
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 18 April 2024
Sec. Microbial Physiology and Metabolism
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1373469
This article is part of the Research TopicNew Insights in Microbial Stress Tolerance MechanismsView all 15 articles
 João M. P. Jorge1†
João M. P. Jorge1† Celso Martins1*†‡
Celso Martins1*†‡ Patrícia Domingos1
Patrícia Domingos1 Tiago M. Martins1
Tiago M. Martins1 Diego O. Hartmann1‡
Diego O. Hartmann1‡ Gustavo H. Goldman1,2
Gustavo H. Goldman1,2 Cristina Silva Pereira1*
Cristina Silva Pereira1*Aspergilli comprise a diversity of species that have been extensively studied due to their catabolic diversity, biotechnological and ecological value, and pathogenicity. An impressive level of structural and functional conservation has been shown for aspergilli, regardless of many (yet) cryptic genomic elements. We have hypothesized the existence of conserved genes responsive to stress in aspergilli. To test the hypothesis of such conserved stress regulators in aspergilli, a straightforward computational strategy integrating well-established bioinformatic tools was used as the starting point. Specifically, five transcriptome-based datasets on exposure to organic compounds were used, covering three distinct Aspergillus species. Among the identified up-regulated genes, only one gene showed the same response in all conditions, AN9181. This gene encodes a protein containing a phenylcoumaran benzylic ether reductase-like domain and a Nitrogen metabolite repressor regulator domain (NmrA). Deletion of this gene caused significant phenotypic alterations compared to that of the parental strain across diverse conditions. Specifically, the deletion of AN9181 raised the mutant’s metabolic activity in different nitrogen sources. The acquired data supports that AN9181 acts by repressing (slowing down) A. nidulans growth when exposed to aromatic compounds in a concentration dependent manner. The same phenotype was observed for amphotericin B. Finally, AN9181 underwent differential upregulation under oxidative stress conditions. Collectively, the data suggest that AN9181, herein assigned as NmrB (Nitrogen Metabolite Repression Regulator B), builds up the genetic machinery of perception of oxidative stress by negatively regulating growth under such conditions.
The aspergilli comprise a diverse group of saprophytic filamentous fungi covering over 200 million years of evolution (Galagan et al., 2005; Gupta et al., 2021) that are found virtually everywhere. Aspergilli have been extensively studied mostly due to high biotechnological value, for example Aspergillus oryzae and A. niger are used for the production of sake and citric acid, respectively (Machida et al., 2008; Behera, 2020), and A. terreus was the first known producer of lovastatin, a cholesterol-lowering statin (Hajjaj et al., 2001; Pearce, 2019). Aspergilli are also important human pathogens, mainly A. fumigatus, which is the major causative agent of aspergillosis (Steenwyk et al., 2020). Recently, this species was included in the list of priority fungal pathogens published by the World Health Organization (World Health Organization, 2022). Despite the extensive knowledge already acquired about aspergilli genomes, many gene functions remain unknown or poorly understood (Galagan et al., 2005).
Fungi show great potential to degrade and/or mineralize various aromatic compounds, including environmental pollutants, a capacity mostly attributed to their impressive catabolic capabilities (Harms et al., 2011; Varela et al., 2015, 2017; Martins et al., 2019). Regardless of efficient degradation, a great stress is imposed; this has been linked to increased pathogenic potential of a fungal community (Martins et al., 2018) and to the production of virulent airborne spores from aspergilli strains in environments polluted with aromatic compounds (Martins et al., 2023). Earlier studies on the genome’s structure and evolution in aspergilli revealed an impressive level of structural and functional conservation and synteny (Galagan et al., 2005; Steenwyk et al., 2020), both in coding and non-coding regions, regardless of evident evolutionary rearrangements. Taking the catabolism of aromatic compounds as an example, in the different available Dikarya genomes, the pathway gene clusters are found often in an array with the peripheral pathway genes (Martins et al., 2019). Such high level of genomic similarity in aspergilli, underlines the hypothesis of the existence of conserved regulatory genes responsive to variable chemical stresses (Galagan et al., 2005; Martins et al., 2019). In the present study, we integrated the transcriptome signatures of different aspergilli (three species) to distinct organic compounds, gathering original and publicly available datasets. To identify genes showing the same response across the different datasets, a straightforward computational strategy that allows comparing transcriptomic-based datasets initially collected in distinct Aspergillus species was used. Only one gene – AN9181, assigned as NmrB (Nitrogen Metabolite Repression Regulator B), showed the same response in all the datasets. The phenotype of A. nidulans single deletion-mutant ∆AN9181 was compared to that of the wild type, covering for example germination fitness, growth rate and susceptibility to distinct chemical stressors. The collected data suggest that NmrB negatively regulates the metabolism of A. nidulans in specific stress conditions.
Bromoquinol (BMQ) was purchased from Alfa Aesar; 6-iodoquinoline (IDQ) and resveratrol (RVT) from TCI Europe; hydrogen peroxide (H2O2) from Merck; dimethyl sulfoxide from Fisher Chemical, and the remaining compounds from Sigma Aldrich, namely pentachlorophenol (PCP), triclosan (TCS), salicylate (Sal), benzo[a]pyrene (BaP), congo red, sodium benzoate, menadione sodium bisulfite (MSB), amphotericin B (Amph B), caspofungin (CSP), itraconazole (ITZ), miconazole (Mic) and 2,7-dichlorofluorescin diacetate (DCFH-DA).
Aspergillus fumigatus CEA17 reference strain was propagated at 37°C in solid complete medium [1% D-glucose, 0.2% Peptone, 0.1% Yeast extract, 0.1% Casamino acids, 50 mL of a 20× salt solution, 0.1% trace elements, 0.1% Vitamin solution, 2% agar, pH 6.5 with NaOH]. The composition of the trace elements, vitamins, and nitrate salts has been described previously (Kafer, 1977).
The Minimal Inhibitory Concentration (MIC) for fungal growth inhibition of IDQ, initially acquired from The Pathogen Box (www.pathogenbox.org/), was defined following the standard methodology implemented by the Clinical and Laboratory Standards Institute (CLSI, 2018). The compound’s antifungal activity was analyzed by serial dilutions using MIC assay (0 to 25 μM) in MOPS buffered RPMI 1640 medium (Sigma-Aldrich), pH 7.0 in 96-wells plates. In each well, a total of 1×104 conidia of A. fumigatus wild-type strain was inoculated. Plates were incubated at 37°C without shaking for 48 h. Non-inoculated controls were done in parallel. All experiments were done in triplicate.
The preparation of total RNA samples for RNA-seq Expression Profiling was as follows. Erlenmeyer flasks (125 mL) were used to inoculate 1×107 spores in 30 mL of Vogel’s Minimal Media (Vogel, 1956) and incubated for 16 h at 37°C, 180 rpm. The medium was then exchanged, and 0, 0.5x MIC (=0.35 μM) or 2x MIC (=1.4 μM) of IDQ was added and incubated for 4 h at 37°C, 180 rpm. Six replicates for each condition were prepared. At the end of the incubation period, the cultures were filtered and frozen immediately in liquid nitrogen. Total RNA from six mycelia per condition were extracted using RNeasy Plant Mini Kit (Qiagen), according to the manufacturer’s protocol, a TissueLyser LT (Qiagen) for cell disruption, and approximately 30 mg of poly(vinylpolypyrrolidone) per sample. RNA quality (integrity) was evaluated using a Nucleic Acid QC - Fragment Analyzer.
For single-end RNA sequencing (RNA-seq), libraries were generated using the Smart-Seq2® mRNA assay (Illumina, Inc.) according to the manufacturer’s instructions. Six samples were indexed and sequenced on the Illumina NextSeq550 (20 M reads per sample). Generated FastQ files were analyzed with FastQC, and any low-quality reads were trimmed with Trimmomatic (20). All libraries were aligned to the corresponding model fungus A. fumigatus A1163 genome assembly (ASM15014v1) with gene annotations from Ensembl Fungi v. 45 using HISAT2 v. 2.1.0 (Kim et al., 2015), and only matches with the best score were reported for each read. All RNA-seq experiments were carried out in three biological replicates. Differential expression analysis was performed using DESeq2 v. 1.24.0 (Love et al., 2014). The genes that showed more than log2 1-fold expression changes with p-adj value < 0.05 were considered as significantly differentially expressed (IDQ dataset in Supplementary Dataset 1).
The transcriptomics datasets were selected based on the following stringent rules: only studies on aspergilli were considered (i), comprising an exposure period lower than six days (ii) to an organic compound displaying broad environmental, biotechnological or health relevance (iii) with a molar mass below 500 g·mol−1. Consequently, we selected five transcriptome-based datasets: the catabolism of the simple aromatic hydrocarbon salicylate (Sal) in A. nidulans (Martins et al., 2021); the mode of action of the polyphenol resveratrol (RVT) in A. flavus (Wang et al., 2015); the degradation of the polycyclic aromatic hydrocarbon benzo[a]pyrene (BaP) in Aspergillus sp. (Loss et al., 2019); the inhibitory effects of the quinoline bromoquinol (BMQ) (Ben Yaakov et al., 2017) and of 6-iodoquinoline (IDQ) in A. fumigatus (see above). The full gene lists (up-regulated only) used in our analysis are available in Supplementary Dataset 2.
