
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 24 April 2024
Sec. Infectious Agents and Disease
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1389859
This article is part of the Research Topic Expanded Genus Brucella: from Taxonomy to Clinical Manifestations and Diagnosis Challenges View all 5 articles
 Zilong Yang1,2†Zili Chai1†Xia Wang1Zehan Zhang1Fengwei Zhang1Fuqiang Kang1Wenting Liu1Hongguang Ren1*Yuan Jin1*Junjie Yue1*
Zilong Yang1,2†Zili Chai1†Xia Wang1Zehan Zhang1Fengwei Zhang1Fuqiang Kang1Wenting Liu1Hongguang Ren1*Yuan Jin1*Junjie Yue1*Some Brucella spp. are important pathogens. According to the latest prokaryotic taxonomy, the Brucella genus consists of facultative intracellular parasitic Brucella species and extracellular opportunistic or environmental Brucella species. Intracellular Brucella species include classical and nonclassical types, with different species generally exhibiting host preferences. Some classical intracellular Brucella species can cause zoonotic brucellosis, including B. melitensis, B. abortus, B. suis, and B. canis. Extracellular Brucella species comprise opportunistic or environmental species which belonged formerly to the genus Ochrobactrum and thus nowadays renamed as for example Brucella intermedia or Brucella anthropi, which are the most frequent opportunistic human pathogens within the recently expanded genus Brucella. The cause of the diverse phenotypic characteristics of different Brucella species is still unclear. To further investigate the genetic evolutionary characteristics of the Brucella genus and elucidate the relationship between its genomic composition and prediction of phenotypic traits, we collected the genomic data of Brucella from the NCBI Genome database and conducted a comparative genomics study. We found that classical and nonclassical intracellular Brucella species and extracellular Brucella species exhibited differences in phylogenetic relationships, horizontal gene transfer and distribution patterns of mobile genetic elements, virulence factor genes, and antibiotic resistance genes, showing the close relationship between the genetic variations and prediction of phenotypic traits of different Brucella species. Furthermore, we found significant differences in horizontal gene transfer and the distribution patterns of mobile genetic elements, virulence factor genes, and antibiotic resistance genes between the two chromosomes of Brucella, indicating that the two chromosomes had distinct dynamics and plasticity and played different roles in the survival and evolution of Brucella. These findings provide new directions for exploring the genetic evolutionary characteristics of the Brucella genus and could offer new clues to elucidate the factors influencing the phenotypic diversity of the Brucella genus.
Brucella is a Gram-negative, facultative intracellular parasitic bacteria. Brucella can infect humans and a variety of animals, causing brucellosis (Cloeckaert et al., 2020). Different Brucella species have unique host preferences, virulence, and pathogenicity (Rajendhran, 2021). B. abortus, B. melitensis, B. suis, and B. canis can be transmitted between humans and animals (Marzetti et al., 2013). B. melitensis is the most common zoonotic Brucella species, followed by B. abortus, B. suis, and B. canis (Cloeckaert et al., 2024). Recent studies have indicated that B. neotomae and B. inopinata may also have the potential for zoonotic transmission, suggesting a potentially wider distribution of zoonotic potential within the Brucella genus (Cloeckaert et al., 2024; Vergnaud et al., 2024). Brucellosis poses significant risks to human health (Franco et al., 2007). Massive outbreaks of brucellosis among livestock can result in significant economic losses in the livestock industry (Hao et al., 2023). Additionally, due to the pathogenicity, high transmissibility, and aerosol transmission of Brucella, there is a potential risk of Brucella being used for the development of bioterrorism agents (Doganay and Doganay, 2013). Genomic analysis revealed a highly conserved genome structure among different Brucella species. The genomic similarity within the Brucella genus is above 97%, and the GC content is approximately 57%. The average genome size of Brucella isolates is 3.3 Mb, consisting of two circular chromosomes of different sizes, approximately 2.1 Mb and 1.2 Mb, respectively (Rajendhran, 2021).
Ochrobactrum are Gram-negative, plant-associated soil bacteria (Lebuhn et al., 2000; Kettaneh et al., 2003). O. anthropi, O. intermedium, and O. pseudintermedium are opportunistic pathogens that can occasionally infect immunocompromised humans (Leclercq et al., 2019). The Ochrobactrum genome spans from 4.7 Mb to 8.3 Mb (Teyssier et al., 2005). Most Ochrobactrum genomes contain two circular chromosomes and multiple plasmids (Teyssier et al., 2005).
The phylogenetic relationships between Brucella and Ochrobactrum are extremely close. The phylogenetic analysis of Brucella and Ochrobactrum by Leclercq et al. indicated that the genus Ochrobactrum was divided into two branches, with one branch mainly composed of environmental species, while the other branch encompassed the entire genus Brucella and opportunistic pathogens (primarily O. anthropi, O. intermedium, and O. pseudintermedium). All Brucella species formed a single genomic species within the Ochrobactrum branch (Leclercq et al., 2019). Recently, Hördt et al. proposed that the Ochrobactrum genus and the Brucella genus should be merged into a single genus for the following two reasons. First, phylogenetic analysis based on 16S rRNA indicated that the entire Brucella genus was nested within the Ochrobactrum genus. Second, despite differences in virulence between Brucella and Ochrobactrum, other genera, such as Mycobacterium, Burkholderia, and Yersinia, also contain populations with different risk levels (Hordt et al., 2020).
Based on the aforementioned whole-genome phylogenetic analysis results and related proposals, the genera Brucella and Ochrobactrum have been merged into a single genus, Brucella (Centers for Disease Control and Prevention, 2022; DSMZ, 2024). The Brucella genus is currently comprised of two major categories: the facultative intracellular parasitic Brucella (formerly known as the Brucella genus) and the extracellular Brucella (formerly known as the Ochrobactrum genus) (Aljanazreh et al., 2023). The intracellular Brucella are further divided into classical intracellular Brucella and nonclassical intracellular Brucella. Classical intracellular Brucella consists of the six initially identified species and newer species such as B. ceti, B. pinnipedialis, B. microti, and B. papionis. Nonclassical intracellular Brucella, more distantly related phylogenetically to classic species, includes isolates from red foxes, Australian rodents, several frog species, rays, and human cases. These nonclassical intracellular Brucella represent several potential new species, with B. inopinata isolated from human cases being the sole representative (Cloeckaert et al., 2020).
In this study, we conducted a comparative genomic analysis of the latest Brucella genus utilizing publicly available genome data from the NCBI Genome database. The main objective of this study was to investigate the genetic evolutionary characteristics of the latest Brucella genus and elucidate the relationship between the genomic composition and prediction of phenotypic traits of Brucella.
With time more Brucella genomes are constantly becoming available. In this study, Brucella genomes with “complete” and “chromosome” assembly levels were collected from the Genome database of NCBI (accessed on 11 November 2022). After quality control was performed using CheckM 1.2.1 (Parks et al., 2015), 255 genomes with completeness greater than 90% were retained for subsequent analysis. The sample information of the 255 genomes was collected (Supplementary Figure S1; Supplementary Table S1). These genomes were obtained from 244 strains of 14 Brucella species and 11 strains that have not yet been identified. Moreover, these strains were collected from 2001 to 2022 in multiple countries, including Italy, China, Norway, and others. Additionally, these Brucella strains were isolated from a wide range of animal hosts and environments, such as humans, water buffaloes, cattle, pigs, dogs, sheep, and so on. The genomes of B. sp. 09RB8471 (GCA_001971625.1) and B. melitensis 6144 (GCA_023796775.1) were separately compared for collinearity with the genome of B. melitensis 16M (GCA_000007125.1) using Mauve 20150226 (Darling et al., 2004), and chromosome numbering errors in these two genomes were corrected (Supplementary Figure S2).
To ensure comparability, 255 Brucella genomes were annotated using Prokka 1.14.6 (Seemann, 2014). EggNOG-mapper 2.1.5 was utilized for functional annotation and for providing COG (clusters of orthologous groups) information for all the genes (Cantalapiedra et al., 2021). Virulence factor genes (VFGs) and antibiotic resistance genes (ARGs) in the Brucella genome were identified using the Virulence Factors of Pathogenic Bacteria Database (VFDB) (accessed on 27 March 2023) (Liu et al., 2022) and the Comprehensive Antibiotic Resistance Database (CARD) (version: card-data.3.2.9) (Alcock et al., 2023), respectively.
Gene families in the pangenome of 255 Brucella strains were identified using OrthoFinder 2.5.4 (-S diamond-M msa-T fasttree) (Emms and Kelly, 2015; Emms and Kelly, 2019). Core gene families (distributed in all 255 genomes), accessory gene families (distributed in at least 2 genomes, at most 254 genomes), and strain-specific genes (distributed only in one genome) were identified based on the OrthoFinder results. The pangenome growth curve was modeled using Heaps’ law (y = AxB + C) (Heaps, 1978; Tettelin et al., 2008), where y represents the number of gene families and x represents the number of genomes. Additionally, a 0–1 matrix file representing the presence/absence pattern of gene families was generated based on the results of OrthoFinder. To obtain a pan tree file, clustering analysis was conducted on the 0–1 matrix file using the R package “pheatmap” (clustering_method = “ward. D2,” clustering_distance = “manhattan”). Moreover, to compare the core tree and pan tree topology, the normalized matching-cluster (nMC) and normalized Robinson–Foulds (nRF) scores were calculated using TreeCmp 2.0-b76 (Bogdanowicz et al., 2012). These two scores range from 0 to 1, with higher values indicating greater differences in the topology between the two trees. Finally, a tanglegram of the two trees was generated using the R package “ape.”
The average nucleotide identity (ANI) values of the pairwise comparison of 255 Brucella genomes were calculated using Pyani 0.2.12 (−m ANIm) (Pritchard et al., 2016). The digital DNA–DNA hybridization (dDDH) values between each of the 255 genomes and B. melitensis 16M were calculated using GGDC 3.0 (Meier-Kolthoff et al., 2013, 2022). The dDDH values of pairwise comparison of the 25 reference genomes from different Brucella species were also calculated using GGDC 3.0. A phylogenetic tree of the 255 Brucella strains was constructed based on the core single-copy orthologous genes. Falsochrobactrum ovis DSM 26720 (GCA_003259955.1) was selected as an outgroup. Specifically, all orthologous groups within the 255 Brucella genomes and one outgroup genome were identified using OrthoFinder 2.5.4. Then, all the core single-copy orthologous gene sequences were aligned using Muscle 3.8.1551 (Edgar, 2021). The alignment products were subsequently trimmed using the Trimal 1.4.rev15 (Capella-Gutierrez et al., 2009). Subsequently, AMAS.py (Borowiec, 2016) was used to concatenate all the alignment results. Finally, using the concatenated multiple sequence alignment file, a maximum likelihood (ML) tree was built using Iqtree 2.0.3 (−m MFP-B 1000 --bnni-T AUTO --seed 123) (Minh et al., 2020). The phylogenetic tree was visualized using ITOL (Letunic and Bork, 2021).
HGTector 2.0b3 (Zhu et al., 2014) was used to identify horizontal transfer gene families (HTGFs) in 255 Brucella genomes that have transferred from various organisms outside the family Brucellaceae to the genus Brucella. Brucella was utilized as the self-group, and Brucellaceae was utilized as the close group.
Insertion sequences (ISs) in 255 Brucella genomes were identified using Isfinder (Siguier et al., 2006). Prophages were detected using PHASTER (Arndt et al., 2016). Integrative conjugative elements (ICEs) and integrative mobilizable elements (IMEs) were identified using ICEfinder (Liu et al., 2019).
Xiaowen Yang et al. and Jagadesan Sankarasubramanian et al. separately conducted studies on the pangenomes of eight (Yang et al., 2016) and 11 (Sankarasubramanian et al., 2017) intracellular Brucella species, respectively. The 255 strains in our study included not only 10 species of intracellular Brucella but also four species of extracellular Brucella. Therefore, compared to those in the aforementioned two studies, the pangenome in our study more comprehensively reflected the species diversity and genetic polymorphisms of the Brucella genus. A total of 9,091 gene families were identified in the pangenome analysis, including 1,404 core gene families, 4,928 accessory gene families, and 2,759 strain-specific genes (Figure 1A; Supplementary Table S2). Several Brucella strains harbored more accessory gene families or strain-specific genes than did the other strains, resulting in a high proportion of accessory gene families and strain-specific genes in the pangenome (Figure 1B). In particular, B. intermedia, B. pseudintermedia, B. anthropi, and B. pseudogrignonensis harbored more accessory gene families and strain-specific genes. With the inclusion of additional genomes, the pangenome expanded continuously (Figure 1C), following Heaps’ law (y = AxB + C). Notably, the parameter B was greater than 0; thus, the pangenome in this study was open, suggesting that a large number of genomes need to be analyzed to fully characterize the complete gene pool of the Brucella genus. The open pangenome reflected the genetic diversity of the Brucella genus, indicating the possibility of genetic exchange among the gene pool of Brucella and the external environment. In contrast, the core genome exhibited the opposite trend (Figure 1C), as depicted by a fitted model (y = AeBx + C), in which the size gradually decreased with the addition of new genomes.
