 Miyabi Sakai1
Miyabi Sakai1 Masafumi Yohda
Masafumi Yohda Issay Narumi
Issay Narumi
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 28 May 2024
Sec. Extreme Microbiology
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1387296
This article is part of the Research TopicInsights in Extreme Microbiology: 2023View all 9 articles
Deinococcus spp. are known for their radiation resistance, toxic compound removal, and production of valuable substances. Therefore, developing gene expression systems for Deinococcus spp. is crucial in advancing genetic engineering applications. To date, plasmid vectors that express foreign genes in D. radiodurans and D. geothermalis have been limited to plasmid pI3 and its derivatives. In contrast, plasmid vectors that express foreign genes in D. grandis include plasmid pZT23 and its derivatives. In this study, we developed a new system for the stable introduction and retention of expression plasmids for D. grandis. Two cryptic plasmids were removed from the wild-type strain to generate the TY3 strain. We then constructed a shuttle vector plasmid, pGRC5, containing the replication initiation region of the smallest cryptic plasmid, pDEGR-3, replication initiation region of the E. coli vector, pACYC184, and an antibiotic resistance gene. We introduced pGRC5, pZT23-derived plasmid pZT29H, and pI3-derived plasmid pRADN8 into strain TY3, and found their coexistence in D. grandis cells. The quantitative PCR assay results found that pGRC5, pZT29H, and pRADN8 had relative copy numbers of 11, 26, and 5 per genome, respectively. Furthermore, we developed a new plasmid in which the luciferase gene was controlled by the promoter region, which contained radiation-desiccation response operator sequences for D. grandis DdrO, a stress response regulon repressor in D. grandis, hence inducing gene expression via ultraviolet-C light irradiation. These plasmids are expected to facilitate the removal and production of toxic and valuable substances, in D. grandis, respectively, particularly of those involving multiple genes.
When genes capable of removing persistent or harmful substances or genes involved in the intracellular uptake of chemical elements are introduced or expressed in radioresistant bacteria using genetic engineering methods, persistent and harmful substances contained in radionuclide-contaminated waste can be removed. Therefore, the genetic engineering of radioresistant bacteria is helpful as a bioremediation technology for radionuclide removal (Gabani and Singh, 2013; Gerber et al., 2015; Li et al., 2021).
Deinococcus spp. are the most studied radioresistant bacteria and can be genetically engineered; Deinococcus radiodurans is the most researched species. It has a multiploid genome (4–10 copies per cell) and a high natural transformation potential (Moseley and Setlow, 1968; Hansen, 1978; Harsojo et al., 1981; Ithurbide et al., 2020). The radiation resistance of D. radiodurans is 250 times that of Escherichia coli and approximately 1,000 times that of most vertebrates. This ability to withstand high radiation levels is primarily attributed to its adequate protein protection by the antioxidant defenses and exceptionally high DNA repair capacity (Cox and Battista, 2005; Ishino and Narumi, 2015; Lim et al., 2019; Daly, 2023; Sadowska-Bartosz and Bartosz, 2023).
Some examples of bioremediation techniques using D. radiodurans include expression of a toluene dioxygenase gene from Pseudomonas putida in D. radiodurans to convert persistent toluene into an easily degradable substance (Lange et al., 1998). A divalent mercury ion reduction gene from E. coli was expressed in D. radiodurans to reduce divalent mercury ions to the less toxic and volatile metallic mercury (Brim et al., 2000). Using D. radiodurans as a host, a nonspecific acid phosphatase gene from Salmonella enterica Typhi or an alkaline phosphatase gene from Sphingomonas sp. was expressed on a plasmid to precipitate uranium from a uranyl nitrate solution (Appukuttan et al., 2006). An endoglucanase gene from Bacillus pumilus was expressed on plasmids to contribute to the bioremediation of cellulosic waste in radioactive environments (Telang et al., 2014). A synthetic gene that encoded a phytochelatin analog or a metallothionein gene from Synechococcus sp. was expressed on a plasmid to improve the cadmium accumulation capacity (Chaturvedi and Archana, 2014). A nickel/cobalt transporter gene from Rhodopseudomonas palustris or Novosphingobium aromaticivorans was expressed on plasmids to improve the efficiency of radioactive cobalt elimination from radioactively contaminated water (Gogada et al., 2015). Another example involves the expression of a divalent mercury ion reduction gene derived from E. coli a plasmid using Deinococcus geothermalis as a host; divalent mercury ions were reduced to less toxic and volatile metallic mercury (Brim et al., 2003).
Plasmid vectors for foreign gene expression using D. radiodurans or D. geothermalis as hosts were limited to D. radiodurans-E. coli shuttle vector pI3 and its derived plasmids, which were developed using replicons of plasmid pUE10 from the D. radiodurans strain Sark and an E. coli vector (Masters and Minton, 1992; Meima and Lidstrom, 2000). It has been reported that pI3-based plasmids are also replicate in Deinococcus deserti (Dulermo et al., 2009).
D. grandis is a radioresistant bacterium isolated from carp intestines in Hino, Japan. A host vector system has been developed for this bacterium (Satoh et al., 2009). In the D. grandis-E. coli shuttle vector pZT23 and its derived plasmids, the replicon of the cryptic plasmid pUE30 from Deinococcus radiopugnans functions in D. grandis. To investigate a specific gene function in D. grandis using the developed host-vector system, a plasmid complementation study was conducted using a host strain of D. grandis with the deletion of the rodZ gene (Morita et al., 2019). However, examples of plasmids with replicons other than pUE30 replicating in D. grandis have not been reported.
Unlike D. radiodurans, D. grandis has the rod-shape related genes, mreBC and rodAZ, and exhibits a rod-shaped morphology (Morita and Nishida, 2018). MreB, the bacterial ancestor of eukaryotic actin, functions as a scaffold for the assembly of cell wall synthesis machinery (van den Ent et al., 2001; Pande et al., 2022). MreC, a membrane-spanning protein with a single transmembrane domain, is required for correct localization of the MreB filament (Kruse et al., 2005). RodA is an integral membrane protein that is involved in the translocation of the lipid II peptidoglycan precursors across the cytoplasmic membrane (den Blaauwen et al., 2008; Sieger et al., 2013). RodZ is a transmembrane protein that directly interacts with the bacterial tubulin homolog FtsZ, and recruits MreB to the divisome (Ago and Shiomi, 2019). In general, rod-shaped microbes have a larger surface area-to-volume ratio than cocci (Harris and Theriot, 2018), giving them an advantage as hosts for bioremediation. D. grandis is also known to form enlarged spheroplasts with large periplasmic spaces under certain culture conditions, and these enlarged spheroplasts fuse to form giant cells with multiple cytoplasms (Nishino et al., 2018, 2019;Morita et al., 2019; Nishino and Nishida, 2019). The large periplasmic spaces have attractive potential as reaction sites for bioremediation and production of valuable substances. This peculiar property motivated the development of a new host-vector system for D. grandis.
When DNA regions with the same sequence are present on multiple plasmids, homologous recombination reactions occur between plasmids at a high frequency because of the action of relevant repair proteins, complicating the stable maintenance of various plasmids within bacteria (Masters et al., 1991; Daly et al., 1994). In addition, the DNA fragment size that can be stably incorporated into a plasmid vector is generally limited to approximately 10 kb (Leonardo and Sedivy, 1990; Siguret et al., 1994). Hence, expression plasmids that incorporate large DNA fragments are considered unstable in host cells. Therefore, when transforming a host with multiple genes, it is desirable to use multiple plasmid vectors. However, the repertoire of known plasmid vectors available for D. grandis was limited (Satoh et al., 2009).
Deinococcus grandis ATCC43672T and D. radiodurans ATCC13939T were purchased from the American Type Culture Collection. E. coli strain JM109 was purchased from Takara Bio. Inc. (Shiga, Japan) (Table 1). D. grandis and D. radiodurans were grown in tryptone glucose yeast (TGY) broth containing 0.5% Bacto Tryptone (BD, NJ, United States), 0.1% glucose, 0.3% Bacto Yeast Extract (BD), or on TGY agar supplemented with 1.5% Bacto Agar (BD) at 30°C, unless otherwise mentioned. E. coli was grown in LB broth-Lennox or LB Agar-Lennox (BD) at 37°C. The following antibiotics were added as needed: for D. grandis, chloramphenicol (3 μg/mL), hygromycin B (50 μg/mL), streptomycin (2 μg/mL); for E. coli, chloramphenicol (15 μg/mL), hygromycin B (100 μg/mL), streptomycin (17.5 μg/mL) or spectinomycin (100 μg/mL).
Table 1. Strains and plasmids used in this study.
Genomic DNA was extracted from D. grandis and D. radiodurans using a FastDNA SPIN Kit (MP Biomedicals, CA, United States) and a FastPrep 24 Instrument Version 4 (MP Biomedicals). PCR was performed using Tks Gflex DNA polymerase (Takara Bio. Inc.) or AmpliTaq Gold 360 DNA polymerase (Thermo Fisher Scientific, MA, United States). PCR was performed as per manufacturer’s instructions. Oligonucleotide primers used in this study are listed in Supplementary Table 1. PCR products were purified using a Gel/PCR Extraction Kit (Nippon Genetics Co., Ltd., Tokyo, Japan).