The COCOA strategy uses a set of established bioinformatics tools (Figure 1A) as detailed below. First, we performed the curation and validation of the five selected datasets by reprocessing the raw data to obtain the gene counts using the HISAT2 methodology (Kim et al., 2015) and identifying the differentially expressed genes using the DESeq2 R-based package (Love et al., 2014). Then, a full protein-translated genome orthology transformation to A. nidulans (the “receiver species”) was performed for eight distinct aspergilli available in the FungiDB database: A. aculeatus ATCC16872, A. flavus NRRL3357, A. fumigatus A1163, A. nidulans FGSCA4, A. niger ATCC13496, A. sydowii CBS593.65, A. terreus NIH2624, and A. versicolor CBS583.65 (Stajich et al., 2012). For the orthology analyzes, we used OrthoFinder (Emms and Kelly, 2015, 2017, 2019) because of its proven excellent performance compared to other orthology tools (Emms and Kelly, 2015). The use of these additional genomes than those strictly necessary adds robustness to the construction of gene trees, resulting in better discrimination and stringency levels between orthogroups (Emms and Kelly, 2015, 2017, 2019; Martins et al., 2020). We transposed the list of the up-regulated genes in the five datasets to the corresponding A. nidulans orthologues. After that, the co-expressed genes were identified using the online tool InteractiVenn (Heberle et al., 2015). Finally, we performed a whole-genome protein domain analysis for the protein-translated genome of the “receiver species,” aiming to obtain hints on the putative functions played by the co-expressed genes, necessary when a functional annotation is lacking.
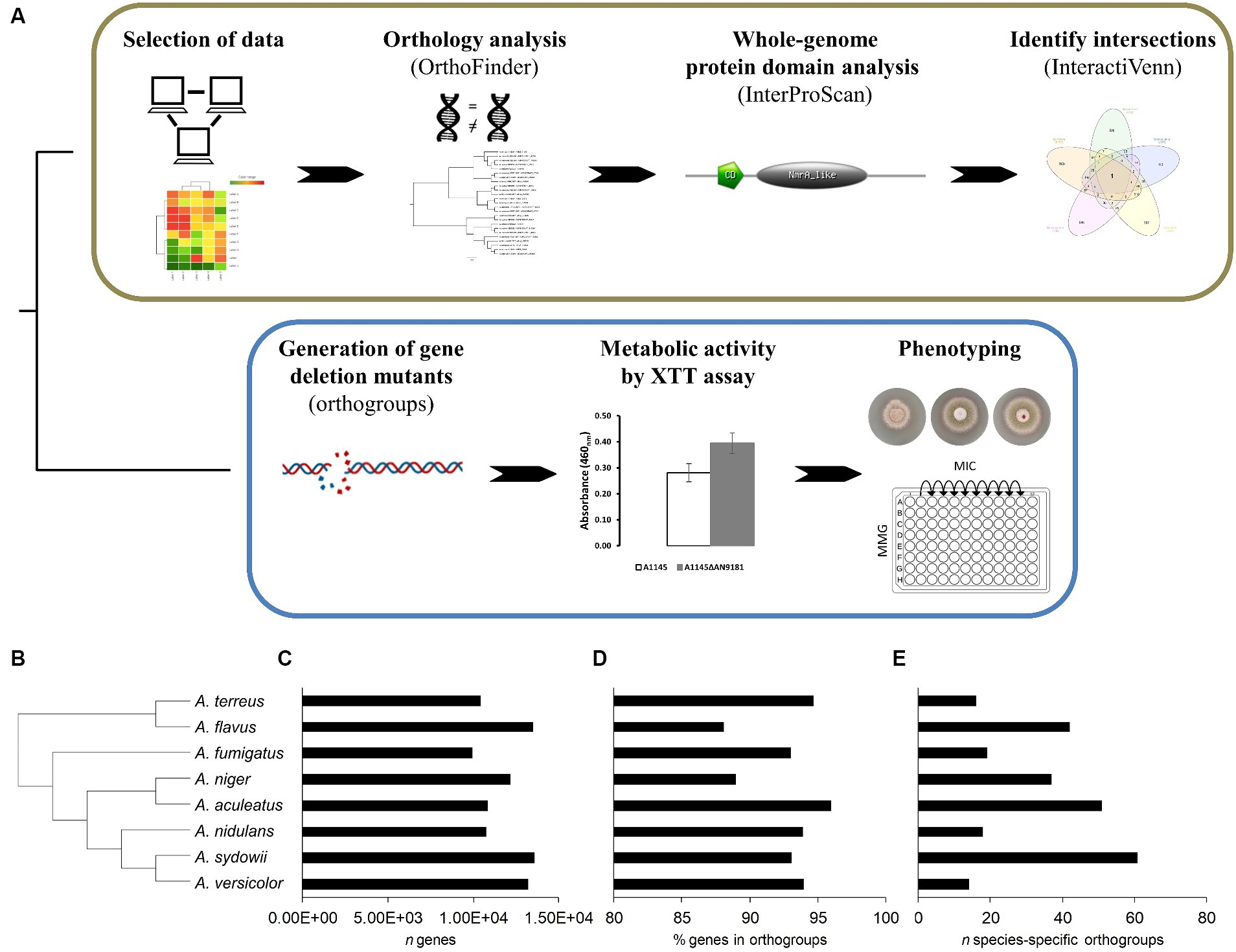
Figure 1. Schematic overview of the COmpregensive CO-expression Analysis (COCOA) strategy developed in this work. (A) – the iterative use of the well-established bioinformatics tools OrthoFinder, InterProScan and InteractiVenn upon selection of suitable and meaningful transcriptome-based datasets is displayed. Laboratory assays implemented to understand the biological relevance of the gene identified by COCOA. Results of the orthology analysis performed using the genomes of eight Aspergillus spp. (B) – the species tree, (C) – the number of genes of each analyzed genome, (D) the percentage of genes in orthogroups and (E) the number (n) of species-specific orthogroups are depicted.
If not mentioned, assays used minimal medium with glucose (10 g·L−1) and nitrate (MMGN), as follows: thiamine (0.01 g·L−1), 5% (v/v) nitrate salts solution [NaNO3 (120.0 g·L−1), KCl (10.4 g·L−1), MgSO4·7H2O (10.4 g·L−1) and KH2PO4 (30.4 g·L−1)], 0.1% (v/v) trace elements solution [ZnSO4·7H2O (22.0 g·L−1); H3BO3 (11.0 g·L−1); MnCl2·4H2O (5.0 g·L−1); FeSO4·7H2O (5.0 g·L−1); CoCl2·6H2O (1.7 g·L−1); CuSO4·5H2O (1.6 g·L−1); Na2MoO4·2H2O (1.5 g·L−1), and Na4EDTA (50.0 g·L−1)], and the pH was adjusted to 6.5 with NaOH. Whenever needed, the media was jellified with 1.5% agar (solid cultivation). The MMG is similar to that described above except that sodium nitrate was removed. Most functional assays used MMG supplemented or not with a defined amount of a N source as mentioned in the results section. Moreover, in all comparative assays of mutant versus parental strain, the medium was supplemented with the essential nutrients: uracil (1.12 g·L−1), uridine (1.22 g·L−1), riboflavin (2.5 mg·L−1) and pyridoxine (0.05 mg·L−1).
EC50 levels of each organic compound were assessed on basis of hyphal radial growth rate for the strain A. nidulans FGSC A4 grown in petri dishes (55 mm) containing the MMGN (jellified) and supplemented with either BaP (1.0–3.5 mM); BMQ (0.1–1.0 mM); Sal (75–400 mM); IDQ (0.002–1.0 mM); RVT (0.005–1.5 mM); or sodium benzoate (25–200 mM). Controls without the organic compounds were also made. The assay was carried out by inoculating 2×105 conidia into the center of the plate, and incubate at 30°C for 120 h. Radial growth of mycelia (colony diameter in cm) was measured using a Vernier caliper (error ± 0.05 mm). The plates that did not displayed visible growth were visualized under microscope to confirm growth inhibition. The EC50 values were calculated using the XLSTAT tool (Microsoft Excel) with the Gompertz model.
For the targeted gene expression analysis of the two genes of the AN9181 orthogroup, Aspergillus nidulans (5×105 spores·mL−1) was pre-grown during 24 h in 12-well plates (2 mL of MMGN per well) at 30°C with gentle agitation (100 rpm), in the dark. After the pre-growth phase, the selected organic compounds were added to the cultures at a defined concentration (Time 0). The concentrations used were as follows: 0.2 mM for BaP [similar to the concentration used by Loss et al. (2019)]; 0.5 mM for BMQ (half of the minimal inhibitory concentration described by Ben Yaakov et al. (2017); 0.3 mM for PCP (concentration previously tested before by Varela et al. (2017), and 0.2 mM for TCS to match the range used for the other organic compounds. For the gene expression analysis of AN9181 in the presence of H2O2, A1145 strain (1×106 spores·mL−1) was pre-grown during 140 h in 12-well plates (2 mL of MMG per well) at 30°C with gentle agitation (100 rpm), in the dark. After the pre-growth phase, H2O2 was added to the cells at a defined concentration (1.0 –4.0 mM) and incubated at 30°C for 30 min. Controls without addition of H2O2 were grown as well. Mycelia samples were collected from the 12-well plates, and immediately frozen using liquid nitrogen until further analysis.
Total RNA extraction and cDNA synthesis were performed using the RNeasy Plant Mini Kit (Qiagen) and the iScript cDNA Synthesis Kit (Bio-Rad), and the RT-qPCR performed as previously reported (Martins et al., 2020). The oligonucleotide pairs for specific A. nidulans genes (Supplementary Table 1) were designed using the Primer-Blast web tool (www.ncbi.nlm.nih.gov/tools/primer-blast/), and purchased from STAB Vida Lda. (Portugal). The RT-qPCR analysis was performed in a CFX96 Thermal Cycler (Bio-Rad), using the SsoFast EvaGreen Supermix (Bio-Rad), 250 nM of each oligonucleotide and the cDNA template equivalent to 10 ng of total RNA, at a final volume of 10 μL per well, in at least three biological replicates. The PCR conditions were as follows: enzyme activation at 95°C for 30 s; 40 cycles of denaturation at 95°C for 5 s, and annealing/extension at 60°C for 15 s; and the melting curve obtained from 65 to 95°C, consisting of 0.5°C increments for 5 s. Data were analyzed with the CFX Manager software (Bio-Rad). In more detail, the expression of each gene was taken as the relative expression compared to the time zero (before incubation with the tested compounds) or as the relative expression compared to the no addition of the stressor. The expression of all target genes was normalized by the expression of the histone H2B gene (AN3469) (Supplementary Dataset 3), used as the internal control. Statistical analyzes used the XL-STAT (Addinsoft) software, and multiple Student’s t-tests. Differences in gene expression with a p-value below 0.05 were considered statistically significant.
