 Jian-Cai Mao
Jian-Cai Mao Miao Yan†
Miao Yan† Jun-Hua Li
Jun-Hua Li Hao-Jie Wang
Hao-Jie Wang- Hami Melon Research Center, Xinjiang Academy of Agricultural Sciences, Urumqi, China
Bidirectional trans-kingdom RNA silencing, a pivotal factor in plant-pathogen interactions, remains less explored in plant host-parasite dynamics. Here, using small RNA sequencing in melon root systems, we investigated microRNA (miRNA) expression variation in resistant and susceptible cultivars pre-and post-infection by the parasitic plant, broomrape. This approach revealed 979 known miRNAs and 110 novel miRNAs across 110 families. When comparing susceptible (F0) and resistant (R0) melon lines with broomrape infection (F25 and R25), 39 significantly differentially expressed miRNAs were observed in F25 vs. F0, 35 in R25 vs. R0, and 5 in R25 vs. F25. Notably, two miRNAs consistently exhibited differential expression across all comparisons, targeting genes linked to plant disease resistance. This suggests their pivotal role in melon’s defense against broomrape. The target genes of these miRNAs were confirmed via degradome sequencing and validated by qRT-PCR, ensuring reliable sequencing outcomes. GO and KEGG analyses shed light on the molecular functions and pathways of these differential miRNAs. Furthermore, our study unveiled four trans-kingdom miRNAs, forming a foundation for exploring melon’s resistance to broomrape.
1 Introduction
Broomrape (Phelipanche aegyptiaca Pers) is a parasitic plant known for inflicting significant economic losses in melon production. This holoparasite attaches itself to the root of the melon plants and establishes a connection with the vascular tissue to absorb nutrients. This parasitic interaction profoundly impairs host development and substantially diminishes melon yields (Bao et al., 2017). The attachment between broomrape and its host plant is facilitated through specialized structures called haustorium, which enable bidirectional movement of photosynthates and other essential macromolecules between the host and parasite, including microRNAs (miRNAs) (Bao et al., 2017). miRNAs, a class of endogenous single-stranded non-coding RNAs spanning 20–40 nt, are widespread in plants (Nizampatnam et al., 2015; Song et al., 2019; Cui et al., 2020). They play an important regulatory role in plant growth, development, hormone signal transduction, and stress response. Remarkably, miRNAs can even exert regulatory influence over gene expression in different species, highlighting their capacity for cross-species regulation (Guo et al., 2017, 2021). In addition, some miRNAs exhibit the ability to traverse between host and parasite plants, resulting in the reverse silencing of target genes, leading to the phenomenon of “trans-kingdom RNA silencing.” This intriguing process involves the transfer of miRNAs and subsequent reverse silencing of target genes, thus influencing gene expression across the plant-parasite interface (Zhang et al., 2016a,b).
The phenomenon of trans-kingdom RNA silencing was first discovered in the interaction between plants and fungi. Notably, Botrytis cinerea demonstrated the ability to transfer a repertoire of small RNAs into plant cells, thereby triggering the silencing of immune response genes across diverse hosts, including Arabidopsis and tomato (Weiberg et al., 2013). Subsequent investigations unveiled the mechanism by which pathogenic small RNAs, interfacing with Arabidopsis AGO1 and harnessing the host RNAi machinery, silence pivotal host genes such as MAPK kinases related to cell wall integrity and genes involved in reactive oxygen species accumulation (Weiberg et al., 2013). This strategic employment of small RNAs enabled the pathogen to seize control of host immune responses, thereby facilitating successful invasion (Zhang et al., 2016a,b, 2022). The trans-kingdom miRNAs represent an innovative class of effector molecules adept at quelling host immune response. Notably, the miRNA (Bc-siR37) from Bostalospora cinerea was significantly enriched in the host Arabidopsis during the early stage of invasion. Bc-siR37 targeted a minimum of 15 genes, including WRKY transcription factors and cell wall modifying enzymes in Arabidopsis (Wang B. et al., 2017). Trans-kingdom miRNAs regulate host target genes to help pathogens successfully invade and colonize. Similarly, in the context of wheat stripe rust, Pst-mi1R1 was identified as a key pathogenic factor of the pathogen, which perturbs the host by targeting the gene SM638 in the genome of the host wheat. Partial silencing of SM638 in wheat significantly reduced the resistance to wheat stripe rust in resistant varieties (Wang M. et al., 2017). Recent results show have extended the scope of trans-kingdom RNA silencing to parasitic plants, exemplified by Cuscuta campestris. Endogenous miRNAs from this parasitic plant can be transported to the host Arabidopsis thaliana, guiding and modulating the parasitization process by regulating the expression of different target genes in the host (Hudzik et al., 2023). This study underscore the intricate involvement of miRNAs in the host immune pathway, shedding light on a previously uncharted aspect of parasitic interactions.
The trans-kingdom RNAi constitutes a pivotal mechanism in bolstering plant defenses against pathogens, exhibiting bidirectional transmission capabilities between host and pathogen. Trans-kingdom RNAi vectors were constructed using Host-Induced Gene Silencing (HIGS) to silence pathogen virulence-associated target genes, thereby allowing the host to acquire resistance against the pathogen. Guo et al. (2017, 2021) used HIGS technology to produce genetically stable transgenic cotton. Herein, the VdH1 gene of Verticillium dahlia, pivotal for nuclear integrity, was effectively silenced, causing a reduction in virulence due to the loss of nucleus formation in the pathogen. This study is the first to demonstrate the potential of HIGS technology in plant disease resistance research (Zhang et al., 2016a,b). In a similar vein, Govindarajulu et al. (2015) harnessed HIGS technology to produce transgenic lettuce resistant to Bremia lactucae. By attenuating a key lettuce gene through RNAi, resistance against the pathogen was realized (Zhang et al., 2016a,b). Likewise, Chen et al. (2016) yielded transgenic wheat with enhanced resistance against Fusarium graminearum through HIGS-mediated silencing of the FcGls1 gene, impairing mycelial cell wall integrity. These remarkable instances offer valuable insights for crop breeding via inter-species RNAi applications.
Evidently, trans-kingdom miRNAs exhibit a propensity for horizontal transfer across several species, such as from humans to malignant malaria-causing insects (Plasmodium falciparum) (LaMonte et al., 2012); from bacteria to insects (LaMonte et al., 2012); from parasitic plants to host plants (Shahid et al., 2018; Hudzik et al., 2023); from plant to insect (Ibrahim et al., 2011); from fungal pathogens to plants (Weiberg et al., 2013), from plant-to-fungal pathogens (Cai et al., 2018); and even reciprocal transfers between plants and insects (Wang et al., 2016). However, a notable gap exists in the exploration of transferable miRNAs in melon plants resistante to the parasitic broomrape. This research aims to unravel the intriguing implications of this novel defense strategy within the interaction between melon and broomrape. By delving into the dynamics of trans-kingdom RNAi, it aspires to shed light on the broader landscape of cross-border RNAi application in plant disease management. Furthermore, this research holds the potential to guide the evolution of pathogenic bacteria as miRNA targets, thereby advancing innovative strategies for combatting broomrape in melon cultivation.
