 Shuangshuang Wan1,2Peijun You1Qikai Shi1Hui Hu1,2Lu Zhang3
Shuangshuang Wan1,2Peijun You1Qikai Shi1Hui Hu1,2Lu Zhang3 Leyang Chen4
Leyang Chen4 Ziyi Wu4Shan Lin1,5Xiaojun Song6Yongneng Luo1,2Yaxuan Wang7Feng Ju3
Ziyi Wu4Shan Lin1,5Xiaojun Song6Yongneng Luo1,2Yaxuan Wang7Feng Ju3 Dazhi Jin1,2*†
Dazhi Jin1,2*† Yu Chen1,2*†
Yu Chen1,2*†- 1School of Laboratory Medicine, Hangzhou Medical College, Hangzhou, China
- 2Key Laboratory of Biomarkers and In Vitro Diagnosis Translation of Zhejiang Province, Hangzhou, China
- 3Environmental Microbiome and Biotechnology Laboratory, School of Engineering, Westlake University, Hangzhou, China
- 4School of Clinical Medicine, Hangzhou Medical College, Hangzhou, China
- 5TEDA Institute of Biological Sciences and Biotechnology, Nankai University, Tianjin, China
- 6Laboratory Medicine Center, Department of Clinical Laboratory, Zhejiang Provincial People’s Hospital, Hangzhou Medical College, Hangzhou, China
- 7National Center for Clinical Laboratories, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing Hospital/National Center of Gerontology, Beijing, China
Introduction: Clostridioides difficile infection (CDI), as well as its etiology and pathogenesis, have been extensively investigated. However, the absence of suitable CDI animal models that reflect CDI symptoms and the associated gut microbiome changes in humans has limited research progress in this field. Thus, we aimed to investigate whether Mongolian gerbils, which present a range of human pathological conditions, can been used in studies on CDI. Methods: In this study, we infected Mongolian gerbils and two existing CDI model animals, mice and hamsters, with the hypervirulent ribotype 027 C. difficile strain, and comparatively analyzed changes in their gut microbiome composition via 16S rRNA gene sequencing.
Methods: In this study, we infected Mongolian gerbils and two existing CDI model animals, mice and hamsters, with the hypervirulent ribotype 027 C. difficile strain, and comparatively analyzed changes in their gut microbiome composition via 16S rRNA gene sequencing.
Results: The results obtained showed that C. difficile colonized the gastrointestinal tracts of the three rodents, and after the C. difficile challenge, C57BL/6J mice did not manifest CDI symptoms and their intestines showed no significant pathological changes. However, the hamsters showed explosive intestinal bleeding and inflammation and the Mongolian gerbils presented diarrhea as well as increased infiltration of inflammatory cells, mucus secretion, and epithelial cell shedding in their intestinal tissue. Further, intestinal microbiome analysis revealed significant differences with respect to intestinal flora abundance and diversity. Specifically, after C. difficile challenge, the Firmicutes/Bacteroidetes ratio decreased for C57BL/6J mice, but increased significantly for Mongolian gerbils and hamsters. Furthermore, the abundance of Proteobacteria increased in all three models, especially in hamsters, while that of Verrucomicrobia only increased significantly in C57BL/6J mice and Mongolian gerbils. Our results also indicated that differences in the relative abundances of Lactobacillaceae and Akkermansia were primarily responsible for the observed differences in response to C. difficile challenge.
Conclusion: Based on the observed responses to C. difficile challenge, we concluded for the first time that the Mongolian gerbil could be used as an animal model for CDI. Additionally, the taxa identified in this study may be used as biomarkers for further studies on CDI and to improve understanding regarding changes in gut microbiome in CDI-related diseases.
1 Introduction
Clostridioides difficile is a gram-positive spore-forming obligate anaerobic bacterium. In patients with long-term use of antibiotics or low immunity, the balance of intestinal flora is disrupted, such that C. difficile adheres to host intestinal epithelial cells, rapidly proliferates the intestinal niche, and produces toxins, such as C. difficile toxin A [TcdA] and C. difficile toxin B [TcdB] as well as binary toxins. These toxins trigger a cascade of cellular events that lead to fluid secretion, inflammation, and tissue damage, which bring about the symptoms of C. difficile infection (CDI), such as pseudomembranous colitis, toxic megacolon, and even death (Juul et al., 2018; Zhou et al., 2019; Rao and Malani, 2020).
CDI and its etiology and pathogenesis have been extensively investigated (Wydau-Dematteis et al., 2018; Castro-Córdova et al., 2021; Luo et al., 2022). Specifically, the progression of CDI has been studied using various animal species, including hamsters, guinea pigs, rabbits, mice, and rats. Among these animals, hamsters have been most widely used in CDI research (Borriello et al., 1987; Best et al., 2012). However, they often develop fulminant cecitis and ultimately die within 1–3 days following challenge with C. difficile. This phenomenon is rarely observed in the clinical course of CDI in humans, in whom CDI shows a more gradual onset, with death being rare (Abutaleb and Seleem, 2021; Singh et al., 2022). Furthermore, a mouse model of antibiotic-induced C. difficile-associated colitis that presents CDI symptoms similar to those in humans, including diarrhea, pseudomembrane formation, variable disease severity, and recovery of disease after vancomycin therapy, has been established. Thus, it serves as an important tool for exploring CDI pathogenesis (Wu et al., 2022). However, mouse models do not always show overt clinical signs of CDI, despite remaining colonized with C. difficile (Reeves et al., 2011). The Mongolian gerbil (Meriones unguiculatus) is an efficient and cost-effective rodent that shows several features which are absent in mice or rats, including relatively low intragastric pH, sensory specializations, and social patterns, similar to humans (Sheh and Fox, 2013; Zorio et al., 2019). Their use in the study of several pathogenic diseases is also very common. For example, studies involving the use of Mongolian gerbils as a model of infection have shown that Helicobacter pylori-infected Mongolian gerbils develop high levels of gastric inflammation and often present with gastric adenocarcinoma or ulceration (Beckett et al., 2018). In contrast, H. pylori can only induce mild inflammation in many mouse models, and it is difficult in establishing infection in mice (Kabir et al., 1997; Lee et al., 1997). However, it is still unclear whether Mongolian gerbils can be used as a novel model. Gut microbes modulate nutrition and immune function and are associated with a growing number of health and disease states (Sharon et al., 2014; Lynch and Pedersen, 2016). For example, in the healthy state, the structure and diversity of the human gut microbiome is relatively stable (Lloyd-Price et al., 2017). However, in the diseased state, this balance is significantly dysregulated. In recent decades, changes in gut microbiome structure have been increasingly recognized as holding promise with respect to advancing CDI research. Antibiotic exposure leading to microbiome disruption and overgrowth of C. difficile is a major risk factor for the development of CDI (Vakili et al., 2020a). It has also been demonstrated that CDI is associated with severe gut microbiome disruption (Buffie et al., 2012; Zhang et al., 2022). CDI-related structural and functional changes in gut microbiome have been investigated in human, and there were significant differences on the composition of gut microbiome between patients with CDI and healthy persons (Berkell et al., 2021). Thus, it was reasonable that change in gut microbiome was recognized as a biomarker to predict development of CDI. Moreover, structural changes in gut microbiome have also been used to comparatively analyze the features for CDI between rodents and human, indicating that CDI similarly led to microbiome dysbiosis of animal (Gu et al., 2020). Nevertheless, it has been still unclear whether changes in gut microbiome configuration were associated with diverse outcomes in different animals after C. difficile challenge. Therefore, in this study, we experimented with three rodent-based CDI models (mice, hamsters, and Mongolian gerbils) and 16S rRNA gene sequencing and analyzed changes in their gut microbiome structure, owing to C. difficile challenge.
2 Materials and methods
2.1 Bacterial strain and growth conditions
Clostridioides difficile strain 4118, obtained from American Type Culture Collection (www.atcc.org), was cultured anaerobically at 37°C on brain heart infusion agar (Oxoid Ltd., Basingstoke, UK) supplemented with 1.5% (w/v) glucose, 0.1% (w/v) L-cysteine, 1.5% (w/v) agar, and 0.1% (w/v) sodium taurocholate. Thereafter, the strain was inoculated into 500 mL of tryptone yeast broth containing 3% tryptone, 2% yeast extract, and 0.1% sodium thioglycolate and further cultured anaerobically at 37°C for 7 days, after which spores were harvested via centrifugation performed at 1,500 g and 4°C for 20 min, washed six times with ddH2O, and resuspended in phosphate buffered saline (PBS). Next, heat-shocking was performed at 65°C for 30 min to remove bacteria. The spores were then 10-fold diluted and anaerobically cultured at 37°C for 48 h after, which the number of spores was calculated. C. difficile was isolated from stool samples from the control and infected rodents, and four toxin genes (tcdA, tcdB, cdtA, and cdtB) were identified as previously reported (Indra et al., 2008; Griffiths et al., 2010).
2.2 Animals
Male C57BL/6 J mice (age, 6–8 weeks) were purchased from Shanghai Laboratory Animal Co., Ltd. (Shanghai, China). Male golden hamsters (age, 6–8 weeks) were purchased from Beijing Weitong Lihua Laboratory Animal Technology Co., Ltd. (Beijing, China), and male Mongolian gerbils (age, 10–12 weeks) were purchased from the Experimental Animal Center of Hangzhou Medical College, China. The animals were housed at the Laboratory Animal Center at Hangzhou Medical College (23°C ± 2°C room temperature, 55% ± 5% relative humidity, and a 12 L:12D photoperiod), at a density of three animals per cage in a specific pathogen-free animal facility. The experimental protocols were approved by the Animal Care and Use Committee of the abovementioned institution (approval number SYXK 2017-0013). Furthermore, all the animal experiments were performed in accordance with Animal Research: Reporting of In Vivo Experiments guidelines (Percie Du Sert et al., 2020). The mice were euthanized 5 days after anesthesia administration via the intraperitoneal injection of chloral hydrate (375 mg/kg of body weight).