The gene AN9181 was replaced with Aspergillus fumigatus pyrG gene (pyrGAfu) in Aspergillus nidulans A1145 and A1149, both auxotrophic strains (pyrG−). Deletion cassettes combining the 5′ and 3′-flanking regions of each target gene with pyrGAfu were obtained by fusion PCR and were used to transform A. nidulans A1145 or A1149 protoplasts and plated onto selective media to generate single-deletion mutant strains (Supplementary Table 2). Isolated transformants were cultivated on the selective media for three generations to assure stable mutations. In greater detail, the deletion cassettes were constructed using a fusion PCR protocol. Six primers (P1-P6) were designed, based on sequences from the Aspergillus Genome Database (www.aspgd.org), and analyzed using the NetPrimer web tool (www.premierbiosoft.com/NetPrimer/AnalyzePrimer.jsp) (listed in Supplementary Table 3). PCR reactions were performed in a T100 Thermal Cycler (Bio-Rad; conditions in Supplementary Table 4). A. fumigatus pyrG was amplified from plasmid pCDS60 (FGSC, Kansas City, MO, United States) using primers CDS164 and CDS165. Flanking fragments upstream and downstream of the gene were amplified with primer pairs P1/P3 and P4/P6, respectively, using genomic DNA from A. nidulans A4 as template. The final cassette was produced by fusing the flanking regions with the A. fumigatus pyrG using nested primers P2 and P5 for the target gene. PCR products were cleaned with NZYGelPure kit (NZYTech). Then, to produce transformable protoplasts, A. nidulans mycelia grown overnight from 108 conidia in 50 mL MMGN with the appropriate nutritional supplements (30°C, 90 rpm), recovered by centrifugation and washed (0.6 M MgSO4), were digested in 20 mL enzymatic mix (300 mg lysing enzymes from Trichoderma harzianum, 150 μL β-glucuronidase from bovine liver, type B-1, and 150 mg Driselase from Basidiomycetes sp., all from Sigma-Aldrich) in osmotic medium (1.2 M MgSO4·7H2O and 10 mM sodium phosphate buffer pH 6.5; final pH adjusted to 5.8 with Na2HPO4) for 20 h at 30°C, 90 rpm. The protoplasts suspension was overlaid (2:1) with trapping buffer (0.6 M sorbitol and 100 mM Tris–HCl pH 7.0), and then recovered by centrifugation (1,500 g, 4°C, 15 min, swing-bucket rotor), washed three times with 10 mL of ST10 buffer (1.2 M sorbitol and 10 mM Tris–HCl pH 7.5) and finally resuspended in 1 mL of the same buffer and incubated overnight at 4°C. In the next day, the protoplasts were collected by centrifugation (1,000 g, at 4°C, 2 min), and resuspended in 700 μL cold STC buffer (1.2 M sorbitol, 10 mM Tris–HCl pH 7.5 and 10 mM CaCl2). The obtained protoplasts (100 μL) were then transformed by mixing with the cleaned fusion PCR product (10 μL), subsequently adding freshly filtered polyethylene glycol (PEG) solution (25% (w/v) in STC buffer; 50 μL) and kept in an ice bath for 25 min. Then, additional PEG solution was added (1 mL), gently mixed using a micropipette and placed at room temperature for 25 min. For the single mutants A1145ΔAN9181 and A1149ΔAN9181, 100 μL of the transformation mix were plated onto a selective medium containing glucose (5.0 g·L−1), yeast extract (5.0 g·L−1), sucrose (342.3 g·L−1), riboflavin (2.5 mg·L−1), pyridoxin (0.05 mg·L−1), 0.1% (v/v) trace elements solution (see above), and 1.5% agar. The selective plates were incubated for 3 to 4 days at 30°C. All transformants were morphologically identical to the parental strain. Two isolated A1145ΔAN9181 transformants (randomly selected) were streaked onto complete medium plates containing glucose (10.0 g·L−1), peptone (2.0 g·L−1), yeast extract (1.0 g·L−1), casein hydrolysate (1.0 g·L−1), 5% (v/v) nitrate salts solution (see above), 0.1% (v/v) trace elements solution, riboflavin (2.5 mg·L−1), pyridoxin (0.05 mg·L−1), pH 6.5, and incubated at 30°C for 4 days. Three generations of each transformant were grown to assure stable mutations and then grown again in MMG without uracil and uridine to confirm the prototrophy to the compounds of the mutant strains (Supplementary Figure 1).
DNA from each transformant was extracted with Quick-DNA™ Fungal/Bacterial Microprep Kit (Zymo Research), and diagnostic PCR was performed with primers P1 and P6 for each gene. Based on amplicon size (3,222 bp for WT and 3,904 bp for A1145ΔAN9181, Supplementary Figure 2A), it was possible to confirm the correct gene replacement of the transformants. To obtain further confirmation, the PCR products were digested for 1 h with the restriction enzymes ApaI or KpnI selected to give differential digestion patterns for mutant and wild-type strains. Supplementary Figure 2B show the products of diagnostic PCR and restriction enzymes digestion for the A1145ΔAN9181 selected strain.
Cell viability was evaluated using the XTT assay in 96-well plates (200 μL per well) using Malt Extract medium or MMG supplemented with 10 mM N source, and an inoculum of 1×106 spores·mL−1, and incubated at 30°C for 24 h (triplicates). After, 10 μL of a solution containing 4.6 mg·mL−1 of XTT and 0.104 mg·mL−1 of menadione was added to each well, incubated for further 2 h, and the absorbance measured (460 nm).
The strains’ spore germination fitness, i.e., number of spores that are able to form a colony, was evaluated by spreading 100 spores onto solid media, then counting the colony forming units (CFUs) daily, during five days (triplicates). The radial growth diameter of each strain (inoculum: 1 μL of a suspension of 2×108 spores·mL−1) in solid MMG, supplemented or not with specific compounds (viz. congo red, H2O2, MSB, Amph B, CSP and ITZ) was measured after five days of incubation (30°C, dark, triplicates). The MIC of each antifungal was determined using the micro-broth dilution method (MMG, 96-well plates, 200 μL per well), testing specific concentration ranges for Amph B (250–550 mg·L−1), CSP (60–120 mg·L−1) and ITZ (0.2–0.5 mg·L−1). An inoculum of 1×106 spores·mL−1 was used, and the plates were incubated at 30°C for 48 h. Negative controls were done in parallel. The lowest concentration that showed no growth under microscopic observation was considered the MIC.
Intracellular ROS was quantified using DCFH-DA. Cultures were grown in MMG (140 h, 30°C); then 2.5 μg·mL−1 of DCFH-DA was added and incubated (30°C, 30 min). H2O2 was added to the cells at increasing concentrations, from 1 mM to 4 mM, and the incubation step repeated. The cell suspension was disrupted in a TissueLyzer LT (Qiagen) with a metal bead at a maximum speed (3 cycles of 1 min). The fluorescence intensity of the supernatant (recovered by centrifugation: 12000 g, 10 min) was measured using a Tecan Infinite M Nano+ Microplate (Männedorf, Switzerland) as follows: excitation length 485/9; emission 528/20; optics, top; read speed, normal; gain, 89; number of flashes, 25; integration time, 40 μs. The fluorescence intensity (per mycelia dry weight) was normalized against the control (no H2O2 added).
The orthology analysis (Figure 1A) revealed a total of 11,992 orthogroups among the eight analyzed Aspergillus spp. genomes (Supplementary Dataset 4). Among these, only 258 were species-specific orthogroups, comprising 0.7% of the total number of genes. Figure 1B displays the phylogenetic relations computed by the orthology analysis, and Figures 1C–E provide for each species the total number of genes, the percentage of genes in orthogroups and the number of species-specific orthogroups, respectively. The number of species-specific orthogroups (Figure 1E) is not necessarily correlated with the percentage of genes in orthogroups (Figure 1D) nor the genome size (Figure 1C). For instance, A. versicolor possesses the third larger genome among the eight analyzed Aspergillus spp. yet displays one of the highest percentages of genes present in orthogroups but the lowest number of species-specific orthogroups. On the other hand, A. sydowii, which possesses a genome size comparable to that of A. versicolor and only a slightly lower percentage of genes belonging to orthogroups, is the species presenting the higher number of species-specific orthogroups. This is an indication of a higher occurrence of gene duplication events (Emms and Kelly, 2015) in A. sydowii compared to other aspergilli, a feature also visible in A. aculeatus and A. flavus. Finally, A. fumigatus and A. nidulans present similarly sized genomes, as well as a comparable percentage of genes included in orthogroups and number of species-specific orthogroups.
Upon transposing the differentially expressed genes (up-regulated) from the five transcriptomic datasets analyzed to the corresponding orthologues in the genome of A. nidulans (Supplementary Dataset 2) we analyzed the set of genes present in at least three, four or five transcriptome-based datasets on exposure to selected organic compounds. We observed that, out of the 241 genes present in at least three datasets, several are predicted to be transporters or secondary metabolism-related genes (Supplementary Dataset 5). Twenty-two genes are present in at least four datasets (Table 1), of which a single gene, AN9181, is present in all five datasets (Figure 2A). This gene is part of an orthogroup that contains two genes in A. nidulans, being the other one AN8970.