2 Materials and methods
2.1 Plant materials and manipulations
The resistant line K1-6 and sensitive line K2-7 melon cultivars are preserved in Xinjiang Academy of Agricultural Sciences (Figure 1). Broomrape seeds, identified as Phelipanche aegyptiaca Pers, were collected in the Kashgar region of Xinjiang. The pots (Diameter: 16 cm, Height: 12.5 cm) were filled with a vermiculite-sand mixture (1:1) and infused with 0.02 g of broomrape seeds. Each pot was planted with 1–2 melon seeds, and this setup was repeated 10 times for each treatment. The pots were placed randomly in a greenhouse (Temperature 28°C, light/darkness = 16: 8), receiving regular watering. Root samples from the parasitized melon were collected at 0 days (non-infectious treatment) and 25 days after infection for the resistant line K1-6 (R0 and R25) and sensitive line K2-7 (F0 and F25). Parasitic broomrape samples were taken for degradome sequencing to find target genes. Each sample was repeated 3 times, rapidly frozen in liquid nitrogen and stored at-80°C. The blank control was induced by strigolactone in Petri dishes and sampled after broomrape germination.

Figure 1. Resistant and susceptible lines were inoculated for 25 days. K1-6 is resistant line and K2-7 is susceptible line, the red box indicates the parasitic broomrape.
2.2 Construction of small RNA library
The broomrape samples were sent to Genepioneer Biotechnologies for small RNA sequencing. Total RNA was extracted using the RNA Easy Fast (DP452), and small RNA libraries were prepared using the TruSeq Small RNA Sample Prep Kits (Illumina, San Diego, United States) according to standard procedures. After library preparation, the constructed libraries were sequenced using Illumina Hiseq 2000/2500 with a sequence reading length of 1 × 50 bp.
2.3 miRNA sequencing and data processing
The initial sequencing data was aligned with the reference genome, with subsequent filtration to compare with the broomrape genomes. The remaining data were analyzed using the miRNA data analysis software ACGT101-miR (LC Sciences, Houston, Texas, United States). The analysis process of the software is as follows: elimination of 3′ junction and irrelevant sequences to get clean data; Length screening, filtering out data with baseline length at 18–25 nt in the clean data; assessment against various RNA databases (mRNA, RFam, Repbase) to exclude non-miRNA data, retaining valid sequences for further analysis.
2.4 miRNA identification
miRNA identification includes both known and novel miRNAs. We compared effective data with plant miRNA precursors and maturing sequences in the miRbase database. This comparison involved BLAST analysis of unique sequences spanning 18–25 nucleotides against a specific species precursor in miRBase (miRBase 21.0; http://www.mirbase.org/), in order to identify known miRNA as well as novel miRNA.
2.5 Degradome sequencing identifies cross-border miRNA target genes
2.5.1 Destruction group building
The samples were sent to Genepioneer Biotechnologies for degradome sequencing. The proposed library steps are as follows: miRNA capture by magnetic beads, adaptor ligation; mixed reverse transcription of Biotinylated Random Primers and mRNA; PCR amplification, after completing the whole library preparation, the constructed library was sequenced with Illumina Hiseq 2000/2500, and the sequencing read length was 1 × 50 bp at the single end.
2.6 Degradome data analysis and target gene prediction
The raw data obtained from sequencing were processed through a series of data processing to obtain comparable sequenced pairs for subsequent analysis. The comparable pair sequences were compared with the melon cDNA database sequences to generate the degradome density files. The target gene mRNA sequences paired with the small RNA sequences of sequenced species were predicted by the Target finder target gene prediction software. The Allen Score was used to evaluate the degree of base complementarity between miRNA and target gene binding sites, indicating the binding site pairing penalty (where lower values indicate better pairing).
2.7 Target gene prediction
The target genes corresponding to melon miRNAs predicted by Target finder software based on the principle of base complementary pairing. The mRNAs in the generated degradome density files were combined and analyzed to identify the target genes of the miRNAs. Functional annotation of the target genes was performed by GO enrichment analysis and KEGG enrichment analysis.
2.8 qRT-PCR
Total RNA was digested by DNase I and cDNA were synthesized using Rrans Script Green miRNA Two-Step qRT-PCR SuperMix. Differential miRNAs were randomly chosen for validation, and primers were designed accordingly. Each sample underwent triplicate repetitions to determine relative expression of the target miRNA, H2O was used as a negative control and CmADP was used as a reference gene. The primers used in RT-PCR are listed in Supplementary Table S1.
3 Results
3.1 sRNA sequencing in melon varieties resistant or sensitive to broomrape
Here, using small RNA sequencing in melon root systems, we investigated miRNA expression variation in resistant and susceptible cultivars pre-and post-infection by the parasitic plant, broomrape (Figure 1). The results showed that a total of 12 melon sRNA libraries were prepared for sequencing on the Illumina SE50 platform. The integrity of the sequencing data was notably robust, with each of the four samples yielding a range of clean data reads (ReadSum) spanning from 11,023,148 to 11,977,704, and a clean date base (BaseSum) ranging from 562,180,582 to 610,862,904. These data were characterized by outstanding quality, as evidenced by Q20 > 98% and Q30 > 96% for each sample and GC content with a consistent range from 52.99 to 54.27% (Table 1). The above results indicate that the quality of the raw data obtained from sequencing meets the requirements for subsequent analysis.
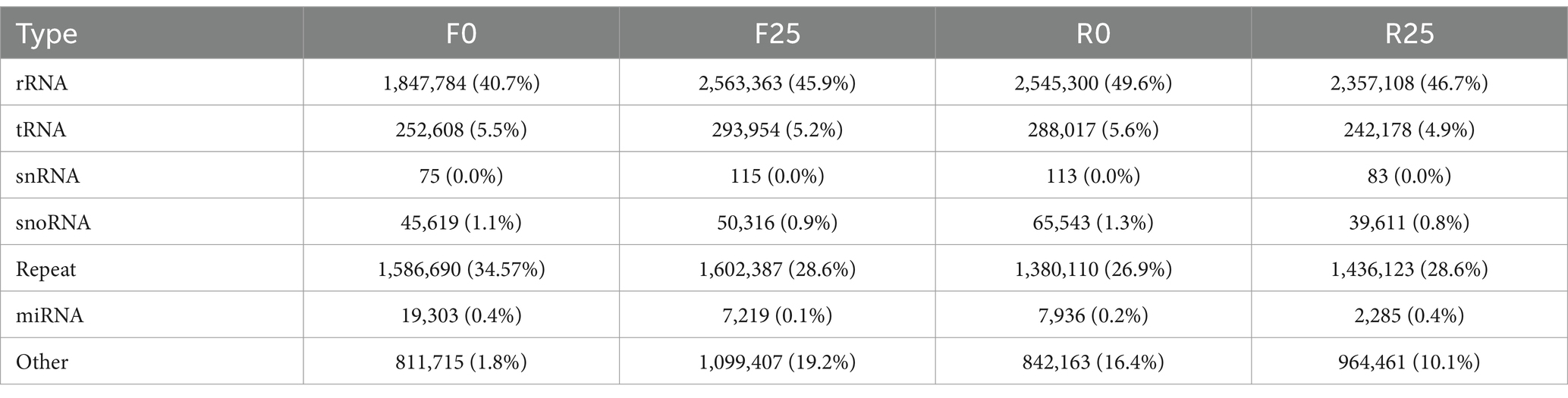
Table 1. sRNA classification annotation statistics.