2.3 Induction of Clostridioides difficile challenge and monitoring of animals
All the animals (mice, hamsters, and Mongolian gerbils) were randomized into groups with corresponding average body weights to establish the necessary durations of antibiotic pretreatment. The control and challenge groups for each species comprised five animals. Furthermore, the fresh antibiotic mixture, which comprised kanamycin (0.4 mg/mL), gentamicin (0.035 mg/mL), metronidazole (0.215 mg/mL), colistin (850 U/mL), and vancomycin (0.045 mg/mL), was purchased from Sigma–Aldrich (St. Louis, MO, United States). Once this mixture was prepared, it was added to the drinking water of the animals, which was replaced daily for 3 days. After this 3-day antibiotic pretreatment, the animals were further treated with clindamycin (10 mg/kg) for 1 day, as described previously (Chen et al., 2008) and then challenged with 5 × 104 colony-forming units of C. difficile strain 4,118 via oral gavage. The animals were then monitored for signs of disease, such as diarrhea, wet tail, bow back, malaise, and weight loss. After 5 days, the animals were euthanized, and their stool samples and intestinal tissues were collected for analysis.
2.4 Detection of TcdB toxins in stool samples via ELISA
TcdB toxins in stool samples from the control and infected rodents were detected via ELISA. The collected stool samples were suspended in 10% (wt/vol) PBS (pH 7.4). Thereafter, the fecal suspensions were seeded into 96-well plates and incubated at 4°C overnight. This was followed by washing with PBST (PBS pH 7.4 + 0.05% tween-20) after which the plate was blocked with 0.5% BSA at 37°C for 2 h. TcdB polyclonal antibodies (List Biological Laboratories Campbell, CA, United States; Chicken IgY, 1:1000 dilution ratio) were used for the first hybridization process, while goat anti-chicken IgY secondary antibody and horse radish peroxidase (Invitrogen, Carlsbad, CA, USA; 1:5000 dilution ratio) were used for the second hybridization. The ELISA-TMB Chromogenic Reagent kit (Sangon, Shanghai, China) was used to determine the absorbance at 450 nm. Pure TcdB toxin (List Biological Laboratories) was used as the positive control, and standard curves were generated.
2.5 Histopathological analysis
Resected ileal loops were fixed in 4% formaldehyde, buffered with PBS, and embedded in paraffin. Next, the tissue sections were deparaffinized, stained with hematoxylin and eosin, and subjected to histological analysis. Tissue injuries were scored by two blinded pathologists, who independently graded inflammation according to five grades based on the degrees of chronic inflammation and tissue damage, as described previously (Rugge et al., 2021).
2.6 Extraction and amplification of genomic DNA
Total genomic DNA was extracted from 0.25 g of intestinal contents using the Dneasy® PowerSoil® Pro Kit (QIAGEN, Hilden, Germany), according to the manufacturer’s protocol. DNA concentration and purity were verified via electrophoresis using 1% agarose gel. Furthermore, the genomic DNA was diluted to 1 ng/Μl using sterile water, and the 16S V3–V4 region of the 16S rRNA gene was amplified using specific primers (341F-5′-CCT AYG GGR BGC ASC AG-3′ and 805R-5′-GAC TAC HVG GGT ATC TAA TCC-3′). All the PCR assays were performed using 15 μL of PCR Master Mix (New England Biolabs, Ipswich, MA, United States), 2 μM forward and reverse primers, and approximately 15 ng of template DNA. The PCR program was as follows: denaturation at 94°C for 3 min, 5 cycles of denaturation at 94°C for 30 s, annealing at 45°C for 20 s, elongation at 65°C for 30 s, 20 cycles of denaturation at 94°C for 20 s, annealing at 55°C for 20 s, elongation at 72°C for 30 s, and 72°C for 5 min. The PCR products obtained were purified using the QIAGEN Gel Extraction Kit (QIAGEN).
2.7 16S rRNA gene sequence analysis
The TruSeq DNA PCR Free sample preparation kit (Illumina, San Diego, CA, United States) was used to generate the sequencing library, and an index code was added to acquire operational taxonomic unit (OTU) clusters (sequences with ≥97% similarity were assigned to the same OTU). Taxonomic assignment in terms of 16S rRNA was performed based on the SILVA 132 reference database1 using UCLUST2 at a 90% threshold. Furthermore, taxonomic assignment based on the ribosomal internal transcribed spacer region was performed using the UNITE reference database (https://unite.ut.ee/, version 01.12.2017) in BLAST at an e-value threshold of 0.001. Furthermore, the determination of phylogenetic relationships based on OTUs, differences in dominant species between different samples (groups), and multiple sequence alignments were carried out using PyNAST software version 1.2 (University of Colorado, Boulder, CO, United States).
2.8 Microbial community diversity and functional analysis
Mothur3 was used to determine parameters related to the complexity and diversity of the intestinal microbiota of the animals, such as abundance-based coverage estimators (ACE) and Chao 1 richness. Simpson, Shannon, and beta diversity indices on both weighted and unweighted UniFrac were also calculated using QIIME software. Cluster analysis was performed using principal component analysis, principal coordinate analysis, and non-metric multidimensional scaling (NMDS). Furthermore, linear discriminant analysis effect size (LefSe) was performed using LefSe software, and the filter value for the linear discriminant analysis (LDA) score was set to 2. Furthermore, to investigate the functions and pathways involved with C. difficile challenge in the test animals, Kyoto Encyclopedia of Genes and Genomes (KEGG) functions were predicted using Tax4Fun software,4 and intergroup differences were analyzed in terms of functional abundance differences among the samples.
2.9 Statistical analysis
All data were presented as the mean ± SEM and were compared by performing t-test using GraphPad Prism software version 8 (San Diego, CA, United States). p < 0.05 was considered statistically significant. Furthermore, p-values relative to the control group (*p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001) were determined via one-way analysis of variance (ANOVA).
3 Results
3.1 Construction of three rodent models with Clostridioides difficile challenge
Three rodent models with C. difficile challenge were established using the classical intragastric administration method, as shown in Figure 1A. Before challenge, the C. difficile 4,118 strains with four toxin genes were not detected in stool samples of three rodents (Supplementary Figure S1A). On the second day after intragastric C. difficile administration, the stool samples were collected and cultured for C. difficile. The results showed that C. difficile 4,118 strains were obtained from stool samples of three rodents, and four toxin genes were positive (Supplementary Figure S1B). TcdB was also detected in all stool samples (Figure 1B). This observation indicated that C. difficile successfully existed in the intestines of the three rodents and produced the corresponding virulence factors. Based on changes in the body weights of the three rodents and their survival proportions, we found that both Mongolian gerbils and hamsters were susceptible to C. difficile, which showed weight loss (Figure 1C). Specifically, all the Mongolian gerbils challenged with C. difficile exhibited diarrhea (Supplementary Figure S2D). Furthermore, the survival rate of the hamsters was significantly lower than those of the gerbils and mice (Figure 1C). Our results also indicated that no diarrhea in mice challenged with C. difficile (Supplementary Figure S2B), and their weights did not change significantly relative to the weights of the controls (Figure 1C).
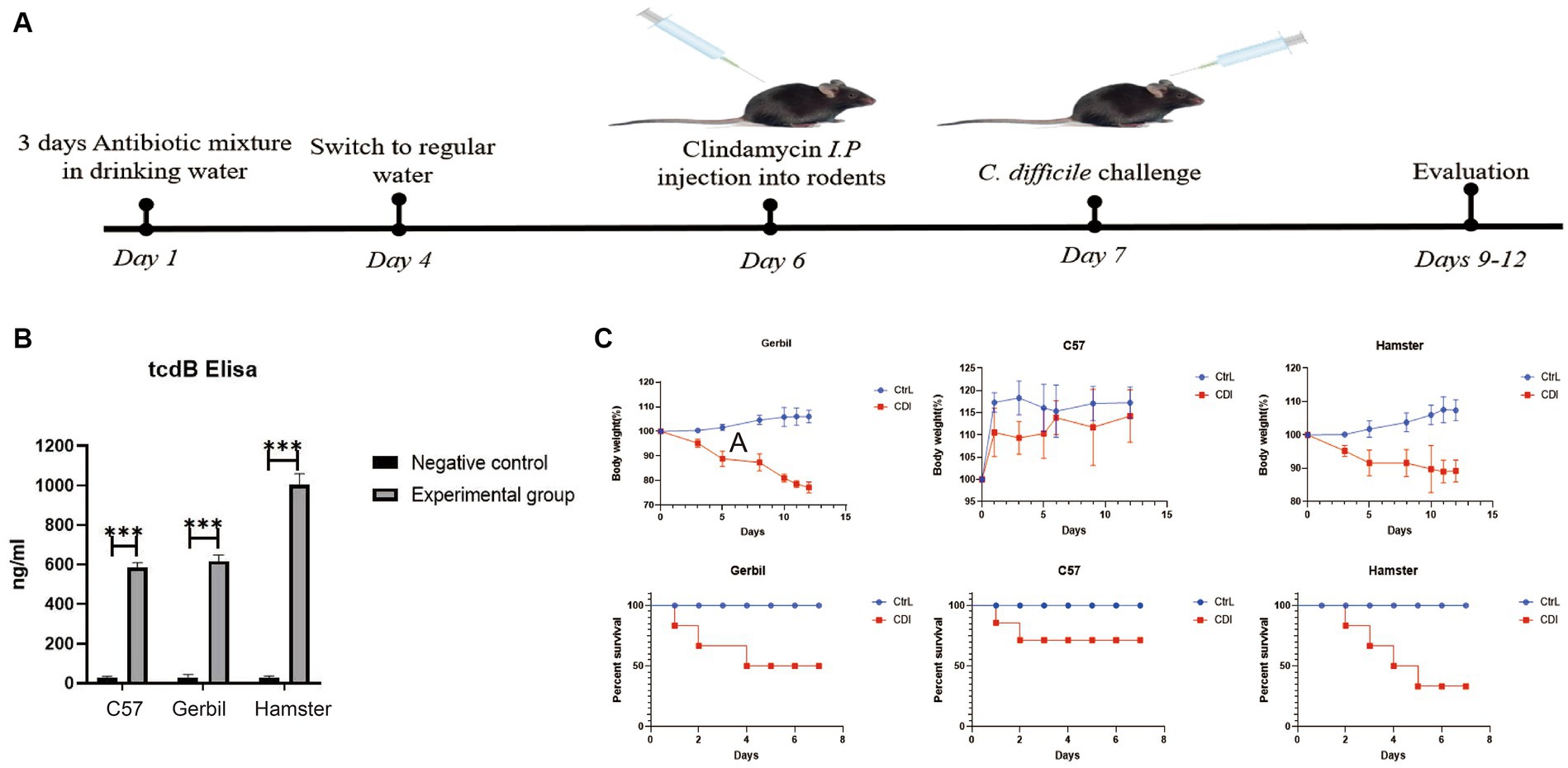
Figure 1. Evaluation of three rodent models with C. difficile challenge. (A) Schematic representation of animal models with C. difficile challenge. (B) TcdB content of stool samples from three rodents determined via ELISA. (C) Changes in body weight and survival proportions after C. difficile challenge. C57, C57Bl/6 J mouse; gerbil, Mongolian gerbil.
