolones-resistant Mycobacterium tuberculosis clinical isolates. Antimicrob. Agents Chemother. 58, 3270–3275. doi: 10.1128/AAC.02066-13
Farhat, M. R., Jacobson, K. R., Franke, M. F., Kaur, D., Murray, M., and Mitnick, C. D. (2017). Fluoroquinolones resistance mutation detection is equivalent to culture-based drug sensitivity testing for predicting multidrug-resistant tuberculosis treatment outcome: a retrospective cohort study. Clin. Infect. Dis. 65, 1364–1370. doi: 10.1093/cid/cix556
Ginsburg, A. S., Hooper, N., Parrish, N., Dooley, K. E., Dorman, S. E., Booth, J., et al. (2003). Fluoroquinolones resistance in patients with newly diagnosed tuberculosis. Clin. Infect. Dis. 37, 1448–1452. doi: 10.1086/379328
Gröschel, M. I., Owens, M., Freschi, L., Vargas, R., Marin, M. G., Phelan, J., et al. (2021). GenTB: a user-friendly genome-based predictor for tuberculosis resistance powered by machine learning. Bioinformatics. 13, 138. doi: 10.1101/2021.03.27.437319
Heyckendorf, J., Andres, S., Köser, C. U., Olaru, I. D., Schön, T., Sturegård, E., et al. (2018). What is resistance? Impact of phenotypic versus molecular drug resistance testing on therapy for multi- and extensively drug-resistant tuberculosis. Antimicrob. Agents Chemother. 62, e01550–e01517. doi: 10.1128/AAC.01550-17
Ho, J., Jelfs, P., and Sintchenko, V. (2014). Fluoroquinolones resistance in non-multidrug-resistant tuberculosis—a surveillance study in New South Wales, Australia, and a review of global resistance rates. Int. J. Infect. Dis. 26, 149–153. doi: 10.1016/j.ijid.2014.03.1388
Hu, Y., Mathema, B., Wang, W., Kreiswirth, B., Jiang, W., and Xu, B. (2011). Population-based investigation of fluoroquinoloness resistant tuberculosis in rural eastern China. Tuberculosis 91, 238–243. doi: 10.1016/j.tube.2011.03.001
Jabbar, A., Phelan, J. E., de Sessions, P. F., Khan, T. A., Rahman, H., Khan, S. N., et al. (2019). Whole genome sequencing of drug resistant Mycobacterium tuberculosis isolates from a high burden tuberculosis region of north West Pakistan. Sci. Rep. 9:14996. doi: 10.1038/s41598-019-51562-6
Kayalı, R. A., Özkan, S. A., and Biçmen, C.Erer OF (2021). The relation between the emergence of fluoroquinolones resistance and fluoroquinolones exposure in new cases of active pulmonary tuberculosis. Turk. Thorac. J. 22:2002272. doi: 10.5152/TurkThoracJ.2021.19128
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. doi: 10.48550/arXiv.1303.3997
Lin, H., Dyar, O. J., Rosales-Klintz, S., Zhang, J., Tomson, G., Hao, M., et al. (2016). Trends and patterns of antibiotic consumption in Shanghai municipality, China: a 6 year surveillance with sales records, 2009–14. J. Antimicrob. Chemother. 71, 1723–1729. doi: 10.1093/jac/dkw013
Maruri, F., Guo, Y., Blackman, A., van der Heijden, Y. F., Rebeiro, P. F., and Sterling, T. R. (2021). Resistance-conferring mutations on whole-genome sequencing of fluoroquinolones-resistant and-susceptible Mycobacterium tuberculosis isolates: a proposed threshold for identifying resistance. Clin. Infect. Dis. 72, 1910–1918. doi: 10.1093/cid/ciaa496
Meehan, C. J., Goig, G. A., Kohl, T. A., Verboven, L., Dippenaar, A., Ezewudo, M., et al. (2019). Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat. Rev. Microbiol. 17, 533–545. doi: 10.1038/s41579-019-0214-5
Miotto, P., Tessema, B., Tagliani, E., Chindelevitch, L., Starks, A. M., Emerson, C., et al. (2017). A standardised method for interpreting the association between mutations and phenotypic drug resistance in Mycobacterium tuberculosis. Eur. Respir. J. 50:1701354. doi: 10.1183/13993003.01354-2017
Nguyen, L.-T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nikolayevskyy, V., Niemann, S., Anthony, R., Van Soolingen, D., Tagliani, E., Ködmön, C., et al. (2019). Role and value of whole genome sequencing in studying tuberculosis transmission. Clin. Microbiol. Infect. 25, 1377–1382. doi: 10.1016/j.cmi.2019.03.022