The length distribution statistics were performed on the clean reads. The distribution of fragment lengths serves as an illuminating marker to identify small RNA species. miRNAs were usually concentrated at 21–22 nt, as shown in Figure 2A. The small RNA lengths of the samples were significantly concentrated at 21 nt. In addition, an insightful examination of the first point base preference in clean reads was conducted. A robust preference for the base pair U at the initial position was pronounced in the context of 21 nt fragments (Figure 2B). The results with the annotation of small RNAs are shown in Table 1.
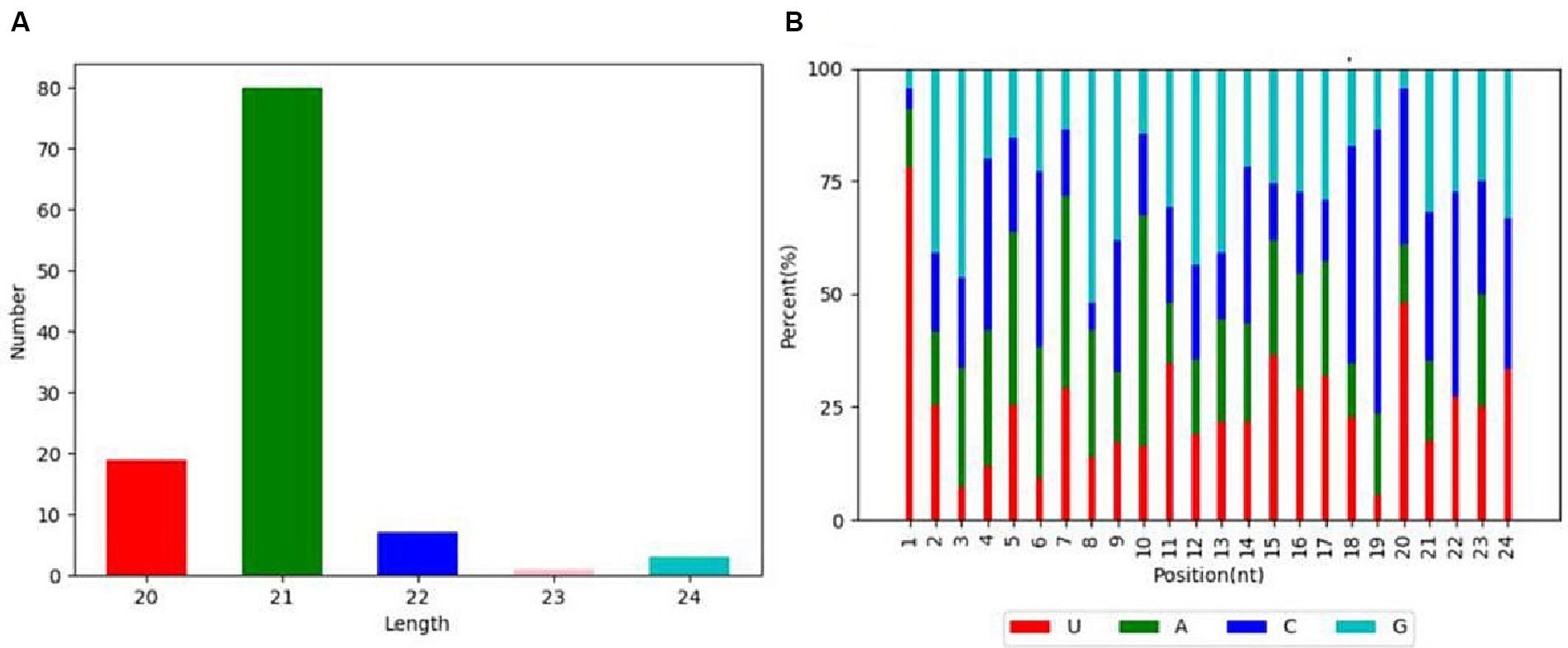
Figure 2. miRNA length distribution and nucleotide bias at each position. (A) The length distribution chart of identified miRNA. (B) Distribution of bases at each miRNA site.
3.2 miRNA identification
The clean reads after removing the sequences of non-miRNAs were compared with plant miRNA precursors and mature sequences in the miRBase database. A total of 979 known miRNAs and 110 novel miRNAs were identified, which belong to 111 miRNA families. These miRNA families contain miRNA members ranging from 1–10. Notably, the MIR156 family emerged as the most populous (Figure 3), harboring an array of members such as cme-MIR156a, cme-MIR156b and cme-MIR156c. These miRNAs, closely linked with defense mechanisms and the accumulation of secondary metabolites in host plants, underscore their significance within this context.
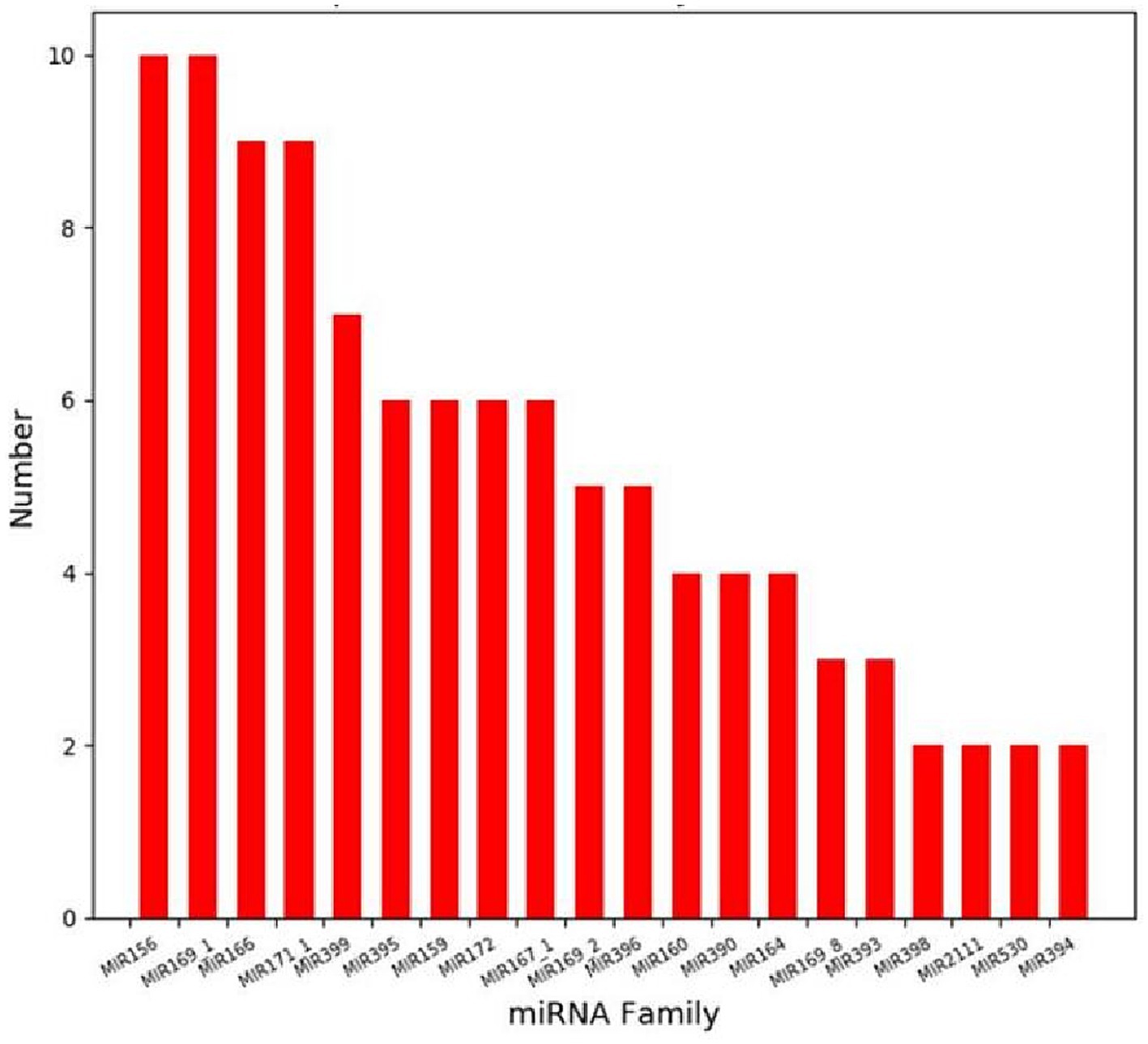
Figure 3. Top 20 miRNA family distribution.