3.2 Intestinal histopathology of the three model animals after Clostridioides difficile challenge
Intestinal histopathology changes were detected in all the rodents (Figure 2). The intestinal mucosal cells of the animals in the three control groups were closely packed and were clearly structured, without hyperemia, edema, and inflammatory infiltration. However, after infection, the hamsters exhibited pathological changes, including explosive intestinal bleeding, and quickly died owing to severe enterocolitis. C57BL/6J mice did not manifest diarrhea. Furthermore, the pathological changes in their intestine were not significant; infection only resulted in the irregular arrangement of myometrium cells, with loose cell arrangement and limited inflammatory cell infiltration. Mongolian gerbils exhibited characteristics that increased infiltration of inflammatory cells into intestinal tissues, including neutrophils, increased mucus secretion and significant epithelial cell shedding. This observation suggested that histopathology in Mongolian gerbil is a typical histologic feature of CDI.
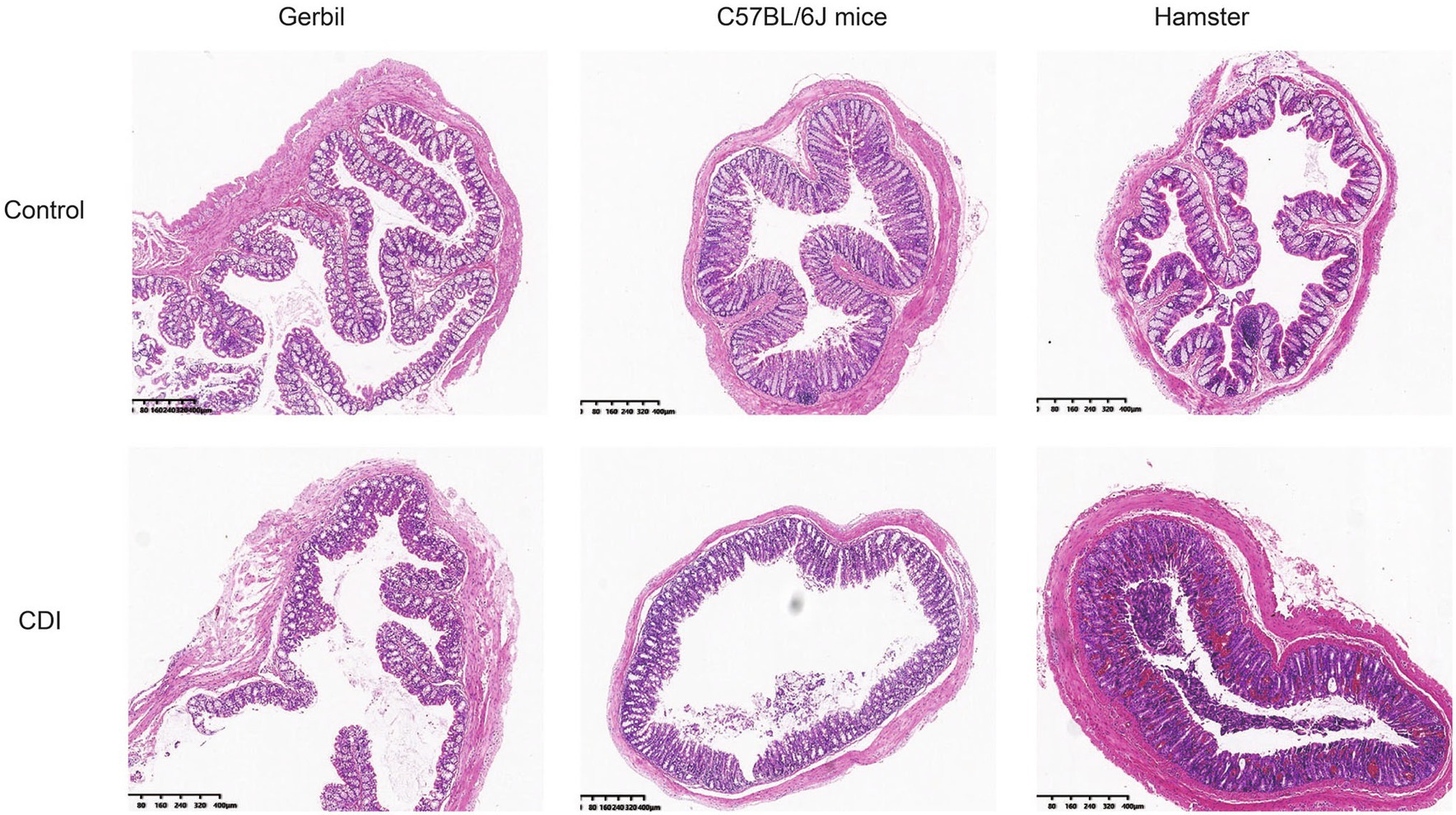
Figure 2. Intestinal histopathology after C. difficile challenge in three rodent models. Histopathologic examination of the colon of Mongolian gerbils, mice, and hamsters after C. difficile challenge; Gerbil, Mongolian gerbil.
3.3 Comparative analysis of intestinal microbial diversity in the three rodent models before and after Clostridioides difficile challenge
Thirty intestinal contents were collected from six rodent groups (five CDI-treated rodents and five control rodents for each species) and subjected to microbiota community analysis using 16S rRNA amplicon sequencing. After quality control, 2,020,374 sequences (40,146 reads per sample) were included for downstream analysis. The overall beta-diversity of gut microbiome signatures in the three challenged and three control rodent groups was analyzed via NMDS. The results revealed different community structures for the gut microbiome in each of the six groups. Samples with high community structure similarities were clustered together, whereas samples with large community structure differences were separated. The NMDS results further showed a separation between the three rodent CDI-treated and control groups and, notably, revealed that the least distance existed between the gut microbiomes of the challenged mouse and gerbil (Figure 3A). This finding suggests that the gut microbiome of mice and Mongolian gerbils possibly became similar following CDI. Furthermore, alpha-diversity indices showed higher phylogenetic diversity in the challenged groups than their corresponding control groups. Notably, hamster gut microbiome showed the greatest changes following C. difficile challenge, Mongolian gerbils, and lastly mice (Figure 3B). The Chao1 and Shannon indices obtained were consistent with the phylogenetic diversity results (Figures 3C–E).
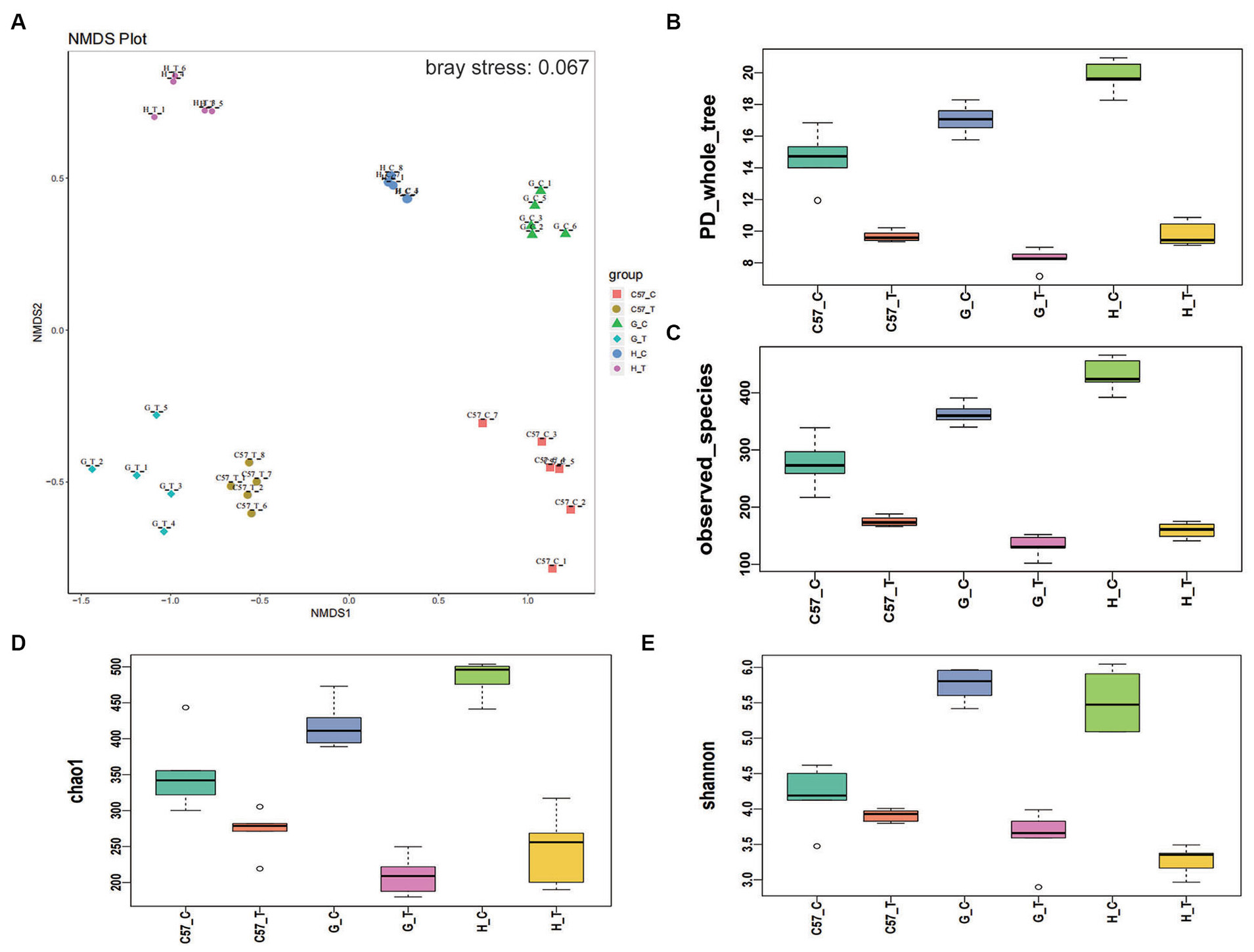
Figure 3. Differences in alpha- and beta-diversity indices between rodent groups after C. difficile challenge. Boxplots of group differences between alpha-diversity and beta-diversity indices: (A) NMDS, (B) phylogenetic diversity whole tree, (C) observed species, (D) Chao1, and (E) Shannon. C, control; C57, C57Bl/6 J mouse; G, Mongolian gerbil; H, hamster; T, treated (i.e., challenged with C. difficile), NMDS, non-metric multidimensional scaling.