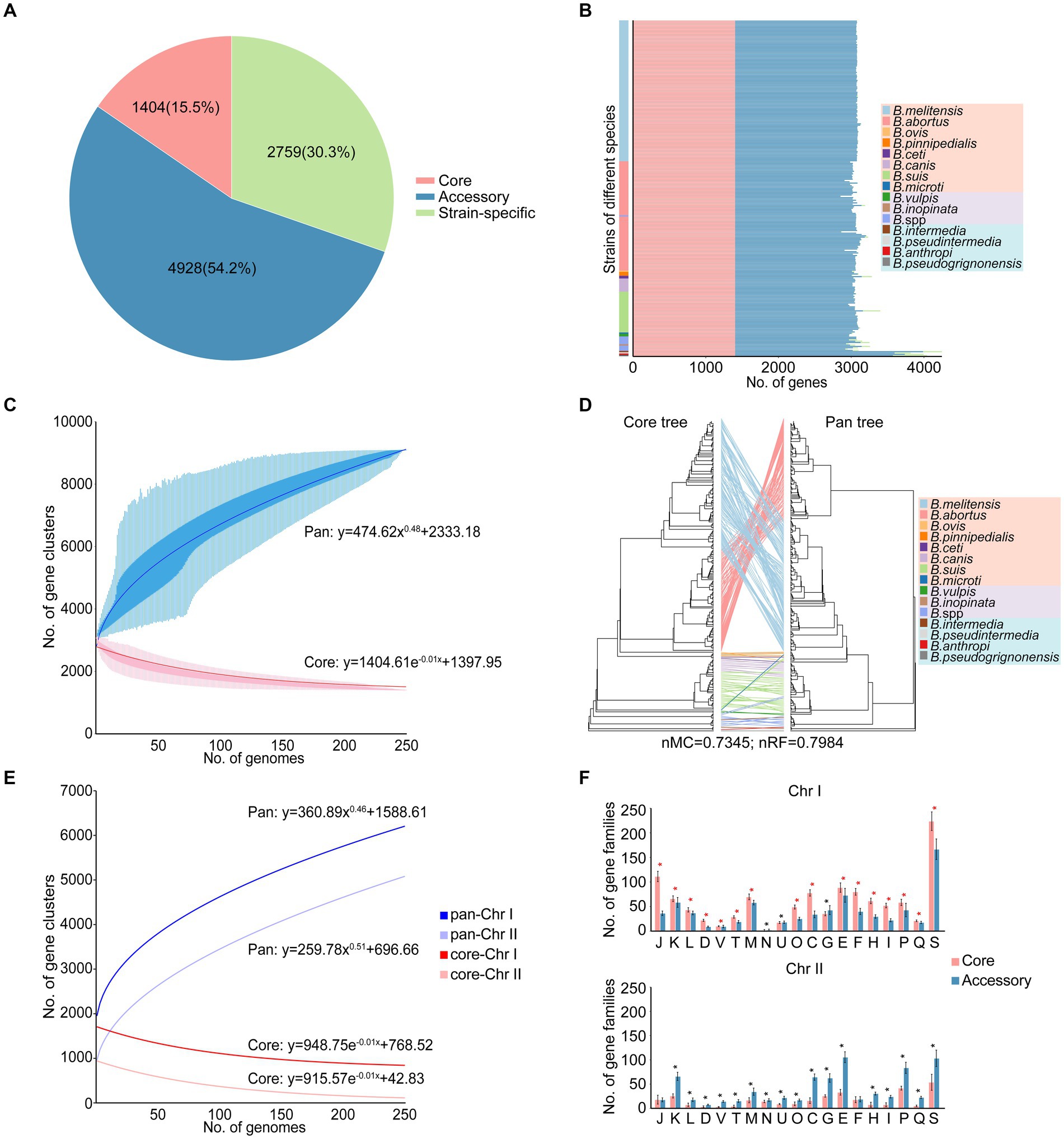
Figure 1. Characteristics of the pangenome of the Brucella genus. (A) The pangenome composition. (B) The numbers of core gene families, accessory gene families and strain-specific genes in each strain. (C) The accumulation curves of the pangenome and core genome of the Brucella genus. The blue curve shows an open pangenome; the deduced pangenome size was y = 474.62x0.48 + 2333.18. The red curve shows the core genome, and the deduced core genome size was y = 1404.61e–0.01x + 1397.95. (D) The tanglegram shows the differences in the topological structures between the core tree and the pan tree. A core tree was created based on core single-copy orthologues. The pan tree was created based on a binary panmatrix (gene presence/absence matrix). (E) The accumulation curves of the pan-Chr I and pan-Chr II of the Brucella genus. The dark color represents Chr I, while the light color represents Chr II. (F) The distribution and COG functional characteristics of the core and accessory gene families on Chr I and Chr II. * t–test p < 0.05.
To elucidate the impact of core gene families, accessory gene families, and strain-specific genes on the phylogeny of the Brucella genus, a core tree and a pan tree were constructed and compared (Figure 1D). The nMC and nRF scores were computed to assess the differences in tree topology between the core tree and the pan tree. In this study, the nMC and nRF scores were 0.7345 and 0.7984, respectively, indicating substantial disparities between the core tree and pan tree of the Brucella genus. The presence of a large number of accessory gene families and strain-specific genes in the open pangenome of the Brucella genus led to significant differences between the core tree and the pan tree. Furthermore, on the core tree and pan tree, extracellular Brucella and intracellular Brucella were significantly separated. Classical intracellular Brucella and nonclassical intracellular Brucella were separated from each other on the core tree, but there was a crossover phenomenon on the pan tree. This indicates a certain difference between extracellular Brucella and intracellular Brucella in core-pangenome assemblies.
The genomes of Brucella species are composed of two chromosomes. Pan-chromosome analyses were conducted on different chromosomes of Brucella. Based on Heaps’ law (y = AxB + C), the openness of chromosome II (Chr II) was greater than that of chromosome I (Chr I) (BChr II > BChr I) (Figure 1E), suggesting that Chr II may be more dynamic than Chr I. The two chromosomes of Brucella are considered to have different evolutionary origins, and the smaller Chr II could be derived from a mega-plasmid possessing many dispensable genes (Bavishi et al., 2010). The functional and distribution characteristics of the core and accessory gene families on Chr I and Chr II were further explored, respectively (Figure 1F). Many core and accessory gene families were distributed on Chr I, with the core gene families being dominant, indicating that Chr I of Brucella was relatively conserved and stable. Except for the core gene families associated with “N: cell motility,” “U: intracellular trafficking, secretion, and vesicular transport,” and “G: carbohydrate transport and metabolism,” the abundance of core gene families related to other functions was significantly higher than that of accessory gene families on Chr I (t-test, p < 0.05). In contrast, the accessory gene families were dominant on Chr II, indicating that Chr II of Brucella was more dynamic and plastic and served as the primary reservoir for genetic diversity in the Brucella genus. Except for the accessory gene families associated with “J: translation, ribosomal structure and biogenesis” and “F: nucleotide transport and metabolism,” the abundance of accessory gene families related to other functions was significantly higher than that of core gene families on Chr II (t-test, p < 0.05).
To further explore the genetic diversity of the Brucella genus, a comparison was conducted on the genome sizes and GC contents of different Brucella species. It was evident that, with an average size of 4.69 Mb and an average of 4,327 protein-coding genes per genome, the genomes of extracellular Brucella species, such as B. pseudogrignonensis, B. anthropi, B. pseudintermedia, and B. intermedia, were significantly larger than those of facultative intracellular Brucella species (Figure 2A; Supplementary Table S1). The average genome size of facultative intracellular Brucella was 3.36 Mb, with an average of 2,971 protein-coding genes per genome. Furthermore, the genomic GC content of extracellular Brucella was significantly different from that of facultative intracellular Brucella (Figure 2A, Supplementary Table S1). The genomic GC content of facultative intracellular Brucella was highly conserved, ranging from 57.1 to 57.3%. However, different extracellular Brucella species had significant variations in genomic GC content, with the highest being 57.96% (B. pseudintermedia strain ASAG-D25) and the lowest being 53.71% (B. pseudogrignonensis strain K8). In summary, there were significant differences in genome size and GC content between facultative intracellular Brucella and extracellular Brucella. The genomes of different facultative intracellular Brucella species were conservative, while greater heterogeneity was observed among the genomes of different extracellular Brucella species.
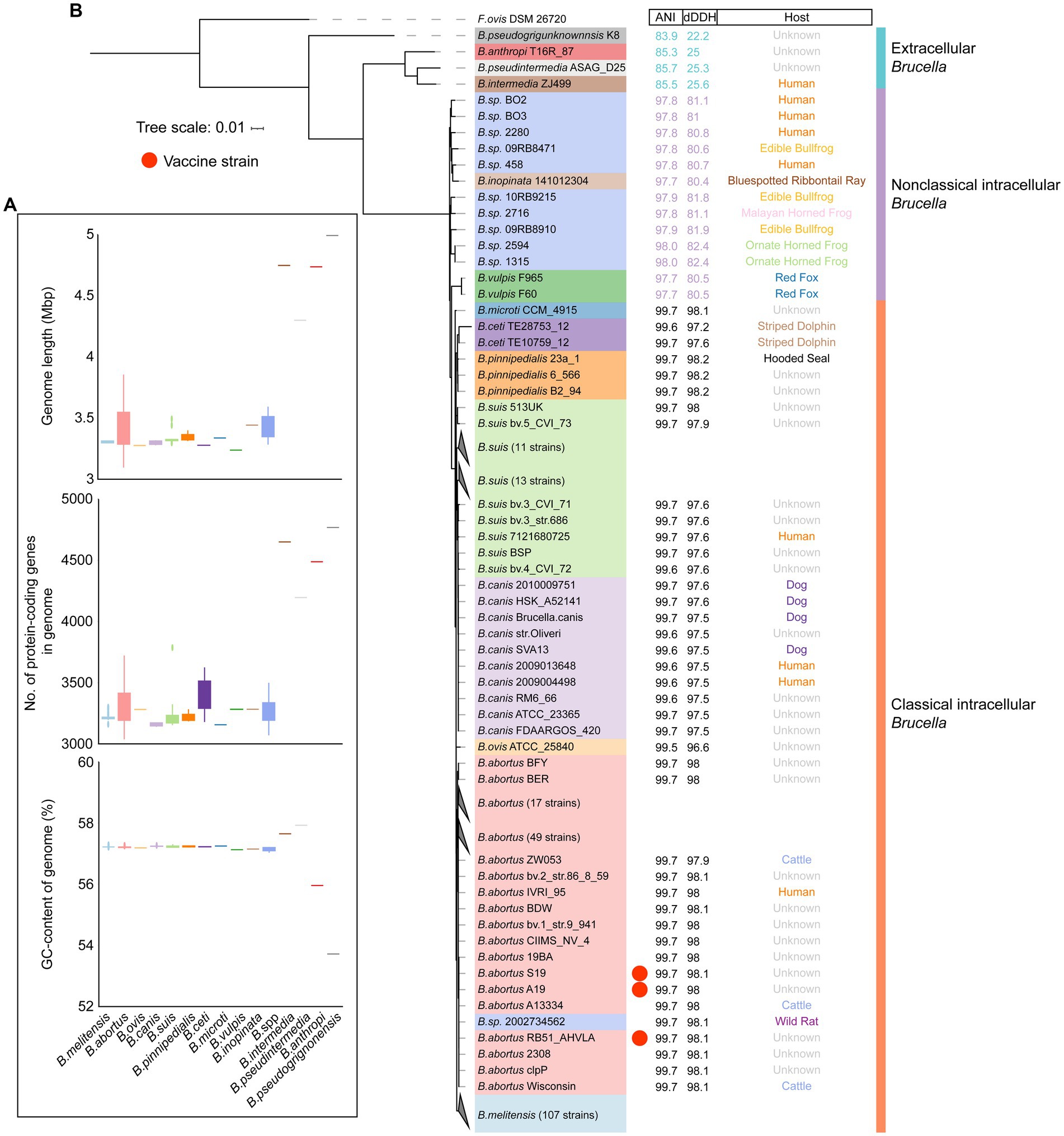
Figure 2. The genomic features and phylogenetic relationships of different Brucella species. (A) The genome length, number of protein-coding genes, and GC content of different Brucella species. (B) Phylogenomic tree showing the relationships between different Brucella species. The tree was created based on core single-copy orthologues using the maximum likelihood method in IQTREE and visualized using ITOL. Falsochrobactrum ovis DSM 26720 was used as an outgroup to root the tree. The strains from different Brucella species are differentiated using different colors. The ANI and dDDH values, and the host of each strain are displayed on the three columns.
