 Peter W. Kopkowski
Peter W. Kopkowski Zhongge Zhang
Zhongge Zhang Milton H. Saier Jr.
Milton H. Saier Jr.
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 11 April 2024
Sec. Infectious Agents and Disease
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1388522
This article is part of the Research Topic Mobile DNA Element-Driven Evolution of Bacterial Pathogens View all 13 articles
The bglGFB operon in Escherichia coli K-12 strain BW25113, encoding the proteins necessary for the uptake and metabolism of β-glucosides, is normally not expressed. Insertion of either IS1 or IS5 upstream of the bgl promoter activates expression of the operon only when the cell is starving in the presence of a β-glucoside, drastically increasing transcription and allowing the cell to survive and grow using this carbon source. Details surrounding the exact mechanism and regulation of the IS insertional event remain unclear. In this work, the role of several DNA-binding proteins in how they affect the rate of insertion upstream of bgl are examined via mutation assays and protocols measuring transcription. Both Crp and IHF exert a positive effect on insertional Bgl+ mutations when present, active, and functional in the cell. Our results characterize IHF’s effect in conjunction with other mutations, show that IHF’s effect on IS insertion into bgl also affects other operons, and indicate that it may exert its effect by binding to and altering the DNA conformation of IS1 and IS5 in their native locations, rather than by directly influencing transposase gene expression. In contrast, the cAMP-CRP complex acts directly upon the bgl operon by binding upstream of the promoter, presumably altering local DNA into a conformation that enhances IS insertion.
Since their discovery, transposable elements (“jumping genes”) have been studied in prokaryotic and eukaryotic models. In both types of organisms, they are best known for their ability to insert into variable locations within an organism’s genome (McClintock, 1950). In some cases, transposition takes place upstream of or within a structural gene, which may cause a change in protein expression for the former and loss of gene function for the latter (Whiteway et al., 1998; Zhang and Saier, 2009a,b).
One of the best characterized examples of this phenomenon is IS (Insertion Sequence) element insertion into the bglGFB operon, which is not expressed in wild-type (WT) Escherichia coli K-12 strain BW25113. Both the pathogenic and non-pathogenic forms of E. coli contain this operon, and to our knowledge it is never expressed in WT. Binding of the global histone-like nucleoid structuring (H-NS) protein at two sites on either side of the otherwise active bgl promoter is the most important factor in silencing transcription of the bgl operon by a strong repression mechanism (Schnetz, 1995; Dole et al., 2004; Lam et al., 2022). Data published by other groups indicate that the upstream and downstream H-NS binding sites exhibit synergy with each other (Nagarajavel et al., 2007). Building upon this discovery, recent data from our group suggest the formation of a repression loop structure that blocks access of RNA polymerase to the bgl promoter (Lam et al., 2022; Tran et al., 2022).
The first gene in the operon, bglG, contains the downstream H-NS binding site within its coding region and is itself flanked by two Rho-independent terminators, limiting the amount of RNA transcript made of bglG as well as the two other downstream genes, bglF and bglB (Figure 1; Mahadevan and Wright, 1987; Schnetz and Rak, 1988). BglG is a homodimer that binds to its own transcript to prevent early termination, allowing transcription to continue and thereby promoting expression of the entire operon (Amster-Choder and Wright, 1992). BglG also has other functions including the positive regulation of insertional and non-insertional Bgl+ mutations, although how it accomplishes these functions has yet to be elucidated (Zhang et al., 2022). The bglF gene immediately follows bglG’s downstream terminator and encodes a membrane-integrated protein responsible for the uptake and concomitant phosphorylation of β-glucosides via a phosphotransferase (PTS)-dependent mechanism (Fox and Wilson, 1968). Thus, BglF passes a phosphoryl group from HPr or a BglG monomer to the incoming β-glucoside concomitant with transport (Chen et al., 1997), marking it for hydrolysis of the aglycone from the glucose-phosphate moiety by BglB, the operon’s third and final gene product (Prasad et al., 1973), and this sugar-P feeds directly into glycolysis. Since phosphorylated BglG is in equilibrium with phosphorylated BglF, transfer of the phosphoryl group from BglG to BglF causes the dephosphorylation of BglG, allowing it to dimerize into its active anti-termination configuration. BglG can be phosphorylated on two specific histidyl residues, one that promotes antitermination and the other which prevents antitermination (Görke and Rak, 1999; Rothe et al., 2012).
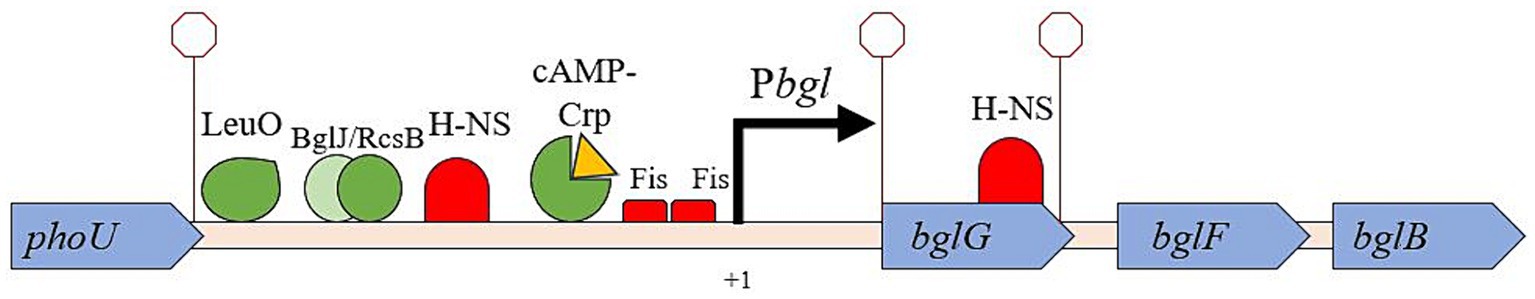
Figure 1. The bgl operon with relevant DNA-binding protein sites and structural features. The phoU/bglG intergenic region is where IS insertion occurs, upstream of the promoter. Several DNA binding proteins are known to bind to the bglGFB regulatory region and influence transcription. A major focus of this work is to determine whether protein binding in this region affects the rate of insertion of IS1 and/or IS5.
While repression of bgl operon expression by H-NS is strong, preventing almost 100% of the maximal transcription rate, transposition of an IS element may occur upstream of the bgl promoter (Reynolds et al., 1981). The vast majority of IS elements that insert into this area are either IS1 or IS5, and both elements insert in either orientation (forward or backward) within an area spanning ~200 bp (Reynolds et al., 1981). This insertional event eliminates repression of bgl operon expression by H-NS (Lopilato and Wright, 1990; Singh et al., 1995), increases bgl operon expression several hundred-fold (Lam et al., 2022), and allows cells to grow, divide and form colonies using β-glucosides as their sole carbon source (Prasad and Schaefler, 1974). This state corresponds to a “Bgl+” phenotype.
Two details about this insertional event remain of great interest. First, Bgl+ mutations of any class, insertional or otherwise, only occur when E. coli cells are starving in the presence of a β-glucoside, which can be freely taken in and used after activation of the bgl operon via mutation or IS insertion. Second, the rate of insertion into bgl under these circumstances is much higher than the random mutation rate for E. coli (Hall, 1998).
Since mutations are widely considered by the scientific community to be random events rather than environmentally directed, the above facts indicate that the bgl operon is operating under an additional, fundamentally novel layer of regulation that is not well understood. Several paradigms like bgl exist, where insertional events occurring under specific circumstances of cellular stress lead to a phenotype which relieves that stress. These operons deserve further study so that the mechanisms of their regulation may be incorporated into today’s accepted models of mutation and evolution.
As noted above, bgl is one of several operons in E. coli that are preferential sites for insertion of IS elements. These sites are commonly found in the operon’s promoter region, have high Gibbs free energy signatures, and exhibit an increase in IS insertion frequency when the bacterial cell is experiencing a specific type of stress related to the operon’s function (Humayun et al., 2017). These so-called superhelical stress-induced duplex destabilization (SIDD) sites are under study by our group and others to determine whether insertion into a SIDD site is directed by environmental conditions, as well as to uncover whether SIDDS evolved specifically to allow IS-mediated operon activation (Humayun et al., 2017).
Since the hydrogen bonds between individual base pairs are relatively weak in a SIDD site, the opening of the DNA may be what allows IS insertion to occur there with increased frequency. This would support previous unpublished findings that H-NS lowers the rate of insertion into the bgl operon (Z.Z., unpublished data). When H-NS binds at or near the SIDD site, it may prevent it from opening to a conformation that allows for easy insertion of an IS element. It is therefore important to identify what roles, if any, that DNA-binding proteins have upon transposition into the sequences on or near their binding sites. In pursuit of this goal, we studied several DNA-binding proteins to better understand their roles in regulating the insertion of IS elements and the phenomena of “adaptive” or “directed” mutations in general. Adding to the body of knowledge of how transposons and their movements are regulated is vital for proper understanding of the “mobilomes” that are present in almost every life form on Earth.
Integration Host Factor (IHF) is a heterodimeric global histone-like DNA-binding protein involved in several cellular functions including transcriptional regulation, DNA recombination, and chromosome compaction (Goosen and van de Putte, 1995; Engelhorn and Geiselmann, 1998). After binding to the minor groove of the double helix, it significantly bends the DNA at least 160° per dimer (Sugimura and Crothers, 2006). Under starvation conditions, IHF concentrations rise 4–8-fold in E. coli (Bushman et al., 1985; Ditto et al., 1994) and has been implicated in the induction of proteins related to carbon starvation (Sclavi et al., 2007). Also of interest is IHF’s role in the transposition of Mu prophage in Pseudomonas putida. In vitro studies of IHF binding to the Mu promoter suggest that IHF assists in formation of a transposase complex that may facilitate excision via supercoiling relief (Surette et al., 1989; Allison and Chaconas, 1992). In vivo, IHF is not required for Mu transposition, but has a dual effect on Mu transposase transcription. IHF binds to the end of the Mu transposase promoter region and activated transcription of the transposase while simultaneously relieving repression by H-NS at a downstream site (van Ulsen et al., 1996). Multiple groups have shown that a Gly to Glu mutation at the 62nd amino acid in IhfA causes loss of IHF’s ability to bind to its DNA consensus sequence but does not prevent dimerization with IhfB (Granston and Nash, 1993; Hales et al., 1994).
Crp is a DNA-binding protein that relies upon cyclic AMP (cAMP) to become activated (Kolb et al., 1993). It is a global transcriptional regulator that affects expression of almost 200 genes in E. coli (Fic et al., 2009). Most applicable to this work, cAMP-Crp is generally involved in the positive regulation of genes concerned with the metabolism of carbon sources (Deutscher, 2008; Görke and Stülke, 2008) and is a necessary factor in the activation of bgl. Even if repression by H-NS is relieved, bgl remains transcriptionally inactive and unable to grow using only β-glucosides, unless activated Crp is present (Mukerji and Mahadevan, 1997; Gulati and Mahadevan, 2000).
In this work, we provide evidence that two DNA-binding proteins affect the rate of IS insertion into bgl: the cAMP-Crp complex and Integration Host Factor (IHF). Both proteins are positive regulators of insertion; that is, their presences and abundances maintain or increase the frequencies of IS-related Bgl+ mutations observed in WT cells. The effect of IHF is not specific to bgl but applies to several other operons into which IS elements insert in response to environmental stresses. Our results suggest that binding of IHF to known sites in IS1 and IS5 does not influence transposase gene transcription, and is therefore likely to influence DNA conformation, transpososome formation and/or energetics at the site of transposition.
The first phase of our research was to determine which genes, if any, demonstrated a clear effect on the frequency of IS insertion into bgl upon deletion. Figures 2A,B show data gathered from several single deletion strains over a 10-day period. The full list of deletion strains tested is tabulated in Supplementary Table S1. Genes were selected for study based on the DNA-binding abilities of their gene products and were loosely separated into two groups: the global histone-like DNA-binding proteins and operon-specific DNA-binding proteins with known binding sites within the bgl control region. Colony PCR was performed on the mutant colonies of each strain, using primers that flank the Pbgl regulatory region into which IS1 and IS5 are known to insert (Supplementary Table S2). Of the deletion strains tested, ∆ihfA was selected for further study, since it caused the greatest change in the frequency of Bgl+ mutant appearances compared to WT (Figure 2A). Further confirmatory Bgl+ mutation assays were performed on WT and ∆ihfA to precisely ascertain the difference in mutant incidence between them. These results again showed an approximately eight-fold drop in total Bgl+ mutants in ∆ihfA, and a more than 11-fold drop in insertional mutants (Figures 2C,D). Deletion of ihfB, encoding IHF’s other subunit IhfB, showed the same effect as ∆ihfA (Figures 2C,D). To determine whether the ∆ihfA mutation prevented cell multiplication but not necessarily insertion, IS5 was inserted upstream of bgl in its usual position in the ∆ihfA mutant. This strain grew as quickly on salicin media as WT Bgl+ cells. This indicates that ∆ihfA acts specifically to lower the IS insertion frequency rather than hinder the growth of Bgl+ mutants after insertion takes place. These observations led us to consider the mechanism of IHF’s role in IS insertion, as well as its specificity for the bgl operon.