3.4 Analysis of microbial community composition differences among the three rodent CDI models before and after Clostridioides difficile challenge
To further analyze the relationship between intestinal flora and CDI, we comparatively explored the dynamic alterations in the composition of the intestinal microbiome in the C57BL/6 J mouse, hamster, and Mongolian gerbil models before and after challenge with C. difficile. At the phylum level, we observed seven bacterial phyla, of which the four most abundant were Firmicutes, Bacteroidetes, Proteobacteria, and Verrucomicrobia, which jointly contributed to an average sequence ratio of approximately 90%. Actinobacteria, Epsilonbacteraeota, Patescibacteria, and Tenericutes only accounted for approximately 6% of the sequences (Figure 4). The gut microbiomes in the three rodent models appeared to be dominated by Firmicutes, followed by Bacteriodetes, while the relative abundances of Proteobacteria, Verrucomicrobia, and Actinobacteria were very low, ranging from 0.03 to 7.8%. Furthermore, after challenge with C. difficile, the relative abundance of Firmicutes in C57BL/6 J mice decreased significantly but was almost unchanged in Mongolian gerbils and hamsters. We also noted that after challenge with C. difficile, the relative abundance of Bacteriodetes in the C57BL/6J mice increased but decreased in the two other species, especially in hamsters, in which Bacteroidetes were hardly detected. The Firmicutes to Bacteriodetes ratio also decreased in C57BL/6 J mice but increased in the other two rodent models, especially in hamsters. Furthermore, the three rodent models, especially hamsters, showed an increase in the relative abundance of Proteobacteria, while the relative abundance of Verrucomicrobia showed a significant increase in C57BL/6 J mice and Mongolian gerbils but was very low in hamsters (undetectable). The relative abundance of Actinobacteria in C57BL/6 J mice and gerbils increased but basically remained unchanged in hamsters.
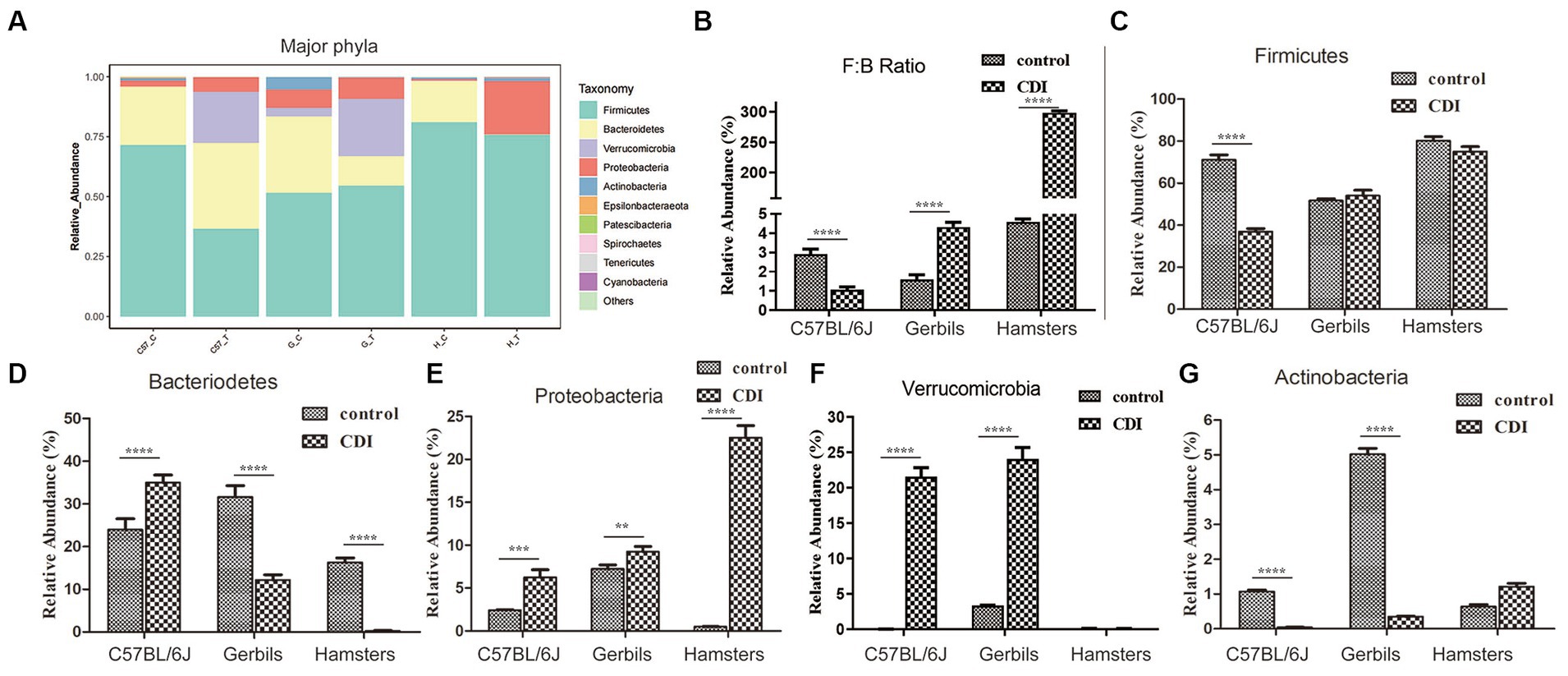
Figure 4. Differences in the relative abundances of major gut bacterial phyla after C. difficile challenge. (A) Relative abundances of the major phyla in each sample. (B–G) Box-plots representing the mean relative abundances of the Firmicutes and Bacteriodetes ratio, Firmicutes, Bacteriodetes, Proteobacteria, Verrucomicrobia, and Actinobacteria in each sample, respectively. p-values were determined via ANOVA versus control conditions (**p < 0.01, ***p < 0.001, and ****p < 0.0001); Gerbils, Mongolian gerbil.
Consistent with phylum-level data, analyses at the family and genus levels showed that the relative abundances of family and genus varied greatly between the different host species and their controls following CDI. For instance, prior to challenge with C. difficile, Firmicutes in C57BL/6 J mice appeared to be dominated by the family Lactobacillaceae (and the genus Lactobacillus) followed by Lachnospiraceae (and the genus NK4A136_group) (Figures 5A,B). Furthermore, in Mongolian gerbils, Firmicutes predominantly comprised Lachnospiraceae (and the genus NK4A136_group) and Lactobacillaceae (and the genus Lactobacillus), followed by Ruminococcaceae and Erysipelotrichaceae, whereas in hamsters, Firmicutes predominantly comprised Erysipelotrichaceae (and the genera Ileibacterium and Allobaculum) followed by Lachnospiraceae (and the genus NK4A136_group), Lactobacillaceae (and the genus Lactobacillus), and Ruminococcaceae (and the genus Blautia). In the three rodent control groups, Bacteriodetes predominantly comprised the family Muribaculaceae. However, the detection rates for families or genera belonging to Proteobacteria or Verrucomicrobia in the three control groups were very low. After challenge with C. difficile, Firmicutes in C57BL/6J mice predominantly comprised OTUs belonging to the family Lachnospiraceae, followed by Erysipelotrichaceae. Notably, compared with the control C57BL/6 J mice, the relative abundances of the genera Blautia and Lachnoclostridium were higher in challenged mice, while those of the genera Lactobacillus and NK4A136_group were significantly lower. In Mongolian gerbils, the family Lactobacillaceae was the most abundant among Firmicutes. Compared with the corresponding control gerbil group, the relative abundances of genera such as Lactobacillus, Blautia, and Lachnoclostridium were higher, while that of the genus NK4A136_group was very low (undetectable). In hamsters, Firmicutes predominantly comprised OTUs belonging to the family Lactobacillaceae, followed by Erysipelotrichaceae and Lachnospiraceae. Similar to the results obtained for gerbils, compared with the corresponding controls, the genera Lactobacillus and Blautia were more abundant in challenged Mongolian gerbils, while the genus NK4Ahamsters136_group was undetectable. In contrast, the family Erysipelotrichaceae and genera Ileibacterium and Allobaculum were highly abundant in challenged gerbils. Bacteroidetes in C57BL/6J mice and Mongolian gerbils after C. difficile challenge predominantly comprised the family Bacteroidaceae (and the genus Bacteroides), followed by Tannerellaceae. The family Muribaculaceae was undetectable in all three models after C. difficile challenge. Notably, the relative abundances of the families Bacteroidaceae (and the genus Bacteroides) and Tannerellaceae were higher in C57BL/6 J mice than in Mongolian gerbils.