A 263-bp DNA fragment containing the promoter for the DNA damage response repressor-encoding ddrO gene was PCR-amplified using D. grandis genomic DNA as a template and treated with KpnI and NcoI. In addition, using the pNL1.1[Nluc] vector (Promega, WI, United States) as a template, a 564-bp DNA fragment of the luciferase gene from deep sea shrimp Oplophorus gracilirostris was PCR-amplified and treated with NcoI and XhoI. These were ligated to the KpnI-XhoI site of pKatAAD2 to construct plasmid pRNKAAD. Fragments upstream and downstream of the DEIGR_500008 gene were PCR-amplified using D. grandis genomic DNA as a template (769 bp and 770 bp, respectively); they were mixed and treated with the restriction enzymes EcoRI, KpnI, PstI, and HindIII. In addition, a PCR-amplified fragment of 3,025 bp containing the Nluc and streptomycin resistance gene (aad), using the plasmid pRNKAAD as a template, was treated with KpnI and PstI. These three treated fragments were ligated to the EcoRI-HindIII site of pUC19 to construct the plasmid pΔ500008LA (Supplementary Figure 1). Plasmids used in this study are listed in Table 1.
To generate D. grandis strain TY1, wild-type strain was incubated at 40°C for 48 h with shaking and then spread on TGY agar. Ten colonies formed after incubation at 30°C for 48 h were randomly selected, inoculated into TGY broth, and incubated at 30°C for 24 h.
To generate D. grandis strain TY3, wild-type strain was transformed with PCR products that were amplified with primers pKat-FP2 and pKat-RP (Supplementary Table 1) using pΔ500008LA as a template to generate strain TY1Nluc. The strain was cultured in TGY broth supplemented with 5 μg/mL rifampicin at 40°C for 48 h with shaking, diluted 100-fold, inoculated in the same medium, and cultured at 40°C for 72 h with shaking. The culture was then spread on TGY agar and incubated at 30°C for 48 h. Luciferase-nonproductive and streptomycin-sensitive colonies were selected from TGY agar, inoculated into TGY broth, and incubated at 30°C for 24 h.
Using the E. coli vector pACYC184 (Chang and Cohen, 1978) as a template, a 933-bp DNA fragment containing the p15A replicon was PCR-amplified and treated with SphI and KpnI. Plasmid pKatAAD2 (Satoh et al., 2006) was also treated with SphI and KpnI to obtain a 981-bp DNA fragment. These DNA fragments were ligated to yield the plasmid pKatAAD2L. A 2,022-bp DNA fragment containing the replicon of plasmid pDEGR-3 of D. grandis was then PCR-amplified, treated with KpnI, and ligated to the KpnI site of pKatAAD2 to construct plasmid pGRC5 (Supplementary Figure 2).
A 2,109-bp DNA fragment of the shuttle vector pZT29 (Satoh et al., 2009), which contained the replicon of D. radiopugnans plasmid pUE30 and that of E. coli vector pUC19 treated with KpnI, was ligated to a 2,492-bp DNA fragment of plasmid pKatHPH4 (Satoh et al., 2006) treated with KpnI, to construct plasmid pZT29H. In this plasmid, the chloramphenicol resistance gene, a marker gene of pZT29, was replaced with a hygromycin resistance gene (Supplementary Figure 3).
Using plasmid pRADN1 (Ohba et al., 2005) as a template, a 3,164-bp DNA fragment containing the pUE10 replicon was PCR-amplified and treated with XhoI and EcoRV. The plasmid pKatCAT5 (Satoh et al., 2009) was treated with XhoI and EcoRV. These fragments were ligated to construct a plasmid pRADN7. Using the E. coli vector pGBM5 as a template, a 1,997-bp fragment containing the replicon was then PCR-amplified and treated with XhoI and HindIII. Plasmid pRADN7 was treated with XhoI and HindIII to obtain a 4,063-bp DNA fragment. These DNA fragments were ligated to construct a plasmid pRADN8 (Supplementary Figure 4).
Escherichia coli cells were transformed with plasmids using an ECM 399 Electroporation System (BTX, MA, United States). D. grandis was transformed with plasmids using the calcium chloride method based on previous studies. Streptomycin was used as a selection marker for the pGRC5-transformants, chloramphenicol for the pRADN8-transformants, and hygromycin B for the pZT29H-transformants.
Plasmids from the E. coli transformants were extracted as per the standard protocol of the FastGene Plasmid Mini Kit (Nippon Genetics). Transformant cultures of D. grandis (20 mL) were collected via centrifugation and resuspended in buffer mP1 (Nippon Genetics) containing 10 mg/mL lysozyme. After incubating the suspension at 37°C for 30 min, plasmid DNA was extracted using the FastGene Plasmid Mini Kit, as per manufacturer’s instructions.
Quantitative PCR (qPCR) was performed to determine the copy number of shuttle vectors in D. grandis transformants essentially as described (Lee et al., 2006). Total DNA was extracted using the SPINeasy DNA Kit for Microbiome (MP Biomedicals). dnaA was selected as a target gene on the D. grandis chromosome. A 101-bp dnaA fragment was PCR-amplified and ligated with TA-cloning vector pGEM-T (Promega) to yield pDgra-dnaA. This plasmid was used to produce standard curves for the dnaA target. pDEGR-3 rep, pUE10 rep, and pUE30 rep were selected as targets for pGRC5, pRADN8, and pZT29H, respectively. Plasmid copy number was calculated as the ratio of quantified rep to dnaA. Oligonucleotide primers for qPCR analysis are listed in Supplementary Table 1. qPCR was carried out using a QuantStudio 1 Real-time PCR System (ThermoFisher Scientific) with PowerTrack SYBR Green Master Mix (ThermoFisher Scientific) according to the manufacturer’s instructions. The final reaction volume was 20 μL. Standard cycling mode was taken as thermal protocol. A tenfold serial dilution series of plasmids containing dnaA or rep was prepared ranging from 1 × 10−5 to 1 × 10−9 ng/reaction and standard curves displaying the cycle threshold parameter (Ct) values plotted against the log of the initial DNA concentration were generated. Plasmid copies were calculated based on the amplicon size of the target genes and the weight of 1 bp (1.095 × 10−12 ng). The relative copy numbers of dnaA and rep genes in samples were determined from qPCR analysis using extracted total DNA with reference to the standard curves.
Plasmid copy number of pRAND8 in D. radiodurans was also determined using qPCR. In this case, a 118-bp D. radiodurans dnaA fragment was PCR-amplified and ligated with TA-cloning vector pGEM-T to yield pDrad-dnaA, and this plasmid was used to produce standard curves for the dnaA target.
Using plasmid pZT90 (Satoh et al., 2009) as a template, a 152-bp fragment containing the D. radiodurans groES minimal promoter region was PCR-amplified and treated with KpnI and NcoI. This DNA fragment was inserted into the KpnI-NcoI site of pRNKAAD to construct pGNKAAD (Supplementary Figure 5). Next, three plasmids were constructed to evaluate the plasmid stability in the transformants without selection pressure. (1) A 669-bp fragment (groEp-Nluc cassette) containing the D. radiodurans groES minimal promoter and deep sea shrimp luciferase gene amplified using pGNKAAD as a template was treated with BamHI and PstI and inserted into the SalI-HindIII site of the shuttle vector pGRC5 to yield the luciferase gene expression plasmid pGRC5GN. (2) The PCR-amplified groEp-Nluc cassette (667 bp) using pGNKAAD as a template was treated with SalI and HindIII and inserted into the SalI-HindIII site of shuttle vector pRADN8 to yield pRADN8GN. (3) The PCR-amplified groEp-Nluc cassette (669 bp), using pGNKAAD as a template, was treated with BamHI and XbaI and inserted into the BamHI-XbaI site of shuttle vector pZT29H to yield pZT29HGN (Supplementary Figure 6).
Evaluation of plasmid stability was performed as described previously (Satoh et al., 2009). D. grandis transformants (strain TY3 carrying pGRC5GN, pRADN8, or pZT29HGN) were cultivated for 24 h in TGY broth supplemented with antibiotics. Cultures were diluted 4,096-fold in TGY broth without antibiotic addition and cultivated for 24 h. Following cell division through 12 generations, cultures were again diluted 4,096-fold in TGY broth. This procedure was repeated until cell division reached 48 generations. At each dilution step, ten milliliters of the culture were transferred to flat-bottomed microplates with 5 mm diameter, and luciferase activity was determined using a Nano-Glo Luciferase Assay System (Promega). Chemiluminescence signals were captured using a LumiCube Chemiluminescence Imaging System (Liponics, Tokyo, Japan) with the Digital Photo Professional 4 software (ver. 4.10.40, CANON) for 5 s at the optimal ISO sensitivity and signal intensities were measured using the JustTLC software (ver. 4.6.3, Sweday, Stockholm, Sweden). Relative chemiluminescence intensity was calculated by setting the chemiluminescence intensity at the 0 generation to 1.
