 Anwar A. Kalalah
Anwar A. Kalalah Sara S. K. Koenig
Sara S. K. Koenig James L. Bono
James L. Bono Joseph M. Bosilevac
Joseph M. Bosilevac Mark Eppinger
Mark Eppinger- 1Department of Molecular Microbiology and Immunology, University of Texas at San Antonio, San Antonio, TX, United States
- 2South Texas Center for Emerging Infectious Diseases (STCEID), San Antonio, TX, United States
- 3U.S. Department of Agriculture (USDA), Agricultural Research Service (ARS), U.S. Meat Animal Research Center, Clay Center, NE, United States
Shiga toxin (Stx)-producing Escherichia coli (STEC) of non-O157:H7 serotypes are responsible for global and widespread human food-borne disease. Among these serogroups, O26, O45, O103, O111, O121, and O145 account for the majority of clinical infections and are colloquially referred to as the “Big Six.” The “Big Six” strain panel we sequenced and analyzed in this study are reference type cultures comprised of six strains representing each of the non-O157 STEC serogroups curated and distributed by the American Type Culture Collection (ATCC) as a resource to the research community under panel number ATCC MP-9. The application of long- and short-read hybrid sequencing yielded closed chromosomes and a total of 14 plasmids of diverse functions. Through high-resolution comparative phylogenomics, we cataloged the shared and strain-specific virulence and resistance gene content and established the close relationship of serogroup O26 and O103 strains featuring flagellar H-type 11. Virulence phenotyping revealed statistically significant differences in the Stx-production capabilities that we found to be correlated to the strain’s individual stx-status. Among the carried Stx1a, Stx2a, and Stx2d phages, the Stx2a phage is by far the most responsive upon RecA-mediated phage mobilization, and in consequence, stx2a + isolates produced the highest-level of toxin in this panel. The availability of high-quality closed genomes for this “Big Six” reference set, including carried plasmids, along with the recorded genomic virulence profiles and Stx-production phenotypes will provide a valuable foundation to further explore the plasticity in evolutionary trajectories in these emerging non-O157 STEC lineages, which are major culprits of human food-borne disease.
1 Introduction
Shiga toxin (Stx)-producing Escherichia coli (STEC) are distinguished from other E. coli pathovars (Kaper et al., 2004) by the production of a phage-borne cytotoxin (Smith et al., 2014; Krüger and Lucchesi, 2015; Zuppi et al., 2020) that is toxigenic toward renal endothelial (Obrig and Karpman, 2012) and intestinal epithelial cells (Schüller, 2011). Escherichia coli are historically classified by their variation in somatic O- and flagellar H-antigens (Orskov et al., 1977). Serotype O157:H7 is the dominant causative agent of STEC disease in the U.S. (Riley et al., 1983; Eppinger et al., 2011, 2013; Sanjar et al., 2014; Rusconi et al., 2016). However, the incidence of non-O157 infections that feature different somatic antigens has been steadily increasing in recent years (Gould et al., 2013; Vishram et al., 2021; Glassman et al., 2022; Tarr et al., 2023). Among these, emerging serogroups O26, O45, O103, O111, O121, and O145 account for the majority of clinical non-O157 STEC infections in the US and are colloquially referred to as the “Big Six” (Eklund et al., 2001; Johnson et al., 2006; Bettelheim, 2007; Hadler et al., 2011; Hegde et al., 2012; Gould et al., 2013; Vishram et al., 2021). Disease in humans can progress to life-threatening complications, such as hemolytic uremic syndrome (HUS) and ultimately renal failure (Karmali et al., 1983; Majowicz et al., 2014). The disease has been linked to the amount and subtype of toxin produced (Donohue-Rolfe et al., 2000; Russo et al., 2016). STEC can harbor one or multiple Stx-bacteriophages featuring different combinations of stx-suballeles (Krüger and Lucchesi, 2015; Rusconi et al., 2016) that can also form hybrid toxins (Skinner et al., 2014). The most potent cytopathic toxins, Stx2a and Stx2d (Fuller et al., 2011; Hauser et al., 2020; McNichol et al., 2021), are prevalent in the Big Six serogroups (Jinnerot et al., 2020), and a strain’s Stx-status is shaped by the dynamic Stx-phage acquisition, rather than by a common evolutionary history (Cowley et al., 2019; Nyong et al., 2020). Mobilization of Stx-prophages is triggered by diverse abiotic and biotic cues (Pacheco et al., 2012; Pacheco and Sperandio, 2012), and is required to produce toxin causing adverse toxigenic effects in murine STEC models (Nguyen and Sperandio, 2012; Tyler et al., 2013; Baumler and Sperandio, 2016; Balasubramanian et al., 2019; Rodríguez-Rubio et al., 2021). Triggering the RecA-dependent SOS-response with sublethal doses of mitomycin C (MMC) constitutes a major pathway of Stx−phage mobilization and is routinely used in public health laboratories to assess the pathogenic potential (Kimmitt et al., 2000). Besides Stx, another major virulence determinant is the locus of enterocyte effacement (LEE) packaged into a pathogenicity island, which encodes a type III secretion system (T3SS) along with its associated effectors, the outer membrane adhesin intimin (eae) and the translocated receptor (tir; Franzin and Sircili, 2015). The majority of the Big Six serogroups also carry serogroup-specific virulence plasmids along with an diverse array of additional plasmids (Caprioli et al., 2005; Ogura et al., 2009). In this study, we report the complete genomes and comprehensive analyses of the pathogenome composition along with Stx-production pathotypes of a Big Six reference strain panel representing each of the non-O157 STEC serogroups curated and distributed by the American Type Culture Collection (ATCC). The gathered pathogen information and recorded virulence traits provide a foundation to further elucidate the make-up and the evolutionary boundaries of these emerging non-O157 STEC.
2 Materials and methods
2.1 Bacterial strains analyzed in this study
Panel MP-9, a representative collection of clinical emerging non-O157 STEC strains, colloquially referred to as the “Big Six,” was obtained from the American Type Culture Collection (ATCC).1 Strains are of serotypes O26:H11 (BAA-2196), O45:H2 (BAA-2193), O103:H11 (BAA-2215), O111:H8 (BAA-2440), O121:H19 (BAA-2219), and O145:NM (BAA-2192). Isolates were sequenced to closure, and the culture’s virulence was profiled in this study. Accessions for genomic reads, assembled annotated chromosomes and plasmids along with strain-associated metadata are provided in Table 1 and Supplementary Table S1.
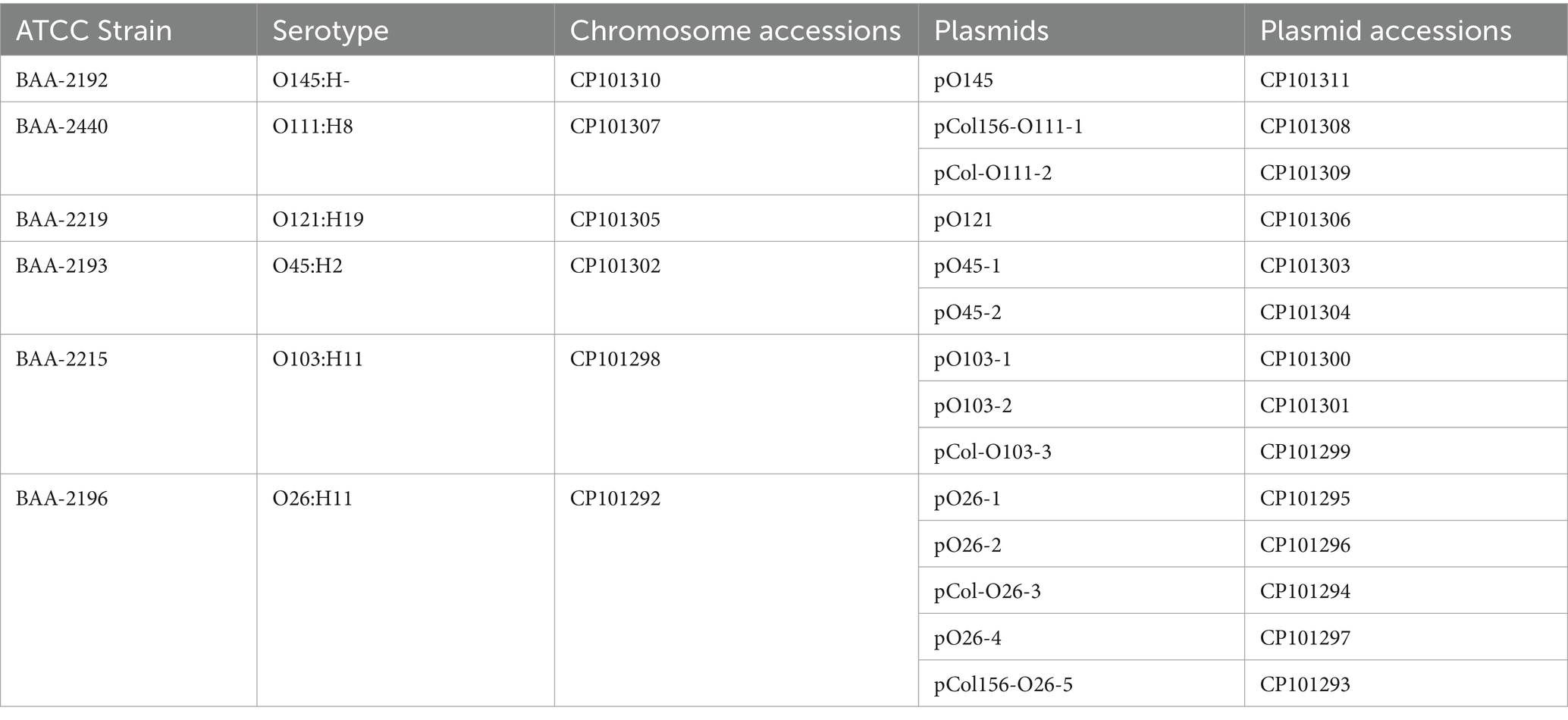
Table 1. Molecules and accessions.
2.2 Genome sequencing, assembly, and annotation
Strains were cultured overnight at 37°C with shaking at 220 rpm in lysogeny broth (LB; Thermo Fisher Scientific, Asheville, NC, United States). To maximize total genomic DNA (gDNA) yields, bacterial overnight cultures were diluted to OD600 of 0.03 in fresh LB medium and grown at 37°C with shaking at 220 rpm to mid-log phase (OD600 ~ 0.5). Total gDNA was extracted using the Qiagen Genomic-tip 100/G Kit (Qiagen, Inc., Valencia, CA, United States) according to the manufacturer’s instructions. Genomic DNA was subjected to both long-read (Oxford Nanopore, Oxford, United Kingdom) and short-read (Illumina, Inc., San Diego, CA, United States) sequencing. For long-read Nanopore sequencing, gDNA was diluted to a concentration of 1.5 μg in 46 μL of nuclease-free water. The library was prepared using the Ligation Sequencing Kit (SQK-LSK109) with the Native Barcoding Expansion 1–12 (EXP-NBD104) according to the manufacturer’s instructions and sequenced on a MinION with the R10.3 SpotON Flow Cell (FLO-MIN111). Paired-end short-read libraries were prepared with the Illumina Nextera XT DNA Library Preparation Kit and sequenced on the MiSeq platform using the MiSeq reagent Kit (v3) with 600-cycles. Sequence reads in the fastq format were imported into Galaxy v.22.05 (Community, 2022). Default parameters were used for all software unless specified otherwise. Quality control of fastq files was assessed using FastQC (v.0.74 + Galaxy0).2 Nanopore and Illumina reads were used for hybrid assembly using Unicycler assembler (v.0.5.0 + Galaxy1; Wick et al., 2017). The chromosomal dnaA and plasmid repA genes, if applicable, were designated as the zero point of the closed molecules prior to annotation using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP; Tatusova et al., 2016).
2.3 Pathogenome make-up and visualization
Chromosomes and plasmids were comprehensively analyzed and visualized in Blast Ring Image Generator BRIG (v.0.95; Alikhan et al., 2011) and MAUVE (v.2.4.1; Darling et al., 2008, 2010). Serotypes in the assembled genomes were confirmed in silico using the EcOH database (Ingle et al., 2016) in ABRicate (Galaxy v.1.0.1)3 with options—minid 80—mincov 80 (Community, 2022). Average nucleotide identities (ANI) using the E. coli strain BAA-2196 (O26:H11) chromosome as designated reference were calculated with FastANI (Galaxy v.1.3), based on MinHash mapping (Jain et al., 2018). Chromosomal repeats were identified with FindRepeats (v.1.8.2 + Galaxy1; Kurtz et al., 2004; Petkau et al., 2017). Virulence and antibiotic resistance genes (ARGs) were identified using VFDB (Liu et al., 2022) and ResFinder4 (Florensa et al., 2022), respectively. Boundaries and locations of intact, partial, or remnant prophages were identified using PHASTER (Zhou et al., 2011; Arndt et al., 2016) and MAUVE (v.2.4.1; Darling et al., 2008, 2010), followed by manual curation with BLASTn/p against the non-redundant NCBI databases (Camacho et al., 2009). Toxin subtypes of the carried Stx-bacteriophages were recorded in silico as described elsewhere by blastn of the carried toxins against an stx suballele database (Scheutz et al., 2012; Ashton et al., 2015; Carrillo et al., 2016). The EHEC phage replication unit (eru) subtype was assigned as described in Llarena et al. (2021) and Fagerlund et al. (2022) and Stx-prophages genomes were visualized in Easyfig (v.2.2.2; Sullivan et al., 2011). Mechanistics of phage insertion can create direct repeats (DR) and insertion sites were investigated for direct repeats (DR) and attachments sites (att) using NUCmer (v.4.0.0rc1 + Galaxy2) and BLASTn (Camacho et al., 2009). Lytic phage loci in ΦStx- and non-ΦStx-prophages were identified with Prophage Hunter (Song et al., 2019). Insertion sequence (IS) elements were identified and curated using ISEScan (v.1.7.2.3 + Galaxy0; Xie and Tang, 2017). Integrons were surveyed with Integron Finder (v.2.0.2 + Galaxy1; Néron et al., 2022). Genomic islands (GI) were detected with IslandViewer4 (Bertelli et al., 2017, 2018; Bertelli and Brinkman, 2018). Plasmid incompatibility groups were identified and analyzed with MOB-Typer (v.3.0.3 + Galaxy0; Robertson and Nash, 2018).
2.4 Shiga toxin and intimin subtyping
For toxin subtyping, the stx genes were aligned to a multifasta file comprised of all currently published stx-suballele nucleotide sequences (Scheutz et al., 2012; Carrillo et al., 2016; Bai et al., 2018; Yang et al., 2020) with BLASTn (Camacho et al., 2009). Heatmaps of cataloged genes were generated with iTol (v.6.8.1; Letunic and Bork, 2021). LEE islands were identified starting from the LEE1 operon gene espG to the espF gene in LEE4, and their comparative analysis was conducted and visualized using GeneSpy (Garcia et al., 2019). We determined the subtypes by aligning the intimin genes to the 27 currently published subtype sequences of eae in GenBank (α1-2, β1-3, γ, δ, ε1-4, ζ1 and 3, η1-2, θ1-2, ι1-2, κ, μ, ν, ξ, ο, π, ρ, σ; Supplementary Table S2) using BLASTn (Camacho et al., 2009).
