 Xing Ma1
Xing Ma1 Xiuyan Zhang2
Xiuyan Zhang2 Paul Stodghill1,3
Paul Stodghill1,3 Renee Rioux4†
Renee Rioux4† Smita Shrestha4
Smita Shrestha4 Brooke Babler5
Brooke Babler5 Hannah Rivedal6
Hannah Rivedal6 Kenneth Frost7
Kenneth Frost7 Jianjun Hao2
Jianjun Hao2 Gary Secor8
Gary Secor8 Bryan Swingle1,3*
Bryan Swingle1,3*- 1Plant Pathology and Plant-Microbe Biology Section, School of Integrative Plant Science, Cornell University, Ithaca, NY, United States
- 2School of Food and Agriculture, University of Maine, Orono, ME, United States
- 3Emerging Pests and Pathogens Research Unit, United States Department of Agriculture-Agricultural Research Service, Robert W. Holley Center, Ithaca, NY, United States
- 4Department of Plant Pathology, University of Wisconsin-Madison, Madison, WI, United States
- 5Wisconsin Seed Potato Certification Program, Department of Plant Pathology, University of Wisconsin-Madison, Middleton, WI, United States
- 6Forage Seed and Cereal Research Unit, United States Department of Agriculture-Agricultural Research Service, Corvallis, OR, United States
- 7Department of Botany and Plant Pathology and Hermiston Agricultural Research and Extension Center, Oregon State University, Hermiston, OR, United States
- 8Department of Plant Pathology, North Dakota State University, Fargo, ND, United States
Introduction: Soft rot Pectobacteriaceae (SRP) bacteria are globally dispersed pathogens that cause significant economic loss in potato and other crops. Our understanding of the SRP species diversity has expanded in recent years due to advances and adoption of whole-genome sequence technologies. There are currently 34 recognized SRP species that belong to the Dickeya and Pectobacterium genera.
Methods: We used whole-genome sequencing based analysis to describe the current distribution and epidemiology of SRP isolated from diseased potato samples obtained from commercial potato cropping systems in the United States. Our primary objectives in the present study were to: (1) identify the species of these SRP isolates recovered from potato samples across 14 states in the US, (2) describe the variation among SRP isolates from various US locations and track their temporal changes, and (3) evaluate the evolutionary relationships among these SRP isolates to deduce their source. We collected 118 SRP strains from diseased potato plants and tubers in 14 states between 2015 and 2022.
Results: We identified three Dickeya and eight Pectobacterium species from diseased potato samples. Dickeya dianthicola, Pectobacterium parmentieri, P. carotovorum, and P. versatile appeared to be the predominant species, constituting 83% of the isolates. Furthermore, all D. dianthicola strains studied here as well as 90% of US D. dianthicola isolates sequenced to date exhibit significant clonality.
Discussion: The prevalence of this specific group of D. dianthicola, temporally and geographically, aligns with the occurrence of blackleg and soft rot outbreaks in the northeastern US after 2014. The genomic diversity observed in P. parmentieri implies multiple introductions to the US from at least four distinct sources, earlier than the arrival of the predominant group of D. dianthicola. In contrast, P. carotovorum and P. versatile appear to be widespread, long-term endemic strains in the US.
Introduction
Soft rot Pectobacteriaceae (SRP) bacteria are phytopathogenic bacteria that belong to the family Pectobacteriaceae and produce extracellular enzymes that break down plant cell walls and give the bacteria access to nutrients within plant cells. SRP bacteria are rod-shaped, gram-negative, facultatively anaerobic bacteria that were previously classified under the genus Erwinia. They were recently reclassified into the genera Pectobacterium and Dickeya (Hauben et al., 1998; Gardan et al., 2003; Czajkowski et al., 2015) and subdivided into 22 Pectobacterium and 12 Dickeya species (Home - Taxonomy - NCBI (nih.gov)). This reorganization, including the naming of many new species of SRP has been facilitated by advances in whole genome sequencing-based methods such as average nucleotide identity (ANI) and digital DNA–DNA hybridization (dDDH) (Zhang et al., 2016; Tian et al., 2016; Parkinson et al., 2014; Hugouvieux-Cotte-Pattat et al., 2019; Hugouvieux-Cotte-Pattat and Van Gijsegem, 2021; Pedron et al., 2019; Zhou et al., 2022; Oulghazi et al., 2019; Dees et al., 2017; Pasanen et al., 2020). Solving the nomenclature can help stakeholders understand differences between the SRP groups in regards to the host ranges, virulence variances, and their origins (Toth et al., 2021).
Given their wide host range, most SRP species can infect potato (Solanum tuberosum) and cause diseases (Ma et al., 2007; Toth et al., 2011). Potato diseases caused by SRP pathogens are named based on the symptoms and the plant parts affected. Soft rot affects potato tubers, causing the tuber flesh to become macerated to a soft consistency that may have a foul odor and darken when exposed to air. Blackleg occurs in stems during the early stages of growth when inky stem rot occurs at the soil line as SRP cells move from sprouting seed tubers to stems. Aerial stem rot occurs when SRP bacteria enter through the natural openings or wounds and causes rotting symptoms on potato shoots and leaves (Stevenson et al., 2001).
Several factors make diseases caused by SRP difficult to control. There are no commercial potato varieties with high-level resistance, so control is limited to seed sanitation practices, including seed treatment and seed certification programs that help to reduce the pathogen levels in seed materials (Czajkowski et al., 2011). However, SRP bacteria have the ability to cause latent, asymptomatic infections that can prevent seed certification scouts from detecting the SRP-related symptoms and getting an accurate assessment of the levels of disease in the crop. Other supplemental practices include field selection, cultivation management, and optimization of storage conditions (van der Wolf et al., 2021).
The major source of SRP inoculum for potato field infection is believed to be carried by latent infected seed potatoes (Perombelon, 1974; Perombelon and Kelman, 1980; Perombelon, 2002; Czajkowski et al., 2011). These latent infections can remain dormant until environmental cues, such as moisture, temperature, or anaerobic conditions stimulate the bacteria to resume virulent growth. But seed tubers are not the only inoculum sources. Some of the SRP bacteria can become soilborne and survive up to 6 months in ideal soil environments (Czajkowski et al., 2011). SRP bacteria can move in soil to neighboring tubers and roots by ground water (Czajkowski et al., 2010) and can also be carried by aerosols in rain for several hundred meters before deposition (Czajkowski et al., 2011). These abilities enable the transmission of SRP bacteria to other plants in the same or adjacent fields.
Since 2014, widespread outbreaks of blackleg and soft rot diseases on potatoes have been documented in the northeast and midwest US (Jiang et al., 2016; Charkowski, 2018; Ma et al., 2018; Ma et al., 2021b; Cai et al., 2018; Rosenzweig et al., 2016; Curland et al., 2021; Patel et al., 2019). In New York State these diseases have been mainly caused by P. parmentieri, P. versatile, and D. dianthicola (Ma et al., 2018; Ma et al., 2021b). In addition to New York, D. dianthicola has been detected in Northeast, Mid-Atlantic, and Midwest of the US as well as in two Canadian provinces (Charkowski, 2018; Jiang et al., 2016; Cai et al., 2018; Rosenzweig et al., 2016). A 2015–2016 investigation on multiple potato growing regions determined that the D. dianthicola and P. parmentieri are the predominant species among many other SRP species discovered in the US (Curland et al., 2021). In the present study, we sequenced the genomes of 118 SRP isolates between 2015 and 2022 from 14 potato growing states across the US to: (1) Determine the species of these SRP isolates, (2) Understand the genetic variation in SRP isolates from different locations and their changes over time, (3) Assess the evolutionary relationships between these SRP isolates to infer epidemiological insights.
Materials and methods
Bacterial strains and culture conditions
Bacterial strains were isolated from field samples originating in 14 states across the US by researchers working in each region (Supplementary Figure S1) using standard methods as described previously (Ma et al., 2021b). These field samples were collected ad hoc by growers, extension agents, or researcher from commercial and research plots. Usually, one isolate from each sample was collected except the isolates from Wisconsin samples. Among the Wisconsin samples, strain UW021-1, -3, -4, -5, -6, -7, -8, and -9 were isolated from one stem sample, UW021-11, 17, and 19 were isolated from another stem sample in the same growing season. Bacteria were shipped overnight to Cornell University in stab cultures. The bacteria were temporarily stored at 4°C before they were recovered in Lysogeny Broth (LB) broth (Sambrook et al., 1989; Bertani, 2004) and incubated at 28°C with shaking at 240 RPM overnight for DNA extraction and two copies of each isolate were preserved in 16% glycerol at −80°C.
Genomic DNA extraction
We extracted bacterial genomic DNA from 1 mL overnight LB cultures using either the phenol-chloroform method (Booher et al., 2015; Ma et al., 2021b), Wizard SV Genomic DNA Purification kit (Toll-Riera et al., 2016) (#A2360, Promega, Madison, WI), or ZymoBiomics DNA Miniprep Kit (#D4300, Zymo Research, Irvine, CA). DNA concentrations were determined with a Nanodrop One at 600 nm (Thermo Fisher, Waltham, MA).
Whole-genome sequencing
DNA Libraries were prepared by the Institute of Biotechnology at Cornell University from bacterial gDNA using Illumina Nextera DNA prep kits with six PCR cycles. Illumina MiSeq Nano 2×150-bp sequencing was conducted on this library to calculate dilution factors and pooled to ensure that each sample was evenly sequenced. Size selection was used to remove small inserts before genome sequencing.
Whole-genome sequencing for the bacterial isolates was conducted using short-read Illumina platforms in three batches. The New York isolates recovered before 2017 (labeled starting with NY14, NY15, and NY17) were sequenced at Cornell Institute of Biotechnology as detailed in the previous study (Ma et al., 2021b), using the Illumina NextSeq 500 platform with the high-output kit, producing up to 400 M single-end 75-bp reads, which consists of 30 Gbp data (referred to as Sequencing platform A, n = 43). The remaining strains, if not specified, were sequenced in the second batch at Azenta Life Science (South Plainfield, NJ) using the Illumina HiSeq platform in 2021. One flowcell can generate up to 2 × 150-bp 350 M paired-end reads, or 105 Gbp data (referred to as Sequencing platform B, n = 62). Additionally, isolates (JB133A, JB88B, JB94B, 15A12, 15A54, 16A18, 16A44, 18B42, 18B43, ME175, UMSS2, UMSS4, and UMSS5) were sequenced in a 2022 batch at Cornell Institute of Biotechnology using the NextSeq 550 platform with mid-output kit producing up to 200 M paired-end 2 × 150-bp reads, equal to 60 Gbp data (referred to as Sequencing platform C thereafter, n = 13).
Genome sequence assembly and quality assessment
Genome sequencing raw reads were demultiplexed by the sequencing facilities. We downloaded them to BioHPC servers at Cornell University and checked MD5 hashtags for integrity. The single-end 75 bp reads were cleaned and assembled as described previously (Ma et al., 2021b). The paired end 2×150 bp reads were analyzed using a customized pipeline1 to clean and assemble reads. This pipeline containerized Unicycler v0.5.0 and other applications to streamline the assembly procedure. In the pipeline, the raw reads were trimmed to remove adaptors and low-quality reads with fastp v0.23.4 (Chen et al., 2018). The trimmed reads were processed by Unicycler v0.5.0 (Wick et al., 2017) to build draft assemblies. The draft assemblies were corrected with trimmed Illumina short reads by NextPolish v4.1 (Hu et al., 2020) to generate polished assemblies. The polished assemblies were annotated using Prokka v1.14.5 (Seemann, 2014). We checked the completeness of each genome assembly using BUSCO 5.5.0 (Seppey et al., 2019) in the genome mode with the enterobacterial lineage database (enterobacterales_odb10).
