
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 24 May 2024
Sec. Microbial Physiology and Metabolism
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1397314
 Xinyu Shi1†
Xinyu Shi1† Hao Yan2†Fang Yuan3,4†Guoying Li4
Hao Yan2†Fang Yuan3,4†Guoying Li4 Jingfang Liu2Chunli Li2Xiaobin Yu3*
Jingfang Liu2Chunli Li2Xiaobin Yu3* Zilong Li2*Yunping Zhu1*
Zilong Li2*Yunping Zhu1* Weishan Wang2,5*
Weishan Wang2,5*Transglutaminase (EC 2.3.2.13, TGase), an enzyme that catalyzes the formation of covalent cross-links between protein or peptide molecules, plays a critical role in commercial food processing, medicine, and textiles. TGase from Streptomyces is the sole commercial enzyme preparation for cross-linking proteins. In this study, we revealed that the SOS response repressor protein LexA in Streptomyces mobaraensis not only triggers morphological development but also enhances TGase synthesis. The absence of lexA significantly diminished TGase production and sporulation. Although LexA does not bind directly to the promoter region of the TGase gene, it indirectly stimulates transcription of the tga gene, which encodes TGase. Furthermore, LexA directly enhances the expression of genes associated with protein synthesis and transcription factors, thus favorably influencing TGase synthesis at both the transcriptional and posttranscriptional levels. Moreover, LexA activates four crucial genes involved in morphological differentiation, promoting spore maturation. Overall, our findings suggest that LexA plays a dual role as a master regulator of the SOS response and a significant contributor to TGase regulation and certain aspects of secondary metabolism, offering insights into the cellular functions of LexA and facilitating the strategic engineering of TGase overproducers.
TGase, also known as transglutaminase (Enzyme commission number: EC 2.3.2.13, TGase), is a transferase enzyme that catalyzes cross-linking within or between protein molecules. It forms connections between proteins and amino acids and facilitates the hydrolysis of glutamine residues within a protein molecule (Gajsek et al., 2019; Bellio et al., 2020). TGase has significant applications in the pharmaceutical (Doti et al., 2020) and food industries. In the food industry, TGase is used to crosslink minced meat, resulting in the formation of whole meat and improving the quality of meat products (Santhi et al., 2017). It is also utilized in treating minced meat and ice cream to enhance food densification and overall taste (Gajsek et al., 2019). Currently, microbial-derived transglutaminase (TGase) is widely employed in industrial applications (Zhang et al., 2023). TGase can be derived from animals, fish, and microorganisms (Schneider et al., 2020). Among these sources, microbial strains, particularly Streptomyces mobaraensis, are globally recognized and approved by the U.S. Food and Drug Administration (FDA) for use as safe cross-linking agents in common foods (Kuraishi et al., 2001). Therefore, it is crucial to enhance the industrial productivity of S. mobaraensis TGase. Researchers worldwide are actively pursuing strategies such as screening high-yielding strains, optimizing growth media (Ceresino et al., 2018) and fermentation conditions, utilizing mutation breeding, and employing genetic engineering tools to enhance TGase production. Genetic engineering tools include methods such as replacing inducible promoters with constitutive promoters, using fusion signal peptides, modifying promoters, and implementing intracellular expression with constitutive promoters (Liu et al., 2016; Javitt et al., 2017; Mu et al., 2018). These approaches collectively aim to increase both the expression level and production efficiency of TGase. The synthesis and maturation of TGase in S. mobaraensis have been well studied. Specifically, the TGase-activating protease (TAMEP) catalyzes the cleavage of 41 amino acids at the N-terminal end of a zymogen of microbial transglutaminase (pro-MTG), resulting in the active intermediate with additional N-terminal tetrapeptide (Phe-Arg-Ala-Pro, FRAP-MTG). Subsequently, the tripeptidyl aminopeptidase of S. mobaraenesis (SM-TAP) cleaves FRAP-MTG, removing the N-terminal tetrapeptide and forming the mature and active MTG (Zotzel et al., 2003).
Streptomyces TGase, an important enzyme in the food industry, plays a significant role in improving food texture. Since the first discovery of bacterial TGase in 1989, microbial fermentation has been the main source of commercial TGase products. Increasing TGase yield has become a major focus of current research. Additionally, low-temperature stress has been found to effectively enhance TGase yield by influencing the mycelial differentiation of Streptomyces spp. (Chen et al., 2013).
During fermentation, TGase activation can be enhanced by eliminating the zymogen region with proteases or by facilitating secretion with the inclusion of cetyltrimethylammonium bromide (CTAB) (Fatima et al., 2019). Furthermore, it has been discovered that Streptomyces strains with increased production efficiency and safety can be obtained through random mutagenesis and targeted genetic modification, making them potential candidates for industrial TGase production (Yin et al., 2021). In the case of S. mobaraensis, TGase is expressed extracellularly as Pro-TG and is subsequently activated by a TGase-activated protease (TAP). Research suggests that TGase production can be increased by regulating the TAP activity of Streptomyces filamentosus. NH4+ has been shown to promote TGase activation (Yin et al., 2022). The understanding of the regulatory network governing TGase biosynthesis is still limited. At least 109 potential transcription factors that may interact with the tga promoter were identified through DNA affinity capture assay (Liu et al., 2023). Most of them have not been verified experimentally.
The protein LexA plays a crucial role as a transcriptional regulator in the SOS response. When DNA sustains severe damage, the SOS system is activated and inhibits LexA synthesis. This leads to the initiation of DNA damage repair (Janion, 2008). The lexA gene is present in all prokaryotes and is highly conserved. In addition to regulating the SOS response, the gene is involved in various biological functions, including repression of SOS pathway under non-stress conditions (Srivastava et al., 2023), and undergoes autoproteolysis in the setting of stress, resulting in derepression of SOS genes (Mo et al., 2014), mutation-induced alterations in cellular phenotypes, and modulation of pathogenicity in numerous bacteria (Walter et al., 2014). Current research on LexA proteins has focused mainly on understanding their molecular cleavage mechanisms, interactions with RecA proteins, conformational characteristics, and presence in cells. For example, in Streptomyces coelicolor, LexA directly inhibits the biosynthesis of actinomycin (Iqbal et al., 2012; Liu et al., 2013), while in Streptomyces venezuelae, LexA is involved in the regulation of DNA damage stress (Stratton et al., 2022).
Our investigation revealed a potential regulatory role of LexA in TGase synthesis in S. mobaraensis. Through a comprehensive analysis using differential protein expression analysis, we observed a positive correlation between LexA levels and TGase expression. To elucidate the functional aspects of LexA, we conducted experiments that confirmed its influence on developmental differentiation as well as its positive regulation of TGase expression in S. mobaraensis. Our results demonstrated that the overexpression of lexA improved TGase production. These findings highlight the diverse roles of LexA, implicating its involvement in essential cellular processes and the enhancement of TGase synthesis.
The bacterial strains and plasmids utilized in this study are listed in Supplementary Table S1, and the corresponding primers used are listed in Supplementary Table S2. S. mobaraensis and Escherichia coli were cultured in ISP2 and LB, respectively. Phenotyping of S. mobaraensis mutants was conducted on ISP2 solid medium. Conventional TGase production utilizes an insoluble fermentation medium with the following basic fermentation formula: glycerol 2%, peptone 2%, soya flour 2%, yeast 0.5%, corn syrup 0.55%, K2HPO4 0.4%, KH2PO4 0.2%, MgSO4 0.2%, and (NH4)2SO4 (Yin et al., 2021). The fermentation conditions involved maintaining the temperature at 30°C and agitating the mixture at 250 rpm. TSB soluble fermentation medium was selected for mycelia cultivation to accurately assess biomass in S. mobaraensis.
To generate a lexA deletion mutant, two fragments flanking lexA were amplified from the genomic DNA of S. mobaraensis NBRC 13819 strain. The 5′ flanking region, spanning 2,175 base pairs (positions −2,160 to +15 relative to the lexA start codon), and the 3′ flanking region, spanning 2,222 base pairs (positions +769 to +2,991), were amplified using primers SXY11/SXY12 and SXY13/SXY14, respectively (Supplementary Figure S1A).