2.5 MLST schemas and phylogenetic analyses
The assembled ATCC MP-9 genomes along with E. coli strains EC4115 (O157:H7; Eppinger et al., 2011) and K-12 substrain MG1655 (Blattner et al., 1997) were imported into SeqSphere+ (v.8.3; Ridom GmbH, Münster, Germany) for gene-by-gene alignment, allele calling, and comparison (Jünemann et al., 2013). MLST typing was performed using targeted and whole genome schemas developed for E. coli (Foley et al., 2009; Zhou et al., 2020). We determined the Sequence Type (ST) by applying the 7-gene ST Achtman schema (Zhou et al., 2020). Allele sequences for the 7 genes (adk, fumC, gyrB, icd, mdh, purA, and recA) were accessed on the EnteroBase website5 and imported into Ridom SeqSphere+. A core genome (cg) MLST schema was developed using the closed chromosome of K-12 substrain MG1655 (GenBank accession U00096; Riley et al., 2006) as seed as previously described (Díaz et al., 2021). Core and accessory MLST targets were identified according to the inclusion/exclusion criteria of the SeqSphere+ Target Definer. The allele information from the targeted seven-gene schema and the defined core genome gene of the panel strains were used to establish phylogenetic hypotheses using the minimum-spanning method (Kruskal, 1956; Francisco et al., 2009) with default settings in Ridom SeqSphere+ (v.8.3).
2.6 Growth of cultures in LB and under phage mobilizing condition in LB + MMC
Strains were cultured overnight (o/n) at 37°C with shaking at 220 rpm in LB. Overnight LB cultures were diluted to an OD600 of 0.03 in fresh LB media, grown to early-log phase (OD600~0.3) at 37°C, and then subdivided into two subcultures, LB and LB + MMC. Triggering the RecA-dependent SOS-response with MMC constitutes a major pathway of Stx−phage mobilization (Kimmitt et al., 2000). Subculture LB + MMC was supplemented with MMC (Sigma-Aldrich, Saint Louis, MO, United States) at a final concentration of 0.5 μg/mL to mobilize the carried prophages, while subculture LB was used to evaluate spontaneous prophage mobilization. To confirm phage mobilization in MMC-treated cultures, growth curves were recorded in a 96-well plate (Corning 3,370, Corning Inc., Corning, NY, United States) on a BioTek Synergy H1 plate reader (BioTek Instruments, Inc., Winooski, VT, United States) recording OD600 values for 6 h at 10 min intervals. All experiments were executed in two biological replicates.
2.7 Virulence phenotypes
2.7.1 PCR experiments
Primers and PCR-conditions are provided in Supplementary Table S3. LB and LB + MMC subcultures were grown for 6 h at 37°C with shaking at 220 rpm and then centrifuged at 5,000 g for 10 min: (1) Cell pellets were used to determine stx-transcripts levels, while (2) the supernatants were used to enumerate ΦStx-phage copies, targeting the phage-borne stx loci as follows: (1) Expression of stx genes RNA was purified using the PureLink RNA Mini kit (Invitrogen, Waltham, MA, United States), and RNA quantity and quality were measured with the NanoDrop ND-1000 Spectrophotometer (Thermo Fisher Scientific, Waltham, MA, United States). Total RNA was treated with amplification grade DNase I (Invitrogen, Waltham, MA, United States), and reverse transcribed using the RevertAid H Minus First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, United States). The stx-RT-qPCR was performed on the StepOne Real-Time PCR System (Applied Biosystems, Foster City, CA, United States) using the GoTaq qPCR Master Mix (Promega, Madison, WI, United States). (2) Enumeration of ΦStx1- and ΦStx2-phage copies Supernatants were filtered through low-protein-binding 0.22-μm-pore-size membrane filters (Millex-GP; Merck Millipore Ltd., Burlington, MA, United States), followed by DNase I (Invitrogen, Waltham, MA, United States) treatment for 15 min to remove bacterial gDNA. Lysate phage DNA was isolated using the QIAamp DNA Mini Kit (Qiagen Inc., Valencia, CA, United States), and eluted with 50 μL nuclease-free water. Phage numbers were determined by stx-qPCR on the StepOne Real-Time PCR System (Applied Biosystems, Foster City, CA, United States) using the GoTaq qPCR Master Mix (Promega, Madison, WI, United States). Standard curves for the stx transcripts and ΦStx-phage copy numbers were calculated using gBlocks (Integrated DNA Technologies (IDT), Coralville, Iowa, United States) in the RT-qPCR and qPCR experiments, respectively.
2.7.2 Stx-production pathotypes
The Stx-production phenotypes of the cultures were determined by recording the Stx titers through Enzyme-Linked ImmunoSorbent Assay (ELISA) under both spontaneous and MMC-induced conditions. Overnight (o/n) cultures were diluted to an OD600 of 0.03 and grown to early-log phase (OD600~0.3) in replenished LB media at 37°C. At this stage, cultures were split and incubated at 37°C for 6 h under non-induced and induced (0.5 μg/mL MMC) conditions. Toxin production was measured after harvesting 5 mL of each culture for parallel processing. To lyse bacterial cells and release produced Stx, cultures were treated with polymyxin B (Sigma-Aldrich, Saint Louis, MO, United States; 6 mg/mL 37°C, 10 min). Supernatants were collected after centrifugation (3,500 rpm, 10 min), filtered through 0.22 μm low protein-binding membrane filters (Millex-GP; Merck Millipore Ltd., Burlington, MA, United States) and diluted to measurable concentrations. Stx-production was measured using the Premier EHEC kit (Meridian Bioscience, Cincinnati, OH, United States) following the manufacturer’s instructions. Titers were calculated using a standard curve generated from serial dilutions of purified Stx2a (BEI Resources, NR-4478). Statistical significance was determined using Prism (v.9.5.0; GraphPad Software, San Diego, CA, United States). A two-way ANOVA with Sidak’s multiple comparisons test was used to compare non-induced to MMC-induced conditions for each strain. Strain-to-strain comparisons were performed with a one-way ANOVA with Tukey’s multiple comparisons test assessing each condition.
3 Results
3.1 Pathogenome composition of MP-9 panel strains
In this study, we sequenced and comprehensively analyzed the pathogenomes and virulence traits of six non-O157 STEC strains. Strain panel MP-9 was obtained from ATCC, which is comprised of six strains representing each of the non-O157 STEC serogroups, colloquially referred to as the “Big Six” (Eklund et al., 2001; Johnson et al., 2006; Bettelheim, 2007; Hadler et al., 2011; Hegde et al., 2012; Gould et al., 2013; Vishram et al., 2021; Supplementary Table S1). STEC genomes house an extensive and partly repetitive phage complement that hampers assembly into closed genomes (Goldstein et al., 2019; Jaudou et al., 2022). In response, we applied a long- and short read sequencing hybrid strategy (Nyong et al., 2020; Allué-Guardia et al., 2022) that allowed us to provide the high-quality closed genomes, including carried plasmids (Figures 1, 2; Supplementary Figures S1, S2). The chromosomes have an average nucleotide identity of 98.8%, with a range from 97.6% to 99.8%, indicative of the substantial conserved chromosomal backbone of E. coli (Rasko et al., 2008; Jain et al., 2018). The chromosome size in this panel ranges from 5,288,508 to 5,840,137 bp with an average GC-content of 50.65%. When compared to non-pathogenic E. coli strain K-12 substrain MG1665, these STEC strains carry at least 648,833 bp of additional genetic information. Genome statistics along with strain-associated metadata are provided in Supplementary Table S1. In Figure 1, we compared the chromosomes using strain BAA-2196 (O26:H11) as the designated reference. In comparison to E. coli strain K-12, the Big Six strains acquired multiple mobile genome elements (MGE), including the hallmark ΦStx-prophages, which are major contributors of STEC genome evolution and diversification (Lawrence and Ochman, 1998; Rasko et al., 2008; Robins-Browne et al., 2016). Individual comparisons referenced to each of the strains can be found in Supplementary Figure S1. The mobilome on the chromosomes consisting of prophages, genomic islands, and IS elements contributes 22.4% to 28.7% of sequence information, in line with the assessment in other STEC (Perna et al., 2001; Delannoy et al., 2017; Supplementary Table S4). Neither chromosomal nor plasmid-borne integrons were detected. The prophages account for 13.9 to 21.2% of the chromosome, followed by genomic islands (5.4 to 6.9%), and IS elements (0.8 to 2.5%). If plasmid-carried IS elements are considered, the percentage of IS elements increases by 1.2 to 2.7%. The IS elements in this panel showed variations in both prevalence and numbers (Supplementary Table S4). ISEScan detected 726 IS elements and categorized them into 16 known families and 40 clusters, indicative of the plasticity present in these non-O157 STEC (Supplementary Figure S3). Eight of the 40 clusters were present in the six isolates, though their respective numbers between the strains vary considerably. We further note that BAA-2196 (O26:H11) and BAA-2215 (O103:H11) strains feature similar copy numbers in shared IS clusters distinct from the remainder of strains indicative of their close relationship (Iguchi et al., 2012; Ju et al., 2012; Supplementary Figure S3; Supplementary Table S4). Thirteen elements of the IS3-168 cluster were found in each of BAA-2196 (O26:H11) and BAA-2215 (O103:H11), compared to an average of 55 copies in other strains. Inversely, the IS66-46 cluster was found to have 48 and 36 copies in BAA-2196 and BAA-2215, respectively, while other strains carry an average of eight copies. Further, eight clusters are strain-specific, and 24 clusters are present in a subset of strains. This may suggest different dynamics in the propagation of these elements.
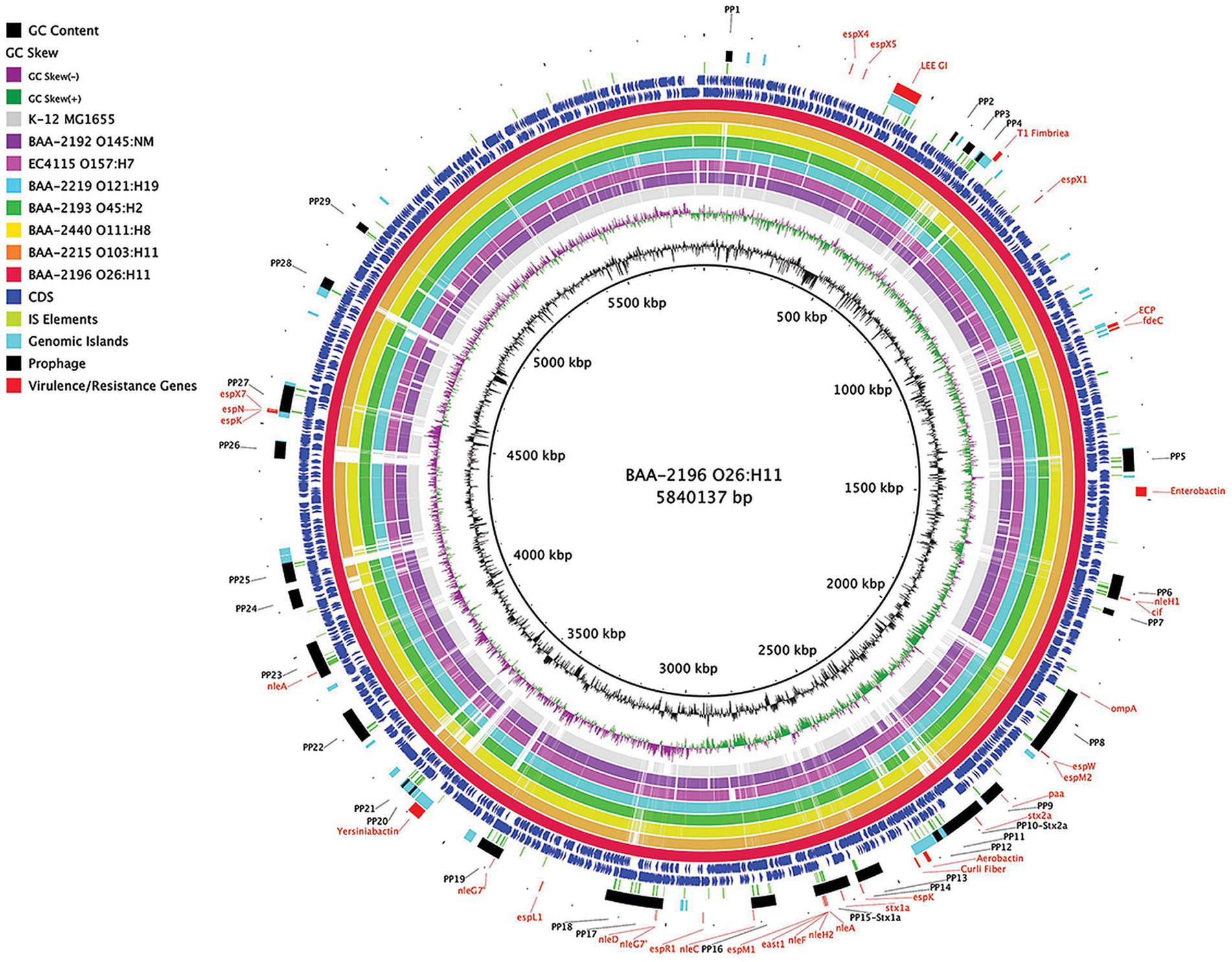
Figure 1. Comparison of ATCC MP-9 panel genomes BRIG comparison of six sequenced strains, along with Escherichia coli strains O157:H7 EC4115 and strain K-12 substrain MG1665, referenced to the 5,840,137 bp chromosome of BAA-2196 (O26:H11). CDS are presented on the +/−strands as blue arrows and functional annotations for virulence genes and other loci of interest are highlighted as shown in the legend. Query genomes are color-coded, and the order plotted in the circle reflects the inferred phylogenomic relationships.