A 263-bp promoter fragment for the DNA damage response repressor-encoding ddrO gene was PCR-amplified using D. grandis genomic DNA and treated with XhoI and NcoI. In addition, a 564-bp fragment of the deep sea shrimp luciferase gene was PCR-amplified using the pNL1.1[Nluc] vector (Promega) and treated with NcoI and HindIII. These were ligated to the XhoI-HindIII site of pGRC5 to construct the plasmid pRDR-Nluc. A PCR product containing the D. radiodurans groES minimal promoter and its downstream region containing the firefly luciferase gene luc + (groEp-luc+) cloned into pZTGL93 (Satoh et al., 2009) was treated with EcoRI and SacI, and then inserted into the EcoRI-SacI site of pRDR-Nluc to yield the dual-luciferase gene expression plasmid pRDR-NlucD (Supplementary Figure 7). This plasmid was used to transform the D. grandis strain TY3 carrying plasmid pZT29H.
The culture of D. grandis transformants (strain TY3 carrying pRDR-NlucD and pZT29H; 1 mL) grown in TGY broth supplemented with streptomycin and hygromycin for 18 h was washed with 1 mL of 10 mM sodium phosphate buffer (pH 7.0) and resuspended in 0.4 mL of the same buffer. The suspensions were transferred to a 35-mm diameter polystyrene dish (Model 3,000–035, AGC Techno Glass Co., Ltd., Shizuoka, Japan) and irradiated at room temperature at constant UV irradiation of 4.2 J/m2/s using four UV lamps. To 0.1 mL of the irradiated suspension, a 25-fold volume of TGY broth supplemented with streptomycin and hygromycin was added and incubated at 30°C for 4.5 h. Every 30 min, 10 μL of the culture was withdrawn and luciferase activity was measured using the Nano-Glo Dual-Luciferase Reporter Assay System (Promega), as described in Section 2.8. Relative reporter activity was calculated as the ratio of chemiluminescence from the experimental reporter (Nluc) to that from the control reporter (luc+). Normalized relative reporter activity was calculated via setting the relative reporter activity at 0 min for each dose to 1.
The data of the plasmid copy number estimation in Section 2.7 was reported as an average of four qPCR assays. The data of the relative chemiluminescence intensity in Section 2.8 was presented as mean ± standard error (n = 9). The data of the normalized relative reporter activity in Section 2.9 was presented as mean ± standard error (n = 3).
Various methods have been developed to delete plasmids from microorganisms (Trevors, 1986; Spengler et al., 2006). In this study, we attempted to culture D. grandis at 40°C, the upper permissive temperature limit for D. grandis, as describes in Section 2.4. The presence of the plasmid pDEGR-3 in bacteria was confirmed using agarose gel electrophoresis. The plasmid was detected in all isolates regardless of incubation at 40°C (Supplementary Figure 8).
Figure 1 shows the results of agarose gel electrophoresis for the presence of PCR products derived from the glutamine synthetase gene (glnN) located on the chromosome of D. grandis, DEIGR_400031, DEIGR_400086, and DEIGR_400127 genes located in pDEGR-PL, and the DEIGR_500007 gene located in pDEGR-3. PCR fragments using primer pairs to amplify the glnN gene (Figure 1, lanes 1 and 6) and genes located at pDEGR-3 (Figure 1, lanes 5 and 10) were detected in both bacterial isolates that were grown at 40°C and those at 30°C. In contrast, three PCR fragments using primer pairs that amplified different DNA regions of pDEGR-PL were detected in the control genome extracted from the bacterial isolate cultured at 30°C (Figure 1, lanes 2–4) but not in that cultured at 40°C (Figure 1, lanes 7–9). Upon culturing at 40°C, the bacterial isolate lost the plasmid pDEGR-PL but retained pDEGR-3, leading to its designation as D. grandis strain TY1.
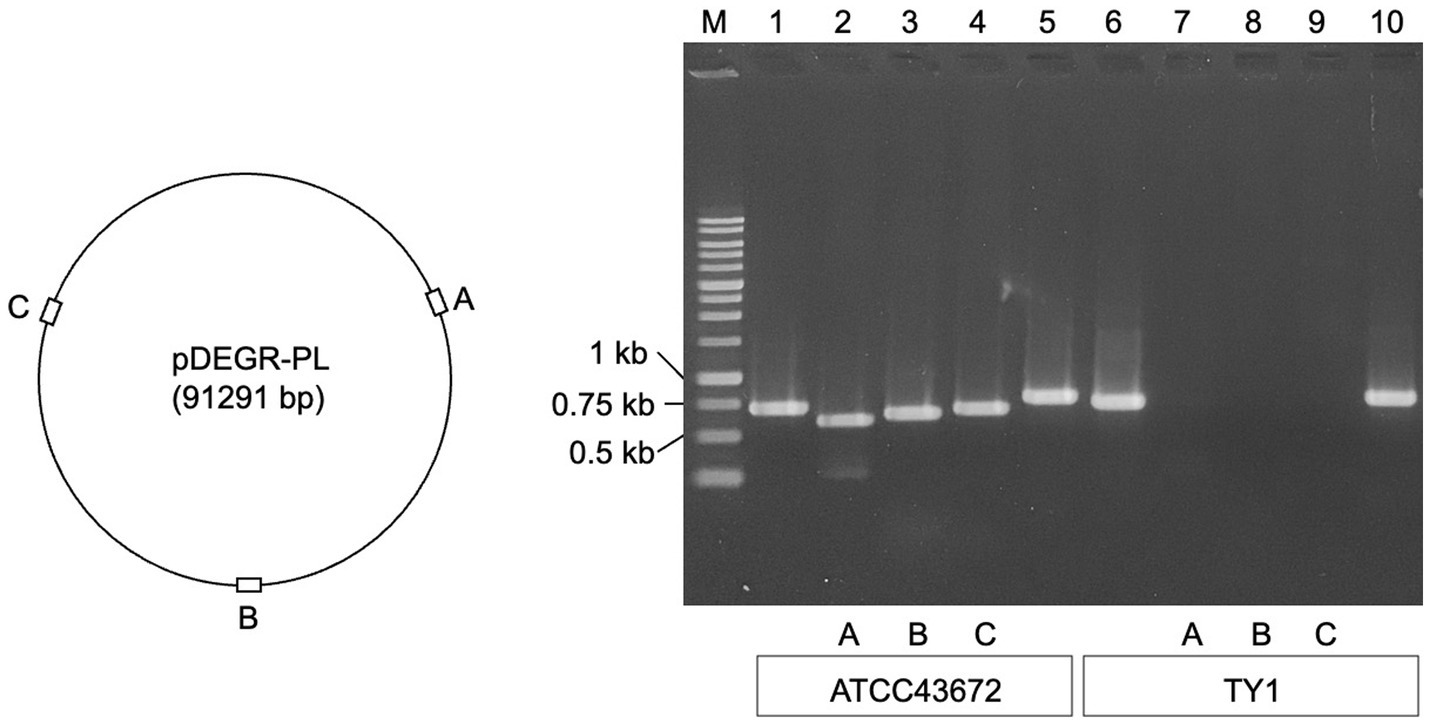
Figure 1. Confirmation of cryptic plasmids pDEGR-PL and pDEGR-3 in D. grandis. (Left) Relative locations of DEIGR_400031 (A), DEIGR_400086 (B), and DEIGR_400127 (C) in pDEGR-PL. (Right) PCR product detection using agarose gel electrophoresis. M, 1 kb ladder marker (Nippon Genetics Co., Ltd.); lanes 1 and 6, PCR products amplifying the glnN gene located on D. grandis chromosome; lanes 2 and 7, PCR products amplifying DEIGR_400031 (A); lanes 3 and 8, PCR products amplifying DEIGR_400086 (B); lanes 4 and 9, PCR products amplifying DEIGR_400127 (C); lanes 5 and 10, PCR products amplifying DEIGR_500007 located on pDEGR-3. In lanes 1 to 5, genomic DNA extracted from ATCC43672 (wild type) was used as the PCR template; in lanes 6 to 10, genomic DNA extracted from D. grandis strain TY1 was used as the PCR template. The oligonucleotide primer sets used are listed in Supplementary Table 1.
In addition to high-temperature incubation, rifampicin can eliminate plasmids from microorganisms (Johnston and Richmond, 1970; McHugh and Swartz, 1977). In this experiment, the addition of these chemical agents to the culture medium was combined with incubation at high temperature. To facilitate selecting pDEGR-3 deletion strains from the D. grandis TY1 strain, the DEIGR_500008 gene present in pDEGR-3 was replaced with a luciferase gene from deep sea shrimp and a streptomycin resistance gene to generate the D. grandis strain TY1Nluc (Section 2.4). Then, luciferase-nonproductive and streptomycin-sensitive colonies were selected as described in Section 2.4. The results of agarose gel electrophoresis are shown in Supplementary Figure 9. This result indicates that the bacterial isolate lacked pDEGR-3Δ8LA of 8,961 bp that was present in strain TY1Nluc and designated as D. grandis strain TY3.