The lexA deletion vector ΔlexA was constructed by ligating fragments amplified with SXY11/SXY14 into the EcoRI/HindIII sites of the pKC1132 plasmid (Bierman et al., 1992), and then transformed into NBRC 13819 protoplasts (Macneil and Klapko, 1987). Transformants on RM14 (Macneil and Klapko, 1987) regeneration medium were transferred to ISP2 plates for sporulation. Spores were spread on apramycin-containing ISP2 plates, which were incubated at 28°C for 2 days. pKC1132 is unable to replicate itself in Streptomyces. Therefore, only a single-crossover mutant with p∆lexA integrated into the chromosome was able to grow. Single-crossover mutants were incubated on ISP2 plates without apramycin for two rounds of sporulation, and colonies of double-crossover mutants were selected and verified by PCR analysis using the primers SXY3F/SXY3R (flanking the exchange region) and SXY4F/SXY4R (within the lexA deletion region) (Supplementary Figure S1B). Amplification with primers SXY3F/SXY3R produced a 4.5 kb band in WT DNA, while ΔlexA DNA yielded only a 3.7 kb band. With primers SXY4F/SXY4R, only WT DNA exhibited the expected 274 bp band (Supplementary Figure S1C). This process generated the lexA gene deletion mutant ΔlexA, which is characterized by the absence of lexA.
To achieve the overexpression of lexA, a 786 bp fragment of the lexA coding region and the strong promoter kasO* were amplified from DNA from S. mobaraensis NBRC 13819 and pDR4-K* (Wang et al., 2013) using the primers SXY21/SXY22 and SXY23/SXY24. The two fragments were digested with BamHI/EcoRI and HindIII/BamHI and then ligated simultaneously into EcoRI and HindIII sites of pSET152 to generate lexA overexpression vector pOlexA (Supplementary Figure S1D), which was then transformed into NBRC 13819 and TBJ3 to construct lexA overexpression strain OlexA and OlexATBJ3. For the complementation of ΔlexA, the amplification product containing the lexA coding region and its native promoter were ligated with the EcoRI/HindIII-digested pSET152 to create lexA-complemented vector pClexA, which was then introduced into ∆lexA to construct complemented strain ClexA (Supplementary Figure S1E).
The lexA coding region (261 amino acids) was amplified from S. mobaraensis NBRC 13819 using the primers SXY31/SXY32 and cloned and inserted into the pET-28a (+) plasmid to generate pET-lexA. The expression plasmids were introduced into E. coli Rosetta (DE3), and the recombinant E. coli were cultured in Luria-Bertani medium supplemented with 100 μg/mL kanamycin and 100 μg/mL chloramphenicol overnight at 37°C with shaking at 250 rpm. Subsequently, the seed culture was transferred to 1 L of LB medium containing equivalent antibiotic concentrations, and the inoculum size was adjusted to 1%. The culture was continued at 37°C for 3 h. At an OD600 of 0.8, the inducer isopropyl β-D-1 thiogalactolate was added at a final concentration of 100 μM, and induction was carried out at 16°C for 19 h. Bacteria containing recombinant protein were harvested, disrupted in lysis buffer (300 mM NaCl, 50 mM NaH2PO4, 10 mM, Imidazole) by sonication on ice, and centrifuged. His6-tagged recombinant protein in supernatant was purified on a column packed with Ni2+-NTA agarose beads (Bio-Works; Sweden) and eluted from resin by lysis buffer plus 200 mM imidazole. The purified His6-LexA were dialyzed against dialysis buffer (10 mM Tris-HCl, 1 mM EDTA, 80 mM NaCl, 4% glycerol, 20 mM β-mercaptoethanol, pH 7.5) to eliminate imidazole, and stored at −80°C until use.
PCR was used to amplify the DNA sequence of the promoter region of the target gene (primers are listed in Supplementary Table S2), and the obtained product was used to prepare the electrophoretic mobility shift assay (EMSA) probe. 0 ng, 100 ng, 200 ng His6-LexA were coincubated with the 25 nM EMSA probe in a 20 μL binding system (binding buffer: 30 mM KCl, 10 mM Tris-HCl, 0.4 mM DTT, 2 mM MgCl2, 0.2 mM EDTA, 2% glycerol) for 30 min at 30°C. This was followed by electrophoresis on an 8% native polyacrylamide gel at 0°C. After 40 min of constant voltage electrophoresis at 100 V, 1% GoldenView was added to pure water, mix in a horizontal shaker for 15 min with the gel, and then DNA is detected using gel imaging (Tanon-5200; China).
For RNA isolation, S. mobaraensis strains were grown on ISP2 solid medium or in liquid TSB medium. Samples were collected from three distinct Streptomyces cultures at different time intervals and subsequently pulverized in liquid nitrogen. Total RNAs were extracted utilizing Trizol reagent (Tiangen; China). Following extraction, crude RNA samples underwent treatment with DNase I (TaKaRa; Japan) to eliminate any potential genomic DNA contaminants. Reverse transcription of total RNA and subsequent reverse transcription real-time quantitative polymerase chain reaction (RT-qPCR) analysis were performed as described previously (Yan et al., 2020). Primer pairs listed in Supplementary Table S2 were utilized for RT-qPCR amplification. DNase I-treated RNA samples, which did not undergo reverse transcription, served as negative controls to validate the absence of DNA contamination. The transcription levels of each gene were normalized relative to the internal control gene 16S rRNA (LOCUS_r00030) using the comparative Ct method. The expression value of each gene at the initial time point was set as 1. Gene expression analyses were performed in three biological replicates.
The activity of TGase was measured by hydroxamate assay (Oteng-Pabi and Keillor, 2013). The quantification of hydroxamate was determined by measuring the absorbance at 525 nm of the red complex formed with FeCl3-TCA (trichloroacetic acid) (Tao et al., 2024). Twenty microliters of fermentation broth supernatant was transferred into the 1.5 mL eppendorf tube. For the blank control, first add 200 μL of reagent B, react at 37°C for 10 min, and then add 200 μL reagent A. Pipette 200 μL of the reaction solution into a 96-well plate, and use a multifunctional microplate reader (EN 60825-1:2007; PerkinElmer, United States) to measure the absorbance of the supernatant at 525 nm. For the tested sample, 200 μL reagent A was added, and the reaction was carried out at 37°C for 10 min. After adding 200 μL reagent B to terminate the reaction, the absorbance of the supernatant was measured at 525 nm. Reagent A: 12.114 g trishydroxylaminomethane (Tris), 3.474 g hydroxylamine hydrochloride, 1.537 g reduced glutathione (GSH), 1.687 g substrate (Nα-Cbz-Gln-Gly), and adjust the pH to 6.0, add water to make the volume to 500 mL. Reagent B: 3 mol/L HCl, 12% trichloroacetic acid, 5% FeCl3 dissolved in 0.1 mol/L HCl, mix the three solutions in equal amounts and mix evenly with a magnetic stirrer.
To measure the dry weight of the cells, the strain was initially cultured in seed medium for 22 h and then transferred to fermentation medium. Seed medium (w/v): 2% glycerol, 2% peptone, 0.5% yeast powder, 0.2% K2HPO4 and 0.2% MgSO4. Adjust pH to 7.0. Fermentation medium (w/v): 2% glycerol, 2% peptone, 0.5% yeast powder, 2% soy protein powder, 0.2% KH2PO4, 0.4% K2HPO4, 0.2% CaCO3 and 0.2% MgSO4 adjusted to pH 7.0. Mycelium samples were collected at 8 h intervals and dried overnight at 80°C until complete evaporation of water. The dry weight was determined using an electronic scale (dry weight = sample tube weight − empty tube weight; n = 3) (Yin et al., 2021).
Mycelial phenotypes were examined using scanning electron microscopy (Hitachi SU8010; Japan). The samples were cut into 1 cm*1 cm pieces by the inoculation shovel and fixed in 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH = 7.2) for 8 h. Subsequently, the samples were washed three times with ultrapure water (8 min per immersion). Dehydration was achieved by treating the samples with ethanol solutions of increasing concentrations (30, 50, 70, 85, 95, and 100%), with each immersion lasting 15 min. Finally, critical point drying was performed, followed by gold spraying and observation using a Hitachi Cold-Field Emission Scanning Electron Microscope (Xie et al., 2023).
To analyze the differential proteins expressed in S. mobaraensis NBRC 13819 and TBJ3 strains, fermentation was conducted in the 7.5 L fermenter (New Brunswick BioFlo®/CelliGen® 115, Germany) at 30°C. The rotation speed was related to dissolved oxygen and set to 300–600 rpm. The dissolved oxygen threshold was 1.8 mg/L. One millilitre of bacterial cells were collected at 12, 27 and 42 h for differential protein expression analysis. Sample processing, liquid chromatography-mass spectrometry/mass spectrometry (LC-MS/MS), and data standardization was performed by the Novogene company (Beijing, China) as described previously (Qiu et al., 2024). Differentially expressed proteins were listed in Supplementary Table S3.
