 Elena Bello-López1
Elena Bello-López1 Ana Sofía Escobedo-Muñoz1
Ana Sofía Escobedo-Muñoz1 Gabriela Guerrero2
Gabriela Guerrero2 Ariadnna Cruz-Córdova3Elvira Garza-González4
Ariadnna Cruz-Córdova3Elvira Garza-González4 Rigoberto Hernández-Castro5
Rigoberto Hernández-Castro5 Patricia Lozano Zarain6
Patricia Lozano Zarain6 Rayo Morfín-Otero7
Rayo Morfín-Otero7 Patricia Volkow8
Patricia Volkow8 Juan Xicohtencatl-Cortes3
Juan Xicohtencatl-Cortes3 Miguel A. Cevallos1*
Miguel A. Cevallos1*- 1Universidad Nacional Autónoma de México, Centro de Ciencias Genómicas, Programa de Genómica Evolutiva, Cuernavaca, Mexico
- 2Universidad Nacional Autónoma de México, Centro de Ciencias Genómicas, Unidad de Análisis Bioinformáticos, Cuernavaca, Mexico
- 3Unidad de Enfermedades Infecciosas, Laboratorio de Investigación en Bacteriología Intestinal, Hospital Infantil de México Federico Gómez, Ciudad de México, Mexico
- 4Universidad Autónoma de Nuevo León, Facultad de Medicina/Hospital Universitario Dr. José Eleuterio González, Departamento de Bioquímica y Medicina Molecular, Monterrey, Mexico
- 5Departamento de Ecología de Agentes Patógenos, Hospital General Dr. Manuel Gea González, Ciudad de México, Mexico
- 6Benemérita Universidad Autónoma de Puebla, Instituto de Ciencias, Centro de Investigaciones en Ciencias Microbiológicas, Laboratorio de Microbiología Hospitalaria y de la Comunidad, Puebla, Mexico
- 7Instituto de Patología Infecciosa y Experimental, Universidad de Guadalajara, Guadalajara, Mexico
- 8Instituto Nacional de Cancerología, Departamento de Enfermedades Infecciosas, Ciudad de México, Mexico
Acinetobacter pittii has increasingly been associated with several types of hospital-acquired severe infections. Genes implicated in carbapenem resistance, tigecycline resistance, or genes encoding extended spectrum cephalosporinases, such as blaADC, are commonly found in isolates implicated in these infections. A. pittii strains that are pandrug resistant have occasionally been identified. Food for human consumption, animals and plants are environmental sources of this pathogen. An alarming situation is that A. pitti has been identified as responsible for outbreaks in different regions worldwide. In this study, 384 genomes of A. pittii were analyzed, comprising sequences from clinical and non-clinical origins from 32 countries. The objective was to investigate if clinical strains possess genetic traits facilitating hospital adaptation. Results indicate significant genomic variability in terms of size and gene content among A. pittii isolates. The core genome represents a small portion (25–36%) of each isolate’s genome, while genes associated with antibiotic resistance and virulence predominantly belong to the accessory genome. Notably, antibiotic resistance genes are encoded by a diverse array of plasmids. As the core genome between environmental and hospital isolates is the same, we can assume that hospital isolates acquired ARGs due to a high selective pressure in these settings. The strain’s phylogeographic distribution indicates that there is no geographical bias in the isolate distribution; isolates from different geographic regions are dispersed throughout a core genome phylogenetic tree. A single clade may include isolates from extremely distant geographical areas. Furthermore, strains isolated from the environment or animal, or plant sources frequently share the same clade as hospital isolates. Our analysis showed that the clinical isolates do not already possess specific genes, other than antibiotic-resistant genes, to thrive in the hospital setting.
Introduction
The constant increase in bacterial strains capable of resisting a wide range of antibiotics imposes a serious problem for human health. Multidrug-resistant (MDR) bacterial pathogens are responsible for 15–50% of hospital-acquired infections worldwide (Aloke and Achilonu, 2023). Unfortunately, the problem is increasing annually. MDR bacteria are responsible for at least 700,000 deaths annually, but it is projected that 10 millions of deaths will occur by 2050 (O’Neill, 2016). With these numbers in mind, the WHO has developed a list of antibiotic-resistant pathogens to channel research leading to the discovery of new antibiotics and the development of new therapies. The most problematic bacterial pathogens within the hospital setting are known as ESKAPE microorganisms, which are known as Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, A. baumannii, Pseudomonas aeruginosa, and Enterobacter species. Nonetheless, carbapenem-resistant A. baumannii is the WHO priority pathogen for the research and development of new antibiotics (Tacconelli et al., 2018), and it is no surprise, considering the number of outbreaks it causes annually worldwide and its mortality rate, especially in intensive care unit patients (Falagas and Rafailidis, 2007; Mohd Sazlly Lim et al., 2019; Cornejo-Juárez et al., 2020; Antimicrobial Resistance Collaborators, 2022).
Importantly, other members of the Acinetobacter genus could follow the same path as A. baumannii. Almost one-third of the species within the Acinetobacter genus are associated with hospital-acquired infections (Nemec, 2022). Aside from A. baumannii, A. pittii is becoming a dangerous emergent pathogen. This microorganism is frequently isolated from environmental sources (He and Wan, 2021; Izotova et al., 2021; Jiang et al., 2022), such as from food for human consumption offered in the markets (Carvalheira et al., 2017a,b), and a wide range of animals, including dogs, cats, birds, frogs, head lice, fishes, and humans. In these hosts, A. pittii is frequently found as an infection agent (Li et al., 2017; Boumbanda-Koyo et al., 2020; Wang et al., 2020; Verma et al., 2021; Cevallos et al., 2022; Leelapsawas et al., 2022; Bello-López et al., 2024).
In recent years, A. pittii has gradually been more frequently linked to different kinds of hospital-acquired infections, such as bloodstream infections, ventilator-associated pneumonia, and wound infections (Horrevorts et al., 1995; Brasiliense et al., 2019; Tian et al., 2022). Isolates involved in hospital-acquired infections frequently carry genes involved in carbapenem resistance, such as NDM-1, blaOXA58, blaGIM-1, blaTEM, or blaVIM-2 (Chapartegui-González et al., 2022; Wunderlich et al., 2023; Tobin et al., 2024); genes involved in tigecycline resistance (Qian et al., 2023); or genes encoding extended-spectrum cephalosporinases, such as blaADC (Acinetobacter-derived cephalosporinases) (Hujer et al., 2005; Périchon et al., 2014). Moreover, a pandrug-resistant A. pittii strain has been isolated from a patient with severe pneumonia, in a Chinese hospital (Yang et al., 2021).
A sign of concern is that A. pitti has been identified as responsible for several outbreaks in different regions worldwide (Horrevorts et al., 1995; Idzenga et al., 2006). Furthermore, in some hospitals from Japan, France and Germany, it has been reported that A. pittii is the most common isolated Acinetobacter species (Schleicher et al., 2013; Pailhoriès et al., 2018; Kiyasu et al., 2020). All these observations drove us to study A. pittii closely and identify all those changes at the genomic level that are enabling it to become a worrying emerging pathogen.
To properly assess the characteristics that could render A. pittii as an emerging hospital-acquired pathogen, they must be contrasted with what we know about A. baumannii. A. baumannii is a well-established nosocomial pathogen. Most hospital-acquired infections caused by A. baumannii are attributed to a limited set of strains, known as international clones. These strains often exhibit resistance to a wide range of antibiotics, a feature crucial for their expansion and success in the hospital environment (Shelenkov et al., 2023). Genes involved in antibiotic resistance are linked to an ample variety of mobile genetic elements (Noel et al., 2022).
Recent research has revealed that A. baumannii strains isolated from non-nosocomial sources, such as grasslands, poultry, livestock, or wastewater, belong to new clones not closely associated with international clones. In other words: the international clones and the environmental isolates are grouped in different clades, strongly suggesting a differentiation between hospital-acquired isolates and those from other origins (Hamidian et al., 2022; Mateo-Estrada et al., 2022, 2023). Moreover, environmental-origin strains generally possess fewer antibiotic-resistance genes compared to international clones (Hamidian et al., 2022; Mateo-Estrada et al., 2022, 2023).
These observations also suggest that strains belonging to the international clones already possess specializations that enable them to survive and thrive in the hospital environment. This also implies that members of the international clones have little contact with environmental strains and prefer to exchange genetic material among themselves. A clear sign of their adaptation to the hospital setting is that members of the international clones easily spread between hospitals and even continents via infected patients; once a clone invades a hospital, eradicating it becomes extremely difficult (Peleg et al., 2008; Zarrilli et al., 2009; Birgand et al., 2014; Cornejo-Juárez et al., 2020; Higgins et al., 2021). Another important feature for survival in the hospital setting is that A. baumannii isolates can resist desiccation and frequently form biofilms (Lucidi et al., 2024).
With this in mind, we analyzed a total of 384 high quality genomic sequences. 352 downloaded from NCBI, and 32 A. pittii strains sequenced by us. 29 of them are nosocomial strains isolated from 7 hospitals, six Mexicans and one Honduran hospital. We also sequenced three strains isolated from animals. The collection included A. pittii isolates samples from 32 countries. Here, we show that the A. pittii genome varies widely in terms of genome size and gene content. The core genome is a small fraction (25 to 36%) of the genome of each A. pittii isolate. Importantly, most of the genes involved in antibiotic resistance or virulence are part of the accessory genome. The majority of the genes involved in antibiotic resistance are encoded by a vast range of plasmids. The strains’ phylogeographic distribution indicates no geographical bias in the isolate distribution; isolates from different geographic regions—even continents—are dispersed throughout a core genome phylogenetic tree. A single clade may include isolates from extremely distant geographical areas. Furthermore, strains isolated from the environment or animal/plant sources frequently share the same clade as hospital isolates. Our analysis showed that the clinical isolates do not already possess specific genes, other than antibiotic-resistant genes, to thrive in the hospital setting.
Results
Phylogeographic distribution of A. pittii isolates
To analyze the geographical distribution from a phyletic perspective, we constructed a maximum likelihood phylogenetic tree with 352 high-quality A. pittii genome assemblies downloaded from NCBI plus 29 genome sequences from Mexican and Honduran hospital-acquired isolates and three genome sequences obtained from animals (Table 1, Supplementary Table S1). We used an A. baumannii genome as the outgroup sequence. Importantly, these sequences came from 32 countries from very different regions; nevertheless, the collection had a high sample bias considering that most of the genomes came from three countries: China, Germany, and the United States. Moreover, most of the genome sequences were obtained from clinical isolates (82%). The collection also contained strains obtained from non-clinical samples: soil/water (24 strains), plants (2 strains), animals (17 strains), fomites (4 strains) and from the International Space Station (21 strains).