Figure 2 shows the genetic organization of the D. grandis cryptic plasmid, pDEGR-3. The plasmid contained nine putative open reading frames, of which the protein encoded by DEIGR_500002 (Accession No. GAQ24009) was considered to be the pDEGR −3 replication initiation protein for the following reasons: Figure 3 shows a comparison of this protein with the replication initiation protein of the cryptic plasmid pUE30 from D. radiopugnans (Accession No. BAH03365), and that of cryptic plasmid pUE10 of D. radiodurans strain Sark (Accession No. AAF44040). The amino acid identity between the replication initiation proteins of pDEGR-3 and pUE30 was 31.7%, and that between the replication initiation proteins of pDEGR-3 and pUE10 was 26.3%. The GC% of a 139-bp non-coding region between DEIGR_500009 (methyltransferase) and DEIGR_500001 (hypothetical protein) was 35.5% and more AT-rich than other plasmid regions because the GC% of the entire pDEGR-3 was 64.9%. The AT-rich region is considered a putative replication origin (Figure 2). Therefore, in this study, the DNA region containing the AT-rich region and DEIGR_500002 was PCR-amplified as a replicon of pDEGR-3 and used to construct a shuttle vector.
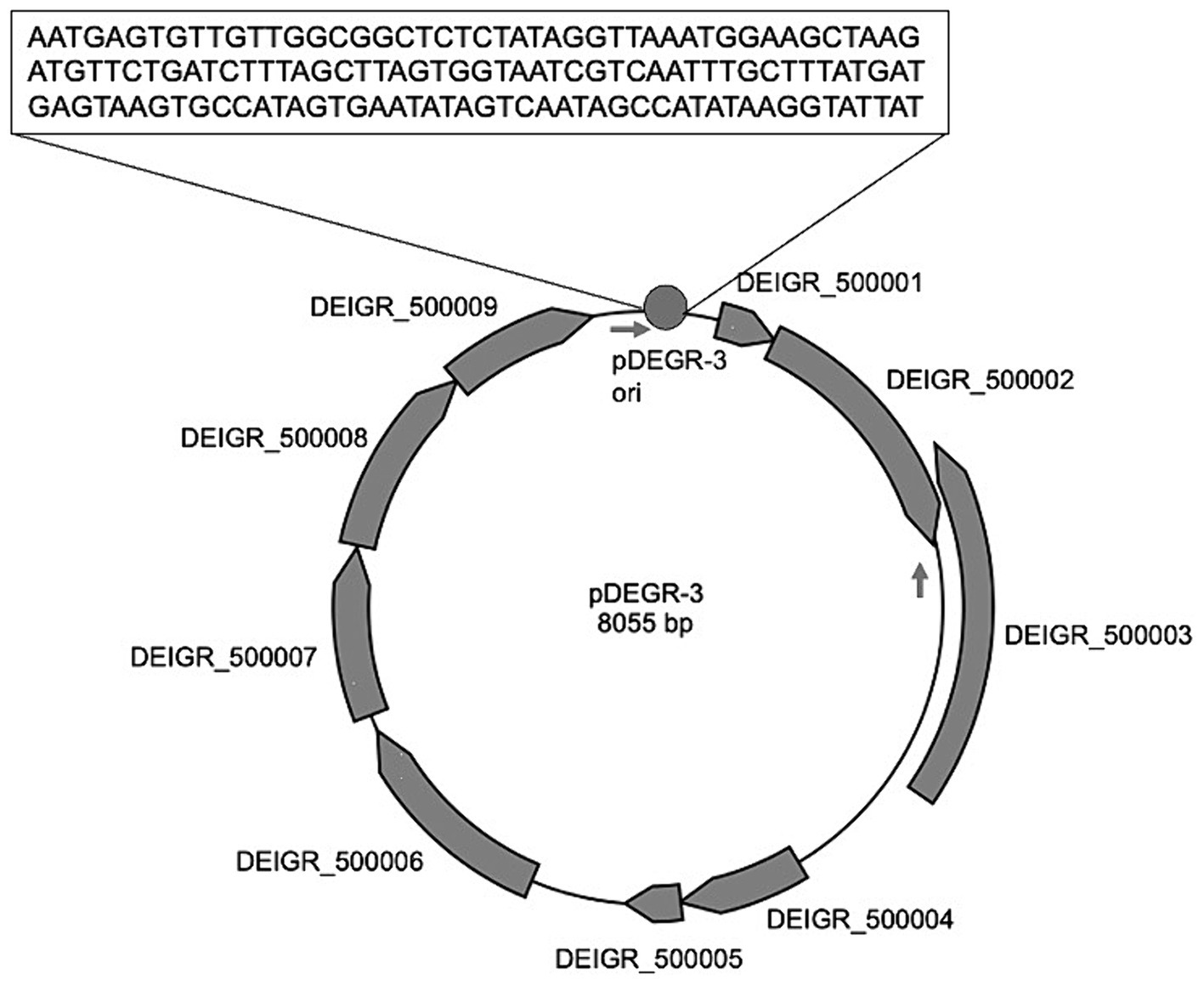
Figure 2. Genetic organization of pDEGR-3. Nine potential open reading frames in pDEGR-3 are depicted as gray boxes; PCR primer (DEGR3F1Kpn and DEGR3R1Kpn) positions for pDEGR-3 replicon amplification are displayed by arrows. Putative pDEGR-3 replication origin is represented as a gray circle. The 139-bp DNA sequence of the putative pDEGR-3 replication origin that shows low %G + C (35%) is presented in a box.

Figure 3. Maximum matching of amino acid sequences of putative replication initiation proteins encoded by two different cryptic plasmids. The upper sequence is from D. grandis cryptic plasmid pDEGR-3 and the lower sequences are from different cryptic plasmids of D. radiopugnans (A) and D. radiodurans Sark (B). Sequence alignment gaps are indicated by dashes and the numbers represent coordinates of each protein. Asterisks indicate conserved amino acid residues between the two proteins. Maximum matching was determined using the GENETYX-MAC software version 22.0.1 (GENETYX Co., Ltd., Tokyo, Japan).
The E. coli-D. grandis shuttle vector pGRC5 was constructed as follows: First, plasmid pKatAAD2L was constructed via ligating plasmid pKatAAD2, which expressed a streptomycin resistance gene (aad) controlled by the D. radiodurans catalase gene promoter (katp), with the replicon of the E. coli vector pACYC184. A DNA fragment containing the replicon of pDEGR-3 of D. grandis was PCR-amplified and ligated to pKatAAD2L to yield pGRC5. The second E. coli-D. grandis shuttle vector, pZT29H, was constructed via changing the selection marker chloramphenicol resistance gene (cat) in the plasmid pZT29 to a hygromycin resistance gene (hyg). The third E. coli-D. grandis shuttle vector pRADN8 was constructed as follows: First, plasmid pKatCAT5, which could express a chloramphenicol resistance gene (cat) by the D. radiodurans catalase gene promoter (katp), and the replicon (pUE10 rep) of the D. radiodurans Sark strain plasmid pUE10 to yield pRAND7. The plasmid was ligated to E. coli vector pGBM5 replicon to yield pRADN8. Structures of the three shuttle vectors are shown in Figure 4, and their DNA sequence information is shown in Supplementary Figures 10–12. Plasmid profiles in D. grandis transformants carrying the three shuttle vectors are shown in Supplementary Figure 13. pGRC5 was mainly detected as a monomer in D. grandis. The dimer forms of pZT29H and pRADN8 were also detected (Supplementary Figure 13A). Each of the plasmids tested had a single XhoI digestion site (Figure 4); the size of the linear plasmids after XhoI digestion was in good agreement with the estimated size of monomers (Supplementary Figure 13B).

Figure 4. Structure of E. coli-D. grandis shuttle vectors. Plasmid pGRC5 consists of the p15A replicon (p15A ori) from E. coli vector pACYC184, the replication initiation protein-coding gene from D. grandis cryptic plasmid pDEGR-3 (pDEGR-3 rep), the putative pDEGR-3 replication origin (pDEGR-3 ori), and a streptomycin resistance gene (aad). The streptomycin resistance gene is controlled by the modified D. radiodurans catalase promoter (katp). pRADN8 contains the pSC101 replicon (pSC101 ori and rep) from E. coli vector pGBM5, the initiation protein-coding gene from D. radiodurans Sark cryptic plasmid pUE10 (pUE10 rep), the putative pUE10 replication origin (pUE10 ori) and a chloramphenicol resistance gene (cat). The chloramphenicol resistance gene is controlled by the modified D. radiodurans catalase promoter (katp). pZT29 contains the replicon of E. coli vector pUC19 (pUC19 ori), the initiation protein-coding gene from D. radiopugnans cryptic plasmid pUE30 (pUE30 rep), the putative pUE30 replication origin (pUE30 ori), and a hygromycin resistance gene (hyg). The hygromycin resistance gene is controlled by the modified D. radiodurans catalase promoter (katp).