To further assess the phylogenetic relationships among the different Brucella species, the core genome phylogenetic tree was constructed for 255 Brucella strains (Figure 2B). Within the extracellular Brucella, a separate branch was formed by B. pseudogrignonensis. B. intermedia, B. pseudintermedia, and B. anthropi clustered together. All the intracellular Brucella species clustered together. These results are consistent with the findings of Leclercq et al. They found that the entire Brucella genus was comprised of two major branches. One branch encompassed extracellular Brucella not involved in opportunistic infections, such as B. pseudogrignonensis. The other branch contained opportunistic pathogenic extracellular Brucella species (including B. intermedia, B. pseudintermedia, and B. anthropi) alongside intracellular Brucella capable of causing brucellosis (Leclercq et al., 2019). By calculating the ANI and dDDH values between each Brucella strain and the reference strain B. melitensis 16M, significant differences were identified among extracellular Brucella, nonclassical intracellular Brucella, and classical intracellular Brucella (Figure 2B). The ANI values between the extracellular Brucella strains and the reference strain B. melitensis 16M ranged from 83.9 to 85.7%. The ANI values between nonclassical intracellular Brucella strains and the reference strain B. melitensis 16M ranged from 97.7 to 98%. The ANI values between classical intracellular Brucella strains and the reference strain B. melitensis 16M were greater than 99%. The dDDH values exhibited the same characteristics as the ANI values (Figure 2B; Supplementary Figure S3). The dDDH values between extracellular Brucella strains and the reference strain B. melitensis 16M ranged from 22.2 to 25.3%. The dDDH values between nonclassical intracellular Brucella strains and the reference strain B. melitensis 16M ranged from 80.4 to 82.4%. The dDDH values between classical intracellular Brucella strains and the reference strain B. melitensis 16M were greater than 95%. ANI and dDDH values are widely used for prokaryotic taxonomic identification (Sentausa and Fournier, 2013). An ANI value greater than 96% or a DDH value greater than 70% was considered to indicate a single species (Konstantinidis and Tiedje, 2005; Goris et al., 2007). However, these two thresholds were not applicable for species identification within the Brucella genus. Nonetheless, the ANI and dDDH values still revealed significant genomic differences between extracellular Brucella, nonclassical intracellular Brucella, and classical intracellular Brucella (Supplementary Figure S4; Supplementary Figure S5).
Subsequently, the phylogenetic positions of all the undefined Brucella strains were examined. B. sp. 2002734562 clustered together with other B. abortus strains (Figure 2B), suggesting that B. sp. 2002734562 may be a member of B. abortus. The branch closest to B. sp. 2002734562 was occupied by B. abortus RB51-AHVLA (Supplementary Figure S3). B. abortus 2308, B. abortus S19, and B. abortus A19 were also close to the branch of B. sp. 2002734562. B. abortus RB51-AHVLA, B. abortus S19, and B. abortus A19 are vaccine strains of B. abortus (Refai, 2002; Moriyón et al., 2004; Wang et al., 2022). B. abortus 2308 is a virulent strain of B. abortus (Kellerman et al., 1970). Moreover, the other undefined strains of nonclassical Brucella exhibited a close phylogenetic relationship with B. inopinata (Figure 2B; Supplementary Figure S3). B. sp. 1315, B. sp. 2594, B. sp. 09RB8910, B. sp. 2716, and B. sp. 10RB9215 clustered together. Interestingly, these five strains were all isolated from frogs or toads. B. sp. 09RB8471 was also isolated from a frog and clustered together with B. inopinata 141012304 and 4 undefined strains isolated from humans. Although these 10 undefined strains and B. inopinata were separated into two clades, considering that the ANI and dDDH values of the members from the two clades were generally similar, we inferred that these undefined strains may come from the same species, such as B. inopinata.
Horizontal gene transfer (HGT) refers to the transfer of genetic material between different organisms that are not related to the parent or offspring. HGT is an important mechanism in bacterial evolution that can promote the diversity and adaptability of bacterial populations (Jain et al., 1999; Ochman et al., 2000). In our study, the open pangenome of the Brucella genus contained a large number of accessory gene families and strain-specific genes, suggesting that HGT may have played an important role in the evolution of Brucella. Using HGTector, a total of 261 horizontal transfer gene families (HTGFs) in 255 Brucella genomes were identified, which had been transferred from various organisms outside the family Brucellaceae to the genus Brucella (Supplementary Table S3). All genomes of different Brucella species were found to contain HTGFs (Figure 3A). Variations were found in the number of HGTFs among different Brucella species (Figure 3B). The highest number of HTGFs was found in the genome of B. pseudogrignonensis, followed by B. intermedia, with 73 and 58 HTGFs, respectively. The genomes of B. ovis, B. vulpis, and B. inopinata contained fewer HTGFs, averaging 21–25 HTGFs each. The genomes of other Brucella species each had an average of 33–44 HTGFs.
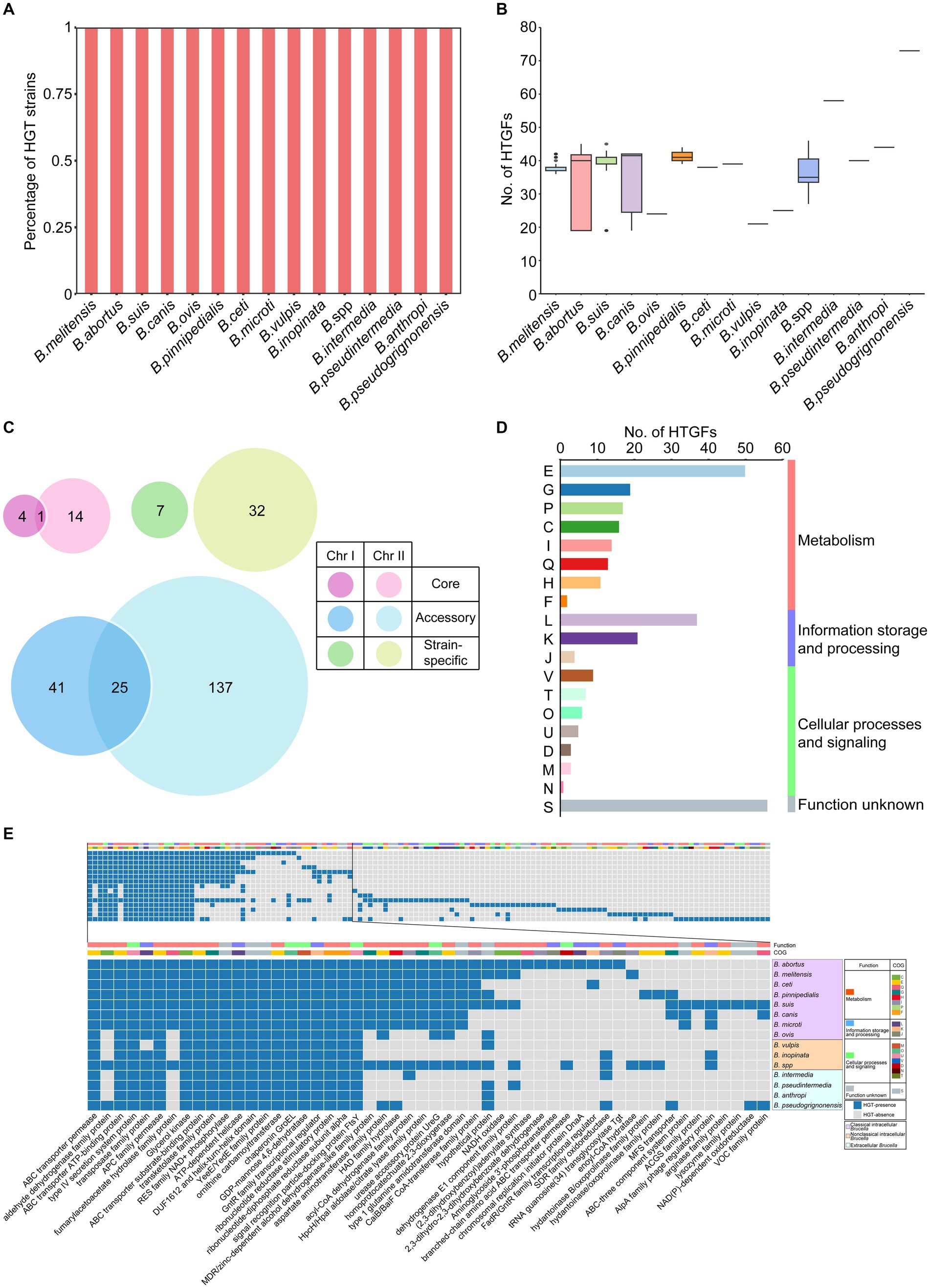
Figure 3. Characteristics of HGT within the Brucella genus. (A) The percentage of strains with HTGFs of different Brucella species. (B) The number of HTGFs carried by each genome of different Brucella species. (C) The distribution of HTGFs on two chromosomes and the pangenome of Brucella. (D) COG categories of HTGFs. (E) The products encoded by horizontal transfer genes of different Brucella species.
Subsequently, the distribution of the 261 HTGFs in the genome and their relationships with the pangenome of the Brucella genus were further analyzed (Figure 3C). Among the 261 HTGFs, 52 (19.9%) were exclusively distributed on Chr I, 183 (70.1%) were exclusively distributed on Chr II, and the remaining 26 (10%) were distributed on both chromosomes. Furthermore, 19 (7.3%) were core gene families, 203 (77.8%) were accessory gene families, and the remaining 39 (14.9%) were strain-specific genes. The size of Chr II of the Brucella genus was significantly smaller than that of Chr I (Rajendhran, 2021). However, Chr II contained more HTGFs than Chr I, suggesting that Chr II of the Brucella genus might have experienced more HGT events than Chr I. HGT is an important mechanism for bacterial evolution and adaptation (De la Cruz and Davies, 2000). Using Alien_hunter, Anish Bavishi et al. found that the level of HGT on Chr II of B. melitensis 16M and B. anthropi ATCC 49188 was greater than that on Chr I (Bavishi et al., 2010). Our study examined HGT in the whole Brucella genus, aligning with Anish Bavishi et al.’s findings, and showing a higher HGT level in Chr II compared to Chr I in Brucella. Anish Bavishi et al. proposed a plasmid-mediated origin for Chr II as the preferred model explaining this bias of HGT (Bavishi et al., 2010). In this model, a resident plasmid would acquire essential gene clusters through intragenomic gene transfer from Chr I and HGT from other species, leading to the formation of Chr II. This accessory chromosome (Chr II) may then acquire new genes via various mechanisms like HGT. With Chr I housing crucial housekeeping genes, Chr II likely evolved as an adaptive reservoir under selection pressure to accommodate different ecological niches and nutritional challenges and played a vital role in Brucella’s evolution and adaptation.
To further elucidate the functional characteristics of these HTGFs, the COG information of these gene families was analyzed. These HTGFs were involved in diverse biological functions (Figure 3D). Nearly half (48%) of the HTGFs were associated with metabolism. Approximately one-fifth (21%) of the HTGFs were related to information storage and processing. Twelve percent of the HTGFs were related to cellular processes and signaling. However, the functions of the remaining 19% of the HTGFs are still unclear. In particular, the proportion of HTGFs associated with “E: amino acid transport and metabolism” was the highest (19%). The proportion of HTGFs associated with “L: replication, recombination, and repair” was the second highest (14%). Brucella are amino acid prototrophs that can grow with ammonium and contain many amino acid/peptide/polyamine uptake genes (Ronneau et al., 2016). Our research revealed that HGT played a significant role in the acquisition of essential biological functions such as amino acid transport and metabolism in the Brucella genus.
The functional characteristics of the HTGFs were further compared between different Brucella species. These 261 HTGFs were found to encode a total of 134 different proteins by a BLAST search using the NCBI nr database (Figure 3E; Supplementary Figure S6). The HTGFs associated with gene products such as ABC transporter, type IV secretion system proteins, APC family permease, and 15 other types were widely distributed throughout the Brucella genus (Figure 3E). The fact that different Brucella species shared these HTGFs suggested that relevant HGT events may have occurred in the ancient ancestor of the Brucella genus. The HTGFs associated with the transposase family proteins were distributed in all Brucella species except for B. vulpis. These HTGFs may represent historical traces of previous HGT in Brucella, and they may also provide a certain material basis for Brucella genomes to undergo additional HGT.