Resolve species identity with ANI and dDDH
ANI scores were computed by Referenceseeker v1.8.0 with GTDB and RefSeq databases accessed on 3/31/2021 (Schwengers et al., 2019) for the query strain to determine its genus. The query strain was aligned in FastANI v1.33 against a set of reference group strain database (Jain et al., 2018). This customized database consisted of 580 RefSeq SRP bacterial assemblies (Supplementary Table S1). The comparison results were ranked from the high ANI score to low. The reference strains that have higher than 95% similarity were examined to determine the species of the query strain.
We identified one isolate (1950-12), for which taxonomy could not be resolved by ANI analysis (i.e., no ANI score above 95% to any genome sequence in RefSeq database, accessed on June 27, 2022). We used the Type (Strain) Genome Server (TYGS, ggdc.dsmz.de) to calculate the dDDH score for isolate 1950-12 (Meier-Kolthoff et al., 2022). TYGS uses their algorithms to estimate the real DDH scores based on the query and the reference genome sequences. The Formula d6 (a.k.a. GGDC Formula 2) was chosen for the dDDH analyses because it is independent of genome length and, therefore, useful for incomplete genome assemblies. Strains above the cut-off of 70% similarity were considered the same species. The dDDH between 1950-12 and P. aroidearum L6 (NZ_CP065044.1) was computed with the Genome-to-Genome Distance Calculator.2
Pathogenicity bioassay
We cultured a loopful of bacterial colony in 5 mL of LB broth and incubated overnight at 28°C with shaking at 240 rpm to reach 109 CFU/mL. We made 5 mm deep holes on yellow flesh potato tubers with a 2 mm diameter sterile wooden applicator and placed 10 μL of inoculum in the hole. For mock controls, 10 μL sterile LB was applied. The potatoes were incubated at 28°C overnight. We cut a longitudinal section of tuber through the inoculation points to observe the maceration symptoms. We picked a loopful of macerated tissue and diluted it in 1 mL of sterile water. A new loop was used to streak the resulting solution on CVP medium. Single colonies that can form pits were selected on CVP after 24 h incubation at 28°C. Colony PCR or PCR from bacterial genomic DNA was conducted with primers dnaXf/dnaXr (Slawiak et al., 2009; Ma et al., 2018). The PCR products were sanger sequenced and we compared these sequences with their genome assembly. The bioassays were conducted twice.
Phylogenetic analysis
The files in GenBank format for each bacterial genome assembly were generated by the Prokka v1.14.5 annotation tool in the customized pipeline previously mentioned. Additionally, we downloaded the GenBank formatted files (with suffix “.gbff”) for each reference genome of Pectobacterium and Dickeya species in the RefSeq database on NCBI websites (Genome - NCBI - NLM (nih.gov)). These files were used as input files in a get_homologues pipeline (Vinuesa et al., 2018) with the “-t all -e -D -X” arguments to call single copy orthologous gene clusters, where, argument “-t all” specified the gene must present in all taxa, argument “-e” makes the search only find single-copy genes, “-D” enables PFAM-domain scanning, “-X” uses DIAMOND (Buchfink et al., 2015) instead of the regular BLASTP in protein searches for faster aligning speed. The get_homologues pipeline used bidirectional best-hits algorithms to find single-copy homolog genes and compile them into gene clustering files. These files were subsequently processed with the get_phylomarkers pipeline v2.2.6 (Vinuesa et al., 2018). From all the discovered single orthologue genes, get_phylomarkers pipeline can filter the loci by the following three conditions: (1) The genes can pass the Pairwise Homoplasy Index (Phi) tests, indicating a low possibility of recombination; (2) The gene with a gene tree that can pass the kdetree test indicating it does not have an outlier tree topology; (3) The genes or with high Shimodaira-Hasegawa approximate likelihood ratio test branch support values. The remaining genes were concatenated to construct a maximum likelihood tree were constructed by IQ-TREE v2.0 (Nguyen et al., 2015).
Single nucleotide polymorphism (SNP) analyses were performed at the intraspecies level for each of the four species with the most isolates including all published strains from the RefSeq database (accessed on 12/22/2023) on NCBI. Snippy v4.6.0 (Seemann, 2015) was used to call variances compared to the NCBI reference genome of the working species. In the procedure, the application will use as input one reference genome and the query genomes, including the genomes generated in this study and the published genomes of the same species in the NCBI RefSeq database. The query genomes were mapped onto the reference genome, individually. Then, Snippy identified SNPs from the comparison between the query genome and the reference genome. The SNP calling results were combined for all strains under the same species and aligned in full length SNP sequences. The aligned file was analyzed with IQ-TREE v2.2.2.6 to construct a maximum likelihood tree with the “-B 1000” argument for 1,000 replications of ultrafast bootstrapping.
Results
De novo genome assembly quality assessment
We assembled a collection of 118 SRP isolates from 14 US states that have important roles in the US potato production systems, including seed tuber production and testing, and commercial potato production for fresh market and processing. We purified genomic DNA and submitted samples for library generation and short reads sequencing as described in Materials and Methods. The raw reads from the sequencing facility were used to produce de novo contig assemblies for each isolate.
We used sequencing coverage of raw reads, total nucleotide length, BUSCO score, L50, and N50 of the genome assemblies to assess the sequencing depths and assembly qualities. These statistics of sequencing data and assembly quality were documented in Supplementary Tables S2, S3, and described in Supplementary material. Before submitting to GenBank, genome assemblies were filtered to remove sequences shorter than 200 bp. The 15th contig from 1950-32 was split into two contigs to remove adaptor sequence found at positions 343–389 of the original assembly. All assembled and annotated genomes are available at GenBank in NCBI. See Supplementary Table S3 for accession number for each genome.
Species identification
In this study, we computed ANI scores based on their de novo assembled genomes to classify the 118 SRP isolates from 14 US states at the species level (detailed information in Supplementary Table S4). Most states have fewer than 10 isolates except Wisconsin with 14 isolates and New York with 52 isolates (Table 1). The SRP isolates were classified into 12 species, specifically, nine Pectobacterium and three Dickeya species based on their ANI results (Table 2). P. carotovorum (19.5% of the total), P. parmentieri (24.6%), P. versatile (19.5%), and D. dianthicola (19.5%) were the most frequently isolated species, which together make up 83.1% of all the isolates recovered in this study (Table 2). We also identified one P. parvum, NY1749A, and one P. aroidearum, 1950-12. This is likely the first time P. parvum and P. aroidearum were found associated with disease potato in New York State and Hawaii, respectively.
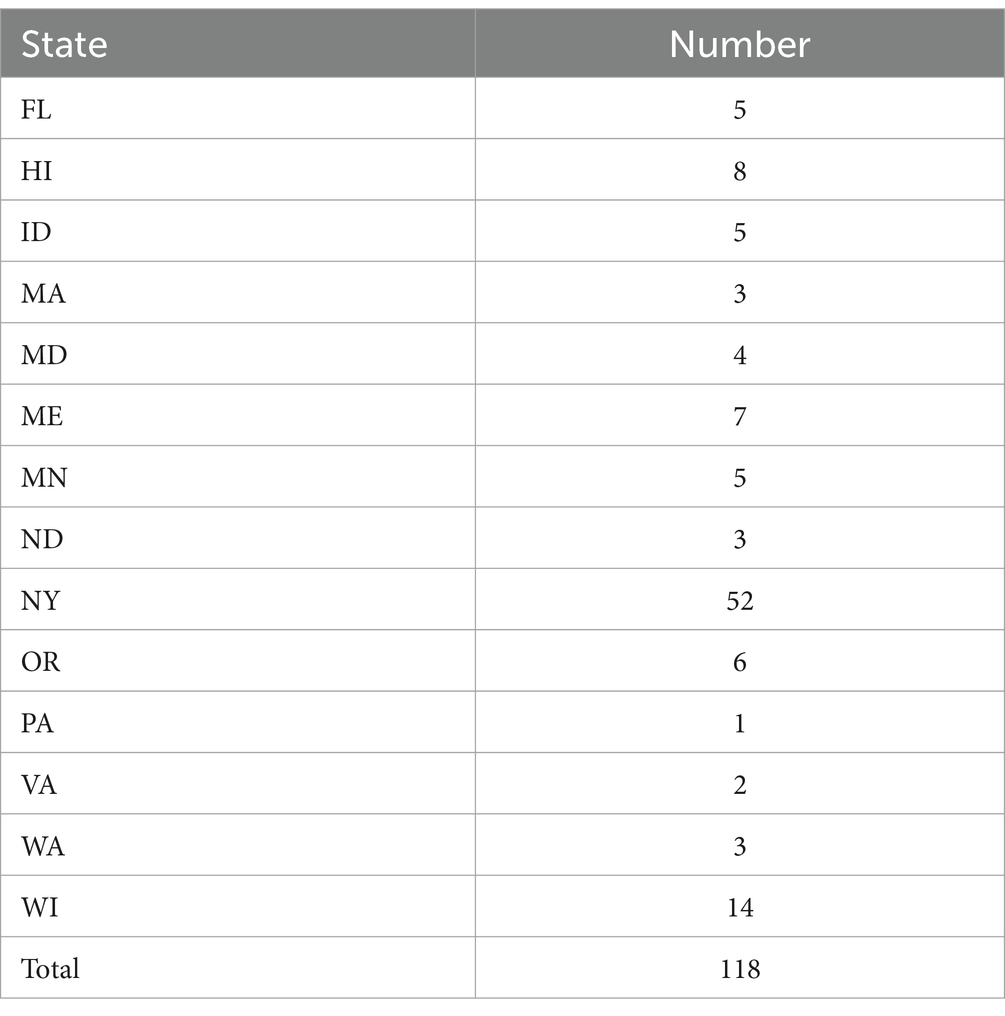
Table 1. Number of isolates from each state.
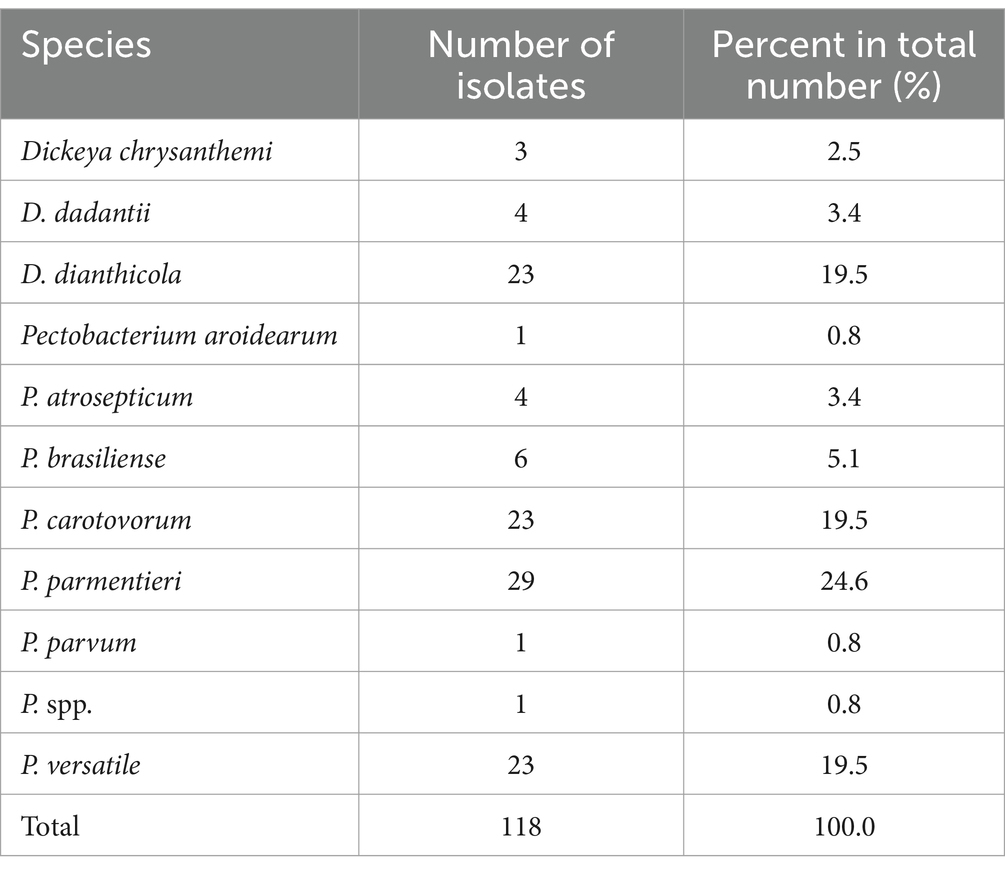
Table 2. Number of isolates of each species.