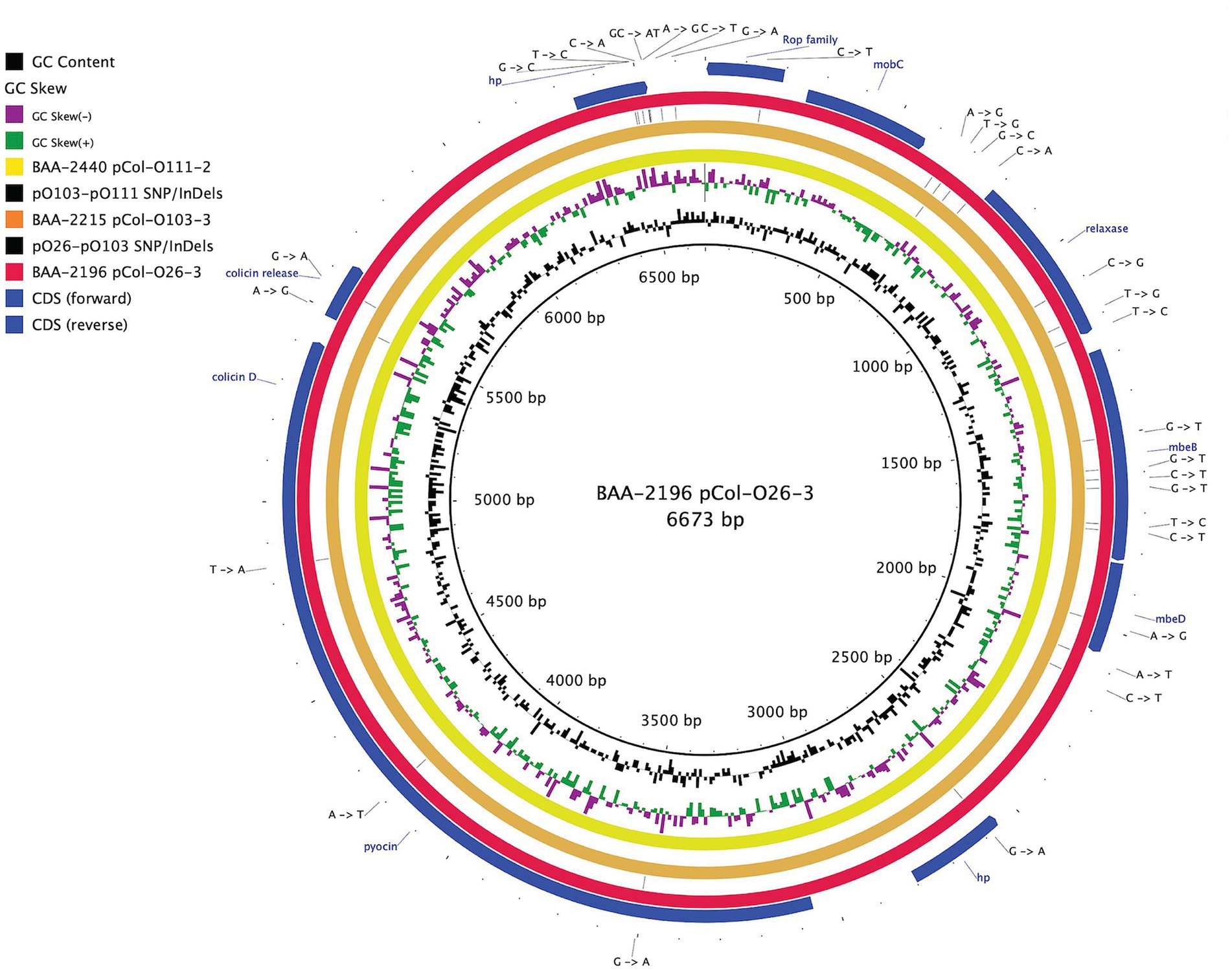
Figure 2. Comparison of a shared colicin plasmid BRIG comparison of a shared colicinogenic plasmid l present in serotypes O26:H11, O103:H11, and O111:H8 and referenced to the 6,673 bp plasmids of BAA-2196 pCol-O26-3. The plasmids are differentiated by a total of 30 SNPs and InDels. CDS are presented on the +/− strands as blue arrows. Query plasmids are plotted according to the strain’s inferred phylogenomic relationships.
3.2 Phylogenomic relatedness of ATCC MP-9 strains
The mobilome is comprised of prophages, genomic islands, IS elements, and plasmids, which evolve at different rates and can be acquired and secondarily lost and thus are often not indicative of evolutionary relationships. To investigate the phylogenomic boundaries of the individual strains, we established a phylogenomic framework inferred from targeted MLST and core genome MLST (cgMLST; Figure 3). As expected for this heterogenous set of serotypes, the strains belong to distinct STs with a total of 14,340 allelic changes and 926 InDels (Figure 3; Supplementary Table S5). Their shared inventory was computed at 4,304 genes comprised of 3,148 core and 908 accessory loci, indicative of the extended conserved E. coli backbone (Abu-Ali et al., 2009; Lim et al., 2010; Eppinger et al., 2011; Yin et al., 2015). High-resolution core genome MLST typing revealed a close phylogenetic relationship of serogroup O26:H11 and O103:H11 strains, as previously suggested by MLST- and genome-wide single nucleotide polymorphisms (SNPs)-based analyses for these serogroups carrying flagellar antigens H2 and H11 (Iguchi et al., 2012; Ju et al., 2012). ST-21 (BAA-2196 O26:H11) and ST-723 (BAA2215 O103:H11) are only separated in their fumC allele and 357 allelic changes in the cgMLST analysis (Figure 3). This intimate relationship is reflected in the isolates’ shared chromosomal and mobilome inventories, such as virulence genes, prophages, and LEE island organization, as discussed below.
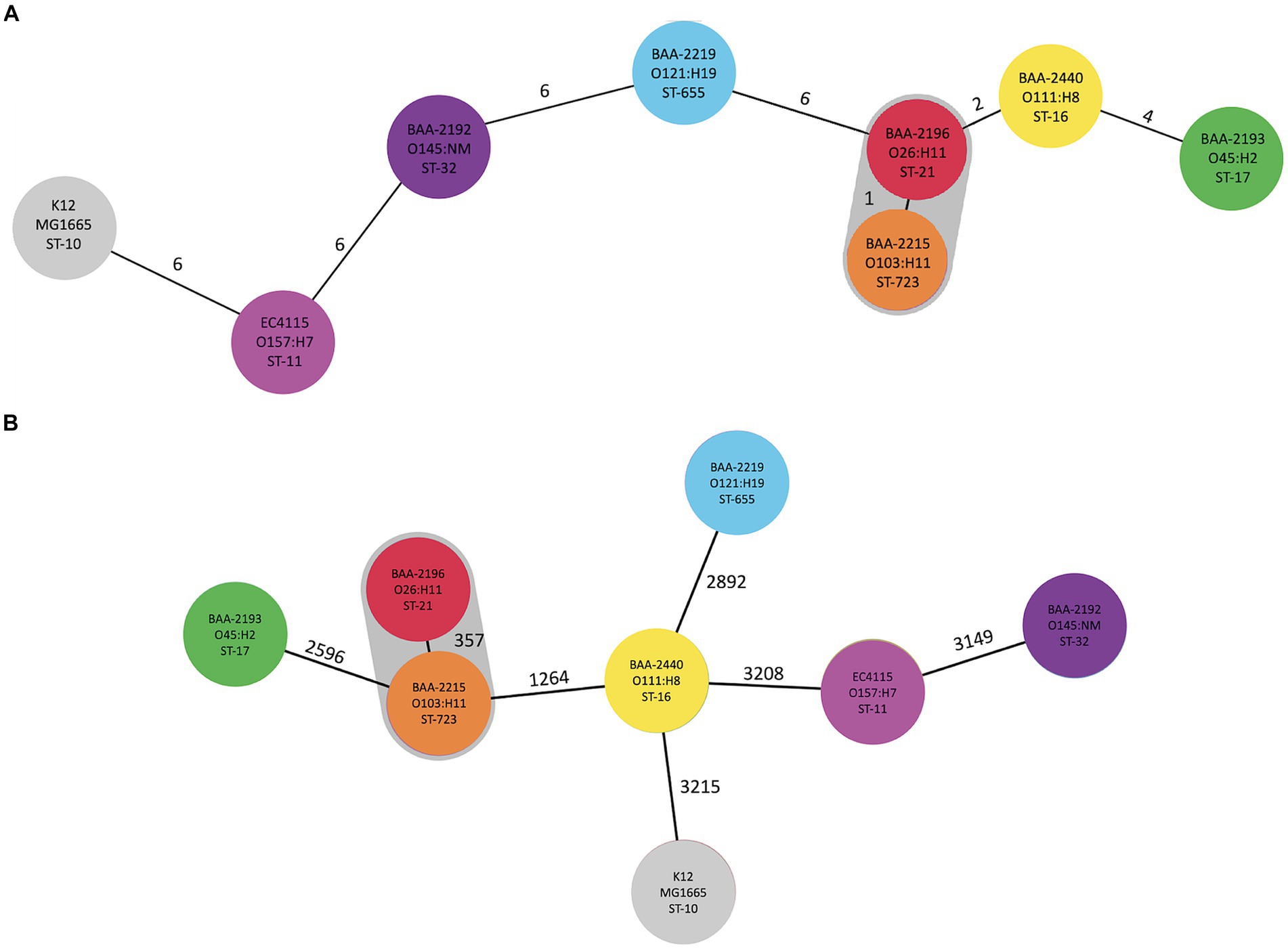
Figure 3. Phylogenomic position of ATCC-MP9 strains The relatedness of panel strains, including O157:H7 strain EC4115, was determined using MLST in Ridom SeqSphere+: (A) targeted seven-gene MLST accessed in EnteroBase. Numbers on connecting branches indicate the number of genes with differing allele status, and (B) cgMLST-based phylogeny using the closed chromosome of E. coli strain K12 subst. MG1655 as seed. The shared gene inventory was determined at 4,304 genes, according to the inclusion/exclusion criteria of the SeqSphere+ Target Definer and is comprised of 3,148 core and 908 accessory loci. Colors denote ST-classifications established in (A).
3.3 Comprehensive analyses of plasmid content and function
The hybrid-sequencing strategy further identified 14 functionally and phylogenetically diverse plasmids that range in size from 5,176 to 93,980 bp and belong to four incompatibility groups (Supplementary Figure S2; Supplementary Table S1). STEC often carry plasmids that contribute diverse virulence determinants (Kaper et al., 2004; Pilla and Tang, 2018). Virulence plasmids coding for hemolysin (hlyCABD), adhesin (toxB), and serine protease (espP) were found in all strains, except in strain BAA-2440 O111:H8 (Tatsuno et al., 2001; Kaper et al., 2004; Caprioli et al., 2005; Tozzoli et al., 2005; Johnson and Nolan, 2009; Figure 4). Colicins are synthesized to gain an advantage in the shared niche and are toxic to other bacterial strains (Cascales et al., 2007). Three strains, BAA-2440, BAA-2196, and BAA-2215, contained colicinogenic plasmids. Strain BAA-2440 O111:H8 codes for colicins E3 and D on plasmids pCol156-O111-1 and pCol-O111-2, respectively. The latter is phylogenetically related to plasmids pCol-O26-3 and pCol-O111-2 exhibiting a highly conserved plasmid backbone differentiated from each other by 30 SNPs and InDels (Figure 2). A Blastn query against the NCBI non-redundant database found related plasmids in Big Six serogroups O26, O103, and O111, and STEC serogroups O104, O157, and O165, among others, as shown in Supplementary Table S4 (Ogura et al., 2009; Yan et al., 2015; Sekizuka et al., 2019; Amadio et al., 2021). Strain BAA-2196 O26:H11 carries plasmid pO26-4, which is a multidrug-resistant plasmid encoding three ARGs (sul2, aph(6)-Ib, and aph(3″)-Ib) conferring resistance to sulfonamide and aminoglycosides (Hammerum et al., 2006; Bean et al., 2009; Messele et al., 2022; Supplementary Figure S2A; Supplementary Table S4). This broad host range plasmid shares high nucleotide similarity (>99%) and coverage (>99%) to plasmids found in E. coli, Shigella sp., Citrobacter freundii and Klebsiella pneumoniae (Iguchi et al., 2009; Ye et al., 2010; Kyle et al., 2012; Liu et al., 2015; AbuOun et al., 2021; Supplementary Table S4).
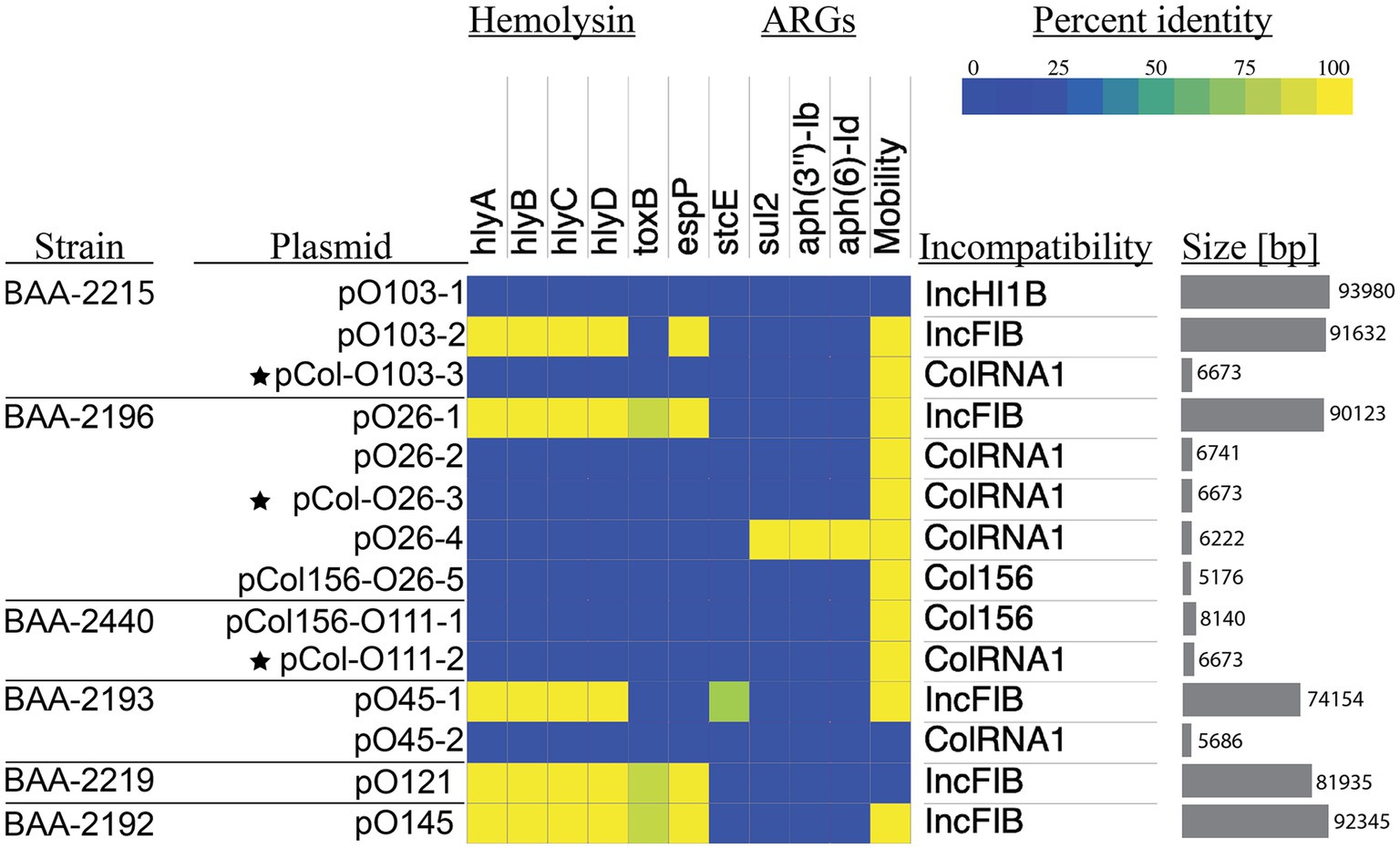
Figure 4. Prevalence and distribution of plasmid-borne virulence determinants Percentage identities of virulence and antimicrobial resistance genes identified in VFDB and ResFinder are visualized in a heatmap. The Plasmid incompatibility group and predicted mobility were determined with MobTyper. The shared colicinogenic plasmid pCol present in the serotype O26:H11, O103:H11, and O111:H8 strains is indicated with a star.