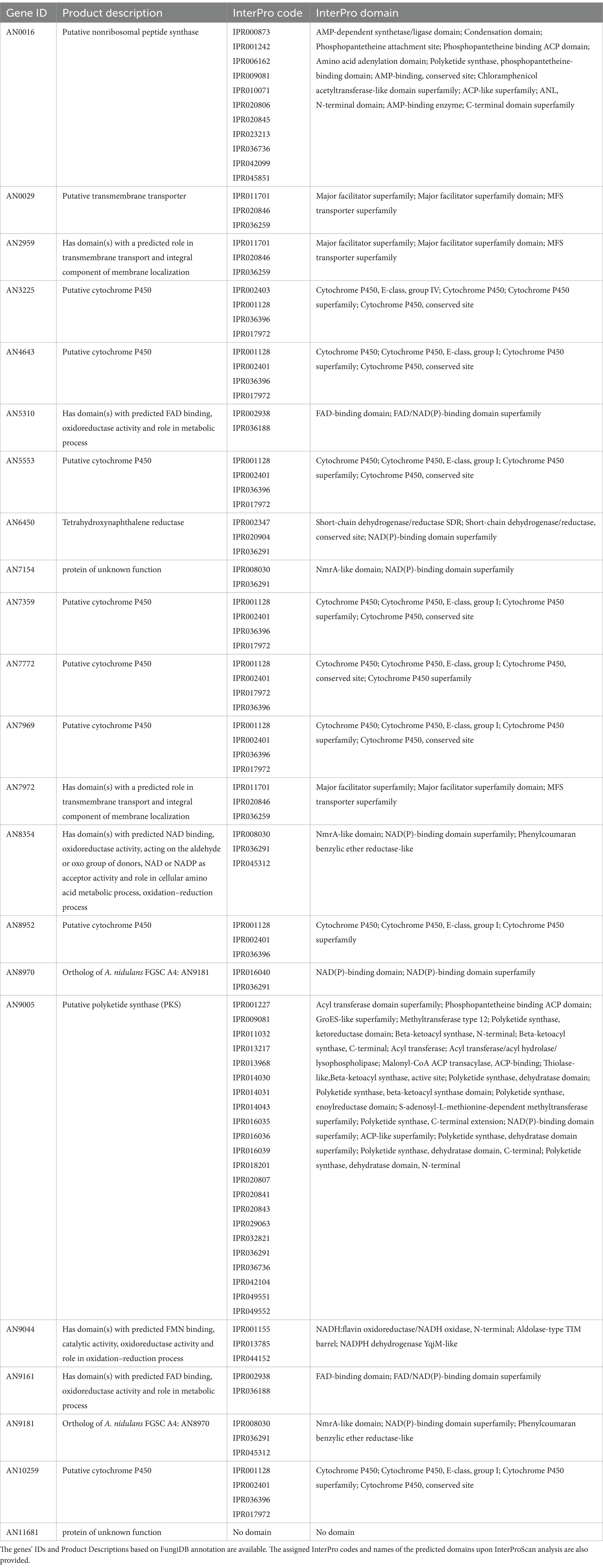
Table 1. List of genes present in at least four distinct transcriptome-based datasets out of the five analyzed using the COCOA strategy.
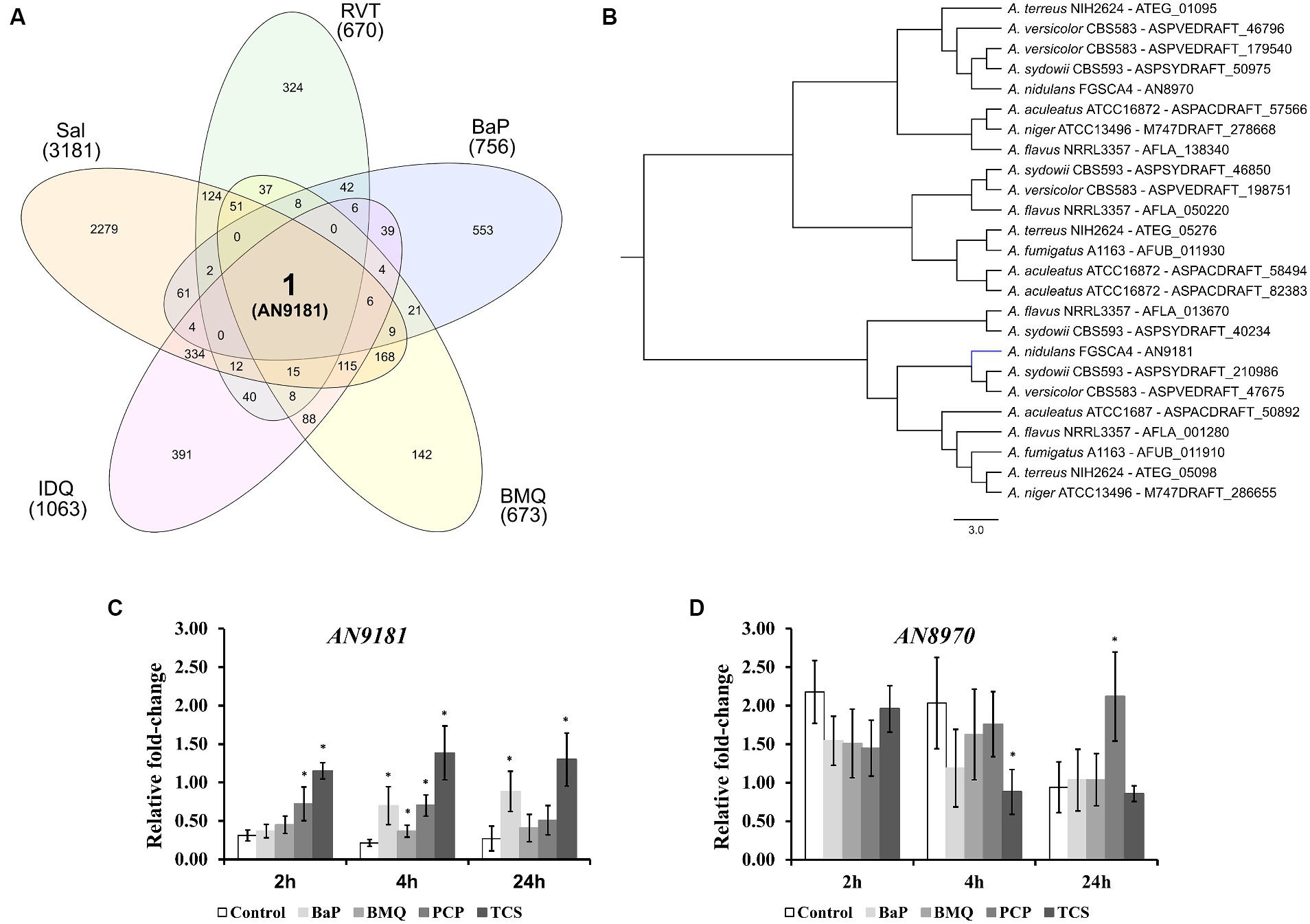
Figure 2. Different expressed genes in the five transcriptomic datasets. (A) – upon orthology transformation of the transcriptome-based datasets to the “receiver species” Aspergillus nidulans, AN9181 was the only gene found consistently up-regulated in all the analyzed datasets. BaP (Benzo[a]pyrene); BMQ (Bromoquinoline); Sal (Sodium Salicylate); IDQ (6-iodoquinoline) and RVT (Resveratrol). (B) – the AN9181 orthogroup comprises 25 genes present in the genomes of the eight orthology-analyzed Aspergillus species, which are separated into two distinct clusters in the gene-tree obtained upon OrthoFinder analysis. (C,D) – targeted gene expression analysis (assessed by RT-qPCR) is displayed for AN9181 (C) and AN8970 (D).
The AN9181 orthogroup (gene tree displayed in Figure 2B) comprises 25 genes across the eight analyzed aspergilli genomes, namely four in A. aculeatus, four in A. flavus, two in A. fumigatus, two in A. nidulans (as mentioned), two in A. niger, four in A. sydowii, three in A. terreus, and four in A. versicolor. The existence of the AN9181 orthogroup is therefore highly conserved in these aspergilli. The presence of four genes belonging to the AN9181 orthogroup in A. sydowii, A. aculeatus and A. flavus reinforces the indication of higher occurrence of gene duplication events in these species. This is an indication that the AN9181 orthogroup, like many other regulatory genes, is undergoing rapid evolution in Aspergillus sp. as a response to environmental changes and adaptive lifestyles. This plasticity is often associated with the evolution/transition to pathogenic lifestyles in aspergilli (Rokas, 2022). The 25 genes that compose the AN9181 orthogroup are largely uncharacterized with neither relevant information in FungiDB and nor predicted protein interactions. However, the orthologous gene cip1 of Candida albicans is involved in oxidative stress response, being regulated by the transcription factor Cap1p (Wang et al., 2006; Znaidi et al., 2009). This past evidence raises the hypothesis that AN9181 participates in stress responses.
Through a computational protein domain analysis (InterProScan), we observed that the AN9181 encodes a protein containing a phenylcoumaran benzylic ether reductase-like domain (IPR045312) and a Nitrogen metabolite repressor regulator (NmrA)-like domain (IPR008030), while AN8970 encodes a protein containing a NAD(P)-binding domain (IPR016040). A deletion mutant of padA, that encodes a protein also carrying a NmrA-like domain of Dictyostelium discoideum, showed to be more sensitive to ammonia than the wild-type (Núñez-Corcuera et al., 2008). Moreover, disruption of this gene resulted in phenotypic defects in development and growth, namely, the thermosensitive mutant allele padA− showed poor and null growth at permissive and restrictive temperatures, respectively. Deletion of the NmrA gene in A. flavus reduced growth in several nitrogen (N) sources, but increased conidia and sclerotia production (Han et al., 2016). This mutant strain produces less aflatoxin when cultivated in glutamine and alanine supplemented media, and shows reduced virulence and increased sensitivity in response to rapamycin and methyl methanesulfonate, but not in response to the osmotic stressors NaCl and sorbitol (Han et al., 2016).