The genome-wide scan of S. mobaraensis NBRC 13819 used the PREDetector 3.1 website.1 The sequences used to calculate LexA conserved motifs are listed in Supplementary Table S4. The S. mobaraensis NBRC 13819 genome information of the strain was downloaded from NCBI. The score of the target genes is cut at 9. The predicted target genes are listed in the Supplementary Table S5.
Using the original strain (S. mobaraensis NBRC 13819) and the TGase industrial strain TBJ3 for fermentation, the culture broth samples were collected every 3 h to analyze the accumulation of TGase. The results showed that TGase production was significantly greater in the TBJ3 strain than NBRC 13819 in the supernatant (Figure 1A). Concurrently, growth curves, determined by measuring cell dry weight, indicated that the growth cycles of WT and TBJ3 were essentially identical, suggesting that the observed difference in TGase production was not attributed to variations in biomass (Supplementary Figure S2A). Further analysis of the TGase activity profiles of the WT and TBJ3 strains revealed that the TGase activity of TBJ3 was 3-fold greater than that of the WT at the 40 h maturation point, when all Pro-TGase forms TGase (Figure 1B).
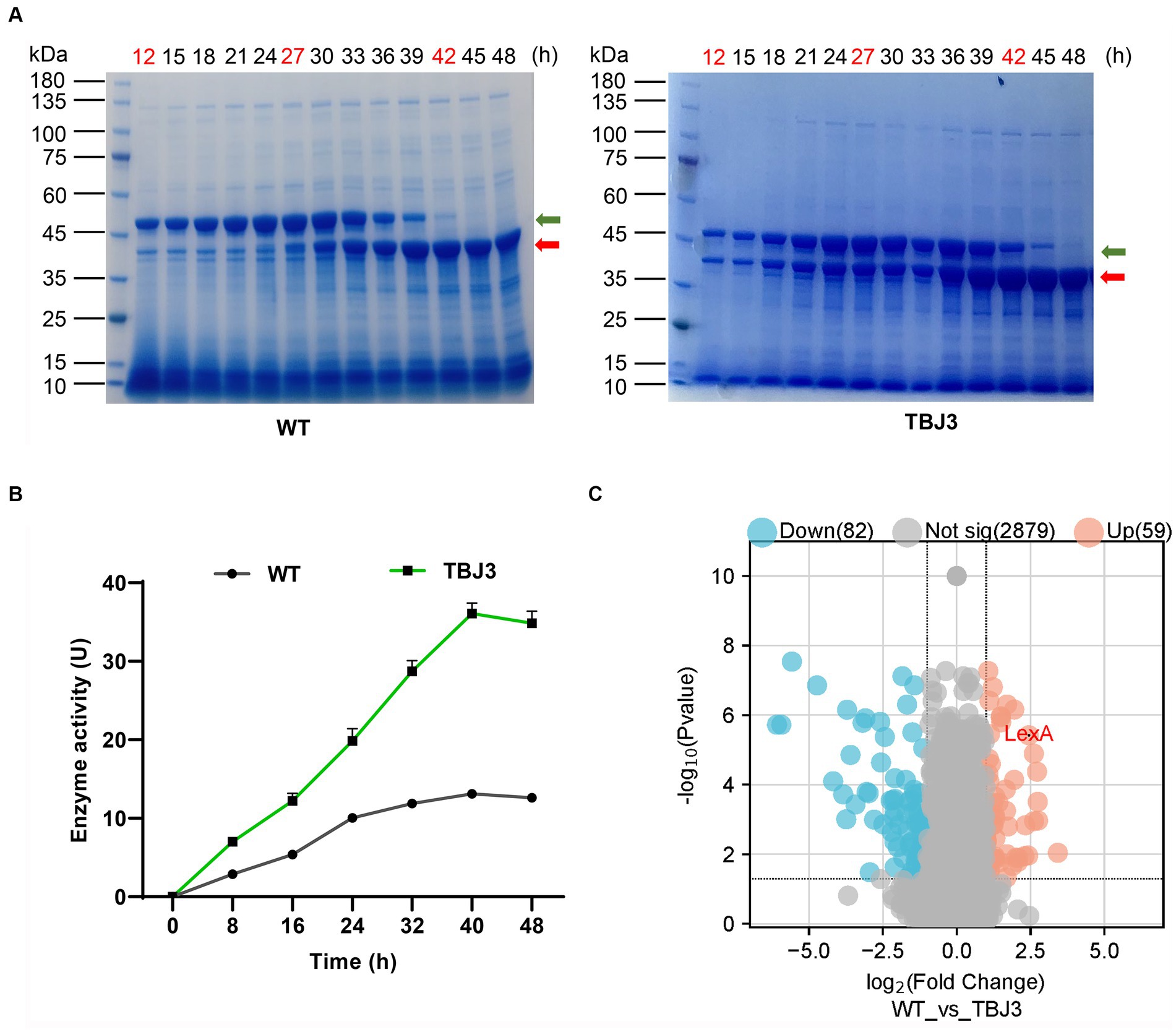
Figure 1. Differential protein expression analysis of type and industrial Streptomyces mobaraensis. (A) SDS-PAGE analysis of Sm-TGase. Protein Marker: M. Left panel: changes in TGase protein expression (yield) in the fermentation broth of the departure strain (WT) at different time points. The panels represent samples of the fermentation broth of the departure strain at different fermentation times. Right panel: changes in TGase protein expression (yield) in the fermentation broth of the high-yielding strain (TBJ3) at different time points. The numbers represent fermentation samples of high-yielding strains at different fermentation times. The time points marked in red represent the samples were used for differential protein expression analysis. The green arrow indicates the bands of Pro-TGase and the red arrow indicates the bands of TGase. (B) Dry cell weight growth curves and enzyme activity curves of the WT and TBJ3 strains. (C) Volcano plots comparing the comparative proteomics of the departure strain and the high-yielding strain at 27 h. The red dots indicate upregulated genes, and blue dots indicate downregulated genes. Data in (B) are shown as the mean ± SD (n = 3 biological replicates).
In addition, to investigate the functional genes influencing TGase synthesis, the bacterial cells were selected at 12 h (logarithmic or log phase), 27 h (stationary phase), and 42 h (lag phase) (Supplementary Figure S2B) for the differential protein expression analysis. The results revealed significant alterations in protein expression between the TBJ3 and WT strains at all three examined time points, with 59 proteins upregulated and 82 downregulated (Figure 1C and Supplementary Table S3). Notably, among the proteins exhibiting substantial upregulation, LexA—an essential transcription factor associated with the regulation of DNA damage repair stress—displayed remarkable fold increases of 4.73, 5.43, and 5.78, respectively, in TBJ3. Based on the significant up-regulation of LexA in TBJ3, we hypothesize that LexA is involved in regulating TGase production.
To elucidate the role of LexA in S. mobaraensis, we generated a lexA deletion mutant strain (ΔlexA) through homologous double exchange. Additionally, an OlexA overexpression strain of lexA was constructed using the integrative vector pSET152 and the potent kasO*p promoter. To restore the lexA gene, the complementation strain ClexA was generated based on the integrating vector pSET152.
In shake flask fermentation experiments, we analyzed the fermentation broth collected at 40 h using SDS-PAGE. The results revealed a significant reduction in TGase production in ΔlexA, a notable increase in OlexA, and a substantial return to wild-type levels in ClexA following lexA complementation (Figures 2A,B). These findings strongly suggest the involvement of LexA in regulating TGase synthesis. Moreover, the overexpression of lexA not only compensates for the reduced TGase production observed in ΔlexA but also significantly surpasses the wild-type levels. This finding underscores the potential effectiveness of lexA overexpression as a strategy to enhance TGase production in S. mobaraensis.