Only one Pectobacterium strain, isolate 1950-15, did not exceed the 95–96% threshold with any published SRP species. The highest ANI scores associated with 1950-15 were obtained from the genomes of P. aroidearum MY7 (94.2%) and other ten P. aroidearum strains from the RefSeq database, all above 94%, followed by P. colocasium (92.3%) and P. brasiliense (91.8%). In the dDDH test, the highest dDDH score was from the comparison between the query isolate and P. aroidearum L6 with 54.6% similarity followed by P. colocasium LJ1 with 46.0% similarity, followed by the score with P. brasiliense LMG21371 of 42.7%. None of the dDDH scores were above the 70% threshold for species assignment. These results suggest that isolate 1950-15 cannot be classified into the same species with any published reference strains in the RefSeq database using computational and genomic methods.
The pathogenicity of P. aroidearum, 1950-12, and the unknown Pectobacterium strain 1950-15 were confirmed with tuber bioassay to complete Koch’s Postulates (Figure 1). Both strains caused extensive macerated tissue surrounding the inoculation points. The same bacteria were reisolated from the macerated tissue and verified by sequencing their partial dnaX gene from two replications. No SRP were isolated from negative controls.
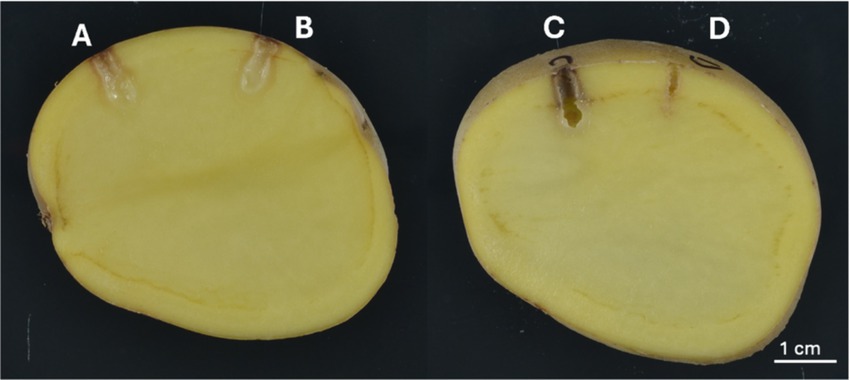
Figure 1. Soft rot symptoms induced in pathogenicity tests on potato tubers. (A) P. aroidearum 1950-12, (B) Pectobacterium sp. 1950-15, (C) D. dianthicola ME23, (D) negative control. Scale bar is equal to 1 cm.
Distribution of SRP bacteria across the US
In this study, D. dianthicola was only found in four eastern states of the US: New York, Pennsylvania, Maryland, and Florida (Figure 2). P. parmentieri was identified in samples from Maine, New York, Wisconsin, Oregon, and Hawaii. Of these states, New York and Wisconsin had relatively higher numbers of P. parmentieri isolates. P. versatile was found in samples from six states: New York, Massachusetts, Maine, Wisconsin, Washington, and Oregon. P. carotovorum had the most widespread distribution from nine states: Maine, New York, Maryland, Wisconsin, Minnesota, North Dakota, Idaho, Washington, and Hawaii. Six P. brasiliense isolates were from five states: New York, Florida, Wisconsin, Idaho, and Hawaii. We identified four P. atrosepticum isolates from New York, North Dakota, and Washington. Three D. chrysanthemi are from Wisconsin, Idaho, and Hawaii. We did not isolate D. chrysanthemi in any state on the east coast. Four D. dadantii were found in Florida and Hawaii. The only P. aroidearum was isolated from Hawaii. The only P. parvum strain was found in New York, which has not been previously reported there. The strain 1950-15 with unknown species was found in Hawaii (Figure 2).
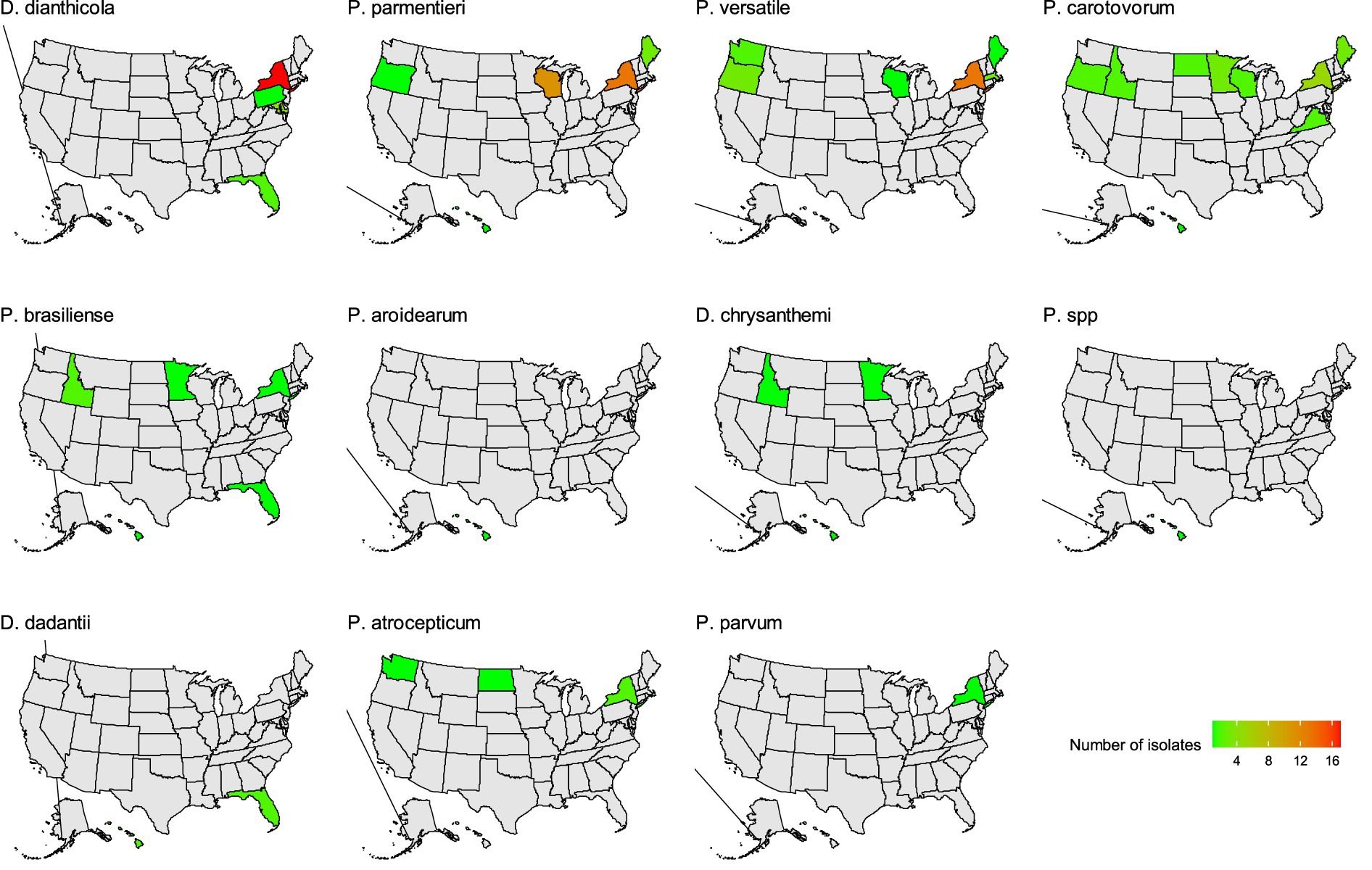
Figure 2. The distribution of soft rot Pectobacteriaceae species identified in this study. Green and red color indicates the number of each species identified. Gray color indicates no isolate was obtained from that state.
The number of each SRP species isolated from each state was plotted by year in Figure 3. Our observation of disease and isolation of SRP was sporadic in most states. It is difficult to know why the numbers of SRP varied from year to year, but we suspect that it may be related to several factors, including the incidence of disease in the area, access to fields, awareness of widespread regional outbreaks, and interest in obtaining a diagnosis. For example, there were widespread outbreaks of D. dianthicola in the Northeast that began around 2014, this led to an increase in interest in managing this new pathogen and an increase in samples received for diagnosis, particularly in New York. The number of isolates submitted by growers in New York decreased over the 7 year period of this study. The numbers of D. dianthicola and P. parmentieri isolates detected dropped sharply after 2016 in New York. The decrease in SRP isolates during this time was most likely due to fewer outbreaks caused by D. dianthicola in the Northeast and decreasing interest in managing this pathogen. We did not find any D. dianthicola after 2018 in all 14 states. P. versatile was consistently found in many states since 2018. Many P. parmentieri were isolated in 2017 from Wisconsin from two samples. We isolated eight SRP isolates in Hawaii samples in 2016. These Hawaiian isolates are very diverse with at least seven species, namely, D. chrysanthemi, D. dadantii, P. aroidearum, P. brasiliense, P. carotovorum, P. parmentieri, and an undetermined Pectobacterium species (Figure 3).