3.3 miRNA differential expression analysis
Employing high-throughput sequencing, the systematic exploration of differential gene expression during biotic stress affords a rapid avenue for the annotation, analysis, and prediction of target genes, thereby facilitating a deeper comprehension of the intricate molecular mechanisms underpinning plant resistance to biotic stress. Our differential expression miRNA analysis for F0 and F25 revealed a total of 39 differentially expressed miRNAs, comprising 32 miRNAs significantly up-regulated and 7 significantly down-regulated (Figures 4A,D). For R0 and R25, we observed 35 differentially expressed miRNAs, with 29 displaying upregulated expression and 6 evincing downregulated expression (Figures 4B,D). A comparative analysis between R25 and F25 unveiled 5 differentially expressed miRNAs, comprising 4 up-regulated and 1 down-regulated (Figures 4C,D).
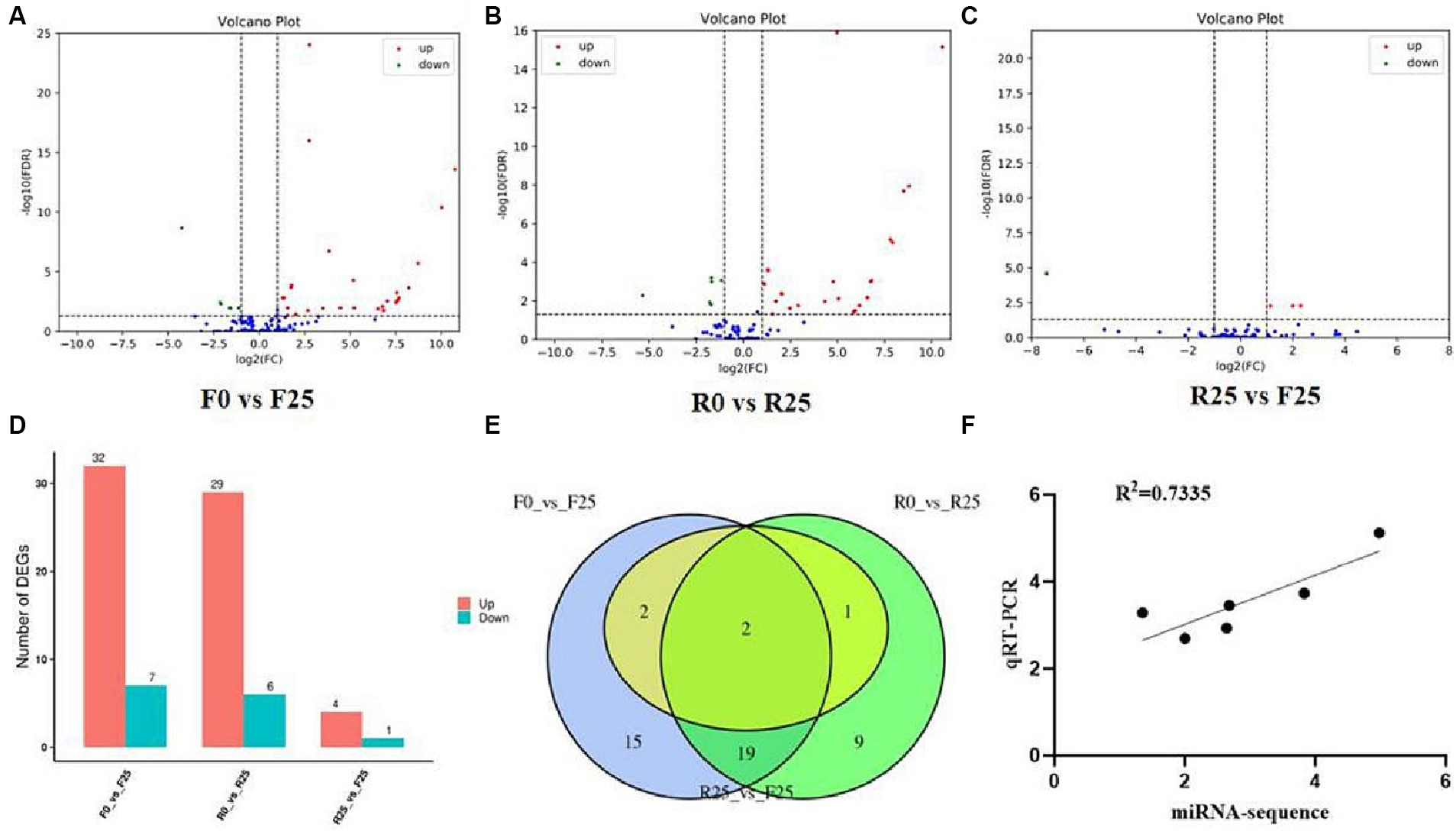
Figure 4. Differential expression of miRNA volcanic plot and Venn diagram. (A) miRNA volcano plot of sensitive line at 0 and 25 days after inoculation. (B) miRNA volcano plot of resistant line at 0 and 25 days after inoculation. (C) miRNA volcano plot of differences between sensitive and resistant lines after 25 days of inoculation. (D) Histogram of differential miRNA statistics. (E) Venn diagram of differentially expressed miRNAs. (F) Correlation analysis between qRT-PCR and miRNA sequencing results. The horizontal coordinate of the A–C plots indicates the change of miRNA expression ploidy in melon after being parasitized by broomrape; the vertical coordinate indicates the significance level of expression difference. The expression of up-regulated miRNA is represented by red dots, and the down-regulated miRNA by green dots indicates that the blue dots have not changed significantly.
Furthermore, in order to elucidate the mechanism of resistance to Broomrape, this study focused on the differences in miRNA expression before and after broomrape infection in K1-6 (R25 VS R0) and in K2-7 (F25 VS F0). The results showed 22 miRNAs exhibiting consistent significant differential expression trends in R25 VS R0 and F25 VS F0, comprising 4 miRNAs down-regulated and 18 up-regulated in both comparisons. Interestingly, two miRNAs, namely cme-miR408 and cme-miR398a, displayed up-regulation across R25 VS R0, F25 VS F0 and R25 VS F25 comparisons (Figure 4E).