Table 1. Strains sequenced in this work.
The genome size of A. pittii varied greatly: The smallest genome was 3.57 Mb, and the largest genome was 4.5 Mb. In concordance, the number of proteins encoded in the A. pittii genomes studied here also showed a high degree of variation, ranging from 3,149 to 4,198. The A. pittii, genes which are from strict core genomes consist of 1,102 families of orthologous proteins, and they are involved mainly in (A) Translation, ribosomal structure, and biogenesis. (B) Amino acid transport and metabolism. (C) Energy production and conversion. (D) Coenzyme transport and metabolism, and (E) Cell wall, membrane a and envelope biogenesis.
This observation also implied that the strict core genome of each isolate represented between 25 and 36% of the genome. The genome size and the number of proteins they encoded did not depend on the isolation source (Figure 1). The pangenome of the A. pittii isolates analyzed here consisted of 32,019 proteins, indicating its ecological versatility. To evaluate the diversity of A. pittii, we analyzed SNVs of 279 core genes that did not exhibit recombination signals after eliminating paralogs. This analysis revealed that A. pittii was highly diverse; the average pairwise distance was 1,698 SNVs, and the largest difference between the two strains was 4,214 SNVs (Supplementary Figure S1).
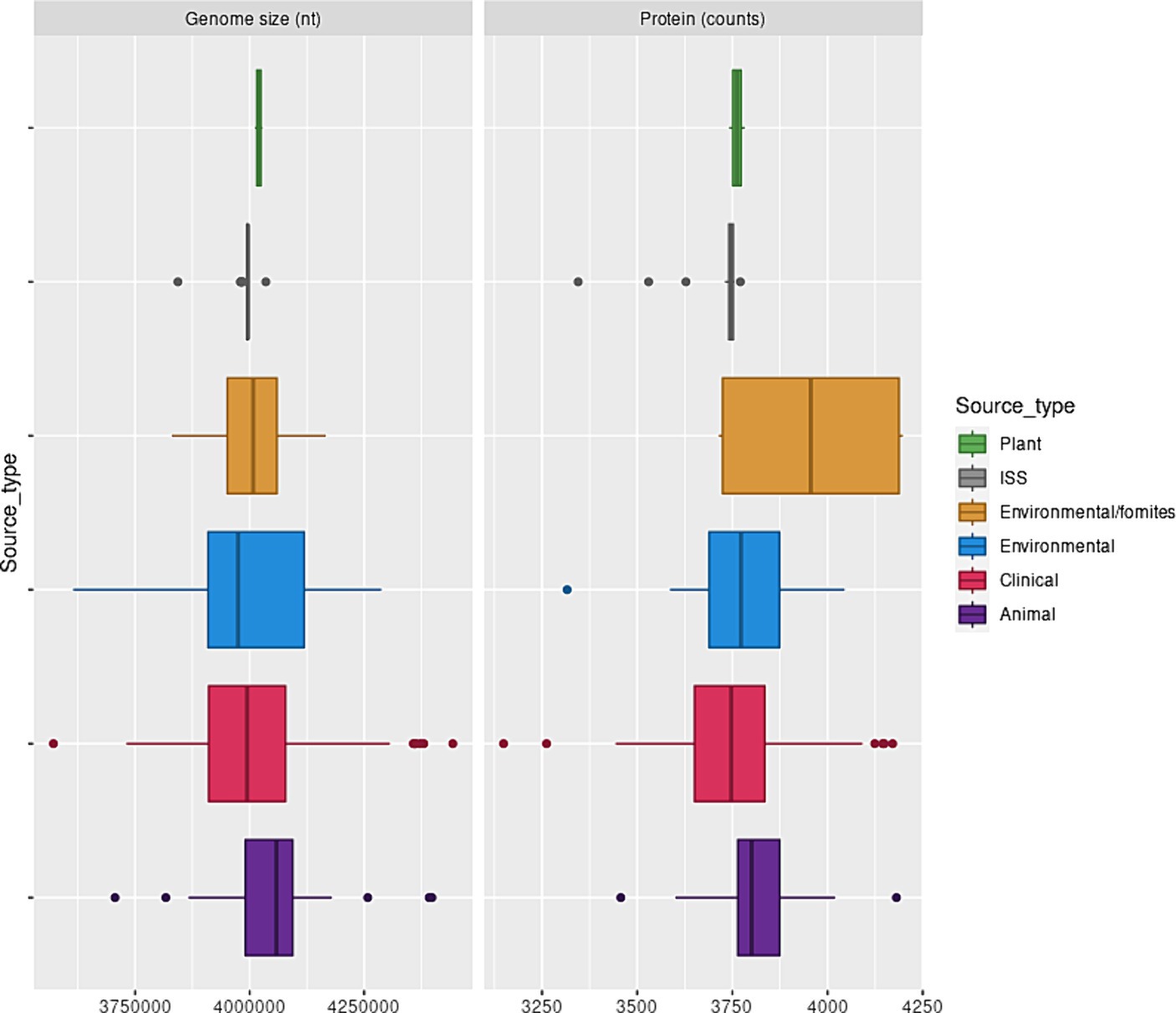
Figure 1. A. pittii genome sizes and protein counts. Box and whisker plot showing the distribution of A. pittii genome sizes and protein counts considering isolation source. Clinical: Median, 3994436.0; Mean, 4001563.8; sd, 134450.5. Animal: Median, 4058504.5; Mean, 4062697.7; sd, 175490.6. Environmental: Median, 3974814.5; Mean, 3991148.0; sd, 158,914. Environmental/Fomites: Median, 4007841.0; Mean, 4002936.0; sd, 138086.1. ISS: Median, 3995985.0; Mean, 3988077.52; sd, 34882.89. Plant: Median, 4,019,960; Mean, 4,019,960; sd, 119.89.
Two important observations could be drawn from the tree topology presented in Figure 2 and Supplementary Figure S2. First, the distribution of the isolates did not have a geographical bias; isolates from distinct geographical regions, even continents, were interspersed along the tree. The same clade could include isolates from distant geographical regions. For example, strains UKK-0145 (Turkey), ABC (India), ABBL019 (USA), WCHAP00001, WCHAP00003, and WCHAP00021 (China) were grouped in the same clade. The only exception we found was that the strains isolated from the International Space Station comprised a single clade, indicating recent clonal expansion. These observations indicated that the A. pittii isolates were subject to frequent intercontinental introductions. In other words, A. pittii did not seem to have obvious migration barriers. The second noteworthy observation was that we did not find specific clones associated with specific habitats. Moreover, clinical isolates frequently shared the same clade as strains obtained from environmental and/or animal or plant sources. One possible interpretation of this observation was that all strains of A. pittii could be pathogenic if they encountered the right host, usually immunocompromised, and the route to invade it.
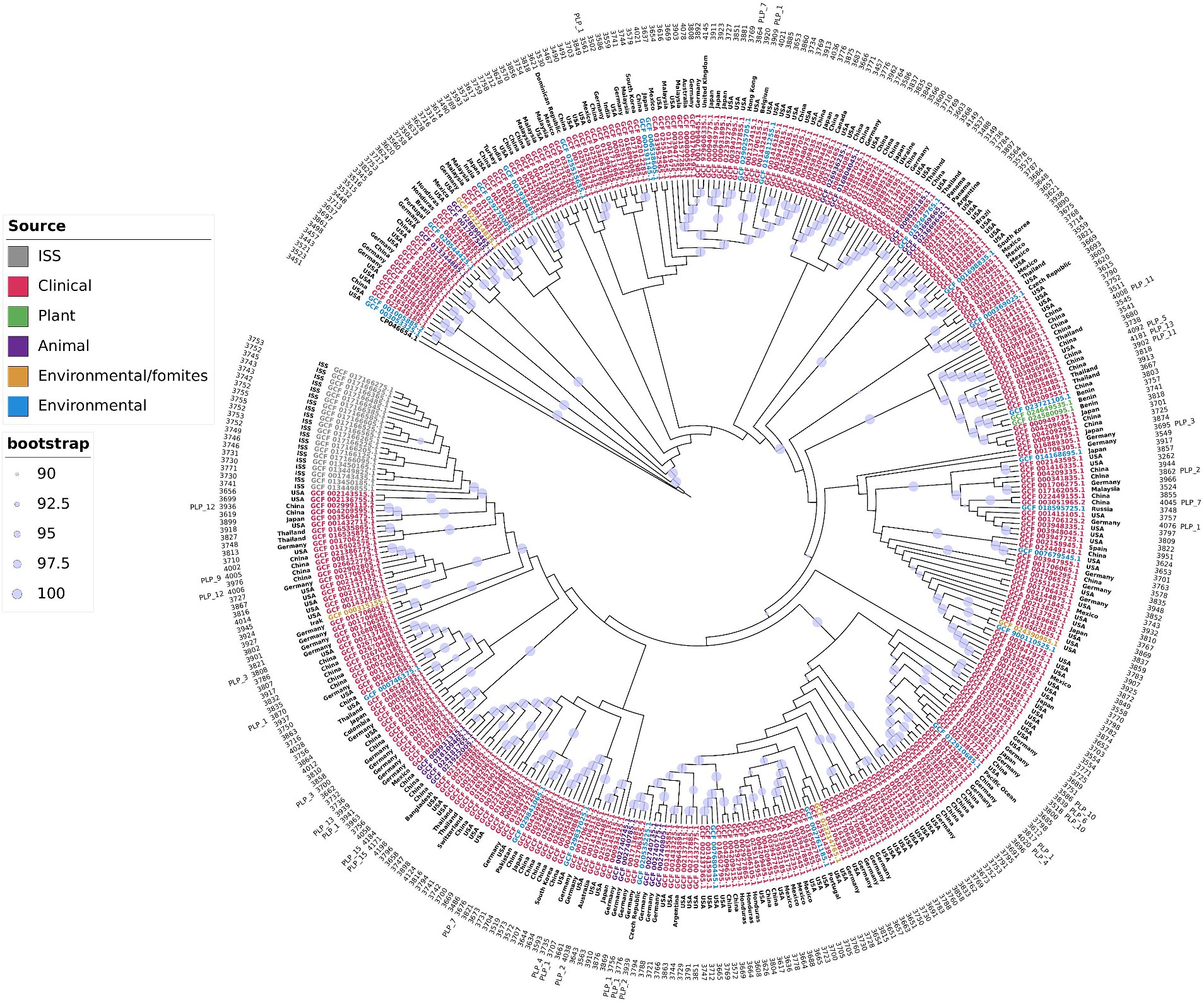
Figure 2. A. pittii core genome phylogenetic tree. Maximum likelihood phylogenetic tree of A. pittii constructed with strict core genome genes of isolates obtained from clinical, environmental, environmental fomites, animal, plant, and International Space Station (ISS) sources. A. baumannii (CP046654.1) was used as outgroup. External circle indicates strains with plasmids belonging to a plasmid lineage (PLP). Numbers in the next inner circle shows the number of proteins of each isolate. The next circle shows the country of origin of each isolate. The Maximum likelihood phylogenetic tree considering distances is presented in Supplementary Figure S2.