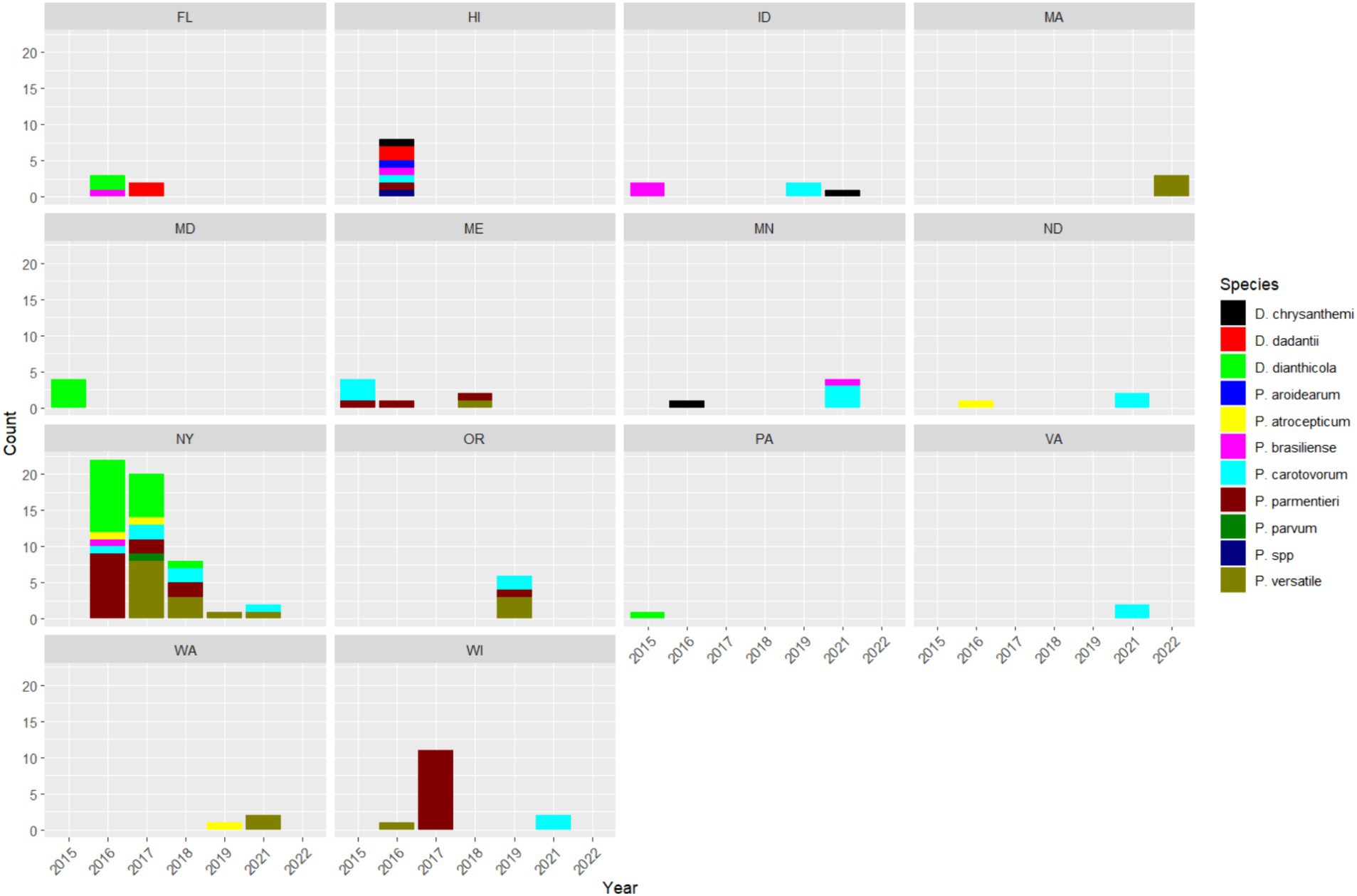
Figure 3. Counts of SRP species identified in each state between 2015 and 2022. The SRP species are represented by different colors.
Phylogeny and epidemiology
We constructed a phylogenetic tree to examine the evolutionary relationships among our isolates and reference strains using 499 single copy orthologous genes shared by the isolates in this study and reference strains from the RefSeq database curated by NCBI (Figure 4). The phylogenetic tree separates the Pectobacterium and Dickeya isolates and reference strains into two distinct clades. The Dickeya clade has generally longer branches than the Pectobacterium clade, which indicates more evolutionary changes per nucleotide after splitting from their closest common ancestor (Figure 4). The isolates, whose species identified by ANI, always clustered with the reference strain in a monophyletic clade, which agrees with the ANI results (Figure 4).

Figure 4. Phylogenetic relationships of soft rot Pectobacteriaceae strains from this study and reference strains of Pectobacterium and Dickeya based on the concatenated 499 single copy orthologue genes shared by the 118 potato isolates, a dahlia isolate NY1897, 21 Pectobacterium reference strains, and 12 Dickeya reference strains. The query isolates are labeled with strain ID, followed by the state and the year of isolation; the reference strains are labeled with their complete scientific names. Monophyletic clades that contain reference strains and query isolates were labeled with their species names. Bootstrap values were calculated from 1,000 replications of UFBoot2 bootstrapping. The length of the scale bar indicates 0.2 change per nucleotide site.
We conducted SNP analyses for our four most frequently isolated species (D. dianthicola, P. parmentieri, P. versatile, and P. carotovorum) to examine the subspecies phylogenetic relationships between our isolates and strains from the RefSeq database. This method was chosen because the accumulation of SNPs among the individuals in these species should produce a very high-resolution phylogeny and allow us to examine the progress of diversification and to test whether we can detect evidence of transmission between potato production regions (Wilson, 2012). Overall, we identified 96,190 SNPs in D. dianthicola group, 74,070 SNPs in P. parmentieri, 337,091 SNPs in P. carotovorum, and 281,515 SNPs in P. versatile.
All the D. dianthicola strains from the present study form a monophyletic clade represented by the reference strain ME23 (Ma et al., 2019) (Figure 5). This clade also includes most northeastern US outbreak strains such as the Delaware strain, DE440, West Virginia strain, WV516, Maine strains ME23, ME30, ME21T, New York strains 16LI01, 16LI02, 16LI04, Virginia strains 16JP03, 16JP05, Massachusetts strain 16MA15T, New Jersey strains 16MB01, Pennsylvania strain PA24 (Ge et al., 2021b) and all sequenced New York isolates (Figure 5). However, a few Northeastern strains are not inside this clade. The New Jersey strain 16NJ11, Pennsylvania strain 16PA07, and a Maine water isolate 59 W formed an independent clade. Another New Jersey strain 16NJ12 groups with some European strains (Figure 6) (Ge et al., 2021b). Interestingly, two non-potato strains from New York, NY1897 (isolated from dahlia) and 67-19 (isolated from impatiens) (Liu et al., 2021), do not belong to any of the three clades (Figure 5).
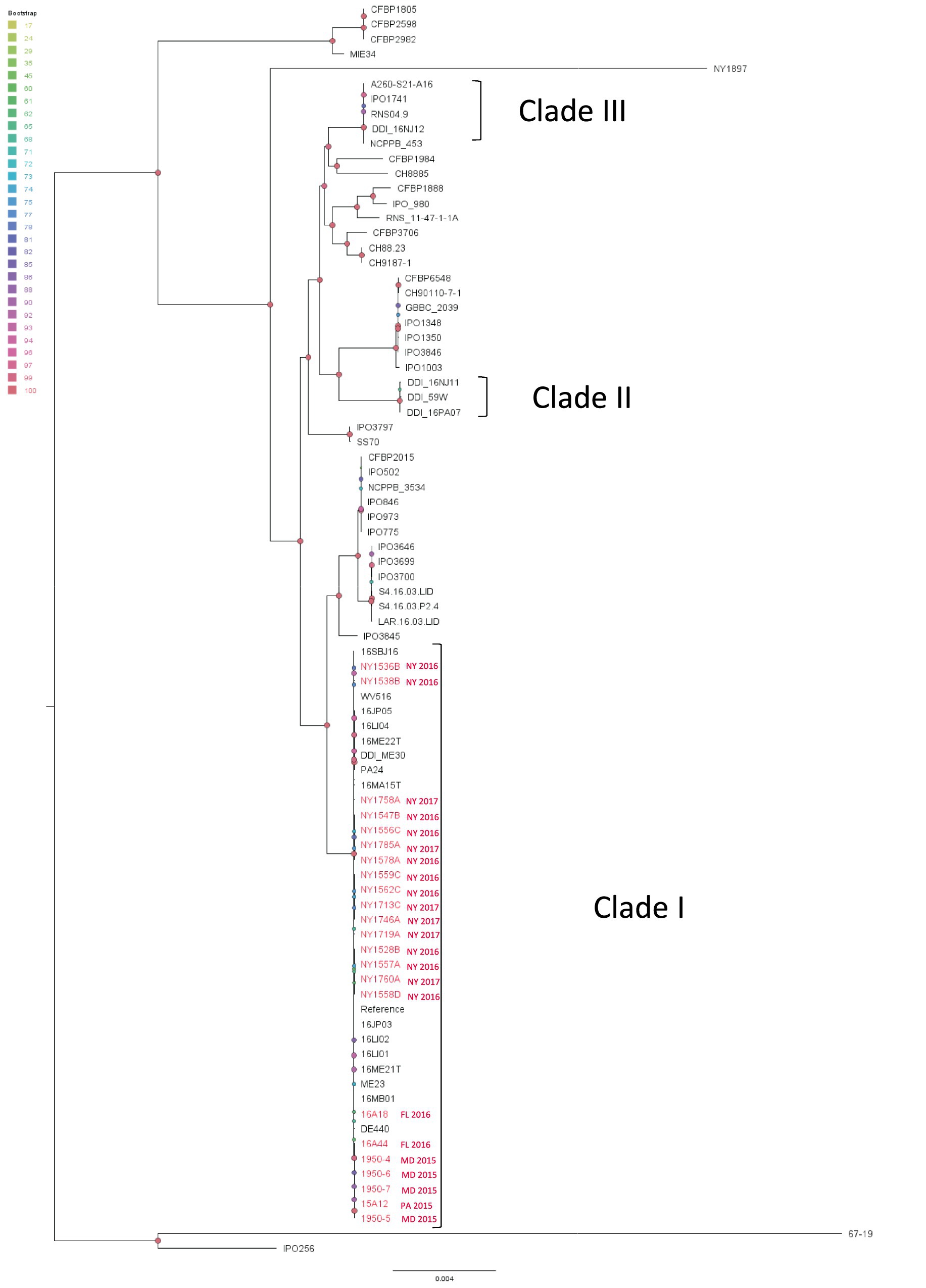
Figure 5. Single nucleotide polymorphism (SNP) based phylogenetic relationship between the Dickeya dianthicola isolates from this study (red taxa) and those from the RefSeq database, NCBI (black taxa). The query genomes were compared with the reference genome to identify the SNPs. ME23 was chosen as the reference genome in the SNP analysis. Bootstrap values were calculated from 1,000 replications of UFBoot2 bootstrapping. The length of the scale bar indicates 0.004 change per nucleotide site.