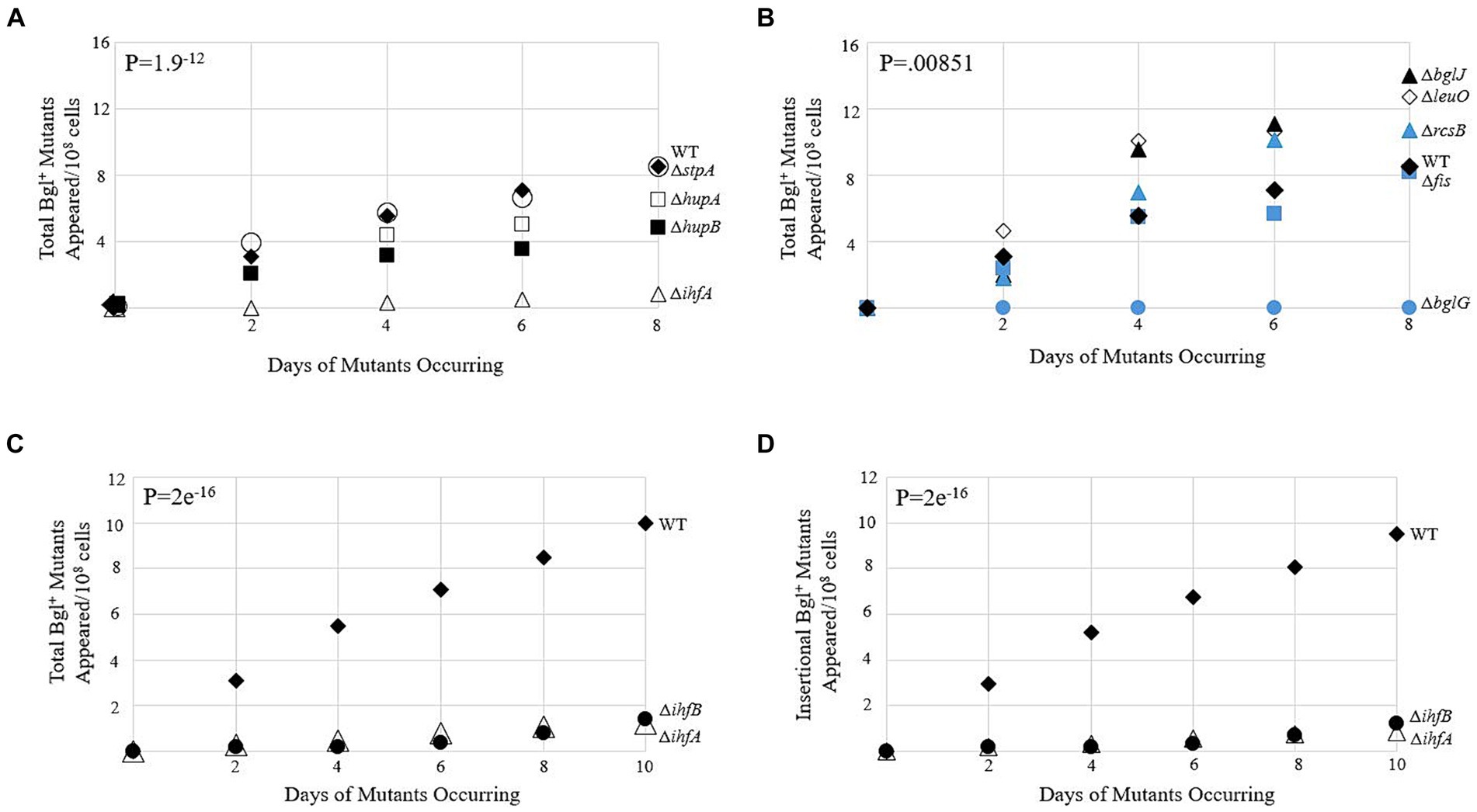
Figure 2. Bgl+ mutation frequencies due to deletion of genes encoding DNA-binding proteins. (A,B) Appearances of total Bgl+ mutants over time. Mutation assays were carried out on M9 + 0.5% salicin plates as described in Materials and Methods. Strains are loosely grouped in A and B based on their global nature (A) or more specific binding to bgl. (B,C) Effects of deleting ihfA and ihfB on appearance of total Bgl+ mutants. (D) Effects of deleting ihfA and ihfB on appearance of insertional Bgl+ mutants. Colony PCR using primers flanking the region of bgl insertion was performed to differentiate between insertional and non-insertional colonies. WT = ♦; ΔihfA = ∆; ΔbglG = ●; ΔstpA = ○; ΔhupA = □; ΔhupB = ■; ΔbglJ = ▲; ΔleuO = ◊; ΔrcsB = ▲; Δfis = ■; ΔihfB = ●.
The ∆ihfA background was transferred to the backgrounds Ptet-G and Iq-G, which constitutively express bglG at the intS locus, leaving the native bgl operon intact. Our objective was to observe how IHF’s observed effect would interact with increased BglG levels, which has been previously shown to increase the rate of Bgl+ insertional and non-insertional mutations (Zhang et al., 2022). The results presented in Figures 3A,B show that the stimulatory effect of increased bglG expression on insertion frequency counteracts ∆ihfA’s negative effect on the same. As with the WT background, deletion of ihfA in either background led to a greater than 10-fold drop in insertional mutants. In the case of Ptet-G, the total mutants fell by four-fold when ihfA was deleted, and a more than 10-fold decrease was observed among insertional mutants. These data further solidify BglG’s effect on insertional mutation rate and confirm our results on ihfA deletion, which appears to mitigate both the insertional and non-insertional mutant incidences in the Ptet-G strain, but not the Iq-G strain.
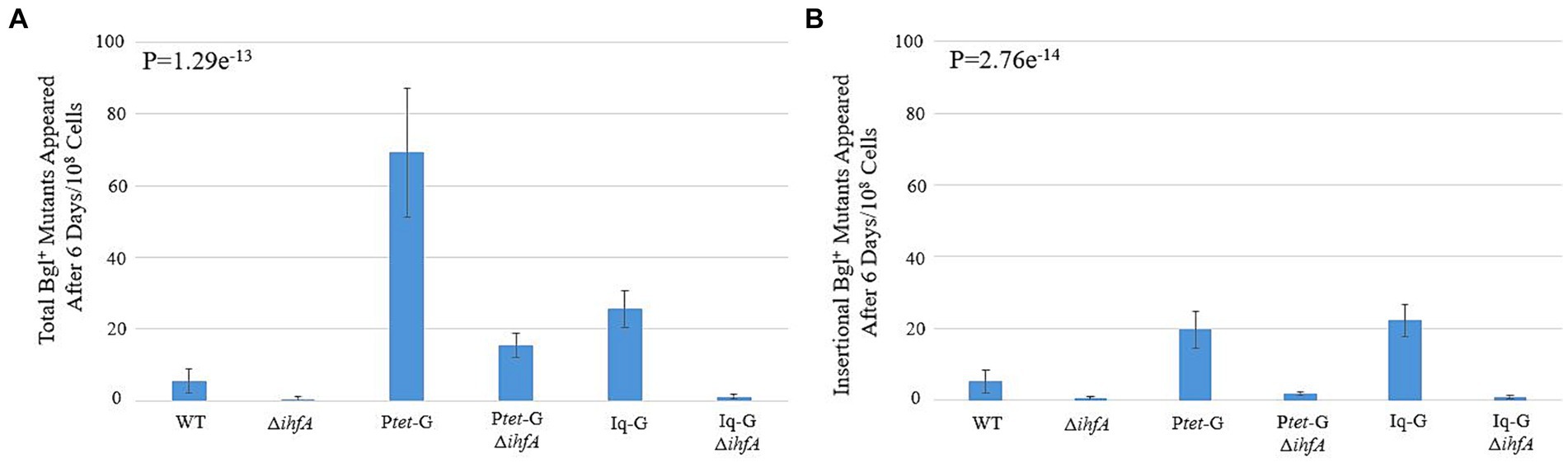
Figure 3. Effect of ihfA deletion combined with increased bglG expression on Bgl+ mutations. (A) Effect of deleting ihfA on total Bgl+ mutations in the cells over expressing bglG. (B) Effect of deleting ihfA on insertional Bgl+ mutations in the cells over expressing bglG. Bgl+ mutation assays were performed as previously described. Two bglG overexpression strains, Ptet-G (Ptet driving bglG at the intS locus) and Iq-G (lacIq driving bglG at the intS locus), were used together with or without the ihfA deletion. This experiment serves to determine if the ihfA deletion has similar inhibitory impact on IS1/IS5 insertions in the cells with higher levels of BglG. Our previous study showed that more BglG produced leads to greater insertional frequencies (Zhang et al., 2022). Colony PCR was used to differentiate insertional and non-insertional Bgl+ mutants.
Since the IHF protein has a significantly positive effect on IS insertion upstream of the bgl promoter, we decided to determine if changing the amounts of both IhfA and IhfB subunits would have a different effect. PK01, a strain expressing both the ihfA and ihfB genes in the chromosome using the constitutive Ptet promoter, was therefore constructed and subjected to a mutation assay as previously described. Interestingly, a decrease in both total and insertional mutants was observed after 10 days (Supplementary Figures S1A,B). We consider several possible reasons for why this occurred (see Discussion).
PK01_R was constructed, containing the tetR gene. tetR encodes the Ptet repressor protein TetR, which is most effective in the absence of its inducer, any one of several tetracycline derivatives. Our goal was to see if different levels of Ptet induction would reveal a similar trend as that already observed with normal PK01. Supplementary Figures S1C,D show the results of a mutation assay as described, but with the addition of several different concentrations of the Tet inducer anhydro-tetracycline (aTC). As expected, the increase of IhfA and IhfB led to more Bgl+ mutations. The number of mutants that appeared was like the previous assay at levels of maximal aTC induction (~30 μM), supporting the results of Supplementary Figure S1B.
To create a strain expressing both IHF subunits at even higher levels, a pZA31 plasmid containing Ptet-driven ihfA and ihfB was constructed and then transformed into wild type BW25113 and its counterpart containing the Tet repressor gene tetR. These new strains, called PK02 and PK02_R, respectively, were compared to each other and an isogenic control (WT Rf) in a mutation assay. All three strains behaved very similarly (Figures 4A,B), suggesting that the increased transcription of both IHF subunits did little to influence the insertion rate. To confirm this, PK02_R was plated on M9 salicin media containing differential levels of the Tet promoter’s inducer aTC (Figures 4C,D). The amount of inducer present did not appear to cause a corresponding change in total or insertional Bgl+ mutants, as consistent with the findings in Figures 4A,B.