The genes AN9181 and AN8970 are in separated clusters within the AN9181 orthogroup gene tree (Figure 2B). This observation is consistent with the fact that they possess distinct functional domains (Gabaldón and Koonin, 2013), and therefore, they are not paralogs. Though AN9181 was the initial candidate, we also evaluated the gene expression of AN8970 (same orthogroup) aiming to understand which one would be functionally relevant to regulate stress responses. Therefore, the expression profiles of either gene composing the AN9181 orthogroup (AN9181 and AN8970) were evaluated after 2, 4 and 24 h of exposure to four selected organic compounds in a medium containing a poor N source. The organic compounds comprise two which were found in the selected transcriptome datasets: benzo[a]pyrene (BaP), and bromoquinol (BMQ), and two additional aromatic halogenated based compounds, namely pentachlorophenol (PCP) and triclosan (TCS), classified as persistent organic pollutant and contaminant of emergent concern, respectively (Varela et al., 2017). The last two compounds were tested due to their frequent association to soil and water contamination (Czaplicka, 2004; Morgan et al., 2015), and past studies showing that either compound increased the production of virulent aspergilli conidia within soil colonizing fungi (Martins et al., 2023). The AN9181 revealed lower expression levels compared to AN8970 (Figures 2C,D). However, compared to control conditions, a significant increase in the expression levels of AN9181 was systematically noticed after 4 h of exposure to all the tested organic compounds, as well as in additional time-points, namely after 2 h to PCP, 2 and 24 h to TCS, and 24 h to BaP (Figure 2C). Recently, AN9181 was also found to be up-regulated after fungal growth in a nitrate minimal medium supplemented with either cadmium chloride, congo red or amphotericin B (Antal et al., 2020). In contrast to AN9181, the expression profiles of AN8970 were similar between the control and upon exposure to organic compounds, except for PCP at 24 h and for TCS 4 h (Figure 2D). At the experimental conditions used, the gene expression analysis did not support the idea of a concerted action of the two genes of the AN9181 orthogroup in A. nidulans. Based on these results, we focused the remaining analyzes on the AN9181.
To better understand the functional roles of AN9181, this gene was deleted and functionally analyzed in A. nidulans. The colony morphology of the parental strain and the A1145ΔAN9181 mutant (hereafter referred to as ΔAN9181) on Malt extract agar (MEA) medium were similar (Figures 3A,B). After five days of growth, the colony diameters of either strain in MMG containing a high amount of a non-preferred (i.e., poor) N source were also comparable (i.e., 71 mM sodium nitrate, Supplementary Figure 3). The conidia viability of either strain (i.e., germination fitness) was measured directly by counting the numbers of CFUs. CFUs were similar for both strains when germinated in the rich medium MEA (Figure 3C) and MMG supplemented with either a superior (i.e., rich) N source (10 mM of ammonium sulfate) (Figure 3D) or a non-preferred N source at high concentration (71 mM of sodium nitrate) (Supplementary Figure 4A). However, the ΔAN9181 conidial viability in MMG supplemented with 10 mM sodium nitrate or no added N source were 1.7-fold and 1.5-fold higher, respectively, compared to the parental strain (Figures 3E,F). This result suggests that AN9181 strongly influences the fitness of conidia germinating in medium having low availability of poor nitrogen sources.
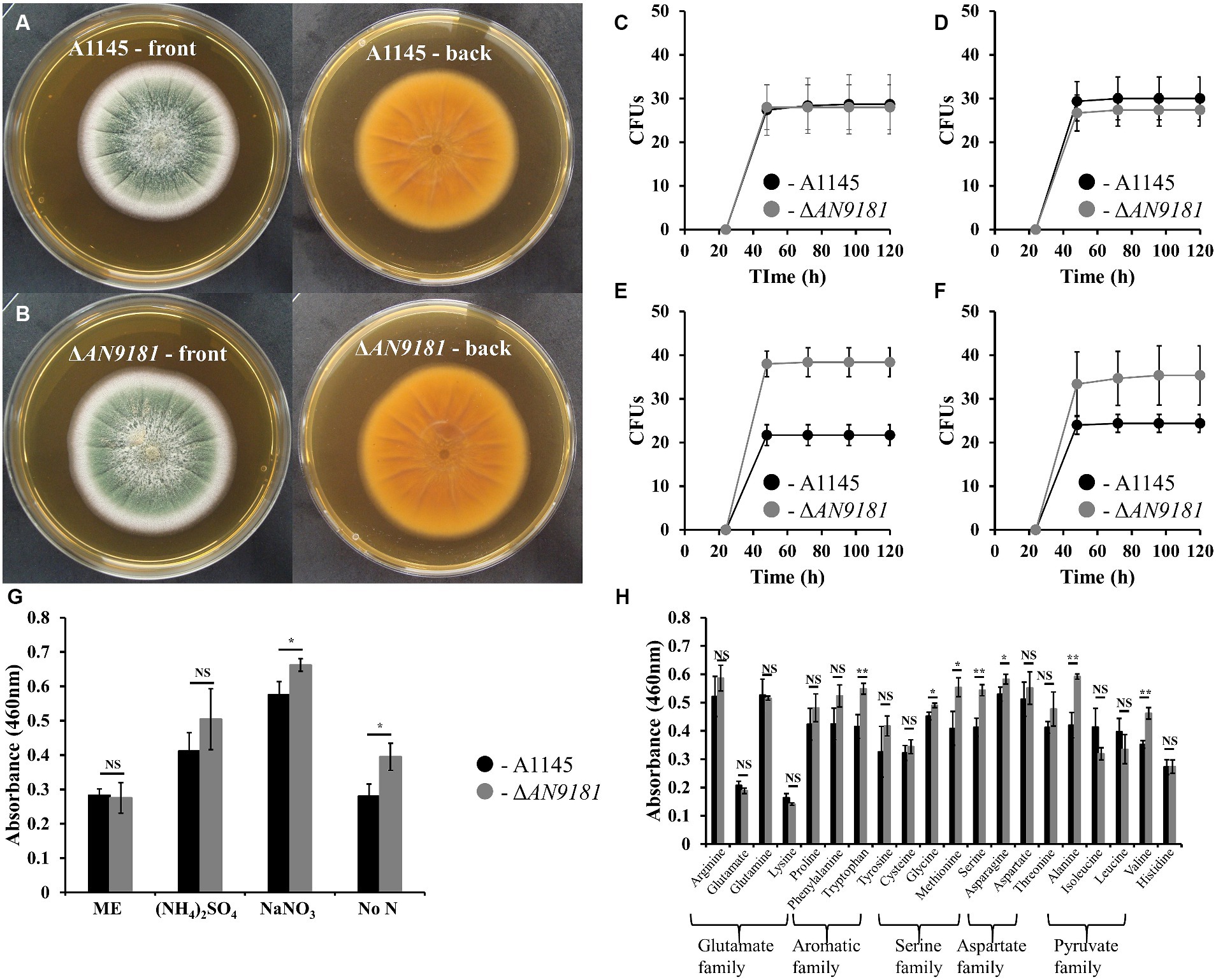
Figure 3. Phenotype features of A1145 and ΔAN9181. (A,B) – the morphology and pigmentation of A1145 strain (A) and ΔAN9181 strain (B) after cultivation at 30°C for five days in Malt Extract Agar. (C–F) – colony forming units of A1145 (black) and ΔAN9181 (gray) strains plated on Malt Extract Agar (C); minimal medium glucose supplemented with 10 mM nitrogen from different sources (NH4)2SO4 (D); 10 mM NaNO3 (E); no nitrogen added (F). (G,H) – cell viability and proliferation measured by XTT assay. A1145 (black) and ΔAN9181 (gray) strains were grown in Malt Extract or in minimal medium glucose supplemented with 10 mM nitrogen from the different nitrogen sources (G) including amino acids (H). Values and error bars represent the mean and the standard deviation of triplicates. Significant differences (Student’s t-test) are marked with asterisks [*). * p ≤ 0.05; ** p ≤ 0.01, and *** p ≤ 0.001. “NS” means non-significant.
The AN9181 encodes for a protein containing a NmrA-like domain. NmrA is a negative transcriptional regulator of several fungi, involved in the post-translational modulation of the GATA transcription factor AreA (Hensel et al., 1998). In A. nidulans, areA regulates the activation of genes involved in the utilization of a broad range of N sources: in the presence of rich N sources, e.g., ammonium and glutamine, NmrA binds to AreA preventing nitrogen catabolic gene expression; contrarily in the presence of nitrate, NmrA and AreA dissociation occurs, hence genes involved in the utilization of alternative nitrogen sources are activated (Han et al., 2016). Based on this, we questioned if AN9181 influences the utilization of different N sources. Specifically, we measured the cellular metabolic activity of both the ΔAN9181 and the parental strain in distinct media. Measurements were similar in both strains grown in media having rich N sources (i.e., MEA and ammonium sulfate) (Figure 3G) and a high availability of sodium nitrate (poor alternative N source) (Supplementary Figure 4B). However, the mutant showed a significant increase in metabolic activity when grown in media with a low availability of a poor N source (10 mM sodium nitrate) or no added N source (Figure 3G). Another mutant - #2ΔAN9181, randomly selected, was used for validation purposes. This mutant showed consistently similar metabolic activity in media having rich N sources and increased metabolic activity in media with low availability of a poor N source or no added N source (Supplementary Figure 5). In addition, the ΔAN9181 and the parental strain were tested in MMG supplemented with 10 mM N of each of the 20 proteinogenic amino acids. Compared to the parental strain, the mutant strain showed higher metabolic activity in tryptophan (aromatic); serine, glycine and methionine (serine family); asparagine (aspartate family); and alanine and valine (pyruvate family) (Figure 3H). Collectively the results suggest that AN9181 influences the utilization of N sources (hence also spore germination fitness) in a nitrogen type specific manner. Overall, the mutant phenotype suggests that AN9181 participates in the regulation of nitrogen catabolism in A. nidulans, resembling NmrA negative regulation of N utilization in many rich N sources. Further assays are however needed to better understand AN9181 regulatory network in the context of nitrogen utilization.