O’Donnell, M. R., Larsen, M. H., Brown, T. S., Jain, P., Munsamy, V., Wolf, A., et al. (2019). Early detection of emergent extensively drug-resistant tuberculosis by flow cytometry-based phenotyping and whole-genome sequencing. Antimicrob. Agents Chemother. 63, e01834–e01818. doi: 10.1128/AAC.01834-18
Phelan, J. E., O’Sullivan, D. M., Machado, D., Ramos, J., Oppong, Y. E. A., Campino, S., et al. (2019). Integrating informatics tools and portable sequencing technology for rapid detection of resistance to anti-tuberculous drugs. Genome Med. 11:41. doi: 10.1186/s13073-019-0650-x
Ruiz, J. (2003). Mechanisms of resistance to quinolones: target alterations, decreased accumulation and DNA gyrase protection. J. Antimicrob. Chemother. 51, 1109–1117. doi: 10.1093/jac/dkg222
Satta, G., Lipman, M., Smith, G. P., Arnold, C., Kon, O. M., and McHugh, T. D. (2018). Mycobacterium tuberculosis and whole-genome sequencing: how close are we to unleashing its full potential? Clin. Microbiol. Infect. 24, 604–609. doi: 10.1016/j.cmi.2017.10.030
Schwalb, A., Cachay, R., Meza, E., Cáceres, T., Blackman, A., Maruri, F., et al. (2021). Fluoroquinolones susceptibility in first-line drug-susceptible M. tuberculosis isolates in Lima, Peru. BMC. Res. Notes 14:413. doi: 10.1186/s13104-021-05832-0
Siddiqui, S., Brooks, M. B., Malik, A. A., Fuad, J., Nazish, A., Bano, S., et al. (2019). Evaluation of GenoType MTBDR plus for the detection of drug-resistant Mycobacterium tuberculosis on isolates from Karachi, Pakistan. PLoS ONE 14:e0221485. doi: 10.1371/journal.pone.0221485
Sjoberg, D. D., Whiting, K., Curry, M., Lavery, J. A., and Larmarange, J. (2021). Reproducible summary tables with the gtsummary package. R Journal 13, 570–580. doi: 10.32614/RJ-2021-053
Tagliani, E., Anthony, R., Kohl, T. A., De Neeling, A., Nikolayevskyy, V., Ködmön, C., et al. (2021). Use of a whole genome sequencing-based approach for Mycobacterium tuberculosis surveillance in Europe in 2017–2019: an ECDC pilot study. Eur. Respir. J. :57. doi: 10.1183/13993003.02272-2020
Takiff, H. E., and Feo, O. (2015). Clinical value of whole-genome sequencing of Mycobacterium tuberculosis. Lancet Infect. Dis. 15, 1077–1090. doi: 10.1016/S1473-3099(15)00071-7
van der Werf, M. J., and Ködmön, C. (2019). Whole-genome sequencing as tool for investigating international tuberculosis outbreaks: a systematic review. Front. Public Health 7:87. doi: 10.3389/fpubh.2019.00087
Walker, T. M., Merker, M., Kohl, T. A., Crook, D. W., Niemann, S., and Peto, T. E. A. (2017). Whole genome sequencing for M/XDR tuberculosis surveillance and for resistance testing. Clin. Microbiol. Infect. 23, 161–166. doi: 10.1016/j.cmi.2016.10.014
Walker, T. M., Miotto, P., Köser, C. U., Fowler, P. W., Knaggs, J., Iqbal, Z., et al. (2022). The 2021 WHO catalogue of Mycobacterium tuberculosis complex mutations associated with drug resistance: a genotypic analysis. Lancet Microbe 3, e265–e273. doi: 10.1016/S2666-5247(21)00301-3
Wang, J.-Y., Hsueh, P.-R., Jan, I.-S., Lee, L.-N., Liaw, Y.-S., Yang, P.-C., et al. (2006). Empirical treatment with a fluoroquinolones delays the treatment for tuberculosis and is associated with a poor prognosis in endemic areas. Thorax 61, 903–908. doi: 10.1136/thx.2005.056887
Witney, A. A., Cosgrove, C. A., Arnold, A., Hinds, J., Stoker, N. G., and Butcher, P. D. (2016). Clinical use of whole genome sequencing for Mycobacterium tuberculosis. BMC Med. 14:46. doi: 10.1186/s12916-016-0598-2
Wood, D. E., and Salzberg, S. L. (2014). Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15, 1–12. doi: 10.1186/gb-2014-15-3-r46
World Health Organization (2020). WHO consolidated guidelines on tuberculosis. Module 4: treatment-drug-resistant tuberculosis treatment, Geneva: World Health Organization.