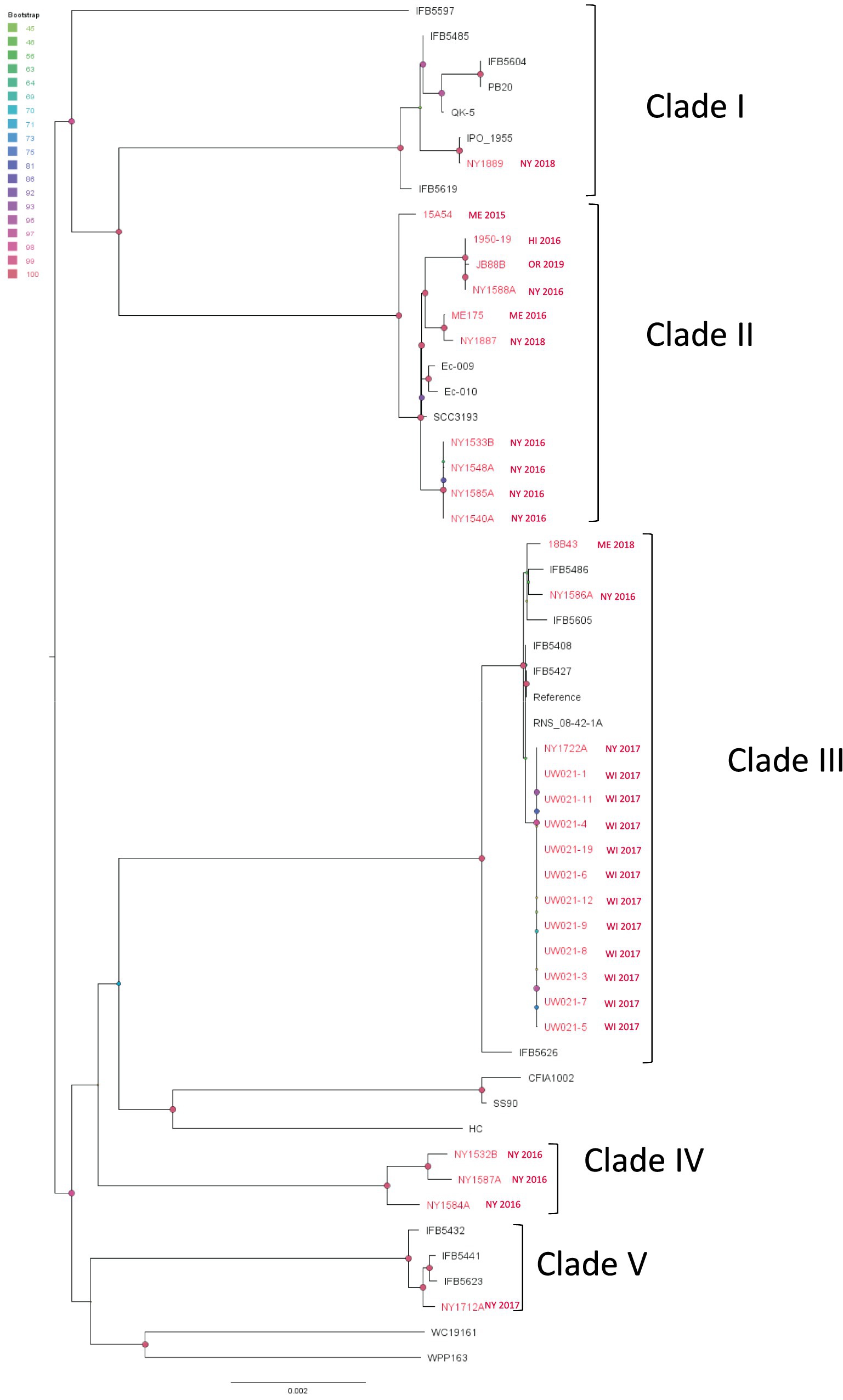
Figure 6. Single nucleotide polymorphism (SNP) based phylogenetic relationship between the Pectobacterium parmentieri strains from this study (red taxa) and from the NCBI RefSeq database (black taxa). The query genomes were compared with the reference genome to identify the SNPs. IFB5427 was chosen as the reference in the SNP analysis. Bootstrap values were calculated from 1,000 replications of UFBoot2 bootstrapping. The length of the scale bar indicates 0.002 change per nucleotide site.
From the SNP analysis of P. parmentieri isolates, we observed that strains in Clade I, II, III, and V were grouped with strains isolated from different continents. And one clade (Clade IV) contains only New York strains (Figure 6). In Clade II, we saw that strains from the east and west coast of the US as well as from New Zealand (Ec-009, Ec-010) and Finland (SCC3193) diverged from a recent ancestor. Note that Clade II isolates from Wisconsin (UW021-XX isolates) were all obtained from only two diseased potato samples (eight from a stem sample and three from a tuber sample). Those Wisconsin isolates are almost clonal along with a New York strain NY1722A. This Wisconsin clade exhibited minimal evolutionary changes from other US and European isolates within Clade II, which implicated a widespread global distribution of P. parmentieri strains of this clade. Likewise, in Clade III, strains from New York, Wisconsin, Maine, Belgium (IFB5486), Poland (IFB5605 IFB5608, IFB5427), and France (RNS_08-42-1A) shared a recent common ancestor. Another group that has pandemic features is Clade V, which includes strains from New York State and Poland (IFB5432, IFB5441, IFB5623). Additionally, the 2018 New York strain NY1889 is almost identical to IPO_1995, collected in 2002 with unknown origin (Figure 6). These observations suggest that some strains of P. parmentieri were introduced to the US from at least five different sources.
The SNP phylogenetic tree of P. carotovorum shows that there is more variation among these isolates compared to the D. dianthicola and P. parmentieri isolates. However, there were six cases of two to three nearly clonal P. carotovorum isolates from the same state and same year forming small clusters (Figure 7). The evolutionary changes among those small clusters are significant, as indicated by the long branches between these clusters. This pattern suggests this species has become endemic to many different areas with localized spread within the same year. The isolates from our study (red taxa in Figure 7) did not cluster with any published strains, except 1950-33 (isolated from Minnesota potato in 2021), which has a recent common ancestor with LMG_2410 (isolated from United Kingdom cucumber in 1957) (Figure 7). This finding suggests the intercontinental transmission and a possible host jump might have occurred to this cluster. However, more historic and modern strains are needed to test the hypothesis.
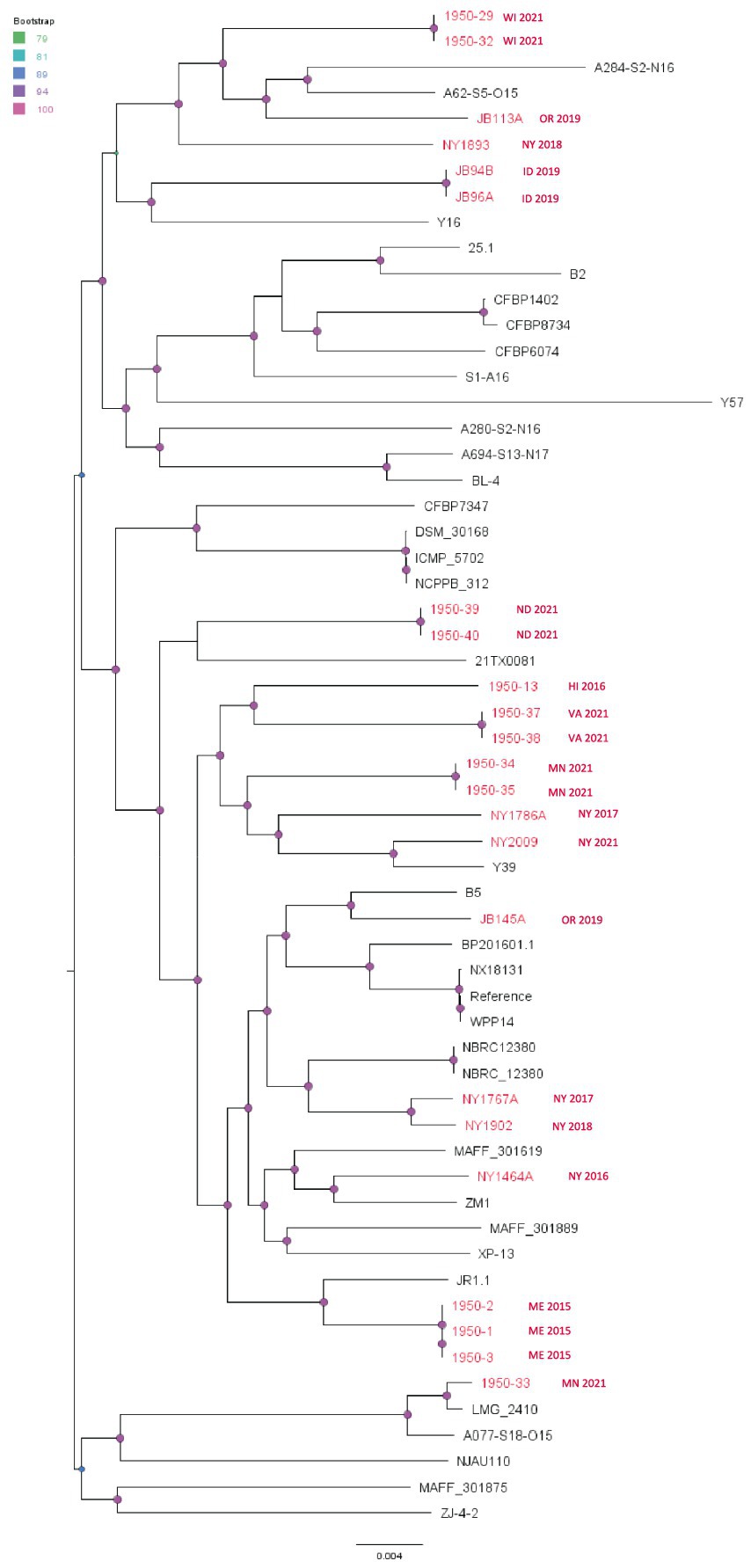
Figure 7. Single nucleotide polymorphism (SNP) based phylogenetic relationship between the Pectobacterium carotovorum strains from this study (red taxa) from the RefSeq database, NCBI. The query genomes were compared with the reference genome to identify the SNPs. WPP14 was chosen as the reference in the SNP analysis. Bootstrap values were calculated from 1,000 replications of UFBoot2 bootstrapping. The length of the scale bar indicates 0.004 change per nucleotide site.
The P. versatile SNP analysis showed similar patterns as found for P. carotovorum. Most of our isolates did not cluster in large clades with recent divergence, but some isolates from the same location and year seem to be clonal (Figure 8). Generally, the branch lengths among different strains are long, which indicates long independent evolutionary histories among those strains. We did not see any strain emerge as a dominant genotype. Therefore, the phylogenetic relationship of P. versatile appears to follow a pattern of endemic populations (Figure 8).
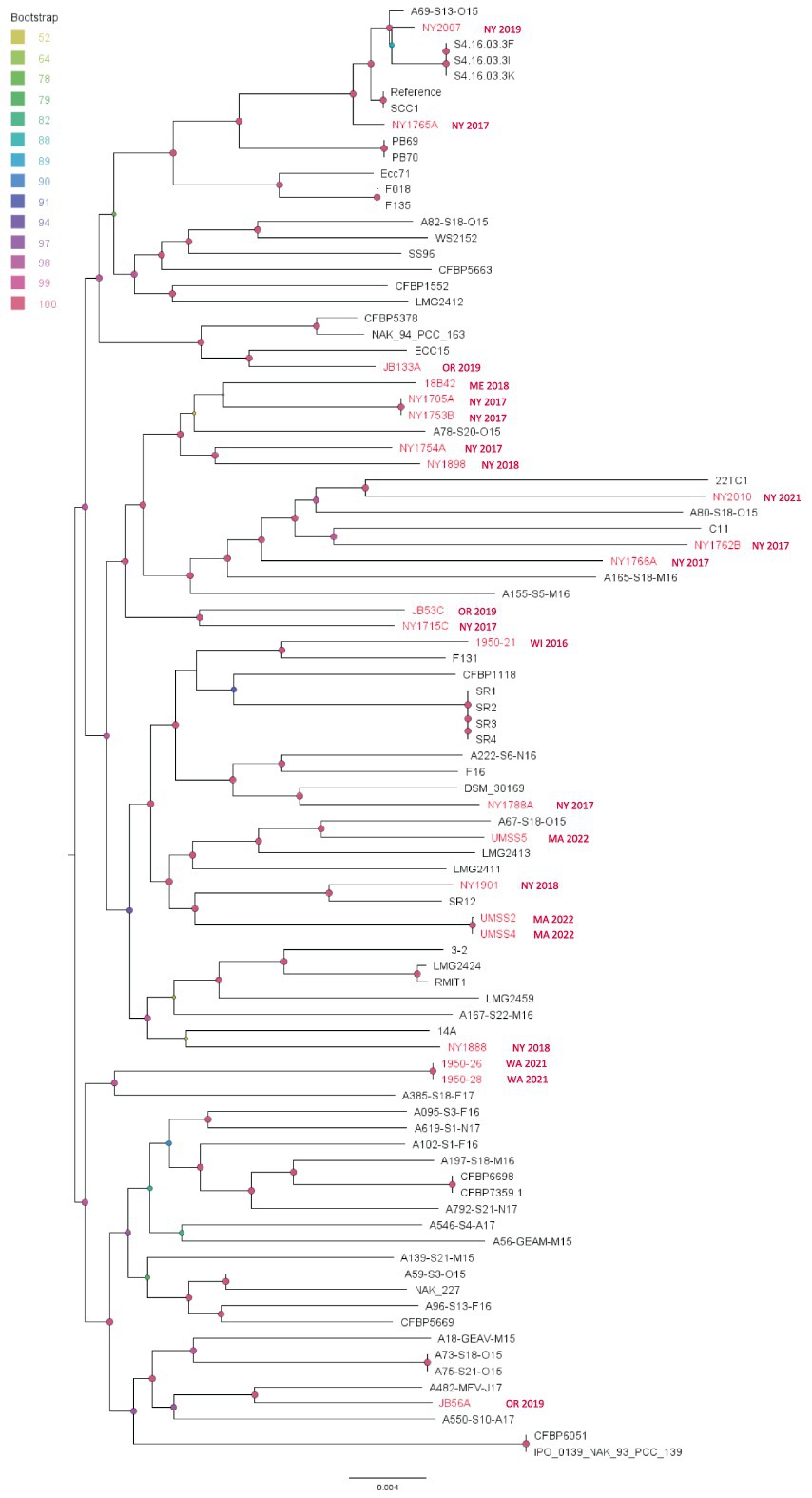
Figure 8. Single nucleotide polymorphism (SNP) based phylogenetic relationships between the Pectobacterium versatile strains from this study (red taxa) and from the NCBI RefSeq database, NCBI (black taxa). Bootstrap values were calculated from 1,000 replications of UFBoot2 bootstrapping. The length of the scale bar indicates 0.004 change per nucleotide site. SCC1 was chosen as the reference in the SNP analysis.