To verify the accuracy of miRNA sequencing data, a random selection of 6 miRNAs, including cme-miR408, cme-miR398a, cme-miR394a, cme-miR397, cme-miR477a and cme-miR160a, was subject to quantitative real-time PCR (qRT-PCR) analysis. The results showed a high degree of correlation between the qRT-PCR and sequencing data of the selected miRNAs, indicating that the miRNA sequencing data in this experiment were authentic and reliable (Figure 4F).
3.4 Degradome sequencing for identification of miRNA target genes
In order to gain deeper insights into the role of melon miRNAs in response to broomrape infestation, this study used degradome sequencing to identify the target genes of miRNAs. The degradome library was constructed from a mixed sample of F0, F25, R0 and R25. The data was compared to a quality control to predict the target genes of miRNAs. A total of 172 miRNA-targeted pairs were detected. In a bid to validate the predictive accuracy of these miRNA-target interactions, a judicious selection of 5 pairs of miRNA-targets for subsequent validation, including cme-miR398a-MELO3C026431, cme-miR408-MELO3C008424, cme-miR477b-MELO3C026237, cme-miR160a-MELO3C011372, and cme-miR162-MELO3C031048. The results showed a compelling negative correlation between the expression trends of miRNAs and their corresponding target genes, indicating that the miRNA target genes found by degradome were reliable (Figure 5).
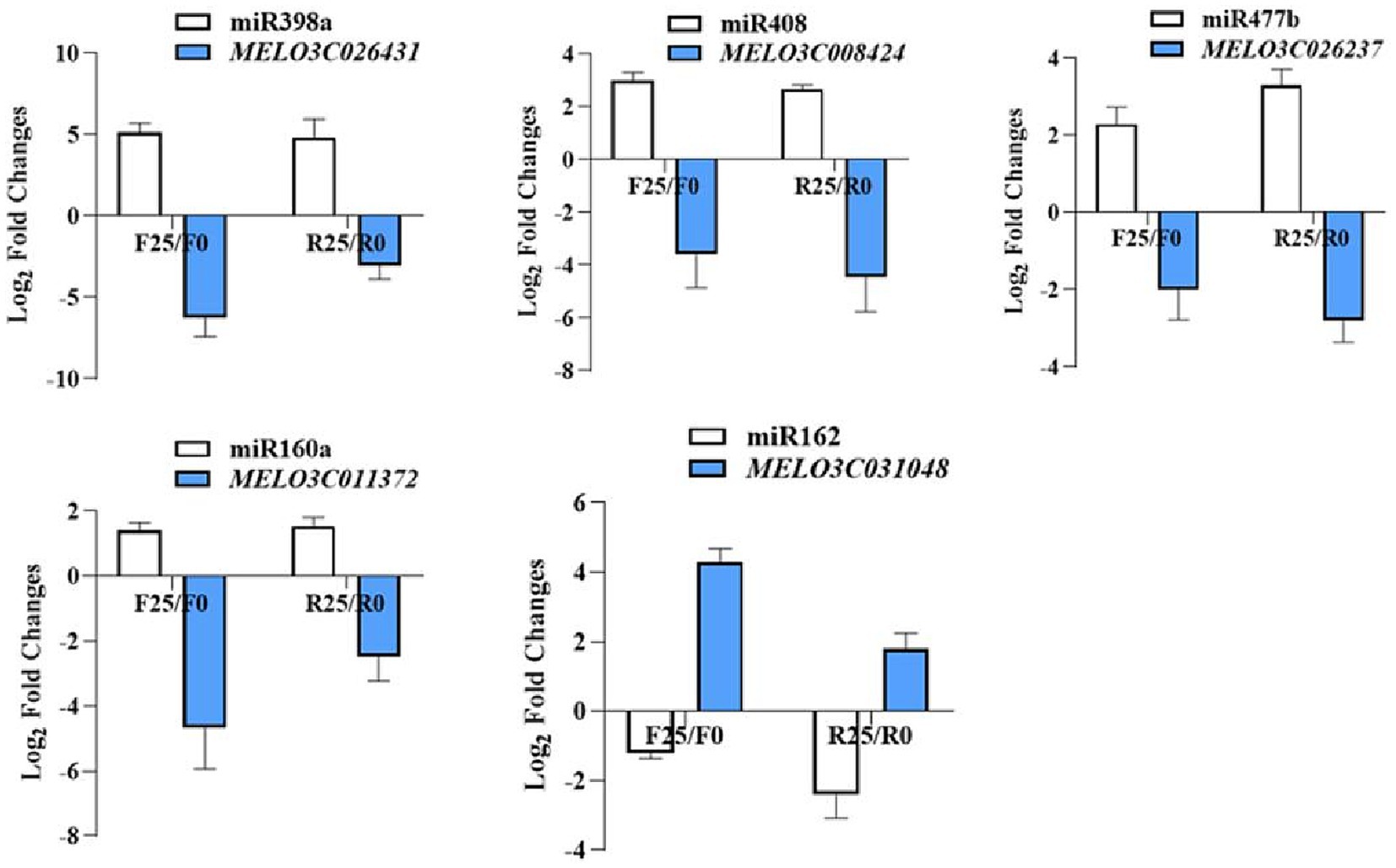
Figure 5. The verify of miRNA-target gene expression by qRT-PCR.
3.5 Functional enrichment of differential miRNA target genes
The miRNA target genes identified by degradome sequencing were subject to GO enrichment analysis. The corresponding GO annotations were classified into three categories according to biological process, cellular component and molecular functions, enriching 432, 341, and 156 target genes, respectively. The target genes were significantly enriched in cellular and metabolic processes in the biological process category. In addition, the target genes were significantly enriched in cells, cell parts and binding within the cellular component and molecular functions domain (Figure 6A).