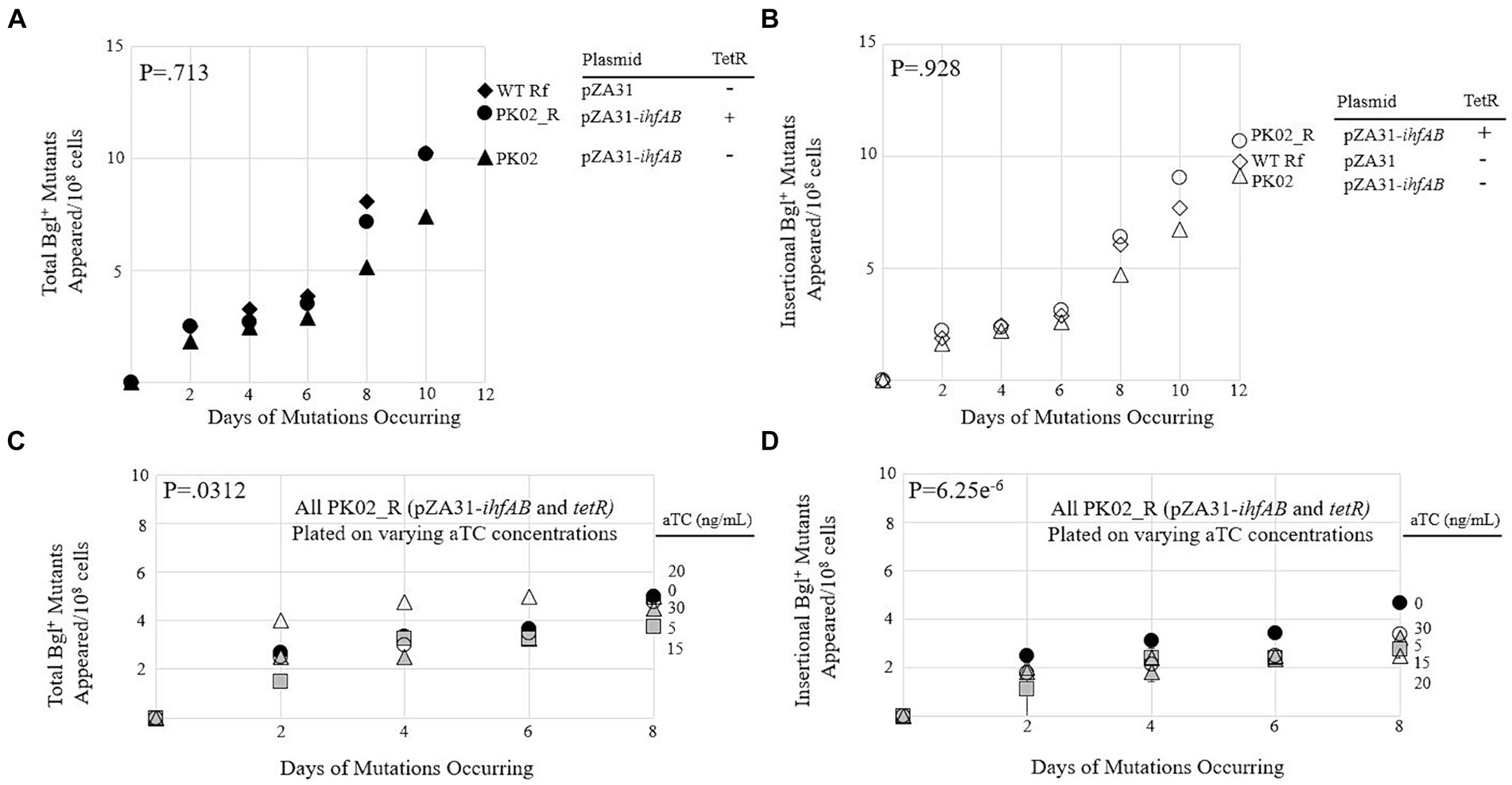
Figure 4. Overexpression of ihfA and ihfB on a plasmid slightly lowers the IS insertional rate into bgl. (A) Effect of IhfA and IhfB overproduction on total Bgl+ mutations. (B) Effect of IhfA and IhfB overproduction on insertional Bgl+ mutations. (C) Effect of titrating ihfA and ihfB expressions on a plasmid on total Bgl+ mutations. (D) Effect of titrating ihfA and ihfB expressions on a plasmid on insertional Bgl+ mutations. For (C,D), mutation assays were performed as previously described on M9 salicin plates containing aTC at 0 to 30 ng/mL over a 10-day period. 0aTC = ●; 5aTC = ▲; 15aTC = ■; 20aTC = ∆; 30aTC = ○.
Our next goal was to establish a mechanism by which IHF exerts its effect on the expression of bgl. If ∆ihfA’s effect on the frequency of Bgl+ insertional mutations is due to the inability of IHF binding to the bgl upstream region, then LacZ assays could show a difference in transcriptional activity when IHF is present versus when it is absent. The constructs we devised for determining this are shown in Supplementary Figure S2. The native bgl operon was left intact to maintain normal induction by β-glucosides. A second, altered bgl promoter and its upstream region were positioned in front of the native lacZ gene in place of its usual promoter. This new construct, Pbgl-Z, can measure the promoter activity of this second bgl operon. A similar construct, Pbgl-G-Z, contained the bglG gene between Pbgl and lacZ for the purpose of determining operon activity. Figures 5A,B show the lacZ activity results, which demonstrated a ~ 2-fold change in promoter activity but no significant change in operon activity between WT and ∆ihfA. We repeated these experiments using a BglB assay, which functions similarly in practice to the LacZ assay. However, this assay uses PNP-Glucoside (PNPG) as its substrate and measures activity of the bgl operon directly, rather than having to rely on additional constructs and the lacZ gene. The results of the BglB assay confirmed those of the LacZ experiments, showing no increase or decrease in bgl activity (Figure 5C). We suspected that any potential change may not be detectable due to the natural low levels of bgl activity; to investigate this possibility, the ∆Pbgl-G strains were constructed. ∆Pbgl-G strains are similar to the strains originally used for BglB but exhibit a 200-fold increase in operonic activity due to the removal of the bglG gene, both of its flanking terminator sequences, and the downstream H-NS binding site. To see if ∆Pbgl-G would lead to observable differences between strains, BglB assays were run again using several previously used strains now in a ∆Pbgl-G background. No large change was observed across the board, leading us to reject the notion that specific binding of IHF significantly affects transcription of bgl (Figure 5D).
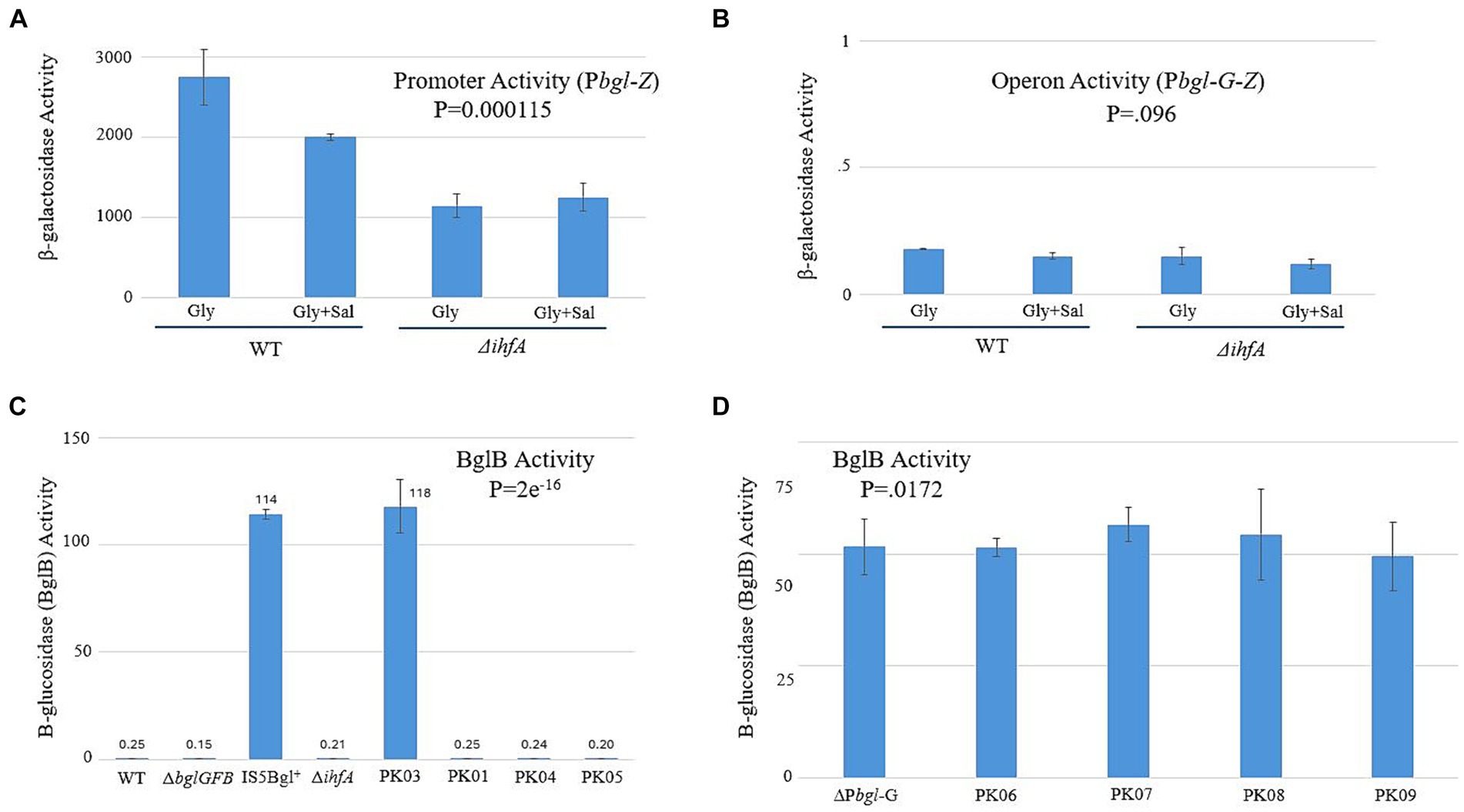
Figure 5. Changing IHF levels does not have a significant effect on bgl operon transcriptional activity. LacZ and BglB assays were carried out as previously described (Materials and Methods). (A) The bglGFB promoter (Pbgl) activities determined using the LacZ assay. (B) The bglGFB operon (Pbgl-bglG) activities determined using LacZ assay. (C) The bglGFB operon activities were determined via the BglB assay. PK01 = Ptet-ihfA_Ptet-ihfB; PK03 = ∆ihfA IS+; PK04 = ∆ihfA in G50; PK05 = Ptet-ihfA_Ptet-ihfB in G50. (D) The bglGFB operon activities in strain ∆Pbgl-G determined using the BglB assay. Strain ∆Pbgl-G was deleted for the bglG gene plus its flanking terminators and is Bgl+. PK06 = ∆ihfA in ∆Pbgl-G; PK07 = ∆fis in ∆Pbgl-G; PK08 = ∆ihfA and ∆fis in ∆Pbgl-G; PK09 = Ptet-ihfA_Ptet-ihfB in ∆Pbgl-G.
Since our LacZ data did not reveal a substantial change in transcriptional activity within the context of bgl, we hypothesized that ∆ihfA does not affect bgl directly but may exert its effect on insertion upstream of bgl by a nonspecific means. If IHF, being nonspecific, is important for IS insertion in general, then it may be involved in the upstream process of IS excision from other locations in the genome. In this situation, other operons into which IS1 and IS5 insert would also experience a change in insertion frequency upon deletion of ihfA. To explore this possibility, a ∆ihfA strain was tested alongside an isogenic WT strain in mutation assays corresponding to several other operons where preferential IS insertion into their respective SIDD sites have been previously documented (McCalla et al., 1978; Chen et al., 1989; Whiteway et al., 1998; Zhang et al., 2010, 2017; Humayun et al., 2017). Agar plates were made with the intention of promoting insertion into the glpFK, fucAO, and flhDC operons, and were inoculated with a number of cells specific to the paradigm tested. The results for these three operons are presented in Figures 6A–C, and all three showed a significant decrease in insertional frequency, just as observed for bgl.
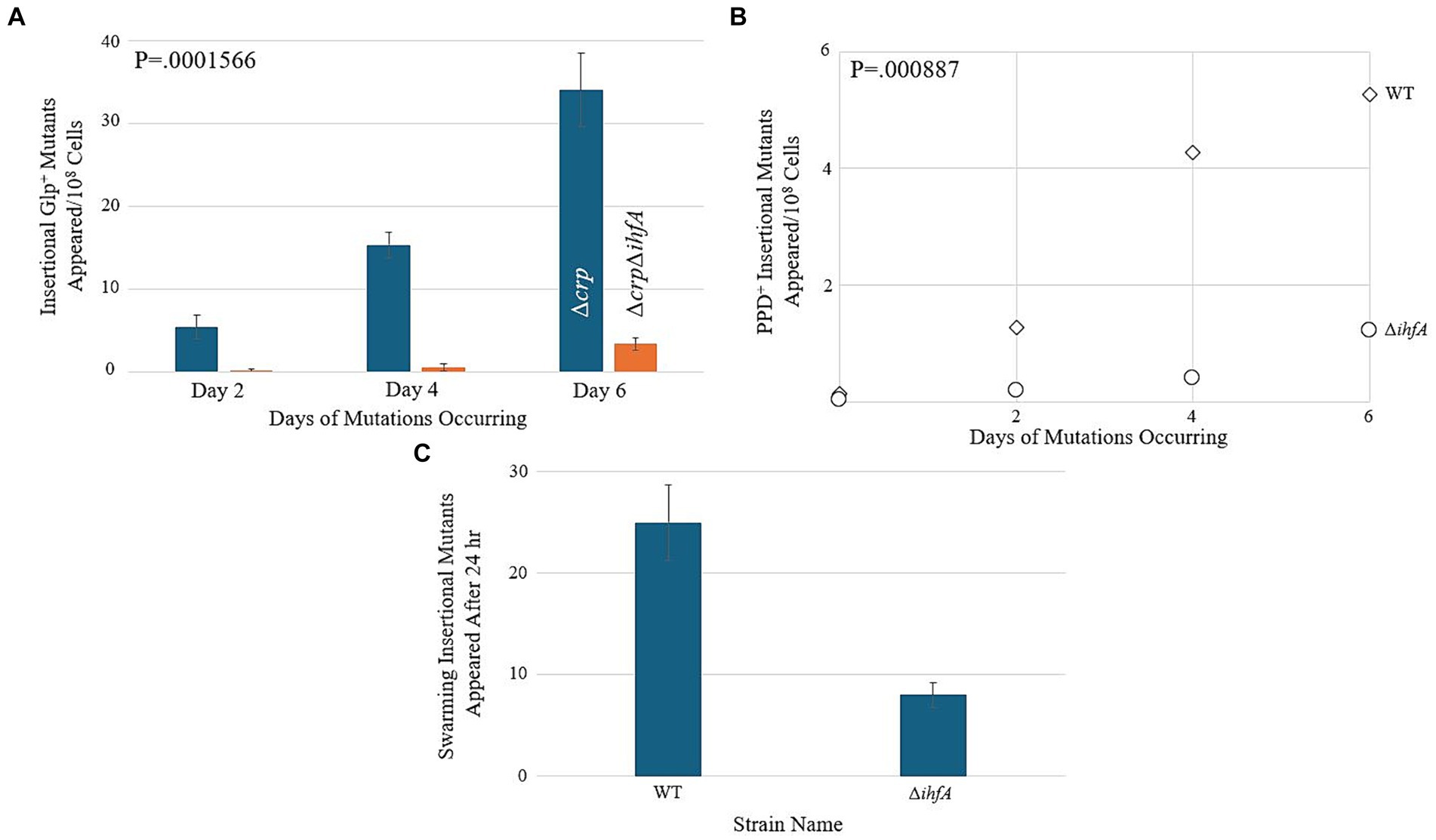
Figure 6. Deletion of ihfA affects IS element insertion into other operons. (A) Effect of ihfA deletion on Glp+ mutations. A ∆crp mutant and a ∆crp ∆ihfA double mutant were subjected to the Glp+ mutation assay on M9 + 0.5% glycerol media as previously described. (B) Effect of ihfA deletion on PPD+ mutations. WT and ∆ihfA strains were subjected to the PPD+ mutation assay on M9 + 1% propanediol media. (C) Effect of ihfA deletion on swarming mutations. WT and ∆ihfA were subjected to the swarming mutation assay on 0.3% LB agar media.