Relative plasmid copy number was estimated using qPCR assay as described in Section 2.7. The results showed that the relative plasmid copy numbers of pGRC5, pZT29H, and pRADN8 in D. grandis transformants were 11, 26, and 5 per genome, respectively (Supplementary Table 2).
In D. grandis, DNA fragments (groEp-Nluc cassette) with the deep sea shrimp luciferase gene placed downstream of D. radiodurans groES minimum promoter were inserted into pGRC5, pZT29H, and pRADN8 to express foreign genes and construct expression plasmids pGRC5GN, pZT29HGN, and pRADN8GN, as described in Section 2.8. The expression plasmid profiles in D. grandis are shown in Supplementary Figure 14. In lane 1, a band was observed at 4,545 bp derived from pGRC5GN, indicating that the plasmid with pDEGR-3 replicon was present only as a monomer in D. grandis strain TY3, similar to the results for pGRC5 shown in Supplementary Figure 13A. In lane 2, bands were observed at 5,250 bp derived from pZT29HGN monomer and 10,500 bp derived from pZT29H dimer. Lane 3 shows a band at 6,683 bp derived from the pRADN8GN monomer and a larger band indicating the plasmid’s multimers. The size of the linear plasmids after XhoI digestion was in good agreement with the estimated size of monomers (Supplementary Figure6, 14B).
To determine whether the shuttle vectors could stably replicate under non-selective conditions, D. grandis transformants carrying pGRC5GN, pZT29HGN, and pRADN8GN were grown in non-selective broth until 48 generations by repeated dilution and incubation, and relative chemiluminescence intensity was determined as described in Section 2.8. As shown in Figure 5, D. grandis transformants carrying pGRC5GN exhibited constant luminescence intensity until 48 generations, whereas the luminescence intensity of D. grandis transformants carrying pZT29HGN or pRADN8GN decreased as the generation progressed. This result was in good agreement with the analysis of plasmid retention in culture under non-selective conditions using agarose gel electrophoresis (Supplementary Figure 14B).
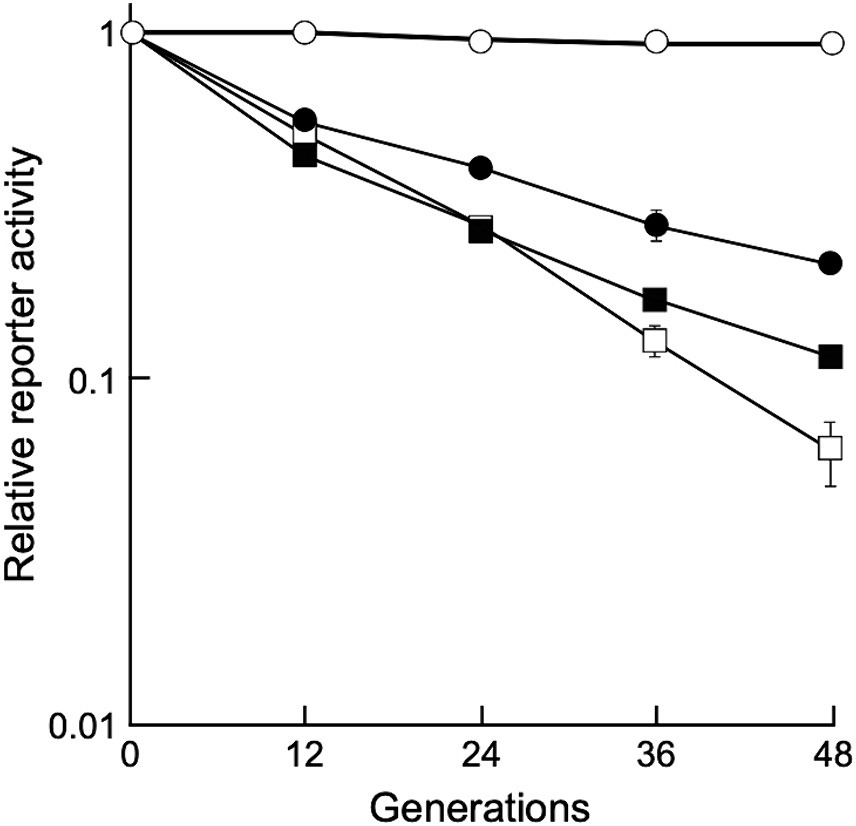
Figure 5. Stability of shuttle vectors under non-selective conditions. Values represent the relative reporter activity at the 0 generation as 1 (mean ± standard error, n = 9). Symbols: open circles, D. grandis strain TY3 carrying pGRC5GN; open squares, D. grandis strain TY3 carrying pZT29HGN; closed circles, D. grandis strain TY3 carrying pRADN8GN; closed squares, D. radiodurans strain ATCC13939 carrying pRADN8GN.
To determine gene expression induction via UV irradiation in D. grandis, a dual-luciferase reporter plasmid, pRDR-NlucD, was constructed as described in Section 2.9. This plasmid is based on shuttle vector pGRC5. It contains the deep sea shrimp luciferase gene (Nluc) with D. grandis ddrO promoter (ddrOp) and firefly luciferase gene (luc+) with the D. radiodurans groES minimal promoter (groEp). This plasmid was introduced into TY3, which already contained pZT29H, and luciferase activity was monitored after UV irradiation. Figure 6 shows the normalized relative reporter activity in the pRDR-NlucR-transformants exposed to different UV-C doses. When irradiated with 20 J/m2 of UV-C, the maximum value (approximately 1.5 times the value immediately after irradiation) was observed 1 h post-irradiation incubation; at 50 J/m2, (approximately 4.9 times the value immediately after irradiation) 2 h post-irradiation incubation; at 150 J/m2, (approximately 9.8 times the value immediately after irradiation) 3.5 h post-irradiation incubation (Figure 6). Thus, using pRDR-NlucD enabled gene expression induction in TY3 via UV irradiation and revealed the irradiation dose dependence of gene expression levels.
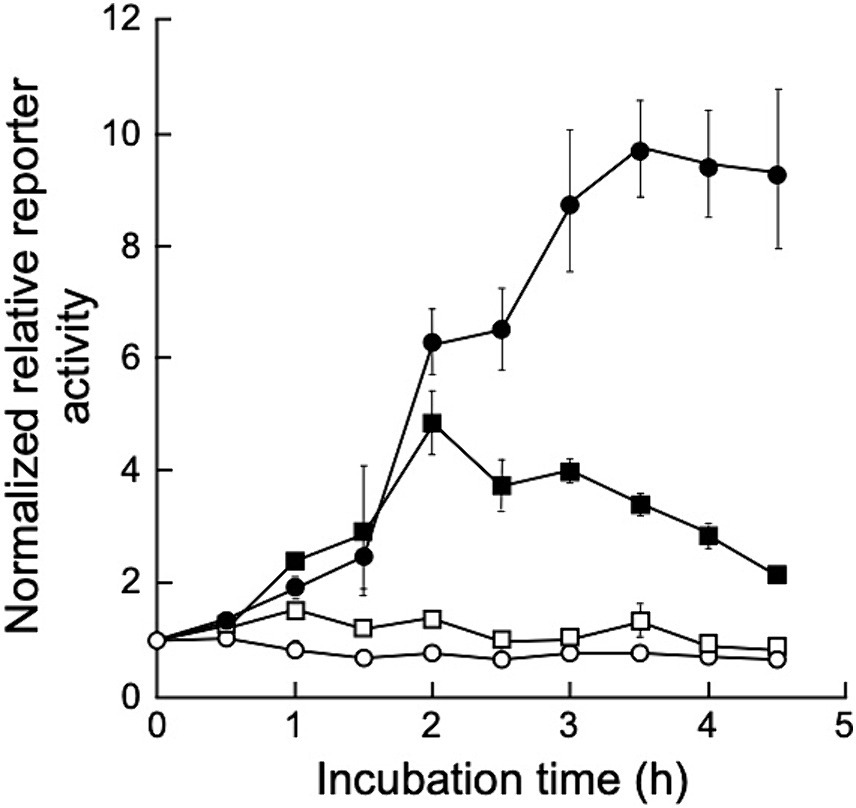
Figure 6. Normalized relative reporter activity in pRDR-NlucR-transformants exposed to different UV-C doses. Values represent the relative reporter activity normalized to activity at the 0-time point for each dose as 1 (mean ± standard error, n = 3). Symbols: open circles, 0 J/m2; open squares, 20 J/m2; closed squares, 50 J/m2; closed circles, 150 J/m2.