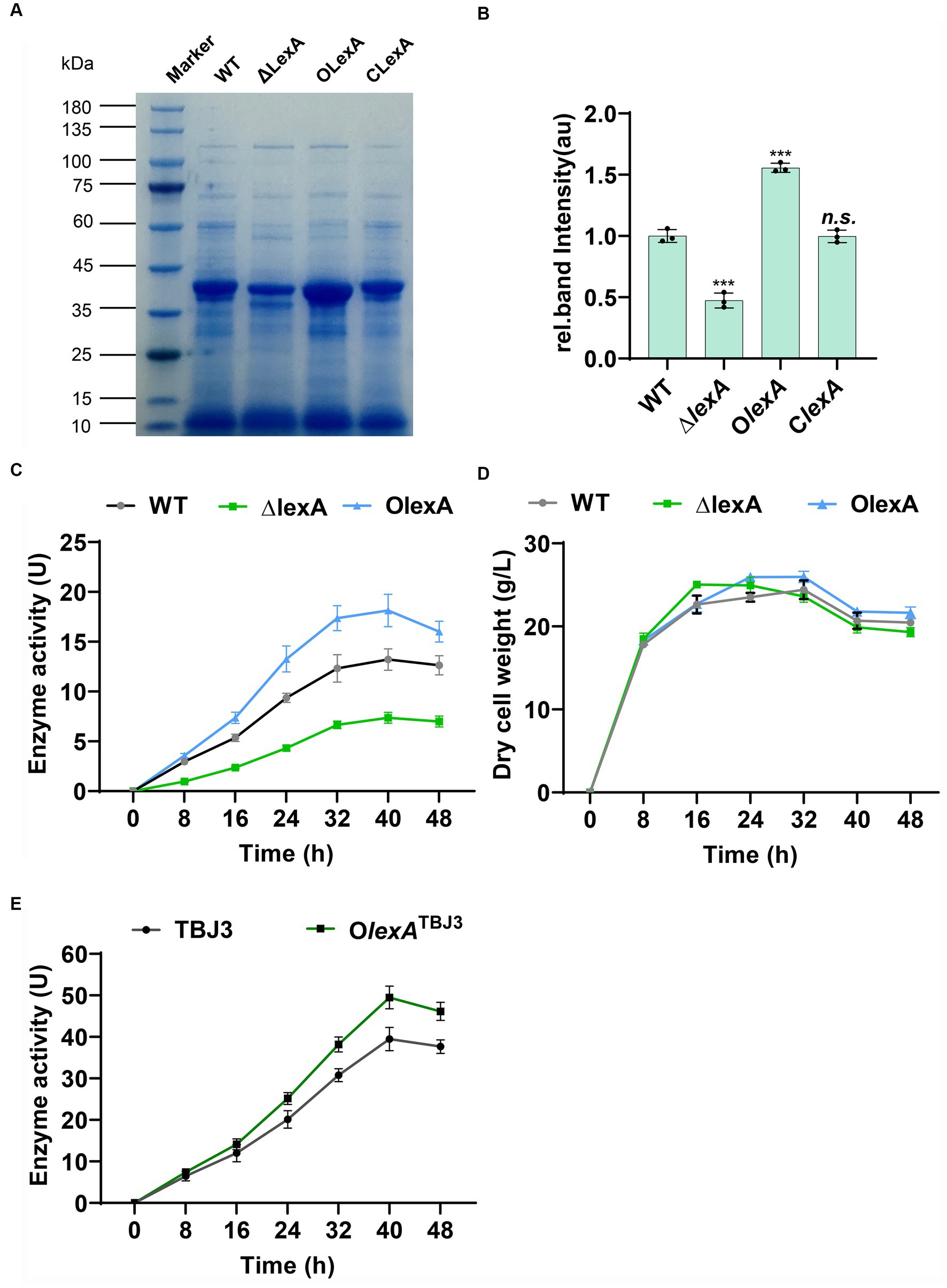
Figure 2. Analysis of enzyme production in lexA-related strains. (A) SDS–PAGE analysis of the WT, ∆lexA, OlexA, and ClexA strains after 40 h of culture. Numbers marked in red are samples sent for differential protein expression analysis. (B) Grayscale quantitative analysis of WT, ∆lexA, OlexA, and ClexA after 45 h of culture. The precise data of protein quantification was acquired via grayscale analysis utilizing ImageJ (National Institutes of Health, USA) software. (C) Enzyme activity curves of the WT, ∆lexA, and OlexA strains. (D) Dry cell weight growth curves of the WT, ∆lexA, and OlexA strains. (E) Enzyme activity curves of the TBJ3 and OlexATBJ3 strains. Data in (C, D, E) are shown as the mean ± SD (n=3 biological replicates). Two-tailed Student’s t test was used in (B) to analyze the statistical significance (n.s. p > 0.05; ***p < 0.001).
To determine the positive regulation of TGase production by LexA without impacting bacterial growth, we conducted enzyme activity and growth profile analyses on the WT, ΔlexA, and OlexA strains. At 40 h of TGase maturation, OlexA exhibited a 38% increase in TGase enzyme activity compared to that of the WT, while the TGase activity of the ΔlexA mutant decreased by 62% (Figure 2C) and the TGase activity of the ClexA mutant basically can be restored to the WT (Supplementary Figure S3A). The growth curves, determined by measuring the cell dry weight of lexA-associated strains, indicated that OlexA, ΔlexA and ClexA do not influence the growth of the bacterium (Figure 2D; Supplementary Figure S3B). These findings confirm that LexA regulates TGase expression without affecting the growth of S. mobaraensis. To further increase the TGase production in industrial strain TBJ3, we overexpressed lexA in TBJ3 to obtain strain OlexATBJ3. By measuring the enzyme activity curve, we found that overexpressing lexA, the TGase activity increased by nearly 25% in 40 h (Figure 2E), indicating that regulating the lexA expression level is an effective strategy to increase TGase production.
In order to investigate the effect of LexA on morphological differentiation of S. mobaraensis, OlexA, ClexA, and ΔlexA were incubated on ISP2 solid media for 5 days at 30°C, with WT serving as the control strain. Bacterial growth was observed to assess morphological changes. The results demonstrated that OlexA exhibited earlier sporulation, initiating on the first day, while the WT formed mature spores by the second day. In contrast, ΔlexA produced fewer spores even by the fifth day, indicating a bald phenotype. Remarkably, the ClexA phenotype was restored to that of the wild type (Figure 3A). Further scanning electron microscopy revealed that by day 2, the WT exhibited a substantial number of filamentous features on the sprocket, whereas the ΔlexA strain presented a relatively cleaner appearance, with only a few filamentous filaments at the initial stage (Figure 3B). These findings strongly suggest that LexA plays a regulatory role in the morphological differentiation of S. mobaraensis.

Figure 3. Effect of the lexA gene on the phenotype of S. mobaraensis. (A) Phenotypes of the WT, LexA deletion mutant (ΔlexA), complementation (ClexA), and overexpression (OlexA) strains grown on ISP2 agar media at 30°C. (B) Scanning electron microscopy images of the WT and ΔlexA strains.
To further understand the regulatory mechanism of LexA, mycelia from the WT, ΔlexA, and OlexA strains fermented for 16 h (logarithmic or log phase), 32 h (stationary phase), or 48 h (death phase) were harvested in shake flasks. RNA extraction was performed, and changes in the transcript levels of tga, the gene encoding TGase, were assessed through RT-qPCR. The results revealed a significant downregulation of the tga transcript in the ΔlexA strain compared to that in the WT, while a substantial upregulation was observed in the OlexA strain relative to that in the WT (Figure 4A). This indicates that LexA positively regulates TGase expression at the transcriptional level.