The phylogenetic tree presented here was constructed from genes that constituted the core genome of this species but as stated above, represented a small percentage of the genes A. pittii genome. To determine whether the accessory genome, the largest fraction of the genome of this bacterium, could be used to group strains by geography or by isolation source, we constructed a phylogenetic tree based on a matrix of the presence and absence of genes. This phylogenetic tree (Supplementary Figure S3) shows that excluding the genomes obtained from the International Space Station, the strains were not associated with a location or an isolation source.
Antibiotic resistance genes
To identify the genes related to acquired antibiotic present in A. pittii genomes, we consulted the Comprehensive Antibiotic Resistance Database (CARD) (Alcock et al., 2020; Florensa et al., 2022). The result of this analysis is presented in Supplementary Figure S4. However, we did the same exercise to determine in which fraction of the genome (core or accessory) the genes were located. We found only four genes in the core genome with matches in this database: The small multidrug resistance (SMR) antibiotic efflux pump AbeS, the resistance-nodulation-cell division (RND) antibiotic efflux pump AdeF, the intrinsic peptide antibiotic-resistant Lps, and the blaADC beta-lactamase. Nevertheless, it is important to consider that all strains possess a blaOXA gene, but they are different enough to belong to distinct families of orthologous proteins. The most common blaOXA genes belong to the blaOXA-213 family (blaOXA500). On the other hand, we obtained 71 matches in the CARD using the A. pittii accessory genome as a query (Supplementary Table S2). As expected, many of these genes were encoded in plasmids (Table 2).
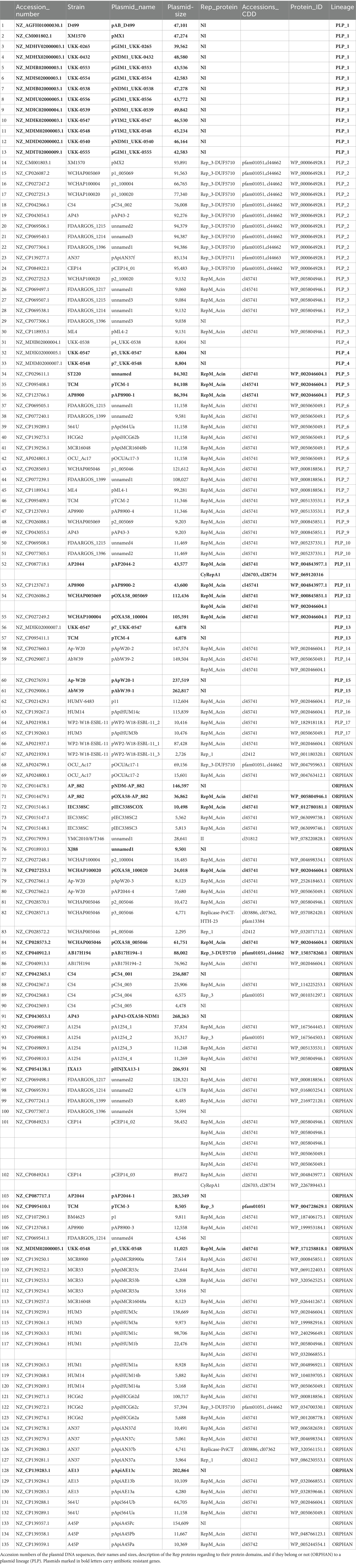
Table 2. A. pittii plasmids.
Virulence genes
To identify genes involved in virulence, we searched the Virulence Factor Database using the protein sequences encoded in the A. pittii strict core genome as a query. Surprisingly, only 15 of these proteins matched this database (Supplementary Table S3A). These genes are involved in immune modulation, biofilm formation, adherence, effector delivery, and regulation. We performed the same analysis using the A. pittii proteins encoded in the accessory genes as a query, and we found 81 matches with the VFDB (Supplementary Table S3B).
The A. pittii plasmids
In the collection of high-quality A. pittii genome sequences, we found 135 plasmids with a wide size range, ranging from 2,295 bp to 283,349 bp. To evaluate their relationships, we grouped the plasmids according to their average nucleotide identities (ANI). With this strategy, we identified 17 plasmid lineages (PLP_1 to PLP_17). PLP_1, the largest lineage, contained 13 plasmids with sizes ranging from 39.5 to 48.5 kb. However, nine were isolated from diverse German locations between 2011 and 2014. The second-largest lineage, PLP_2, contained 11 plasmids, and PLP_3 had six members. The remaining plasmid lineages contained two or three members. However, 70 (51.4%) of these plasmids were orphans (Table 2).
The 135 A. pittii plasmids had, as a group, 6,882 genes; however, 4,673 (67.9%) of them encoded proteins annotated as hypothetical. These plasmids also possessed 665 transposase genes. We identified members of the 21 transposase families. The family with the greatest number of members was the IS3 family, with 227 members, followed by the IS5 family (144 members) and the IS6 family (71 members). This observation suggests that transposable elements have played a crucial role in the transfer of antibiotic resistance genes between plasmids and between plasmids and chromosomes.
The diversity of replication-initiation proteins encoded by the A. pittii plasmid was limited. Of the 111 Rep proteins identified in the plasmid collection, 86 belonged to the RepM_Acin superfamily (cl45741) according to the Conserved Domain Database classification (Lu et al., 2020). Interestingly, all plasmids encoding this Rep protein were adjacent to or near a gene or relics of a gene encoding an uncharacterized protein but were annotated as a plasmid replication DNA-binding protein (rep_pAB02_ORF2 superfamily). The next most abundant proteins, with 14 representatives, were Rep proteins with two protein domains: Rep_3 and DUF5710. Additionally, we found three Rep proteins having only a Rep_3 domain, two with a Rep_1 domain, and two containing the CyRepA1 domain. Finally, we detected two Rep proteins, one with an II domain and the other with Replicase-PriCT-HTH-23 domains. Nevertheless, we could not identify genes encoding replication initiator proteins in 33 (24.2%) plasmids from our collection (Table 2). Among the plasmids with no identifiable Rep genes were all members of the PLP_1, PLP_4, PLP_13, and PLP_15 plasmid lineages and 12 orphan plasmids.
Plasmids belonging to the same plasmid lineage encoded identical Rep proteins; for example, members of the PLP_2 plasmid lineage encoded Rep proteins with a protein identification number (protein_id) WP_000064928.1, or members of the PLP_3 lineage had Rep genes encoding proteins with the same id (WP_000064928.1). Similarly, PLP_5 contained Rep genes encoding WP_002046604.1 proteins. This situation is not rare in plasmids, as this has been described for many A. baumannii plasmids (Salgado-Camargo et al., 2020). Six of the A. pittii plasmids possessed genes encoding more than one Rep protein, which were usually not identical (different protein_id). The only exception was plasmid pCEP14_02, which encoded five Rep proteins with two different protein_ids (WP_005804946.1 and WP_005065049.1) (Table 2).
To evaluate the potential host range of the A. pittii plasmids, we searched for identical Rep proteins encoded by other plasmids. Our results, summarized in Supplementary Table S4, suggested that the host range of the A. pittii plasmids varied widely; some seemed to have a very narrow host range because we found only identical proteins encoded in other A. pittii plasmids. This was the case for the plasmids pIEC338SC2, pIEC338SC3, pA1254_1, and pApW20-3. In contrast, other plasmids appeared to be capable of replicating in an ample variety of Acinetobacter species, such as members of the PLP_2 and PLP_3 plasmid lineages. Moreover, we also found that some plasmids, such as the members of PLP_5, PLP_9, and PLP_14, shared identical Rep proteins with plasmids belonging to other genera, such as Flavobacterium johnsoniae, Salmonella enterica, and Klebsiella pneumoniae.
For the same purpose, we used the 135 A. pittii plasmids under study as queries in BLASTn searches, looking for plasmids with a DNA sequence identity equal to or greater than 95% and a query coverage of at least 75% in other Acinetobacter species. We found that 19 A. pittii plasmids were closely related to other Acinetobacter species, including A. cumulans, A. chinensis, A. nosocomialis, A. baumannii, A. ursingii, A. wuhouensis, A. defluvii, and A. septicus. Interestingly, 18 of the 19 plasmids that matched the plasmids of other Acinetobacter species carried antibiotic-resistance genes (Table 3). This observation suggested that non-baumannii Acinetobacter species are important reservoirs of antibiotic-resistance genes.