The growth of ΔAN9181 strain in the presence of each organic compound initially covered in the investigated transcriptome datasets was tested. To standardize conditions, all growth assays used MMG supplemented with the compounds under test at their determined EC50 values (Supplementary Table 5). In the presence of BaP, BMQ and IDQ no differences in growth were observed between the parental and the mutant strain. On the contrary, in media supplemented with sodium salicylate or resveratrol, the ΔAN9181 strain grows more than the parental strain (Figure 4A). The cultivation conditions varied from those of the initial studies, including medium composition, time and temperature, as well as the concentration of the aromatic compounds. To test if the latter was influencing the phenotype, we tested the mutant’s growth in media supplemented with increasing concentrations of two selected aromatic compounds. Salicylate is degraded via the catechol branch of the 3-oxoadipate pathway in A. nidulans (Martins et al., 2015). The Ascomycota Phomopsis liquidambari degrades resveratrol into 3,5-dihydroxybenzaldehyde and 4-hydroxybenzaldehyde, which are subsequently oxidized to 3,5-dihydroxybenzoic acid and 4-hydroxybenzoic acid, respectively (Abo-Kadoum et al., 2022). The latter, is an intermediate of the protocatechuate branch of the 3-oxoadipate pathway used by A. nidulans to degrade benzoate. Therefore, it is possible that resveratrol and benzoate pathways are interconnected in A. nidulans, thus the phenotype was tested in increasing concentrations of sodium salicylate and sodium benzoate (the last instead of resveratrol). The results showed that the ΔAN9181 strain growth-phenotype is indeed concentration dependent for sodium salicylate and for sodium benzoate: a clear phenotype is noticed for concentrations >150 mM and > 55 mM, respectively (i.e., concentrations equal or above the EC50 determined for these compounds; see Supplementary Table 5) (Figures 4B,C).
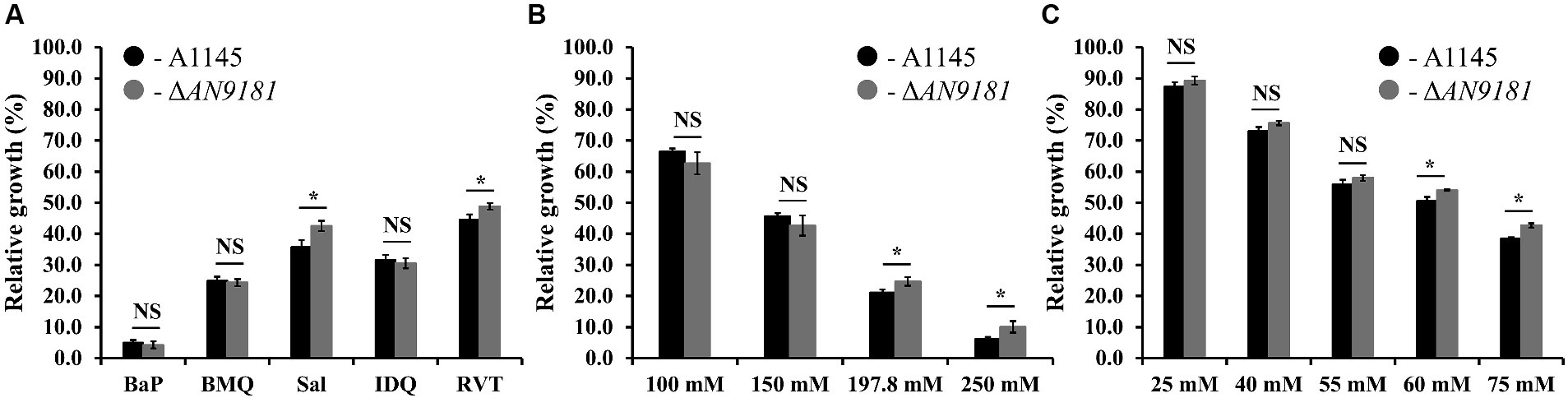
Figure 4. Phenotype features of A1145 and ΔAN9181. (A–C) relative growth of the A1145 (black) and ΔAN9181 (gray) strains grown in minimal medium glucose (solid) supplemented with the EC50 concentrations of each organic compound compared to the control condition (no chemical addition). BaP (Benzo[a]pyrene, 1.61 mM); BMQ (5-Bromoquinoline, 0.2 mM); Sal (Sodium Salicylate, 197.8 mM); IDQ (6-iodoquinoline, 0.2 mM) and RVT (Resveratrol, 0.45 mM) (A); and with increasing concentrations of sodium salicylate (B) or sodium benzoate (C). Values and error bars represent the mean and the standard deviation of triplicates. Significant differences (Student’s t-test) are marked with asterisks [*). *p ≤ 0.05; **p ≤ 0.01, and ***p ≤ 0.001. “NS” means non-significant.
The absence of a phenotype in three (out of five) of the organic compounds herein tested (Figure 4A) is likely related to the fact that the used concentrations were below the threshold to cause major stress effect under the utilized cultivation conditions. Moreover, the response is also influenced by the regulation of nitrogen catabolism as analyzed above. The observation that the growth phenotype of the ΔAN9181 strain is sodium salicylate and sodium benzoate concentration dependent, obvious for concentrations above the corresponding EC50 values, is consistent with the working hypothesis that AN9181 regulates stress responsive metabolism.
Recently, it was reported that AN9181 underwent up-regulation during A. nidulans exposure to congo red or amphotericin B grown in a nitrate minimal medium (Antal et al., 2020). This result expands the regulatory impact of AN9181 under growth in stress conditions. We evaluated if AN9181 deletion influences the susceptibility to congo red and to different antifungal drugs: amphotericin B (targets the cell membrane), caspofungin (targets the cell wall), and itraconazole (inhibits the synthesis of ergosterol, altering the cell membrane permeability). The results showed that ΔAN9181 strain susceptibility to amphotericin B is lower compared to the parental strain (Figure 5A). On the contrary, compared to the parental strain, the susceptibility of the mutant strain was similar and higher to caspofungin and itraconazole, respectively (Figure 5A). Similar results were attained with the A1149ΔAN9181 compared to its corresponding parental strain (Supplementary Figure 6A), notwithstanding that the magnitude of the susceptibility decrease to amphotericin B was more obvious. The determined MICs of each antifungal for the ΔAN9181 mutant strain and its parental strain validate these results (Table 2). Specifically, the MIC of amphotericin B for the ΔAN9181 mutant strain increase 1.4-fold compared to that of the parental strain (Table 2). The determined MICs of itraconazole and caspofungin were similar for the mutant and the parental strains (liquid media), regardless of the mutant’s higher susceptibility to itraconazole (solid media). Differences in the growth phenotypes between liquid and solid media have been reported before (Nichols et al., 2011; Martins et al., 2015). These differences can be related to changes in the drug bioavailability and also carbon availability. We also reassessed the growth phenotype of the strains in the presence of congo red (Figure 5A). A significant decrease in the susceptibility of ΔAN9181 mutant strain compared to the parental strain was noticed. The MIC for congo red in either strain is higher than 256 mg·L−1, regardless that the upper inhibitory limit could not be precisely determined due to the strong red color of the media. All tests were conducted in the same growth media, hence the observed differential responses to the antifungal compounds cannot be simply explained by the regulation of nitrogen utilization by AN9181.
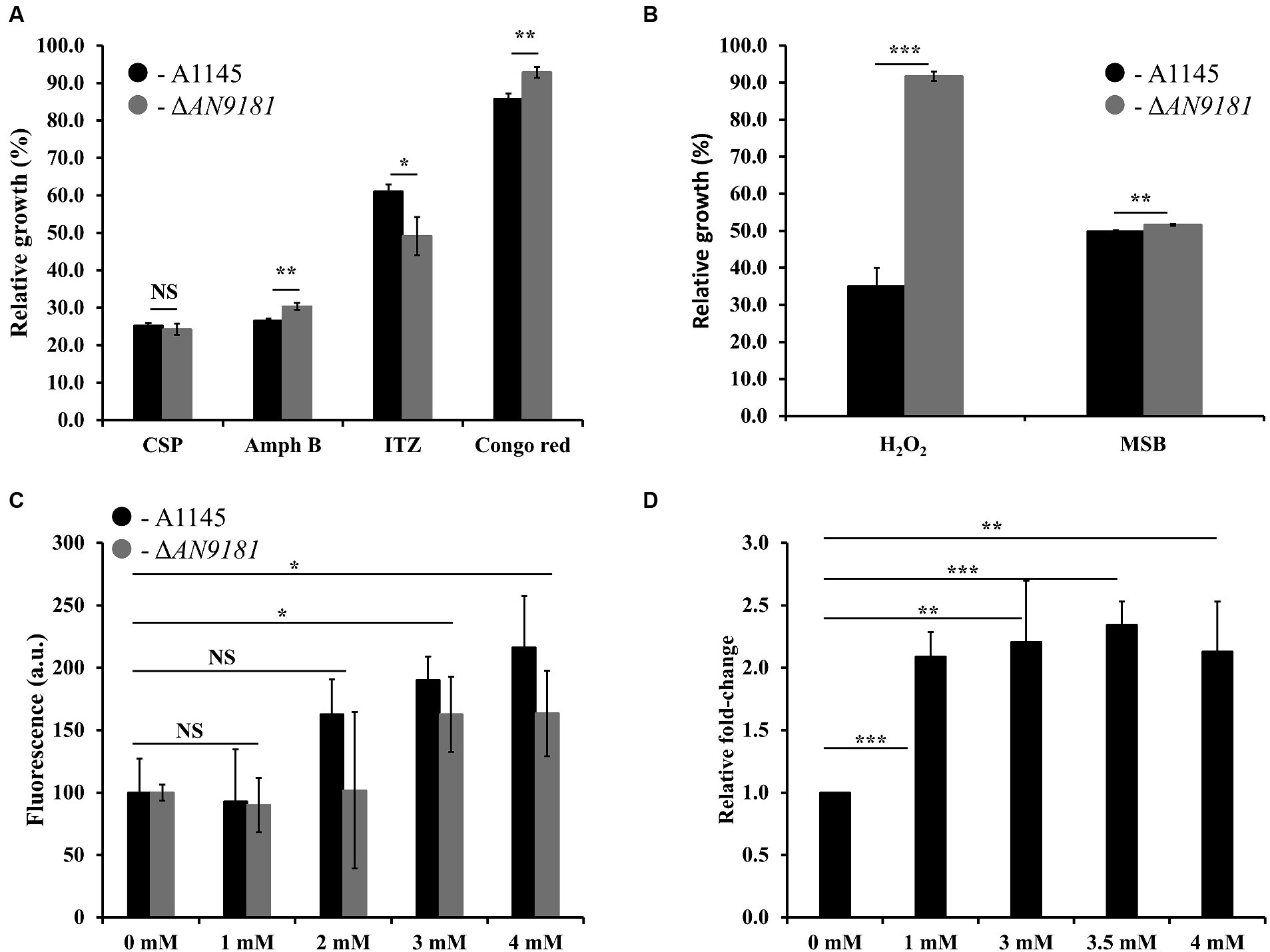
Figure 5. Phenotype features of A1145 and ΔAN9181. (A,B) relative growth of the A1145 (black) and ΔAN9181 (gray) strains grown in minimal medium glucose (solid) supplemented with Caspofungin (0.5 mg·L−1), Amphotericin B (150 mg·L−1), Itraconazole (0.01 mg·L−1), Congo Red (0.01 mM), hydrogen peroxide (3.5 mM) and menadione sodium bisulfite (0.1 mM) compared to the control condition (no chemical addition). Values and error bars represent the mean and the standard deviation of triplicates. (C) Intracellular ROS production, as indicated by DCFH-DA, quantified for the A1145 (black) and ΔAN9181 (gray) strains in the presence of different concentrations of H2O2. The fluorescence intensity per biomass dry weight amount of the control was defined as 100. Values and error bars represent the mean and the standard deviation of at least five replicates. (D) targeted gene expression analysis (assessed by RT-qPCR) is displayed for AN9181. Fold change relative to the control (without H2O2 addition) and normalized to AN3469. Significant differences (Student’s t-test) are marked with asterisks [*). *p ≤ 0.05; **p ≤ 0.01, and ***p ≤ 0.001. “NS” means non-significant.