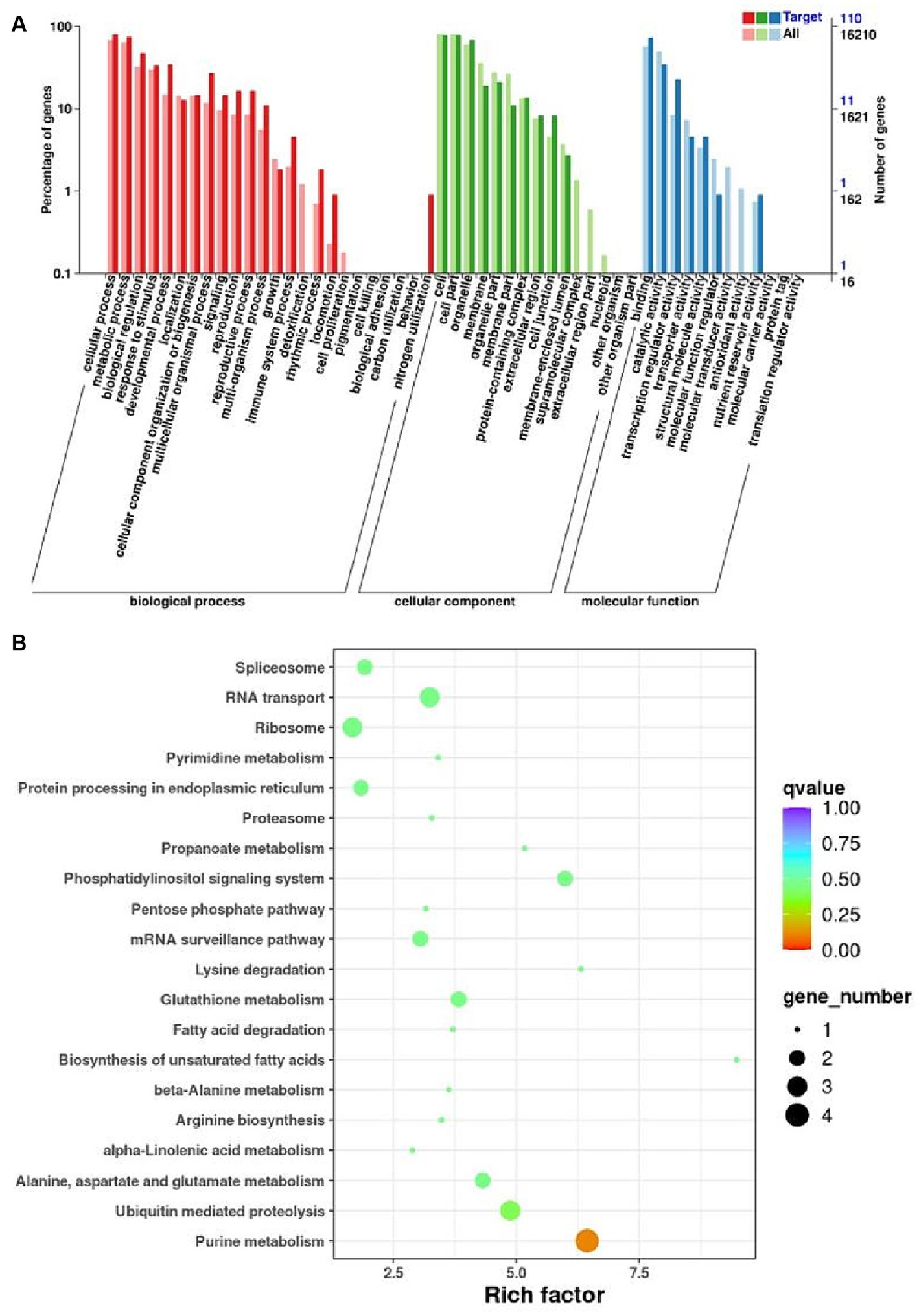
Figure 6. Functional annotation of the target genes of the differential expressed miRNAs. (A) GO enrichment analysis of differentially expressed miRNA target genes. The horizontal coordinate is the GO function entry and the vertical coordinate is the number of differential genes. (B) Analysis of KEEG enrichment pathway for differentially expressed miRNA target genes. Each row in the graph represents a KEGG pathway. The horizontal coordinate is the enrichment factor.
To glean further insights into the intricate pathways associated with melon miRNAs’ response to broomrape infestation, a comparative analysis was pursued by aligning the miRNA-target genes with the KEGG database. Target genes are enriched in phosphatidylinositol signaling and purine metabolism, which are important pathways involved in the defense response of melon. The results underscore the importance of miRNA regulation in orchestrating melon’s defense response to broomrape infestation (Figure 6B).
3.6 Transferable miRNA screening
We identified a total of four known miRNAs transferred from melon to broomrape through screening of the transferable miRNA, which were cme-miR156a, cme-m0671-5p, cme-m0738-3p, and cme-miR398a. These miRNAs are mainly involved in structural constituent of cell wall, transposon protein, glutamate receptor and cysteine-rich repeat secretory protein (Table 2). From the functions of these transferred miRNAs, it is speculated that the miRNAs transferred from melon to broomrape may inhibit lepidote growth by affecting the biosynthesis of lepidote miRNAs.
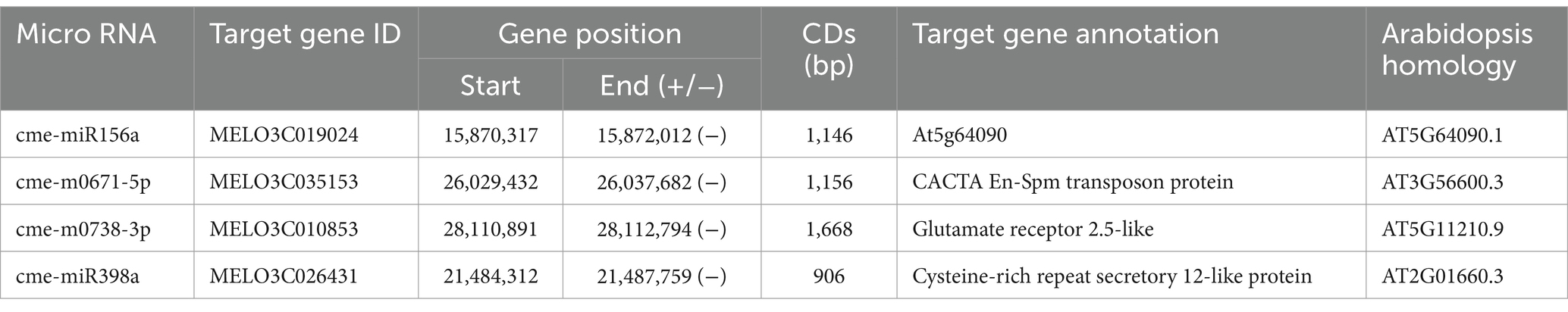
Table 2. Transferable miRNA and target gene annotation information.
4 Discussion
miRNAs represent a class of non-coding RNAs pivotal for regulatory functions that exert critical influences in almost all aspects of plant growth and development. Additionally, they play important regulatory roles in response to environmental variables such as light and nutrients, and both biotic and abiotic stresses (Budak et al., 2015; Shriram et al., 2016; Aslam et al., 2022; Kajla et al., 2023). The advent of small RNA sequencing, based on high-throughput sequencing, has effectively dismantled barriers in miRNA research, facilitating a more incisive exploration of their biological functions. While some reports have used small RNA sequencing to study melon miRNAs, the context of broomrape resistance in melon remains elusive. In this study, a total of 979 known miRNAs and 110 novel miRNAs were identified, belonging to 111 miRNA families. Intriguingly, the MIR156 family emerged as the most prolific, associated with plant resistance and accumulation of secondary metabolites (Cui et al., 2014; Wang, 2014; Rahimi et al., 2022). The results imply a potentially pivotal role for this miRNA family in the resistance of melon to broomrape infestation.