3.4 Comprehensive analyses of virulence determinants and Stx-status
The prevalence of the identified chromosomal and phage- and plasmid-borne virulence genes revealed a considerable plasticity in the individual virulence complement. We surveyed chromosomes for virulence and resistance loci and analyzed their prevalence and distribution (Figure 5; Supplementary Table S6). In total, we identified 149 chromosomal virulence genes of which 113 are shared by all strains (Figure 5). The latter includes the phage-borne stx, along with genes that make up the LEE including its effectors (Moon et al., 1983; Mellies et al., 1999; Jores et al., 2004; Kaper et al., 2004; Sadiq et al., 2014; Franzin and Sircili, 2015). The strains feature four distinct siderophore types that facilitate iron acquisition in the iron limiting condition of mammalian hosts (Ratledge and Dover, 2000; Skaar, 2010; Caza and Kronstad, 2013; Sheldon et al., 2016). All strains possess enterobactin (ent), widely distributed in E. coli (Cox et al., 1970; Garcia et al., 2011; Mey et al., 2021). Yersiniabactin (ybt) and hydroxamate aerobactin (iuc) are present in the phylogenetically related strains BAA-2196 (O26:H11) and BAA-2215 (O103:H11; Iguchi et al., 2012; Ju et al., 2012; Figure 3). Hydroxamate aerobactin is also found in strains BAA-2440 (O111:H8) and BAA-2192 (O145:NM), and the heme utilization operon (chu) in BAA-2192 (O145:NM; Figure 5). We note here that siderophores such as ybt and chu have been proposed biomarkers for serotypes O26, O157, and O145 (Pasquali et al., 2018; Jarocki et al., 2019; Carbonari et al., 2022). Antimicrobial-resistant STEC, though uncommon, have been isolated from humans, animals, and food (Eppinger et al., 2011; Amézquita-López et al., 2016; Mukherjee et al., 2017; Greig et al., 2023; Lee et al., 2023). The ATCC MP-9 strains do not carry any chromosomal antimicrobial resistance loci other than the efflux pump gene mdf(A) (Edgar and Bibi, 1997), found in most E. coli (Ahmed et al., 2020; Moser et al., 2021; Zhou et al., 2022; Awosile et al., 2023; Lee et al., 2023).
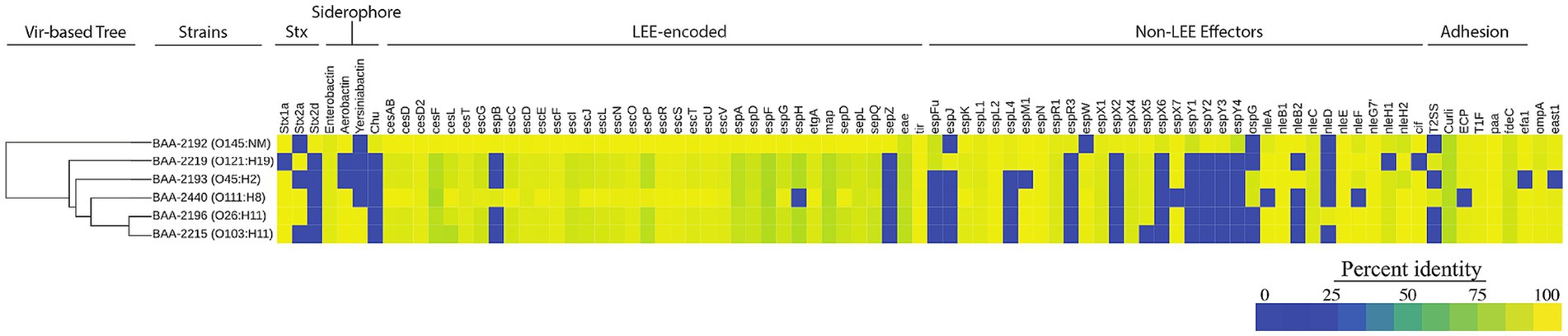
Figure 5. Prevalence and distribution of chromosomal virulence determinants Percentage identities for each virulence gene identified in VFDB are visualized in a heatmap. The panel strains encode a total of 149 distinct virulence genes of which 113 are shared. Among these are toxin suballeles stx1a, stx2a, and stx2d, the LEE genomic island, and siderophores, among others. The strain order reflects the inferred phylogenomic relationships. The hierarchical clustering of virulence genes based on their pair-wise distance is shown on the left.
3.5 Comprehensive analysis of Stx-phages
Carriage of ΦStx-prophages is a virulence hallmark of STEC; and genomes can contain multiple ΦStx-prophages in diverse stx-suballele combinations (Huang et al., 1987; Eppinger et al., 2011; Krüger and Lucchesi, 2015; Rusconi et al., 2016; Allué-Guardia et al., 2022). Stx is a key virulence factor responsible for the severe symptoms associated with STEC infections such as HUS (Karmali et al., 1983). The panel strains carry either one or two ΦStx-prophages featuring suballeles stx1a, stx2a, and stx2d (Figures 5, 6). Two suballeles have been associated with elevated cytotoxicity, stx2a (Fuller et al., 2011; Hauser et al., 2020; Pinto et al., 2021) and stx2d (McNichol et al., 2021). Suballele stx2a was found alone (BAA-2219 O121:H19) or in combination with stx1a (BAA-2196 O26:H11, BAA-2440 O111:H8). Two strains carry stx1a only (BAA-2215 O103:H11, BAA-2193 O45:H2), or in combination with stx2d (BAA-2192 O145:NM). As evident in the comparison of the individual subtypes (Figure 6), the prophages show a high degree of genomic plasticity, in particular upstream of the toxin locus, important for regulation and replication (Unkmeir and Schmidt, 2000; Yin et al., 2015). Variability in these regions has been linked to strain-level differences in Stx-production (Herold et al., 2004; Smith et al., 2014; Ogura et al., 2015; Yin et al., 2015; Llarena et al., 2021; Rodríguez-Rubio et al., 2021; Fagerlund et al., 2022; Zhang et al., 2022; Yano et al., 2023). In total, seven chromosomal sites are occupied (Supplementary Table S4), some of which are known ΦStx-phage targets (Serra-Moreno et al., 2007; Eppinger et al., 2011; Bonanno et al., 2015; Rusconi et al., 2016; Allué-Guardia et al., 2022). The ΦStx-phage integrases have undergone evolution that allows them to target distinct insertion sites. Stx-phages tend to primarily integrate at a specific site; however, the integrase demonstrates the capacity to detect alternate insertion sites for integration if the preferred site is already occupied or absent (Groth and Calos, 2004; Serra-Moreno et al., 2007; Casjens and Hendrix, 2015; Henderson et al., 2021). ΦStx2a phages are inserted into either arginine tRNA argW or NAD(P) H dehydrogenase wrbA, and the ΦStx1a phage, in analogy to some ΦStx2a in wrbA, or alternatively in peptide chain release factor prfC, outer membrane protein ompW, the tRNA-dihydrouridine synthase dusA, or tmRNA ssrA, while the ΦStx2d phage is disrupting the spermidine uptake gene potC (Figure 6). As evident in the occupation status of wrbA by either ΦStx1 or ΦStx2, there is no association between toxin suballele and insertion sites in line with previous observation (Groth and Calos, 2004; Serra-Moreno et al., 2007; Steyert et al., 2012; Henderson et al., 2021).
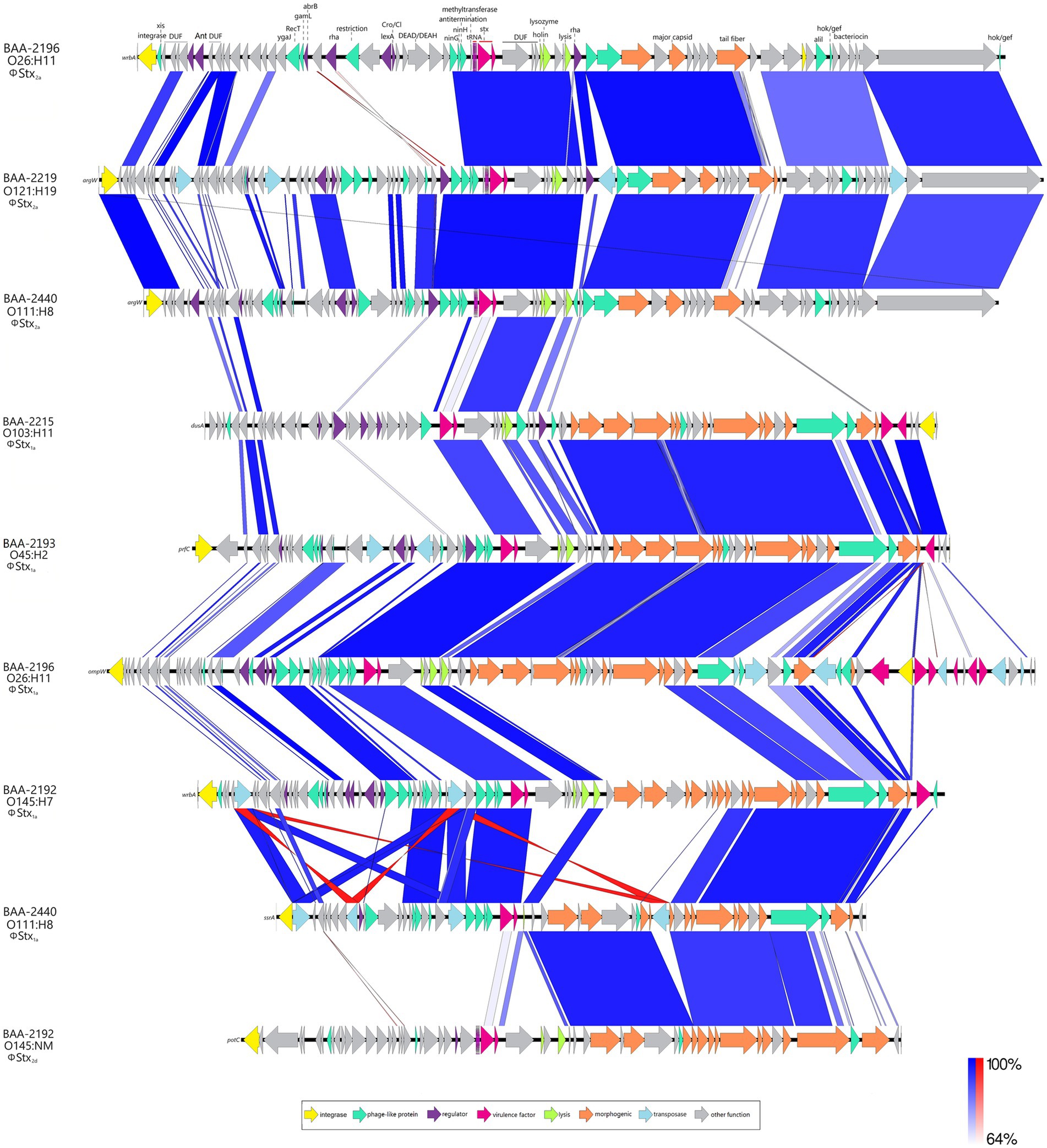
Figure 6. Comparison of Stx-prophages BLASTn-based comparison of architectures and content of carried ΦStx2a-, ΦStx1a-, and ΦStx2d-prophages. For uniform annotation, we inferred the annotation from the curated ΦStx2a-prophage genome of strain BAA-2196 (O26:H11).
3.6 Comprehensive analyses of the locus of enterocyte effacement
Carriage of the LEE pathogenicity island is responsible for the development of the characteristic attaching and effacing (A/E) lesions (Jerse et al., 1990; McDaniel et al., 1995; Sperandio et al., 1998; Schmidt, 2010; Stevens and Frankel, 2014; Franzin and Sircili, 2015). It is organized into polycistronic operons, LEE1 to 5, encoding T3SS components and regulators, chaperones, and effectors (Jerse et al., 1990; Mellies et al., 1999; Kirsch et al., 2004; Schmidt, 2010). Among the LEE-encoded proteins is intimin (Eae), an outer membrane adhesin that mediates the intimate bacterial attachment to the host’s intestinal cells (Supplementary Table S2). We detected eae subtypes β, ε, γ, and θ, and further located the respective boundaries of the islands (Figure 7). The LEE operon organization is conserved with minor rearrangements in BAA-2440 O111:H8 at espG/rorf1 as previously described in the O111:H- serotype (Ogura et al., 2009). As evident in Figure 7, the LEEs of β-eae + strains BAA-2196 O26:H11 and BAA-2215 O103:H11 exhibit syntenic organization and inventory, again suggesting a close relationship as established by our cgMLST analyses (Figure 3).
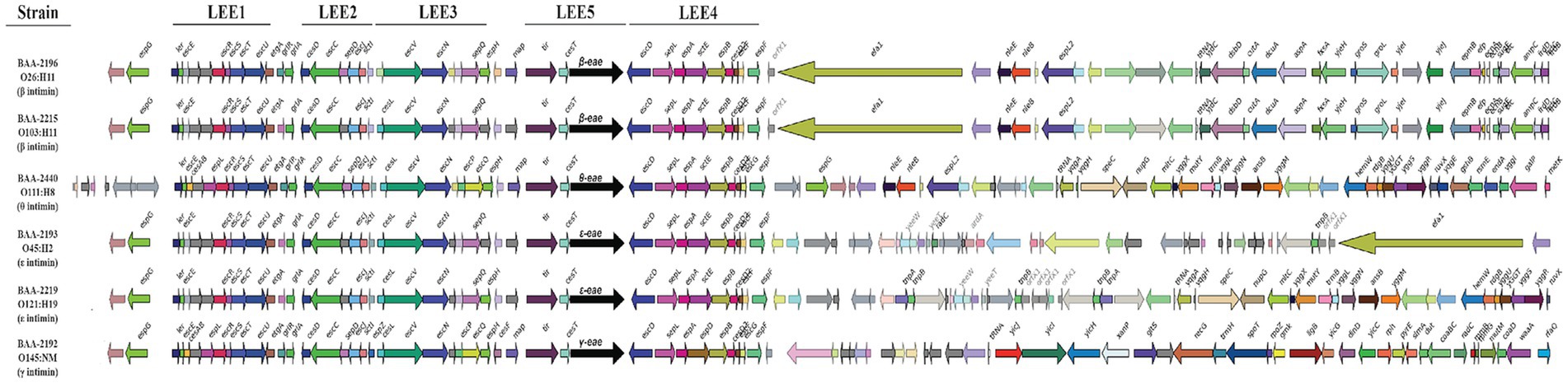
Figure 7. Comparison of LEE islands The complete LEE islands were extracted and compared in GeneSpy. Genes are colored according to their nucleotide homologies. The organization of the LEE1 to 5 operons is indicated above. The order of strains reflects their inferred phylogenomic position and is mirrored in the LEE island organization.