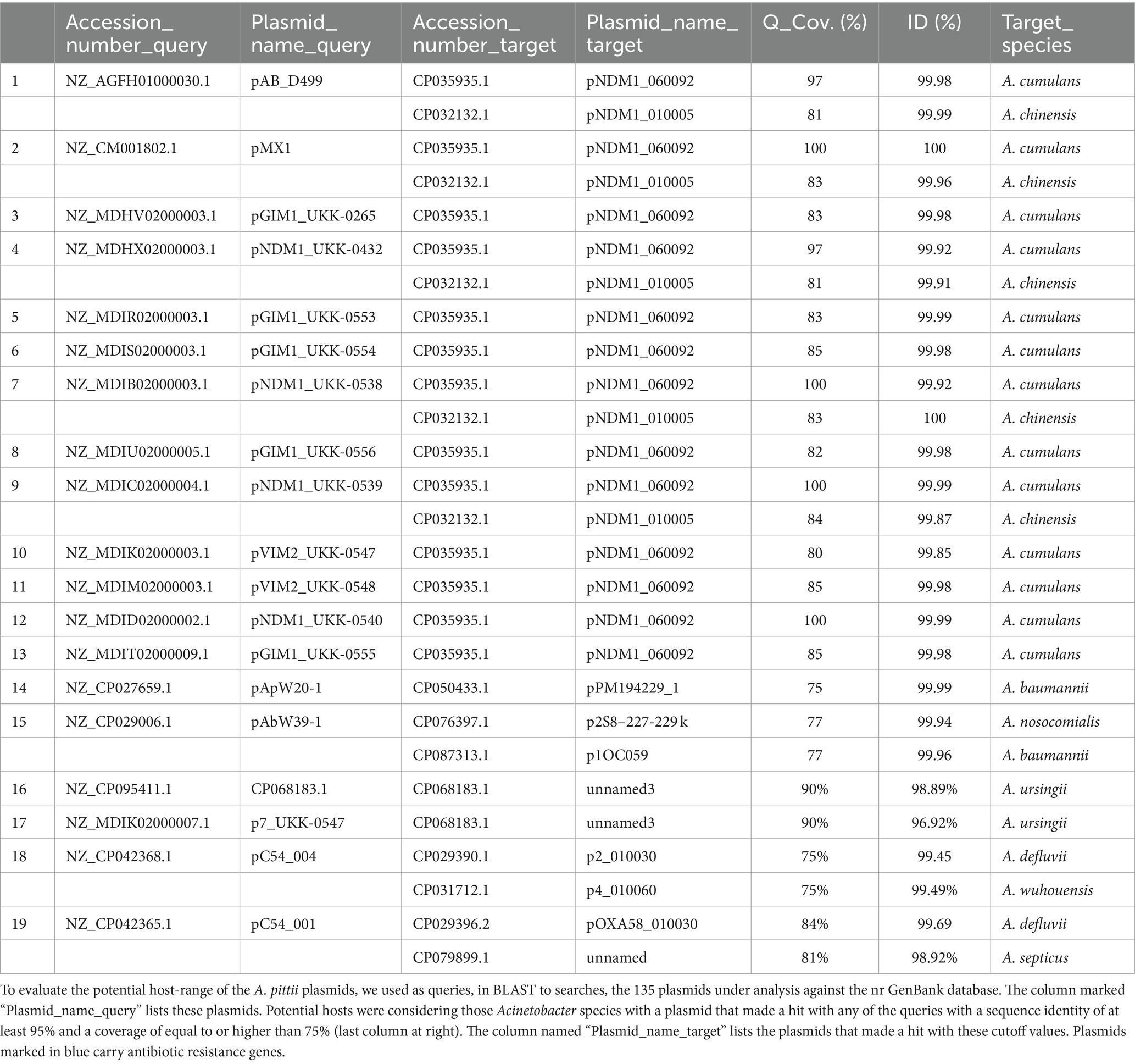
Table 3. A. pittii plasmids host-range.
A. pittii plasmids are crucial in disseminating antibiotic resistance genes; 30.1% of the plasmids studied here carried genes involved in antibiotic resistance. All members of lineages PLP_1, PLP_5, PLP_11, PLP_12, PLP_13, and PLP_15 carried antibiotic resistance genes, but orphan plasmids also functioned in the spread of antibiotic resistance genes, considering that 16 of the 70 orphan plasmids carried genes involved in antibiotic resistance (Table 2). Importantly, some plasmids, such as those belonging to the plasmid lineages PLP_5, PLP_9, and PLP_14, had a very wide potential host range that included other pathogens, such as A. baumannii, K. pneumoniae, and S. enterica.
Table 4 lists all the antibiotic resistance genes found in the A. pittii plasmids. Genes involved in aminoglycoside resistance were the most common. However, genes conferring resistance to carbapenems, such as blaNDM1, blaNDM44, blaOXA58, blaOXA72, blaGIM1, and blaVIM1, were also found. The number of antibiotic resistance genes encoded in plasmids varied widely; some, such as pAB_D499 or p7_UKK-0548, carried only one antibiotic resistance gene, while others contained up to 12 different genes involved in antibiotic resistance, such as plasmid pC54_001. Frequently, antibiotic resistance genes were near IS sequences, indicating the role of transposable elements in the dispersion of antibiotic resistance.
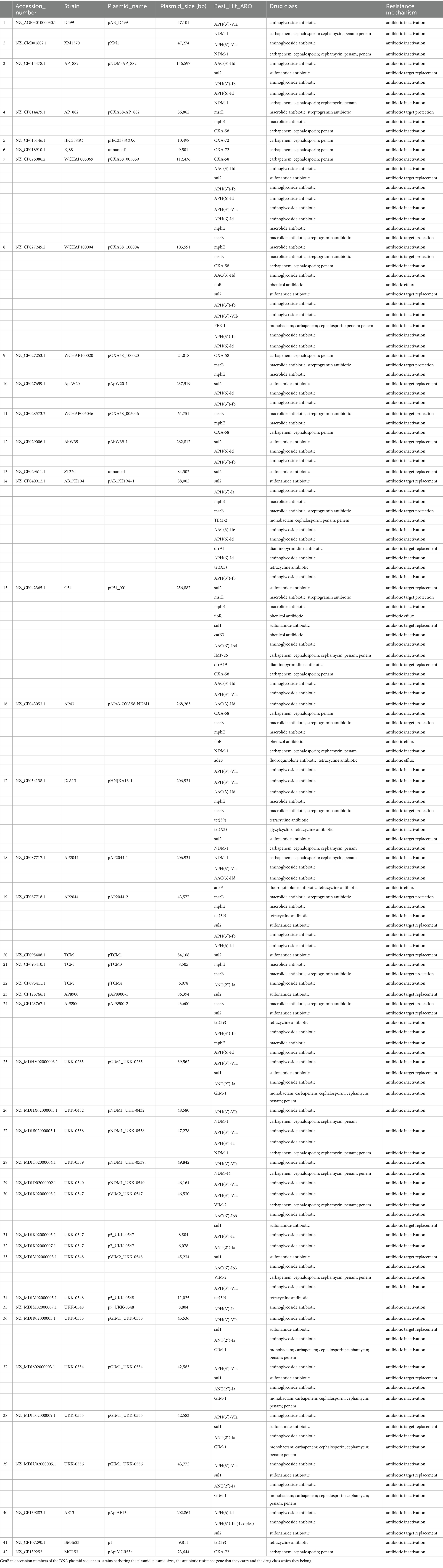
Table 4. A. pittii plasmids carrying antibiotic resistance genes.
Analysis of the distribution of the plasmids grouped into lineages (PLP_1 to PLP_17) revealed that members of the same lineage were scattered throughout the core genome phylogenetic tree (Figure 2) (Table 2). This was evidence that these genetic elements were subject to horizontal transfer events. However, none had a complete set of genes required for conjugation. The plasmid with the greatest number of genes involved in the synthesis of a Type IV secretion system (T4SS) required for conjugation was the plasmid pNDM1_UKK-0432, which contained 10 of the 12 core proteins that Gram-negative bacteria require for this process (Costa et al., 2021). Thirty-seven plasmids had MobA/MobL-type proteins, suggesting that they were potentially mobilizable.
Interestingly, a search of the gene annotations of the A. pittii genomes revealed that most did not contain genes encoding Type IV secretion systems. A few of these genes, except those mentioned above, contained, at most, 5 genes involved in the synthesis and function of this secretion apparatus.
Phylogenomic analysis at the local level
To examine the population dynamics more closely at the local level, we sequenced and analyzed the genomes of 29 clinical isolates obtained from Third-level hospitals: Two are in Mexico City, three are in different Mexican states (provinces), and one in Honduras. We also sequenced a strain isolated from a red-lored parrot and two A. pittii strains obtained from a Panamanian frogs, one of the genomes sequences was completed and closed (Cevallos et al., 2022; Bello-López et al., 2024). Depending on their position in a core-genome phylogenetic tree, we obtained the complete and closed genome sequences of 14 isolates representing different clades. The ANI values of 28 genome sequences, when contrasted with the A. pittii type strain, were greater than 95%, confirming that they belonged to A. pittii. However, four strains, MCR8900, AbaK, HUM6, and AN51, exhibited ANI values in a range from 92 to 95%. Among named Acinetobacter species, A. pittii had the closest ANI to these four strains, indicating that they did not belong to A. pittii as a species but were closely related to it. For this reason, we will call them A. pittii-like, provisionally (A. Nemec, personal communication).
To evaluate the relationships and phylogeography of the strains from Mexico and Honduras, we constructed a maximum likelihood phylogenetic tree of the core genome (Figure 3). We included 36 complete and closed A. pittii genome sequences in this tree. These genome sequences were obtained from strains isolated in 14 countries and from environmental, animal, and human (clinical) sources.
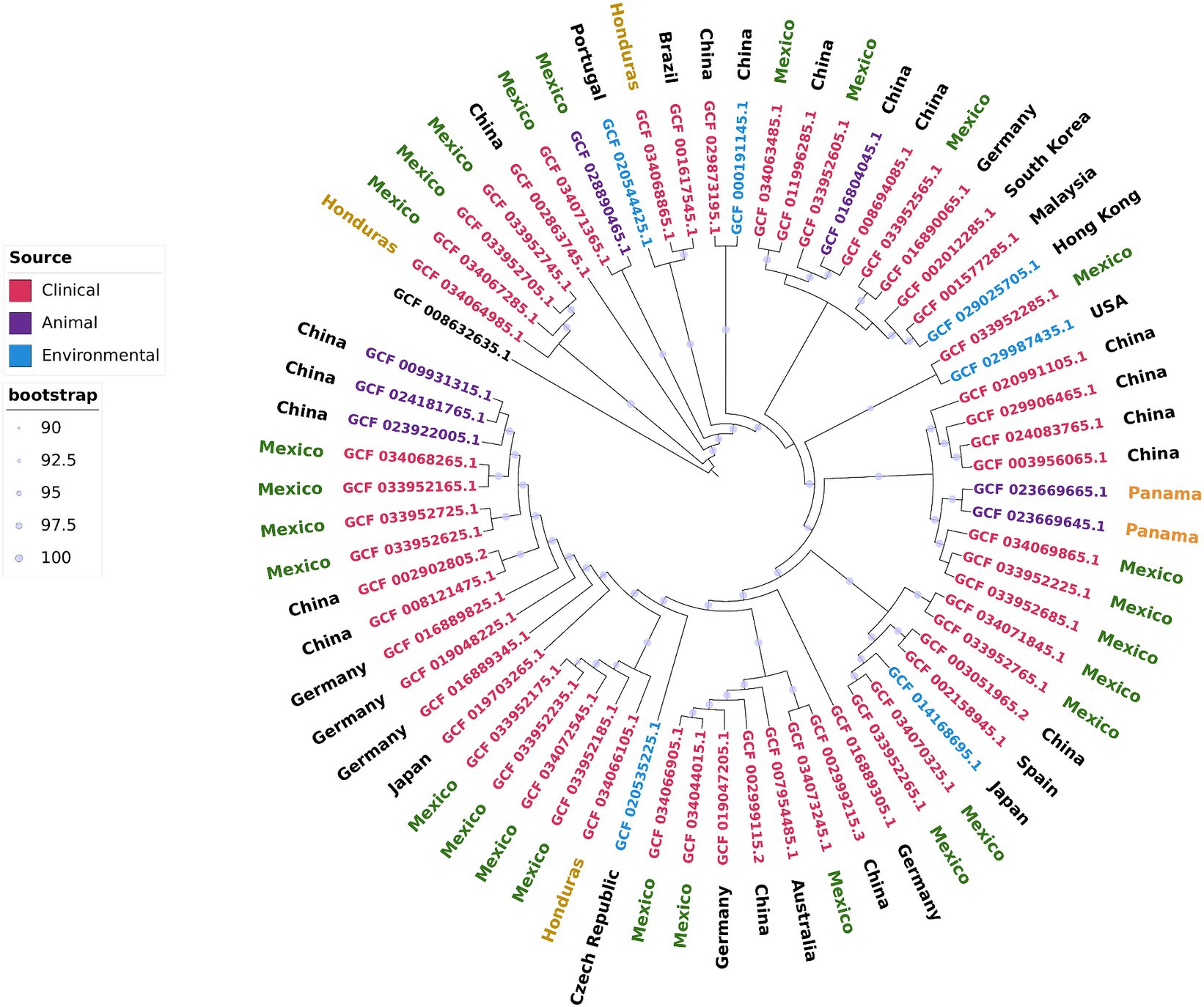
Figure 3. Core genome maximum likelihood phylogenetic tree including the complete and closed A. pittii genomes downloaded from NCBI and those sequenced in this work. A. baumannii (CP046654.1) was used as outgroup.