As when testing bgl, these experiments compared ∆ihfA to the WT strain. However, the parameters of each experiment differed depending on the specific operon being tested. Interestingly, all the operons examined showed a significant decrease in the insertional mutant appearance rate of the ∆ihfA counterpart, suggesting that the dependency of this process on IHF affects several operons into which insertions occur, and consequently, it may affect an early step (IS excision or transposition) of the IS1 or IS5 element itself.
Since our view of IHF’s effect proved to be relevant to other operons, the question remained: What is the mechanism of the IHF effect on transposition? First, we tested whether the DNA-binding ability of IHF was the cause of the observed multi-operon effect. A mutation assay was performed comparing WT and ∆ihfA to two other strains. These strains have Ptet-driven ihfA at the intS locus, but here the ihfA gene product contains a G-E substitution at the 62nd position (referred to as G62E). The product of this ihfA mutant loses its DNA-binding function but is still able to dimerize with IhfB (Granston and Nash, 1993; Hales et al., 1994). The ihfA_G62E construct was placed into WT as well as the deletion strain for ihfA at its native position (∆ihfA), yielding PK10 and PK11. If binding to DNA was important for IHF’s effect, then PK11 would show similar insertion rates as ∆ihfA, while PK10 would have binding and nonbinding IhfA and therefore would not experience as drastic of a change. Figures 7A,B show the results of a bgl mutation assay, conducted as previously described. The strains acted according to expectations, with PK10 having fewer mutants than WT, but more than its ∆ihfA counterpart PK11.
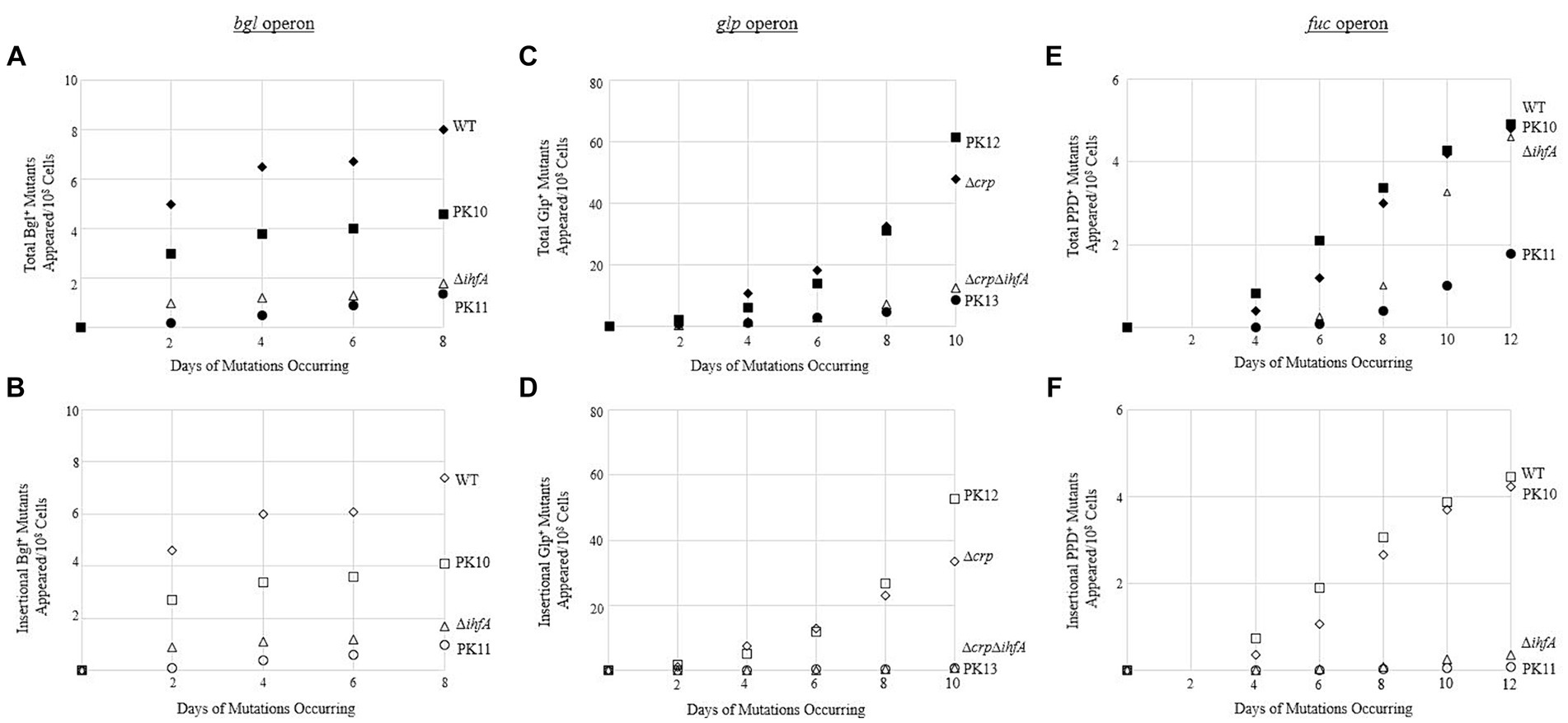
Figure 7. Binding of IHF is necessary to effectively exert its positive effect on IS insertion upstream of the bgl operon. (A,B) Negative effect of expressing ihfA_G62E on total Bgl+ mutations (A) or on insertional Bgl+ mutations (B). (C,D) Negative effect of expressing ihfA_G62E on total Glp+ mutations (C) or on insertional Glp+ mutations (D). (E,F) Negative effect of expressing ihfA_G62E on total PPD+ mutations (E) or on insertional PPD+ mutations (F). IhfA with the G62E mutation loses the DNA binding property but is still able to dimerize with IhfB. The dimers consisting of IhfA_G62E and IhfB are incapable of DNA binding. WT and Δcrp = ♦ total and ◊ insertional; PK10 and PK12 = ■ total and □ insertional; ΔihfA and ΔcrpΔihfA = ▲ total and ∆ insertional; PK11 and PK13 = ● total and ○ insertional.
To confirm these results, the G62E constructs were also used in mutation assays for the glpFK and fucAO operons as previously described; the results of these are in Figures 7C–F. The G62E strains act similarly with respect to other operons as with bgl; in all operons tested, the G62E mutant generated around the same number of mutations as the WT if the native ihfA gene was intact. However, the G62E mutation in the ∆ihfA deletion mutant still caused a drastic drop to the same level of ∆ihfA or lower.
Research conducted by other laboratory groups had demonstrated that IHF binding sites are present at both ends of IS1 and on one end of IS5 (hereafter referred to as IS5C; IS5A is the end without an IHF binding site) (Gamas et al., 1987; Prentki et al., 1987; Muramatsu et al., 1988). We therefore decided to test whether the presence and/or binding of IHF causes a change in the level of transcription of either or both IS elements. This was accomplished by placing the promoter regions for IS1 and both ends of IS5 (containing the Inverted Repeats to which IHF binds) directly before the native lacZ gene (Figure 8A). This construct was placed in the WT, ∆ihfA, Ptet-driven ihfA at the intS locus, and Ptet-driven ihfA G62E at the intS locus (containing the G-E substitution at the 62nd position that abolishes DNA-binding ability of IHF). The results for each reporter are presented in Figures 8B–D. Overall, no significant change was observed, regardless of the genetic background. Since IHF is known to bind to either end of IS1 and to IS5C, these results strongly suggest that IHF exerts its effect via direct binding to IS1 and IS5 without altering the expression level of either IS element.
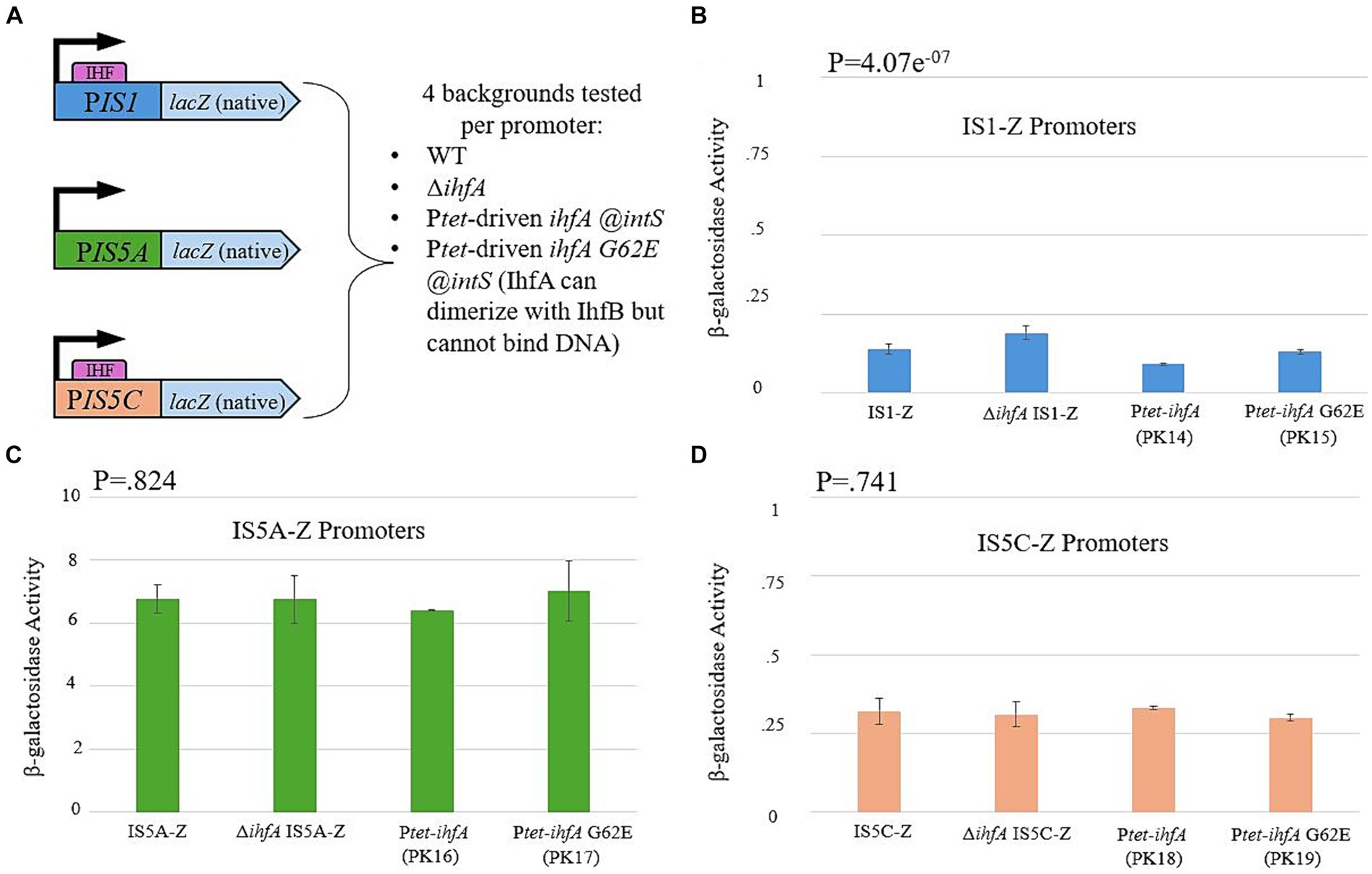
Figure 8. IHF does not influence transcriptional activity of IS1 or IS5. (A) A graph showing the construction of each LacZ reporter, which uses the promoter region of the 5′ end of IS1 and both ends of IS5 (PIS5A and PIS5C). (B) The IS1 transposase promoter (PIS1) activity in various genetic backgrounds. (C) The ins5A transposase promoter (PIS5A) activity in various genetic backgrounds. (D) The ins5BC operon promoter (PIS5C) activity in various genetic backgrounds.