3.7 Comparison of Stx-virulence pathotypes
The actual disease outcome cannot be predicted from in silico virulence profiling, considering the complex interactions between infective agent, the host microbiota (Pruimboom-Brees et al., 2000; Gamage et al., 2006; Nguyen and Sperandio, 2012), and the infected patient (Wong et al., 2000; Dundas et al., 2001; Gould et al., 2009; Foster, 2013). Induction efficiency of the Stx-phages is positively correlated to Stx-production (Muniesa et al., 2004; Loś et al., 2009; Del Cogliano et al., 2018) and thus mobilization of Stx-phages is used as a means to assess the conferred pathogenic potential (Karch et al., 1999; Eppinger et al., 2022; Miyata et al., 2023). For the panel cultures, we recorded Stx-production traits under non-induced culture growth in LB and under phage mobilizing conditions by adding sublethal doses of MMC to the standard LB medium (Figure 8). In all cultures, toxin production was significantly elevated when grown in phage-inducing LB + MMC media. LB titers were undistinguishable between the cultures. In contrast, we observed culture-level differences in Stx-production capabilities upon MMC treatment. More specifically, we noted a correlation of Stx-levels to the respective stx status patterns of the strains. The class of ΦStx2a phages carry a highly potent cytotoxin (Fuller et al., 2011; Hauser et al., 2020; Pinto et al., 2021) and are known to mobilize upon activation of the SOS-response (Bonanno et al., 2016; Zhang et al., 2019; Eppinger et al., 2022). In consequence, the Stx titers of the three stx2a + isolates were all found exacerbated (Figure 8). Strain BAA-2219 (O121:H19), carrying only stx2a, is the highest-level producer followed by stx1a/stx2a + strains BAA-2440 (O111:H8) and BAA-2196 (O26:H11). Significantly lower and comparable titers were found in the remainder of strains: stx1 strains BAA-2215 (O103:H11) and BAA-2193 (O45:H2), as well as stx1a/stx2d strain BAA-2192 (O145:NM). One caveat using this methodology is that it cannot distinguish between the contribution of individual ΦStx-phages to the overall Stx titer (Skinner et al., 2014). For this reason, we further investigated the mobilization of individual ΦStx-phages and resulting stx expression in the three strains that co-harbor ΦStx1a, ΦStx2a, and ΦStx2d phages (Supplementary Figure S4). Both phages carried by stx1a/stx2a + strains BAA-2196 (O26:H11) and BAA-2440 (O111:H8) respond to MMC treatment (Supplementary Figure S4). In the latter, ΦStx2a copies and stx2a transcripts exceed the respective ΦStx1a numbers in both media, while in strain BAA-2196 the stx1a and stx2a transcript copies are comparable under non-induced growth in LB. In contrast, only the ΦStx1a phage is significantly mobilized in stx1a/stx2d + strain BAA-2192 (O145:NM), and in consequence stx1a transcripts surpass stx2d copies upon MMC induction, while stx2d copies are more abundant under non-induced growth in LB. Our observations suggest a considerable heterogeneity in ΦStx-phage mobilization, even within the same ΦStx-phage subtype (Muniesa et al., 2004; Yano et al., 2023). Overall, we observed a positive correlation between phage mobilization, toxin transcript levels, and produced titers (Figure 8; Supplementary Figure S4); in analogy to other studies (de Sablet et al., 2008; Bielaszewska et al., 2012).
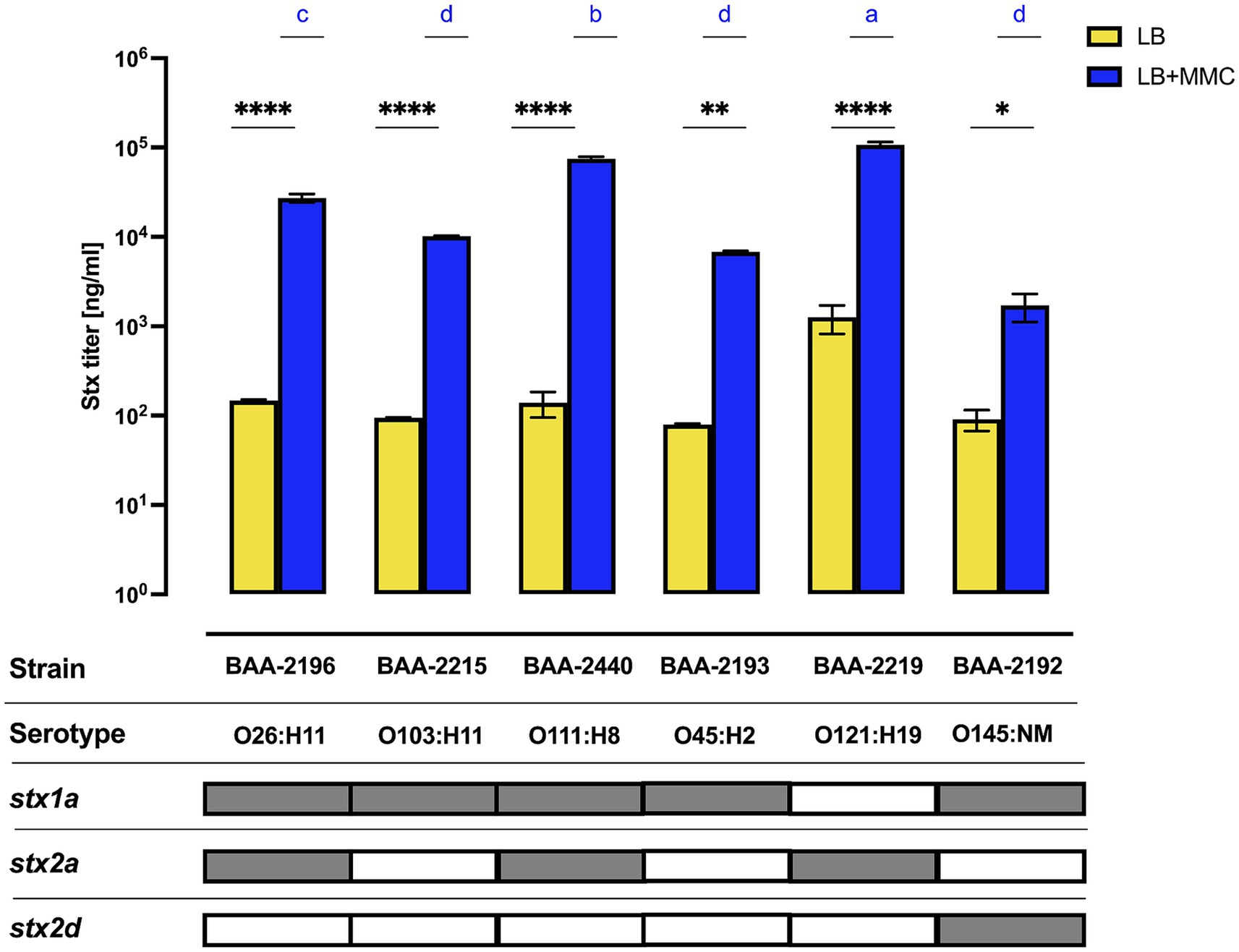
Figure 8. Variability in Stx-production The concentration of Stx1 and Stx2 produced under non-induced and MMC-induced phage mobilizing conditions was quantified by ELISA. Differences between the non-induced and MMC-induced phage mobilizing conditions for each strain were assessed using a two-way ANOVA, followed by Sidak’s multiple comparisons. Statistical significance is denoted as *p < 0.05; **p < 0.005; ***p < 0.0005; and ****p < 0.00005. For strain–strain comparison under MMC-induced conditions, differences in toxin concentration are indicated by letters (a–d), with “a” denoting the highest concentration group, in a descending order determined by a one-way ANOVA with Tukey’s multiple comparisons test.
4 Discussion and conclusions
Non-O157 STEC are a heterogenous group of isolates. The clinically most relevant serogroups, O26, O103, O111, O45, O121, and O145, are colloquially referred to as the “Big Six” due to the rising incidence of human infections. Integration of genome and virulence information for these emerging lineages is critical to improve risk assessment, biosurveillance, and prevention strategies (Franz et al., 2014; Eppinger and Cebula, 2015; Sadiq et al., 2015; Rusconi and Eppinger, 2016). Our study of these ATCC reference type cultures, comprised of six strains representing each of the non-O157 Big Six serogroups, can only provide a glimpse into the genome composition and virulence features. Our future efforts are directed to profile larger strain sets, anchored by the here presented genomes, in an attempt to capture the extent of plasticity found in the emerging human pathogenic Big Six serogroups. Comprehensive analyses of this panel highlight the distinct ΦStx-phage subtypes and their dissimilar phage mobilization patterns, likely associated with the plasticity of regulator regions relevant for replication (Ogura et al., 2015; Llarena et al., 2021; Allué-Guardia et al., 2022; Fagerlund et al., 2022), and intimately linked to Stx-production and Stx-conferred virulence. The different plasmid types and gene contents, including colicin types E3 and D and several antibiotics resistance determinants, provide only a glimpse into the genomic plasticity that can be found in this heterogenous panel of non-O157 STECs (Cortimiglia et al., 2020). Production of colicins and antibiotic resistance are major drivers of microbial evolution (Feldgarden and Riley, 1999; Leekitcharoenphon et al., 2021). Fitness effects mediated by colicins and antibiotic resistance determinants will impact a strain’s individual evolutionary trajectory, and we note that antibiotic resistance and thus pathogenic potential among all STEC serogroups has increased over time and calls for enhanced biosurveillance (Mukherjee et al., 2021). The availability of closed high-quality genomes and carried plasmids of representative Big Six strains, along with insight into their pathogenome make-up and Stx-virulence pathotypes provides a foundation for the research community to broadly explore common and lineage-specific characteristics and evolutionary trajectories of these globally emerging human pathogenic non-O157 STEC lineages.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
AK: Writing – review & editing, Data curation, Formal analysis, Investigation, Validation, Visualization. SK: Writing – review & editing, Formal analysis, Investigation, Project administration, Validation. JaB: Writing – review & editing, Formal analysis, Investigation, Resources. JoB: Writing – review & editing, Formal analysis, Investigation, Resources. ME: Writing – original draft, Writing – review & editing, Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Software, Supervision, Visualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award Number SC1GM135110, and the South Texas Center for Emerging Infectious Diseases (STCEID). This work received computational support from the High-Performance Computing Cluster (HPCC) operated by Tech Solutions at UTSA.
Acknowledgments
The use of product and company names is necessary to accurately report the methods and results; however, the United States Department of Agriculture (USDA) neither guarantees nor warrants the standard of the products, and the use of names by the USDA implies no approval of the product to the exclusion of others that may also be suitable. The USDA is an equal opportunity provider and employer. We would like to thank Felix Borrego for assistance with data visualization.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1364026/full#supplementary-material
Footnotes
1. ^https://www.atcc.org/products/mp-9
2. ^http://www.bioinformatics.babraham.ac.uk/projects/fastqc
3. ^https://github.com/tseemann/ABRicate
4. ^https://cge.cbs.dtu.dk/services/ResFinder/
5. ^https://enterobase.warwick.ac.uk/species/ecoli/download_7_gene
References
Abu-Ali, G. S., Lacher, D. W., Wick, L. M., Qi, W., and Whittam, T. S. (2009). Genomic diversity of pathogenic Escherichia coli of the EHEC 2 clonal complex. BMC Genomics 10:296. doi: 10.1186/1471-2164-10-296
AbuOun, M., Jones, H., Stubberfield, E., Gilson, D., Shaw, L. P., Hubbard, A. T. M., et al. (2021). A genomic epidemiological study shows that prevalence of antimicrobial resistance in Enterobacterales is associated with the livestock host, as well as antimicrobial usage. Microb Genom 7:630. doi: 10.1099/mgen.0.000630
Ahmed, S., Das, T., Islam, M. Z., Herrero-Fresno, A., Biswas, P. K., and Olsen, J. E. (2020). High prevalence of mcr-1-encoded colistin resistance in commensal Escherichia coli from broiler chicken in Bangladesh. Sci. Rep. 10:18637. doi: 10.1038/s41598-020-75608-2
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Allué-Guardia, A., Koenig, S. S. K., Martinez, R. A., Rodriguez, A. L., Bosilevac, J. M., Feng, P., et al. (2022). Pathogenomes and variations in Shiga toxin production among geographically distinct clones of Escherichia coli O113:H21. Microb Genom 8:796. doi: 10.1099/mgen.0.000796
Amadio, A., Bono, J. L., Irazoqui, M., Larzábal, M., Marques Da Silva, W., Eberhardt, M. F., et al. (2021). Genomic analysis of Shiga toxin-containing Escherichia coli O157:H7 isolated from Argentinean cattle. PLoS One 16:e0258753. doi: 10.1371/journal.pone.0258753
Amézquita-López, B. A., Quiñones, B., Soto-Beltrán, M., Lee, B. G., Yambao, J. C., Lugo-Melchor, O. Y., et al. (2016). Antimicrobial resistance profiles of Shiga toxin-producing Escherichia coli O157 and non-O157 recovered from domestic farm animals in rural communities in northwestern Mexico. Antimicrob. Resist. Infect. Control 5:1. doi: 10.1186/s13756-015-0100-5
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Ashton, P. M., Perry, N., Ellis, R., Petrovska, L., Wain, J., Grant, K. A., et al. (2015). Insight into Shiga toxin genes encoded by Escherichia coli O157 from whole genome sequencing. PeerJ 3:e739. doi: 10.7717/peerj.739
Awosile, B., Fritzler, J., Levent, G., Rahman, M. K., Ajulo, S., Daniel, I., et al. (2023). Genomic characterization of fecal Escherichia coli isolates with reduced susceptibility to Beta-lactam antimicrobials from wild hogs and coyotes. Pathogens 12:929. doi: 10.3390/pathogens12070929
Bai, X., Fu, S., Zhang, J., Fan, R., Xu, Y., Sun, H., et al. (2018). Identification and pathogenomic analysis of an Escherichia coli strain producing a novel Shiga toxin 2 subtype. Sci. Rep. 8:6756. doi: 10.1038/s41598-018-25233-x
Balasubramanian, S., Osburne, M. S., Brinjones, H., Tai, A. K., and Leong, J. M. (2019). Prophage induction, but not production of phage particles, is required for lethal disease in a microbiome-replete murine model of enterohemorrhagic E. coli infection. PLoS Pathog. 15:e1007494. doi: 10.1371/journal.ppat.1007494
Baumler, A. J., and Sperandio, V. (2016). Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535, 85–93. doi: 10.1038/nature18849
Bean, D. C., Livermore, D. M., and Hall, L. M. (2009). Plasmids imparting sulfonamide resistance in Escherichia coli: implications for persistence. Antimicrob. Agents Chemother. 53, 1088–1093. doi: 10.1128/AAC.00800-08
Bertelli, C., and Brinkman, F. S. L. (2018). Improved genomic island predictions with IslandPath-DIMOB. Bioinformatics 34, 2161–2167. doi: 10.1093/bioinformatics/bty095