First, we observed that all the isolates from Mexico were dispersed throughout the phylogenetic tree. Moreover, isolates from the same hospital generally did not cluster in the same clade. The exceptions were strains AN37, AN38, and AN42, which were isolated from the same patient over almost 2 years. These observations indicated that the Mexican hospitals included in our study suffer several independent introductions and that several genetically different isolates are circulating within the country. The second significant observation was that Mexican isolates were closely related to isolates from the Far East (continental China, Taiwan, and Malaysia). These results confirmed only the observation that A. pittii isolates were subject to intercontinental dissemination, as stated above. The third observation was that the hospital-acquired isolates were closely linked to environmental isolates. For example, strain WP2-W18-ESBL-11 isolated from wastewater was closely related to the Mexican hospital-acquired isolates 564 U and 539 U, and in the same sense, strain PHEA-2 obtained from wastewater was also close to INC1094, a hospital-acquired isolate. The fourth observation was that some isolates from Mexico and other hospital-acquired isolates were linked to those obtained from animals, including some that were sick. For instance, strain 978-A_19 was isolated from a red-lored parrot with pneumonia and kept in captivity in Mexico City, and the Chinese strain Ap-W20 was obtained from a sick fish from a farm (Megalobrama amblycephala) (Li et al., 2017; Bello-López et al., 2024). The four strains close to A. pittii were grouped in the same clade, and they likely deserved to be considered new species; however, better characterization is needed before a name can be given to this possible new species.
An analysis of the antibiotic resistance genes of the strains sequenced in this work revealed that all the isolates possessed intrinsic blaOXA genes belonging to the blaOXA213 family. Thirteen also had a blaADC-25-like gene. One strain, HUM8, had three blaOXA genes; one was a member of the blaOXA213 family, and the other two were part of the blaOXA143 family. Strain MCR53 had a plasmid encoding the blaOXA72 gene. Finally, AE13 had six plasmid-encoded genes related to antibiotic resistance. Five were related to aminoglycoside resistance, and another, sul2, was related to sulfonamide resistance (Table 5).
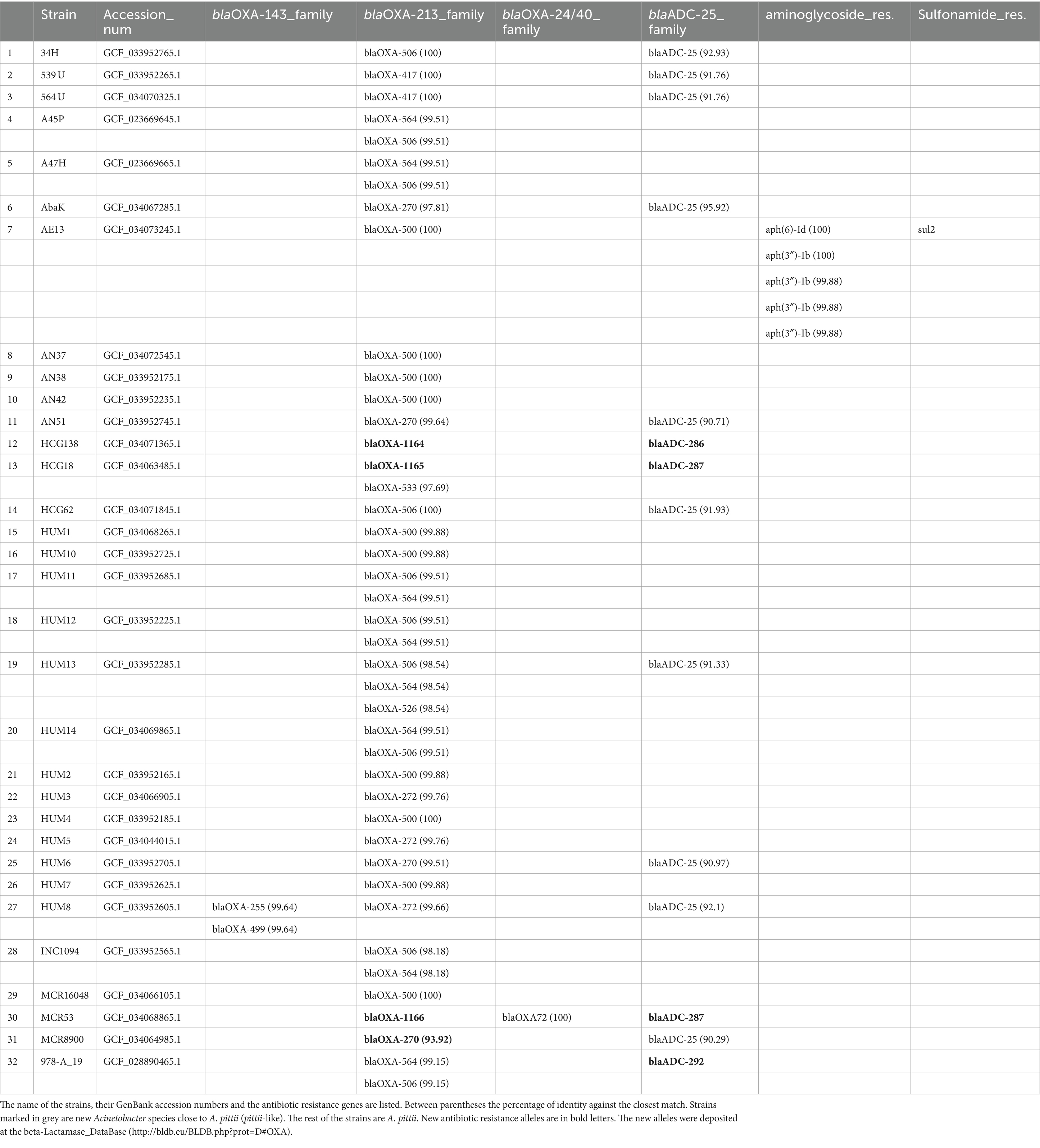
Table 5. Antibiotic resistance genes present on the strains sequenced in this work.
As a group, the genes of these strains, matched 92 virulence genes (from the VFDB database). All shared a common set of 39 virulence genes, but the number of virulence genes per strain varied widely. Noteworthy, the four “pittii-like” strains has less virulence related genes (Figure 4).

Figure 4. Virulence genes present in the strains sequenced in this work. On the left, virulence genes that are found in each of the strains sequenced in this work. On the right, their potential role in virulence. A colored box indicates the presence of the gene.
Discussion
When we started the analysis of A. pittii from a genomic perspective, we thought we would find a structured population where strains coexisting in a given site (geography), environment (i.e., water or soil), or specific hosts (like plants or animals) were going to share alleles and/or specific genes, which in some ways were going to twin them. However, in contrast to what was initially thought, this study demonstrated that A. pittii isolates obtained from both hospital environments and non-clinical settings frequently share the same clade in a phylogenetic tree constructed with the core genome. Additionally, we showed that A. pittii isolates from different geographical regions, even from different continents, may be closely related in the same clade in a core genome-based phylogenetic tree. We also showed that strains obtained in the same hospital might not be closely related, suggesting that hospitals may have suffered multiple independent introductions of genetically distinct groups of this pathogen. These observations indicated that A. pittii lacks evident migratory barriers. In this context, it is important to note that the constant immigration of new genes or alleles into a given population delays or prevents the population from structuring.
The possible migration vectors can be very varied. In the first place, as previously documented, traveling has proven to be an effective route for introducing multidrug-resistant bacteria from one country to another (Birgand et al., 2014; Higgins et al., 2021). Secondly, since it has been possible to isolate A. pittii from a wide range of animals including mammals, amphibians, birds, and fish, some of whom have migratory habits, they could also easily function as migration vectors (Li et al., 2017; Morakchi et al., 2017; Wang et al., 2020; Ying et al., 2020; Cevallos et al., 2022; Leelapsawas et al., 2022). Thirdly, A. pittii has also been isolated from aquatic environments like freshwater and seawater, so these means could facilitate the migration of members of this species from one site to another (Kong et al., 2021; Koskeroglu et al., 2023). These observations raise the possibility of genetic exchange between A. pittii strains from different geographical origins.
Plasmids are efficient vectors for disseminating virulence and antibiotic-resistance genes among bacterial strains within the same species and between different species and genera (San Millan, 2018; MacLean and San Millan, 2019). The capacity of these plasmids to transfer genes will depend mainly on whether they are conjugative or mobilizable, as well as their host range. Here, we observe that a considerable proportion of A. pittii plasmids (51.4%) are classified as orphans, suggesting a potentially wide diversity of plasmids within this species. On the other hand, the rest of the plasmids belong to a few lineages, most composed of only two or three members. This situation differs from what is observed in A. baumannii, where most plasmids belong to a very limited number of lineages (Salgado-Camargo et al., 2020). This disparity may be attributed to more frequent plasmid exchange between A. baumannii strains belonging to international clones already adapted to the hospital environment than those isolated from non-hospital environments.
A close examination of Rep proteins present in A. pittii plasmids suggests that their host range spectrum varies considerably. While some plasmids appear to have a marked specificity, others can replicate in a wide range of Acinetobacter species, including A. baumannii. This analysis also indicates that some of these plasmids may replicate in species outside the Acinetobacter genus (see Supplementary Table S4). However, a recent study revealed that the most common Rep proteins in A. baumannii belong to the Rep_3 superfamily. In contrast, the sample of A. pittii plasmids studied here exhibits Rep genes frequently associated with the RepM_Acin superfamily. These observations suggest that, although many A. pittii plasmids may theoretically replicate in A. baumannii, the exchange of plasmids between these two species remains limited. If exchanges were more frequent, a similar distribution of different Rep families between both species would be expected. Nevertheless, such an exchange is likely to occur at low frequency. By analyzing the DNA sequences of the 135 A. pittii plasmids using BLAST in NCBI, we found that some show significant similarities to plasmids from other Acinetobacter species, including A. baumannii.