To test whether Crp (when activated by cAMP) affects the rate of insertional mutations in the upstream region, two types of mutation assays were conducted. Simply testing a ∆crp mutant is not an option in this case, as Crp is required for expression of the bgl operon. If insertion takes place in a crp deletion mutant, we would be unable to observe it since the colony would not grow. Therefore, we first decided to perform mutation assays using WT on M9 salicin plates with minimal cAMP and compare mutation rates to those growing on plates with an excess of cAMP. The results of this experiment are shown in Figure 9A. They demonstrate no change in insertional mutants as the amount of extracellular cAMP increases.
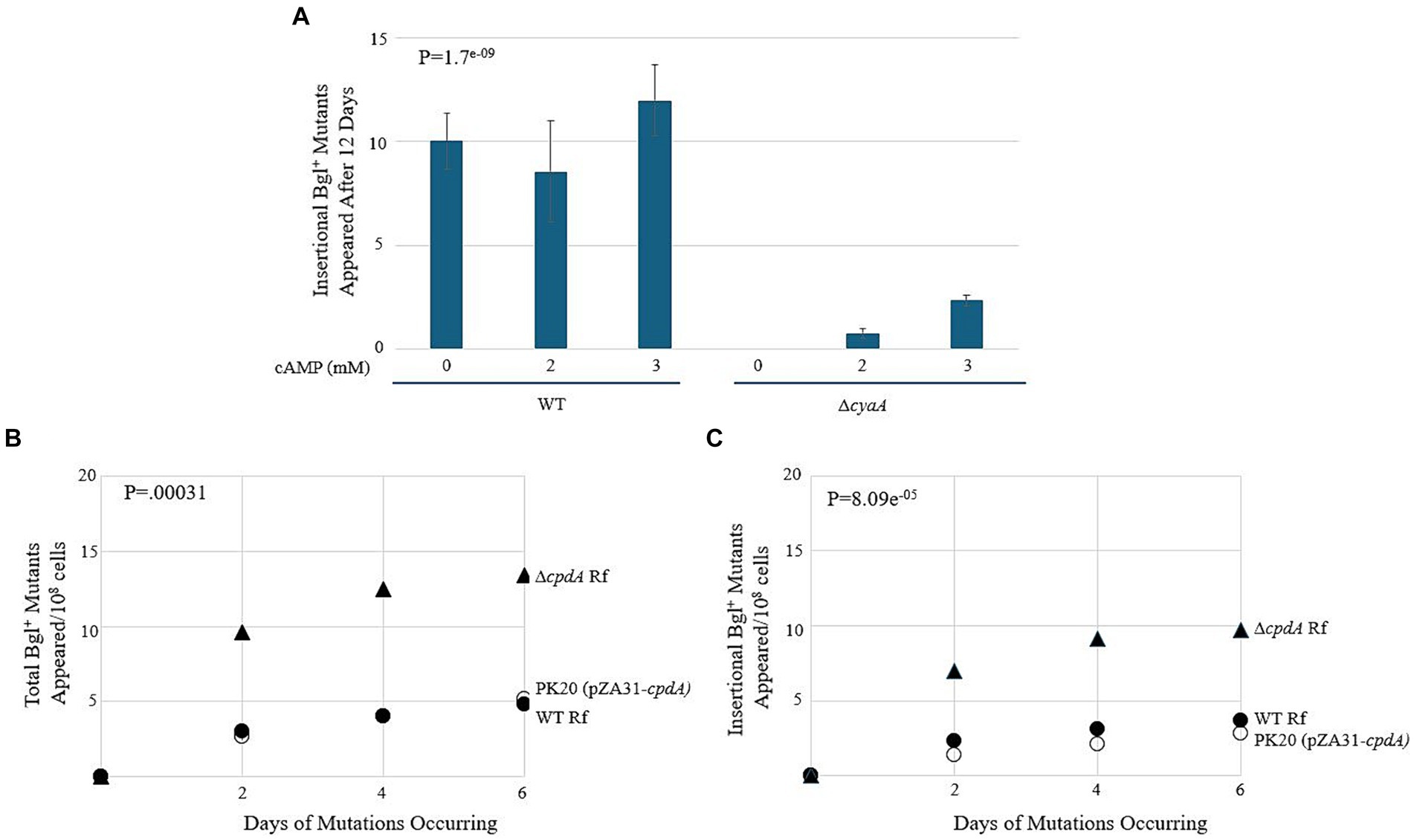
Figure 9. cAMP-Crp is a positive regulator of IS insertion into bgl. (A) Effect of adding external cAMP on insertional Bgl+ mutations in wild type cells and ∆cyaA cells. cAMP was added to M9 + Salicin agar from 0 mM to 3 mM. (B,C) Effects of cpdA deletion and overexpression on total Bgl+ mutations (B) and insertional Bgl+ mutations (C). cpdA encodes a phosphodiesterase which degrades cAMP to 5’-AMP, and in its absence, cAMP levels increase. For overexpression, the strong constitutive tet promoter, Ptet, was used to drive cpdA on the plasmid pZA31. WT Rf = ●; ∆cpdA Rf = ▲; PK20 = ○.
Next, we compared these WT results to those of a cyaA deletion mutant. cyaA encodes adenylate cyclase, and without it, the cells are unable to produce cAMP and must rely on extracellular cAMP provided in the growth media. In Figure 9A, we observe an increase in insertional mutants as the level of extracellular cAMP increases. Interestingly, the number of total colonies was like those observed for the WT strain, suggesting that the lack of cyaA has a significant effect on IS insertion specifically. The next set of experiments used cpdA to control the levels of intracellular cAMP. cpdA encodes a phosphodiesterase which degrades cAMP to 5’-AMP, and in its absence, cAMP levels increase. Similarly, if cpdA is expressed at higher levels, the amount of cAMP, and therefore the amount of active Crp, should decrease. A cpdA deletion mutant was constructed and was given an “empty” pZA31 plasmid for isogeneity, producing ∆cpdA Rf. A pZA31 plasmid containing Ptet-cpdA was electroporated into WT cells, yielding PK20. All three strains were subjected to the bgl mutation assay as previously described. The results are presented in Figures 9B,C. The ∆cpdA mutant showed a more than two-fold increase in insertional mutants compared to the WT strain, and the cpdA overexpression strain PK20 showed a similar number of total colonies as WT, but with only half as many insertional mutants. Together, these data suggest that Crp, when activated by cAMP, is a positive regulator of insertion upstream of the bgl operon. In the absence of cAMP via deletion of cyaA or via increased degradation of cAMP, the insertion rate decreased 2–3-fold.
To observe how loss of ihfA affects mutation frequencies along with differing cAMP-Crp complex levels, the ∆cpdA Rf and PK20 backgrounds were transferred to the ∆ihfA strain. The results of this set of mutation essays are shown in Figures 10A,B. In the absence of IHF, increased Crp-cAMP by deleting cpdA led to a more than 2-fold elevation of Bgl+ mutations. Similarly, decreased Crp-cAMP by overexpressing cpdA led to a 3-fold reduction of Bgl+ mutations in the absence of IHF. Combining both IHF and Crp, these two proteins affect Bgl+ mutations by up to 33-fold (Figures 9B, 10A).
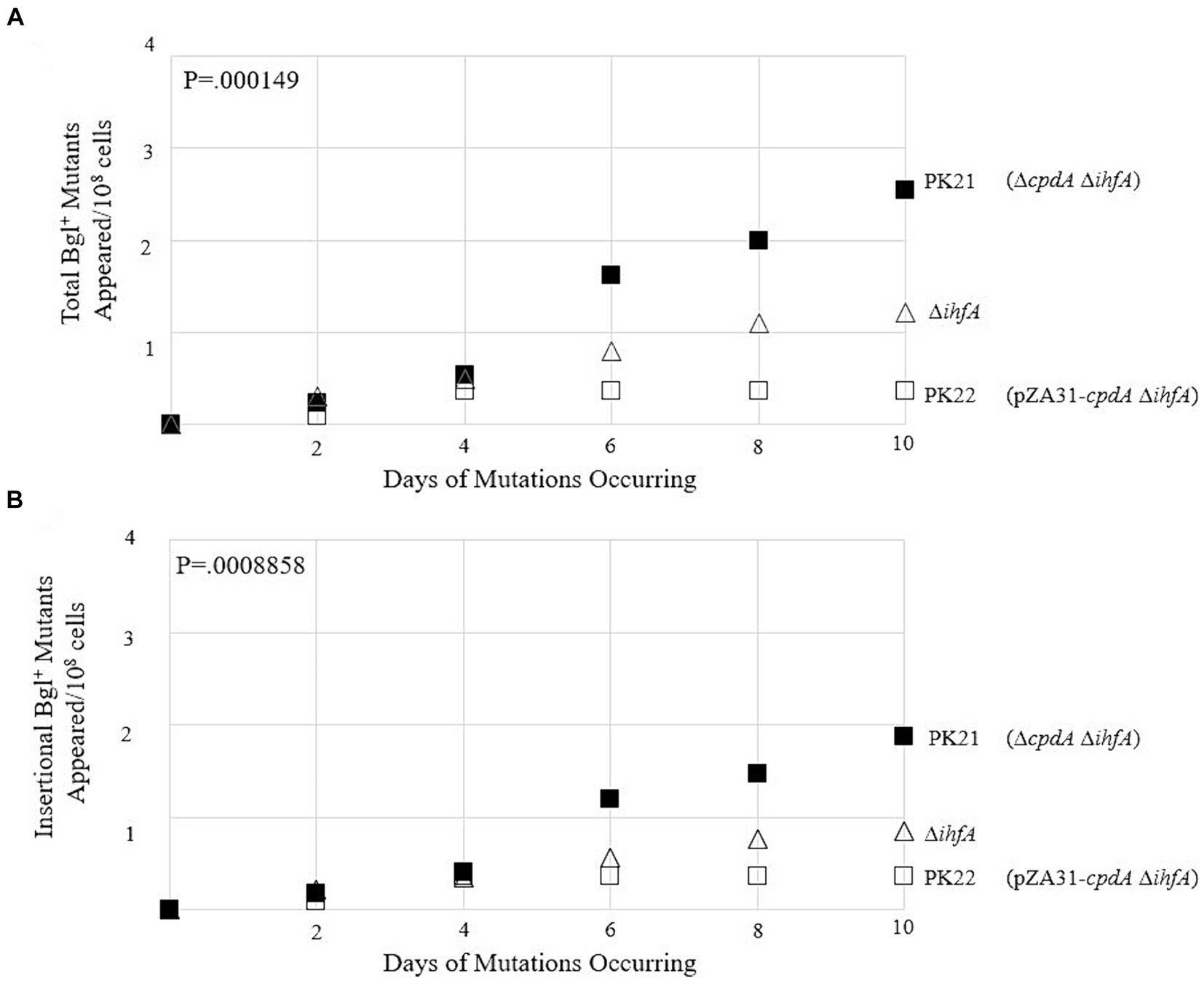
Figure 10. Effects of varying levels of cAMP on Bgl+ mutations in ∆ihfA cells. (A) Effects of cpdA deletion and overexpression on total Bgl+ mutations in ∆ihfA cells. (B) Effects of cpdA deletion and overexpression on insertional Bgl+ mutations in ∆ihfA cells. To elevate cellular cAMP levels, cpdA was deleted. To decrease cellular cAMP levels, cpdA was driven by Ptet on plasmid pZA31.
PK01, which replaced the native promoters of ihfA and ihfB with the constitutive Ptet promoter, had the surprising effect of lowering the insertion rate into bgl by nearly two-fold compared to WT. Additionally, the titratable strain PK02_R, which grows similarly to WT, grew only around half as many colonies when saturated with its inducer aTC. We here provide a few possible explanations for these phenomena. The first and most likely possibility is that the native promoters for ihfA and ihfB are already strong. Since IHF is a global protein with several functions in the cell and with genes in locations adjacent to other important genes, promoters for both subunits may be very strong (Pozdeev et al., 2022). It may be that the promoters driving these subunits are already operating at a higher rate of expression than Ptet itself. Therefore, in an effort to measure the mutational response of a cell by increasing expression, it is possible that replacement by Ptet, while strong, could actually have expressed ihfA and ihfB at a lower rate than the native promoter. Our data on the titratable strains PK01_R and PK02_R are in support of this hypothesis, because even at maximal levels of inductions, the mutation rate remains lower than WT.