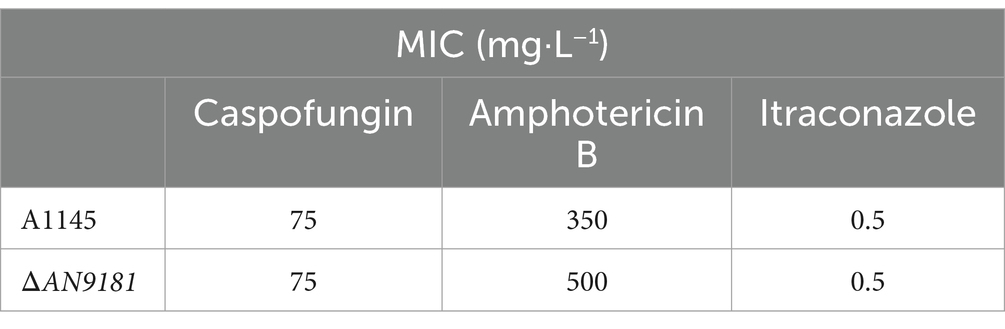
Table 2. MICs of antifungals A1145 and ΔAN9181 strains.
Additionally, the growth phenotype of the strains in the presence of H2O2 and in the presence of the superoxide menadione sodium bisulfite was assessed. The susceptibility of the ΔAN9181 mutant strain compared to the parental strain showed a major and minor decrease in the presence of H2O2 and MSB, respectively (Figure 5B). Using another mutant – #2ΔAN9181 – it was confirmed that the mutant susceptibility to H2O2 was indeed lower than that of the parental strain (Supplementary Figure 6B).
To complement this result, we evaluated the intracellular ROS levels of the parental and the ΔAN9181 mutant strains after exposure to specific compounds. The ROS intracellular levels increased upon exposure of either strain to 3 mM or higher concentrations of H2O2 (Figure 5C). ROS levels increased when the parental strain was exposed to either Amph B or RVT at a concentration where its growth was lower than that of the mutant but slight decreased when exposed to either BMQ and ITZ at concentrations where its growth was similar or higher than that of the mutant, respectively (Supplementary Figure 7). Finally, we observed that the expression of AN9181 in the parental strain underwent a 2-fold increase in the presence of H2O2 (≥ 1 mM) compared to the negative control; a clear indication that the AN9181 is upregulated under oxidative stress (Figure 5D). Collectively the results suggest that AN9181 participates indeed in stress responses upon exposure to different chemical agents leading to oxidative stress.
In this study, we aimed to disclose aspergilli genes showing a conserved response when exposed to different organic compounds. For that, we used mostly peer-reviewed public data in combination with a straightforward computational strategy to test the strength of this scientific hypothesis. Relevant transcriptome-based datasets (Wang et al., 2015; Ben Yaakov et al., 2017; Loss et al., 2019; Martins et al., 2021) (one of which was yet unpublished), initially focusing very different biological questions were selected (five datasets in total). The computational strategy herein used allowed to pinpoint AN9181 as the only gene that showed the same differential upregulation in all five datasets. The AN9181 orthogroup comprises only two genes in A. nidulans: AN9181 and AN8970. These genes are located in different clusters of the AN9181 orthogroup gene tree and possess distinct functional domains. Only AN9181 showed differential upregulation when the fungus was grown in media containing distinct chemical stressors, including two that were used in the previous studies, and two additional toxic aromatic compounds. Therefore, subsequent experimental analyzes focused on the gene AN9181, specifically by studying the phenotype of the generated single-deletion mutant compared to the parental strain. This included measuring the viability of the conidia (i.e., germination fitness), the metabolic profile in different nitrogen sources, the radial growth in the presence of different stress conditions and the minimal inhibitory concentration to different antifungal drugs. The acquired data showed that the deletion of the gene AN9181 lead to higher metabolic activities in different N sources, and a decreased susceptibility to sodium salicylate, resveratrol, H2O2, MSB, congo red and amphotericin B (a clinically relevant antifungal drug). These opening results support the hypothesis that AN9181 is involved in the regulation against different stress responses, including oxidative stress, in aspergilli, impacting also nitrogen utilization. This gene, AN9181, herein assigned as NmrB (Nitrogen Metabolite Repression Regulator B), deserves focused in-deep analysis, especially as it is not only conserved in aspergilli but also in several other fungi (e. g. Candida spp., Fusarium spp., or Neurospora spp., to name a few). The identification of the AN9181 as a putative regulator of stress in aspergilli could not be anticipated, regardless that some of the transcriptome-based datasets used here were generated years ago. This strategy can help to identify (yet) cryptic genetic elements by reusing publicly available peer-reviewed transcriptome-based data from a diversity of scientific fields. It remains unresolved the nature of its regulation but AN9181 apparently is able to slow down growth and metabolic activity in conditions that would be otherwise harmful for the fungus. The absence of orthologs in the human genome (according to OrthoMCL) increases the significance of this inaugural observations, especially in the context of invasive aspergillosis.
The data presented in the study are deposited in the NCBI repository, accession number PRJNA1065793.
JJ: Formal analysis, Investigation, Methodology, Writing – original draft, Writing – review & editing. CM: Conceptualization, Investigation, Methodology, Writing – original draft, Writing – review & editing. PD: Formal analysis, Investigation, Writing – review & editing. TM: Formal analysis, Investigation, Writing – review & editing. DH: Investigation, Writing – review & editing. GG: Funding acquisition, Resources, Writing – review & editing. CS: Conceptualization, Funding acquisition, Project administration, Resources, Supervision, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was financially supported by Fundação para a Ciência e a Tecnologia (FCT) by Project MOSTMICRO ITQB with refs UIDB/04612/2020 and UIDP/04612/2020, LS4FUTURE Associated Laboratory (LA/P/0087/2020) and Project FATE with ref. PDTC/CTA-AMB/6587/2020. JJ is grateful for the working contract PTDC/CTA-AMB/6587/2020. CM and DH are grateful for the fellowships SFRH/BD/118377/2016 and SFRH/BPD/121354/2016. TM is grateful for the working contract financed by national funds under norma transitória D.L. n.° 57/2016.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1373469/full#supplementary-material
Abo-Kadoum, M. A., Abouelela, M. E., al Mousa, A. A., Abo-Dahab, N. F., Mosa, M. A., Helmy, Y. A., et al. (2022). Resveratrol biosynthesis, optimization, induction, bio-transformation and bio-degradation in mycoendophytes. Front. Microbiol. 13:13. doi: 10.3389/fmicb.2022.1010332
Antal, K., Gila, B. C., Pócsi, I., and Emri, T. (2020). General stress response or adaptation to rapid growth in aspergillus nidulans? Fungal Biol. 124, 376–386. doi: 10.1016/j.funbio.2019.10.009
Behera, B. C. (2020). Citric acid from aspergillus Niger: a comprehensive overview. Crit. Rev. Microbiol. 46, 727–749. doi: 10.1080/1040841X.2020.1828815
Ben Yaakov, D., Shadkchan, Y., Albert, N., Kontoyiannis, D. P., and Osherov, N. (2017). The quinoline bromoquinol exhibits broad-spectrum antifungal activity and induces oxidative stress and apoptosis in Aspergillus fumigatus. J. Antimicrob. Chemother. 72, 2263–2272. doi: 10.1093/jac/dkx117
CLSI, Performance standards for antimicrobial susceptibility testing: Approved 28th (2018). Wayne, PA: CLSI.
