 Alba Cuecas
Alba Cuecas M. Julia Barrau
M. Julia Barrau Juan M. Gonzalez
Juan M. Gonzalez- Institute of Natural Resources and Agrobiology, Spanish National Council for Research, IRNAS-CSIC, Sevilla, Spain
Species differentiation and the appearance of novel diversity on Earth is a major issue to understand the past and future of microbial evolution. Herein, we propose the analysis of a singular evolutive example, the case of microorganisms carrying out the process of anammox (anaerobic ammonium oxidation). Anammox represents a singular physiology active on Earth from ancient times and, at present, this group is still represented by a relatively limited number of species carrying out a specific metabolism within the Phylum Planctomycetota. The key enzyme on the anammox pathway is hydrazine dehydrogenase (HDH) which has been used as a model in this study. HDH and rRNA (16S subunit) phylogenies are in agreement suggesting a monophyletic origin. The diversity of this singular phylogenetic group is represented by a few enriched bacterial consortia awaiting to be cultured as monospecific taxa. The apparent evolution of the HDH genes in these anammox bacteria is highly related to the diversification of the anammox clades and their genomes as pointed by phylogenomics, their GC content and codon usage profile. This study represents a clear case where bacterial evolution presents a paralleled genome, gene and species diversification through time from a common ancestor; a scenario that most times is masked by a web-like phylogeny and the huge complexity within the prokaryotes. Besides, this contribution suggests that microbial evolution of the anammox bacteria has followed an ordered, vertical diversification through Earth history and will present a potentially similar speciation fate in the future.
1 Introduction
Anaerobic ammonium oxidation (anammox) (Broda, 1977) is carried out by a specific group of bacteria with great significance in the global biogeochemical N-cycle (Kuypers et al., 2018). Anammox bacteria accounts for a 30–70% of all N2 released into the atmosphere (Lam and Kuypers, 2011; Oshiki et al., 2016; Stein and Klotz, 2016). Thus, anammox bacteria have been widely used in wastewater treatment plants for the removal of fixed nitrogen loads (Kartal et al., 2010; Stein and Klotz, 2016; Li et al., 2020) which indicates a major relevance in today’s world economy and sustainability. So far, the known anammox bacteria belong to the Phylum Planctomycetota (Wang et al., 2019) which includes seven proposed candidate taxa based on 16S rRNA gene sequences: “Candidatus Brocadia,” “Ca. Kuenenia,” “Ca. Jettenia,” “Ca. Scalindua,” “Ca. Anammoximicrobium,” “Ca. Anammoxoglobus” and “Ca. Bathyanammoxibiaceae” (Zhang and Okabe, 2020; Liao et al., 2022; Zhao et al., 2022). Because the culturing as monoespecific culture of these bacteria is arduous, today, most information on these bacteria has been obtained through whole-genome sequencing (WGS), including MAGs (Metagenome Assembled Genomes), of bacterial consortia and assemblages from nature or bioreactors (Liao et al., 2022).
The anammox process involves the use of nitrite or nitric oxide and ammonium to end by releasing N2 to the atmosphere. The enzyme performing at the final step of the anammox pathway is hydrazine dehydrogenase (HDH) which represents the key enzyme of the process and it is present in all anammox-performing bacteria (Maalcke et al., 2016; Akram et al., 2019; Liao et al., 2022). Hydrazine is a key intermediary (Kartal et al., 2011) formed, today, mainly by a biotic process (Dietl et al., 2015; Maalcke et al., 2016) or, in the ancient Earth, potentially by abiotic reactions (Folsome et al., 1981; Jia et al., 2021).
In spite of their relevance, scarce information is available on the evolutionary history of anammox bacteria. A recent study by Liao et al. (2022) suggests the origin of anammox bacteria on Earth around the Great Oxygenation Event, about 2.5 billion years ago. This places anammox bacteria close to the diversification of nitric reductase into the use of NO and O2 as major substrates and the origin of aerobic respiration (Ducluzeau et al., 2008; Santana et al., 2017). The transition from anaerobic respiration and the dominance of anaerobic processes (including denitrification) to an increasing significance of aerobic processes (including aerobic metabolisms) represented a major milestone in the history of the biogeochemical N-cycle on Earth. Specifically, as a consequence of increasing O2 concentration, the diversification of ammonium oxidizing processes could appeared around that time, gaining importance the aerobic ammonium oxidizing microorganisms (including ammonium-oxidizing bacteria, ammonium-oxidizing archaea and commamox species carrying out the nitrification process) and likely limiting the expansion of the anammox bacteria restricted to anoxic niches including the oxygen-free side of anoxic-oxic boundaries. At their initials, anammox appeared to be related to anoxic, oxygen-poor, niches and environments (Santana et al., 2017; Liao et al., 2022). Thus, the anammox bacteria constitute an ancient bacterial group maintaining their singularity through time (Wang et al., 2019; Liao et al., 2022) and preserving their activity in spite of the numerous changes undergone on Earth. At present, anammox bacteria remain as a unified metabolic and phylogenetic bacterial group which are unique in their capability of carrying out the anammox process and directly releasing to the atmosphere around 30–70% of fixed nitrogen forms as N2 (Lam and Kuypers, 2011; Oshiki et al., 2016; Stein and Klotz, 2016).