Another possibility is that this strain is in fact expressing IHF at levels higher than WT, but that this has an overall negative effect on mutation frequency. For example, these increased levels of expression may be toxic to the cell, or the increased binding of IHF to the multitude of its recognized binding sites may have pleiotropic effects that lead to lower growth rates, and consequently less insertion.
A final possibility is related to the fact that IHF levels increase greatly when the cell is starving (Bushman et al., 1985; Ditto et al., 1994). It may be that this increase is regulated by transcription factors affecting the native ihfA/ihfB promoters under starvation conditions such as during the mutation assay. If we change the promoters to Ptet, we may well have inadvertently removed the cell’s normal ability to respond to starvation by upregulating ihfA and ihfB expression, which would also bring IHF levels to a lower-than-expected value.
cAMP-activated Crp, which is known to bind in bgl’s promoter region and positively regulate transcription, was also shown to positively regulate the rate of IS element transposition upstream of bgl. Results of a mutation assay comparing WT with an adenylate cyclase deletion mutant (∆cyaA) showed a decrease in the incidence of insertional colonies, although the overall Bgl+ colony counts remained comparable. Similarly, in backgrounds overexpressing or deleting the cAMP-degrading phosphodiesterase gene, cpdA, the rate of insertion decreased or increased, respectively. Thus, these results reveal a new role for Crp within the context of bgl as a regulator of transposition as well as transcription. Interestingly, previous data from our group have suggested that in the glpFK operon, cAMP-Crp has a negative effect on upstream IS insertion. It appears that while Crp maintains its status as a positive transcriptional regulator in the vast majority of operons which it influences, its role with respect to mutational changes in the bgl operon may be responsive to the sequences and/or DNA structures surrounding its binding site.
Crp has a known binding site in bgl’s promoter region, and this site is close to or overlapping the projected SIDD site into which IS elements insert (Humayun et al., 2017). This novel effect of cAMP-activated Crp presented here likely results from the increased energetic favorability for insertion to take place due to its binding. However, advances in computational programs that analyze energetics would be helpful to confirm this, as the current programs do not account for the binding of proteins to the DNA.
The IS1-lacZ reporter activity in section 2.7 proved to be low, even when compared to the two orientations of the IS5 reporter. After a thorough examination of the sequence of the construct, no mutation was found, leading us to consider other possibilities as to why IS1 exerted such a low operon transcriptional activity compared to IS5. We submit that either the growth conditions were not sufficient to increase IS1’s promoter to higher levels, or the region upstream of the IS1-lacZ gene construct lacks a site for a postulated positive transcription factor for the IS1 promoter. If so, it must be present in the other native locations in the genome where IS1 exists. Both considerations may independently or together explain why IS1’s reporter showed such low activity.
As stated in the Results section, both ends of IS1 and at least one end of IS5 contain IHF binding sites (Gamas et al., 1987; Prentki et al., 1987; Muramatsu et al., 1988). However, LacZ assays measuring the activities of the IS1 and IS5 promoters showed no clear change when the level of IHF binding was altered. Despite this, our other data indicate a non-specific effect of IHF on transposition of IS1 and IS5. Thus, the effect exerted by IHF on these IS elements must be of a nature other than transcriptional. Both IS1 and IS5 have DDE catalytic regions (Ohta et al., 2002; Curcio and Derbyshire, 2003), and may therefore act similarly for the purposes of replicative excision from the E. coli genome. Our IHF mutation assay data supports this; if IHF only affected one element’s transposition and not the other, then only a 2-fold change at best would be expected, and all the insertional colonies would contain the same element, instead of the roughly equal levels of IS1 and IS5 insertion that has been classically observed (Schnetz and Rak, 1988; P.K., unpublished data). Work on other transposons with DDE activity has suggested that IHF plays an important, but not essential, role in the efficient transposition of Tn10 and the Mu bacteriophage (Kobryn et al., 2002; Swinger and Rice, 2004; Gueguen et al., 2005). The role of IHF involves assisting in the conformation of the transposon via binding so that the transposase protein can effectively bind to the transposon ends and then dimerize (or tetramerize in the case of Mu). It has been shown that when IHF is not present, DNA supercoiling has a compensatory effect, allowing for some transposition to occur even when an important conformational factor is not present (Chalmers et al., 1998). Interestingly, Tn10 in vitro experiments mimicking in vivo supercoiling levels in the absence of IHF observed a ~ 10-fold decrease in transposition frequency (called the “basal level” of 27 excision), which is a similar decrease to what we observed in the ihfA deletion mutant (∆ihfA) for the Bgl+ mutation assay (Chalmers et al., 1998; Figure 2A). We utilized in vivo models to support past findings in relation to other transposable elements and draw further similarities between IS elements and other transposons in the DDE transposase family.
In this work, we demonstrate that IHF and Crp are positive regulators of IS1 and IS5 insertion. Without both functional IHF subunits and the ability to bind DNA, a strong decrease in total Bgl+ mutations is observed, and an even larger decrease in insertional Bgl+ mutations. No matter the amount of antiterminator protein BglG, this strong decrease is still observed. This effect extends to other operons where IS1 and IS5 insertion takes place. We show that the effect of IHF is not transcriptional in nature - that is, binding of IHF to the IS element causes no significant change in transposase gene expression. Therefore, we suggest that IHF’s role may instead be related to assisting DNA conformation or transpososome formation at the site of the IS element. We show that increasing the amount of extracellular cAMP increases insertional Bgl+ mutations. Additionally, by increasing intracellular cAMP levels via cpdA deletion, a more than 2-fold increase in insertional Bgl+ mutations is observed. IHF and Crp’s effects are independent of each other, and combining their effects causes an up to 33-fold change in insertional mutations.
CGSC strains JW3964-1, JW0430-3, JW1702-1, and JW3000-1 (E. coli Genetic Stock Center, Yale Univ.) carry the deletion mutations of hupA, hupB, ihfA, and cpdA, respectively. For each of these mutants, a kanamycin resistance (kmr) gene was substituted for the target gene. These mutations were individually transferred to strain BW25113 (wild type/WT) (Datsenko and Wanner, 2000) by P1 transduction, and the kmr gene was subsequently flipped out by pCP20 (Datsenko and Wanner, 2000), yielding deletion mutant strains ΔhupA, ΔhupB, ΔihfA, and ΔcpdA, respectively (Supplementary Table S1). Using P1 transduction, the ihfA mutation was transferred into the crp deletion strain Δcrp (Zhang and Saier, 2009b), yielding the ΔcrpΔihfA double mutant. The same ihfA mutation was transferred into strain ΔcpdA, yielding the ΔcpdAΔihfA double mutant.
The cpdA gene was amplified from BW25113 chromosomal DNA using oligos cpdA-Kpn-F and cpdA-Bam-R (Supplementary Table S2). The PCR products were gel purified, digested with KpnI/BamHI and then ligated into the same sites of pZA31Ptet (Lutz and Bujard, 1997) yielding pZA31-cpdA. pZA31-cpdA was transformed into strains BW23113 and ΔihfA, yielding PK20 and PK22, respectively (Supplementary Table S1). Plasmid pZA31 Ptet-Rf (Levine et al., 2007) carries a random fragment (RF) and was used as a negative control (WT Rf).
Using plasmid pKDT:Ptet (Klumpp et al., 2009) as a template, the cassette “kmr:rrnBT:Ptet,” containing the kmr gene, the rrnB terminator (rrnBT) and the Ptet promoter, was amplified using the primer pair Ptet-ihfA-P1 and Ptet-ihfA-P2 (Supplementary Table S2). Using lambda-red system (Datsenko and Wanner, 2000), the PCR products were integrated into the chromosome of BW25113 to replace the “CCT” nucleotides immediately upstream of the ihfA translational start point. Chromosomal integration was confirmed, first by colony PCR, and subsequently by DNA sequencing. This yielded strain Ptet-ihfA, in which the strong tet promoter drives expression of the ihfA gene. Similarly, the “kmr:rrnBT:Ptet” cassette amplified by Ptet-ihfB-P1 and Ptet-ihfB-P2 (Supplementary Table S2) from pKDT:Ptet was substituted for the ihfB upstream promoter region (−46 to −1 relative to the ihfB translational start point), yielding strain Ptet-ihfB.
To make an ihfA/ihfB double overexpression strain, the kmr gene was first flipped out from strain Ptet-ihfB by pCP20. The cassette of “kmr:rrnBT:Ptet” driving ihfA gene from strain Ptet-ihfA was then transferred to Km-sensitive Ptet-ihfB by P1- transduction, yielding strain PK01 (Supplementary Table S1), in which both ihfA and ihfB are simultaneously driven by Ptet. To titrate expression of ihfA and ihfB, the transcription unit including a constitutively expressed tetR gene and a spectinomycin resistance (spr) marker was transferred to PK03 by P1-transduction as mentioned above, yielding strain PK01_R.
To further increase ihfA expression, a second copy of Ptet driving ihfA was inserted to another chromosomal location. To do so, the kmr:rrnBT:Ptet-ihfA expression cassette was amplified from the genomic DNA of strain Ptet-ihfA using primers intS1-P1 and ihfA2-P2. The products were integrated into the intS site to replace the region between −229 and + 1,101 (relative to the intS translational initiation site). The chromosomal integration was confirmed by colony PCR and subsequently by DNA sequencing, yielding strain Ptet-ihfA @intS.
The Glycine residue at position 62 is required for IhfA to bind to the DNA (Granston and Nash, 1993; Hales et al., 1994). To reduce or abolish IhfA’s DNA-binding ability, this residue was changed to a glutamate residue using fusion PCR. The first part (5′ region) of ihfA was amplified from BW25113 gnomic DNA using ihfA-Kpn-F and ihfA.G62E-R (carrying the G62E alteration). The second part (3′ region) was amplified using ihfA.G62E-F (carrying the G62E alteration and overlapping ihfA.G62E-R) and ihfA-Bam-R. Both products were gel purified and fused together using primers ihfA-Kpn-F and ihfA-Bam-R. The fused products (that is, ihfA.G62E) were gel purified, digested with KpnI and BamHI, and then ligated into the same sites of pKDT_Ptet, yielding pKDT_Ptet-ihfA.G62E. The cassette “kmr:rrnBT:Ptet-ihfA.G62E” was amplified using intS-P1 and ihfA-P2, gel purified and then integrated into the intS site as for Ptet-ihfA @intS. The chromosomal integration was confirmed by colony PCR and subsequently by DNA sequencing, yielding strain PK10, which still maintains the native ihfA but constitutively expresses the modified ihfA.G62E at the intS locus. This cassette was transferred to an ihfA deletion background by P1 transduction, yielding PK11.
To over express ihfA and ihfB simultaneously, both structural genes were first PCR amplified individually from E. coli genomic DNA. After being gel purified, these two DNA fragments were fused together by PCR. There is a 20 bp intergenic region with nucleotide sequences “tctgattAGAGGAaacagct” between these two genes. The capitalized nucleotides refer to the RBS site for ihfB, which is the same as one for ihfA located within Ptet. The fusion ihfA/ihfB products were digested with KpnI and BamHI and then ligated into pZA31Ptet digested with the same enzymes, yielding pZA31-ihfAB, in which ihfA and ihfB are driven by Ptet and both genes have the same RBS site.
The promoter region (−55 to +30 relative to the insA translation initiation site) driving the IS1 transposase gene insAB1 was amplified using IS1p-Xho-F and IS1pBam-R from BW25113 genomic DNA. This region contains all of the upstream region (including the left-hand IR) and the first 10 residues of insA (plus a stop codon TAA). The amplified products were digested with XhoI and BamHI and cloned into the same sites of the default integration vector, pKDT (Klumpp et al., 2009), yielding pKDT-PIS1. The region carrying the kmr, rrnBT and PIS1 (kmr:rrnBT:PIS1) was PCR amplified using oligos IS1- Z-P1 and IS1-Z-P2 (Supplementary Table S2) and then integrated into the chromosomal default strain EQ42 (Klumpp et al., 2009) to replace the lacI gene and the lacZ promoter. The resultant strain carries the kmr:rrnBT:PIS1 cassette followed by lacZ’s ribosomal binding site (RBS) and the lacZ structural gene within the lac locus. After being confirmed by PCR and sequencing, the promoter reporter, PIS1, driving lacZ expression (that is, PIS1-lacZ) was transferred into BW25113 and various genetic backgrounds by P1 transduction. This yielded the IS1 promoter reporter strains IS1-Z, ΔihfA_IS1-Z, PK14, and PK15, respectively.