Bertelli, C., Laird, M. R., Williams, K. P., Simon Fraser University Research Computing Group Lau, B. Y., Hoad, G., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bertelli, C., Tilley, K. E., and Brinkman, F. S. L. (2018). Microbial genomic island discovery, visualization and analysis. Brief. Bioinform. 20, 1685–1698. doi: 10.1093/bib/bby042
Bettelheim, K. A. (2007). The non-O157 Shiga-toxigenic (verocytotoxigenic) Escherichia coli; under-rated pathogens. Crit. Rev. Microbiol. 33, 67–87. doi: 10.1080/10408410601172172
Bielaszewska, M., Idelevich, E. A., Zhang, W., Bauwens, A., Schaumburg, F., Mellmann, A., et al. (2012). Effects of antibiotics on Shiga toxin 2 production and bacteriophage induction by epidemic Escherichia coli O104:H4 strain. Antimicrob. Agents Chemother. 56, 3277–3282. doi: 10.1128/AAC.06315-11
Blattner, F. R., Plunkett, G. 3rd, Bloch, C. A., Perna, N. T., Burland, V., Riley, M., et al. (1997). The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1462. doi: 10.1126/science.277.5331.1453
Bonanno, L., Loukiadis, E., Mariani-Kurkdjian, P., Oswald, E., Garnier, L., Michel, V., et al. (2015). Diversity of Shiga toxin-producing Escherichia coli (STEC) O26:H11 strains examined via stx subtypes and insertion sites of Stx and Esp K bacteriophages. Appl. Environ. Microbiol. 81, 3712–3721. doi: 10.1128/AEM.00077-15
Bonanno, L., Petit, M. A., Loukiadis, E., Michel, V., and Auvray, F. (2016). Heterogeneity in induction level, infection ability, and morphology of Shiga toxin-encoding phages (Stx phages) from dairy and human Shiga toxin-producing Escherichia coli O26:H11 isolates. Appl. Environ. Microbiol. 82, 2177–2186. doi: 10.1128/AEM.03463-15
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Caprioli, A., Morabito, S., Brugère, H., and Oswald, E. (2005). Enterohaemorrhagic Escherichia coli: emerging issues on virulence and modes of transmission. Vet. Res. 36, 289–311. doi: 10.1051/vetres:2005002
Carbonari, C. C., Miliwebsky, E. S., Zolezzi, G., Deza, N. L., Fittipaldi, N., Manfredi, E., et al. (2022). The importance of Shiga toxin-producing Escherichia coli O145:NM[H28]/H28 infections in Argentina, 1998-2020. Microorganisms 10:582. doi: 10.3390/microorganisms10030582
Carrillo, C. D., Koziol, A. G., Mathews, A., Goji, N., Lambert, D., Huszczynski, G., et al. (2016). Comparative evaluation of genomic and laboratory approaches for determination of Shiga toxin subtypes in Escherichia coli. J. Food Prot. 79, 2078–2085. doi: 10.4315/0362-028X.JFP-16-228
Cascales, E., Buchanan, S. K., Duché, D., Kleanthous, C., Lloubès, R., Postle, K., et al. (2007). Colicin biology. Microbiol. Mol. Biol. Rev. 71, 158–229. doi: 10.1128/MMBR.00036-06
Casjens, S. R., and Hendrix, R. W. (2015). Bacteriophage lambda: early pioneer and still relevant. Virology 479-480, 310–330. doi: 10.1016/j.virol.2015.02.010
Caza, M., and Kronstad, J. W. (2013). Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front. Cell. Infect. Microbiol. 3:80. doi: 10.3389/fcimb.2013.00080
Community, T. G. (2022). The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 50, W345–W351. doi: 10.1093/nar/gkac247
Cortimiglia, C., Borney, M. F., Bassi, D., and Cocconcelli, P. S. (2020). Genomic investigation of virulence potential in Shiga toxin Escherichia coli (STEC) strains from a semi-hard raw Milk cheese. Front. Microbiol. 11:629189. doi: 10.3389/fmicb.2020.629189
Cowley, L. A., Dallman, T. J., Jenkins, C., and Sheppard, S. K. (2019). Phage predation shapes the population structure of Shiga-toxigenic Escherichia coli O157:H7 in the UK: an evolutionary perspective. Front. Genet. 10:763. doi: 10.3389/fgene.2019.00763
Cox, G. B., Gibson, F., Luke, R. K., Newton, N. A., O’Brien, I. G., and Rosenberg, H. (1970). Mutations affecting iron transport in Escherichia coli. J. Bacteriol. 104, 219–226. doi: 10.1128/jb.104.1.219-226.1970
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Darling, A. E., Miklos, I., and Ragan, M. A. (2008). Dynamics of genome rearrangement in bacterial populations. PLoS Genet. 4:e1000128. doi: 10.1371/journal.pgen.1000128
De Sablet, T., Bertin, Y., Vareille, M., Girardeau, J.-P., Garrivier, A., Gobert, A. P., et al. (2008). Differential expression of stx2 variants in Shiga toxin-producing Escherichia coli belonging to seropathotypes A and C. Microbiology 154, 176–186. doi: 10.1099/mic.0.2007/009704-0
Del Cogliano, M. E., Pinto, A., Goldstein, J., Zotta, E., Ochoa, F., Fernandez-Brando, R. J., et al. (2018). Relevance of bacteriophage 933W in the development of hemolytic uremic syndrome (HUS). Front. Microbiol. 9:3104. doi: 10.3389/fmicb.2018.03104
Delannoy, S., Mariani-Kurkdjian, P., Webb, H. E., Bonacorsi, S., and Fach, P. (2017). The Mobilome; A major contributor to Escherichia coli stx2-positive O26:H11 strains intra-serotype diversity. Front. Microbiol. 8:1625. doi: 10.3389/fmicb.2017.01625
Díaz, L., Gutierrez, S., Moreno-Switt, A. I., Hervé, L. P., Hamilton-West, C., Padola, N. L., et al. (2021). Diversity of non-O157 Shiga toxin-producing Escherichia coli isolated from cattle from central and southern Chile. Animals 11:2388. doi: 10.3390/ani11082388
Donohue-Rolfe, A., Kondova, I., Oswald, S., Hutto, D., and Tzipori, S. (2000). Escherichia coli O157:H7 strains that express Shiga toxin (Stx) 2 alone are more neurotropic for gnotobiotic piglets than are isotypes producing only Stx1 or both Stx1 and Stx2. J Infect Dis 181, 1825–1829. doi: 10.1086/315421
Dundas, S., Todd, W. T., Stewart, A. I., Murdoch, P. S., Chaudhuri, A. K., and Hutchinson, S. J. (2001). The Central Scotland Escherichia coli O157:H7 outbreak: risk factors for the hemolytic uremic syndrome and death among hospitalized patients. Clin. Infect. Dis. 33, 923–931. doi: 10.1086/322598
Edgar, R., and Bibi, E. (1997). MdfA, an Escherichia coli multidrug resistance protein with an extraordinarily broad spectrum of drug recognition. J. Bacteriol. 179, 2274–2280. doi: 10.1128/jb.179.7.2274-2280.1997
Eklund, M., Scheutz, F., and Siitonen, A. (2001). Clinical isolates of non-O157 Shiga toxin-producing Escherichia coli: serotypes, virulence characteristics, and molecular profiles of strains of the same serotype. J. Clin. Microbiol. 39, 2829–2834. doi: 10.1128/JCM.39.8.2829-2834.2001
Eppinger, M., Almería, S., Allué-Guardia, A., Bagi, L. K., Kalalah, A. A., Gurtler, J. B., et al. (2022). Genome sequence analysis and characterization of Shiga toxin 2 production by Escherichia coli O157:H7 strains associated with a laboratory infection. Front. Cell. Infect. Microbiol. 12:888568. doi: 10.3389/fcimb.2022.888568
Eppinger, M., and Cebula, T. A. (2015). Future perspectives, applications and challenges of genomic epidemiology studies for food-borne pathogens: A case study of Enterohemorrhagic Escherichia coli (EHEC) of the O157:H7 serotype. Gut Microbes 6, 194–201. doi: 10.4161/19490976.2014.969979
Eppinger, M., Daugherty, S., Agrawal, S., Galens, K., Sengamalay, N., Sadzewicz, L., et al. (2013). Whole-genome draft sequences of 26 Enterohemorrhagic Escherichia coli O157:H7 strains. Genome Announc. 1:e0013412. doi: 10.1128/genomeA.00134-12
Eppinger, M., Mammel, M. K., Leclerc, J. E., Ravel, J., and Cebula, T. A. (2011). Genomic anatomy of Escherichia coli O157:H7 outbreaks. Proc. Natl. Acad. Sci. U. S. A. 108, 20142–20147. doi: 10.1073/pnas.1107176108
Fagerlund, A., Aspholm, M., Węgrzyn, G., and Lindbäck, T. (2022). High diversity in the regulatory region of Shiga toxin encoding bacteriophages. BMC Genomics 23:230. doi: 10.1186/s12864-022-08428-5
Feldgarden, M., and Riley, M. A. (1999). The phenotypic and fitness effects of colicin resistance in Escherichia Coli K-12. Evolution 53, 1019–1027. doi: 10.1111/j.1558-5646.1999.tb04517.x
Florensa, A. F., Kaas, R. S., Clausen, P., Aytan-Aktug, D., and Aarestrup, F. M. (2022). ResFinder - an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb Genom 8:748. doi: 10.1099/mgen.0.000748
Foley, S. L., Lynne, A. M., and Nayak, R. (2009). Molecular typing methodologies for microbial source tracking and epidemiological investigations of gram-negative bacterial foodborne pathogens. Infect. Genet. Evol. 9, 430–440. doi: 10.1016/j.meegid.2009.03.004
Foster, D. B. (2013). Modulation of the enterohemorrhagic E-coli virulence program through the human gastrointestinal tract. Virulence 4, 315–323. doi: 10.4161/viru.24318
Francisco, A. P., Bugalho, M., Ramirez, M., and Carriço, J. A. (2009). Global optimal eBURST analysis of multilocus typing data using a graphic matroid approach. BMC Bioinformatics 10:152. doi: 10.1186/1471-2105-10-152
Franz, E., Delaquis, P., Morabito, S., Beutin, L., Gobius, K., Rasko, D. A., et al. (2014). Exploiting the explosion of information associated with whole genome sequencing to tackle Shiga toxin-producing Escherichia coli (STEC) in global food production systems. Int. J. Food Microbiol. 187, 57–72. doi: 10.1016/j.ijfoodmicro.2014.07.002
Franzin, F. M., and Sircili, M. P. (2015). Locus of enterocyte effacement: a pathogenicity island involved in the virulence of enteropathogenic and enterohemorragic Escherichia coli subjected to a complex network of gene regulation. Biomed. Res. Int. 2015:534738. doi: 10.1155/2015/534738
Fuller, C. A., Pellino, C. A., Flagler, M. J., Strasser, J. E., and Weiss, A. A. (2011). Shiga toxin subtypes display dramatic differences in potency. Infect. Immun. 79, 1329–1337. doi: 10.1128/IAI.01182-10
Gamage, S. D., Patton, A. K., Strasser, J. E., Chalk, C. L., and Weiss, A. A. (2006). Commensal bacteria influence Escherichia coli O157:H7 persistence and Shiga toxin production in the mouse intestine. Infect. Immun. 74, 1977–1983. doi: 10.1128/IAI.74.3.1977-1983.2006
Garcia, E. C., Brumbaugh, A. R., and Mobley, H. L. (2011). Redundancy and specificity of Escherichia coli iron acquisition systems during urinary tract infection. Infect. Immun. 79, 1225–1235. doi: 10.1128/IAI.01222-10
Garcia, P. S., Jauffrit, F., Grangeasse, C., and Brochier-Armanet, C. (2019). GeneSpy, a user-friendly and flexible genomic context visualizer. Bioinformatics 35, 329–331. doi: 10.1093/bioinformatics/bty459
Glassman, H., Ferrato, C., and Chui, L. (2022). Epidemiology of non-O157 Shiga toxin-producing Escherichia coli in the province of Alberta, Canada, from 2018 to 2021. Microorganisms 10:814. doi: 10.3390/microorganisms10040814
Goldstein, S., Beka, L., Graf, J., and Klassen, J. L. (2019). Evaluation of strategies for the assembly of diverse bacterial genomes using MinION long-read sequencing. BMC Genomics 20:23. doi: 10.1186/s12864-018-5381-7
Gould, L. H., Demma, L., Jones, T. F., Hurd, S., Vugia, D. J., Smith, K., et al. (2009). Hemolytic uremic syndrome and death in persons with Escherichia coli O157:H7 infection, foodborne diseases active surveillance network sites, 2000-2006. Clin. Infect. Dis. 49, 1480–1485. doi: 10.1086/644621
Gould, L. H., Mody, R. K., Ong, K. L., Clogher, P., Cronquist, A. B., Garman, K. N., et al. (2013). Increased recognition of non-O157 Shiga toxin-producing Escherichia coli infections in the United States during 2000-2010: epidemiologic features and comparison with E. coli O157 infections. Foodborne Pathog. Dis. 10, 453–460. doi: 10.1089/fpd.2012.1401
Greig, D. R., Do Nascimento, V., Olonade, I., Swift, C., Nair, S., and Jenkins, C. (2023). Surveillance of antimicrobial resistant Shiga toxin-producing E. coli O157:H7 in England, 2016–2020. J. Antimicrob. Chemother. 78, 2263–2273. doi: 10.1093/jac/dkad231
Groth, A. C., and Calos, M. P. (2004). Phage integrases: biology and applications. J. Mol. Biol. 335, 667–678. doi: 10.1016/j.jmb.2003.09.082
Hadler, J. L., Clogher, P., Hurd, S., Phan, Q., Mandour, M., Bemis, K., et al. (2011). Ten-year trends and risk factors for non-O157 Shiga toxin-producing Escherichia coli found through Shiga toxin testing, Connecticut, 2000-2009. Clin. Infect. Dis. 53, 269–276. doi: 10.1093/cid/cir377