We attempted to introduce the newly developed shuttle vector into D. radiodurans. As expected, D. radiodurans transformants were generated with pRADN8. Relative plasmid copy numbers of pRADN8 in D. radiodurans transformants were 15 per genome (Supplementary Table 2). D. radiodurans transformants carrying pRADN8GN exhibited approximately 15.5% of the initial luminescence intensity at the 48 generation (Figure 5). Similar to previous studies, we were unable to generate D. radiodurans transformants using pUE30-based shuttle vector pZT29H (Satoh et al., 2009). Likewise, D. radiodurans transformants could not be generated using pGRC5.
We previously analyzed the draft and whole genome sequences of D. grandis (Satoh et al., 2016; Shibai et al., 2019); both are available in public genome databases (GenBank Assembly ID: GCA_001485435.1 and GCA_009177165.1). Table 2 summarizes results of the two analyses. Four circular and three linear contigs were registered in the draft genome analysis; four circular DNAs were registered in the whole genome sequence analysis: chromosome, pDEGR-1, pDEGR-2, and pDEGR-3. Next-generation sequencing analyses were performed using genomic DNA extracted from the D. grandis type strain. However, whole genome sequence analysis did not detect one of the circular contigs (BCMS01000006) found in the draft genome analysis. Previous studies have not described a specific name for this plasmid; hence, it was referred to as pDEGR-PL. The laboratory stock of the D. grandis type strain used in draft genome sequencing was used in this study.
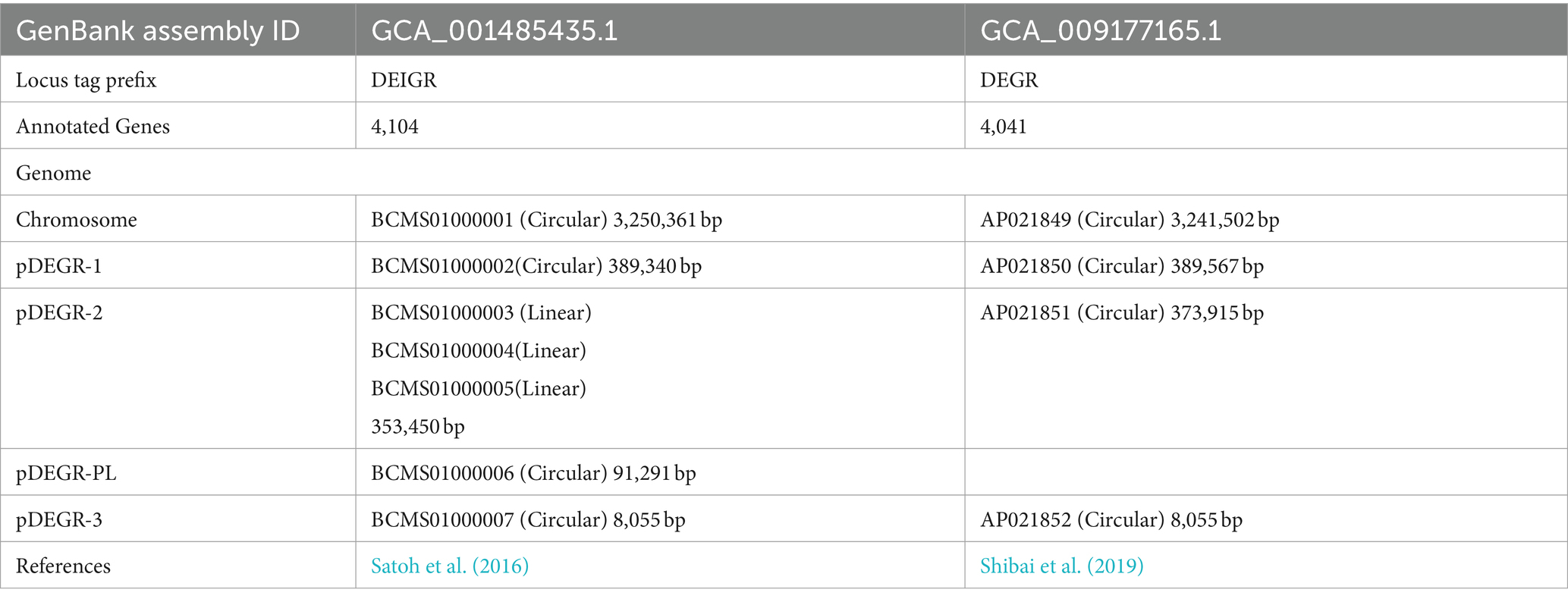
Table 2. Summary of D. grandis genome analysis.
In this study, we generated two strains: TY1 that lacked pDEGR-PL, and TY3 that lacked both pDEGR-PL and pDEGR-3. Plasmid pDEGR-PL was deleted via cultivation at 40°C, the upper limit of the permissible temperature, whereas pDEGR-3 was stable against heat treatment. Plasmid pDEGR-PL was undetected via whole genome sequencing in a previous study (Shibai et al., 2019). As the largest coding DNA sequence of the undetected circular contig was similar to that of the phage tail protein, this circular DNA was assumed to be a mobile genetic factor and thus lost prior to whole-genome sequencing. In this study, pDEGR-PL was believed to be easily cured from the D. grandis type strain owing to the instability of the plasmid against heat. Thus, pDEGR-PL may have been transferred from other psychrophiles to D. grandis.
When multiple plasmid vectors are used, homologous recombination reactions between them must also be suppressed. To overcome this, we constructed a shuttle plasmid as a replication initiation region of three plasmids from E. coli with different nucleotide sequences and three plasmids from Deinococcus spp. ligated together, thereby avoiding plasmid instability owing to nucleotide sequence homology. The replication initiation regions of the three plasmids from E. coli used in this study are known to coexist without interfering with each other (Bartolome et al., 1991; Manen et al., 1997). However, the compatibility of replication initiation regions of the three plasmids from Deinococcus spp. is unclear. In this study, we experimentally demonstrated that, despite the limited homology of these replication initiation proteins (Figure 3), the plasmids did not interfere with each other and could stably coexist in D. grandis (Supplementary Figure 13).
Each shuttle vector carries a different antibiotic resistance gene as a selection marker, which ensures the plasmid presence in the bacteria in a selective culture medium, extraction and purification of plasmids from the bacteria, and introduction of plasmids into the bacteria via transformation. The antibiotic resistance genes in the three shuttle vectors are regulated by the D. radiodurans catalase gene promoter (katp), and this promoter region (123 bp) is the same sequence in the three shuttle vectors. Because homologous recombination between different shuttle vectors did not occur under the experimental conditions, homologous recombination in D. grandis is thought to require a longer homologous DNA size. In a previous study, D. grandis could not be transformed with the pUE10-based shuttle vector pRADN1 (Satoh et al., 2009); however, in the present study, D. grandis was transformed with pRADN8. This difference is because of the different plasmid DNA sequences, and likely depends on the presence or absence of plasmid DNA cleavage by the restriction enzymes present in D. grandis.
pGRC5, pZT29H, and pRADN8 have pUC19-derived multiple cloning sites upstream and downstream of the antibiotic marker gene, and some of these restriction sites can be used as cloning sites. However, in the process of constructing the shuttle vector, additional recognition sites were created at other sites in the plasmid, making some of the restriction sites in the multiple cloning sites unusable as cloning sites. By appropriately modifying the nucleotide sequence of the newly developed shuttle vectors, the unusable cloning sites can be restored in the future.
Among the three plasmids, the copy number per genome for pRADN8 was 5. The propensity of pRADN8 and its derived plasmids to form multimeric forms in D. grandis may be related to its low copy numbers (Field and Summers, 2011; Crozat et al., 2014). The pUE10-based shuttle vector pRAD1 was shown to be unretained and shed by D. radiodurans under culture conditions with no selection pressure (Meima and Lidstrom, 2000). Plasmid pRADN8 is presumed to be unstably retained by D. grandis under culture conditions without selection pressure. In this study, we determined plasmid stability in D. grandis transformants carrying plasmids with the groEp-Nluc cassette (Figure 5). As expected, pGRC5 showed high stability in D. grandis strain TY3, whereas pRADN8 showed low stability.
pUE30-based pZT29H was retained in D. grandis, mainly as a monomer and partly as a dimer. The mode of presence of this plasmid was similar to that of pZT29 in a previous study (Satoh et al., 2009). The pUE30-based shuttle vector pZTGL93 that contains the D. radiodurans groES minimal promoter and firefly luciferase gene has been shown to be stably retained by D. grandis wild-type strain for 48 generations under culture conditions without selection pressure (Satoh et al., 2009). Unlike this, D. grandis strain TY3 transformants carrying pZT29HGN exhibited approximately 6% of the initial luminescence intensity at the 48 generation (Figure 5). We suggest that plasmids present in wild strain but absent in strain TY3, namely pDEGR-PL or pDEGR-3, may encode genes responsible for the stability of pUE30-based shuttle vectors.