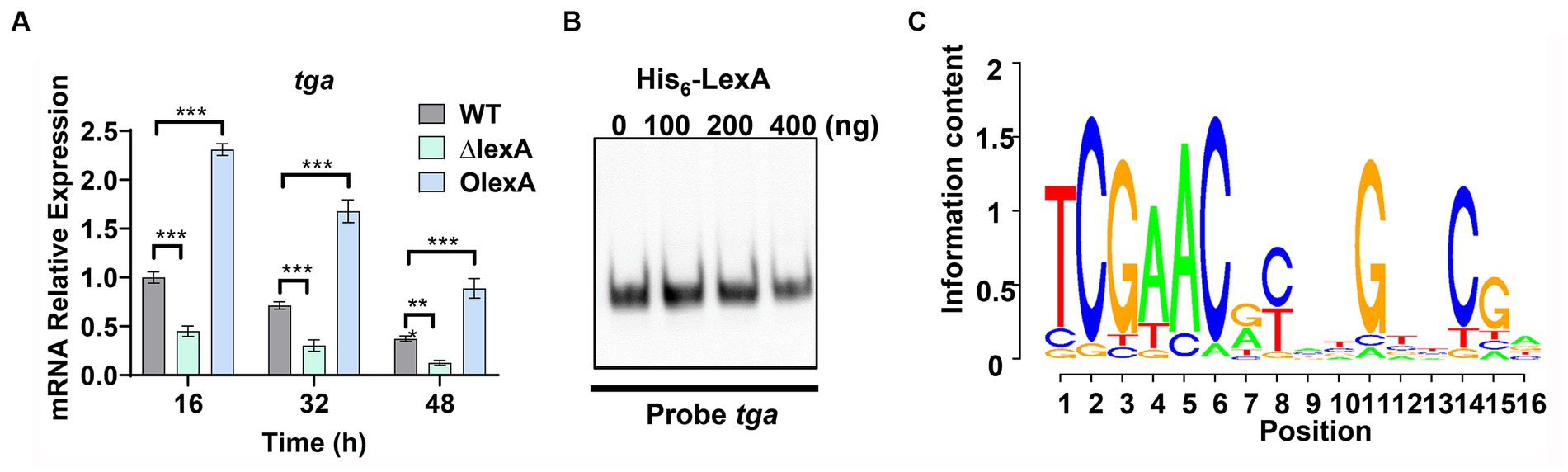
Figure 4. Identification of LexA Target Genes Associated with Development. (A) RT-qPCR analysis of the transcript levels of TGase-encoding genes in the WT, ΔlexA, and OlexA strains after 16, 32, and 48 h of culture. (B) Electrophoretic mobility shift assays (EMSAs) to detect the interaction of His6-LexA with TGase-encoding genes. (C) WebLogo analysis of LexA binding sequences. Data in (A) are shown as the mean ± SD (n = 3 biological replicates). Two-tailed student’s t-test was used in (A) to analyze the statistical significance (**p < 0.01 and ***p < 0.001). Two-tailed Student’s t-test was used in (B) to analyze the statistical significance (n.s. p > 0.05; ***p < 0.001).
To explore the direct regulatory role of LexA in TGase expression, the recombinant protein His6-LexA, which was tagged with His6 at the N-terminus, was heterologously expressed and purified in E. coli (Supplementary Figure S4A). Electrophoretic mobility shift assays (EMSAs) using His6-LexA with a sequence probe of the tga promoter region demonstrated that LexA could not bind to the tga promoter (Figure 4B). This finding suggested that LexA does not directly regulate TGase expression.
In S. venezuelae, the conserved binding motif for LexA is a 16 bp incomplete palindrome sequence (Stratton et al., 2022). The lexA gene in S. mobaraensis is 786 base pairs in length and encodes 281 amino acids. By analyzing 268 Streptomyces species, we found that LexA is present in more than 99% of them. Protein homology analysis revealed that the LexA protein in S. mobaraensis shares high homology (94.8, 96.3, 94.2, and 96.8%) with LexA proteins in S. venezuelae, S. coelicolor, Streptomyces griseus, and Streptomyces avermitilis (Supplementary Figure S5). These genes exhibit complete agreement in their DNA-binding HTH domains, suggesting that LexA has a highly conserved regulatory function across Streptomyces species.
Based on the identified LexA binding sequence in S. venezuelae, we predicted a conserved binding motif for LexA, represented by a 16-nucleotide binding sequence: 5′-TCGAACRNNGNNCGA-3′ (S=C/G; R = G/A; W = A/T; N = A/T/C/G; D = G/A/T) (Figure 4C). Additionally, a genome-wide scan of S. mobaraensis using the PREDetector 3.1 website (see text footnote 1) predicted a total of 157 target genes for LexA with a score greater than 9.0 (Supplementary Table S5). Among the predicted target genes, we identified 32 transcription factors and 5 target genes associated with protein synthesis. For the EMSAs, we randomly selected two transcription factors: marR1 (LOCUS_20350; MarR family transcriptional regulator) and araC1 (LOCUS_63640; AraC family transcriptional regulator). The results demonstrated that LexA effectively binds to the promoter regions of these two genes (Figure 5A). Further analysis of the transcript levels of marR1 and araC1 in the ΔlexA and WT strains by RT-qPCR revealed significant downregulation in the ΔlexA strain compared to the WT strain (Figure 5B). Therefore, LexA directly upregulates the transcript levels of marR1 and araC1, which influences their expression.
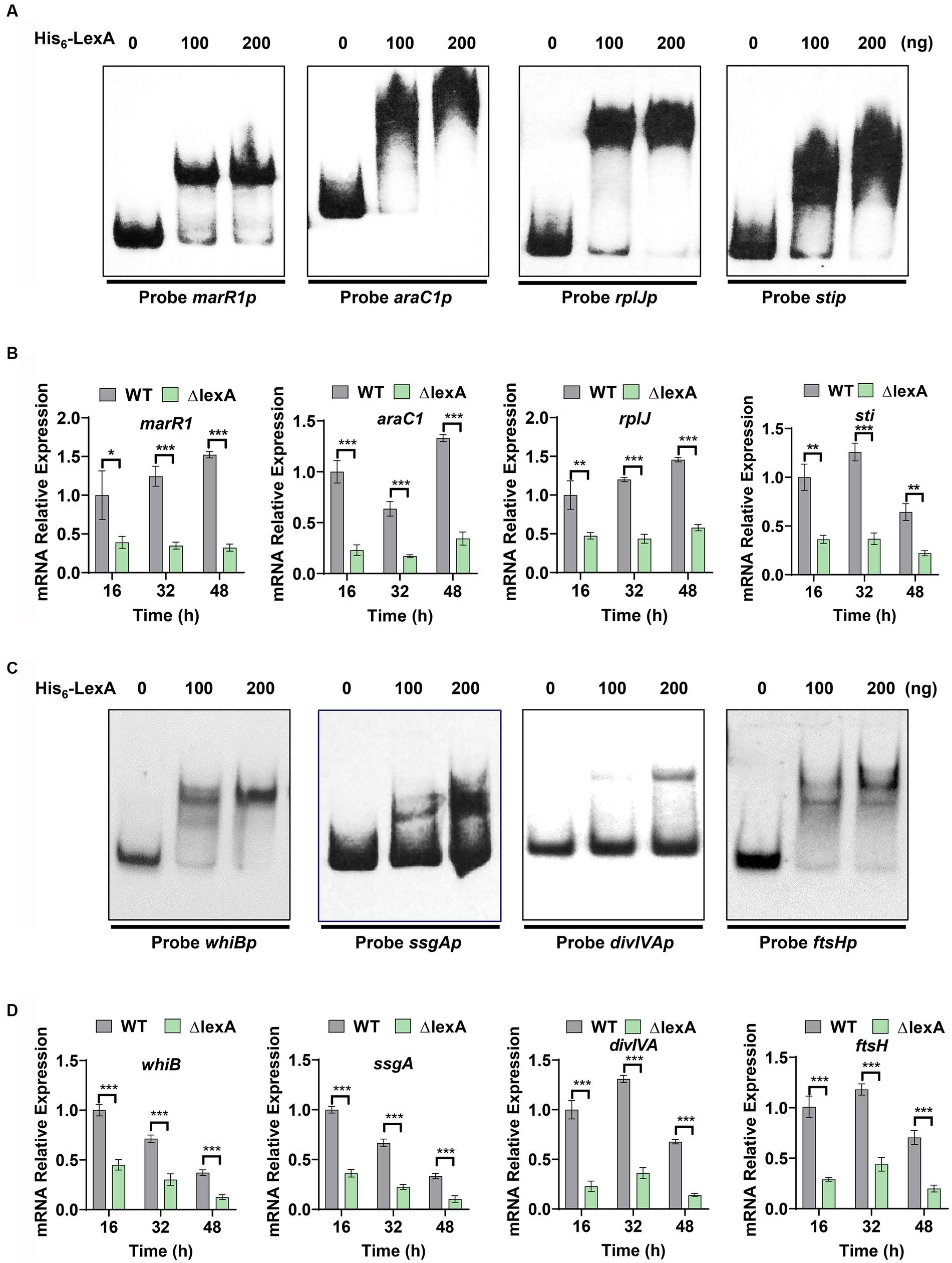
Figure 5. Target identification and regulatory validation of LexA. (A) EMSAs to detect the interaction of His6-LexA with protein synthesis-related probes. Probe marR1 (112 bp), Probe araC1 (110 bp), Probe rplJ (118 bp), Probe sti (111 bp). (B) RT-qPCR analysis of the WT and ΔlexA strains with protein synthesis-related genes after 16, 32, and 48 h of culture. (C) EMSAs to detect the interaction of His6-LexA with probes related to influencing morphology. Probe whiB (123 bp), Probe ssgA (106 bp), Probe divIVA (101 bp), Probe ftsH (117 bp). (D) RT-qPCR analysis of the morphology-related genes in the WT and ΔlexA strains after 16 h, 32 h, and 48 h of culture. Data in (B,D) are shown as the mean ± SD (n = 3 biological replicates). Two-tailed student’s t-test was used in (B,D) to analyze the statistical significance (*p < 0.05, **p < 0.01, and ***p < 0.001).