IS5 carries three open reading frames (ins5A, ins5B, and ins5C). ins5A encodes the main transposase, and it is transcribed from its own promoter located close to the left-hand IR while the divergent ins5B and ins5C genes may form an operon that is driven by another promoter located close to the right-hand IR (Sawers, 2005). The ins5A promoter region (−68 to +30 relative to the ins5A translational initiation site) and the ins5CB promoter region (−207 to +30 relative to the ins5C translational initiation site) were individually cloned into pKDT, yielding pKDT_PIS5A and pKDT_PIS5C, respectively. As for PIS1 described above, PIS5A and PIS5CB each carries the first 10 amino acids from the target gene followed by a stop codon TAA. These promoters were integrated into the lac site on BW25113 chromosome as for PIS1-Z, yielding strains IS5A-Z and IS5C-Z, respectively. Using P1 transductions, these new IS5 promoter lacZ reporters were transferred to other genetic backgrounds by P1 transduction. This yielded the IS5 transposase promoter (PIS5A) reporter strains ΔihfA_IS5A-Z, PK16, and PK17, as well as the IS5C promoter reporter strains ΔihfA_IS5C-Z, PK18, and PK19.
Escherichia coli reporter strains were cultured in 4 mL of LB contained in glass test tubes (1.5 cm in diameter × 15 cm in length) with shaking at 37°C for 8 h. An amount of 30 μL of LB cultures were used to inoculate 3 mL of M63 minimal media in smaller glass tubes (1.2 cm × 12 cm), and the tubes were shaken at 37°C overnight. The carbon sources were 0.5% glycerol, 0.5% salicin, or both. The tubes were rotated at 250 rpm and 37°C, and cell densities (OD600) were measured with a Bio-Rad spectrophotometer. During the exponential growth phase, four samples were collected in the range of OD600 from 0.1 to 1. The samples (roughly 0.3 mL for promoter reporter strains, 0.9 mL for the IS1/5 promoters), and 0.6 mL for operon reporter strains were immediately frozen at −20°C prior to β-galactosidase assays. To measure β-galactosidase activities in bgl promoter reporter strains, 0.8 mL of Z-buffer containing β-mercaptoethanol (2.7 μL/mL) and sodium dodecyl sulfate (SDS) (0.005%) was mixed with 0.2 mL of sample and 25 μL of CHCl3 in test tubes. Alternatively, for bgl operon reporter strains, 0.5 mL of Z-buffer was mixed with 0.5 mL of the sample. The tubes were vortexed twice (each time for 10 s at a constant speed) and incubated in a 37°C water bath until temperature equilibration. A 0.2 mL aliquot of O-nitrophenyl galactoside (ONPG) substrate (4 mg/mL) was then added to each test tube. When a yellow color developed, the reaction was stopped by adding 0.5 mL of 1 M Na2CO3 followed by vortexing. Reaction mixtures were centrifuged (15,000 rpm, 3 min), and the absorbance values of the supernatants were measured at 420 nm and 550 nm. A control tube was run in parallel using M63 salts instead of the test sample. β-galactosidase activity (Miller units) = [(OD420–1.75 × OD550)/(sample volume in mL × time in min)] × 1,000 (Miller, 1972). For a given test strain, the slope of OD600 values versus β-galactosidase activities was referred to as the promoter activity or the operon activity.
To test the effect of IHF on bglGFB transcription, the bgl promoter reporter (Pbgl-Z) and the bgl operon reporter (Pbgl-G-Z) (Tran et al., 2022) were individually transferred to an ihfA deletion background. This yielded strains ∆ihfA_Pbgl-Z and ∆ihfA_Pbgl-G-Z.
β-glucosidase assay was performed as described in our previous study (Lam et al., 2022). Briefly, 20 μL of LB culture was transferred to M63 minimal medium with 0.66% casamino acids (CAA). After overnight growth at 37°C with shaking, the culture was diluted into 6 mL of M63 + 0.66% CAA +0.5% salicin minimal medium at a starting OD600 of 0.025. The cells were grown at 37°C with shaking. Five samples with 0.8 mL each were collected during the late exponential growth phase when the bgl operon was fully induced with an OD600 of above 1.5. The samples were centrifuged at a speed of 5,500 rpm for 2.5 min and the cells were suspended with 1 mL of Z-buffer with 50 μg/mL chloramphenicol.
To measure β-glucosidase (BglB) activities, test samples were warmed in a 37°C water bath, and 200 μL of p-nitrophenyl-β-D-glucoside (PNPG, 8 mg/mL) was added to the cell suspension in Z-buffer. After a visible yellow color appeared, the reaction was terminated by adding 0.5 mL of 1 M Na2CO3 and subsequently vortexing. The reaction mixture was centrifuged, and the absorbance of the reaction mixture was measured at 420 nm and 550 nm. The BglB activity of each sample was calculated using the equation: β-glucosidase (BglB) activity = [1,000 × (OD420 nm – 1.75 × OD550 nm) × Dilution factor]/[OD600 nm × Time of reaction (min) × Volume of sample (mL)]. The activity of the strain was determined by averaging the BglB activities of the samples measured.
β-glucosidase (BglB) assays were conducted in wild type and ∆ihfA backgrounds. To see the IHF effect on the cells with higher operon expression, strains G50 (deleted for the two terminators flanking bglG) and ∆Pbgl-G (deleted for Pbgl and bglG together with two terminators; Lam et al., 2022) was used.
Bgl+ mutation assays were performed on minimal M9 agar plates with 0.5% of a β-glucoside (salicin) as the sole carbon source. Strains to be tested (from single fresh colonies) were cultured in LB liquid medium for approximately 7 h at 37°C, washed twice with carbon source-free M9 salts (M9) and applied onto plates (2 × 107 cells/plate). The plates were then incubated in a 30°C incubator and were examined every 2 days for the appearance of Bgl+ colonies, with each colony representing a new Bgl+ mutation. On these β-glucoside minimal agar plates, any colonies appearing by day 2 were considered to be from Bgl+ cells initially applied onto the plates. They were therefore subtracted from the subsequent measurements. IS1 and IS5 have been previously shown to insert into the same upstream bgl promoter region. Colony PCR using primers flanking this region was used to differentiate between insertional and non-insertional mutants. Mutation frequency was determined in the manner described in Cairns and Foster (1991) and Zhang and Saier (2009b).
For the mutation assays which used differential levels of aTC, the growth media was prepared with different amounts of aTC to obtain several ng/mL concentrations as outlined in Results 2.3.
To determine the effects of other carbon sources on Bgl+ mutations, mutation assays were performed on minimal M9 agar plates with 0.25% glycerol or 1% propanediol as the sole carbon source. To determine the total populations, the cells were washed off the minimal M9 agar plates at relevant time points, serially diluted, and plated onto LB agar plates. To determine Bgl+ populations, appropriate dilutions were applied on M9 + salicin agar plates. The frequencies of Bgl+ mutations were determined as described above for Bgl+ on M9 + salicin agar plates.
Glp + mutation assays were conducted on glycerol M9 minimal agar plates as described in Zhang and Saier (2009b). Strains ∆crp and ∆crp∆ihfA were used for the mutation assays.
Using the wild-type and ΔihfA strains, the swarming mutation assays for the appearance of hyper-swarming mutants (outgrowing subpopulations from the inoculated cells) were carried out using the method of Barker et al. (2004). Briefly, overnight cell cultures in LB media were washed once with M9 salts and diluted to an OD600 of 1.0 prior to use. Two microliters of the cell suspensions were streaked across the centers of LB semisolid (0.3% agar) plates (diameter = 9 cm) using a plastic transfer loop. The plates were incubated at 30°C. The swarming mutants, represented by outgrowths of motile subpopulations from the streaked cells, were counted. The mutation frequency was normalized as outgrowths (mutations) per 9-cm cell streak. Insertional mutants were verified via colony PCR.
The assay for PPD + mutations was conducted by applying cell suspensions from fresh overnight cultures onto propanediol (1%) M9 minimal agar plates (~108 cells/plate). The wild-type and other strains were tested over a period of several days as in the Bgl+ mutation assay. Total populations and mutation frequency were determined as described above under “Bgl+ mutation assays.”
The program R was used for all statistical analyses. p-values were generated to determine statistical significance, with values under 0.05 treated as statistically significant. The strains tested were plated along with known controls for each experimental run. Results shown are from a minimum of 3 separate runs/strain, with each run consisting of several plates. The data reported on the figures are expressed as the mean of a minimum of three runs for each strain at each time point. LacZ activity assay results are expressed as the slope of a line generated from 4 time points/run and a minimum of 2 runs (at least 8 data points total). BglB assay results are expressed as the mean of 12 data points from a minimum of 3 separate runs.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding authors.
PK: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. ZZ: Conceptualization, Data curation, Methodology, Supervision, Writing – original draft, Writing – review & editing. MS: Conceptualization, Funding acquisition, Project administration, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This research was funded by NIH grant GM077402 and private contributions, both to MS.
We thank Arturo Medrano-Soto, Dennis Tran, Katie Lam, and Harry Zhou for their expertise and support in the preparation of this manuscript. We also thank Jialu Huo for his help with statistical analysis.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1388522/full#supplementary-material
Allison, R. G., and Chaconas, G. (1992). Role of the a protein-binding sites in the in vitro transposition of mu DNA. A complex circuit of interactions involving the mu ends and the transpositional enhancer. J. Biol. Chem. 267, 19963–19970. doi: 10.1016/S0021-9258(19)88651-9
Amster-Choder, O., and Wright, A. (1992). Modulation of the dimerization of a transcriptional antiterminator protein by phosphorylation. Science 257, 1395–1398. doi: 10.1126/science.1382312
Barker, C. S., Prüss, B. M., and Matsumura, P. (2004). Increased motility of Escherichia coli by insertion sequence element integration into the regulatory region of the flhD operon. J. Bacteriol. 186, 7529–7537. doi: 10.1128/JB.186.22.7529-7537.2004
Bushman, W., Thompson, J. F., Vargas, L., and Landy, A. (1985). Control of directionality in lambda site specific recombination. Science 230, 906–911. doi: 10.1126/science.2932798
Cairns, J., and Foster, P. L. (1991). Adaptive reversion of a frameshift mutation in Escherichia coli. Genetics 128, 695–701. doi: 10.1093/genetics/128.4.695
Chalmers, R., Guhathakurta, A., Benjamin, H., and Kleckner, N. (1998). IHF modulation of Tn10 transposition: sensory transduction of supercoiling status via a proposed protein/DNA molecular spring. Cell 93, 897–908. doi: 10.1016/S0092-8674(00)81449-X
Chen, Q., Arents, J. C., Bader, R., Postma, P. W., and Amster-Choder, O. (1997). BglF, the sensor of the E. coli bgl system, uses the same site to phosphorylate both a sugar and a regulatory protein. EMBO J. 16, 4617–4627. doi: 10.1093/emboj/16.15.4617
Chen, Y. M., Lu, Z., and Lin, E. C. (1989). Constitutive activation of the fucAO operon and silencing of the divergently transcribed fucPIK operon by an IS5 element in Escherichia coli mutants selected for growth on L-1,2-propanediol. J. Bacteriol. 171, 6097–6105. doi: 10.1128/jb.171.11.6097-6105.1989
Curcio, M. J., and Derbyshire, K. M. (2003). The outs and ins of transposition: from mu to kangaroo. Nat. Rev. Mol. Cell Biol. 4, 865–877. doi: 10.1038/nrm1241
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97, 6640–6645. doi: 10.1073/pnas.120163297
Deutscher, J. (2008). The mechanisms of carbon catabolite repression in bacteria. Curr. Opin. Microbiol. 11, 87–93. doi: 10.1016/j.mib.2008.02.007