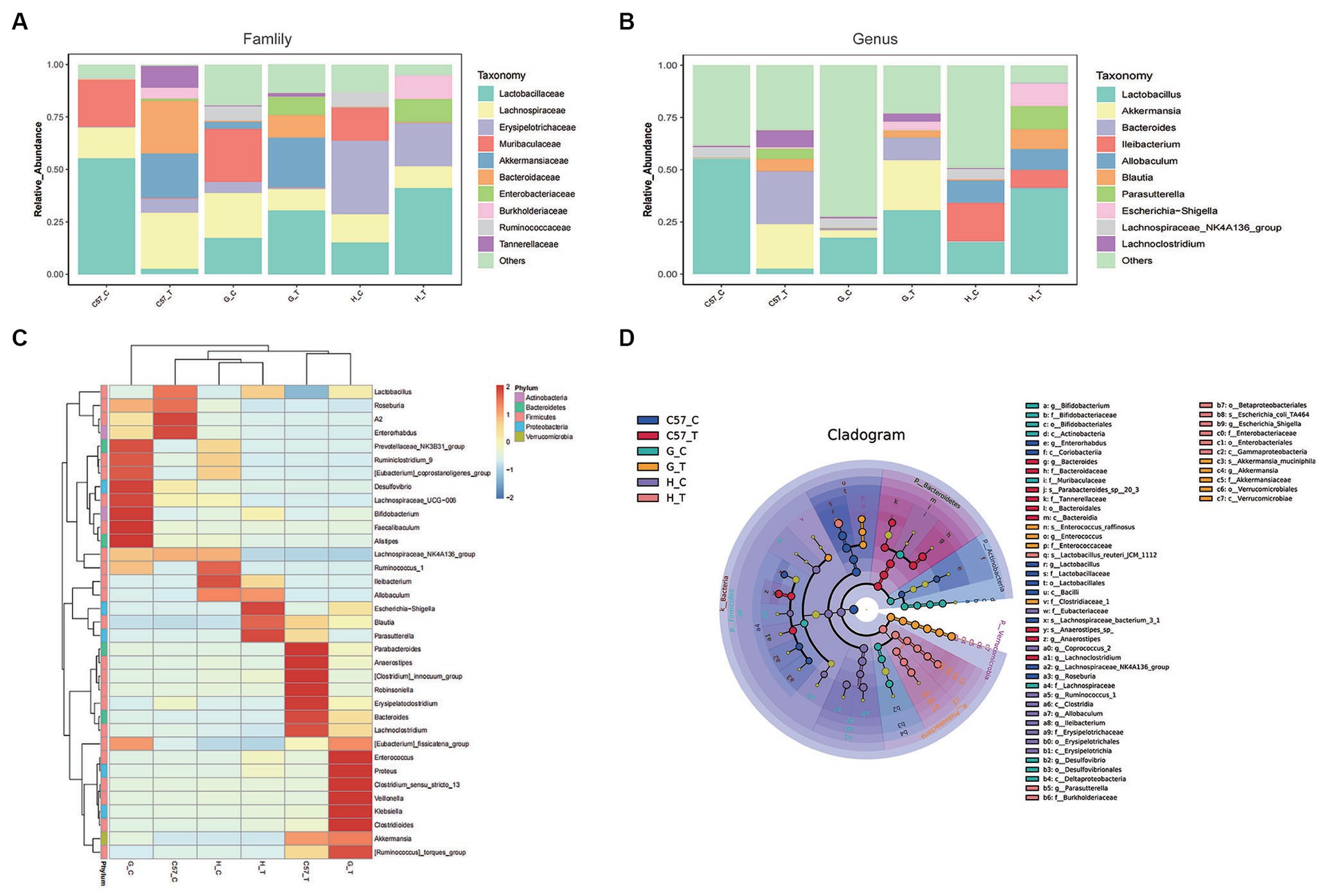
Figure 5. Relative abundance of major gut bacterial families and genera after C. difficile challenge in three rodent models. (A) Bar graphs representing the relative abundances of major families. (B) Bar graphs representing the relative abundances of major genera. (C) Heat-map illustrating the hierarchical clustering of operational taxonomic units (OTUs) in each sample. (D) LEfSe cladogram representing unique bacterial signatures identified in each sample; the corresponding value of the middle heat map represents the Z-value obtained based on the standardized relative abundance of the species in each line; C, control; C57, C57Bl/6 J mouse; G, Mongolian gerbil; H, hamster; T, treated (i.e., challenged with C. difficile).
After challenge with C. difficile, Akkermansia (a major member of the family Verrucomicrobiaceae) showed a high relative abundance in C57BL/6 J mice and Mongolian gerbils but was rarely detected in hamsters (Figures 5A,B). Additionally, the three models challenged with C. difficile predominantly comprised the families Burkholderiaceae (and the genus Parasutterella) and Enterobacteriaceae (and the genus Escherichia−Shigella), and the relative abundances of Parasutterella and Escherichia−Shigella were higher in hamsters than in C57BL/6 J mice and Mongolian gerbils, respectively.
The analysis of the top 35 OTUs detected in the three models via heat map clustering before and after challenge with C. difficile showed similar patterns as reported above. Notably, after challenge with C. difficile, C57BL/6 J, clusters predominantly comprising Blautia, Erysipelatoclostridium, Anaerostipes, Robinsoniella, Lachnoclostridium, Bacteroides, Akkermansia, Parabacteroides, and Parasutterella were observed in C57BL/6J mice (Figure 5C). In Mongolian gerbils, the clusters predominantly comprised Blautia, Erysipelatoclostridium, Anaerostipes, Lachnoclostridium, Bacteroides, Akkermansia, Lactobacillus, Clostridioides, Klebsiella, Escherichia−Shigella, Enterococcus, and Proteus, and in hamsters, they predominantly comprised Lactobacillus, Ileibacterium, Allobaculum, Blautia, Parasutterella, Escherichia−Shigella, Enterococcus, and Proteus. These features were further confirmed via LEfSe analysis of the major OTUs detected in these groups (Figure 5D).
3.5 KEGG analysis of the metabolic functions of the intestinal flora in the three rodent models after Clostridioides difficile challenge
KEGG pathway enrichment analysis showed the involvement of multiple metabolic pathways in C. difficile challenge in the three rodent models. Statistically significant differences were also observed among the three models. Difference analysis at the second classification level performed under the conditions: relative abundance >0.1% of the total observed pathways and threshold p-value = 0.05. The results showed 36, 24, and 31 differential pathways in C57BL/6 J mice, Mongolian gerbils, and hamsters, respectively (Figure 6). Among the three groups, the terms “amino acid metabolism,” “membrane transport,” “metabolism of cofactors and vitamins,” “signal transduction,” “nucleotide metabolism,” “energy metabolism,” and “glycan biosynthesis and metabolism” dominated gut microbiome functions. Notably, upregulation of “glycan biosynthesis and metabolism” was observed in the three groups, while the other pathways were only consistent for Mongolian gerbils and hamsters but not for C57BL/6 J mice. Additionally, in C57BL/6 J mice, “amino acid metabolism,” “metabolism of cofactors and vitamins,” and “signal transduction and energy metabolism” were upregulated, while “membrane transport” and “nucleotide metabolism” were downregulated. The identified pathways in the three groups and their functions at the third classification level are shown in detail in Supplementary Figures S3–S5. We also observed that in C57BL/6 J mice, “cysteine and methionine metabolism,” “two-component system,” “oxidative phosphorylation,” “nitrogen metabolism,” “carbon fixation pathways,” and “sulfur metabolism” were upregulated, while “phosphotransferase system (PTS)” was downregulated, but in Mongolian gerbils and hamsters, “phosphotransferase system (PTS)” remained unchanged or showed trends opposite to those observed in C57BL/6 J mice.
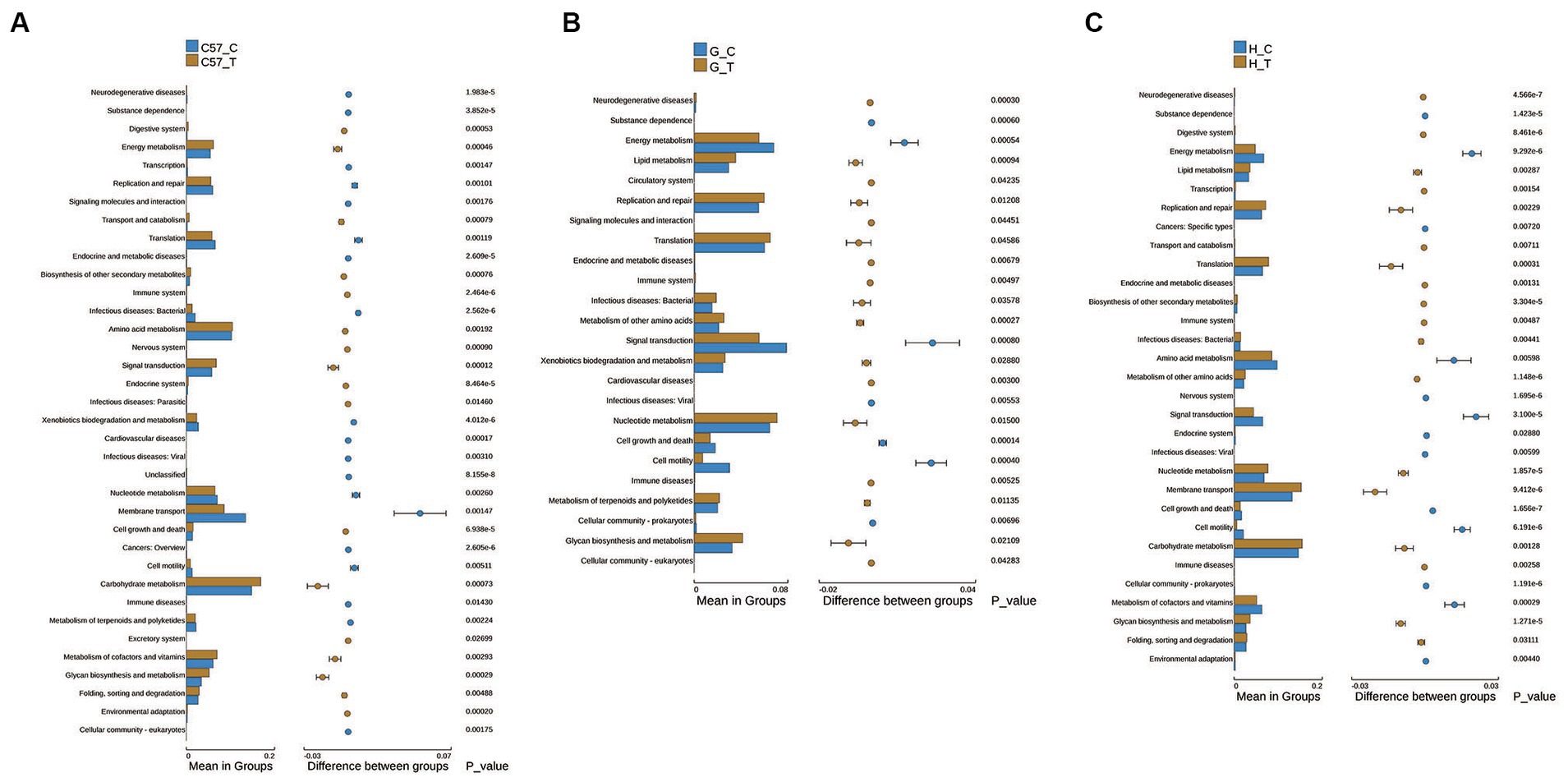
Figure 6. Differential metabolic pathways in functional level 2 determined via KEGG pathway enrichment analysis of the intestinal flora of three rodent models after C. difficile challenge. (A) C57BL/6 J mice, (B) Mongolian gerbils, and (C) Hamsters. The right panel shows the difference in confidence between the groups, with the leftmost and rightmost ends of each circle indicating the lower and upper limits of the 95% confidence interval for the mean difference, respectively. The groups represented using individual-colored circles are those with a high mean. The far-right end of the circle represents the intergroup significance test p-values for the corresponding differential function; C, control; C57, C57Bl/6 J mouse; G, Mongolian gerbil; H, hamster; T, treated (i.e., challenged with C. difficile).