Our analysis also revealed that 28.6% of A. pittii plasmids carry antibiotic-resistance genes, but no evidence of virulence-related genes was found. Furthermore, none of the plasmids encoded a complete set of genes for a type IV secretion system, indicating they are not self-transmissible. However, members of each of the plasmid lineages of A. pittii are dispersed throughout the phylogenetic tree constructed with the core genome (Figure 1), suggesting they are horizontally transferred with the help of other plasmids (mobilization), or using different mechanisms such as transformation, transduction, or through outer membrane vesicles (OMVs) (Rumbo et al., 2011; Chatterjee et al., 2017).
Acquiring plasmids with antibiotic-resistance genes will be crucial in the initial differentiation between environmental isolates and those leading to hospital-acquired infections. This differentiation will reduce the genetic diversity of nosocomial isolates and enhance their fitness in the hospital environment. Other characteristics that will be important in hospital adaptation and that some A. pittii strains already possess include forming biofilms and desiccation resistance. Therefore, we suggest that this organism is taking its first steps to become an emerging nosocomial pathogen and, thus, could be an excellent model for studying this process (Bravo et al., 2018; Chapartegui-González et al., 2022; Wunderlich et al., 2023).
As shown above, A. pittii strains of environmental, animal, or plant origin may be closely related to strains isolated from hospital patients. A first interpretation of this association is that non-hospital strains may be pathogenic if they find an immunocompromised right host and an invading route, as has already been observed for other emerging pathogens. The problem is that although the two strains are closely related by core genome, the number and gene content of their accessory genomes may differ, and these differences may include those genes encoding virulence factors. In fact, we found in the accessory genome genes encoding a phospholipase C (an exotoxin), metabolic factors that allow its persistence in the host, such as genes involved in the synthesis of Acinetobactin (Song and Kim, 2020), genes important in the adhesion to solid surfaces as the type IV pilus (Vo et al., 2023), in the formation of biofilms like the chaperone-usher pili (Csu) (Ahmad et al., 2023), or genes encoding the synthesis machinery of capsular polysaccharides. It should be noted that the ability of A. baumannii and A. pittii to adhere to solid surfaces and form biofilms is not only important in the infection process, but these characteristics could also explain their persistence in the hospital environment (Rajangam and Narasimhan, 2024).
Another way to interpret these observations is that all strains of A. pittii contain a platform of genes that could allow it to infect an immunocompromised patient if a route of infection exists, but for this to occur efficiently, an additional set of virulence genes must be acquired from other strains. Thus, for example, if a strain can infect an animal, it must acquire additional genes to adapt to humans. Given the differences in the number of genes between two given strains, an extensive system of horizontal gene transfer is needed, which cannot be sustained by conjugation alone. As such, transformation could be a more efficient way for this phenomenon to occur. Extensive experimental evidence is needed to support the hypotheses we put forward here.
Materials and methods
Genome sequence collection
In this work, we used two genome sequence sources: first, we downloaded all genome sequences available in the RefSeq and GenBank databases (NCBI) in May 2022. The quality of the genome sequences was evaluated regarding their completeness and degree of contamination with CheckM (Parks et al., 2015). Assemblies with less than 95% of completeness or possessing a contamination rate higher than 5% were eliminated. To evaluate if the downloaded genomes were correctly identified as A. pittii, we calculated the pairwise average nucleotide Average Nucleotide Identity (ANI) of all genome sequences against the type strain (ATCC 19004, accession number: GCF_000369045.1) with pyANI v0.2.9 (Pritchard et al., 2016). Genome sequences with an ANI value less than 95% against the type strain were eliminated from our collection, taking into consideration that the recommended species delineation threshold of ANI is 95–96%. After these filters, we kept a study set of 352 A. pittii genome sequences (Supplementary Table S1). Second, to increase the diversity of our genome collection, we obtained the complete genome sequences of 26 Mexican A. pittii isolates from six different hospitals located in different regions of the country (Table 1). Ten of these genomes were closed and circularized. Also, we also obtained the genome sequences of two Panamanian frogs, one of them was circularized and closed (Cevallos et al., 2022). Additionally, we did the same procedure with the genome sequences of three Honduran hospital-acquired A. pittii isolates.
Genome sequence determinations
Genomic DNA of all strains was extracted from overnight cultures grown overnight at 37°C and 250 rpm in 3 mL of Luria–Bertani broth, using the Genomic DNA purification kit (Thermo-Fisher) following the manufacturer’s instructions, with a small modification: samples were treated with RNAse (10 ng/mL) (Thermo-Fisher) at 37°C, 30 min prior final DNA precipitation. Draft genome sequences of the Mexican strains were obtained by sequencing short read libraries (2 × 150 bp) with the BGISEQ-2000 platform at the Beijing Genomics Institute, China. The Honduran strain’s genome sequences were obtained with an Illumina MiSeq platform with 2 × 300 bp paired-end reads at the Instituto Nacional de Medicina Genómica (INMEGEN, México). Trim Galore v0.6.4, developed by Babrahan Bioinformatics, was used to remove adapters and quality trimming of the sequencing reads.
Genome short-read assemblies were constructed using three algorithms: Velvet 1.2.10 (Zerbino and Birney, 2008), SPAdes 3.9.0 (Bankevich et al., 2012), and ABySS 2.0.1 (Jackman et al., 2017), and with several kmers. The best assembly obtained with each program was selected to obtain a merged and optimized assembly for each strain using Metassembler 1.5 (Wences and Schatz, 2015). Additionally, 14 A. pittii isolates were selected to close and circularize their genome sequences, using an Oxford-Nanopore device (PrometION). Oxford-Nanopore libraries were constructed and sequenced at the Beijing Genomic Institute (China). Adapter sequences were removed using Porechop 0.2.4 and base calling was performed with Guppy 5.1.13v using the high-accuracy base-calling mode. Hybrid assemblies were obtained utilizing Unicycler 0.4.8 (Wick et al., 2017).
Assembly statistics were calculated with Quast (v5.0.2) (Gurevich et al., 2013). The completeness and degree of contamination of these assemblies were evaluated with CheckM (Parks et al., 2015). Finally, genome sequences were annotated with the NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al., 2016). GenBank accession numbers are shown in Table 1.
Virulence and antibiotic-resistance genes identification
To identify genes involved in antibiotic resistance, we consulted the Comprehensive Antibiotic Resistance Database CARD 3.1.4 with RGI (Resistant Gene Identifier) and selected perfect or strict hits (Alcock et al., 2020). For plasmids, we also search the ResFinder database (v4.4.2) at https://www.genomicepidemiology.org (Florensa et al., 2022). The virulence genes were identified by consulting through BLASTp, the Virulence Factor Database (setA) (VFDB), asking for matches with an E value of 0 and a sequence identity of at least 80% (Liu et al., 2022). Only representative genes linked to experimentally validated Virulence Factors are included in the VFDB setA, or core dataset.
Phylogenetic tree construction
The first phylogenetic tree constructed includes all of the strains in our genome collection. The second tree considers the A. pittii Refseq complete genome sequences from NCBI and the strains from Mexico and Honduras that were sequenced by us. To do this, the strict core genome of the strains was obtained using Roary (v3.13.0) (Page et al., 2015), and with these data, maximum likelihood phylogenetic trees were constructed using IQ-TREE (v2.1.4-beta) with a TIM2 + F + R10 substitution mode. We used as an outgroup the genome sequence of A. baumannii ATCC19606 (CP046654.1). Also, we constructed a phylogenetic tree based on the presence/absence of genes of the A. pittii pangenome, using IQ-TREE (v2.1.4-beta) for binary data analysis. Trees were visualized and annotated with iTOL (Letunic and Bork, 2021). To evaluate Single Nucleotide Variants (SNVs), a consensus core-genome of 279 aligned clusters, without evidence of recombination, was obtained using GET_HOMOLOGUES and GET_PHYLOMARKERS (Contreras-Moreira and Vinuesa, 2013; Vinuesa et al., 2018). The SNVs present in the alignments were then evaluated with show-snps which is a utility MUMmer 3.0 software (Kurtz et al., 2004).
Plasmid sequence analysis
In this work, we analyzed the DNA sequences of 135 plasmids obtained from the complete and closed genomes of A. pittii (Table 2). To evaluate their relationships, we first calculated the average nucleotide sequences of all potential plasmid pairs. Then, we sorted the plasmids in two stages: in the first one, the plasmids were grouped based on their nucleotide sequence identities (≥ 95%) with a sequence coverage of 75% or higher. In the second stage, we incorporated new members into a specific the group if the new plasmid had a sequence identity of ≥95% and a sequence coverage of ≥75% with at least one member of the group.
The plasmid replication initiation (Rep) proteins were identified by analyzing the genome annotations provided by NCBI and classifying them according to their protein domains using the CD-search tool at NCBI. To evaluate the potential host range of the plasmids, we search for identical Rep proteins present in other plasmids in other species. We considered potential host species those that shared the same Rep in one of their plasmids.
To identify plasmids similar to those of A. pittii in other Acinetobacter species, we used as probes, in BLASTn searches at NCBI, the DNA sequences of the 135 A. pittii plasmids described in this work. In these searches, we considered those plasmids with a DNA sequence identity of at least 95% and coverage equal to or greater than 70%.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
EB-L: Data curation, Formal analysis, Investigation, Writing – review & editing. AE-M: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – review & editing. GG: Data curation, Formal analysis, Writing – review & editing. AC-C: Data curation, Formal analysis, Writing – review & editing. EG-G: Data curation, Formal analysis, Writing – review & editing. RH-C: Data curation, Formal analysis, Writing – review & editing. PZ: Data curation, Formal analysis, Writing – review & editing. RM-O: Data curation, Formal analysis, Writing – review & editing. PV: Data curation, Formal analysis, Writing – review & editing. JX-C: Data curation, Formal analysis, Writing – review & editing. MC: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Programa de Apoyo a Proyectos de Investigación e Innovación Tecnológica (IN204421), Universidad Nacional Autónoma de México. Postdoctoral researchers were supported by the El Consejo Nacional de Ciencia y Tecnología (1200/94/2020 and 1200/224/2021 to EB-L CVU_469558). EB-L is currently receiving a postdoctoral grant from DGAPA, Universidad Nacional Autónoma de México.