We found that the tga gene transcription level was significantly down-regulated in lexA deletion strain, but the EMSA experimental results showed that LexA was not directly involved in regulating the transcription of the tga gene. This shows that LexA regulates the tga transcription indirectly by regulating other transcription factors. Among our predicted target genes, 32 genes exert regulatory functions, indicating that LexA may indirectly regulate the tga transcription by regulating the expression of these genes. Additionally, we focused on two target genes associated with protein synthesis, namely, rplJ (Gomez-Escribano et al., 2006) (LOCUS_22270; 50S ribosomal protein L10) and sti (LOCUS_52980; membrane protein). The encoded protein, 50S-subunit ribosomal protein L10, plays a vital role in the process of protein translation, while sti encodes a protease inhibitory protein involved in the process of protein degradation. The EMSA results demonstrated that LexA directly interacts with the promoter regions of rplJ and sti (Figure 5A). RT-qPCR experiments confirmed that the transcript levels of rplJ and sti were significantly lower in the ΔlexA mutant than in the WT (Figure 5B). These findings indicate that LexA directly activates the expression of rplJ and sti. Thus, LexA can also affect the TGase production by regulating the processes involved in protein synthesis and degradation.
Plate phenotyping and scanning electron microscopy experiments were conducted to investigate the role of LexA in the maturation of S. mobaraensis spores. From our analysis of predicted target genes, we identified four key genes that are crucial for morphological differentiation: whiB (Davis and Chater, 1992), ssgA (Traag and van Wezel, 2008), divIVA (Passot et al., 2022) and ftsH (Xu et al., 2022) (Supplementary Table S5). To confirm the regulatory impact of LexA on these genes, probes were generated through PCR amplification of their promoter regions (Probe whiBp, ssgAp, divIVAp, and ftsHp). Subsequent EMSA experiments using purified His6-LexA demonstrated specific binding of LexA to the promoter regions of these genes (Figure 5C), further validating their predicted regulation.
To assess the regulatory effects of LexA on the aforementioned four target genes, colonies from both the ∆lexA and WT strains were harvested at 16, 32, and 48 h time points on ISP2 solid media. RNA extraction and RT-qPCR were performed to measure the transcript levels of these genes. Our results consistently showed a significant downregulation of the transcript levels in ∆lexA compared to those in WT at all time points (Figure 5D). These findings strongly indicate that LexA plays a positive role in the developmental differentiation of S. mobaraensis by activating the expression of these key target genes.
Investigating the regulatory network controlling TGase production holds pivotal importance in the development of high-yield genetic engineering strains. Through a meticulous analysis of differential protein expression profiles between two strains exhibiting markedly distinct TGase production levels, we discerned the potential involvement of the global transcription factor LexA in regulation of TGase production. While our investigation revealed that LexA influences the expression of the tga gene at the transcriptional level, direct evidence linking LexA to the tga gene remains elusive. Consequently, we predicted 157 target genes of LexA in S. mobaraensis, encompassing 32 regulatory genes and 5 genes implicated in protein synthesis and degradation. Subsequent validation efforts confirmed that LexA indeed directly regulates the expression of select target genes, thereby exerting multifaceted control over TGase expression. Remarkably, our exploration also unveiled the participation of LexA in modulating the morphological differentiation of S. mobaraensis, underscoring its broad regulatory influence within the organism.
LexA primarily functions as an inhibitory protein in the SOS response system. In response to DNA damage, bacteria initiate a coordinated cellular response governed by the RecA and LexA proteins (Butala et al., 2009). Under normal conditions, LexA suppresses the transcription of genes involved in DNA repair by binding to the SOS box. However, during SOS stress, the accumulation of RecA, ATP, and single-stranded DNA (ssDNA) complexes triggers autocleavage of LexA. As a result, LexA dissociates from target genes, allowing the expression of genes associated with the SOS response (Zhang et al., 2010). LexA also plays a role in regulating mobile genetic elements (MGEs), enabling cells to respond to diverse stresses (Fornelos et al., 2016). Similarly, in S. venezuelae, LexA regulates the core set of SOS genes involved in DNA repair (Stratton et al., 2022).
In addition to responding to SOS, this study elucidates the role of LexA as a global transcription factor indirectly regulating TGase synthesis. While direct regulation of TGase expression by LexA was not directly observed, it is postulated that such regulation might occur through the control of these transcription factors, consequently exerting an indirect impact on TGase synthesis. This revelation not only underscores the multifunctionality of LexA as a global transcription factor but also accentuates the intricacy inherent in this regulatory network. As the main microbial resource for mining and producing secondary metabolites, Streptomyces contains abundant secondary metabolite biosynthetic gene clusters. There may be LexA Box in the clusters, for example, LexA directly inhibit the expression of the intra-cluster regulatory gene actII-ORF4 and regulate the biosynthesis of actinorhodin in S. coelicolor (Liu et al., 2013). Therefore, LexA is a potential target for genetic engineering of Streptomyces.
As multicellular Gram-positive bacteria, Streptomyces has a complex morphological differentiation process. Four related genes were identified as targets of LexA in S. mobaraensis, namely whiB, ssgA, ftsH and divIVA. WhiB and WhiA depend on each other to form heterodimers that jointly inhibit the expression of the cell scaffold protein FilP which located at the top of the hyphae, and activate the expression of key genes ftsZ, ftsW and ftsK in cell division (Lilic et al., 2023). SsgA is involved in identifying a location for developing the septum and germination site (Kumar et al., 2023). FtsH is the membrane-anchored metalloprotease among the AAA+ proteases. FtsH is relatively less studied in Streptomyces, although FtsH is not a key developmental gene of Streptomyces, it has a certain degree of effect on cell differentiation (Xu et al., 2022). DivIVA is located at the tip of the hyphae and is a key structure protein for the polar growth of Streptomyces that enhance polar cell wall synthesis by assembling larger multiprotein complexes (Hempel et al., 2008). LexA promotes spore maturation by directly activating the expression of these genes.
Based on the present findings, we propose a conceptual model of the LexA-mediated regulatory network involved in TGase production and S. mobaraensis development (Figure 6). Overall, this work reveals the molecular mechanism by which LexA positively regulates morphological differentiation and TGase synthesis in S. mobaraensis, expands the understanding of the function and regulatory mechanism of LexA, and lays the foundation for elucidating the complex regulatory network of TGase biosynthesis, which provides us with an in depth and comprehensive understanding of LexA function, and a new direction to promote the engineering construction of TGase high-yielding strains.