4 Discussion
Even though there have been reports of animal models of CDI, significant differences among host species in terms of susceptibility to CDI are apparent, and host responses to CDI vary greatly depending on the animal species (Keel and Songer, 2006). This study is the first to report that Mongolian gerbils can be used as a model for CDI, to comparatively analyze gut microbiome among C57BL/6 J mice, Mongolian gerbils, and hamsters. Notably, the three rodent models presented different symptoms, degrees of intestinal histopathology, and changes in gut microbiome after C. difficile challenge. The hamsters challenged with C. difficile exhibited pathological changes, including explosive intestinal bleeding, and quickly died owing to severe enterocolitis. This result is consistent with those obtained in previous studies (Abutaleb and Seleem, 2021; Singh et al., 2022). C57BL/6 J mice did not manifest diarrhea, and furthermore, the pathological changes in their intestine were not significant. This finding is inconsistent with the previously reported findings (Chen et al., 2008; Wang et al., 2022). Histopathology in Mongolian gerbil is a typical histologic feature of CDI. Changes in their gut microbiome might be the main factor, leading to diverse response after C. difficile challenge.
An increasing number of studies have suggested that changes in gut microbiome composition play an important role in host health status (Zorio et al., 2019; Su et al., 2022). It has also been shown that a highly dense and diverse intestinal microbiome favors CDI prevention. Individuals colonized by C. difficile may not progress directly to symptomatic CDI, depending on the intestinal microenvironment, host immunity, and pathogen-related factors (AbdelKhalek and Narayanan, 2022; Reasoner et al., 2023). Furthermore, patients with CDI generally exhibit a significant loss of microbiota diversity, characterized primarily by the relative abundances of decreased Bacteroides, Firmicutes, Lachnospiraceae, and Ruminococcaceae, accompanied by an increase in the relative abundance of Proteobacteria, Lactobacilliaceae, Enterococcaceae, and Streptococcaceae (Milani et al., 2016; Gonzales-Luna et al., 2023). Additionally, Enterococcus, Lactobacillus, Escherichia coli, and Akkermansia are enriched in patients with CDI, and this enrichment is associated with various metabolic processes (Pérez-Cobas et al., 2014; Vakili et al., 2020a). In our studies, the gut microbiome of Mongolian gerbils after CDI was more similar to that of patients, as shown in decreased relative abundances of Bacteroides, Lachnospiraceae, and Ruminococcaceae, with corresponding increases in the relative abundances of Proteobacteria, Lactobacilliaceae, and Akkermansia. Thus, Mongolian gerbils showed different CDI symptoms than mice and hamasters and might show a similar trend with human in gut microbiota changes after C. difficile challenge. Enterobacteriaceae (Proteobacteria) are enriched in both animals and patients with CDI (Reeves et al., 2011; Schubert et al., 2014). The microbiota in rCDI patients is dominated by Enterobacteriaceae (Collins and Auchtung, 2017). Enterobacteriaceae are capable of thriving in the inflamed intestine and perpetuating continued inflammation (Bäumler and Sperandio, 2016). Thus, Enterobacteriaceae may have growth advantages in the gut environment of patients with CDI. Similar observations were made in this study; Enterobacteriaceae were enriched in Mongolian gerbils and hamsters challenged with C. difficile, and their overgrowth destroyed intestinal permeability and hence worsened colitis. The relative abundance of Enterobacteriaceae was lower in C57BL/6 J mice challenged with C. difficile than in challenged Mongolian gerbils and hamsters, and this animal showed no obvious intestinal tissue damage.
Some studies have shown that species of the family Lactobacillaceae can prevent the growth of pathogens (González-Lozano et al., 2022; Walter and O’toole, 2023). However, Lactobacillaceae was dominant in CDI samples from both humans and mice (Rea et al., 2011; Antharam et al., 2013). In this study, after challenge with C. difficile, we observed a significant decrease in the relative abundance of the Lactobacillaceae family in C57BL/6 J mice, while the Lachnospiraceae family became the most abundant Firmicutes. Contrasting observations were made for Mongolian gerbils and hamsters. These findings may be attributed to species of the family Lactobacillaceae, metabolizing hexoses via homofermentative or heterofermentative metabolism. KEGG functional enrichment analysis further revealed that PTS was decreased in C57BL/6 J mice but increased in Mongolian gerbils and hamsters. Reportedly, heterofermentative species of the family Lactobacillaceae harbor fewer PTS than homofermentative species (Zheng et al., 2015). Additionally, homofermentative species of the family Lactobacillaceae robustly acidify and inhibit the growth of Lachnospiraceae and Muribaculaceae (Lagkouvardos et al., 2019; Brownlie et al., 2022). A decrease in the abundance of Lachnospiraceae, resulting in low butyric acid production, may be associated with the development of ulcerative colitis (Sasaki et al., 2019). Additionally, Muribaculaceae can produce propionate as the main fermentation product, which, reportedly, is associated with gut health and an extended lifespan in mice (Smith et al., 2021). Furthermore, Bacteroides can degrade carbohydrates to form short-chain fatty acids (SCFAs), such as acetate, propionate, and butyrate (Koh et al., 2016). Notably, SCFAs can inhibit intestinal inflammation by upregulating G protein-coupled receptor 43 (GPR43), which facilitates the effects of hepatic portal pulsatile inflammatory factors (Maslowski et al., 2009). A decrease in the relative abundance of Bacteroidetes is associated with irritable bowel syndrome (Rajilić-Stojanović et al., 2011). Therefore, it is possible that, following challenge with C. difficile in Mongolian gerbils and hamsters, homofermentative species of the family Lactobacillaceae produce higher quantities of acid to inhibit the growth of Lachnospiraceae and Muribaculaceae, and the resulting low level of SCFAs may trigger intestinal pathology. However, in C57BL/6 J mice, although the abundance of Muribaculaceae was reduced, Lachnospiraceae and Bacteroides increased SCFA content to protect the intestine. Furthermore, in previous studies, C57BL/6 mice from different vendors had different levels of two different Lactobacillus spp. and had different responses to H. pylori infection, highlighting the importance of husbandry and the environment on the gastrointestinal microbiota profile (Rolig et al., 2013). This may also be the reason that mice are not sensitive to C. difficile challenge.
The changes in the relative abundance of Akkermansia were also interesting in three rodents challenged with C. difficile. In patients with CDI, an increased abundance of Akkermansia is associated with degraded mucin, which gives selective advantage to C. difficile, adhering to mucus (Hernández et al., 2018). Additionally, Akkermansia overrepresentation may reflect an epiphenomenon, leading to enteric mucosal inflammation with increased mucus production (Milani et al., 2016; Vakili et al., 2020b). An elevated abundance of Akkermansia may be a predictive marker for the presence of a CDI (Hernández et al., 2018). In this study, the abundance of Akkermansia also significantly increased in Mongolian gerbils challenged with C. difficile. Akkermansia was overrepresented in infected Mongolian gerbils, which was consistent with human CDI. However, we also observed increased Akkermansia levels in C57BL/6 J mice, and this genus was undetectable in hamsters challenged with C. difficile. A. muciniphila, as one of typical members in Akkermansia, found in the human gut, is a mucin-degrading bacterium that continuously remodels and refreshes the mucus layer, thereby creating a healthy environment for epithelial cells and maintaining intestinal barrier integrity (Everard et al., 2013). It has been reported that A. muciniphila could improve the abundance of Akkermansia, which alleviated intestinal inflammation reduced by CDI through ameliorating the intestinal barrier function and microbiota structure (Wu et al., 2022). This observation suggested that Akkermansia possibly played an important role in intestinal anti-inflammation in C57BL/6 J mice and hamsters challenged with C. difficile. Thus, the functions of Akkermansia might be diverse in three rodents. In Mongolian gerbils, the increased abundance of Akkermansia might be a potential predictive marker for CDI.
The results of this study also showed that some metabolic pathways were altered and, possibly, were related to CDI. Specifically, “glycan biosynthesis and metabolism” was upregulated in the three rodent groups, indicating that after challenge with C. difficile, each rodent model showed increased food utilization, which facilitated adaptation to pathogen infection. In CDI, sulfide, a direct precursor of both methionine and cysteine, plays an important role in sulfur metabolism (Dubois et al., 2016). It has also been shown that the accumulation of cysteine by-products can inhibit toxin production by C. difficile (Gu et al., 2017, 2018). Enhanced sulfur, cysteine, and methionine metabolism can also repress toxin production in C. difficile. Furthermore, the two-component signal transduction system (TCS) can sense and respond to environmental signals and its activation can inhibit sporulation in C. difficile (Kempher et al., 2022). In Mongolian gerbils and hamsters challenged with C. difficile, the reduced TCS could increase spore production, followed by subsequent CDI progression. Finally, our results indicated that the metabolism of cofactors and vitamins was upregulated in C57BL/6 J mice. Reportedly, vitamin B6 has pleiotropic functions in human health, e.g., it showed improved immune system activity (Hellmann and Mooney, 2010). This effect of vitamin B may also be the reason C57BL/6 J mice challenged with C. difficile did not manifest CDI symptoms, inconsistent with the results of the previous studies (Wu et al., 2022). Thus, the genetic backgrounds of mice of the same strain may not be completely consistent, owing to reproductive system differences between countries and regions. There were slight differences in feed formula composition, leading to different mouse intestinal flora; therefore, susceptibility to disease may be highly diverse.
This study had some limitations. First, the intestinal contents were used to infer changes in gut microbiome characteristics with a focus on just microbiome composition and function. Relevant data on metabolomics and transcriptomics might be helpful to further validate our findings. Second, we studied changes of gut microbiome, considering only the effect of C. difficile in this regard. Possibly, other factors, such as host response, affect the gut microbiome of the test animals. Thus, further investigations are required to investigate the role of other factors.