Acknowledgments
We would like to thank to Dr. Luis Lozano-Aguirre for their advice on bioinformatics. Also, we want to thank Ángeles Pérez-Oseguera for her technical support, and Dr. Esteban Gonzalez-Diaz, head of the Epidemiology Department of Hospital Civil de Guadalajara for his support and advice.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1412775/full#supplementary-material
References
Ahmad, I., Nadeem, A., Mushtaq, F., Zlatkov, N., Shahzad, M., Zavialov, A. V., et al. (2023). Csu pili dependent biofilm formation and virulence of Acinetobacter baumannii. NPJ Biofilms Microbiomes 9:101. doi: 10.1038/s41522-023-00465-6
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/nar/gkz935
Aloke, C., and Achilonu, I. (2023). Coping with the ESKAPE pathogens: evolving strategies, challenges and future prospects. Microb. Pathog. 175:105963. doi: 10.1016/j.micpath.2022.105963
Antimicrobial Resistance Collaborators (2022). Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet (London, England) 399, 629–655. doi: 10.1016/S0140-6736(21)02724-0
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bello-López, E., Escobedo-Muñoz, A. S., Hernández-Castro, R., and Cevallos, M. A. (2024). Genome sequence of an Acinetobacter pittii strain obtained from a red-lored parrot with pneumonia. Microbiol. Resour. Announc. 13:e0103823. doi: 10.1128/mra.01038-23
Birgand, G., Armand-Lefevre, L., Lepainteur, M., Lolom, I., Neulier, C., Reibel, F., et al. (2014). Introduction of highly resistant bacteria into a hospital via patients repatriated or recently hospitalized in a foreign country. Clin. Microbiol. Infect. 20, O887–O890. doi: 10.1111/1469-0691.12604
Boumbanda-Koyo, C. S., Mediannikov, O., Amanzougaghene, N., Oyegue-Liabagui, S. L., Imboumi-Limoukou, R. K., Raoult, D., et al. (2020). Molecular identification of head lice collected in Franceville (Gabon) and their associated bacteria. Parasit. Vectors 13:410. doi: 10.1186/s13071-020-04293-x
Brasiliense, D. M., Lima, K. V. B., Pérez-Chaparro, P. J., Mamizuka, E. M., de Oliveira Souza, C., Dutra, L. M. G., et al. (2019). Emergence of carbapenem-resistant Acinetobacter pittii carrying the blaOXA-72 gene in the Amazon region, Brazil. Diagn. Microbiol. Infect. Dis. 93, 82–84. doi: 10.1016/j.diagmicrobio.2018.07.017
Bravo, Z., Chapartegui-González, I., Lázaro-Díez, M., and Ramos-Vivas, J. (2018). Acinetobacter pittii biofilm formation on inanimate surfaces after long-term desiccation. J. Hosp. Infect. 98, 74–82. doi: 10.1016/j.jhin.2017.07.031
Carvalheira, A., Casquete, R., Silva, J., and Teixeira, P. (2017a). Prevalence and antimicrobial susceptibility of Acinetobacter spp. isolated from meat. Int. J. Food Microbiol. 243, 58–63. doi: 10.1016/j.ijfoodmicro.2016.12.001
Carvalheira, A., Silva, J., and Teixeira, P. (2017b). Lettuce and fruits as a source of multidrug resistant Acinetobacter spp. Food Microbiol. 64, 119–125. doi: 10.1016/j.fm.2016.12.005
Cevallos, M. A., Basanta, M. D., Bello-López, E., Escobedo-Muñoz, A. S., González-Serrano, F. M., Nemec, A., et al. (2022). Genomic characterization of antifungal Acinetobacter bacteria isolated from the skin of the frogs Agalychnis callidryas and Craugastor fitzingeri. FEMS Microbiol. Ecol. 98, 1–11. doi: 10.1093/femsec/fiac126
Chapartegui-González, I., Lázaro-Díez, M., and Ramos-Vivas, J. (2022). Genetic resistance determinants in clinical Acinetobacter pittii genomes. Antibiotics (Basel, Switzerland) 11:11. doi: 10.3390/antibiotics11050676
Chatterjee, S., Mondal, A., Mitra, S., and Basu, S. (2017). Acinetobacter baumannii transfers the blaNDM-1 gene via outer membrane vesicles. J. Antimicrob. Chemother. 72, 2201–2207. doi: 10.1093/jac/dkx131
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Cornejo-Juárez, P., Cevallos, M. A., Castro-Jaimes, S., Castillo-Ramírez, S., Velázquez-Acosta, C., Martínez-Oliva, D., et al. (2020). High mortality in an outbreak of multidrug resistant Acinetobacter baumannii infection introduced to an oncological hospital by a patient transferred from a general hospital. PLoS One 15:e0234684. doi: 10.1371/journal.pone.0234684
Costa, T. R. D., Harb, L., Khara, P., Zeng, L., Hu, B., and Christie, P. J. (2021). Type IV secretion systems: advances in structure, function, and activation. Mol. Microbiol. 115, 436–452. doi: 10.1111/mmi.14670
Falagas, M. E., and Rafailidis, P. I. (2007). Attributable mortality of Acinetobacter baumannii: no longer a controversial issue. Crit. Care 11:134. doi: 10.1186/cc5911
Florensa, A. F., Kaas, R. S., Clausen, P. T. L. C., Aytan-Aktug, D., and Aarestrup, F. M. (2022). ResFinder – an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genom. 8, 1–10. doi: 10.1099/mgen.0.000748
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hamidian, M., Maharjan, R. P., Farrugia, D. N., Delgado, N. N., Dinh, H., Short, F. L., et al. (2022). Genomic and phenotypic analyses of diverse non-clinical Acinetobacter baumannii strains reveals strain-specific virulence and resistance capacity. Microb. Genom. 8, 1–19. doi: 10.1099/mgen.0.000765
He, D., and Wan, W. (2021). Phosphate-solubilizing bacterium Acinetobacter pittii gp-1 affects rhizosphere bacterial community to alleviate soil phosphorus limitation for growth of soybean (Glycine max). Front. Microbiol. 12:737116. doi: 10.3389/fmicb.2021.737116
Higgins, P. G., Hagen, R. M., Kreikemeyer, B., Warnke, P., Podbielski, A., Frickmann, H., et al. (2021). Molecular epidemiology of Carbapenem-resistant Acinetobacter baumannii isolates from northern Africa and the Middle East. Antibiot. (Basel, Switzerland) :10. doi: 10.3390/antibiotics10030291
Horrevorts, A., Bergman, K., Kollée, L., Breuker, I., Tjernberg, I., and Dijkshoorn, L. (1995). Clinical and epidemiological investigations of Acinetobacter genomospecies 3 in a neonatal intensive care unit. J. Clin. Microbiol. 33, 1567–1572. doi: 10.1128/jcm.33.6.1567-1572.1995
Hujer, K. M., Hamza, N. S., Hujer, A. M., Perez, F., Helfand, M. S., Bethel, C. R., et al. (2005). Identification of a new allelic variant of the Acinetobacter baumannii cephalosporinase, ADC-7 beta-lactamase: defining a unique family of class C enzymes. Antimicrob. Agents Chemother. 49, 2941–2948. doi: 10.1128/AAC.49.7.2941-2948.2005
Idzenga, D., Schouten, M. A., and van Zanten, A. R. H. (2006). Outbreak of Acinetobacter genomic species 3 in a Dutch intensive care unit. J. Hosp. Infect. 63, 485–487. doi: 10.1016/j.jhin.2006.03.014
Izotova, A. O., Petrova, K. O., Korzhenkov, A. A., Bavtushnyi, A. A., Sidoruk, K. V., Patrusheva, E. V., et al. (2021). Draft De novo genome assembly of Acinetobacter pittii strain VKPM B-3780, a prospective multifunctional bioremediation agent. Microbiol. Resour. Announc. 10:e0055421. doi: 10.1128/MRA.00554-21
Jackman, S. D., Vandervalk, B. P., Mohamadi, H., Chu, J., Yeo, S., Hammond, S. A., et al. (2017). ABySS 2.0: resource-efficient assembly of large genomes using a bloom filter. Genome Res. 27, 768–777. doi: 10.1101/gr.214346.116
Jiang, J., Liang, D., and Hu, Y. (2022). Solid slow-release carbon sources improve the simultaneous nitrification and denitrification processes in low carbon resource wastewater. Bioresour. Technol. 365:128148. doi: 10.1016/j.biortech.2022.128148
Kiyasu, Y., Hitomi, S., Funayama, Y., Saito, K., and Ishikawa, H. (2020). Characteristics of invasive Acinetobacter infection: a multicenter investigation with molecular identification of causative organisms. J. Infect. Chemother. 26, 475–482. doi: 10.1016/j.jiac.2019.12.010
Kong, W., Zhao, C., Gao, X., Wang, L., Tian, Q., Liu, Y., et al. (2021). Characterization and transcriptome analysis of a long-chain n-alkane-degrading strain Acinetobacter pittii SW-1. Int. J. Environ. Res. Public Health. 18, 1–15. doi: 10.3390/ijerph18126365
Koskeroglu, K., Barel, M., Hizlisoy, H., and Yildirim, Y. (2023). Biofilm formation and antibiotic resistance profiles of water-borne pathogens. Res. Microbiol. 174:104056. doi: 10.1016/j.resmic.2023.104056
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Leelapsawas, C., Yindee, J., Nittayasut, N., Chueahiran, S., Boonkham, P., Suanpairintr, N., et al. (2022). Emergence and multi-lineages of carbapenemase-producing Acinetobacter baumannii-calcoaceticus complex from canine and feline origins. J. Vet. Med. Sci. 84, 1377–1384. doi: 10.1292/jvms.22-0276
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, J., Cao, J., Wang, X., Liu, N., Wang, W., and Luo, Y. (2017). Acinetobacter pittii, an emerging new multi-drug resistant fish pathogen isolated from diseased blunt snout bream (Megalobrama amblycephala Yih) in China. Appl. Microbiol. Biotechnol. 101, 6459–6471. doi: 10.1007/s00253-017-8392-4
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi: 10.1093/nar/gkab1107
Lu, S., Wang, J., Chitsaz, F., Derbyshire, M. K., Geer, R. C., Gonzales, N. R., et al. (2020). CDD/SPARCLE: the conserved domain database in 2020. Nucleic Acids Res. 48, D265–D268. doi: 10.1093/nar/gkz991
Lucidi, M., Visaggio, D., Migliaccio, A., Capecchi, G., Visca, P., Imperi, F., et al. (2024). Pathogenicity and virulence of Acinetobacter baumannii: factors contributing to the fitness in healthcare settings and the infected host. Virulence 15:2289769. doi: 10.1080/21505594.2023.2289769
MacLean, R. C., and San Millan, A. (2019). The evolution of antibiotic resistance. Science 365, 1082–1083. doi: 10.1126/science.aax3879
Mateo-Estrada, V., Tyrrell, C., Evans, B. A., Aguilar-Vera, A., Drissner, D., Castillo-Ramirez, S., et al. (2023). Acinetobacter baumannii from grass: novel but non-resistant clones. Microb. Genom. 9, 1–7. doi: 10.1099/mgen.0.001054
Mateo-Estrada, V., Vali, L., Hamouda, A., Evans, B. A., and Castillo-Ramírez, S. (2022). Acinetobacter baumannii sampled from cattle and pigs represent novel clones. Microbiol. Spectr. 10:e0128922. doi: 10.1128/spectrum.01289-22
Mohd Sazlly Lim, S., Zainal Abidin, A., Liew, S. M., Roberts, J. A., and Sime, F. B. (2019). The global prevalence of multidrug-resistance among Acinetobacter baumannii causing hospital-acquired and ventilator-associated pneumonia and its associated mortality: a systematic review and meta-analysis. J. Infect. 79, 593–600. doi: 10.1016/j.jinf.2019.09.012
Morakchi, H., Loucif, L., Gacemi-Kirane, D., and Rolain, J.-M. (2017). Molecular characterisation of carbapenemases in urban pigeon droppings in France and Algeria. J. Glob. Antimicrob. Resist. 9, 103–110. doi: 10.1016/j.jgar.2017.02.010
Nemec, A. (2022). “Acinetobacter” in Bergey’s manual of systematics of Archaea and Bacteria. ed. W. B. Whitman (Wiley), 1–78. doi: 10.1002/9781118960608.gbm01203.pub2
Noel, H. R., Petrey, J. R., and Palmer, L. D. (2022). Mobile genetic elements in Acinetobacter antibiotic-resistance acquisition and dissemination. Ann. N. Y. Acad. Sci. 1518, 166–182. doi: 10.1111/nyas.14918
O’Neill, J. (2016). Tackling drug-resistant infections globally: final report and recommendations. doi:https://doi.org/APO-63983.