In classical intracellular Brucella, the HTGFs of B. abortus and B. suis encoded the most gene products, with 41 types (Figure 3E). The HTGFs of B. ovis encoded the fewest gene products, with 23 types. Different classical intracellular Brucella species commonly contained HTGFs associated with 22 gene products, such as ABC transporter permease, ABC transporter ATP-binding proteins, type IV secretion system proteins, and so on. Compared with other classical intracellular Brucella species, B. ovis lacked HTGFs associated with aldehyde dehydrogenase family proteins, fumarylacetoacetate hydrolase family proteins, and MDR/zinc-dependent alcohol dehydrogenase-like family proteins, and so on. Additionally, the abundance of HTGFs associated with ABC transporter permease, ABC transporter ATP-binding proteins in B. ovis was relatively lower than in other classical intracellular Brucella, and these ABC transport systems were primarily associated with amino acid transport (Supplementary Figure S6). ABC transporters play a significant role in the antibiotic resistance and virulence of bacteria (van Veen and Konings, 1998; Paulley et al., 2007). Jenner et al. reported that B. abortus, B. melitensis, B. suis, B. canis, and B. ovis all contain a large number of ABC transport systems and that B. ovis has relatively fewer ABC transport systems than the other four species, primarily lacking polyamine transporters. Considering that B. ovis does not cause zoonotic infections, Jenner et al. speculated that the absence of ABC transport systems in B. ovis may be related to the virulence of Brucella (Jenner et al., 2009). In our study, HTGFs associated with ABC transport systems were relatively less abundant in B. ovis compared to other classical intracellular Brucella species. The differential distribution of HTGFs associated with ABC transport systems and other functional proteins between B. ovis and other classical intracellular Brucella species may be linked to the significant differences in zoonotic potential and host preferences between them. Additionally, B. suis and B. canis have a close phylogenetic relationship, with B. canis often nested within the branch of B. suis on the phylogenetic tree (Fretin et al., 2008). However, significant differences were found in their HTGFs. The HTGFs associated with ACGS family proteins, arginase family proteins, and lysozyme family proteins were unique to B. suis (Figure 3E). These HTGFs were not found in B. canis.
In nonclassical intracellular Brucella, B. vulpis harbored HTGFs related to 20 gene products, among which those associated with DEAD/DEAH box helicase were unique to B. vulpis (Supplementary Figure S6). B. inopinata harbored HTGFs related to 24 gene products but did not contain any unique HTGFs. The unclassified Brucella strains harbored HTGFs related to 59 gene products, among which those associated with 14 gene products such as FAD-dependent oxidoreductase, GNAT family N-acetyltransferase, and helicase RepA family proteins were specific to these unclassified Brucella strains.
Different extracellular Brucella species had HTGFs that correspond to 34–54 gene products (Supplementary Figure S6). The HTGFs associated with 14 gene products such as the AraC family transcriptional regulator, and M20 aminoacylase family proteins were unique to B. intermedia. The HTGFs related to 10 gene products including ACR3 family arsenite efflux transporter, and LysE family translocator were specific to B. pseudintermedia. The HTGFs associated with 11 gene products such as arginine deiminase, and histidinol dehydrogenase were unique to B. anthropi. Additionally, HTGFs related to 19 gene products like amidohydrolase, and ferrochelatase were specific to B. pseudogrignonensis.
In summary, significant differences in the distribution of HTGFs were found among classical intracellular Brucella, nonclassical intracellular Brucella, and extracellular Brucella. In particular, extracellular Brucella was found to contain a greater number of unique HTGFs than intracellular Brucella. Intracellular Brucella has a host preference and an intracellular lifestyle, which to some extent hinders the occurrence of HGT (Rouli et al., 2015). On the contrary, the free distribution of extracellular Brucella in the environment was more conducive to genetic exchange with other organisms. These findings indicate that HGT enhanced the genetic diversity of Brucella and facilitated the acquisition of various important biological functions.
Mobile genetic elements (MGEs) play a crucial role in HGT as important carriers for transmitting genetic information across cells, individuals, and species (Haudiquet et al., 2022). IS elements, one type of MGE, are small DNA fragments ranging in length from 700 bp to 2,500 bp (Aljanazreh et al., 2023). IS elements play a crucial role in promoting genomic plasticity and genetic variability. Beneficial mutations generated by IS transposition help bacteria overcome environmental challenges and adapt to new ecological niches (Vandecraen et al., 2017). In our study, IS elements were found in all 255 Brucella genomes (Figure 4; Supplementary Table S4). Each Brucella genome contained an average of 12 IS elements. Different Brucella species contained varying quantities of IS elements (Figure 4). The genomes of B. ovis, B. pinnipedialis, B. vulpis, B. inopinata, B. pseudintermedia, B. sp. 2280, and B. sp. 458 contained relatively high numbers of IS elements, with an average of 42. In particular, the genome of B. sp. 2280 had the highest number of IS elements, with as many as 58 IS elements. In contrast, the other Brucella genomes contained fewer IS elements, with an average of 11. IS elements were found on Chr I of all 255 Brucella genomes and were found on Chr II of 250 Brucella genomes (Figure 4). Furthermore, Significant differences were observed in the distribution of IS elements between intracellular and extracellular Brucella. Chr I of intracellular Brucella contained a greater number of IS elements than did Chr II. However, Chr I of extracellular Brucella contained a fewer number of IS elements than did Chr II. In particular, Chr I of B. intermedia, B. pseudintermedia, and B. pseudogrignonensis had fewer IS elements than did Chr II. Chr I and Chr II of B. anthropi contained an equal number of IS elements. The density of IS elements on the two chromosomes of classical intracellular Brucella, nonclassical intracellular Brucella, and extracellular Brucella varied significantly (Supplementary Figure S7). The average distribution density of IS elements on Chr I was significantly higher than that on Chr II in classical and nonclassical intracellular Brucella. Conversely, the average distribution density of insertion sequences on Chr I was significantly lower than that on Chr II in extracellular Brucella.
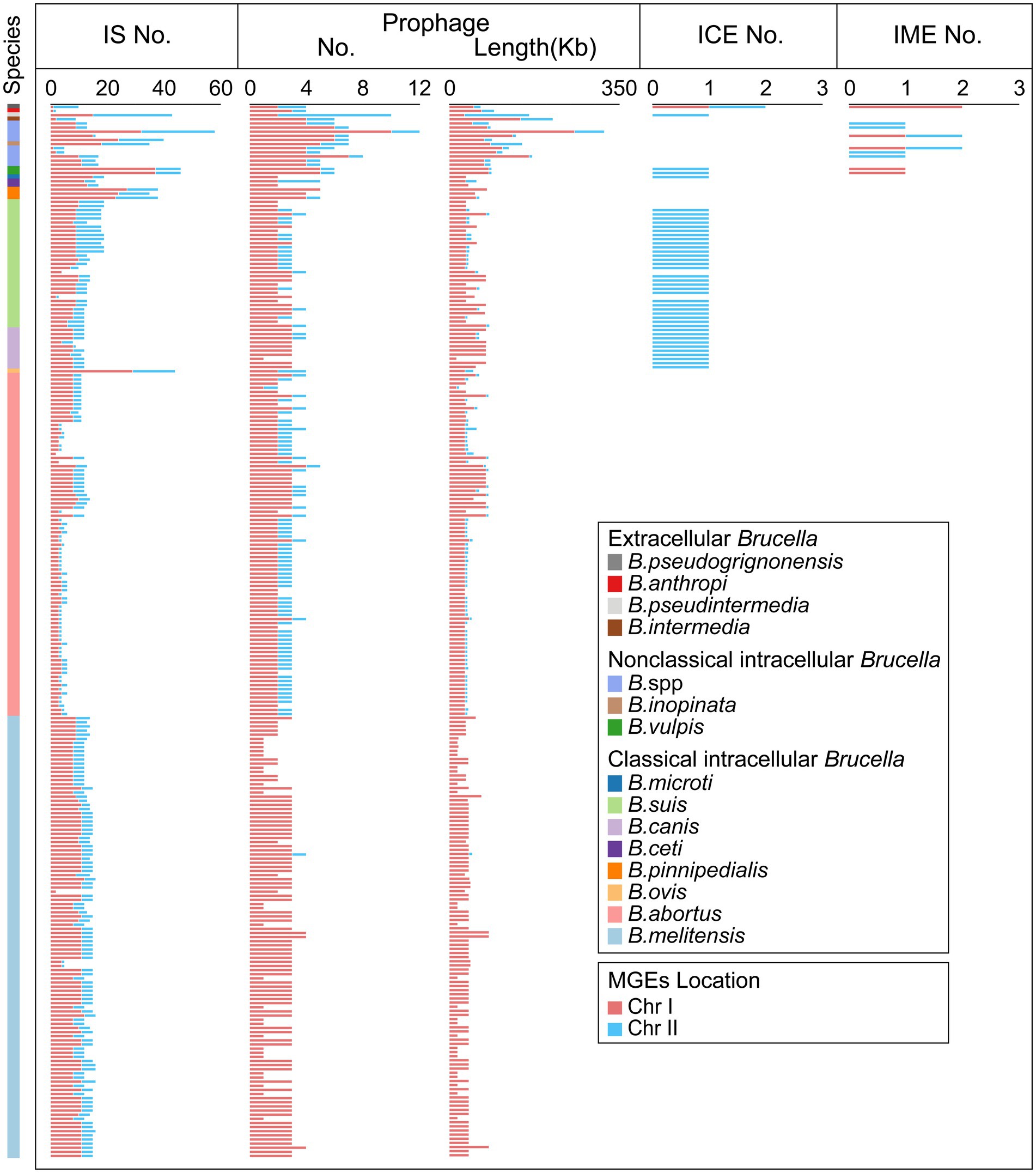
Figure 4. Distribution of MGEs in the Brucella genus. The presence of IS elements, prophages, ICEs, and IMEs, on Chr I and Chr II of each Brucella strain are shown.
The types of all IS elements present in the 255 Brucella genomes were further analyzed. A total of 62 types of IS elements were identified (Figure 5). Intact copies of IS711, ISBm3, ISBm2, IS1953, and IS2020 were present in almost all the genomes of classical intracellular Brucella without any other types of IS elements (Figure 5). ISBm3 and ISBm2 were mostly present as single copies in these genomes. IS1953 and IS2020 were mostly present as double copies in these genomes. The number of IS711 was significantly greater than that of the other four IS elements, with an average of 8 copies per genome. Different species of classical intracellular Brucella possessed unique copy numbers of IS711, and Chr I of these Brucella strains contained more IS711 than Chr II (Supplementary Table S4). B. ovis contained the highest number of IS711, with 26 IS711 present on Chr I and 12 on Chr II. B. pinnipedialis contained the second highest number of IS711, with 21 IS711 present on Chr I and 10 on Chr II. The number of IS711 present in the genomes of other species of classical intracellular Brucella did not exceed 15. Furthermore, the aforementioned five types of IS elements were also present in some genomes of nonclassical intracellular Brucella species, but none of these genomes simultaneously harbored all five types of IS elements. Apart from these five types of IS elements with intact copies, 24 other types of truncated IS elements were distributed in the genomes of nonclassical intracellular Brucella species, with completeness less than 6%. Moreover, none of the genomes of extracellular Brucella contained the five aforementioned types of IS elements. B. anthropi did not possess any unique IS elements. The other three extracellular Brucella species, namely, B. intermedia, B. pseudintermedia, and B. pseudogrignonensis, harbored 6, 19, and 18 unique IS elements, respectively. Among them, ISKpn12, ISPaes1, and ISPye59 were unique to B. pseudintermedia and exhibited completeness levels of 100, 100, and 31.28%, respectively. The completeness of the other IS elements were less than 6%. In summary, the genomes of classical intracellular Brucella strains almost universally contained intact copies of IS711, ISBm3, ISBm2, IS1953, and IS2020. The genomes of nonclassical intracellular Brucella and extracellular Brucella contained residual components of many other types of IS elements, which are completely absent in classical intracellular Brucella species.