World Health Organization (2022a). WHO operational handbook on tuberculosis. Module 4: treatment-drug-resistant tuberculosis treatment, 2022 update, Geneva: World Health Organization.
World Health Organization (2022b). WHO consolidated guidelines on tuberculosis. Module 4: treatment-drug-susceptible tuberculosis treatment, Geneva: World Health Organization.
Wu, X., Tan, G., Sha, W., Liu, H., Yang, J., Guo, Y., et al. (2022). Use of whole-genome sequencing to predict Mycobacterium tuberculosis complex drug resistance from early positive liquid cultures. Microbiol. Spectr. 10, e02516–e02521. doi: 10.1128/spectrum.02516-21
Yang, C., Luo, T., Shen, X., Wu, J., Gan, M., Xu, P., et al. (2017). Transmission of multidrug-resistant Mycobacterium tuberculosis in Shanghai, China: a retrospective observational study using whole-genome sequencing and epidemiological investigation. Lancet Infect. Dis. 17, 275–284. doi: 10.1016/S1473-3099(16)30418-2
Yang, C., Shen, X., Peng, Y., Lan, R., Zhao, Y., Long, B., et al. (2015). Transmission of Mycobacterium tuberculosis in China: a population-based molecular epidemiologic study. Clin. Infect. Dis. 61, 219–227. doi: 10.1093/cid/civ255
Yuen, C. M., Amanullah, F., Dharmadhikari, A., Nardell, E. A., Seddon, J. A., Vasilyeva, I., et al. (2015). Turning off the tap: stopping tuberculosis transmission through active case-finding and prompt effective treatment. Lancet 386, 2334–2343. doi: 10.1016/S0140-6736(15)00322-0
Zhang, Y., Jiang, Y., Yu, C., Li, J., Shen, X., Pan, Q., et al. (2022). Whole-genome sequencing for surveillance of fluoroquinolone resistance in rifampicin-susceptible tuberculosis in a rural district of Shanghai: a 10-year retrospective study. Front. Public Health 10:990894. doi: 10.3389/fpubh.2022.990894
Keywords: fluoroquinolones resistance, drug resistance, whole-genome sequencing, rifampicin-susceptible tuberculosis, hetero-resistance
Citation: Che Y, Lu Y, Zhu Y, He T, Li X, Gao J, Gao J, Wang X, Liu Z and Tong F (2024) Surveillance of fluoroquinolones resistance in rifampicin-susceptible tuberculosis in eastern China with whole-genome sequencing-based approach. Front. Microbiol. 15:1413618. doi: 10.3389/fmicb.2024.1413618
Edited by:
Ning Dong, Zhejiang University, ChinaReviewed by:
Shu-Hua Wang, The Ohio State University, United StatesCopyright © 2024 Che, Lu, Zhu, He, Li, Gao, Gao, Wang, Liu and Tong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhengwei Liu, emh3bGl1QGNkYy56ai5jbg==; Feng Tong, dG9uZ2ZlbmcxMDMxQDE2My5jb20=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.