Hammerum, A. M., Sandvang, D., Andersen, S. R., Seyfarth, A. M., Porsbo, L. J., Frimodt-Møller, N., et al. (2006). Detection of sul1, sul2 and sul3 in sulphonamide resistant Escherichia coli isolates obtained from healthy humans, pork and pigs in Denmark. Int. J. Food Microbiol. 106, 235–237. doi: 10.1016/j.ijfoodmicro.2005.06.023
Hauser, J. R., Atitkar, R. R., Petro, C. D., Lindsey, R. L., Strockbine, N., O’Brien, A. D., et al. (2020). The virulence of Escherichia coli O157:H7 isolates in mice depends on Shiga toxin type 2a (Stx2a)-induction and high levels of Stx2a in stool. Front. Cell. Infect. Microbiol. 10:62. doi: 10.3389/fcimb.2020.00062
Hegde, N. V., Cote, R., Jayarao, B. M., Muldoon, M., Lindpaintner, K., Kapur, V., et al. (2012). Detection of the top six non-O157 Shiga toxin-producing Escherichia coli O groups by ELISA. Foodborne Pathog. Dis. 9, 1044–1048. doi: 10.1089/fpd.2012.1231
Henderson, S. T., Singh, P., Knupp, D., Lacher, D. W., Abu-Ali, G. S., Rudrik, J. T., et al. (2021). Variability in the occupancy of Escherichia coli O157 integration sites by Shiga toxin-encoding prophages. Toxins 13:433. doi: 10.3390/toxins13070433
Herold, S., Karch, H., and Schmidt, H. (2004). Shiga toxin-encoding bacteriophages – genomes in motion. Int. J. Med. Microbiol. 294, 115–121. doi: 10.1016/j.ijmm.2004.06.023
Huang, A., Friesen, J., and Brunton, J. L. (1987). Characterization of a bacteriophage that carries the genes for production of Shiga-like toxin 1 in Escherichia coli. J. Bacteriol. 169, 4308–4312. doi: 10.1128/jb.169.9.4308-4312.1987
Iguchi, A., Iyoda, S., and Ohnishi, M. (2012). Molecular characterization reveals three distinct clonal groups among clinical Shiga toxin-producing Escherichia coli strains of serogroup O103. J. Clin. Microbiol. 50, 2894–2900. doi: 10.1128/JCM.00789-12
Iguchi, A., Thomson, N. R., Ogura, Y., Saunders, D., Ooka, T., Henderson, I. R., et al. (2009). Complete genome sequence and comparative genome analysis of enteropathogenic Escherichia coli O127:H6 strain E2348/69. J. Bacteriol. 191, 347–354. doi: 10.1128/JB.01238-08
Ingle, D. J., Valcanis, M., Kuzevski, A., Tauschek, M., Inouye, M., Stinear, T., et al. (2016). In silico serotyping of E. coli from short read data identifies limited novel O-loci but extensive diversity of O:H serotype combinations within and between pathogenic lineages. Microb Genom 2:e000064. doi: 10.1099/mgen.0.000064
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Jarocki, V. M., Reid, C. J., Chapman, T. A., and Djordjevic, S. P. (2019). Escherichia coli ST302: genomic analysis of virulence potential and antimicrobial resistance mediated by Mobile genetic elements. Front. Microbiol. 10:3098. doi: 10.3389/fmicb.2019.03098
Jaudou, S., Tran, M. L., Vorimore, F., Fach, P., and Delannoy, S. (2022). Evaluation of high molecular weight DNA extraction methods for long-read sequencing of Shiga toxin-producing Escherichia coli. PLoS One 17:e0270751. doi: 10.1371/journal.pone.0270751
Jerse, A. E., Yu, J., Tall, B. D., and Kaper, J. B. (1990). A genetic locus of enteropathogenic Escherichia coli necessary for the production of attaching and effacing lesions on tissue culture cells. Proc. Natl. Acad. Sci. U. S. A. 87, 7839–7843. doi: 10.1073/pnas.87.20.7839
Jinnerot, T., Tomaselli, A. T. P., Johannessen, G. S., Söderlund, R., Urdahl, A. M., Aspán, A., et al. (2020). The prevalence and genomic context of Shiga toxin 2a genes in E. coli found in cattle. PLoS One 15:e0232305. doi: 10.1371/journal.pone.0232305
Johnson, T. J., and Nolan, L. K. (2009). Pathogenomics of the virulence plasmids of Escherichia coli. Microbiol. Mol. Biol. Rev. 73, 750–774. doi: 10.1128/MMBR.00015-09
Johnson, K. E., Thorpe, C. M., and Sears, C. L. (2006). The emerging clinical importance of non-O157 Shiga toxin-producing Escherichia coli. Clin. Infect. Dis. 43, 1587–1595. doi: 10.1086/509573
Jores, J., Rumer, L., and Wieler, L. H. (2004). Impact of the locus of enterocyte effacement pathogenicity island on the evolution of pathogenic Escherichia coli. Int. J. Med. Microbiol. 294, 103–113. doi: 10.1016/j.ijmm.2004.06.024
Ju, W., Cao, G., Rump, L., Strain, E., Luo, Y., Timme, R., et al. (2012). Phylogenetic analysis of non-O157 Shiga toxin-producing Escherichia coli strains by whole-genome sequencing. J. Clin. Microbiol. 50, 4123–4127. doi: 10.1128/JCM.02262-12
Jünemann, S., Sedlazeck, F. J., Prior, K., Albersmeier, A., John, U., Kalinowski, J., et al. (2013). Updating benchtop sequencing performance comparison. Nat. Biotechnol. 31, 294–296. doi: 10.1038/nbt.2522
Kaper, J. B., Nataro, J. P., and Mobley, H. L. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140. doi: 10.1038/nrmicro818
Karch, H., Schmidt, H., Janetzki-Mittmann, C., Scheef, J., and Kroger, M. (1999). Shiga toxins even when different are encoded at identical positions in the genomes of related temperate bacteriophages. Mol. Gen. Genet. 262, 600–607. doi: 10.1007/s004380051122
Karmali, M. A., Petric, M., Lim, C., Fleming, P. C., and Steele, B. T. (1983). Escherichia coli cytotoxin, haemolytic-uraemic syndrome, and haemorrhagic colitis. Lancet 2, 1299–1300. doi: 10.1016/S0140-6736(83)91167-4
Kimmitt, P. T., Harwood, C. R., and Barer, M. R. (2000). Toxin gene expression by Shiga toxin-producing Escherichia coli: the role of antibiotics and the bacterial SOS response. Emerg. Infect. Dis. 6, 458–465. doi: 10.3201/eid0605.000503
Kirsch, P., Jores, J., and Wieler, L. H. (2004). Plasticity of bacterial genomes: pathogenicity islands and the locus of enterocyte effacement (LEE). Berl. Munch. Tierarztl. Wochenschr. 117, 116–129.
Krüger, A., and Lucchesi, P. M. (2015). Shiga toxins and stx phages: highly diverse entities. Microbiology 161, 451–462. doi: 10.1099/mic.0.000003
Kruskal, J. B. (1956). On the shortest spanning subtree of a graph and the traveling salesman problem. Proc. Am. Math. Soc. 7, 48–50. doi: 10.1090/S0002-9939-1956-0078686-7
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Kyle, J. L., Cummings, C. A., Parker, C. T., Quiñones, B., Vatta, P., Newton, E., et al. (2012). Escherichia coli serotype O55:H7 diversity supports parallel acquisition of bacteriophage at Shiga toxin phage insertion sites during evolution of the O157:H7 lineage. J. Bacteriol. 194, 1885–1896. doi: 10.1128/JB.00120-12
Lawrence, J. G., and Ochman, H. (1998). Molecular archaeology of the Escherichia coli genome. Proc. Natl. Acad. Sci. U. S. A. 95, 9413–9417. doi: 10.1073/pnas.95.16.9413
Lee, K. Y., Lavelle, K., Huang, A., Atwill, E. R., Pitesky, M., and Li, X. (2023). Assessment of prevalence and diversity of antimicrobial resistant Escherichia coli from retail meats in Southern California. Antibiotics 12:782. doi: 10.3390/antibiotics12040782
Leekitcharoenphon, P., Johansson, M. H. K., Munk, P., Malorny, B., Skarżyńska, M., Wadepohl, K., et al. (2021). Genomic evolution of antimicrobial resistance in Escherichia coli. Sci. Rep. 11:15108. doi: 10.1038/s41598-021-93970-7
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Lim, J. Y., Yoon, J., and Hovde, C. J. (2010). A brief overview of Escherichia coli O157:H7 and its plasmid O157. J. Microbiol. Biotechnol. 20, 5–14. doi: 10.4014/jmb.0908.08007
Liu, C., Zheng, H., Yang, M., Xu, Z., Wang, X., Wei, L., et al. (2015). Genome analysis and in vivo virulence of porcine extraintestinal pathogenic Escherichia coli strain PCN033. BMC Genomics 16:717. doi: 10.1186/s12864-015-1890-9
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: a general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–d917. doi: 10.1093/nar/gkab1107
Llarena, A. K., Aspholm, M., O’sullivan, K., Wêgrzyn, G., and Lindbäck, T. (2021). Replication region analysis reveals non-lambdoid Shiga toxin converting bacteriophages. Front. Microbiol. 12:640945. doi: 10.3389/fmicb.2021.640945
Loś, J. M., Loś, M., Wegrzyn, G., and Wegrzyn, A. (2009). Differential efficiency of induction of various lambdoid prophages responsible for production of Shiga toxins in response to different induction agents. Microb. Pathog. 47, 289–298. doi: 10.1016/j.micpath.2009.09.006
Majowicz, S. E., Scallan, E., Jones-Bitton, A., Sargeant, J. M., Stapleton, J., Angulo, F. J., et al. (2014). Global incidence of human Shiga toxin-producing Escherichia coli infections and deaths: a systematic review and knowledge synthesis. Foodborne Pathog. Dis. 11, 447–455. doi: 10.1089/fpd.2013.1704
Mcdaniel, T. K., Jarvis, K. G., Donnenberg, M. S., and Kaper, J. B. (1995). A genetic locus of enterocyte effacement conserved among diverse enterobacterial pathogens. Proc. Natl. Acad. Sci. U. S. A. 92, 1664–1668. doi: 10.1073/pnas.92.5.1664
Mcnichol, B. A., Bova, R. A., Torres, K., Preston, L. N., and Melton-Celsa, A. R. (2021). Switching Shiga toxin (Stx) type from Stx2d to Stx2a but not Stx2c alters virulence of Stx-producing Escherichia coli (STEC) Strain B2F1 in streptomycin (Str)-treated mice. Toxins 13:64. doi: 10.3390/toxins13010064
Mellies, J. L., Elliott, S. J., Sperandio, V., Donnenberg, M. S., and Kaper, J. B. (1999). The per regulon of enteropathogenic Escherichia coli: identification of a regulatory cascade and a novel transcriptional activator, the locus of enterocyte effacement (LEE)-encoded regulator (Ler). Mol. Microbiol. 33, 296–306. doi: 10.1046/j.1365-2958.1999.01473.x
Messele, Y. E., Alkhallawi, M., Veltman, T., Trott, D. J., Mcmeniman, J. P., Kidd, S. P., et al. (2022). Phenotypic and genotypic analysis of antimicrobial resistance in Escherichia coli recovered from feedlot beef cattle in Australia. Animals 12:2256. doi: 10.3390/ani12172256
Mey, A. R., Gómez-Garzón, C., and Payne, S. M. (2021). Iron transport and metabolism in Escherichia, Shigella, and Salmonella. EcoSal Plus 9:eESP00342020. doi: 10.1128/ecosalplus.ESP-0034-2020
Miyata, T., Taniguchi, I., Nakamura, K., Gotoh, Y., Yoshimura, D., Itoh, T., et al. (2023). Alteration of a Shiga toxin-encoding phage associated with a change in toxin production level and disease severity in Escherichia coli. Microb Genom 9:935. doi: 10.1099/mgen.0.000935
Moon, H. W., Whipp, S. C., Argenzio, R. A., Levine, M. M., and Giannella, R. A. (1983). Attaching and effacing activities of rabbit and human enteropathogenic Escherichia coli in pig and rabbit intestines. Infect. Immun. 41, 1340–1351. doi: 10.1128/iai.41.3.1340-1351.1983
Moser, A. I., Kuenzli, E., Campos-Madueno, E. I., Büdel, T., Rattanavong, S., Vongsouvath, M., et al. (2021). Antimicrobial-resistant Escherichia coli strains and their plasmids in people, poultry, and chicken meat in Laos. Front. Microbiol. 12:708182. doi: 10.3389/fmicb.2021.708182
Mukherjee, S., Blankenship, H. M., Rodrigues, J. A., Mosci, R. E., Rudrik, J. T., and Manning, S. D. (2021). Antibiotic susceptibility profiles and frequency of resistance genes in clinical Shiga toxin-producing Escherichia coli isolates from Michigan over a 14-year period. Antimicrob. Agents Chemother. 65:e0118921. doi: 10.1128/AAC.01189-21
Mukherjee, S., Mosci, R. E., Anderson, C. M., Snyder, B. A., Collins, J., Rudrik, J. T., et al. (2017). Antimicrobial drug-resistant Shiga toxin-producing Escherichia coli infections, Michigan, USA. Emerg. Infect. Dis. 23, 1609–1611. doi: 10.3201/eid2309.170523
Muniesa, M., Blanco, J. E., De Simón, M., Serra-Moreno, R., Blanch, A. R., and Jofre, J. (2004). Diversity of stx2 converting bacteriophages induced from Shiga-toxin-producing Escherichia coli strains isolated from cattle. Microbiology 150, 2959–2971. doi: 10.1099/mic.0.27188-0
Néron, B., Littner, E., Haudiquet, M., Perrin, A., Cury, J., and Rocha, E. P. C. (2022). IntegronFinder 2.0: identification and analysis of integrons across Bacteria, with a focus on antibiotic resistance in Klebsiella. bioRxiv [Preprint].