Discussion
In this study we used whole-genome sequencing-based methods to analyze 118 potato SRP strains isolated from 14 states across the US. We identified three Dickeya and eight Pectobacterium species. Aside from the unidentified species, the rest are known potato pathogens. Koch’s Postulates have been conducted on P. versatile isolates from New York, Oregon, and Washington States (Ma et al., 2021a; Ma et al., 2021b). In this study we verified the pathogenicity of the P. aroidearum from New York and the unidentified species from Hawaii. Although the bacterial isolates were submitted ad hoc by researchers from different states, this study provide a window into the SRP geographic distribution and their changes over time. Whole-genome sequencing based methods maximize the resolution in population diversity and enable us to infer the possible association of SRP species with diseases in different states.
D. dianthicola has been found in many northeastern and mid-Atlantic states including Delaware, Maine, Maryland, New Jersey, New York, Pennsylvania, Rhode Island, and Virginia. Other states including North Carolina, Michigan, as well as two provinces in Canada also reported D. dianthicola (Ge et al., 2021a; Ma et al., 2021b; Ma et al., 2018; Charkowski, 2018; Cai et al., 2018; Jiang et al., 2016; Patel et al., 2019; Curland et al., 2021). The phylogenetic tree built using 499 single copy orthologous genes showed that the D. dianthicola outbreak strains from the US can be classified into three clades, which agrees with the previous SNP analysis (Ge et al., 2021b). Clade I and Clade III in our study correspond to the Sequencing Type 1 (ST1) and ST5, respectively, from a previous analysis using dnaJ, dnaX, and gyrB gene sequences (Curland et al., 2021). But with higher resolution genomic methods, we found that NY1556C assigned to ST2 in the previous study was retained in Clade I. We identified a D. dianthicola strain from Florida, which echoes the findings of the 2015–2016 investigation that also identified one D. dianthicola in Florida (Curland et al., 2021). To date, D. dianthicola appears restricted to the eastern and midwestern US, we did not identify nor has D. dianthicola been reported to have caused outbreaks in the Western US potato production areas. We are astonished by the transmission efficiency of the almost clonal Clade I strain in the eastern US, most likely facilitated by seed potato trade networks (Czajkowski et al., 2011). D. dianthicola represented by this clade have spread to nine states in the eastern US and we found that about 90% of the sequenced US D. dianthicola strains (RefSeq databased accessed on 12/22/2023) and 100% of the D. dianthicola isolates that we analyzed in this study are the Clade I strain. This clade does not contain any non-US strains, non-potato strains, or evidence of incremental adaptation from other potato strains. Therefore, its origin remains undetermined. However, the sudden prevalence of this clonal strain in the eastern US coincided with the 2014 outbreaks, strongly suggests its emergence or introduction was a significant factor in causing the widespread outbreaks in the eastern US.
P. parmentieri was previously classified under the species P. wasabiae and associated with aerial stem rot of potato in Washington State (Dung et al., 2012). Researchers found P. parmentieri to be a different species using whole-genome sequence methods (Zhang et al., 2016; Khayi et al., 2016). Later, P. parmentieri was recovered from potatoes with soft rot in Minnesota and North Dakota (McNally et al., 2017). The discovery of P. parmentieri in the northeastern US also coincided with the blackleg and soft rot outbreaks there (Ge et al., 2018; Ma et al., 2018; Curland et al., 2021). Our results indicate that the introduction of P. parmentieri to the US growing fields was probably from at least five sources. The SNP analysis suggests that most of the isolates in each clade are unlikely to be clonal except for some Clade II isolates, but even so, the P. parmentieri we isolated across the US have much fewer sequence differences within clades than endemic strains as seen with P. carotovorum and P. versatile. This finding suggests that their introduction into the US occurred multiple times and probably earlier than the 2014 Dickeya outbreaks. But more genome sequences of historic and/or current P. parmentieri strains are needed to test this hypothesis.
P. carotovorum (previously known as Erwinia carotovorum) was one of the earliest described SRP pathogens and, until recently, most SRP isolates were classified as subspecies of P. carotovorum (Skerman et al., 1980). Seven new species were later elevated from former P. carotovorum subspecies, including P. atrosepticum, P. betavasculorum, P. wasabiae, P. actinidiae, P. carotovorum, P. brasiliense, and P. oderiferum (Gardan et al., 2003; Portier et al., 2019). Some species, such as P. aroidearum and P. versatile, that were previously considered as P. carotovorum were elevated to distinct species (Portier et al., 2019; Nabhan et al., 2013). We observed in our study that the modern P. carotovorum strains persist in significant numbers, exhibiting widespread distribution in potato-growing states with high genomic diversity. Given the long history and high diversity, P. carotovorum is a prevalent endemic pathogen in the US.
Although P. versatile was a recently defined species, the earliest known example of a US P. versatile strain was isolated from an iris sample 1946 (Portier et al., 2019). Globally, P. versatile has been reported from diverse vegetable and ornamental hosts in France, United Kingdom, The Netherlands, Morocco, Algeria, Lebanon, Russia, Serbia, Korea, China, and Canada (Portier et al., 2019; Shirshikov et al., 2018; Marković et al., 2022; Park et al., 2023; Su et al., 2023). In the US, P. versatile has been isolated from surface water in Maine and from potatoes in New York, Minnesota, North Dakota, Oregon, and Washington (Ma et al., 2021a; Ma et al., 2021b; Curland et al., 2021). In this study, we isolated P. versatile strains from potato in two more states: Maine and Massachusetts. P. versatile is one of the most isolated species of SRP across the US potato growing regions. The level of genomic diversity within the species suggests its long-term endemic nature.
P. brasiliense appears to have a global distribution. It has been reported from potato tubers in Switzerland, Poland, Russia, Kenya, Turkey, China, and Japan (Onkendi and Moleleki, 2014; Ozturk and Aksoy, 2016; Fujimoto et al., 2017; Voronina et al., 2019; de Werra et al., 2015; Waleron et al., 2015; Zhao et al., 2018). In the US, P. brasiliense has been recently reported on potatoes from Minnesota, Pennsylvania, and Hawaii (Boluk et al., 2020; Zhang et al., 2023; Curland et al., 2021). The same species has also been isolated from diseased sugar beets in North Dakota and Minnesota (Secor et al., 2016). Our study discovered that P. brasiliense is infecting potatoes in four additional states: New York, Florida, Wisconsin, and Idaho.
Strains of D. chrysanthemi were previously isolated in Minnesota (Curland et al., 2021). In addition to Minnesota, we also found strains in Idaho and Hawaii. Compared to Curland and colleague’s study, we did not find any P. polaris and P. punjabense. But we found a single strain of P. parvum and, notably, it is the first time this species was isolated in New York. P. parvum is a recently named new species closely related to P. polaris (Pasanen et al., 2020). Some of P. parvum strains have been in the French Collection for Plant-Associated Bacteria since 1969 (Portier et al., 2020). These SRP strains were only observed in small numbers and their presence fluctuated from year to year.
We reported one strain of P. aroidearum from Hawaii. P. aroidearum is a pathogen considered to prefer monocotyledon hosts but can also infect dicotyledons (Nabhan et al., 2013). It has been isolated in Asia, South America, and South Africa (Moraes et al., 2017; Li et al., 2022; Tang et al., 2021; Chen et al., 2020; Nabhan et al., 2013). But most of P. aroidearum strains were not isolated from potato samples except one report from Lebanon (Moretti et al., 2016). This is the first time that P. aroidearum has been found associated with disease in potatoes on US soil and we have conducted pathogenicity tests and complete Koch’s Postulates on this isolate (Figure 1). Interestingly, the Dickeya species that cause diseases on monocots (e.i. D. zeae, D. parazeae, and D. oryza) form a clade, which agrees with the previous studies (Yishay et al., 2008).
In conclusion, our study reveals that, across a wide range of US potato growing regions, we identified three Dickeya species and eight Pectobacterium species in diseased potatoes between 2015 and 2022. Among which, D. dianthicola, P. parmentieri, P. carotovorum, and P. versatile were the most prevalent species, making up to 83% of the isolates. All D. dianthicola from this study and 90% sequenced US D. dianthicola are so far almost clonal. The prevalence of this group of D. dianthicola temporally and geographically coincided with the blackleg and soft rot outbreaks between 2014 and 2018 in the northeastern US. The genomic diversity of P. parmentieri suggests that some strains were introduced earlier than D. dianthicola to the US from at least five different sources. In contrast, P. carotovorum and P. versatile seem to be widespread endemic species in the US.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
XM: Conceptualization, Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. XZ: Resources, Writing – review & editing. PS: Software, Writing – review & editing. RR: Resources, Writing – review & editing. SS: Resources, Writing – review & editing. BB: Resources, Writing – review & editing. HR: Resources, Writing – review & editing. KF: Resources, Writing – review & editing. JH: Resources, Writing – review & editing. GS: Resources, Writing – review & editing. BS: Conceptualization, Funding acquisition, Project administration, Supervision, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was partially funded by the National Institute for Food and Agriculture, Specialty Crops Research Initiative (2017-51181-26827). The sampling and isolation conducted at University of Wisconsin-Madison was also funded by Specialty Crop Block Grant and Wisconsin Potato and Vegetable Growers Association under the project title “Development of next-generation detection methods and improved biological understanding for tuber necrotic viruses in Wisconsin seed potato production.”