In this study, small RNA sequencing was performed to identify and quantify the expression of miRNAs in the root systems of susceptible and resistant melon lines before and after being infected by broomrape. The differential expression profiles were constructed by comparing the miRNA expression of resistant and susceptible strains. The Venn diagrams unveiled noteworthy distinctions, with cme-miR398a and cme-miR408 standing out as significantly divergent across all examined combinations. It is worth stating that while we identified the differentially expressed miRNAs in resistant and susceptible lines by small RNA sequencing, the target genes of these miRNAs and their regulatory functions remain unknown, which can be well addressed by degradome sequencing. In comparison, predictive target genes based on sequence complimentarity often entail high rates of false positives. Alternately, experimental approaches, though comprehensive, suffer from limitations of expense and time. In this context, degradome sequencing presents an elegant solution. The degradome sequencing technology is based on the principle that plant miRNAs can shear target mRNAs. The intersection of the sheared mRNA fragments are collected and sequenced by high-throughput sequencing technology, thereby allowing the prediction of miRNA target genes by Target finder software. The degradome sequencing effectively overcome the drawbacks and defects of the previous methods for identifying miRNA targets. The target genes of these two miRNAs encode cysteine-rich repeat secretory protein and copper-transporting ATPase. A recent study showed that both proteins are associated with the defense response of the organisms (Tao et al., 2021). Therefore, we speculate that these two miRNAs and their target genes play an important role in the resistance of melon to broomrape infestation.
It is well documented that there is a complex transfer of substances between the host and the parasitic organism, including miRNA, and that these substances have essential effects on both parties after the transfer. miRNAs can be transferred through the host to the parasitic plant and improves the host resistance by RNA interference. It has been shown that Cuscuta campestris transfers a large number of miRNAs to the host Arabidopsis and Benthamiana, which may function as virulence factors during parasitization as cross-species regulators of host gene expression. However, the extent of long-distance miRNA movement during broomrape parasitization has remained a question. The results of this study show that certain miRNAs are transferred from resistant melon varieties to broomrape. One of the screened transfer miRNAs, cme-miR156a, is a very conserved small non-coding RNAs in plants, and its target gene family is the SPL (SQUAOSA promoter binding like) family (Matthew et al., 2002; Bonnet et al., 2010). The miR156-SPL pathway plays a regulatory role in several plant growth and developmental processes, including phytohormone signaling and stress response (Wang, 2014; Lei et al., 2016; Miao et al., 2019). Therefore, it is hypothesized that the melon can initiate defense response through the overexpression and transfer of miR156 to broomrape, thus affecting the infestation process of broomrape. In addition, miR398a is mainly involved in the negative regulation of dismutase genes (Raichur et al., 2007; Zidenga et al., 2012). When melon was infected by broomrape, the expression of cme-miR398a in the roots was elevated, indicating that cme-miR398a may increase ROS content by inhibiting the activity of reactive oxygen species (ROS) enzymes in the cells at the infected site, which eventually resulted in an allergic necrotic response and prevented broomrape infestation.
In this study, a combination of small RNA sequencing and degradome sequencing was used to identify the differentially expressed miRNAs and their target genes between susceptible and resistant lines when infested by broomrape and to explore the defense response of melon against broomrape. In addition, 4 transferable miRNAs were identified in the analysis of melon and broomrape interactions. This comprehensive investigation into the novel defense strategy that underpins melon’s interaction with broomrape furnishes valuable reference insights, with implications extending to the broader application of cross-border RNA interference in the realm of plant disease control.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
J-CM: Writing – original draft, Writing – review & editing. MY: Writing – review & editing. J-HL: Funding acquisition, Writing – review & editing. J-YY: Writing – original draft. H-JW: Funding acquisition, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Public Welfare Basic Scientific Research Funds of Xinjiang Academy of Agricultural Sciences (Grant no. KY2022002), and National Natural Science Foundation of China (Grant no. 32260758).
Acknowledgments
The authors are grateful to BL (Xinjiang Academy of Agricultural Sciences) for his suggestions to this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1408926/full#supplementary-material
References
Aslam, M., Fakher, B., and Qin, Y. (2022). Big role of small RNAs in female gametophyte development. Int. J. Mol. Sci. 23:1979. doi: 10.3390/ijms23041979
Bao, Y. Z., Yao, Z. Q., Cao, X. L., Peng, J. F., Xu, Y., Chen, M. X., et al. (2017). Transcriptome analysis of Phelipanche aegyptiaca seed germination mechanisms stimulated by fluridone, TIS108, and GR24. PLoS One 12:e187539. doi: 10.1371/journal.pone.0187539
Bonnet, E., He, Y., Billiau, K., and Van de Peer, Y. (2010). TAPIR, a web server for the prediction of plant microRNA targets, including target mimics. Bioinformatics 26, 1566–1568. doi: 10.1093/bioinformatics/btq233
Budak, H., Kantar, M., Bulut, R., and Akpinar, B. A. (2015). Stress responsive miRNAs and isomiRs in cereals. Plant Sci. 235, 1–13. doi: 10.1016/j.plantsci.2015.02.008
Cai, Q., Qiao, L., Wang, M., He, B., Lin, F. M., Palmquist, J., et al. (2018). Plants send small RNAs in extracellular vesicles to fungal pathogen to silence virulence genes. Science 360, 1126–1129. doi: 10.1126/science.aar4142
Chen, W., Kastner, C., Nowara, D., Oliveira-Garcia, E., Rutten, T., Zhao, Y., et al. (2016). Host-induced silencing of Fusarium culmorum genes protects wheat from infection. J. Exp. Bot. 67, 4979–4991. doi: 10.1093/jxb/erw263
Cui, L. G., Shan, J. X., Shi, M., Gao, J. P., and Lin, H. X. (2014). The miR156-SPL9-DFR pathway coordinates the relationship between development and abiotic stress tolerance in plants. Plant J. 80, 1108–1117. doi: 10.1111/tpj.12712
Cui, L., Zheng, F., Wang, J., Zhang, C., Xiao, F., Ye, J., et al. (2020). MiR156a-targeted SBP-box transcription factor SlSPL13 regulates inflorescence morphogenesis by directly activating SFT in tomato. Plant Biotechnol. J. 18, 1670–1682. doi: 10.1111/pbi.13331
David-Schwartz, R., Runo, S., Townsley, B., Machuka, J., and Sinha, N. (2008). Long-distance transport of mRNA via parenchyma cells and phloem across the host-parasite junction in Cuscuta. New Phytol. 179, 1133–1141. doi: 10.1111/j.1469-8137.2008.02540.x