Czaplicka, M. (2004). Sources and transformations of chlorophenols in the natural environment. Sci. Total Environ. 322, 21–39. doi: 10.1016/j.scitotenv.2003.09.015
Emms, D. M., and Kelly, S. (2015). OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 16:157. doi: 10.1186/s13059-015-0721-2
Emms, D. M., and Kelly, S. (2017). STRIDE: species tree root inference from gene duplication events. Mol. Biol. Evol. 34, 3267–3278. doi: 10.1093/molbev/msx259
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
Gabaldón, T., and Koonin, E. V. (2013). Functional and evolutionary implications of gene orthology. Nat. Rev. Genet. 14, 360–366. doi: 10.1038/nrg3456
Galagan, J. E., Calvo, S. E., Cuomo, C., Ma, L. J., Wortman, J. R., Batzoglou, S., et al. (2005). Sequencing of aspergillus nidulans and comparative analysis with a. fumigatus and A. oryzae. Nature 438, 1105–1115. doi: 10.1038/nature04341
Gupta, S. K., Srivastava, M., Osmanoglu, Ö., Xu, Z., Brakhage, A. A., and Dandekar, T. (2021). Aspergillus fumigatus versus genus aspergillus: conservation, adaptive evolution and specific virulence genes. Microorganisms 9:2014. doi: 10.3390/microorganisms9102014
Hajjaj, H., Niederberger, P., and Duboc, P. (2001). Lovastatin biosynthesis by aspergillus terreus in a chemically defined medium. Appl. Environ. Microbiol. 67, 2596–2602. doi: 10.1128/AEM.67.6.2596-2602.2001
Han, X., Qiu, M., Wang, B., Yin, W. B., Nie, X., Qin, Q., et al. (2016). Functional analysis of the nitrogen metabolite repression regulator gene nmrA in aspergillus flavus. Front. Microbiol. 7:1794.
Harms, H., Schlosser, D., and Wick, L. Y. (2011). Untapped potential: exploiting fungi in bioremediation of hazardous chemicals. Nat. Rev. Microbiol. 9, 177–192. doi: 10.1038/nrmicro2519
Heberle, H., Meirelles, G. V., da Silva, F. R., Telles, G. P., and Minghim, R. (2015). InteractiVenn: a web-based tool for the analysis of sets through Venn diagrams. BMC Bioinform. 16, 1–7. doi: 10.1186/s12859-015-0611-3
Hensel, M., Arst Jr, H. N., Aufauvre-Brown, A., and Holden, D. W. (1998). The role of the Aspergillus fumigatus areA gene in invasive pulmonary aspergillosis. Mol. Gen. Genet. 258, 553–557. doi: 10.1007/s004380050767
Kafer, E. (1977). Meiotic and mitotic recombination in aspergillus and its chromosomal aberrations. Adv. Genet. 19, 33–131.
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Loss, E. M. O., Lee, M. K., Wu, M. Y., Martien, J., Chen, W. P., Amador-Noguez, D., et al. (2019). Cytochrome P450 monooxygenase-mediated metabolic utilization of benzo[a]pyrene by aspergillus species. MBio. 10:e00558–197. doi: 10.1128/mBio.00558-19
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Machida, M., Yamada, O., and Gomi, K. (2008). Genomics of aspergillus oryzae: learning from the history of Koji mold and exploration of its future. DNA Res. 15, 173–183. doi: 10.1093/dnares/dsn020
Martins, T. M., Hartmann, D. O., Planchon, S., Martins, I., Renaut, J., and Silva Pereira, C. (2015). The old 3-oxoadipate pathway revisited: new insights in the catabolism of aromatics in the saprophytic fungus aspergillus nidulans. Fungal Genet. Biol. 74, 32–44. doi: 10.1016/j.fgb.2014.11.002
Martins, C., Hartmann, D. O., Varela, A., Coelho, J. A. S., Lamosa, P., Afonso, C. A. M., et al. (2020). Securing a furan-based biorefinery: disclosing the genetic basis of the degradation of hydroxymethylfurfural and its derivatives in the model fungus aspergillus nidulans. Microb. Biotechnol. 13, 1983–1996. doi: 10.1111/1751-7915.13649
Martins, T. M., Martins, C., Guedes, P., and Silva Pereira, C. (2021). Twists and turns in the salicylate catabolism of aspergillus terreus, revealing new roles of the 3-Hydroxyanthranilate pathway. mSystems 6:e00230–20. doi: 10.1128/mSystems.00230-20
Martins, T. M., Martins, C., and Silva Pereira, C. (2019). Multiple degrees of separation in the central pathways of the catabolism of aromatic compounds in fungi belonging to the Dikarya sub-kingdom. Adv. Microb. Physiol. 75, 177–203. doi: 10.1016/bs.ampbs.2019.07.003
Martins, C., Piontkivska, D., Mil-Homens, D., Guedes, P., Jorge, J. M. P., Brinco, J., et al. (2023). Increased production of pathogenic, airborne fungal spores upon exposure of a soil Mycobiota to chlorinated aromatic hydrocarbon pollutants. Microbiol. Spectr. 11:e0066723. doi: 10.1128/spectrum.00667-23
Martins, C., Varela, A., Leclercq, C. C., Núñez, O., Větrovský, T., Renaut, J., et al. (2018). Specialisation events of fungal metacommunities exposed to a persistent organic pollutant are suggestive of augmented pathogenic potential. Microbiome 6:208. doi: 10.1186/s40168-018-0589-y
Morgan, M., Jones, P., and Sobus, J. (2015). Short-term variability and predictors of urinary pentachlorophenol levels in Ohio preschool children. Int. J. Environ. Res. Public Health 12, 800–815. doi: 10.3390/ijerph120100800
Nichols, R. J., Sen, S., Choo, Y. J., Beltrao, P., Zietek, M., Chaba, R., et al. (2011). Phenotypic landscape of a bacterial cell. Cell 144, 143–156. doi: 10.1016/j.cell.2010.11.052
Núñez-Corcuera, B., Serafimidis, I., Arias-Palomo, E., Rivera-Calzada, A., and Suarez, T. (2008). A new protein carrying an NmrA-like domain is required for cell differentiation and development in Dictyostelium discoideum. Dev. Biol. 321, 331–342. doi: 10.1016/j.ydbio.2008.06.027
Pearce, C. J. (2019). Review of new and future developments in microbial biotechnology and bioengineering: aspergillus system properties and applications. J. Nat. Prod. 82:1051. doi: 10.1021/acs.jnatprod.9b00211
Rokas, A. (2022). Evolution of the human pathogenic lifestyle in fungi. Nat. Microbiol. 7, 607–619. doi: 10.1038/s41564-022-01112-0
Stajich, J. E., Harris, T., Brunk, B. P., Brestelli, J., Fischer, S., Harb, O. S., et al. (2012). FungiDB: an integrated functional genomics database for fungi. Nucleic Acids Res. 40, D675–D681. doi: 10.1093/nar/gkr918
Steenwyk, J. L., Mead, M. E., Knowles, S. L., Raja, H. A., Roberts, C. D., Bader, O., et al. (2020). Variation among biosynthetic gene clusters, secondary metabolite profiles, and cards of virulence across aspergillus species. Genetics 216, 481–497. doi: 10.1534/genetics.120.303549
Varela, A., Martins, C., Núñez, O., Martins, I., Houbraken, J. A., Martins, T. M., et al. (2015). Understanding fungal functional biodiversity during the mitigation of environmentally dispersed pentachlorophenol in cork oak forest soils. Environ. Microbiol. 17, 2922–2934. doi: 10.1111/1462-2920.12837
Varela, A., Martins, C., and Silva Pereira, C. (2017). A three-act play: pentachlorophenol threats to the cork oak forest soils mycobiome. Curr. Opin. Microbiol. 37, 142–149. doi: 10.1016/j.mib.2017.06.007
Vogel, H. J. (1956). A convenient growth medium for Neurospora (medium N). Microb. Genet. Bull. 13, 42–43.
Wang, Y., Cao, Y. Y., Jia, X. M., Cao, Y. B., Gao, P. H., Fu, X. P., et al. (2006). Cap1p is involved in multiple pathways of oxidative stress response in Candida albicans. Free Radic. Biol. Med. 40, 1201–1209.
Wang, H., Lei, Y., Yan, L., Cheng, K., Dai, X., Wan, L., et al. (2015). Deep sequencing analysis of transcriptomes in aspergillus flavus in response to resveratrol. BMC Microbiol. 15:182. doi: 10.1186/s12866-015-0513-6
World Health Organization. WHO fungal priority pathogens list to guide research, development and public health action, I. 978-92-4-006024-1, Editor. (2022).
Keywords: organic chemicals, stress responses, transcriptomics, aspergilli, phenylcoumaran benzylic ether reductase-like domain (IPR045312), NmrA-like domain (IPR008030), global regulators, oxidative stress
Citation: Jorge JMP, Martins C, Domingos P, Martins TM, Hartmann DO, Goldman GH and Silva Pereira C (2024) NmrB (AN9181) expression is activated under oxidative stress conditions acting as a metabolic repressor of Aspergillus nidulans. Front. Microbiol. 15:1373469. doi: 10.3389/fmicb.2024.1373469
Edited by:
Z. Petek Cakar, Istanbul Technical University, TürkiyeReviewed by:
Vineet Kumar, The University of Texas at Austin, United StatesCopyright © 2024 Jorge, Martins, Domingos, Martins, Hartmann, Goldman and Silva Pereira. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Celso Martins, Y2Vsc28ubWFydGluc0B1bmlsLmNo; Cristina Silva Pereira, c3BlcmVpcmFAaXRxYi51bmwucHQ=
‡Present addresses: Celso Martins, Center for Integrative Genomics, Faculty of Biology and Medicine, University of Lausanne, Lausanne, Switzerland
Diego O. Hartmann, Sea4Us – Biotecnologia e Recursos Marinhos S.A., Porto de Pesca da Baleeira, Armazém 8, Sagres, Portugal
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.