Acknowledgments
We would like to show special gratitude to Professor Keith Perry for his contributions to this project. We are grateful to Sandra Menasha (Cornell Cooperative Extension) for identifying and collecting diseased samples and communicating with growers. We thank Zhongmeng Bao, Zichu Yang, Haibi Wang, Kensy Rodriguez-Herrera, and Fatemeh Ekbataniamiri at Cornell University for facilitating our research and reviewing the manuscript. We thank Jenna Rach who worked with Dr. Rioux at University of Wisconsin-Madison, Department of Plant Pathology. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the United States Department of Agriculture. USDA is an equal opportunity provider and employer.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1403121/full#supplementary-material
Footnotes
References
Bertani, G. (2004). Lysogeny at mid-twentieth century: P1, P2, and other experimental systems. J. Bacteriol. 186, 595–600. doi: 10.1128/JB.186.3.595-600.2004
Boluk, G., Arizala, D., Ocenar, J., Mokwele, J., Silva, J., Dobhal, S., et al. (2020). First report of Pectobacterium brasiliense causing soft rot on Brassica oleracea var. sabellica in Hawaii, United States. Plant Dis. 104:2721. doi: 10.1094/PDIS-04-20-0701-PDN
Booher, N. J., Carpenter, S. C. D., Sebra, R. P., Wang, L., Salzberg, S. L., Leach, J. E., et al. (2015). Single molecule real-time sequencing of Xanthomonas oryzae genomes reveals a dynamic structure and complex TAL (transcription activator-like) effector gene relationships. Microb. Genom. 1:e000032. doi: 10.1099/mgen.0.000032
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cai, W., Srivastava, S. K., Stulberg, M. J., Nakhla, M. K., and Rascoe, J. (2018). Draft genome sequences of two Dickeya dianthicola isolates from potato. Genome Announc. 6, e00115–e00118. doi: 10.1128/genomeA.00115-18
Charkowski, A. O. (2018). The changing face of bacterial soft-rot diseases. Annu. Rev. Phytopathol. 56, 269–288. doi: 10.1146/annurev-phyto-080417-045906
Chen, L., Lin, P., and Huang, C. (2020). First report of Pectobacterium aroidearum causing soft rot disease of white Calla lily in Taiwan. Plant Dis. 104:563. doi: 10.1094/PDIS-07-19-1462-PDN
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Curland, R. D., Mainello, A., Perry, K. L., Hao, J., Charkowski, A. O., Bull, C. T., et al. (2021). Species of Dickeya and Pectobacterium isolated during an outbreak of blackleg and soft rot of potato in northeastern and north Central United States. Microorganisms 9:1733. doi: 10.3390/microorganisms9081733
Czajkowski, R., de Boer, W. J., Velvis, H., and van der Wolf, J. M. (2010). Systemic colonization of potato plants by a soilborne, green fluorescent protein-tagged strain of Dickeya sp. biovar 3. Phytopathology 100, 134–142. doi: 10.1094/PHYTO-100-2-0134
Czajkowski, R., Pérombelon, M. C. M., Jafra, S., Lojkowska, E., Potrykus, M., van der Wolf, J. M., et al. (2015). Detection, identification and differentiation of Pectobacterium and Dickeya species causing potato blackleg and tuber soft rot: a review. Ann. Appl. Biol. 166, 18–38. doi: 10.1111/aab.12166
Czajkowski, R., Pérombelon, M. C. M., van Veen, J. A., and van der Wolf, J. M. (2011). Control of blackleg and tuber soft rot of potato caused by Pectobacterium and Dickeya species: a review. Plant Pathol. 60, 999–1013. doi: 10.1111/j.1365-3059.2011.02470.x
de Werra, P., Bussereau, F., Keiser, A., and Ziegler, D. (2015). First report of potato blackleg caused by Pectobacterium carotovorum subsp. brasiliensein Switzerland. Plant Dis. 99:551. doi: 10.1094/PDIS-07-14-0742-PDN
Dees, M. W., Lysoe, E., Rossmann, S., Perminow, J., and Brurberg, M. B. (2017). Pectobacterium polaris sp. nov., isolated from potato (Solanum tuberosum). Int. J. Syst. Evol. Microbiol. 67, 5222–5229. doi: 10.1099/ijsem.0.002448
Dung, J. K. S., Johnson, D. A., and Schroeder, B. K. (2012). First report of Pectobacterium wasabiae causing aerial stem rot of potato in Washington state. Plant Dis. 96:1819. doi: 10.1094/PDIS-05-12-0444-PDN
Fujimoto, T., Yasuoka, S., Aono, Y., Nakayama, T., Ohki, T., Sayama, M., et al. (2017). First report of potato blackleg caused by Pectobacterium carotovorum subsp. brasiliense in Japan. Plant Dis. 101:241. doi: 10.1094/PDIS-06-16-0928-PDN
Gardan, L., Gouy, C., Christen, R., and Samson, R. (2003). Elevation of three subspecies of Pectobacterium carotovorum to species level: Pectobacterium atrosepticum sp. nov., Pectobacterium betavasculorum sp. nov. and Pectobacterium wasabiae sp. nov. Int. J. Syst. Evol. Microbiol. 53, 381–391. doi: 10.1099/ijs.0.02423-0
Ge, T. L., Jiang, H. H., Hao, J. J., and Johnson, S. B. (2018). First report of Pectobacterium parmentieri causing bacterial soft rot and blackleg on potato in Maine. Plant Dis. 102:437. doi: 10.1094/PDIS-05-17-0659-PDN
Ge, T., Jiang, H., Johnson, S. B., Larkin, R. P., Charkowski, A. O., Secor, G., et al. (2021a). Genotyping Dickeya dianthicola causing potato blackleg and soft rot outbreak associated with inoculum geography in the United States. Plant Dis. 105, 1976–1983. doi: 10.1094/PDIS-10-20-2138-RE
Ge, T., Jiang, H., Tan, E. H., Johnson, S. B., Larkin, R. P., Charkowski, A. O., et al. (2021b). Pangenomic analysis of Dickeya dianthicola strains related to the outbreak of blackleg and soft rot of potato in the United States. Plant Dis. 105, 3946–3955. doi: 10.1094/PDIS-03-21-0587-RE
Hauben, L., Moore, E. R., Vauterin, L., Steenackers, M., Mergaert, J., Verdonck, L., et al. (1998). Phylogenetic position of phytopathogens within the Enterobacteriaceae. Syst. Appl. Microbiol. 21, 384–397. doi: 10.1016/S0723-2020(98)80048-9
Hu, J., Fan, J., Sun, Z., and Liu, S. (2020). NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255. doi: 10.1093/bioinformatics/btz891
Hugouvieux-Cotte-Pattat, N., Jacot-des-Combes, C., and Briolay, J. (2019). Dickeya lacustris sp. nov., a water-living pectinolytic bacterium isolated from lakes in France. Int. J. Syst. Evol. Microbiol. 69, 721–726. doi: 10.1099/ijsem.0.003208
Hugouvieux-Cotte-Pattat, N., and Van Gijsegem, F. (2021). Diversity within the Dickeya zeae complex, identification of Dickeya zeae and Dickeya oryzae members, proposal of the novel species Dickeya parazeae sp. nov. Int. J. Syst. Evol. Microbiol. 71, 1–10. doi: 10.1099/ijsem.0.005059
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jiang, H. H., Hao, J. J., Johnson, S. B., Brueggeman, R. S., and Secor, G. (2016). First report of Dickeya dianthicola causing blackleg and bacterial soft rot on potato in Maine. Plant Dis. 100:2320. doi: 10.1094/PDIS-12-15-1513-PDN
Khayi, S., Cigna, J., Chong, T. M., Quetu-Laurent, A., Chan, K. G., Helias, V., et al. (2016). Transfer of the potato plant isolates of Pectobacterium wasabiae to Pectobacterium parmentieri sp. nov. Int. J. Syst. Evol. Microbiol. 66, 5379–5383. doi: 10.1099/ijsem.0.001524
Li, L., Yuan, L., Zhao, Y., Shi, Y., Chai, A., Xie, X., et al. (2022). Emergence of bacterial soft rot in calla lily caused by Pectobacterium aroidearum in China. Crop Prot. 152:105854. doi: 10.1016/j.cropro.2021.105854
Liu, Y., Vasiu, S., Daughtrey, M., and Filiatrault, M. J. (2021). First report of Dickeya dianthicola causing blackleg on New Guinea Impatiens (Impatiens hawkeri) in New York state, U.S.a. Plant Dis. 105:1192. doi: 10.1094/PDIS-09-20-2020-PDN
Ma, X., Brazil, J., Rivedal, H. M., Perry, K. L., Frost, K., and Swingle, B. (2021a). First report of Pectobacterium versatile causing potato soft rot in Oregon and Washington. Plant Dis. 106:1292. doi: 10.1094/PDIS-08-21-1635-PDN
Ma, B., Hibbing, M. E., Kim, H. S., Reedy, R. M., Yedidia, I., Breuer, J., et al. (2007). Host range and molecular phylogenies of the soft rot enterobacterial genera Pectobacterium and Dickeya. Phytopathology 97, 1150–1163. doi: 10.1094/PHYTO-97-9-1150
Ma, X., Perna, N. T., Glasner, J. D., Hao, J., Johnson, S., Nasaruddin, A. S., et al. (2019). Complete genome sequence of Dickeya dianthicola ME23, a pathogen causing blackleg and soft rot diseases of potato. Microbiol. Resour. Announc. 8, e01526–e01518. doi: 10.1128/MRA.01526-18
Ma, X., Schloop, A., Swingle, B., and Perry, K. L. (2018). Pectobacterium and Dickeya responsible for potato blackleg disease in New York state in 2016. Plant Dis. 102, 1834–1840. doi: 10.1094/PDIS-10-17-1595-RE
Ma, X., Stodghill, P., Gao, M., Perry, K. L., and Swingle, B. (2021b). Identification of Pectobacterium versatile causing blackleg of potato in New York state. Plant Dis. 105, 2585–2594. doi: 10.1094/PDIS-09-20-2089-RE
Marković, S., Milić Komić, S., Jelušić, A., Iličić, R., Bagi, F., Stanković, S., et al. (2022). First report of Pectobacterium versatile causing blackleg of potato in Serbia. Plant Dis. 106:312. doi: 10.1094/PDIS-06-21-1128-PDN
McNally, R. R., Curland, R. D., Webster, B. T., Robinson, A. P., and Ishimaru, C. A. (2017). First report of blackleg and tuber soft rot of potato caused by Pectobacterium parmentieri in Minnesota and North Dakota. Plant Dis. 101:PDIS-04-17-0608-PDN. doi: 10.1094/PDIS-04-17-0608-PDN
Meier-Kolthoff, J. B., Carbasse, J. S., Peinado-Olarte, R. L., and Göker, M. (2022). TYGS and LPSN: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes Nucleic Acids Res. 50 (D1), D801-D807.