In this study, we established a Mongolian gerbil CDI model for the first time. The changes in the gut microbiome during challenge reflected the important role of the gut microbial community in the three rodent models analyzed. After C. difficile challenge, the three rodents showed a significant loss of microbiota diversity. The high abundance of species of the family Lactobacillaceae led to the production of higher quantities of acids to inhibit the growth of Lachnospiraceae and Muribaculaceae, leading to decreases in the levels of SCFAs and triggering intestinal pathology. Moreover, a decrease in the relative abundance of Bacteroidetes and an increase in that of Proteobacteria could also disrupt intestinal permeability and worsen colitis. Therefore, these phyla may be used as potential disease biomarkers or drug targets in the future. In addition, the increase in the Akkermansia population in both C57BL/6 J mice and Mongolian gerbils might result in different outcomes. Taken together, changes in gut microbiome constituted a key factor responsible for inducing the symptoms observed in the three rodent models following C. difficile challenge.
Data availability statement
The original contributions presented in the study are publicly available. This data can be found at: https://www.ncbi.nlm.nih.gov/bioproject/; PRJNA1014570.
Ethics statement
The animal study was approved by Laboratory Animal Center, Hangzhou Medical College. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
SW: Writing – original draft, Conceptualization, Data curation, Investigation, Methodology, Writing – review & editing. PY: Investigation, Writing – review & editing, Data curation, Methodology, Validation. QS: Writing – review & editing, Data curation, Methodology, Validation. HH: Methodology, Writing – review & editing, Formal analysis, Investigation, Software. LC: Writing – review & editing, Data curation, Formal analysis, Software, Validation. ZW: Software, Writing – review & editing, Data curation, Formal analysis, Validation. SL: Writing – review & editing, Methodology, Data curation, Investigation, Supervision. XS: Formal analysis, Writing – review & editing, Validation, Methodology, Software. YL: Writing – review & editing, Formal analysis, Data curation, Validation, Writing – original draft. YW: Resources, Writing – review & editing, Formal analysis, Software. DJ: Writing – original draft, Conceptualization, Funding acquisition, Investigation, Methodology, Supervision, Writing – review & editing, Resources. YC: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Software, Supervision, Validation, Writing – original draft, Writing – review & editing. LZ: Data curation, Investigation, Writing – review & editing. FJ: Data curation, Investigation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Medical Science and Technology Project of Zhejiang Province (2021KY639), the Natural Science Foundation of Zhejiang Province (LQ20H280002 and LXZ22H300001), the National Natural Science Foundation of China (82372299), the Scientific Research Fund of Zhejiang Provincial Education Department (Y201942763 and Y202045376), and the Basic Research Projects of Hangzhou Medical College (KYZD202004, KYQN202002, and KYYB2023003).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1368194/full#supplementary-material
Footnotes
References
Abdelkhalek, A., and Narayanan, S. K. (2022). Comparison between symptomatic and asymptomatic mice after Clostridioides difficile infection reveals novel inflammatory pathways and contributing microbiota. Microorganisms 10:2380. doi: 10.3390/microorganisms10122380
Abutaleb, N. S., and Seleem, M. N. (2021). In vivo efficacy of auranofin in a hamster model of Clostridioides difficile infection. Sci. Rep. 11:7093. doi: 10.1038/s41598-021-86595-3
Antharam, V. C., Li, E. C., Ishmael, A., Sharma, A., Mai, V., Rand, K. H., et al. (2013). Intestinal dysbiosis and depletion of butyrogenic bacteria in Clostridium difficile infection and nosocomial diarrhea. J. Clin. Microbiol. 51, 2884–2892. doi: 10.1128/jcm.00845-13
Bäumler, A. J., and Sperandio, V. (2016). Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535, 85–93. doi: 10.1038/nature18849
Beckett, A. C., Loh, J. T., Chopra, A., Leary, S., Lin, A. S., Mcdonnell, W. J., et al. (2018). Helicobacter pylori genetic diversification in the Mongolian gerbil model. PeerJ 6:e4803. doi: 10.7717/peerj.4803
Berkell, M., Mysara, M., Xavier, B. B., Van Werkhoven, C. H., Monsieurs, P., Lammens, C., et al. (2021). Microbiota-based markers predictive of development of Clostridioides difficile infection. Nat. Commun. 12:2241. doi: 10.1038/s41467-021-22302-0
Best, E. L., Freeman, J., and Wilcox, M. H. (2012). Models for the study of Clostridium difficile infection. Gut Microbes 3, 145–167. doi: 10.4161/gmic.19526
Borriello, S. P., Ketley, J. M., Mitchell, T. J., Barclay, F. E., Welch, A. R., Price, A. B., et al. (1987). Clostridium difficile--a spectrum of virulence and analysis of putative virulence determinants in the hamster model of antibiotic-associated colitis. J. Med. Microbiol. 24, 53–64. doi: 10.1099/00222615-24-1-53
Brownlie, E. J. E., Chaharlangi, D., Wong, E. O., Kim, D., and Navarre, W. W. (2022). Acids produced by lactobacilli inhibit the growth of commensal Lachnospiraceae and S24-7 bacteria. Gut Microbes 14:2046452. doi: 10.1080/19490976.2022.2046452
Buffie, C. G., Jarchum, I., Equinda, M., Lipuma, L., Gobourne, A., Viale, A., et al. (2012). Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 80, 62–73. doi: 10.1128/iai.05496-11
Castro-Córdova, P., Mora-Uribe, P., Reyes-Ramírez, R., Cofré-Araneda, G., Orozco-Aguilar, J., Brito-Silva, C., et al. (2021). Entry of spores into intestinal epithelial cells contributes to recurrence of Clostridioides difficile infection. Nat. Commun. 12:1140. doi: 10.1038/s41467-021-21355-5
Chen, X., Katchar, K., Goldsmith, J. D., Nanthakumar, N., Cheknis, A., Gerding, D. N., et al. (2008). A mouse model of Clostridium difficile-associated disease. Gastroenterology 135, 1984–1992. doi: 10.1053/j.gastro.2008.09.002
Collins, J., and Auchtung, J. M. (2017). Control of Clostridium difficile infection by defined microbial communities. Microbiol. Spectr. 5:BAD-0009-2016. doi: 10.1128/microbiolspec.BAD-0009-2016
Dubois, T., Dancer-Thibonnier, M., Monot, M., Hamiot, A., Bouillaut, L., Soutourina, O., et al. (2016). Control of Clostridium difficile physiopathology in response to cysteine availability. Infect. Immun. 84, 2389–2405. doi: 10.1128/iai.00121-16
Everard, A., Belzer, C., Geurts, L., Ouwerkerk, J. P., Druart, C., Bindels, L. B., et al. (2013). Cross-talk between Akkermansia muciniphila and intestinal epithelium controls diet-induced obesity. Proc. Natl. Acad. Sci. USA 110, 9066–9071. doi: 10.1073/pnas.1219451110
Gonzales-Luna, A. J., Carlson, T. J., and Garey, K. W. (2023). Gut microbiota changes associated with Clostridioides difficile infection and its various treatment strategies. Gut Microbes 15:2223345. doi: 10.1080/19490976.2023.2223345
González-Lozano, E., García-García, J., Gálvez, J., Hidalgo-García, L., Rodríguez-Nogales, A., Rodríguez-Cabezas, M. E., et al. (2022). Novel horizons in postbiotics: Lactobacillaceae extracellular vesicles and their applications in health and disease. Nutrients 14:5296. doi: 10.3390/nu14245296
Griffiths, D., Fawley, W., Kachrimanidou, M., Bowden, R., Crook, D. W., Fung, R., et al. (2010). Multilocus sequence typing of Clostridium difficile. J. Clin. Microbiol. 48, 770–778. doi: 10.1128/jcm.01796-09
Gu, W., Li, W., Wang, W., Kuang, D., Zhang, W., Lu, C., et al. (2020). Response of the gut microbiota during the Clostridioides difficile infection in tree shrews mimics those in humans. BMC Microbiol. 20:260. doi: 10.1186/s12866-020-01943-z
Gu, H., Shi, K., Liao, Z., Qi, H., Chen, S., Wang, H., et al. (2018). Time-resolved transcriptome analysis of Clostridium difficile R20291 response to cysteine. Microbiol. Res. 215, 114–125. doi: 10.1016/j.micres.2018.07.003
Gu, H., Yang, Y., Wang, M., Chen, S., Wang, H., Li, S., et al. (2017). Novel cysteine Desulfidase CdsB involved in releasing cysteine repression of toxin synthesis in Clostridium difficile. Front. Cell. Infect. Microbiol. 7:531. doi: 10.3389/fcimb.2017.00531
Hellmann, H., and Mooney, S. (2010). Vitamin B6: a molecule for human health? Molecules 15, 442–459. doi: 10.3390/molecules15010442
Hernández, M., De Frutos, M., Rodríguez-Lázaro, D., López-Urrutia, L., Quijada, N. M., and Eiros, J. M. (2018). Fecal microbiota of toxigenic Clostridioides difficile-associated Diarrhea. Front. Microbiol. 9:3331. doi: 10.3389/fmicb.2018.03331
Indra, A., Huhulescu, S., Schneeweis, M., Hasenberger, P., Kernbichler, S., Fiedler, A., et al. (2008). Characterization of Clostridium difficile isolates using capillary gel electrophoresis-based PCR ribotyping. J. Med. Microbiol. 57, 1377–1382. doi: 10.1099/jmm.0.47714-0
Juul, F. E., Garborg, K., Bretthauer, M., Skudal, H., Øines, M. N., Wiig, H., et al. (2018). Fecal microbiota transplantation for primary Clostridium difficile infection. N. Engl. J. Med. 378, 2535–2536. doi: 10.1056/NEJMc1803103