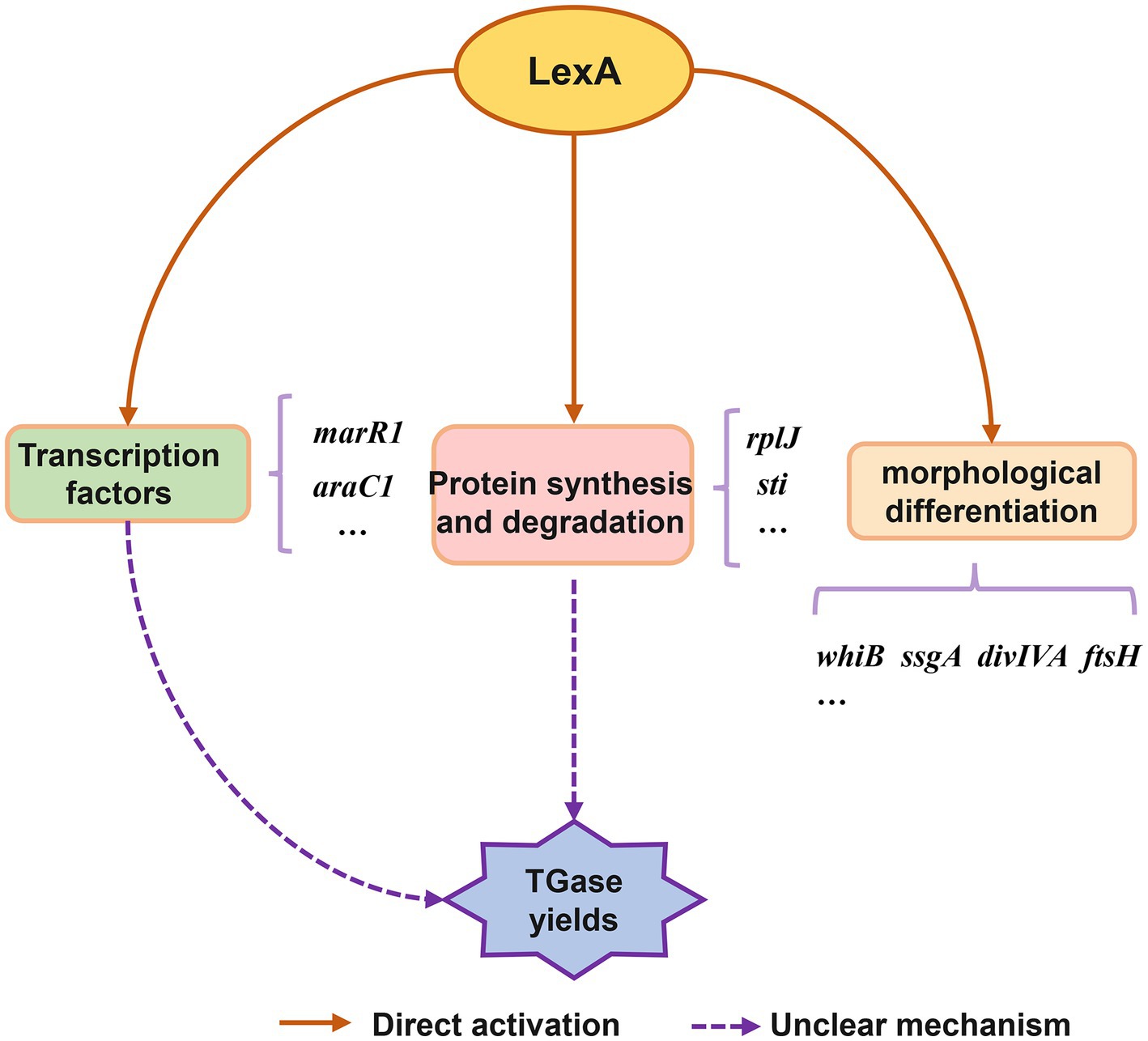
Figure 6. Conceptual model of regulatory role of LexA in control of TGase production and S. mobaraensis development.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
XS: Writing – review & editing, Writing – original draft, Software, Methodology, Investigation, Formal analysis, Data curation. HY: Writing – original draft, Supervision, Software, Methodology, Investigation, Data curation, Writing – review & editing, Resources, Funding acquisition. FY: Project administration, Writing – review & editing, Supervision, Resources, Funding acquisition. GL: Writing – review & editing, Resources, Funding acquisition. JL: Writing – review & editing, Formal analysis, Data curation. CL: Writing – review & editing, Formal analysis, Data curation. XY: Writing – review & editing, Supervision. ZL: Writing – original draft, Project administration, Resources, Investigation, Conceptualization, Writing – review & editing, Supervision, Funding acquisition. YZ: Validation, Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Funding acquisition. WW: Data curation, Writing – review & editing, Writing – original draft, Supervision, Resources, Project administration, Investigation, Funding acquisition, Conceptualization.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Natural Science Foundation of China 32100066 to HY.
The authors are grateful to Yiming Biological (Jiangsu, China) for its financial support.
FY and GL were employed by Jiangsu Yiming Biological Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer LB declared a past co-authorship with the author WW to the handling editor.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1397314/full#supplementary-material
Bellio, P., Mancini, A., Di Pietro, L., Cracchiolo, S., Franceschini, N., Reale, S., et al. (2020). Inhibition of the transcriptional repressor LexA: withstanding drug resistance by inhibiting the bacterial mechanisms of adaptation to antimicrobials. Life Sci. 241:117116. doi: 10.1016/j.lfs.2019.117116
Bierman, M., Logan, R., O’Brien, K., Seno, E. T., Rao, R. N., and Schoner, B. E. (1992). Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116, 43–49. doi: 10.1016/0378-1119(92)90627-2
Butala, M., Zgur-Bertok, D., and Busby, S. J. (2009). The bacterial LexA transcriptional repressor. Cell. Mol. Life Sci. 66, 82–93. doi: 10.1007/s00018-008-8378-6
Ceresino, E. B., de Melo, R. R., Kuktaite, R., Hedenqvist, M. S., Zucchi, T. D., Johansson, E., et al. (2018). Transglutaminase from newly isolated Streptomyces sp. CBMAI 1617: production optimization, characterization and evaluation in wheat protein and dough systems. Food Chem. 241, 403–410. doi: 10.1016/j.foodchem.2017.09.010
Chen, K., Zhang, D., Liu, S., Wang, N. S., Wang, M., Du, G., et al. (2013). Improvement of transglutaminase production by extending differentiation phase of Streptomyces hygroscopicus: mechanism and application. Appl. Microbiol. Biotechnol. 97, 7711–7719. doi: 10.1007/s00253-012-4614-y
Davis, N. K., and Chater, K. F. (1992). The Streptomyces coelicolor whiB gene encodes a small transcription factor-like protein dispensable for growth but essential for sporulation. Mol. Gen. Genet. 232, 351–358. doi: 10.1007/BF00266237
Doti, N., Caporale, A., Monti, A., Sandomenico, A., Selis, F., and Ruvo, M. (2020). A recent update on the use of microbial transglutaminase for the generation of biotherapeutics. World J. Microbiol. Biotechnol. 36:53. doi: 10.1007/s11274-020-02829-y
Fatima, S. W., Tiwari, R., and Khare, S. K. (2019). Utilization of agro-industrial waste for production of transglutaminase from Streptomyces mobaraensis. Bioresour. Technol. 287:121391. doi: 10.1016/j.biortech.2019.121391
Fornelos, N., Browning, D. F., and Butala, M. (2016). The use and abuse of LexA by mobile genetic elements. Trends Microbiol. 24, 391–401. doi: 10.1016/j.tim.2016.02.009
Gajsek, M., Jancic, U., Vasic, K., Knez, Z., and Leitgeb, M. (2019). Enhanced activity of immobilized transglutaminase for cleaner production technologies. J. Clean. Prod. 240:118218. doi: 10.1016/j.jclepro.2019.118218
Gomez-Escribano, J. P., Liras, P., Pisabarro, A., and Martin, J. F. (2006). An rplKDelta29-PALG-32 mutation leads to reduced expression of the regulatory genes ccaR and claR and very low transcription of the ceaS2 gene for clavulanic acid biosynthesis in Streptomyces clavuligerus. Mol. Microbiol. 61, 758–770. doi: 10.1111/j.1365-2958.2006.05266.x
Hempel, A. M., Wang, S. B., Letek, M., Gil, J. A., and Flardh, K. (2008). Assemblies of DivIVA mark sites for hyphal branching and can establish new zones of cell wall growth in Streptomyces coelicolor. J. Bacteriol. 190, 7579–7583. doi: 10.1128/JB.00839-08
Iqbal, M., Mast, Y., Amin, R., Hodgson, D. A., STREAM ConsortiumWohlleben, W., et al. (2012). Extracting regulator activity profiles by integration of de novo motifs and expression data: characterizing key regulators of nutrient depletion responses in Streptomyces coelicolor. Nucleic Acids Res. 40, 5227–5239. doi: 10.1093/nar/gks205
Janion, C. (2008). Inducible SOS response system of DNA repair and mutagenesis in Escherichia coli. Int. J. Biol. Sci. 4, 338–344. doi: 10.7150/ijbs.4.338
Javitt, G., Ben-Barak-Zelas, Z., Jerabek-Willemsen, M., and Fishman, A. (2017). Constitutive expression of active microbial transglutaminase in Escherichia coli and comparative characterization to a known variant. BMC Biotechnol. 17:23. doi: 10.1186/s12896-017-0339-4
Kumar, P., Khushboo Rajput, D., and Dubey, K. K. (2023). Insights into the mechanism of mycelium transformation of Streptomyces toxytricini into pellet. FEMS Microbes 4:xtad017. doi: 10.1093/femsmc/xtad017
Kuraishi, C., Yamazaki, K., and Susa, Y. (2001). Transglutaminase: its utilization in the food industry. Food Rev. Int. 17, 221–246. doi: 10.1081/Fri-100001258
Lilic, M., Holmes, N. A., Bush, M. J., Marti, A. K., Widdick, D. A., Findlay, K. C., et al. (2023). Structural basis of dual activation of cell division by the actinobacterial transcription factors WhiA and WhiB. Proc. Natl. Acad. Sci. U.S.A. 120:e2220785120. doi: 10.1073/pnas.2220785120