Nguyen, Y., and Sperandio, V. (2012). Enterohemorrhagic E. coli (EHEC) pathogenesis. Front. Cell. Infect. Microbiol. 2:90. doi: 10.3389/fcimb.2012.00090
Nyong, E. C., Zaia, S. R., Allué-Guardia, A., Rodriguez, A. L., Irion-Byrd, Z., Koenig, S. S. K., et al. (2020). Pathogenomes of atypical non-shigatoxigenic Escherichia coli NSF/SF O157:H7/NM: comprehensive Phylogenomic analysis using closed genomes. Front. Microbiol. 11:619. doi: 10.3389/fmicb.2020.00619
Obrig, T. G., and Karpman, D. (2012). Shiga toxin pathogenesis: kidney complications and renal failure. Curr. Top. Microbiol. Immunol. 357, 105–136. doi: 10.1007/82_2011_172
Ogura, Y., Mondal, S. I., Islam, M. R., Mako, T., Arisawa, K., Katsura, K., et al. (2015). The Shiga toxin 2 production level in enterohemorrhagic Escherichia coli O157:H7 is correlated with the subtypes of toxin-encoding phage. Sci. Rep. 5:16663. doi: 10.1038/srep16663
Ogura, Y., Ooka, T., Iguchi, A., Toh, H., Asadulghani, M., Oshima, K., et al. (2009). Comparative genomics reveal the mechanism of the parallel evolution of O157 and non-O157 enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 106, 17939–17944. doi: 10.1073/pnas.0903585106
Orskov, I., Orskov, F., Jann, B., and Jann, K. (1977). Serology, chemistry, and genetics of O and K antigens of Escherichia coli. Bacteriol. Rev. 41, 667–710. doi: 10.1128/br.41.3.667-710.1977
Pacheco, A. R., Curtis, M. M., Ritchie, J. M., Munera, D., Waldor, M. K., Moreira, C. G., et al. (2012). Fucose sensing regulates bacterial intestinal colonization. Nature 492, 113–117. doi: 10.1038/nature11623
Pacheco, A. R., and Sperandio, V. (2012). Shiga toxin in enterohemorrhagic E.Coli: regulation and novel anti-virulence strategies. Front. Cell. Infect. Microbiol. 2:81. doi: 10.3389/fcimb.2012.00081
Pasquali, F., Palma, F., Trevisani, M., Parisi, A., Lucchi, A., Cesare, A., et al. (2018). Whole genome sequencing based typing and characterisation of Shiga-toxin producing Escherichia coli strains belonging to O157 and O26 serotypes and isolated in dairy farms. Ital J Food Saf 7:7673. doi: 10.4081/ijfs.2018.7673
Perna, N. T., Plunkett, G. 3rd, Burland, V., Mau, B., Glasner, J. D., Rose, D. J., et al. (2001). Genome sequence of enterohaemorrhagic Escherichia coli O157:H7. Nature 409, 529–533. doi: 10.1038/35054089
Petkau, A., Mabon, P., Sieffert, C., Knox, N. C., Cabral, J., Iskander, M., et al. (2017). SNVPhyl: a single nucleotide variant phylogenomics pipeline for microbial genomic epidemiology. Microbial Genomics 3:116. doi: 10.1099/mgen.0.000116
Pilla, G., and Tang, C. M. (2018). Going around in circles: virulence plasmids in enteric pathogens. Nat. Rev. Microbiol. 16, 484–495. doi: 10.1038/s41579-018-0031-2
Pinto, G., Sampaio, M., Dias, O., Almeida, C., Azeredo, J., and Oliveira, H. (2021). Insights into the genome architecture and evolution of Shiga toxin encoding bacteriophages of Escherichia coli. BMC Genomics 22:366. doi: 10.1186/s12864-021-07685-0
Pruimboom-Brees, I. M., Morgan, T. W., Ackermann, M. R., Nystrom, E. D., Samuel, J. E., Cornick, N. A., et al. (2000). Cattle lack vascular receptors for Escherichia coli O157:H7 Shiga toxins. Proc. Natl. Acad. Sci. U. S. A. 97, 10325–10329. doi: 10.1073/pnas.190329997
Rasko, D. A., Rosovitz, M. J., Myers, G. S., Mongodin, E. F., Fricke, W. F., Gajer, P., et al. (2008). The pangenome structure of Escherichia coli: comparative genomic analysis of E. coli commensal and pathogenic isolates. J. Bacteriol. 190, 6881–6893. doi: 10.1128/JB.00619-08
Ratledge, C., and Dover, L. G. (2000). Iron metabolism in pathogenic bacteria. Annu. Rev. Microbiol. 54, 881–941. doi: 10.1146/annurev.micro.54.1.881
Riley, M., Abe, T., Arnaud, M. B., Berlyn, M. K., Blattner, F. R., Chaudhuri, R. R., et al. (2006). Escherichia coli K-12: a cooperatively developed annotation snapshot--2005. Nucleic Acids Res. 34, 1–9. doi: 10.1093/nar/gkj405
Riley, L. W., Remis, R. S., Helgerson, S. D., Mcgee, H. B., Wells, J. G., Davis, B. R., et al. (1983). Hemorrhagic colitis associated with a rare Escherichia coli serotype. N. Engl. J. Med. 308, 681–685. doi: 10.1056/NEJM198303243081203
Robertson, J., and Nash, J. H. E. (2018). MOB-suite: software tools for clustering, reconstruction and typing of plasmids from draft assemblies. Microb Genom 4:206. doi: 10.1099/mgen.0.000206
Robins-Browne, R. M., Holt, K. E., Ingle, D. J., Hocking, D. M., Yang, J., and Tauschek, M. (2016). Are Escherichia coli Pathotypes still relevant in the era of whole-genome sequencing? Front. Cell. Infect. Microbiol. 6:141. doi: 10.3389/fcimb.2016.00141
Rodríguez-Rubio, L., Haarmann, N., Schwidder, M., Muniesa, M., and Schmidt, H. (2021). Bacteriophages of Shiga toxin-producing Escherichia coli and their contribution to pathogenicity. Pathogens 10:404. doi: 10.3390/pathogens10040404
Rusconi, B., and Eppinger, M. (2016). “Whole genome sequence typing strategies for enterohaemorrhagic Escherichia coli of the O157: H7 serotype” in The handbook of microbial bioresources. eds. V. K. Gupta, G. D. Sharma, M. G. Tuohy, and R. Gaur (Wallingford UK: CABI), 616–633.
Rusconi, B., Sanjar, F., Koenig, S. S., Mammel, M. K., Tarr, P. I., and Eppinger, M. (2016). Whole genome sequencing for genomics-guided investigations of Escherichia coli O157:H7 outbreaks. Front. Microbiol. 7:985. doi: 10.3389/fmicb.2016.00985
Russo, L. M., Melton-Celsa, A. R., and O’brien, A. D. (2016). Shiga toxin (Stx) type 1a reduces the Oral toxicity of Stx type 2a. J Infect Dis 213, 1271–1279. doi: 10.1093/infdis/jiv557
Sadiq, S. M., Hazen, T. H., Rasko, D. A., and Eppinger, M. (2014). EHEC genomics: past, present, and future. Microbiol Spectr 2:Ehec-0020-2013. doi: 10.1128/microbiolspec.EHEC-0020-2013
Sadiq, M., Hazen, T., Rasko, D., and Eppinger, M. (2015). “Enterohemorrhagic Escherichia coli genomics: past, present, and future” in Enterohemorrhagic Escherichia coli and other Shiga toxin producing E. coli. eds. V. Sperandio and C. J. Hovde. 1st ed (Washington, DC: ASM Press), 55–71.
Sanjar, F., Hazen, T. H., Shah, S. M., Koenig, S. S., Agrawal, S., Daugherty, S., et al. (2014). Genome sequence of Escherichia coli O157:H7 Strain 2886-75, associated with the first reported case of human infection in the United States. Genome Announc. 2:13. doi: 10.1128/genomeA.01120-13
Scheutz, F., Teel, L. D., Beutin, L., Pierard, D., Buvens, G., Karch, H., et al. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. doi: 10.1128/JCM.00860-12
Schmidt, M. A. (2010). LEEways: tales of EPEC, ATEC and EHEC. Cell. Microbiol. 12, 1544–1552. doi: 10.1111/j.1462-5822.2010.01518.x
Schüller, S. (2011). Shiga toxin interaction with human intestinal epithelium. Toxins 3, 626–639. doi: 10.3390/toxins3060626
Sekizuka, T., Lee, K., Kimata, K., Isobe, J., Kuroda, M., Iyoda, S., et al. (2019). Complete genome sequence of an Enterohemorrhagic Escherichia coli O111:H8 Strain recovered from a large outbreak in Japan associated with consumption of raw beef. Microbiol Resour Announc 8:19. doi: 10.1128/MRA.00882-19
Serra-Moreno, R., Jofre, J., and Muniesa, M. (2007). Insertion site occupancy by stx2 bacteriophages depends on the locus availability of the host strain chromosome. J. Bacteriol. 189, 6645–6654. doi: 10.1128/JB.00466-07
Sheldon, J. R., Laakso, H. A., and Heinrichs, D. E. (2016). Iron acquisition strategies of bacterial pathogens. Microbiol Spectr 4:2015. doi: 10.1128/microbiolspec.VMBF-0010-2015
Skaar, E. P. (2010). The Battle for Iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6:e1000949. doi: 10.1371/journal.ppat.1000949
Skinner, C., Patfield, S., Stanker, L. H., Fratamico, P., and He, X. (2014). New high-affinity monoclonal antibodies against Shiga toxin 1 facilitate the detection of hybrid Stx1/Stx2 in vivo. PLoS One 9:e99854. doi: 10.1371/journal.pone.0099854
Smith, J. L., Fratamico, P. M., and Gunther, N. W. T. (2014). Shiga toxin-producing Escherichia coli. Adv. Appl. Microbiol. 86, 145–197. doi: 10.1016/B978-0-12-800262-9.00003-2
Song, W., Sun, H. X., Zhang, C., Cheng, L., Peng, Y., Deng, Z., et al. (2019). Prophage hunter: an integrative hunting tool for active prophages. Nucleic Acids Res. 47, W74–W80. doi: 10.1093/nar/gkz380
Sperandio, V., Kaper, J. B., Bortolini, M. R., Neves, B. C., Keller, R., and Trabulsi, L. R. (1998). Characterization of the locus of enterocyte effacement (LEE) in different enteropathogenic Escherichia coli (EPEC) and Shiga-toxin producing Escherichia coli (STEC) serotypes. FEMS Microbiol. Lett. 164, 133–139. doi: 10.1111/j.1574-6968.1998.tb13078.x
Stevens, M. P., and Frankel, G. M. (2014). The locus of enterocyte effacement and associated virulence factors of Enterohemorrhagic Escherichia coli. Microbiol Spectr 2:Ehec-0007-2013. doi: 10.1128/microbiolspec.EHEC-0007-2013
Steyert, S. R., Sahl, J. W., Fraser, C. M., Teel, L. D., Scheutz, F., and Rasko, D. A. (2012). Comparative genomics and stx phage characterization of LEE-negative Shiga toxin-producing Escherichia coli. Front. Cell. Infect. Microbiol. 2:133. doi: 10.3389/fcimb.2012.00133
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Tarr, G. A. M., Rounds, J., Vachon, M. S., Smith, K., Medus, C., and Hedberg, C. W. (2023). Differences in risk factors for transmission among Shiga toxin-producing Escherichia coli serogroups and stx profiles. J. Infect. 87, 498–505. doi: 10.1016/j.jinf.2023.10.017
Tatsuno, I., Horie, M., Abe, H., Miki, T., Makino, K., Shinagawa, H., et al. (2001). toxB gene on pO157 of enterohemorrhagic Escherichia coli O157:H7 is required for full epithelial cell adherence phenotype. Infect. Immun. 69, 6660–6669. doi: 10.1128/IAI.69.11.6660-6669.2001
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tozzoli, R., Caprioli, A., and Morabito, S. (2005). Detection of toxB, a plasmid virulence gene of Escherichia coli O157, in enterohemorrhagic and enteropathogenic E. coli. J. Clin. Microbiol. 43, 4052–4056. doi: 10.1128/JCM.43.8.4052-4056.2005
Tyler, J. S., Beeri, K., Reynolds, J. L., Alteri, C. J., Skinner, K. G., Friedman, J. H., et al. (2013). Prophage induction is enhanced and required for renal disease and lethality in an EHEC mouse model. PLoS Pathog. 9:e1003236. doi: 10.1371/journal.ppat.1003236
Unkmeir, A., and Schmidt, H. (2000). Structural analysis of phage-borne stx genes and their flanking sequences in Shiga toxin-producing Escherichia coli and Shigella dysenteriae type 1 strains. Infect. Immun. 68, 4856–4864. doi: 10.1128/IAI.68.9.4856-4864.2000
Vishram, B., Jenkins, C., Greig, D. R., Godbole, G., Carroll, K., Balasegaram, S., et al. (2021). The emerging importance of Shiga toxin-producing Escherichia coli other than serogroup O157 in England. J. Med. Microbiol. 70:1375. doi: 10.1099/jmm.0.001375
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Wong, C. S., Jelacic, S., Habeeb, R. L., Watkins, S. L., and Tarr, P. I. (2000). The risk of the hemolytic-uremic syndrome after antibiotic treatment of Escherichia coli O157:H7 infections. N. Engl. J. Med. 342, 1930–1936. doi: 10.1056/NEJM200006293422601
Xie, Z., and Tang, H. (2017). ISEScan: automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 33, 3340–3347. doi: 10.1093/bioinformatics/btx433
Yan, X., Fratamico, P. M., Bono, J. L., Baranzoni, G. M., and Chen, C. Y. (2015). Genome sequencing and comparative genomics provides insights on the evolutionary dynamics and pathogenic potential of different H-serotypes of Shiga toxin-producing Escherichia coli O104. BMC Microbiol. 15:83. doi: 10.1186/s12866-015-0413-9
Yang, X., Bai, X., Zhang, J., Sun, H., Fu, S., Fan, R., et al. (2020). Escherichia coli strains producing a novel Shiga toxin 2 subtype circulate in China. Int. J. Med. Microbiol. 310:151377. doi: 10.1016/j.ijmm.2019.151377
Yano, B., Taniguchi, I., Gotoh, Y., Hayashi, T., and Nakamura, K. (2023). Dynamic changes in Shiga toxin (Stx) 1 transducing phage throughout the evolution of O26:H11 Stx-producing Escherichia coli. Sci. Rep. 13:4935. doi: 10.1038/s41598-023-32111-8
Ye, C., Lan, R., Xia, S., Zhang, J., Sun, Q., Zhang, S., et al. (2010). Emergence of a new multidrug-resistant serotype X variant in an epidemic clone of Shigella flexneri. J. Clin. Microbiol. 48, 419–426. doi: 10.1128/JCM.00614-09
Yin, S., Rusconi, B., Sanjar, F., Goswami, K., Xiaoli, L., Eppinger, M., et al. (2015). Escherichia coli O157:H7 strains harbor at least three distinct sequence types of Shiga toxin 2a-converting phages. BMC Genomics 16:733. doi: 10.1186/s12864-015-1934-1
Zhang, Y., Liao, Y. T., Salvador, A., Sun, X., and Wu, V. C. H. (2019). Prediction, diversity, and genomic analysis of temperate phages induced from Shiga toxin-producing Escherichia coli strains. Front. Microbiol. 10:3093. doi: 10.3389/fmicb.2019.03093
Zhang, J., Yu, D., Dian, L., Hai, Y., Xin, Y., and Wei, Y. (2022). Metagenomics insights into the profiles of antibiotic resistome in combined sewage overflows from reads to metagenome assembly genomes. J. Hazard. Mater. 429:128277. doi: 10.1016/j.jhazmat.2022.128277
Zhou, Z., Alikhan, N. F., Mohamed, K., Fan, Y., and Achtman, M. (2020). The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 30, 138–152. doi: 10.1101/gr.251678.119
Zhou, Y., Liang, Y., Lynch, K. H., Dennis, J. J., and Wishart, D. S. (2011). PHAST: a fast phage search tool. Nucleic Acids Res. 39, W347–W352. doi: 10.1093/nar/gkr485
Zhou, W., Lin, R., Zhou, Z., Ma, J., Lin, H., Zheng, X., et al. (2022). Antimicrobial resistance and genomic characterization of Escherichia coli from pigs and chickens in Zhejiang, China. Front. Microbiol. 13:1018682. doi: 10.3389/fmicb.2022.1018682
Keywords: Shiga toxin (Stx)-producing Escherichia coli (STEC), non-O157 big six serogroups, whole genome sequencing and typing (WGST), phylogenomics, virulence phenotyping
Citation: Kalalah AA, Koenig SSK, Bono JL, Bosilevac JM and Eppinger M (2024) Pathogenomes and virulence profiles of representative big six non-O157 serogroup Shiga toxin-producing Escherichia coli. Front. Microbiol. 15:1364026. doi: 10.3389/fmicb.2024.1364026
Edited by:
Axel Cloeckaert, Institut National de recherche pour l’agriculture, l’alimentation et l’environnement (INRAE), FranceReviewed by:
Sabine Delannoy, Agence Nationale de Sécurité Sanitaire de l’Alimentation, de l’Environnement et du Travail (ANSES), FranceMiklos Fuzi, Independent researcher, Budapest, Hungary
Ana Hurtado, Animalien Osasuna, NEIKER-Instituto Vasco de Investigación y Desarrollo Agrario, Spain
David W. Ussery, University of Arkansas for Medical Sciences, United States
Copyright © 2024 Kalalah, Koenig, Bono, Bosilevac and Eppinger. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Eppinger, bWFyay5lcHBpbmdlckB1dHNhLmVkdQ==