Kabir, A. M., Aiba, Y., Takagi, A., Kamiya, S., Miwa, T., and Koga, Y. (1997). Prevention of Helicobacter pylori infection by lactobacilli in a gnotobiotic murine model. Gut 41, 49–55. doi: 10.1136/gut.41.1.49
Keel, M. K., and Songer, J. G. (2006). The comparative pathology of Clostridium difficile-associated disease. Vet. Pathol. 43, 225–240. doi: 10.1354/vp.43-3-225
Kempher, M. L., Morris, S. C., Shadid, T. M., Menon, S. K., Ballard, J. D., and West, A. H. (2022). Response regulator CD1688 is a negative modulator of sporulation in Clostridioides difficile. J. Bacteriol. 204:e0013022. doi: 10.1128/jb.00130-22
Koh, A., De Vadder, F., Kovatcheva-Datchary, P., and Bäckhed, F. (2016). From dietary Fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165, 1332–1345. doi: 10.1016/j.cell.2016.05.041
Lagkouvardos, I., Lesker, T. R., Hitch, T. C. A., Gálvez, E. J. C., Smit, N., Neuhaus, K., et al. (2019). Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 7:28. doi: 10.1186/s40168-019-0637-2
Lee, A., O'rourke, J., De Ungria, M. C., Robertson, B., Daskalopoulos, G., and Dixon, M. F. (1997). A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterology 112, 1386–1397. doi: 10.1016/s0016-5085(97)70155-0
Lloyd-Price, J., Mahurkar, A., Rahnavard, G., Crabtree, J., Orvis, J., Hall, A. B., et al. (2017). Strains, functions and dynamics in the expanded human microbiome project. Nature 550, 61–66. doi: 10.1038/nature23889
Luo, J., Yang, Q., Zhang, X., Zhang, Y., Wan, L., Zhan, X., et al. (2022). TFPI is a colonic crypt receptor for TcdB from hypervirulent clade 2 C. difficile. Cell 185, 980–994.e15. doi: 10.1016/j.cell.2022.02.010
Lynch, S. V., and Pedersen, O. (2016). The human intestinal microbiome in health and disease. N. Engl. J. Med. 375, 2369–2379. doi: 10.1056/NEJMra1600266
Maslowski, K. M., Vieira, A. T., Ng, A., Kranich, J., Sierro, F., Yu, D., et al. (2009). Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 461, 1282–1286. doi: 10.1038/nature08530
Milani, C., Ticinesi, A., Gerritsen, J., Nouvenne, A., Lugli, G. A., Mancabelli, L., et al. (2016). Gut microbiota composition and Clostridium difficile infection in hospitalized elderly individuals: a metagenomic study. Sci. Rep. 6:25945. doi: 10.1038/srep25945
Percie Du Sert, N., Hurst, V., Ahluwalia, A., Alam, S., Avey, M. T., Baker, M., et al. (2020). The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. PLoS Biol. 18:e3000410. doi: 10.1371/journal.pbio.3000410
Pérez-Cobas, A. E., Artacho, A., Ott, S. J., Moya, A., Gosalbes, M. J., and Latorre, A. (2014). Structural and functional changes in the gut microbiota associated to Clostridium difficile infection. Front. Microbiol. 5:335. doi: 10.3389/fmicb.2014.00335
Rajilić-Stojanović, M., Biagi, E., Heilig, H. G., Kajander, K., Kekkonen, R. A., Tims, S., et al. (2011). Global and deep molecular analysis of microbiota signatures in fecal samples from patients with irritable bowel syndrome. Gastroenterology 141, 1792–1801. doi: 10.1053/j.gastro.2011.07.043
Rao, K., and Malani, P. N. (2020). Diagnosis and treatment of Clostridioides (Clostridium) difficile infection in adults in 2020. JAMA 323, 1403–1404. doi: 10.1001/jama.2019.3849
Rea, M. C., Dobson, A., O'sullivan, O., Crispie, F., Fouhy, F., Cotter, P. D., et al. (2011). Effect of broad-and narrow-spectrum antimicrobials on Clostridium difficile and microbial diversity in a model of the distal colon. Proc. Natl. Acad. Sci. USA 108, 4639–4644. doi: 10.1073/pnas.1001224107
Reasoner, S. A., Fazili, I. F., Bernard, R., Parnell, J. M., Sokolow, A. G., Thomsen, K. F., et al. (2023). Prevalence, risk factors, and sequelae of asymptomatic Clostridioides difficile colonization in children with cystic fibrosis. J. Cyst. Fibros. 22, 884–887. doi: 10.1016/j.jcf.2022.12.014
Reeves, A. E., Theriot, C. M., Bergin, I. L., Huffnagle, G. B., Schloss, P. D., and Young, V. B. (2011). The interplay between microbiome dynamics and pathogen dynamics in a murine model of Clostridium difficile infection. Gut Microbes 2, 145–158. doi: 10.4161/gmic.2.3.16333
Rolig, A. S., Cech, C., Ahler, E., Carter, J. E., and Ottemann, K. M. (2013). The degree of Helicobacter pylori-triggered inflammation is manipulated by preinfection host microbiota. Infect. Immun. 81, 1382–1389. doi: 10.1128/iai.00044-13
Rugge, M., Sacchi, D., Genta, R. M., Zanco, F., Guzzinati, S., Pizzi, M., et al. (2021). Histological assessment of gastric pseudopyloric metaplasia: intra-and inter-observer consistency. Dig. Liver Dis. 53, 61–65. doi: 10.1016/j.dld.2020.09.003
Sasaki, K., Inoue, J., Sasaki, D., Hoshi, N., Shirai, T., Fukuda, I., et al. (2019). Construction of a model culture system of human colonic microbiota to detect decreased Lachnospiraceae abundance and Butyrogenesis in the Feces of ulcerative colitis patients. Biotechnol. J. 14:e1800555. doi: 10.1002/biot.201800555
Schubert, A. M., Rogers, M. A., Ring, C., Mogle, J., Petrosino, J. P., Young, V. B., et al. (2014). Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. MBio 5, e01021–e01014. doi: 10.1128/mBio.01021-14
Sharon, G., Garg, N., Debelius, J., Knight, R., Dorrestein, P. C., and Mazmanian, S. K. (2014). Specialized metabolites from the microbiome in health and disease. Cell Metab. 20, 719–730. doi: 10.1016/j.cmet.2014.10.016
Sheh, A., and Fox, J. G. (2013). The role of the gastrointestinal microbiome in Helicobacter pylori pathogenesis. Gut Microbes 4, 505–531. doi: 10.4161/gmic.26205
Singh, S. B., Miesel, L., Kramer, S., Xu, L., Li, F., Lan, J., et al. (2022). Nocathiacin, Thiazomycin, and polar Analogs are highly effective agents against toxigenic Clostridioides difficile. J. Nat. Prod. 85, 1141–1146. doi: 10.1021/acs.jnatprod.2c00093
Smith, B., Miller, R., and Schmidt, T. (2021). MuribaculaceaeGenomes assembled from metagenomes suggest genetic drivers of differential response to acarbose treatment in mice. mSphere 6:e0085121. doi: 10.1128/msphere.00851-21
Su, X., Gao, Y., and Yang, R. (2022). Gut microbiota-derived tryptophan metabolites maintain gut and systemic homeostasis. Cells 11:2296. doi: 10.3390/cells11152296
Vakili, B., Fateh, A., Asadzadeh, A. H., Sotoodehnejadnematalahi, F., and Siadat, S. D. (2020a). Characterization of gut microbiota in hospitalized patients with Clostridioides difficile infection. Curr. Microbiol. 77, 1673–1680. doi: 10.1007/s00284-020-01980-x
Vakili, B., Fateh, A., Asadzadeh, A. H., Sotoodehnejadnematalahi, F., and Siadat, S. D. (2020b). Intestinal microbiota in elderly inpatients with Clostridioides difficile infection. Infect. Drug. Resist. 13, 2723–2731. doi: 10.2147/idr.S262019
Walter, J., and O'toole, P. W. (2023). Microbe profile: the Lactobacillaceae. Microbiology (Reading) 169:1414. doi: 10.1099/mic.0.001414
Wang, S., Deng, W., Li, F., Xiang, L., Lv, P., and Chen, Y. (2022). Treatment with butyrate alleviates dextran sulfate sodium and Clostridium difficile-induced colitis by preventing activity of Th17 cells via regulation of SIRT1/mTOR in mice. J. Nutr. Biochem. 111:109155. doi: 10.1016/j.jnutbio.2022.109155
Wu, Z., Xu, Q., Gu, S., Chen, Y., Lv, L., Zheng, B., et al. (2022). Akkermansia muciniphila ameliorates Clostridioides difficile infection in mice by modulating the intestinal microbiome and metabolites. Front. Microbiol. 13:841920. doi: 10.3389/fmicb.2022.841920
Wydau-Dematteis, S., El Meouche, I., Courtin, P., Hamiot, A., Lai-Kuen, R., Saubaméa, B., et al. (2018). Cwp19 is a novel lytic Transglycosylase involved in stationary-phase autolysis resulting in toxin release in Clostridium difficile. MBio 9:e00648. doi: 10.1128/mBio.00648-18
Zhang, Y., Saint, F. A., and Feng, H. (2022). The development of live biotherapeutics against Clostridioides difficile infection towards reconstituting gut microbiota. Gut Microbes 14:2052698. doi: 10.1080/19490976.2022.2052698
Zheng, J., Ruan, L., Sun, M., and Gänzle, M. (2015). A genomic view of lactobacilli and Pediococci demonstrates that phylogeny matches ecology and physiology. Appl. Environ. Microbiol. 81, 7233–7243. doi: 10.1128/aem.02116-15
Zhou, Y., Mao, L., Yu, J., Lin, Q., Luo, Y., Zhu, X., et al. (2019). Epidemiology of Clostridium difficile infection in hospitalized adults and the first isolation of C. difficile PCR ribotype 027 in Central China. BMC Infect. Dis. 19:232. doi: 10.1186/s12879-019-3841-6
Keywords: Clostridioides difficile , rodent models, Mongolian gerbils, gut microbiome, Lactobacillaceae , Akkermansia
Citation: Wan S, You P, Shi Q, Hu H, Zhang L, Chen L, Wu Z, Lin S, Song X, Luo Y, Wang Y, Ju F, Jin D and Chen Y (2024) Gut microbiome changes in mouse, Mongolian gerbil, and hamster models following Clostridioides difficile challenge. Front. Microbiol. 15:1368194. doi: 10.3389/fmicb.2024.1368194
Edited by:
Axel Cloeckaert, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
Jozsef Soki, University of Szeged, HungaryMingsong Kang, Canadian Food Inspection Agency (CFIA), Canada
Copyright © 2024 Wan, You, Shi, Hu, Zhang, Chen, Wu, Lin, Song, Luo, Wang, Ju, Jin and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dazhi Jin, amluZEBobWMuZWR1LmNu; Yu Chen, eWNoZW5AaG1jLmVkdS5jbg==
†These authors have contributed equally to this work