Liu, G., Chater, K. F., Chandra, G., Niu, G., and Tan, H. (2013). Molecular regulation of antibiotic biosynthesis in Streptomyces. Microbiol. Mol. Biol. Rev. 77, 112–143. doi: 10.1128/MMBR.00054-12
Liu, S., Wang, M., Du, G. C., and Chen, J. (2016). Improving the active expression of transglutaminase in by promoter engineering and codon optimization. BMC Biotechnol. 16:75. doi: 10.1186/s12896-016-0304-7
Liu, X., Wang, D., Zhang, Y., Zhuang, X., and Bai, L. (2023). Identification of multiple regulatory genes involved in TGase production in Streptomyces mobaraensis. Eng. Microbiol. 3:100098. doi: 10.1016/j.engmic.2023.100098
Macneil, D. J., and Klapko, L. M. (1987). Transformation of Streptomyces-avermitilis by plasmid DNA. J. Ind. Microbiol. 2, 209–218. doi: 10.1007/Bf01569542
Mo, C. Y., Birdwell, L. D., and Kohli, R. M. (2014). Specificity determinants for autoproteolysis of LexA, a key regulator of bacterial SOS mutagenesis. Biochemistry 53, 3158–3168. doi: 10.1021/bi500026e
Mu, D., Lu, J., Qiao, M., Kuipers, O. P., Zhu, J., Li, X., et al. (2018). Heterologous signal peptides-directing secretion of Streptomyces mobaraensis transglutaminase by Bacillus subtilis. Appl. Microbiol. Biotechnol. 102, 5533–5543. doi: 10.1007/s00253-018-9000-y
Oteng-Pabi, S. K., and Keillor, J. W. (2013). Continuous enzyme-coupled assay for microbial transglutaminase activity. Anal. Biochem. 441, 169–173. doi: 10.1016/j.ab.2013.07.014
Passot, F. M., Cantlay, S., and Flardh, K. (2022). Protein phosphatase SppA regulates apical growth and dephosphorylates cell polarity determinant DivIVA in Streptomyces coelicolor. Mol. Microbiol. 117, 411–428. doi: 10.1111/mmi.14856
Qiu, S., Yang, B., Li, Z., Li, S., Yan, H., Xin, Z., et al. (2024). Building a highly efficient Streptomyces super-chassis for secondary metabolite production by reprogramming naturally-evolved multifaceted shifts. Metab. Eng. 81, 210–226. doi: 10.1016/j.ymben.2023.12.007
Santhi, D., Kalaikannan, A., Malairaj, P., and Arun Prabhu, S. (2017). Application of microbial transglutaminase in meat foods: a review. Crit. Rev. Food Sci. Nutr. 57, 2071–2076. doi: 10.1080/10408398.2014.945990
Schneider, H., Deweid, L., Avrutina, O., and Kolmar, H. (2020). Recent progress in transglutaminase-mediated assembly of antibody-drug conjugates. Anal. Biochem. 595:113615. doi: 10.1016/j.ab.2020.113615
Srivastava, A., Kumar, A., Biswas, S., Kumar, R., Srivastava, V., Rajaram, H., et al. (2023). Gamma (gamma)-radiation stress response of the cyanobacterium Anabaena sp. PCC7120: regulatory role of LexA and photophysiological changes. Plant Sci. 326:111529. doi: 10.1016/j.plantsci.2022.111529
Stratton, K. J., Bush, M. J., Chandra, G., Stevenson, C. E. M., Findlay, K. C., and Schlimpert, S. (2022). Genome-wide identification of the LexA-mediated DNA damage response in Streptomyces venezuelae. J. Bacteriol. 204:e0010822. doi: 10.1128/jb.00108-22
Tao, Y., Xu, J., Zhang, N., Jiao, X., Yan, B., Zhao, J., et al. (2024). Unraveling the binding mechanisms of transglutaminase and substrate subjected to microwaves: molecular docking and molecular dynamic simulations. Food Chem. 443:138568. doi: 10.1016/j.foodchem.2024.138568
Traag, B. A., and van Wezel, G. P. (2008). The SsgA-like proteins in actinomycetes: small proteins up to a big task. Antonie Van Leeuwenhoek 94, 85–97. doi: 10.1007/s10482-008-9225-3
Walter, B. M., Rupnik, M., Hodnik, V., Anderluh, G., Dupuy, B., Paulic, N., et al. (2014). The LexA regulated genes of the Clostridium difficile. BMC Microbiol. 14:88. doi: 10.1186/1471-2180-14-88
Wang, W., Li, X., Wang, J., Xiang, S., Feng, X., and Yang, K. (2013). An engineered strong promoter for streptomycetes. Appl. Environ. Microbiol. 79, 4484–4492. doi: 10.1128/AEM.00985-13
Xie, H., Ruan, J. Y., Bu, Q. T., Li, Y. P., Su, Y. T., Zhao, Q. W., et al. (2023). Transcriptional regulation of the fidaxomicin gene cluster and cellular development in Actinoplanes deccanensis YP-1 by the pleiotropic regulator MtrA. Microbiol. Spectr. 11:e0270223. doi: 10.1128/spectrum.02702-23
Xu, W., Gao, W., Bu, Q., and Li, Y. (2022). Degradation mechanism of AAA+ proteases and regulation of Streptomyces metabolism. Biomolecules 12:1848. doi: 10.3390/biom12121848
Yan, H., Lu, X., Sun, D., Zhuang, S., Chen, Q., Chen, Z., et al. (2020). BldD, a master developmental repressor, activates antibiotic production in two Streptomyces species. Mol. Microbiol. 113, 123–142. doi: 10.1111/mmi.14405
Yin, X., Li, Y., Zhou, J., Rao, S., Du, G., Chen, J., et al. (2021). Enhanced production of transglutaminase in Streptomyces mobaraensis through random mutagenesis and site-directed genetic modification. J. Agric. Food Chem. 69, 3144–3153. doi: 10.1021/acs.jafc.1c00645
Yin, X., Rao, S., Zhou, J., Du, G., Chen, J., and Liu, S. (2022). Improved productivity of Streptomyces mobaraensis transglutaminase by regulating zymogen activation. Front. Bioeng. Biotechnol. 10:878795. doi: 10.3389/fbioe.2022.878795
Zhang, W., Hedayati, S., Tarahi, M., Can Karaca, A., Hadidi, M., Assadpour, E., et al. (2023). Advances in transglutaminase cross-linked protein-based food packaging films; a review. Int. J. Biol. Macromol. 253:127399. doi: 10.1016/j.ijbiomac.2023.127399
Zhang, A. P., Pigli, Y. Z., and Rice, P. A. (2010). Structure of the LexA-DNA complex and implications for SOS box measurement. Nature 466, 883–886. doi: 10.1038/nature09200
Keywords: LexA, streptomyces, TGase, transcription factor, regulation
Citation: Shi X, Yan H, Yuan F, Li G, Liu J, Li C, Yu X, Li Z, Zhu Y and Wang W (2024) LexA, an SOS response repressor, activates TGase synthesis in Streptomyces mobaraensis. Front. Microbiol. 15:1397314. doi: 10.3389/fmicb.2024.1397314
Edited by:
Yuriy Rebets, Explogen LLC, UkraineReviewed by:
Sukhithasri Vijayrajratnam, Washington University in St. Louis, United StatesCopyright © 2024 Shi, Yan, Yuan, Li, Liu, Li, Yu, Li, Zhu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaobin Yu, eGJ5dUBqaWFuZ25hbi5lZHUuY24=; Zilong Li, bGl6bEBpbS5hYy5jbg==; Yunping Zhu, emh1eXBAdGguYnRidS5lZHUuY24=; Weishan Wang, d2FuZ3dzQGltLmFjLmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.