At present, microbial phylogeny and evolution present high complexity due to the huge microbial diversity existing on Earth (Curtis et al., 2002). In addition, numerous processes are leading to horizontal gene transfer (HGT) in the prokaryotes, such as mobile genetic elements, viruses, transposition events, among others (Johnston et al., 2014; Cuecas et al., 2017; Abe et al., 2020). Consequently, microbial phylogeny is often represented by web-like trees (Doolittle, 2000; Hug et al., 2016) showing an increased level of complexity to account for the frequent HGT events occurring through evolution among the prokaryotes (Goldman and Kaçar, 2022). Anammox can be selected as a singular group maintaining its uniqueness through evolution from ancient Earth. Thus, the anammox bacteria, and specifically their key HDH enzyme-encoding genes, can be an excellent case study for the analysis of gene, genome and species divergence through evolutionary history with perspectives to future diversification. The aim of this study is to analyze the divergence among the anammox bacteria as a unique metabolic and phylogenetic bacterial group that remains relatively independent of the rest of prokaryotes, as suggested by the related evolution of the HDH genes and the anammox bacterial genomes.
2 Data sources and methods
A blastp search was carried out at the NCBI1 against the non-redundant protein database (nr) using as reference the protein sequence of the enzyme hydrazine dehydrogenase from Ca. Kuenenia stuttgartiensis CSTR1 (Accession number QII14076.1). HDH gene sequences satisfying more than 85% coverage, 70% identity and an available genome sequence (either from WGS or MAG, >2 Mbp) were selected for further analysis. The Phylogenomic analysis was performed using get_HOMOLOGUES with the bidirectional best-hit algorithm (Contreras-Moreira and Vinuesa, 2013). Sequences were aligned using ClustalW (Thompson et al., 1994). Phylogenies were generated with MEGA X (Kumar et al., 2018) using the Neighbor-Joining method and the Poisson model. Bootstrap values are expressed as percentages corresponding to 1,000 trials. GC content and frequency of codon usage were calculated using own software. Non-metric multidimensional scaling (NMDS) using Bray distances was performed using PAST (Hammer and Harper, 2001) on the frequency of codon usage of genes and genomes.
3 Phylogeny
From the discovery of the anammox bacteria in the early 1990s, a major preliminary taxonomic classification of Candidatus (Ca.) taxa and the clades within the anammox bacteria have been envisioned from the analysis of 16S rRNA gene sequencing (Kuenen, 2008; Liao et al., 2022). Due to the lack of monospecific cultures in this singular bacterial group, those 16S rRNA gene sequences have been retrieved from bacterial enrichments, consortia and assemblages either from natural environments or the bioreactor assays pursuing an increased of the anammox process with views aimed to applications mostly related to waste treatments. Most additional information on anammox bacteria have been based on whole genome sequencing (WGS) or metagenomic assembled genomes (MAGs) from those sources of anammox bacteria (Kuenen, 2008; Liao et al., 2022).
Based on 16S rRNA gene sequence analysis, the anammox bacteria have been classified within the Phylum Planctomycetota and into a total of seven major groups, candidate to future taxa: “Candidatus Brocadia,” “Ca. Kuenenia,” “Ca. Jettenia,” “Ca. Scalindua,” “Ca. Anammoximicrobium,” “Ca. Anammoxoglobus” and “Ca. Bathyanammoxibiaceae” (Zhang and Okabe, 2020; Lodha et al., 2021; Liao et al., 2022; Zhao et al., 2022). This uniqueness of anammox bacteria among the broad prokaryotic diversity suggests certain independence and evolutive isolation between the anammox bacteria and all other prokaryotes, at least, in relationship to their genome history. Besides, this uniqueness represents an evidence to propose a monophyletic view for this bacterial group, also in agreement to previous reports (Kuenen, 2008; Liao et al., 2022).