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pailhoriès, H., Tiry, C., Eveillard, M., and Kempf, M. (2018). Acinetobacter pittii isolated more frequently than Acinetobacter baumannii in blood cultures: the experience of a French hospital. J. Hosp. Infect. 99, 360–363. doi: 10.1016/j.jhin.2018.03.019
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Peleg, A. Y., Seifert, H., and Paterson, D. L. (2008). Acinetobacter baumannii: emergence of a successful pathogen. Clin. Microbiol. Rev. 21, 538–582. doi: 10.1128/CMR.00058-07
Périchon, B., Goussard, S., Walewski, V., Krizova, L., Cerqueira, G., Murphy, C., et al. (2014). Identification of 50 class D β-lactamases and 65 Acinetobacter-derived cephalosporinases in Acinetobacter spp. Antimicrob. Agents Chemother. 58, 936–949. doi: 10.1128/AAC.01261-13
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Qian, C., Ma, Z., Feng, L., Guo, W., Han, Y., Zhang, Y., et al. (2023). Emergence of tet(X2) in Acinetobacter pittii confers clinical resistance to tigecycline. J. Antimicrob. Chemother. 78, 1543–1546. doi: 10.1093/jac/dkad133
Rajangam, S. L., and Narasimhan, M. K. (2024). Current treatment strategies for targeting virulence factors and biofilm formation in Acinetobacter baumannii. Future Microbiol., 1–21. doi: 10.2217/fmb-2023-0263
Rumbo, C., Fernández-Moreira, E., Merino, M., Poza, M., Mendez, J. A., Soares, N. C., et al. (2011). Horizontal transfer of the OXA-24 carbapenemase gene via outer membrane vesicles: a new mechanism of dissemination of carbapenem resistance genes in Acinetobacter baumannii. Antimicrob. Agents Chemother. 55, 3084–3090. doi: 10.1128/AAC.00929-10
Salgado-Camargo, A. D., Castro-Jaimes, S., Gutierrez-Rios, R.-M., Lozano, L. F., Altamirano-Pacheco, L., Silva-Sanchez, J., et al. (2020). Structure and evolution of Acinetobacter baumannii plasmids. Front. Microbiol. 11:1283. doi: 10.3389/fmicb.2020.01283
San Millan, A. (2018). Evolution of plasmid-mediated antibiotic resistance in the clinical context. Trends Microbiol. 26, 978–985. doi: 10.1016/j.tim.2018.06.007
Schleicher, X., Higgins, P. G., Wisplinghoff, H., Körber-Irrgang, B., Kresken, M., and Seifert, H. (2013). Molecular epidemiology of Acinetobacter baumannii and Acinetobacter nosocomialis in Germany over a 5-year period (2005-2009). Clin. Microbiol. Infect. 19, 737–742. doi: 10.1111/1469-0691.12026
Shelenkov, A., Akimkin, V., and Mikhaylova, Y. (2023). International clones of high risk of Acinetobacter Baumannii-definitions, history, properties and perspectives. Microorganisms 11, 1–22. doi: 10.3390/microorganisms11082115
Song, W. Y., and Kim, H. J. (2020). Current biochemical understanding regarding the metabolism of acinetobactin, the major siderophore of the human pathogen Acinetobacter baumannii, and outlook for discovery of novel anti-infectious agents based thereon. Nat. Prod. Rep. 37, 477–487. doi: 10.1039/c9np00046a
Tacconelli, E., Carrara, E., Savoldi, A., Harbarth, S., Mendelson, M., Monnet, D. L., et al. (2018). Discovery, research, and development of new antibiotics: the WHO priority list of antibiotic-resistant bacteria and tuberculosis. Lancet Infect. Dis. 18, 318–327. doi: 10.1016/S1473-3099(17)30753-3
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tian, C., Xing, M., Fu, L., Zhao, Y., Fan, X., and Wang, S. (2022). Emergence of uncommon KL38-OCL6-ST220 carbapenem-resistant Acinetobacter pittii strain, co-producing chromosomal NDM-1 and OXA-820 carbapenemases. Front. Cell. Infect. Microbiol. 12:943735. doi: 10.3389/fcimb.2022.943735
Tobin, L. A., Cain, A. K., Djordjevic, S. P., and Hamidian, M. (2024). Transposons carrying the aacC2e aminoglycoside and blaTEM Beta-lactam resistance genes in Acinetobacter. Microb. Drug Resist. 1–17. doi: 10.1089/mdr.2023.0299
Verma, A., Carney, K., Taylor, M., Amsler, K., Morgan, J., Gruszynski, K., et al. (2021). Occurrence of potentially zoonotic and cephalosporin resistant enteric bacteria among shelter dogs in the central and south-central Appalachia. BMC Vet. Res. 17:313. doi: 10.1186/s12917-021-03025-2
Vinuesa, P., Ochoa-Sánchez, L. E., and Contreras-Moreira, B. (2018). GET_PHYLOMARKERS, a software package to select optimal orthologous clusters for Phylogenomics and inferring Pan-genome phylogenies, used for a critical Geno-taxonomic revision of the genus Stenotrophomonas. Front. Microbiol. 9:771. doi: 10.3389/fmicb.2018.00771
Vo, N., Sidner, B. S., Yu, Y., and Piepenbrink, K. H. (2023). Type IV pilus-mediated inhibition of Acinetobacter baumannii biofilm formation by phenothiazine compounds. Microbiol. Spectr. 11:e0102323. doi: 10.1128/spectrum.01023-23
Wang, X., Li, J., Cao, X., Wang, W., and Luo, Y. (2020). Isolation, identification and characterisation of an emerging fish pathogen, Acinetobacter pittii, from diseased loach (Misgurnus anguillicaudatus) in China. Antonie Van Leeuwenhoek 113, 21–32. doi: 10.1007/s10482-019-01312-5
Wences, A. H., and Schatz, M. C. (2015). Metassembler: merging and optimizing de novo genome assemblies. Genome Biol. 16:207. doi: 10.1186/s13059-015-0764-4
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wunderlich, A., Xanthopoulou, K., Wille, J., Wohlfarth, E., Gerson, S., Kaase, M., et al. (2023). Carbapenem resistance in Acinetobacter pittii isolates mediated by metallo-β-lactamases. J. Antimicrob. Chemother. 78, 488–496. doi: 10.1093/jac/dkac418
Yang, L., Dong, N., Xu, C., Ye, L., and Chen, S. (2021). Emergence of ST63 Pandrug-resistant Acinetobacter pittii isolated from an AECOPD patient in China. Front. Cell. Infect. Microbiol. 11:739211. doi: 10.3389/fcimb.2021.739211
Ying, C.-P., Jiang, M., You, L., Tan, J.-H., Yang, Y.-P., Wang, Y.-P., et al. (2020). Variations and potential factors of gut prokaryotic microbiome during spawning migration in Coilia nasus. Curr. Microbiol. 77, 2802–2812. doi: 10.1007/s00284-020-02088-y
Zarrilli, R., Giannouli, M., Tomasone, F., Triassi, M., and Tsakris, A. (2009). Carbapenem resistance in Acinetobacter baumannii: the molecular epidemic features of an emerging problem in health care facilities. J. Infect. Dev. Ctries. 3, 335–341. doi: 10.3855/jidc.240
Keywords: ESKAPE, infection, antibiotic-resistance genes, virulence, evolution
Citation: Bello-López E, Escobedo-Muñoz AS, Guerrero G, Cruz-Córdova A, Garza-González E, Hernández-Castro R, Zarain PL, Morfín-Otero R, Volkow P, Xicohtencatl-Cortes J and Cevallos MA (2024) Acinetobacter pittii: the emergence of a hospital-acquired pathogen analyzed from the genomic perspective. Front. Microbiol. 15:1412775. doi: 10.3389/fmicb.2024.1412775
Edited by:
Axel Cloeckaert, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
Milena Milakovic Obradovic, Ludwig Maximilian University of Munich, GermanyJose Ramos-Vivas, Universidad Europea del Atlántico, Spain
Itziar Chapartegui-González, Karolinska Institutet (KI), Sweden
Piyali Chatterjee, United States Department of Veterans Affairs, United States
Lucía Gallego, University of the Basque Country, Spain
Copyright © 2024 Bello-López, Escobedo-Muñoz, Guerrero, Cruz-Córdova, Garza-González, Hernández-Castro, Zarain, Morfín-Otero, Volkow, Xicohtencatl-Cortes and Cevallos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Miguel A. Cevallos, bWFjQGNjZy51bmFtLm14