In this study, we constructed pRDR-NlucD, in which the luciferase gene was controlled by the ddrO promoter in D. grandis. We showed that UV irradiation increased luciferase activity in pRDR-NlucD-transformants (Figure 6). The ddrO promoter region contains an operator sequence called the radiation desiccation-responsive motif (RDRM) that binds to the repressor DdrO protein and represses its gene expression. The DdrO protein is cleaved by the protease activity of the PprI protein activated by DNA damage stress, de-repressing the downstream gene expression (Ishino and Narumi, 2015). This DNA damage stress response mechanism by PprI and DdrO is highly conserved in several Deinococcus spp., including D. grandis, for which genome analysis has been completed (Lim et al., 2019). The dual-luciferase reporter plasmid pRDR-NlucD coexisted with another type of shuttle vector, pZT29H. In the future, cloning genes that affect the DdrO/PprI stress response mechanism into pZT29H could provide a tool to elucidate the molecular mechanisms of the DdrO/PprI-dependent stress response in D. grandis.
In addition to the ddrO promoter, promoters that are upstream of the pprA, ddrA, ddrB, and gyrB genes and have RDRM operator sequences in their vicinity can be used as promoters to induce the expression of foreign genes in D. grandis following UV irradiation (Narasimha and Basu, 2021). Their activity in foreign gene expression in D. grandis should be investigated in the future. Promoters of DR1261, rpmB, and dnaK have been reported to allow constitutively high expression of these genes in D. radiodurans (Chen et al., 2019). Therefore, these promoters should be tested in the future to determine the similarity of their effects on D. grandis. It will also be desirable to develop vectors for the expression of recombinant genes in the periplasm of enlarged D. grandis spheroplasts in the future.
Thus this study reports on the successful development of a novel system that stably introduced and retained expression plasmids for D. grandis. From the wild-type strain, two cryptic plasmids were eliminated to generate the strain TY3. Three shuttle vector plasmids, pGRC5, pRADN8, and pZT29H, were shown to coexist in D. grandis. These plasmids are expected to facilitate the removal of toxic materials and produce useful compounds in D. grandis.
The nucleotide sequences of pGRC5, pZT29H, and pRADN8 has been assigned in the DDBJ/EMBL-Bank/GenBank Accession Nos. LC801619, LC801620, and LC801621, respectively.
MS: Data curation, Investigation, Methodology, Writing – original draft, Writing – review & editing. TS: Investigation, Methodology, Writing – original draft, Writing – review & editing. KK: Investigation, Methodology, Writing – original draft, Writing – review & editing. MY: Conceptualization, Supervision, Writing – original draft, Writing – review & editing. IN: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Part of this research was supported by the Grant-in-Aid for Scientific Research (23 K05015) and the Toyo University Priority Research Promotion Program for Narumi, and by the Support for Pioneering Research Initiated by the Next Generation (SPRING) for MS.
The authors thank Aya Kubo for valuable comments on the manuscript drafts and the National Institute for Genetics, Japan for providing pGBM5. The authors also thank Editage (www.editage.jp) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1387296/full#supplementary-material
Ago, R., and Shiomi, D. (2019). RodZ: a key-player in cell elongation and cell division in Escherichia coli. AIMS Microbiol 5, 358–367. doi: 10.3934/microbiol.2019.4.358
Appukuttan, D., Rao, A. S., and Apte, S. K. (2006). Engineering of Deinococcus radiodurans R1 for bioprecipitation of uranium from dilute nuclear waste. Appl. Environ. Microbiol. 72, 7873–7878. doi: 10.1128/AEM.01362-06
Bartolome, B., Jubete, Y., Martinez, E., and de la Cruz, F. (1991). Construction and properties of a family of pACYC184-derived cloning vectors compatible with pBR322 and its derivatives. Gene 102, 75–78. doi: 10.1016/0378-1119(91)90541-i
Brim, H., McFarlan, S. C., Fredrickson, J. K., Minton, K. W., Zhai, M., Wackett, L. P., et al. (2000). Engineering Deinococcus radiodurans for metal remediation in radioactive mixed waste environments. Nat. Biotechnol. 18, 85–90. doi: 10.1038/71986
Brim, H., Venkateswaran, A., Kostandarithes, H. M., Fredrickson, J. K., and Daly, M. J. (2003). Engineering Deinococcus geothermalis for bioremediation of high-temperature radioactive waste environments. Appl. Environ. Microbiol. 69, 4575–4582. doi: 10.1128/AEM.69.8.4575-4582.2003
Chang, A. C., and Cohen, S. N. (1978). Construction and characterization of amplifiable multicopy DNA cloning vehicles derived from the P15A cryptic miniplasmid. J. Bacteriol. 134, 1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978
Chaturvedi, R., and Archana, G. (2014). Cytosolic expression of synthetic phytochelatin and bacterial metallothionein genes in Deinococcus radiodurans R1 for enhanced tolerance and bioaccumulation of cadmium. Biometals 27, 471–482. doi: 10.1007/s10534-014-9721-z
Chen, A., Sherman, M. W., Chu, C., Gonzalez, N., Patel, T., and Contreras, L. M. (2019). Discovery and characterization of native Deinococcus radiodurans promoters for tunable gene expression. Appl. Environ. Microbiol. 85:e01356-19. doi: 10.1128/AEM.01356-19
Cox, M. M., and Battista, J. R. (2005). Deinococcus radiodurans - the consummate survivor. Nat. Rev. Microbiol. 3, 882–892. doi: 10.1038/nrmicro1264
Crozat, E., Fournes, F., Cornet, F., Hallet, B., and Rousseau, P. (2014). Resolution of multimeric forms of circular plasmids and chromosomes. Microbiol Spectr. 2:PLAS-0025-2014. doi: 10.1128/microbiolspec.PLAS-0025-2014
Daly, M. J. (2023). The scientific revolution that unraveled the astonishing DNA repair capacity of the Deinococcaceae: 40 years on. Can. J. Microbiol. 69, 369–386. doi: 10.1139/cjm-2023-0059
Daly, M. J., Ling, O., and Minton, K. W. (1994). Interplasmidic recombination following irradiation of the radioresistant bacterium Deinococcus radiodurans. J. Bacteriol. 176, 7506–7515. doi: 10.1128/jb.176.24.7506-7515.1994
den Blaauwen, T., de Pedro, M., Nguyen-Distèche, M., and Ayala, J. A. (2008). Morphogenesis of rod-shaped sacculi. FEMS Microbiol. Rev. 32, 321–344. doi: 10.1111/j.1574-6976.2007.00090.x
Dulermo, R., Fochesato, S., Blanchard, L., and De Groot, A. (2009). Mutagenic lesion bypass and two functionally different RecA proteins in Deinococcus deserti. Mol. Microbiol. 74, 194–208. doi: 10.1111/j.1365-2958.2009.06861.x
Field, C. M., and Summers, D. K. (2011). Multicopy plasmid stability: revisiting the dimer catastrophe. J. Theor. Biol. 291, 119–127. doi: 10.1016/j.jtbi.2011.09.006
Gabani, P., and Singh, O. V. (2013). Radiation-resistant extremophiles and their potential in biotechnology and therapeutics. Appl. Microbiol. Biotechnol. 97, 993–1004. doi: 10.1007/s00253-012-4642-7
Gerber, E., Bernard, R., Castang, S., Chabot, N., Coze, F., Dreux-Zigha, A., et al. (2015). Deinococcus as new chassis for industrial biotechnology: biology, physiology and tools. J. Appl. Microbiol. 119, 1–10. doi: 10.1111/jam.12808
Gogada, R., Singh, S. S., Lunavat, S. K., Pamarthi, M. M., Rodrigue, A., Vadivelu, B., et al. (2015). Engineered Deinococcus radiodurans R1 with NiCoT genes for bioremoval of trace cobalt from spent decontamination solutions of nuclear power reactors. Appl. Microbiol. Biotechnol. 99, 9203–9213. doi: 10.1007/s00253-015-6761-4
Hansen, M. T. (1978). Multiplicity of genome equivalents in the radiation-resistant bacterium Micrococcus radiodurans. J. Bacteriol. 134, 71–75. doi: 10.1128/jb.134.1.71-75.1978
Harris, L. K., and Theriot, J. A. (2018). Surface area to volume ratio: a natural variable for bacterial morphogenesis. Trends Microbiol. 26, 815–832. doi: 10.1016/j.tim.2018.04.008
Harsojo,, Kitayama, S., and Matsuyama, A. (1981). Genome multiplicity and radiation resistance in Micrococcus radiodurans. J. Biochem. 90, 877–880. doi: 10.1093/oxfordjournals.jbchem.a133544