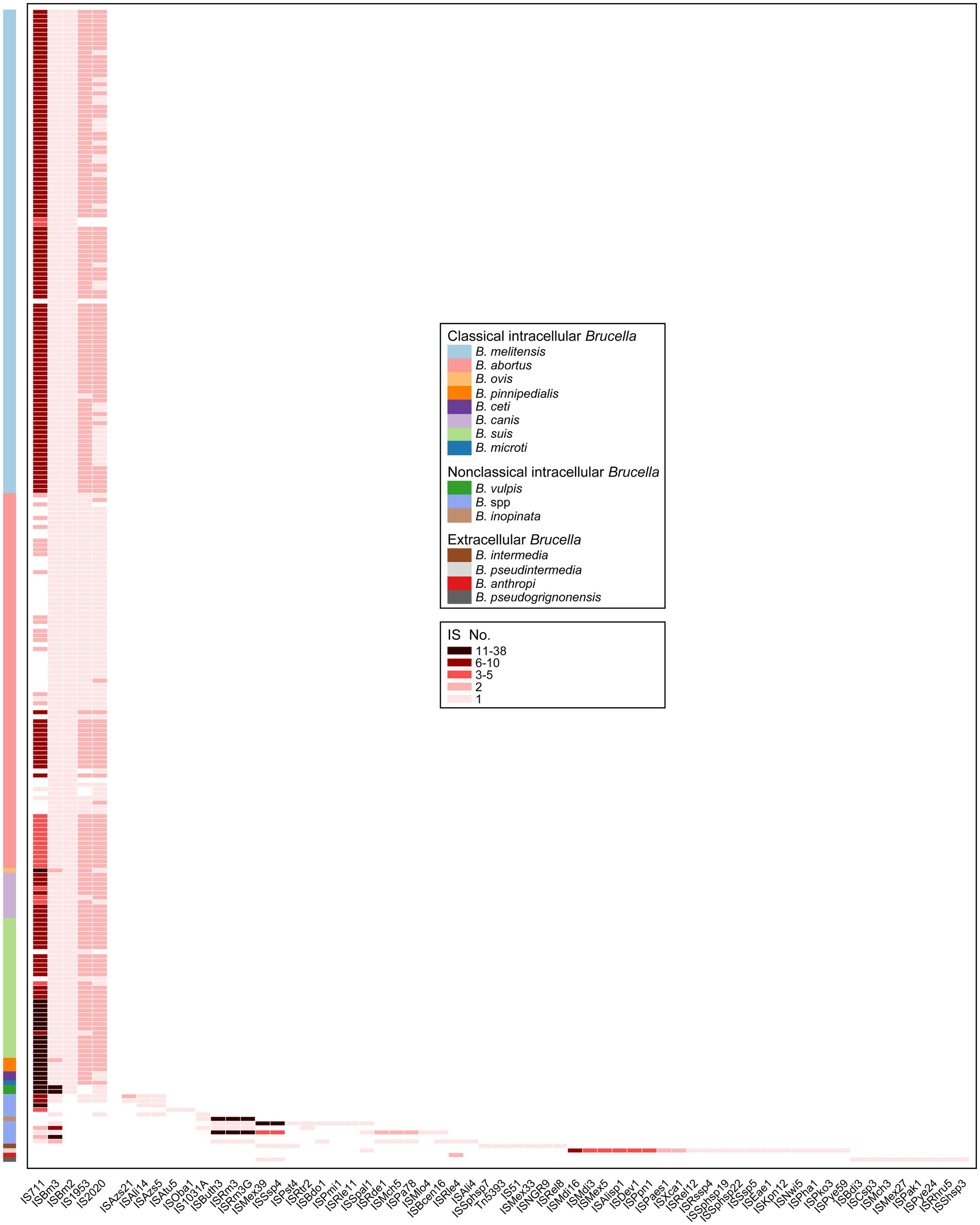
Figure 5. Characteristics of the types and copy numbers of IS elements carried by each genome of different Brucella species.
The residual IS components act as “scars” in the genome and represent traces of numerous ancestral transposition events; these components can provide important insights into the evolutionary history of host genomes (Siguier et al., 2014). The ancient ancestors of the Brucella genus are typically considered a group of free-living bacteria distributed in environments such as soil (Suarez-Esquivel et al., 2020). As the ancestors of the Brucella genus evolved from extracellular to intracellular bacteria, many non-essential genes on their genomes were eliminated through pseudogenization, leading to continuous genome reduction (Liu et al., 2004). IS elements participated in the pseudogenization process by transposition, and they were partially or completely lost as the genome shrank (Siguier et al., 2014). This study found that extracellular Brucella and nonclassical intracellular Brucella contained many residual traces of IS elements, while classical intracellular Brucella completely lacked these residual traces. This may indicate, to better adapt to their respective environments, that classical intracellular Brucella underwent higher levels of pseudogenization and genome reduction compared to nonclassical intracellular Brucella and extracellular Brucella.
The number of IS711 among the same Brucella species was comparatively conserved (Ouahrani et al., 1993). Due to its species-specific nature, IS711 has been utilized for the identification of different Brucella species (Paul et al., 2020). However, in our study, some genomes of B. melitensis, B. abortus, B. suis, and undefined Brucella strains did not contain IS711. Thus, there are limitations to using IS711 for identifying Brucella species, and caution should be exercised when applying this method in practical use. Furthermore, Ocampo-Sosa et al. reported that under laboratory conditions, IS711 in the genomes of B. ovis and B. pinnipedialis exhibited transpositional activity, while no similar phenomenon was detected for B. melitensis or B. abortus (Ocampo-Sosa and Garcia-Lobo, 2008). B. ovis and B. pinnipedialis possessed a significantly high number of IS711 copies, with 38 and 37 copies, respectively. In our study, B. vulpis also had a high number (27) of IS711 copies. Determining whether IS711 in B. vulpis could exhibit transpositional activity under laboratory conditions, similar to that of B. ovis and B. pinnipedialis, requires further exploration.
Prophages, which are a type of MGE, can integrate into the bacterial genome, influencing genetic variation, biological characteristics, and the epidemiology of bacteria (Canchaya et al., 2003; Kaden et al., 2014). In our study, 775 prophages (19 intact prophages, 23 questionable prophages, and 733 incomplete prophages) were identified in 255 Brucella genomes, with an average of 3 prophages per genome (Figure 4, Supplementary Table S5). Except for a few intact prophages, the vast majority of prophages in the Brucella genomes were defective. These defective prophages may represent ancient components of Brucella genomes (Canchaya et al., 2003). Among the 255 Brucella genomes, 147 genomes harbored prophages exclusively on Chr I, including 106 B. melitensis genomes, 15 B. abortus genomes, 14 B. suis genomes, 8 B. canis genomes, 1 B. ceti genome, 2 B. pinnipedialis genomes, and 1 B. microti genome. The remaining 108 genomes harbored prophages on both Chr I and Chr II. Moreover, except for the genomes of B. pseudintermedia ASAG_D25 and B. ceti TE28753-12, Chr I of the remaining 253 genomes harbored more prophages than Chr II, with an average of 3 and 1 prophages, respectively. Chr I and Chr II of B. pseudintermedia ASAG_D25 harbored 2 and 8 prophages, respectively. Chr I and Chr II of B. ceti TE28753-12 harbored 2 and 3 prophages, respectively. Furthermore, the average length of the prophages on Chr I was significantly greater than that on Chr II, specifically 17.3 Kb and 8.8 Kb, respectively. In addition, different Brucella species harbored varying numbers of prophages. The genome of B. sp. 2280 had the highest number of prophages, with up to 12. The genome of B. pseudintermedia ASAG_D25 contained the second highest number of prophages, with up to 10. The genomes of B. vulpis, B. inopinata, B. intermedia, and other undefined Brucella strains had an average of 6 prophages. The remaining Brucella genomes had an average of 3 prophages.
Prophages play significant roles in bacterial evolution and can facilitate the genetic diversity of bacteria (Canchaya et al., 2003). Studies have shown that most prophages in bacterial genomes are defective and in a state of gradual decay (Canchaya et al., 2003). Furthermore, research has found that Brucella’s Chr II contains more pseudogenes than Chr I, indicating a higher degree of genome reduction on Chr II (Zhong et al., 2012). In our study, the majority of prophages in the Brucella genus were found to be incomplete, the number of prophages on Chr II was fewer and their average length was lower compared to Chr I, suggesting that as the two chromosomes of Brucella underwent different degrees of decay, the prophage components gradually diminished, with more severe decay on Chr II. In addition, there were significant differences in the content of prophages among different Brucella species, especially with genomes of nonclassical intracellular Brucella containing a greater number of prophages, indicating a significant relationship between prophages and the genetic diversity of Brucella.
The types of prophages in the Brucella genomes were further analyzed. The 775 prophages were classified into 64 different types according to the bacteriophage genes carried by the prophages (Figure 6). Among these types, 35 were exclusively distributed on Chr I of Brucella, 19 were exclusively distributed on Chr II, and the remaining 10 were distributed on both Chr I and Chr II. Escher_RCS47, Dinoro_vB_DshS_R5C, Rhizob_RHEph10, Pseudo_nickie, Paenib_Tripp, Salmon_SSU5, and Brucel_BiPBO1 were relatively widely distributed in the Brucella genus. Brucel_BiPBO1 had the widest distribution range and was found in at least 10 species, including B. melitensis, B. abortus, B. suis, B. canis, B. ceti, B. pinnipedialis, B. vulpis, B. intermedia, B. pseudintermedia, and some undefined Brucella strains. Escher_RCS47 was exclusively distributed on Chr II of Brucella, while the remaining 6 types were primarily distributed on Chr I. Moreover, the other 57 types of prophages were distributed in specific Brucella species.
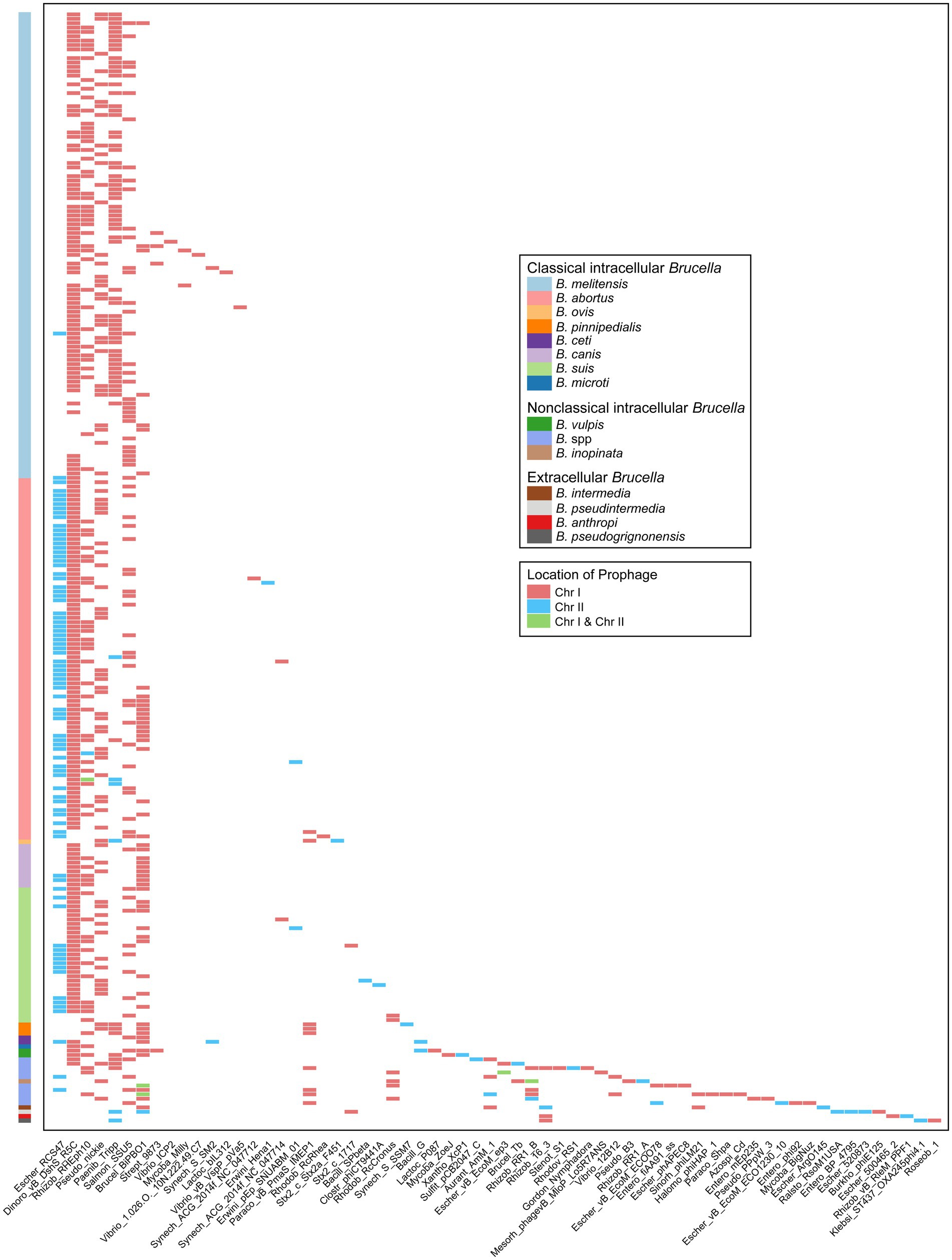
Figure 6. Characteristics of the types and chromosomal locations of prophages carried by the genomes of different Brucella species.