Govindarajulu, M., Epstein, L., Wroblewski, T., and Michelmore, R. W. (2015). Host-induced gene silencing inhibits the biotrophic pathogen causing downy mildew of lettuce. Plant Biotechnol. J. 13, 875–883. doi: 10.1111/pbi.12307
Guo, C., Jiang, Y., Shi, M., Wu, X., and Wu, G. (2021). ABI5 acts downstream of miR159 to delay vegetative phase change in Arabidopsis. New Phytol. 231, 339–350. doi: 10.1111/nph.17371
Guo, C., Xu, Y., Shi, M., Lai, Y., Wu, X., Wang, H., et al. (2017). Repression of miR156 by miR159 regulates the timing of the juvenile-to-adult transition in Arabidopsis. Plant Cell 29, 1293–1304. doi: 10.1105/tpc.16.00975
Hudzik, C., Maguire, S., Guan, S., Held, J., and Axtell, M. J. (2023). Trans-species microRNA loci in the parasitic plant Cuscuta campestris have a U6-like snRNA promoter. Plant Cell 35, 1834–1847. doi: 10.1093/plcell/koad076
Ibrahim, H. M., Alkharouf, N. W., Meyer, S. L., Aly, M. A., Gamal El-Din Ael, K., Hussein, E. H., et al. (2011). Post-transcriptional gene silencing of root-knot nematode in transformed soybean roots. Exp. Parasitol. 127, 90–99. doi: 10.1016/j.exppara.2010.06.037
Kajla, M., Roy, A., Singh, I. K., and Singh, A. (2023). Regulation of the regulators: transcription factors controlling biosynthesis of plant secondary metabolites during biotic stresses and their regulation by miRNAs. Front. Plant Sci. 14:1126567. doi: 10.3389/fpls.2023.1126567
Kim, G., and Westwood, J. H. (2015). Macromolecule exchange in Cuscuta-host plant interactions. Curr. Opin. Plant Biol. 26, 20–25. doi: 10.1016/j.pbi.2015.05.012
LaMonte, G., Philip, N., Reardon, J., Lacsina, J. R., Majoros, W., Chapman, L., et al. (2012). Translocation of sickle cell erythrocyte microRNAs into Plasmodium falciparum inhibits parasite translation and contributes to malaria resistance. Cell Host Microbe 12, 187–199. doi: 10.1016/j.chom.2012.06.007
Lei, K. J., Lin, Y. M., and An, G. Y. (2016). MiR156 modulates rhizosphere acidification in response to phosphate limitation in Arabidopsis. J. Plant Res. 129, 275–284. doi: 10.1007/s10265-015-0778-8
Liu, H., Wang, X., Wang, H. D., Wu, J., Ren, J., Meng, L., et al. (2012). Escherichia coli noncoding RNAs can affect gene expression and physiology of Caenorhabditis elegans. Nat. Commun. 3:1073. doi: 10.1038/ncomms2071
Matthew, W. R., Brenda, J. R., Lee, P. L., Christopher, B. B., Bonnie, B., et al. (2002). Prediction of plant microRNA targets. Cell 110, 513–520. doi: 10.1016/s0092-8674(02)00863-2
Miao, C., Wang, Z., Zhang, L., Yao, J., Hua, K., Liu, X., et al. (2019). The grain yield modulator miR156 regulates seed dormancy through the gibberellin pathway in rice. Nat. Commun. 10:3822. doi: 10.1038/s41467-019-11830-5
Nizampatnam, N. R., Schreier, S. J., Damodaran, S., Adhikari, S., and Subramanian, S. (2015). microRNA160 dictates stage-specific auxin and cytokinin sensitivities and directs soybean nodule development. Plant J. 84, 140–153. doi: 10.1111/tpj.12965
Rahimi, A., Karami, O., Balazadeh, S., and Offringa, R. (2022). MiR156-independent repression of the ageing pathway by longevity-promoting AHL proteins in Arabidopsis. New Phytol. 235, 2424–2438. doi: 10.1111/nph.18292
Raichur, S., Lau, P., Staels, B., and Muscat, G. E. (2007). Retinoid-related orphan receptor gamma regulates several genes that control metabolism in skeletal muscle cells: links to modulation of reactive oxygen species production. J. Mol. Endocrinol. 39, 29–44. doi: 10.1677/jme.1.00010
Shahid, S., Kim, G., Johnson, N. R., Wafula, E., Wang, F., Coruh, C., et al. (2018). MicroRNAs from the parasitic plant Cuscuta campestris target host messenger RNAs. Nature 553, 82–85. doi: 10.1038/nature25027
Shriram, V., Kumar, V., Devarumath, R. M., Khare, T. S., and Wani, S. H. (2016). MicroRNAs as potential targets for abiotic stress tolerance in plants. Front. Plant Sci. 7:817. doi: 10.3389/fpls.2016.00817
Song, X., Li, Y., Cao, X., and Qi, Y. (2019). microRNAs and their regulatory roles in plant-environment interactions. Annu. Rev. Plant Biol. 70, 489–525. doi: 10.1146/annurev-arplant-050718-100334
Tao, Z., Yalong, Z., Dewei, T., Xiaozhong, Z., Xueshi, L., et al. (2021). Rice blast resistance-associated genes based on different RNA-seq resources. Agric. Biotechnol. 10, 1–6,
Wang, J. W. (2014). Regulation of flowering time by the miR156-mediated age pathway. J. Exp. Bot. 65, 4723–4730. doi: 10.1093/jxb/eru246
Wang, B., Sun, Y., Song, N., Zhao, M., Liu, R., Feng, H., et al. (2017). Puccinia striiformis f. sp. tritici microRNA-like RNA 1 (Pst-milR1), an important pathogenicity factor of Pst, impairs wheat resistance to Pst by suppressing the wheat pathogenesis-related 2 gene. New Phytol. 215, 338–350. doi: 10.1111/nph.14577
Wang, M., Weiberg, A., Dellota, E. J., Yamane, D., and Jin, H. (2017). Botrytis small RNA Bc-siR37 suppresses plant defense genes by cross-kingdom RNAi. RNA Biol. 14, 421–428. doi: 10.1080/15476286.2017.1291112
Wang, M., Weiberg, A., Lin, F. M., Thomma, B. P., Huang, H. D., et al. (2016). Bidirectional cross-kingdom RNAi and fungal uptake of external RNAs confer plant protection. Nat. Plants 2:16151. doi: 10.1038/nplants.2016.151
Weiberg, A., Wang, M., Lin, F. M., Zhao, H., Zhang, Z., Kaloshian, I., et al. (2013). Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 342, 118–123. doi: 10.1126/science.1239705
Zhang, T., Jin, Y., Zhao, J. H., Gao, F., Zhou, B. J., Fang, Y. Y., et al. (2016a). Host-induced gene silencing of the target gene in fungal cells confers effective resistance to the cotton wilt disease pathogen Verticillium dahliae. Mol. Plant 9, 939–942. doi: 10.1016/j.molp.2016.02.008
Zhang, B. S., Li, Y. C., Guo, H. S., and Zhao, J. H. (2022). Verticillium dahliae secretes small RNA to target host MIR157d and retard plant floral transition during infection. Front. Plant Sci. 13:847086. doi: 10.3389/fpls.2022.847086
Zhang, T., Zhao, Y. L., Zhao, J. H., Wang, S., Jin, Y., Chen, Z. Q., et al. (2016b). Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2:16153. doi: 10.1038/nplants.2016.153
Zhu, C., Liu, T., Chang, Y. N., and Duan, C. G. (2019). Small RNA functions as a trafficking effector in plant immunity. Int. J. Mol. Sci. 20:2816. doi: 10.3390/ijms20112816
Keywords: trans-kingdom RNAi, melon, broomrape, small RNA sequencing, degradome sequencing
Citation: Mao J-C, Yan M, Li J-H, Yang J-Y and Wang H-J (2024) The role of small RNAs in resistant melon cultivar against Phelipanche aegyptiaca parasitization. Front. Microbiol. 15:1408926. doi: 10.3389/fmicb.2024.1408926
Edited by:
Hongju Zhu, Chinese Academy of Agricultural Sciences, ChinaReviewed by:
Tao Zhang, Chinese Academy of Sciences (CAS), ChinaXingwang Liu, China Agricultural University, China
Shakil Ahmad, Hainan University, China
Copyright © 2024 Mao, Yan, Li, Yang and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao-Jie Wang, d2FuZ2hhb2ppZUB4YWFzLmFjLmNu
†These authors have contributed equally to this work