Ishino, Y., and Narumi, I. (2015). DNA repair in hyperthermophilic and hyperradioresistant microorganisms. Curr. Opin. Microbiol. 25, 103–112. doi: 10.1016/j.mib.2015.05.010
Ithurbide, S., Coste, G., Lisboa, J., Eugénie, N., Bentchikou, E., Bouthier de la Tour, C., et al. (2020). Natural transformation in Deinococcus radiodurans: a genetic analysis reveals the major roles of DprA, DdrB, RecA, RecF, and RecO proteins. Front. Microbiol. 11:1253. doi: 10.3389/fmicb.2020.01253
Johnston, J. H., and Richmond, M. H. (1970). The increased rate of loss of penicillinase plasmids from Staphylococcus aureus in the presence of rifampicin. J. Gen. Microbiol. 60, 137–139. doi: 10.1099/00221287-60-1-137
Kruse, T., Bork-Jensen, J., and Gerdes, K. (2005). The morphogenetic MreBCD proteins of Escherichia coli form an essential membrane-bound complex. Mol. Microbiol. 55, 78–89. doi: 10.1111/j.1365-2958.2004.04367.x
Lange, C. C., Wackett, L. P., Minton, K. W., and Daly, M. J. (1998). Engineering a recombinant Deinococcus radiodurans for organopollutant degradation in radioactive mixed waste environments. Nat. Biotechnol. 16, 929–933. doi: 10.1038/nbt1098-929
Lee, C., Kim, J., Shin, S. G., and Hwang, S. (2006). Absolute and relative QPCR quantification of plasmid copy number in Escherichia coli. J. Biotechnol. 123, 273–280. doi: 10.1016/j.jbiotec.2005.11.014
Leonardo, E. D., and Sedivy, J. M. (1990). A new vector for cloning large eukaryotic DNA segments in Escherichia coli. Biotechnology (N Y) 8, 841–844. doi: 10.1038/nbt0990-841
Li, S., Zhu, Q., Luo, J., Shu, Y., Guo, K., Xie, J., et al. (2021). Application Progress of Deinococcus radiodurans in biological treatment of radioactive uranium-containing wastewater. Indian J. Microbiol. 61, 417–426. doi: 10.1007/s12088-021-00969-9
Lim, S., Jung, J. H., Blanchard, L., and de Groot, A. (2019). Conservation and diversity of radiation and oxidative stress resistance mechanisms in Deinococcus species. FEMS Microbiol. Rev. 43, 19–52. doi: 10.1093/femsre/fuy037
Manen, D., Pougeon, M., Damay, P., and Geiselmann, J. (1997). A sensitive reporter gene system using bacterial luciferase based on a series of plasmid cloning vectors compatible with derivatives of pBR322. Gene 186, 197–200. doi: 10.1016/s0378-1119(96)00702-0
Masters, C. I., and Minton, K. W. (1992). Promoter probe and shuttle plasmids for Deinococcus radiodurans. Plasmid 28, 258–261. doi: 10.1016/0147-619x(92)90057-h
Masters, C. I., Smith, M. D., Gutman, P. D., and Minton, K. W. (1991). Heterozygosity and instability of amplified chromosomal insertions in the radioresistant bacterium Deinococcus radiodurans. J. Bacteriol. 173, 6110–6117. doi: 10.1128/jb.173.19.6110-6117.1991
McHugh, G. L., and Swartz, M. N. (1977). Elimination of plasmids from several bacterial species by novobiocin. Antimicrob. Agents Chemother. 12, 423–426. doi: 10.1128/AAC.12.3.423
Meima, R., and Lidstrom, M. E. (2000). Characterization of the minimal replicon of a cryptic Deinococcus radiodurans SARK plasmid and development of versatile Escherichia coli -D. radiodurans shuttle vectors. Appl. Environ. Microbiol. 66, 3856–3867. doi: 10.1128/AEM.66.9.3856-3867.2000
Morita, Y., and Nishida, H. (2018). The common ancestor of Deinococcus species was rod-shaped. Open Bioinform. J. 11, 252–258. doi: 10.2174/1875036201811010252
Morita, Y., Okumura, M., Narumi, I., and Nishida, H. (2019). Sensitivity of Deinococcus grandis rodZ deletion mutant to calcium ions results in enhanced spheroplast size. AIMS Microbiol 5, 176–185. doi: 10.3934/microbiol.2019.2.176
Moseley, B. E., and Setlow, J. K. (1968). Transformation in Micrococcus radiodurans and the ultraviolet sensitivity of its transforming DNA. Proc. Natl. Acad. Sci. USA 61, 176–183. doi: 10.1073/pnas.61.1.176
Narasimha, A., and Basu, B. (2021). New insights into the activation of radiation desiccation response regulon in Deinococcus radiodurans. J. Biosci. 46:10. doi: 10.1007/s12038-020-00123-5
Nishino, K., Morita, Y., Takahashi, S., Okumura, M., Shiratani, S., Umemura, K., et al. (2018). Enlargement of Deinococcus grandis spheroplasts requires Mg2+ or Ca2+. Microbiology (Reading) 164, 1361–1371. doi: 10.1099/mic.0.000716
Nishino, K., and Nishida, H. (2019). Calcium ion induces outer membrane fusion of Deinococcus grandis spheroplasts to generate giant spheroplasts with multiple cytoplasms. FEMS Microbiol. Lett. 366:fny282. doi: 10.1093/femsle/fny282
Nishino, K., Tsuchikado, R., and Nishida, H. (2019). Sugar enhances outer membrane fusion in Deinococcus grandis spheroplasts to generate calcium ion-dependent extra-huge cells. FEMS Microbiol. Lett. 366:fnz087. doi: 10.1093/femsle/fnz087
Ohba, H., Satoh, K., Yanagisawa, T., and Narumi, I. (2005). The radiation responsive promoter of the Deinococcus radiodurans pprA gene. Gene 363, 133–141. doi: 10.1016/j.gene.2005.07.035
Pande, V., Mitra, N., Bagde, S. R., Srinivasan, R., and Gayathri, P. (2022). Filament organization of the bacterial actin MreB is dependent on the nucleotide state. J. Cell Biol. 221:e202106092. doi: 10.1083/jcb.202106092
Sadowska-Bartosz, I., and Bartosz, G. (2023). Antioxidant defense of Deinococcus radiodurans: how does it contribute to extreme radiation resistance? Int. J. Radiat. Biol. 99, 1803–1829. doi: 10.1080/09553002.2023.2241895
Satoh, K., Ohba, H., Sghaier, H., and Narumi, I. (2006). Down-regulation of radioresistance by LexA2 in Deinococcus radiodurans. Microbiology (Reading) 152, 3217–3226. doi: 10.1099/mic.0.29139-0
Satoh, K., Onodera, T., Omoso, K., Takeda-Yano, K., Katayama, T., Oono, Y., et al. (2016). Draft genome sequence of the Radioresistant bacterium Deinococcus grandis, isolated from freshwater fish in Japan. Genome Announc. 4:e01631-15. doi: 10.1128/genomeA.01631-15
Satoh, K., Tu, Z., Ohba, H., and Narumi, I. (2009). Development of versatile shuttle vectors for Deinococcus grandis. Plasmid 62, 1–9. doi: 10.1016/j.plasmid.2009.01.005
Shibai, A., Satoh, K., Kawada, M., Kotani, H., Narumi, I., and Furusawa, C. (2019). Complete genome sequence of a Radioresistant bacterial strain, Deinococcus grandis ATCC 43672. Microbiol. Resour. Announc. 8:e01226-19. doi: 10.1128/MRA.01226-19
Sieger, B., Schubert, K., Donovan, C., and Bramkamp, M. (2013). The lipid II flippase RodA determines morphology and growth in Corynebacterium glutamicum. Mol. Microbiol. 90, 966–982. doi: 10.1111/mmi.12411
Siguret, V., Ribba, A. S., Cherel, G., Meyer, D., and Pietu, G. (1994). Effect of plasmid size on transformation efficiency by electroporation of Escherichia coli DH5 alpha. BioTechniques 16, 422–426
Spengler, G., Molnar, A., Schelz, Z., Amaral, L., Sharples, D., and Molnar, J. (2006). The mechanism of plasmid curing in bacteria. Curr. Drug Targets 7, 823–841. doi: 10.2174/138945006777709601
Telang, S., Patel, P., Sarangdhar, V., and Donde, S. (2014). Isolation and cloning of the endoglucanase gene from Bacillus pumilus and its expression in Deinococcus radiodurans. 3 Biotech 4, 57–65. doi: 10.1007/s13205-013-0127-3
Trevors, J. T. (1986). Plasmid curing in bacteria. FEMS Microbiol. Lett. 32, 149–157. doi: 10.1111/j.1574-6968.1986.tb01189.x
Keywords: Deinococcus grandis , host-vector system, plasmid, shuttle vector, gene expression, copy number
Citation: Sakai M, Shimosaka T, Katsumata K, Yohda M and Narumi I (2024) Developing a new host-vector system for Deinococcus grandis. Front. Microbiol. 15:1387296. doi: 10.3389/fmicb.2024.1387296
Edited by:
Andreas Teske, University of North Carolina at Chapel Hill, United StatesReviewed by:
Sangyong Lim, Korea Atomic Energy Research Institute (KAERI), Republic of KoreaCopyright © 2024 Sakai, Shimosaka, Katsumata, Yohda and Narumi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Issay Narumi, bmFydW1pQHRveW8uanA=
†Present address: Taichi Shimosaka, Graduate School of Science and Engineering, Saitama University, Shimo-Okubo, Saitama, Japan
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.