Several studies on prophages in the Brucella genus have been published to date. Studies have shown that prophages are related to the genetic diversity of B. canis (Kaden et al., 2014) and B. melitensis (Azam et al., 2016; Abou Zaki et al., 2017). Active phages were successfully induced by Hammerl et al. from intracellular Brucella (Hammerl et al., 2016) and by Jackel et al. from extracellular Brucella (Jackel et al., 2017). In our study, a more comprehensive investigation of prophages of the entire Brucella genus was conducted. The aforementioned five studies employed PHAST (Zhou et al., 2011) for the prediction and analysis of prophages. However, PHASTER was utilized in our study. PHASTER exhibits higher accuracy and sensitivity than PHAST (Arndt et al., 2016). Some Brucella strains from the aforementioned five studies were included, and additional prophages were indeed identified in these strains in our study. For example, Kaden et al. identified an intact Roseobacter prophage (72.5 Kb) on Chr I of the B. canis strain SVA13 (Kaden et al., 2014). In our study, two additional incomplete prophages were found in other regions on Chr I of B. canis strain SVA13, with sizes of 41.2 Kb and 19.5 Kb, respectively. Similar results were found in other strains, including B. melitensis M28 (Azam et al., 2016), B. abortus S19, B. suis 1330, B. sp. BO2 (Hammerl et al., 2016), and B. pseudogrignonensis K8 (Jackel et al., 2017). These new findings further revealed the importance of prophages in enhancing the genetic diversity of the Brucella genus. BiPBO1 (46,877 bp) was the first temperate phage induced from the Brucella genus (Hammerl et al., 2016). In our study, the prophage Brucel_BiPBO1 was widely distributed among various species of intracellular Brucella and extracellular Brucella, the length of which ranged from 7.2 Kb to 41.9 Kb. Some of these Brucel_BiPBO1 prophages may be inducible and deserve further exploration. In addition, some intact prophages were identified in B. abortus BDW, B. suis ZW043, and seven undefined Brucella strains in our study. These intact prophages may also be inducible and deserve further exploration.
Integrative conjugative elements (ICEs) are a type of mobile genetic elements (MGEs) that can be excised, transferred, and integrated into the host genome (Siguier et al., 2014). ICEs are composed of three modules: the integration/excision module, the conjugation module, and the adaptation modules (Partridge et al., 2018). In our study, a total of 43 putative ICEs were identified in 42 Brucella genomes, including 27 B. suis genomes, 10 B. canis genomes, 1 B. microti genome, 2 B. vulpis genomes, 1 B. pseudintermedia genome, and 1 B. pseudogrignonensis genome (Figure 4; Supplementary Table S6). On Chr I of B. pseudogrignonensis K8, an ICE with a length of 84,516 bp was identified, representing 1.69% of the entire genome (Supplementary Table S6). The remaining 42 ICEs distributed on Chr II ranged in length from 169,588 bp to 225,620 bp and accounted for 4.19–5.83% of the entire genome (Supplementary Table S6). Moreover, except for the ICE in the genome of B. suis 2011017258, the GC content of the other 42 ICEs was significantly different from the GC content of their respective genomes, suggesting that these ICEs might have been acquired through HGT. Furthermore, Lavigne et al. found that the IncP island, which carries the CP4-like integrase gene bciA that encodes a functional integrase, was present in the genomes of Brucella suis biovars 1 to 4, B. canis, B. neotomae, and strains isolated from marine mammals but not in B. melitensis, B. abortus, B. ovis, or B. suis biovar 5 (Lavigne et al., 2005). In our study, all the ICEs of B. suis, B. canis, and B. microti harbored a complete IncP island, which accounted for 10% of the entire ICE (Supplementary Table S6). The IncP island may play a crucial role in the horizontal transfer of these ICEs in B. suis, B. canis, and B. microti.
Integrative mobilizable elements (IMEs) are a type of MGE that harbors the genes responsible for excision and integration, without the inclusion of any genes associated with conjugative transfer (Bellanger et al., 2014). In our study, a total of 12 putative IMEs were identified in the genomes of 2 B. vulpis strains, 1 B. pseudogrignonensis strain, and 6 undefined Brucella strains (Figure 4; Supplementary Table S7). The genomes of B. pseudogrignonensis K8, B. sp. 09RB8471, and B. sp. 10RB9215 all contained 2 putative IMEs, while the remaining 6 genomes all contained 1 putative IME. The GC content of these IMEs varied from 51.28 to 62.34% (Supplementary Table S7). The lengths of these IMEs were significantly shorter than those of the ICEs in Brucella genomes, ranging from 2,621 bp to 51,821 bp and accounting for 0.08–1.51% of the genome (Supplementary Table S7).
In addition to B. suis and B. canis being the classical intracellular Brucella, the ICEs and IMEs identified in our study were found predominantly in nonclassical intracellular Brucella and extracellular Brucella. Only the genomes of B. vulpis and B. pseudogrignonensis contained both ICEs and IMEs. These findings collectively revealed that ICEs and IMEs mainly contributed to the genetic diversity of nonclassical intracellular Brucella and extracellular Brucella.
Virulence factors are properties (i.e., gene products) that enable a microorganism to establish itself on or within a host of a particular species and enhance its potential to cause disease (Liu et al., 2022). In our study, a total of 69 VFGs were identified in 255 Brucella genomes (Figure 7; Supplementary Table S8). Among these VFGs, 50 were widely distributed on Chr I of Brucella, while the remaining 19 were primarily distributed on Chr II of Brucella. Several VFGs, including acpXL, bmaB/omaA, and others, were distributed on MGEs in some Brucella genomes. Specifically, wbdA and wboA in B. abortus, B. suis, B. canis, B. ceti, B. pinnipedialis, and B. vulpis, as well as ricA in B. melitensis, B. pinnipedialis, and B. vulpis, were widely distributed on prophages of Chr I. Furthermore, manBcore and manCcore in B. suis, B. canis, and B. microti were mainly distributed on the ICEs of Chr II. The lpxK and waaA genes of B. vulpis were distributed on the IMEs of Chr II.
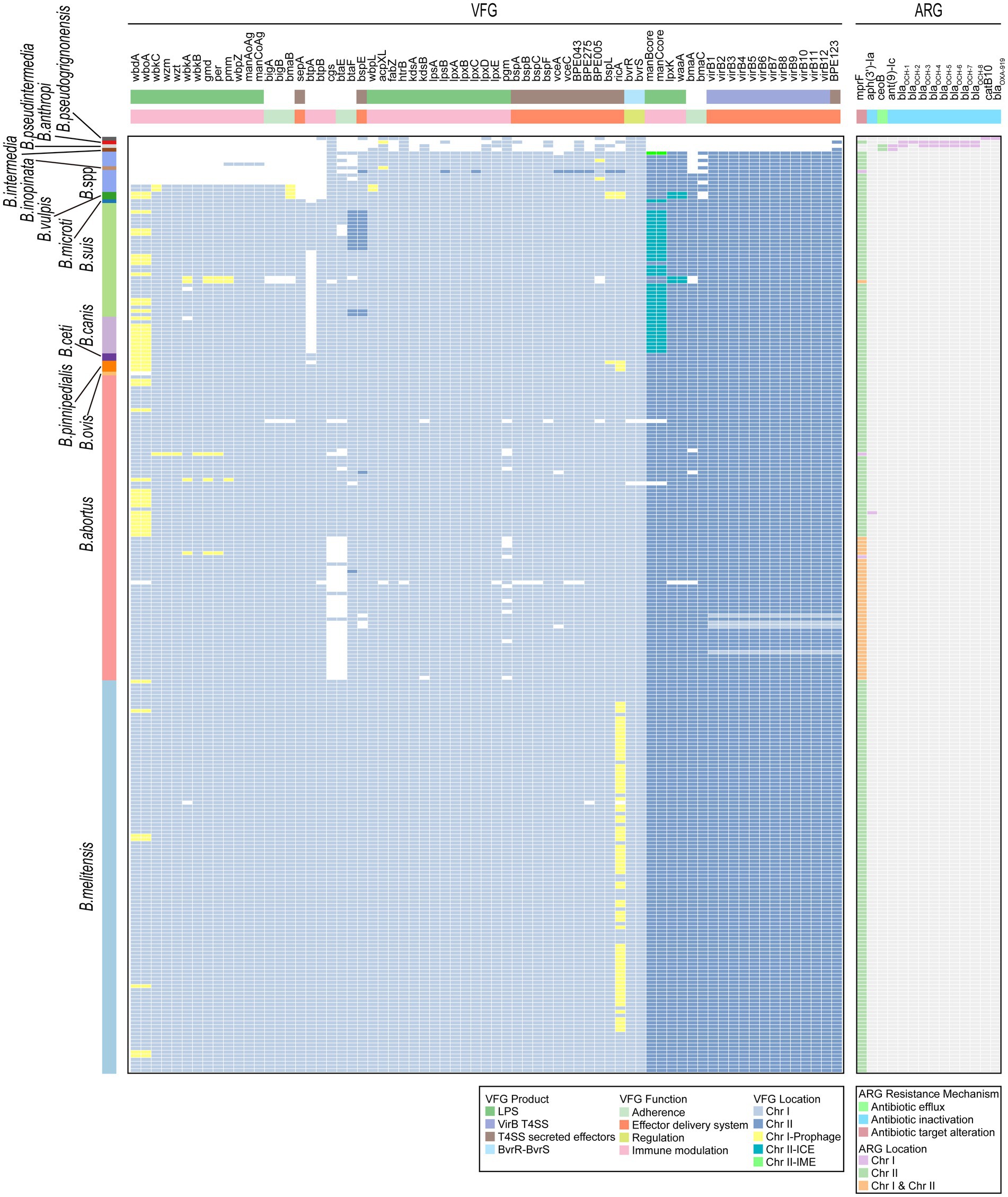
Figure 7. Distribution of VFGs and ARGs in the Brucella genus. Two heatmaps individually show the presence of VFGs and ARGs carried by each Brucella genome. The squares in the heatmaps are colored differently to indicate whether the corresponding VFG or ARG was distributed on the MGEs of Chr I or Chr II. The VFG product information, the resistance mechanisms of ARGs, and the corresponding species of each Brucella strain are also shown.
The classical intracellular Brucella possessed a relatively rich repertoire of VFGs, with each species containing 67–69 different VFGs (Figure 7). B. ovis lacked wbdA and wboA. B. canis did not possess btpA. Among the strains of nonclassical intracellular Brucella, B. vulpis strains, B. sp. 1315, and B. sp. 2594 possessed a considerable number of VFGs, with each strain harboring 65–68 different VFGs (Figure 7). Other nonclassical intracellular Brucella strains possessed fewer VFGs, with each strain containing 46–50 VFGs (Figure 7). In comparison to classical intracellular Brucella strains, these nonclassical intracellular Brucella strains lacked some VFGs associated with adherence and immune modulation (LPS) (Figure 7). The extracellular Brucella strains, which included B. intermedia, B. pseudintermedia, B. anthropi, and B. pseudogrignonensis, harbored the fewest VFGs, with each species possessing only 11–13 VFGs (Figure 7). These VFGs were predominantly located on Chr I of extracellular Brucella, with very few VFGs located on Chr II.
Intracellular Brucella is an important human and veterinary pathogen that is capable of causing brucellosis (Roop et al., 2021). In contrast, extracellular Brucella species represent a diverse group of free-living Brucella, among which only a few species are opportunistic pathogens that occasionally infect humans (Leclercq et al., 2019). In our study, intracellular Brucella possessed significantly more VFGs than did the extracellular Brucella. These VFGs were mainly involved in adherence, host immune modulation, and effector delivery systems and may be key factors leading to the difference in the pathogenicity of intracellular and extracellular Brucella. Additionally, some strains of nonclassical intracellular Brucella lacked a considerable number of VFGs associated with adherence and host immune modulation. These VFGs may cause differences in the pathogenicity between nonclassical intracellular Brucella strains and classical intracellular Brucella strains. Furthermore, although the distribution of VFGs is relatively conserved in classical intracellular Brucella species, B. ovis lacked wbdA and wboA, and B. canis lacked btpA in this study. Both wbdA and wboA are related to the biosynthesis of the lipopolysaccharide O-chain (Prasolova et al., 2023). btpA is related to the regulation of the host immune response (Salcedo et al., 2013). Vizcaino et al. reported the absence of wboA in B. ovis (Vizcaino et al., 2004). To our knowledge, there have been no other reports on the absence of wbdA in B. ovis or the absence of btpA in B. canis, and our research is the first to identify this phenomenon. In summary, different Brucella species exhibit diverse distribution patterns of VFGs, and these polymorphisms are likely closely related to variations in virulence, pathogenicity, and lifestyle among different Brucella species.