Assuming a highly likely monophyletic origin of anammox, the major genes involved in the anammox pathway should show as well a similar evolutive pattern. We are not aware of a phylogeny for the key enzyme of the anammox pathway, HDH (Liao et al., 2022) and our results (Figure 1) suggest a paralleled phylogeny of 16S rRNA gene dendrogram (Kuenen, 2008; Lodha et al., 2021; Liao et al., 2022) and that generated for the HDH gene sequences. This evidence supports a similar evolutive history of the anammox bacteria and their HDH genes. Previously, Harhangi et al. (2012) showed a similar parallelism between the 16S rRNA gene phylogeny and the hydrazine synthase-encoding genes in these bacteria which further support the hypothesis of a similar evolutive history for these bacteria and their major anammox enzyme-encoding genes. Thus, results suggest certain evolutive isolation from other bacterial phyla and even from non-anammox Planctomycetota bacteria (Kuenen, 2008; Liao et al., 2022).
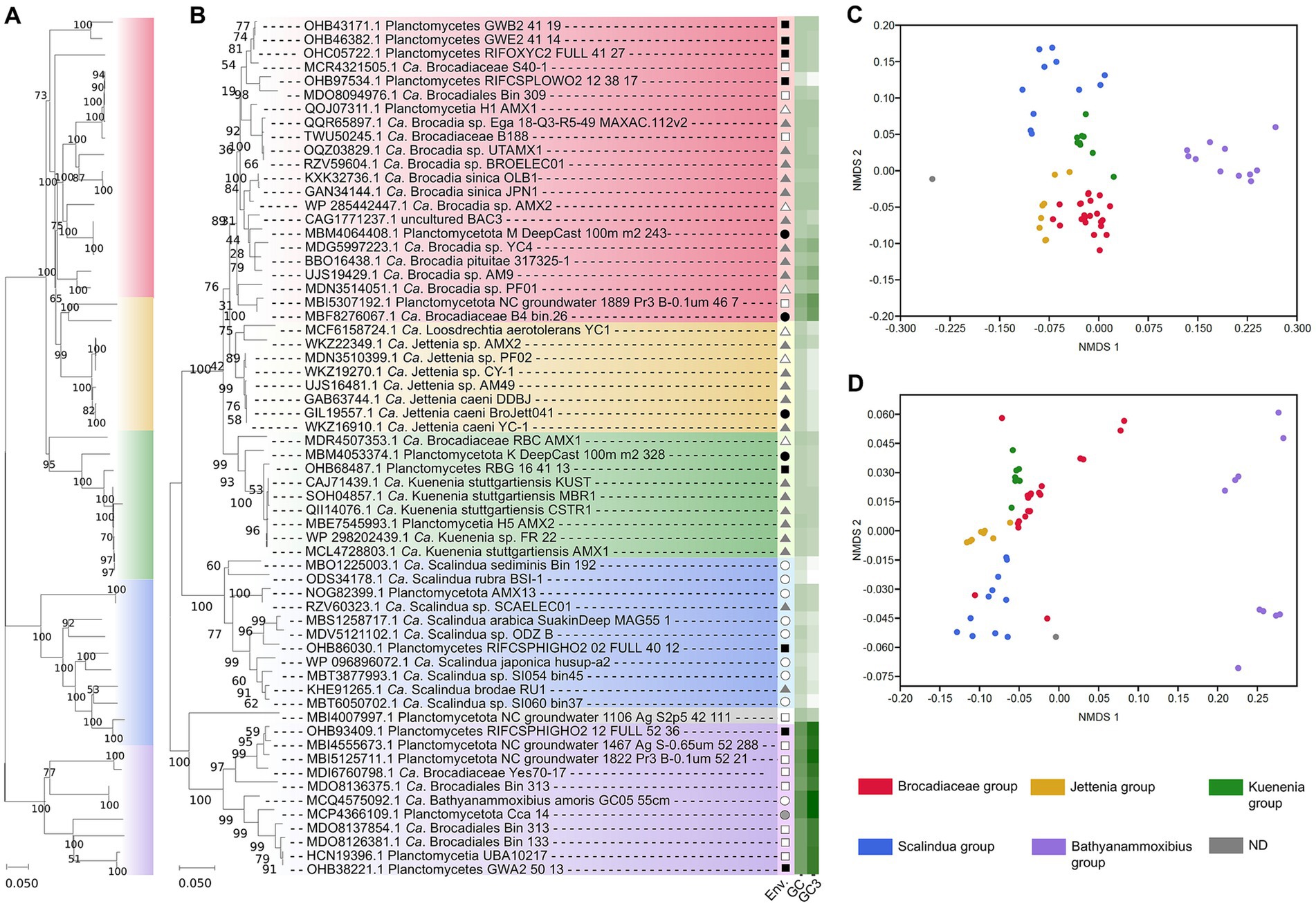
Figure 1. Diversification of anammox bacteria based on phylogenomics (WGS and MAGs) (A) and based on HDH-encoding gene sequences (B). The origin of these sequences is indicated in the column “Env.” by the following symbols: soil (black square), ground water (open square), freshwater (black circle), marine and saline environment (open circle), bioreactors (black triangle), wastewater (open triangle), and animal isolate (gray-filled circle). The GC content (GC) and the GC content at the third codon position (GC3) is indicated in a white (lower values) to green (higher values) color scale. NMDS analyzes show a distinctive distribution of clades and they are based on the frequency of codon usage in HDH-encoding genes (C) and anammox bacterial genomes (D). A total of 52 genome assemblies including 21 orthologs have been used to construct the phylogenomic dendrogram (A). A total of 62 HDH-encoding gene sequences constitute the HDH-based dendrogram (B) with an approximate sequence length of 700 bp. The color code [used in (A–D)] to differentiate the major clades distinguished in the analyzes is shown at the bottom-right. Bootstrap values represent percentages from 1,000 trails.