Moraes, A., Souza, E., Mariano, R., Silva, A., Lima, N., Peixoto, A., et al. (2017). First report of Pectobacterium aroidearum and Pectobacterium carotovorum subsp. brasiliensis causing soft rot of Cucurbita pepo in Brazil. Plant Dis. 101:379. doi: 10.1094/PDIS-08-16-1168-PDN
Moretti, C., Fakhr, R., Cortese, C., De Vos, P., Cerri, M., Geagea, L., et al. (2016). Pectobacterium aroidearum and Pectobacterium carotovorum subsp. carotovorum as causal agents of potato soft rot in Lebanon. Eur. J. Plant Pathol. 144, 205–211. doi: 10.1007/s10658-015-0743-3
Nabhan, S., De Boer, S. H., Maiss, E., and Wydra, K. (2013). Pectobacterium aroidearum sp. nov., a soft rot pathogen with preference for monocotyledonous plants. Int. J. Syst. Evol. Microbiol. 63, 2520–2525. doi: 10.1099/ijs.0.046011-0
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Onkendi, E. M., and Moleleki, L. N. (2014). Characterization of Pectobacterium carotovorum subsp. carotovorum and brasiliense from diseased potatoes in Kenya. Eur. J. Plant Pathol. 139, 557–566. doi: 10.1007/s10658-014-0411-z
Oulghazi, S., Cigna, J., Lau, Y. Y., Moumni, M., Chan, K. G., and Faure, D. (2019). Transfer of the waterfall source isolate Pectobacterium carotovorum M022 to Pectobacterium fontis sp. nov., a deep-branching species within the genus Pectobacterium. Int. J. Syst. Evol. Microbiol. 69, 470–475. doi: 10.1099/ijsem.0.003180
Ozturk, M., and Aksoy, H. (2016). First report of Pectobacterium carotovorum subsp. brasiliense causing blackleg and soft rot of potato in Turkey. J. Plant Pathol. 98:692. doi: 10.4454/JPP.V98I3.055
Park, K.-T., Hong, S.-M., Back, C.-G., Cho, Y.-J., Lee, S.-Y., Ten, L. N., et al. (2023). First report of Pectobacterium versatile as the causal pathogen of soft rot in kimchi cabbage in Korea. Res. Plant Dis. 29, 72–78. doi: 10.5423/RPD.2023.29.1.72
Parkinson, N., DeVos, P., Pirhonen, M., and Elphinstone, J. (2014). Dickeya aquatica sp. nov., isolated from waterways. Int. J. Syst. Evol. Microbiol. 64, 2264–2266. doi: 10.1099/ijs.0.058693-0
Pasanen, M., Waleron, M., Schott, T., Cleenwerck, I., Misztak, A., Waleron, K., et al. (2020). Pectobacterium parvum sp. nov., having a Salmonella SPI-1-like type III secretion system and low virulence. Int. J. Syst. Evol. Microbiol. 70:2440. doi: 10.1099/ijsem.0.004057
Patel, N., Baldwin, A., Patel, R., Kobayashi, D., and Wyenandt, C. (2019). First report of Dickeya dianthicola causing blackleg and soft rot on potato (Solanum tuberosum) in New Jersey, USA. Plant Dis. 103:146. doi: 10.1094/PDIS-05-18-0775-PDN
Pedron, J., Bertrand, C., Taghouti, G., Portier, P., and Barny, M. A. (2019). Pectobacterium aquaticum sp. nov., isolated from waterways. Int. J. Syst. Evol. Microbiol. 69, 745–751. doi: 10.1099/ijsem.0.003229
Perombelon, M. C. M. (1974). The role of the seed tuber in the contamination by Erwinia carotovora of potato crops in Scotland. Potato Res. 17, 187–199. doi: 10.1007/BF02360386
Perombelon, M. C. M. (2002). Potato diseases caused by soft rot erwinias: an overview of pathogenesis. Plant Pathol. 51, 1–12. doi: 10.1046/j.0032-0862.2001.Shorttitle.doc.x
Perombelon, M. C. M., and Kelman, A. (1980). Ecology of the soft rot Erwinias. Annu. Rev. Phytopathol. 18, 361–387. doi: 10.1146/annurev.py.18.090180.002045
Portier, P., Pédron, J., Taghouti, G., Dutrieux, C., and Barny, M.-A. (2020). Updated taxonomy of Pectobacterium genus in the CIRM-CFBP bacterial collection: when newly described species reveal “old” endemic population. Microorganisms 8:1441. doi: 10.3390/microorganisms8091441
Portier, P., Pedron, J., Taghouti, G., Fischer-Le Saux, M., Caullireau, E., Bertrand, C., et al. (2019). Elevation of Pectobacterium carotovorum subsp. odoriferum to species level as Pectobacterium odoriferum sp. nov., proposal of Pectobacterium brasiliense sp. nov. and Pectobacterium actinidiae sp. nov., emended description of Pectobacterium carotovorum and description of Pectobacterium versatile sp. nov., isolated from streams and symptoms on diverse plants. Int. J. Syst. Evol. Microbiol. 69, 3207–3216. doi: 10.1099/ijsem.0.003611
Rosenzweig, N., Steere, L., Kirk, W. W., Mambetova, S., Long, C., Schafer, R., et al. (2016). First report of Dickeya dianthicola and Pectobacterium wasabiae causing aerial stem rot of potato in Michigan, USA. New Dis. Rep. 33:10. doi: 10.5197/j.2044-0588.2016.033.010
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular cloning: A laboratory manual. Cold Spring Harbor, NY: Cold spring Harbor Laboratory Press.
Schwengers, O., Hain, T., Chakraborty, T., and Goesmann, A. (2019). ReferenceSeeker: rapid determination of appropriate reference genomes. J. Open Source Softw. 5:1994. doi: 10.21105/joss.01994
Secor, G. A., Rivera-Varas, V. V., Brueggeman, R. S., Metzger, M. S., Rengifo, J., and Richards, J. K. (2016). First report of field decay of sugar beet caused by Pectobacterium carotovorum subsp. brasiliense in North America. Plant Dis. 100:2160. doi: 10.1094/PDIS-05-16-0588-PDN
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Seemann, T. (2015) snippy: fast bacterial variant calling from NGS reads. (version 4.6.0). Available at: https://github.com/tseemann/snippy/.GoogleScholar
Seppey, M., Manni, M., and Zdobnov, E. M. (2019). “BUSCO: assessing genome assembly and annotation completeness” in Gene Prediction. Methods in Molecular Biology. Ed. Kollmar, M. vol 1962. New York, NY: Humana.
Shirshikov, F. V., Korzhenkov, A. A., Miroshnikov, K. K., Kabanova, A. P., Barannik, A. P., Ignatov, A. N., et al. (2018). Draft genome sequences of new genomospecies “Candidatus Pectobacterium maceratum” strains, which cause soft rot in plants. Genome Announc. 6, e00260–e00218. doi: 10.1128/genomeA.00260-18
Skerman, V. B. D., McGowan, V., and Sneath, P. H. A. (1980). Approved lists of bacterial names. Int. J. Syst. Bacteriol. 30, 225–420.
Slawiak, M., van Beckhoven, J. R., Speksnijder, A. G., Czajkowski, R., Grabe, G., and van der Wolf, J. M. (2009). Biochemical and genetical analysis reveal a new clade of biovar 3 Dickeya spp. strains isolated from potato in Europe. Eur. J. Plant Pathol. 125, 245–261. doi: 10.1007/s10658-009-9479-2
Stevenson, W. R., Loria, R., Franc, G. D., and Weingartner, D. P. (2001). Compendium of potato diseases. St. Paul: American Phytopathological Society, 106.
Su, Y., Li, X., Li, L., Lukianova, A., Tokmakova, A., Chen, C., et al. (2023). Occurrence, characteristics, and qPCR-based identification of Pectobacterium versatile causing soft rot of Chinese cabbage in China. Plant Dis. 107, 2751–2762. doi: 10.1094/PDIS-12-22-2770-RE
Tang, W.-Q., Chang, C.-Y., Lee, Y.-J., and Chu, C.-C. (2021). First report of Pectobacterium aroidearum causing bacterial soft rot of carrot in Taiwan. Plant Dis. 105:695. doi: 10.1094/PDIS-08-20-1824-PDN
Tian, Y., Zhao, Y., Yuan, X., Yi, J., Fan, J., Xu, Z., et al. (2016). Dickeya fangzhongdai sp. nov., a plant-pathogenic bacterium isolated from pear trees (Pyrus pyrifolia). Int. J. Syst. Evol. Microbiol. 66, 2831–2835. doi: 10.1099/ijsem.0.001060
Toll-Riera, M., San Millan, A., Wagner, A., and MacLean, R. C. (2016). The genomic basis of evolutionary innovation in Pseudomonas aeruginosa. PLoS Genet. 12:e1006005. doi: 10.1371/journal.pgen.1006005
Toth, I. K., Barny, M.-A., Czajkowski, R., Elphinstone, J. G., Li, X., Pédron, J., et al. (2021). “Pectobacterium and dickeya: taxonomy and evolution” in Plant Diseases Caused by Dickeya and Pectobacterium Species, 13–37.
Toth, I. K., van der Wolf, J. M., Saddler, G., Lojkowska, E., Hélias, V., Pirhonen, M., et al. (2011). Dickeya species: an emerging problem for potato production in Europe. Plant Pathol. 60, 385–399. doi: 10.1111/j.1365-3059.2011.02427.x
van der Wolf, J. M., De Boer, S. H., Czajkowski, R., Cahill, G., Van Gijsegem, F., Davey, T., et al. (2021). “Management of diseases caused by Pectobacterium and Dickeya species” in Plant diseases caused by Dickeya and Pectobacterium species, 175–214.
Vinuesa, P., Ochoa-Sanchez, L. E., and Contreras-Moreira, B. (2018). GET_PHYLOMARKERS, a software package to select optimal orthologous clusters for Phylogenomics and inferring Pan-genome phylogenies, used for a critical Geno-taxonomic revision of the genus Stenotrophomonas. Front. Microbiol. 9:771. doi: 10.3389/fmicb.2018.00771
Voronina, M., Kabanova, A., Shneider, M., Korzhenkov, A., Toschakov, S., Miroshnikov, K., et al. (2019). First report of Pectobacterium carotovorum subsp. brasiliense causing blackleg and stem rot disease of potato in Russia. Plant Dis. 103:364. doi: 10.1094/PDIS-03-18-0456-PDN
Waleron, M., Waleron, K., and Lojkowska, E. (2015). First report of Pectobacterium carotovorum subsp. brasiliense causing soft rot on potato and other vegetables in Poland. Plant Dis. 99:1271. doi: 10.1094/PDIS-02-15-0180-PDN
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wilson, D. J. (2012). Insights from Genomics into Bacterial Pathogen Populations. doi: 10.1371/journal.ppat.1002874
Yishay, M., Burdman, S., Valverde, A., Luzzatto, T., Ophir, R., and Yedidia, I. (2008). Differential pathogenicity and genetic diversity among Pectobacterium carotovorum ssp. carotovorum isolates from monocot and dicot hosts support early genomic divergence within this taxon. Environ. Microbiol. 10, 2746–2759. doi: 10.1111/j.1462-2920.2008.01694.x
Zhang, Y., Fan, Q., and Loria, R. (2016). A re-evaluation of the taxonomy of phytopathogenic genera Dickeya and Pectobacterium using whole-genome sequencing data. Syst. Appl. Microbiol. 39, 252–259. doi: 10.1016/j.syapm.2016.04.001
Zhang, X., Ma, X., Fan, X., Ge, T., Leiby, R. E., Swingle, B., et al. (2023). First report of Pectobacterium brasiliense causing bacterial blackleg and soft rot of potato in Pennsylvania. Plant Dis. 107:2512. doi: 10.1094/PDIS-09-22-2085-PDN
Zhao, Y., Dou, J., Geng, G., Tian, Y., Fan, J., Li, X., et al. (2018). First report of Pectobacterium carotovorum subsp. brasiliense causing blackleg and stem rot on potato in China. Plant Dis. 102:1653. doi: 10.1094/PDIS-11-17-1779-PDN
Keywords: soft rot and blackleg disease, Pectobacteriacea, Dickeya, whole genome sequencing, potato
Citation: Ma X, Zhang X, Stodghill P, Rioux R, Shrestha S, Babler B, Rivedal H, Frost K, Hao J, Secor G and Swingle B (2024) Analysis of soft rot Pectobacteriaceae population diversity in US potato growing regions between 2015 and 2022. Front. Microbiol. 15:1403121. doi: 10.3389/fmicb.2024.1403121
Edited by:
Quan Zeng, Connecticut Agricultural Experiment Station, United StatesReviewed by:
Iris Yedidia, Agricultural Research Organization (ARO), IsraelLucy N. Moleleki, University of Pretoria, South Africa
Copyright © 2024 Ma, Zhang, Stodghill, Rioux, Shrestha, Babler, Rivedal, Frost, Hao, Secor and Swingle. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bryan Swingle, QnJ5YW4uU3dpbmdsZUB1c2RhLmdvdg==
†Present address: Renee Rioux, BASF Agricultural Solutions, Raleigh, NC, United States