The inappropriate use of antibiotics has promoted a widespread increase in antibiotic resistance in bacteria. The HGT of ARGs encoded on MGEs is one of the key driving factors in this process (Botelho and Schulenburg, 2021). In our study, a total of 14 ARGs were identified in the 255 Brucella genomes (Figure 7; Supplementary Table S9). These ARGs confer antibiotic resistance through mechanisms such as antibiotic efflux, antibiotic inactivation, and antibiotic target alteration. mprF confers resistance to peptide antibiotics. Ant(9)-Ic and aph(3′)-Ia confer resistance to aminoglycoside antibiotics. catB10 confers resistance to phenicol antibiotics. It should be noted that catB10 only exhibits a query coverage of 73% in this study, and this incomplete sequence of catB10 may result in non-functional resistance of Brucella. ceoB confers resistance to aminoglycoside and fluoroquinolone antibiotics. blaOXA-919 confers resistance to penam antibiotics. The remaining 8 ARGs confer resistance to penem, penam, cephamycin, cephalosporin, and monobactam antibiotics. There are nine ARGs that are beta-lactamase genes, including blaOCH-1, blaOCH-2, blaOCH-3, blaOCH-4, blaOCH-5, blaOCH-6, blaOCH-7, blaOCH-8, and blaOXA-919. Moreover, mprF and ceoB were located primarily on Chr II of Brucella, while the remaining 12 ARGs were distributed only on Chr I (Figure 7). Furthermore, except for aph(3′)-Ia and mprF, the remaining 12 ARGs were distributed only among extracellular Brucella. The genome of B. abortus clpP contained both aph(3′)-Ia and mprF. The other intracellular Brucella strains contained only mprF.
In this study, various MGEs in Brucella genomes, including prophages, ICEs, and IMEs, were devoid of any ARGs, suggesting that the role of MGEs in the dissemination of ARGs might be limited in the Brucella genus. Furthermore, all the intracellular Brucella strains commonly harbored only one ARG, mprF. The remaining 12 ARGs were exclusively present in extracellular Brucella, suggesting that these two types of Brucella may have significantly different antibiotic resistance. Intracellular Brucella and extracellular Brucella exhibit distinct survival habits. The parasitism of intracellular Brucella may have limited their acquisition of ARGs through HGT. In contrast, extracellular Brucella species exist mainly in extracellular environments, which facilitates frequent genetic exchange and the acquisition of ARGs. In addition, recent studies have revealed that the resistance of Brucella spp. to various antibiotics, such as rifampicin, azithromycin, tetracycline, and so on, has been increasing (Wareth et al., 2021, 2022; Celik et al., 2023; Rezaei Shahrabi et al., 2023; Beig et al., 2024). These studies indicate an increase in antibiotic resistance in intracellular Brucella, which urgently requires attention. However, the distribution of ARGs within the Brucella genus was found to be very limited in our study. This may be attributed to the limitations of the CARD database that was used in our research, which may lack sufficient information on Brucella ARGs. In addition, more complex mechanisms may also contribute to drug resistance in Brucella.
In this study, a comparative genomic analysis of the Brucella genus was conducted using publicly available genomic data. The Brucella genus was found to possess a dynamic and open pangenome, with Chr II exhibiting a higher level of dynamism, openness, and plasticity than Chr I. On Chr I of Brucella, the core gene families were predominant, while on Chr II, the noncore gene families were predominant. Moreover, Brucella genomes were found to harbor some gene families acquired through HGT; these gene families were distributed mainly on Chr II and consisted primarily of noncore gene families. Furthermore, various MGEs, such as IS elements, prophages, ICEs, and IMEs, exhibited significantly different distribution patterns between Chr I and Chr II of Brucella. Additionally, different species of classical intracellular Brucella, nonclassical intracellular Brucella, and extracellular Brucella exhibited different levels of HGT and different distribution patterns of MGEs, VFGs, and ARGs. These differences among different Brucella species may be of significant importance for the genetic diversity of the Brucella genus, and to some extent could explain the diverse phenotypic characteristics exhibited by Brucella species. Due to limitations of the currently available public genomic data, the dataset used in this study could not encompass all species within the Brucella genus. In particular, the limited number of available genomes from extracellular Brucella and several other species in this study compromised the generality of some results. Thus, incorporating additional Brucella genomes in future research is necessary to acquire a more comprehensive understanding of the genetic evolutionary characteristics of the entire Brucella genus.
The original contributions presented in the study are included in the Supplementary material, further inquiries can be directed to the corresponding authors.
ZY: Conceptualization, Methodology, Software, Visualization, Writing – original draft. ZC: Conceptualization, Methodology, Software, Visualization, Writing – review & editing. XW: Investigation, Resources, Writing – review & editing. ZZ: Software, Validation, Writing – review & editing. FZ: Data curation, Project administration, Writing – review & editing. FK: Formal analysis, Validation, Writing – review & editing. WL: Software, Writing – review & editing. HR: Funding acquisition, Supervision, Writing – review & editing. YJ: Conceptualization, Funding acquisition, Supervision, Writing – review & editing. JY: Conceptualization, Funding acquisition, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Key Research and Development Program of China (No. 2022YFC2600704) and the Research Project from State Key Laboratory of Pathogen and Biosecurity (No. SKLPBS2251). The funders had no role in the study design, data collection and interpretation or the decision to submit the work for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1389859/full#supplementary-material
NCBI, National Center for Biotechnology Information; COG, clusters of orthologous groups; VFGs, virulence factor genes; ARGs, antibiotic resistance genes; VFDB, Virulence Factors of Pathogenic Bacteria Database; CARD, Comprehensive Antibiotic Resistance Database; nMC, normalized matching-cluster; nRF, normalized Robinson–Foulds; ANI, average nucleotide identity; dDDH, digital DNA–DNA hybridization; ML, maximum likelihood; HTGFs, horizontal transfer gene families; IS, insertion sequence; ICEs, integrative conjugative elements; IMEs, integrative mobilizable elements; Chr I, chromosome I; Chr II, chromosome II; HGT, horizontal gene transfer; MGEs, mobile genetic elements.
Abou Zaki, N., Salloum, T., Osman, M., Rafei, R., Hamze, M., and Tokajian, S. (2017). Typing and comparative genome analysis of Brucella melitensis isolated from Lebanon. FEMS Microbiol. Lett. 364:199. doi: 10.1093/femsle/fnx199
Alcock, B. P., Huynh, W., Chalil, R., Smith, K. W., Raphenya, A. R., Wlodarski, M. A., et al. (2023). Card 2023: expanded curation, support for machine learning, and resistome prediction at the comprehensive antibiotic resistance database. Nucleic Acids Res. 51, D690–D699. doi: 10.1093/nar/gkac920
Aljanazreh, B., Shamseye, A. A., Abuawad, A., and Ashhab, Y. (2023). Genomic distribution of the insertion sequence is711 reveal a potential role in brucella genome plasticity and host preference. Infect. Genet. Evol. 112:105457. doi: 10.1016/j.meegid.2023.105457
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). Phaster: a better, faster version of the phast phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Azam, S., Rao, S. B., Jakka, P., NarasimhaRao, V., Bhargavi, B., Gupta, V. K., et al. (2016). Genetic characterization and comparative genome analysis of Brucella melitensis isolates from India. Int J Genomics 2016:3034756. doi: 10.1155/2016/3034756
Bavishi, A., Abhishek, A., Lin, L., and Choudhary, M. (2010). Complex prokaryotic genome structure: rapid evolution of chromosome ii. Genome 53, 675–687. doi: 10.1139/g10-046
Beig, M., Moradkasani, S., Goodarzi, F., and Sholeh, M. (2024). Prevalence of brucella melitensis and Brucella abortus fluoroquinolones resistant isolates: a systematic review and meta-analysis. Vector Borne Zoonotic Dis. 24, 1–9. doi: 10.1089/vbz.2023.0063
Bellanger, X., Payot, S., Leblond-Bourget, N., and Guedon, G. (2014). Conjugative and mobilizable genomic islands in bacteria: evolution and diversity. FEMS Microbiol. Rev. 38, 720–760. doi: 10.1111/1574-6976.12058
Bogdanowicz, D., Giaro, K., and Wróbel, B. (2012). Treecmp: comparison of trees in polynomial time. Evol. Bioinforma. 8:EBO.S9657. doi: 10.4137/ebo.S9657
Borowiec, M. L. (2016). Amas: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4:e1660. doi: 10.7717/peerj.1660
Botelho, J., and Schulenburg, H. (2021). The role of integrative and conjugative elements in antibiotic resistance evolution. Trends Microbiol. 29, 8–18. doi: 10.1016/j.tim.2020.05.011
Canchaya, C., Proux, C., Fournous, G., Bruttin, A., and Brussow, H. (2003). Prophage genomics. Microbiol. Mol. Biol. Rev. 67, 238–276. doi: 10.1128/MMBR.67.2.238-276.2003
Cantalapiedra, C. P., Hernandez-Plaza, A., Letunic, I., Bork, P., and Huerta-Cepas, J. (2021). Eggnog-mapper v2: functional annotation, orthology assignments, and domain prediction at the metagenomic scale. Mol. Biol. Evol. 38, 5825–5829. doi: 10.1093/molbev/msab293
Capella-Gutierrez, S., Silla-Martinez, J. M., and Gabaldon, T. (2009). Trimal: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Celik, E., Kayman, T., Buyuk, F., Gulmez Saglam, A., Abay, S., Akar, M., et al. (2023). The canonical brucella species-host dependency is changing, however, the antibiotic susceptibility profiles remain unchanged. Microb. Pathog. 182:106261. doi: 10.1016/j.micpath.2023.106261
Centers for Disease Control and Prevention. (2022). 12/19/2022: lab update: reclassification of ochrobactrum species into the Brucella genus. Available at: https://www.cdc.gov/locs/2022/12-19-2022-Lab-Update-Reclassification_Ochrobactrum_species_Brucella_genus.html (Accessed April 03, 2024).
Cloeckaert, A., Roop, R. M. 2nd, Scholz, H. C., Whatmore, A. M., and Zygmunt, M. S. (2024). Editorial: Pathogenomics of the genus brucella and beyond, volume ii. Front. Microbiol. 15:1370330. doi: 10.3389/fmicb.2024.1370330
Cloeckaert, A., Vergnaud, G., and Zygmunt, M. S. (2020). Omp2b porin alteration in the course of evolution of brucella spp. Front. Microbiol. 11:284. doi: 10.3389/fmicb.2020.00284
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
De la Cruz, F., and Davies, J. (2000). Horizontal gene transfer and the origin of species: lessons from bacteria. Trends Microbiol. 8, 128–133. doi: 10.1016/s0966-842x(00)01703-0
Doganay, G. D., and Doganay, M. (2013). Brucella as a potential agent of bioterrorism. Recent Pat. Antiinfect. Drug Discov. 8, 27–33. doi: 10.2174/1574891x11308010006
DSMZ. (2024). Genus Brucella. Available at: https://lpsn.dsmz.de/genus/brucella?ACSTrackingID=USCDC_2146-DM96026&ACSTrackingLabel=Lab+Update%3A+Reclassification+of+Ochrobactrum+species+into+the+Brucella+genus&deliveryName=USCDC_2146-DM96026 (Accessed April 03, 2024).
Edgar, R. C. (2021). Muscle v5 enables improved estimates of phylogenetic tree confidence by ensemble bootstrapping. BioRxiv 2021:449169. doi: 10.1101/2021.06.20.449169
Emms, D. M., and Kelly, S. (2015). Orthofinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Emms, D. M., and Kelly, S. (2019). Orthofinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Franco, M. P., Mulder, M., Gilman, R. H., and Smits, H. L. (2007). Human brucellosis. Lancet Infect. Dis. 7, 775–786. doi: 10.1016/S1473-3099(07)70286-4
Fretin, D., Whatmore, A. M., Al Dahouk, S., Neubauer, H., Garin-Bastuji, B., Albert, D., et al. (2008). Brucella suis identification and biovar typing by real-time pcr. Vet. Microbiol. 131, 376–385. doi: 10.1016/j.vetmic.2008.04.003
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Hammerl, J. A., Gollner, C., Al Dahouk, S., Nockler, K., Reetz, J., and Hertwig, S. (2016). Analysis of the first temperate broad host range brucellaphage (bipbo1) isolated from b Inopinata. Front. Microbiol. 7:24. doi: 10.3389/fmicb.2016.00024
Hao, M., Wang, M., Tang, T., Zhao, D., Yin, S., Shi, Y., et al. (2023). Regulation of the gene for alanine racemase modulates amino acid metabolism with consequent alterations in cell wall properties and adhesive capability in brucella spp. Int. J. Mol. Sci. 24:16145. doi: 10.3390/ijms242216145
Haudiquet, M., De Sousa, J. M., Touchon, M., and Rocha, E. P. C. (2022). Selfish, promiscuous and sometimes useful: how mobile genetic elements drive horizontal gene transfer in microbial populations. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 377:20210234. doi: 10.1098/rstb.2021.0234
Heaps, H. S. (1978). Information retrieval: Computational and theoretical aspects. Cambridge, MA: Academic Press, Inc.