Ditto, M. D., Roberts, D., and Weisberg, R. A. (1994). Growth phase variation of integration host factor level in Escherichia coli. J. Bacteriol. 176, 3738–3748. doi: 10.1128/jb.176.12.3738-3748.1994
Dole, S., Nagarajavel, V., and Schnetz, K. (2004). The histone-like nucleoid structuring protein H-NS represses the Escherichia coli bgl operon downstream of the promoter. Mol. Microbiol. 52, 589–600. doi: 10.1111/j.1365-2958.2004.04001.x
Engelhorn, M., and Geiselmann, J. (1998). Maximal transcriptional activation by the IHF protein of Escherichia coli depends on optimal DNA bending by the activator. Mol. Microbiol. 30, 431–441. doi: 10.1046/j.1365-2958.1998.01078.x
Fic, E., Bonarek, P., Gorecki, A., Kedracka-Krok, S., Mikolajczak, J., Polit, A., et al. (2009). cAMP receptor protein from escherichia coli as a model of signal transduction in proteins--a review. J. Mol. Microbiol. Biotechnol. 17, 1–11. doi: 10.1159/000178014
Fox, C. F., and Wilson, G. (1968). The role of a phosphoenolpyruvate-dependent kinase system in beta-glucoside catabolism in Escherichia coli. Proc. Natl. Acad. Sci. USA 59, 988–995. doi: 10.1073/pnas.59.3.988
Gamas, P., Chandler, M. G., Prentki, P., and Galas, D. J. (1987). Escherichia coli integration host factor binds specifically to the ends of the insertion sequence IS1 and to its major insertion hot-spot in pBR322. J. Mol. Biol. 195, 261–272. doi: 10.1016/0022-2836(87)90648-6
Goosen, N., and van de Putte, P. (1995). The regulation of transcription initiation by integration host factor. Mol. Microbiol. 16, 1–7. doi: 10.1111/j.1365-2958.1995.tb02386.x
Görke, B., and Rak, B. (1999). Catabolite control of Escherichia coli regulatory protein BglG activity by antagonistically acting phosphorylations. EMBO J. 18, 3370–3379. doi: 10.1093/emboj/18.12.3370
Görke, B., and Stülke, J. (2008). Carbon catabolite repression in bacteria: many ways to make the most out of nutrients. Nat. Rev. Microbiol. 6, 613–624. doi: 10.1038/nrmicro1932
Granston, A. E., and Nash, H. A. (1993). Characterization of a set of integration host factor mutants deficient for DNA binding. J. Mol. Biol. 234, 45–59. doi: 10.1006/jmbi.1993.1562
Gueguen, E., Rousseau, P., Duval-Valentin, G., and Chandler, M. (2005). The transpososome: control of transposition at the level of catalysis. Trends Microbiol. 13, 543–549. doi: 10.1016/j.tim.2005.09.002
Gulati, A., and Mahadevan, S. (2000). Mechanism of catabolite repression in the bgl operon of Escherichia coli: involvement of the anti-terminator BglG, CRP-cAMP and EIIAGlc in mediating glucose effect downstream of transcription initiation. Genes Cells 5, 239–250. doi: 10.1046/j.1365-2443.2000.00322.x
Hales, L. M., Gumport, R. I., and Gardner, J. F. (1994). Mutants of Escherichia coli integration host factor: DNA-binding and recombination properties. Biochimie 76, 1030–1040. doi: 10.1016/0300-9084(94)90027-2
Hall, B. G. (1998). Activation of the bgl operon by adaptive mutation. Mol. Biol. Evol. 15, 1–5. doi: 10.1093/oxfordjournals.molbev.a025842
Humayun, M. Z., Zhang, Z., Butcher, A. M., Moshayedi, A., and Saier, M. H. (2017). Hopping into a hot seat: role of DNA structural features on IS5-mediated gene activation and inactivation under stress. PLoS One 12:e0180156. doi: 10.1371/journal.pone.0180156
Klumpp, S., Zhang, Z., and Hwa, T. (2009). Growth rate-dependent global effects on gene expression in bacteria. Cell 139, 1366–1375. doi: 10.1016/j.cell.2009.12.001
Kobryn, K., Watson, M. A., Allison, R. G., and Chaconas, G. (2002). The mu three-site synapse: a strained assembly platform in which delivery of the L1 transposase binding site triggers catalytic commitment. Mol. Cell 10, 659–669. doi: 10.1016/S1097-2765(02)00596-8
Kolb, A., Busby, S., Buc, H., Garges, S., and Adhya, S. (1993). Transcriptional regulation by cAMP and its receptor protein. Annu. Rev. Biochem. 62, 749–797. doi: 10.1146/annurev.bi.62.070193.003533
Lam, K. J. K., Zhang, Z., and Saier, M. H. (2022). Histone-like nucleoid structuring (H-NS) protein silences the beta-glucoside (bgl) utilization operon in Escherichia coli by forming a DNA loop. Comput. Struct. Biotechnol. J. 20, 6287–6301. doi: 10.1016/j.csbj.2022.11.027
Levine, E., Zhang, Z., Kuhlman, T., and Hwa, T. (2007). Quantitative characteristics of gene regulation by small RNA. PLoS Biol. 5:e229. doi: 10.1371/journal.pbio.0050229
Lopilato, J., and Wright, A. (1990). “Mechanisms of activation of the cryptic bgl operon of Escherichia coli K-12” in The bacterial chromosome. eds. K. Drilca and M. Riley (Washington DC: American Society for Microbiology), 435–444.
Lutz, R., and Bujard, H. (1997). Independent and tight regulation of transcriptional units in Escherichia coli via the lac R/O, the TetR/O and AraC/I1-I2 regulatory elements. Nucleic Acids Res. 25, 1203–1210. doi: 10.1093/nar/25.6.1203
Mahadevan, S., and Wright, A. (1987). A bacterial gene involved in transcription antitermination: regulation at a rho-independent terminator in the bgl operon of E. coli. Cell 50, 485–494. doi: 10.1016/0092-8674(87)90502-2
McCalla, D. R., Kaiser, C., and Green, M. H. (1978). Genetics of nitrofurazone resistance in Escherichia coli. J. Bacteriol. 133, 10–16. doi: 10.1128/jb.133.1.10-16.1978
McClintock, B. (1950). The origin and behavior of mutable loci in maize. Proc. Natl. Acad. Sci. USA 36, 344–355. doi: 10.1073/pnas.36.6.344
Miller, F. (1972). Glycopeptides of human immunoglobulins. 3. The use and preparation of specific glycosidases. Immunochemistry 9, 217–228. doi: 10.1016/0019-2791(72)90087-0
Mukerji, M., and Mahadevan, S. (1997). Characterization of the negative elements involved in silencing the bgl operon of Escherichia coli: possible roles for DNA gyrase, H-NS, and CRP-cAMP in regulation. Mol. Microbiol. 24, 617–627. doi: 10.1046/j.1365-2958.1997.3621725.x
Muramatsu, S., Kato, M., Kohara, Y., and Mizuno, T. (1988). Insertion sequence IS5 contains a sharply curved DNA structure at its terminus. Mol. Gen. Genet. MGG 214, 433–438. doi: 10.1007/BF00330477
Nagarajavel, V., Madhusudan, S., Dole, S., Rahmouni, A. R., and Schnetz, K. (2007). Repression by binding of H-NS within the transcription unit. J. Biol. Chem. 282, 23622–23630. doi: 10.1074/jbc.M702753200
Ohta, S., Tsuchida, K., Choi, S., Sekine, Y., Shiga, Y., and Ohtsubo, E. (2002). Presence of a characteristic D-D-E motif in IS1 transposase. J. Bacteriol. 184, 6146–6154. doi: 10.1128/JB.184.22.6146-6154.2002
Pozdeev, G., Beckett, M. C., Mogre, A., Thomson, N. R., and Dorman, C. J. (2022). Reciprocally rewiring and repositioning the integration host factor (IHF) subunit genes in Salmonella enterica serovar typhimurium: impacts on physiology and virulence. Microb. Genom. 8:000768. doi: 10.1099/mgen.0.000768
Prasad, I., and Schaefler, S. (1974). Regulation of the beta-glucoside system in Escherchia coli K-12. J. Bacteriol. 120, 638–650. doi: 10.1128/jb.120.2.638-650.1974
Prasad, I., Young, B., and Schaefler, S. (1973). Genetic determination of the constitutive biosynthesis of phospho-β-glucosidase a in Escherichia coli K-12. J. Bacteriol. 114, 909–915. doi: 10.1128/jb.114.3.909-915.1973
Prentki, P., Chandler, M., and Galas, D. J. (1987). Escherichia coli integration host factor bends the DNA at the ends of IS1 and in an insertion hotspot with multiple IHF binding sites. EMBO J. 6, 2479–2487. doi: 10.1002/j.1460-2075.1987.tb02529.x
Reynolds, A. E., Felton, J., and Wright, A. (1981). Insertion of DNA activates the cryptic bgl operon in E. coli K12. Nature 293, 625–629. doi: 10.1038/293625a0
Rothe, F. M., Bahr, T., Stülke, J., Rak, B., and Görke, B. (2012). Activation of Escherichia coli antiterminator BglG requires its phosphorylation. Proc. Natl. Acad. Sci. USA 109, 15906–15911. doi: 10.1073/pnas.1210443109
Sawers, R. G. (2005). Transcript analysis of Escherichia coli K-12 insertion element IS5. FEMS Microbiol. Lett. 244, 397–401. doi: 10.1016/j.femsle.2005.02.019
Schnetz, K. (1995). Silencing of Escherichia coli bgl promoter by flanking sequence elements. EMBO J. 14, 2545–2550. doi: 10.1002/j.1460-2075.1995.tb07252.x
Schnetz, K., and Rak, B. (1988). Regulation of the bgl operon of Escherichia coli by transcriptional antitermination. EMBO J. 7, 3271–3277. doi: 10.1002/j.1460-2075.1988.tb03194.x
Sclavi, B., Beatty, C. M., Thach, D. S., Fredericks, C. E., Buckle, M., and Wolfe, A. J. (2007). The multiple roles of CRP at the complex acs promoter depend on activation region 2 and IHF. Mol. Microbiol. 65, 425–440. doi: 10.1111/j.1365-2958.2007.05797.x
Singh, J., Mukerji, M., and Mahadevan, S. (1995). Transcriptional activation of the Escherichia coli bgl operon: negative regulation by DNA structural elements near the promoter. Mol. Microbiol. 17, 1085–1092. doi: 10.1111/j.1365-2958.1995.mmi_17061085.x
Sugimura, S., and Crothers, D. M. (2006). Stepwise binding and bending of DNA by Escherichia coli integration host factor. Proc. Natl. Acad. Sci. USA 103, 18510–18514. doi: 10.1073/pnas.0608337103
Surette, M. G., Lavoie, B. D., and Chaconas, G. (1989). Action at a distance in mu DNA transposition: an enhancer-like element is the site of action of supercoiling relief activity by integration host factor (IHF). EMBO J. 8, 3483–3489. doi: 10.1002/j.1460-2075.1989.tb08513.x
Swinger, K. K., and Rice, P. A. (2004). IHF and HU: flexible architects of bent DNA. Curr. Opin. Struct. Biol. 14, 28–35. doi: 10.1016/j.sbi.2003.12.003
Tran, D., Zhang, Z., Lam, K. J. K., and Saier, M. H. (2022). Effects of global and specific DNA-binding proteins on transcriptional regulation of the E. coli bgl operon. Int. J. Mol. Sci. 23:10343. doi: 10.3390/ijms231810343
van Ulsen, P., Hillebrand, M., Zulianello, L., van de Putte, P., and Goosen, N. (1996). Integration host factor alleviates the H-NS-mediated repression of the early promoter of bacteriophage mu. Mol. Microbiol. 21, 567–578. doi: 10.1111/j.1365-2958.1996.tb02565.x
Whiteway, J., Koziarz, P., Veall, J., Sandhu, N., Kumar, P., Hoecher, B., et al. (1998). Oxygen-insensitive nitroreductases: analysis of the roles of nfsA and nfsB in development of resistance to 5-nitrofuran derivatives in Escherichia coli. J. Bacteriol. 180, 5529–5539. doi: 10.1128/JB.180.21.5529-5539.1998
Zhang, Z., Kukita, C., Humayun, M. Z., and Saier, M. H. (2017). Environment-directed activation of the Escherichia coliflhdc operon by transposons. Microbiology 163, 554–569. doi: 10.1099/mic.0.000426
Zhang, Z., and Saier, M. H. (2009a). A novel mechanism of transposon-mediated gene activation. PLoS Genet. 5:e1000689. doi: 10.1371/journal.pgen.1000689
Zhang, Z., and Saier, M. H. (2009b). A mechanism of transposon-mediated directed mutation. Mol. Microbiol. 74, 29–43. doi: 10.1111/j.1365-2958.2009.06831.x
Zhang, Z., Yen, M. R., and Saier, M. H. (2010). Precise excision of IS5 from the intergenic region between the fucPIK and the fucAO operons and mutational control of fucPIK operon expression in Escherichia coli. J. Bacteriol. 192, 2013–2019. doi: 10.1128/JB.01085-09
Zhang, Z., Zhou, K., Tran, D., and Saier, M. (2022). Insertion sequence (IS) element-mediated activating mutations of the cryptic aromatic β-glucoside utilization (Bgl GFB) operon are promoted by the anti-terminator protein (BglG) in Escherichia coli. Int. J. Mol. Sci. 23:1505. doi: 10.3390/ijms23031505
Keywords: IS element, insertional mutation, adaptive mutation, Crp, IHF, DNA-binding protein
Citation: Kopkowski PW, Zhang Z and Saier MH (2024) The effect of DNA-binding proteins on insertion sequence element transposition upstream of the bgl operon in Escherichia coli. Front. Microbiol. 15:1388522. doi: 10.3389/fmicb.2024.1388522
Edited by:
Axel Cloeckaert, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
Sung-Jae Lee, Kyung Hee University, Republic of KoreaCopyright © 2024 Kopkowski, Zhang and Saier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence:Milton H. Saier Jr., bXNhaWVyQHVjc2QuZWR1; Zhongge Zhang, enpob25nZ2VAdWNzZC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.