Hordt, A., Lopez, M. G., Meier-Kolthoff, J. P., Schleuning, M., Weinhold, L. M., Tindall, B. J., et al. (2020). Analysis of 1,000+ type-strain genomes substantially improves taxonomic classification of alphaproteobacteria. Front. Microbiol. 11:468. doi: 10.3389/fmicb.2020.00468
Jackel, C., Hertwig, S., Scholz, H. C., Nockler, K., Reetz, J., and Hammerl, J. A. (2017). Prevalence, host range, and comparative genomic analysis of temperate ochrobactrum phages. Front. Microbiol. 8:1207. doi: 10.3389/fmicb.2017.01207
Jain, R., Rivera, M. C., and Lake, J. A. (1999). Horizontal gene transfer among genomes: the complexity hypothesis. Proc. Natl. Acad. Sci. USA 96, 3801–3806. doi: 10.1073/pnas.96.7.3801
Jenner, D. C., Dassa, E., Whatmore, A. M., and Atkins, H. S. (2009). Atp-binding cassette systems of brucella. Comp. Funct. Genomics 2009:354649. doi: 10.1155/2009/354649
Kaden, R., Agren, J., Baverud, V., Hallgren, G., Ferrari, S., Borjesson, J., et al. (2014). Brucellosis outbreak in a Swedish kennel in 2013: determination of genetic markers for source tracing. Vet. Microbiol. 174, 523–530. doi: 10.1016/j.vetmic.2014.10.015
Kellerman, G. D., Foster, J. W., and Badakhsh, F. F. (1970). Comparison of chemical components of cell walls of Brucella abortus strains of low and high virulence. Infect. Immun. 2, 237–243. doi: 10.1128/iai.2.3.237-243.1970
Kettaneh, A., Weill, F. X., Poilane, I., Fain, O., Thomas, M., Herrmann, J. L., et al. (2003). Septic shock caused by Ochrobactrum anthropi in an otherwise healthy host. J. Clin. Microbiol. 41, 1339–1341. doi: 10.1128/JCM.41.3.1339-1341.2003
Konstantinidis, K. T., and Tiedje, J. M. (2005). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 102, 2567–2572. doi: 10.1073/pnas.0409727102
Lavigne, J. P., Vergunst, A. C., Bourg, G., and O’Callaghan, D. (2005). The incp island in the genome of brucella suis 1330 was acquired by site-specific integration. Infect. Immun. 73, 7779–7783. doi: 10.1128/IAI.73.11.7779-7783.2005
Lebuhn, M., Achouak, W., Schloter, M., Berge, O., Meier, H., Barakat, M., et al. (2000). Taxonomic characterization of ochrobactrum sp. isolates from soil samples and wheat roots, and description of Ochrobactrum tritici sp. nov. Int. J. Syst. Evol. Microbiol. 50, 2207–2223. doi: 10.1099/00207713-50-6-2207
Leclercq, S. O., Cloeckaert, A., and Zygmunt, M. S. (2019). Taxonomic organization of the family brucellaceae based on a phylogenomic approach. Front. Microbiol. 10:3083. doi: 10.3389/fmicb.2019.03083
Letunic, I., and Bork, P. (2021). Interactive tree of life (itol) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Liu, Y., Harrison, P. M., Kunin, V., and Gerstein, M. (2004). Comprehensive analysis of pseudogenes in prokaryotes: widespread gene decay and failure of putative horizontally transferred genes. Genome Biol. 5:R64. doi: 10.1186/gb-2004-5-9-r64
Liu, M., Li, X., Xie, Y., Bi, D., Sun, J., Li, J., et al. (2019). Iceberg 2.0: an updated database of bacterial integrative and conjugative elements. Nucleic Acids Res. 47, D660–D665. doi: 10.1093/nar/gky1123
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). Vfdb 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi: 10.1093/nar/gkab1107
Marzetti, S., Carranza, C., Roncallo, M., Escobar, G. I., and Lucero, N. E. (2013). Recent trends in human Brucella canis infection. Comp. Immunol. Microbiol. Infect. Dis. 36, 55–61. doi: 10.1016/j.cimid.2012.09.002
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Goker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Carbasse, J. S., Peinado-Olarte, R. L., and Goker, M. (2022). Tygs and lpsn: a database tandem for fast and reliable genome-based classification and nomenclature of prokaryotes. Nucleic Acids Res. 50, D801–D807. doi: 10.1093/nar/gkab902
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., Von Haeseler, A., et al. (2020). Iq-tree 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Moriyón, I., Grilló, M. J., Monreal, D., González, D., Marín, C., López-Goñi, I., et al. (2004). Rough vaccines in animal brucellosis: structural and genetic basis and present status. Vet. Res. 35, 1–38. doi: 10.1051/vetres:2003037
Ocampo-Sosa, A. A., and Garcia-Lobo, J. M. (2008). Demonstration of is711 transposition in brucella ovis and Brucella pinnipedialis. BMC Microbiol. 8:17. doi: 10.1186/1471-2180-8-17
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
Ouahrani, S., Michaux, S., Sri Widada, J., Bourg, G., Tournebize, R., Ramuz, M., et al. (1993). Identification and sequence analysis of is6501, an insertion sequence in brucella spp.: relationship between genomic structure and the number of is6501 copies. J. Gen. Microbiol. 139, 3265–3273. doi: 10.1099/00221287-139-12-3265
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). Checkm: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Partridge, S. R., Kwong, S. M., Firth, N., and Jensen, S. O. (2018). Mobile genetic elements associated with antimicrobial resistance. Clin. Microbiol. Rev. 31:17. doi: 10.1128/CMR.00088-17
Paul, S., Peddayelachagiri, B. V., Gogoi, M., Nagaraj, S., Ramlal, S., Konduru, B., et al. (2020). Genome-wide unique insertion sequences among five brucella species and demonstration of differential identification of brucella by multiplex pcr assay. Sci. Rep. 10:6368. doi: 10.1038/s41598-020-62472-3
Paulley, J. T., Anderson, E. S., and Roop, R. M. (2007). Brucella abortus requires the heme transporter bhua for maintenance of chronic infection in balb/c mice. Infect. Immun. 75, 5248–5254. doi: 10.1128/IAI.00460-07
Prasolova, O., Krylova, E., Bogomazova, A., Soltynskaya, I., Sklyarov, O., Gordeeva, V., et al. (2023). Russian collection of Brucella abortus vaccine strains: annotation, implementation and genomic analysis. Front. Vet. Sci. 10:1154520. doi: 10.3389/fvets.2023.1154520
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Rajendhran, J. (2021). Genomic insights into brucella. Infect. Genet. Evol. 87:104635. doi: 10.1016/j.meegid.2020.104635
Refai, M. (2002). Incidence and control of brucellosis in the near east region. Vet. Microbiol. 90, 81–110. doi: 10.1016/S0378-1135(02)00248-1
Rezaei Shahrabi, A., Moradkasani, S., Goodarzi, F., Beig, M., and Sholeh, M. (2023). Prevalence of brucella melitensis and Brucella abortus tetracyclines resistance: a systematic review and meta-analysis. Microb. Pathog. 183:106321. doi: 10.1016/j.micpath.2023.106321
Ronneau, S., Moussa, S., Barbier, T., Conde-Alvarez, R., Zuniga-Ripa, A., Moriyon, I., et al. (2016). Brucella, nitrogen and virulence. Crit. Rev. Microbiol. 42, 507–525. doi: 10.3109/1040841X.2014.962480
Roop, R. M., Barton, I. S., Hopersberger, D., and Martin, D. W. (2021). Uncovering the hidden credentials of brucella virulence. Microbiol. Mol. Biol. Rev. 85:19. doi: 10.1128/MMBR.00021-19
Rouli, L., Merhej, V., Fournier, P. E., and Raoult, D. (2015). The bacterial pangenome as a new tool for analysing pathogenic bacteria. New Microbes New Infect. 7, 72–85. doi: 10.1016/j.nmni.2015.06.005
Salcedo, S. P., Marchesini, M. I., Degos, C., Terwagne, M., Von Bargen, K., Lepidi, H., et al. (2013). Btpb, a novel brucella tir-containing effector protein with immune modulatory functions. Front. Cell. Infect. Microbiol. 3:28. doi: 10.3389/fcimb.2013.00028
Sankarasubramanian, J., Vishnu, U. S., Gunasekaran, P., and Rajendhran, J. (2017). Identification of genetic variants of brucella spp. through genome-wide association studies. Infect. Genet. Evol. 56, 92–98. doi: 10.1016/j.meegid.2017.11.016
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sentausa, E., and Fournier, P. E. (2013). Advantages and limitations of genomics in prokaryotic taxonomy. Clin. Microbiol. Infect. 19, 790–795. doi: 10.1111/1469-0691.12181
Siguier, P., Gourbeyre, E., and Chandler, M. (2014). Bacterial insertion sequences: their genomic impact and diversity. FEMS Microbiol. Rev. 38, 865–891. doi: 10.1111/1574-6976.12067
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). Isfinder: the reference Centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Suarez-Esquivel, M., Chaves-Olarte, E., Moreno, E., and Guzman-Verri, C. (2020). Brucella genomics: macro and micro evolution. Int. J. Mol. Sci. 21:749. doi: 10.3390/ijms21207749
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Teyssier, C., Marchandin, H., Masnou, A., Jeannot, J. L., de Buochberg, M. S., and Jumas-Bilak, E. (2005). Pulsed-field gel electrophoresis to study the diversity of whole-genome organization in the genus Ochrobactrum. Electrophoresis 26, 2898–2907. doi: 10.1002/elps.200410323
van Veen, H. W., and Konings, W. N. (1998). The abc family of multidrug transporters in microorganisms. Biochim. Biophys. Acta 1365, 31–36. doi: 10.1016/s0005-2728(98)00039-5
Vandecraen, J., Chandler, M., Aertsen, A., and Van Houdt, R. (2017). The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit. Rev. Microbiol. 43, 709–730. doi: 10.1080/1040841X.2017.1303661
Vergnaud, G., Zygmunt, M. S., Ashford, R. T., Whatmore, A. M., and Cloeckaert, A. (2024). Genomic diversity and zoonotic potential of Brucella neotomae. Emerg. Infect. Dis. 30, 155–158. doi: 10.3201/eid3001.221783
Vizcaino, N., Caro-Hernandez, P., Cloeckaert, A., and Fernandez-Lago, L. (2004). DNA polymorphism in the omp25/omp31 family of brucella spp.: identification of a 1.7-kb inversion in brucella cetaceae and of a 15.1-kb genomic island, absent from Brucella ovis, related to the synthesis of smooth lipopolysaccharide. Microbes Infect. 6, 821–834. doi: 10.1016/j.micinf.2004.04.009
Wang, S., Zhao, X., Sun, K., Bateer, H., and Wang, W. (2022). The genome sequence of Brucella abortus vaccine strain a19 provides insights on its virulence attenuation compared to Brucella abortus strain 9-941. Gene 830:146521. doi: 10.1016/j.gene.2022.146521
Wareth, G., Dadar, M., Ali, H., Hamdy, M. E. R., Al-Talhy, A. M., Elkharsawi, A. R., et al. (2022). The perspective of antibiotic therapeutic challenges of brucellosis in the middle east and north african countries: current situation and therapeutic management. Transbound. Emerg. Dis. 69, e1253–e1268. doi: 10.1111/tbed.14502
Wareth, G., El-Diasty, M., Abdel-Hamid, N. H., Holzer, K., Hamdy, M. E. R., Moustafa, S., et al. (2021). Molecular characterization and antimicrobial susceptibility testing of clinical and non-clinical brucella melitensis and Brucella abortus isolates from Egypt. One Health 13:100255. doi: 10.1016/j.onehlt.2021.100255
Yang, X., Li, Y., Zang, J., Li, Y., Bie, P., Lu, Y., et al. (2016). Analysis of pan-genome to identify the core genes and essential genes of Brucella spp. Mol. Genet. Genomics 291, 905–912. doi: 10.1007/s00438-015-1154-z
Zhong, Z., Wang, Y., Xu, J., Chen, Y., Ke, Y., Zhou, X., et al. (2012). Parallel gene loss and acquisition among strains of different Brucella species and biovars. J. Microbiol. 50, 567–574. doi: 10.1007/s12275-012-2022-8
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J., and Wishart, D. S. (2011). Phast: a fast phage search tool. Nucleic Acids Res. 39, W347–W352. doi: 10.1093/nar/gkr485
Keywords: Brucella, pangenome, horizontal gene transfer, mobile genetic elements, virulence factors, antibiotic resistance genes
Citation: Yang Z, Chai Z, Wang X, Zhang Z, Zhang F, Kang F, Liu W, Ren H, Jin Y and Yue J (2024) Comparative genomic analysis provides insights into the genetic diversity and pathogenicity of the genus Brucella. Front. Microbiol. 15:1389859. doi: 10.3389/fmicb.2024.1389859
Edited by:
Georgia Vrioni, National and Kapodistrian University of Athens, GreeceReviewed by:
Jian Yang, Chinese Academy of Medical Sciences and Peking Union Medical College, ChinaCopyright © 2024 Yang, Chai, Wang, Zhang, Zhang, Kang, Liu, Ren, Jin and Yue. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongguang Ren, bioren@163.com; Yuan Jin, jinyuan@bmi.ac.cn; Junjie Yue, yue_junjie@126.com
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.