At present, the anammox bacteria shows a fairly independent phylogeny with scarce HGT influence (so far unreported) from other bacteria, unlike most other bacterial phyla (Cuecas et al., 2017; Sheinman et al., 2021; Sengupta and Azad, 2022). This singularity within the complexity of bacterial phylogeny at present time represents an additional piece of evidence in support of a monophyletic origin of the anammox-performing bacteria previously shown on the basis of 16S rRNA (Kuenen, 2008; Lodha et al., 2021; Liao et al., 2022) and in this study through the HDH-encoding gene sequences and phylogenomics (Figure 1).
A monophyletic origin for the anammox suggests the existence, at some time point, of a last common ancestor (LCA) for the anammox bacteria. The LCA for the anammox bacteria has been proposed to be dated back to around 2.5 billion years (Liao et al., 2022). This ancient date suggests a major relevance of anammox throughout our planet history and their potential contribution to the biogeochemical processes on Earth. In addition, current biotechnological interests, specifically related to the transformation of fixed nitrogen forms during waste treatments highlights the importance of these bacteria for a sustainable economy.
4 Genes and genomes divergence
Analyzes of GC content and codon usage in the HDH-encoding genes and their genomes (mostly MAGs) (Figure 1) suggests equivalent evolutive patterns of divergence differentiating similar clades. Non-metric multidimensional scaling analyses show that codon usage in the HDH-encoding genes and their genome sequences present related divergence because similar grouping is obtained. Codon usage of the HDH-encoding genes and the available anammox genome annotated sequences suggest that a paralleled evolutive divergence is taking place leading to differentiating clades (mainly corresponding to the proposed candidate genera) within the anammox bacteria. The results (Figure 1) suggest that the anammox bacteria are slowly and progressively diverging which can be observed from the different clades detected in this singular group. Similar diversification can be observed when analyzing, using a similar procedure, the HDH-encoding genes. These observations suggest that a key anammox gene and the anammox bacterial genomes show a similar evolutive divergence along Earth history. This parallelism might imply that similar or random evolutive mechanisms influence the differentiation of the genomes of these candidate bacterial taxa and their key enzyme (HDH)-encoding gene. If this is the case, which needs to be confirmed through further analyzes, the evolutive fate of the anammox bacteria and their key enzyme (HDH) might diverge progressively following specific whole-genome wide trends (to be determined) likely governed by singular environmental and specific niche requirements.
Among the anammox bacteria, different niche preferences have been reported for the different anammox clades within the group (Lam and Kuypers, 2011; Zhang and Okabe, 2020; Zhao et al., 2023) (Figure 1). The ecological distribution of anammox bacteria presents a strong dependence on the environment (Oshiki et al., 2016; Zhang and Okabe, 2020). Anammox are commonly found at the oxic-anoxic interface in a variety of environments (Jensen et al., 2008; Seuntjens et al., 2018; Liao et al., 2022). Salinity is a factor affecting anammox distribution. For example, “Ca. Scalindua” dominate saline environments (Sonthiphand et al., 2014) and “Ca. Brocadia” and “Ca. Kuenenia” have been mostly located to non-salty environments such as soils and freshwater systems (Zhang and Okabe, 2020). The anammox bacteria also show niche differentiation among clades. Temporal and spatial differential distribution have been reported for “Ca. Brocadia” and “Ca. Kuenenia.” The characteristic dependency of anammox bacteria on fixed nitrogen sources (nitrite and ammonium) determines species distribution (van der Star et al., 2007; Pitcher et al., 2011) as well as the availability of additional C sources (Zhao et al., 2022, 2023).
Although the anammox bacteria show a number of clades when attempting phylogenetic analyzes either on their genes or genomes, assuming the long evolutive time (ca. 2.5 billion years) required to show these differences suggests a fairly slow progression towards the divergence of the anammox clades adapting to specific niches. The uniqueness of the anammox process and the relative independence of this phylogenetic group from other bacteria could suggest than the evolutive fate of the anammox might be fairly conserved and maintained with a minor and slow changing rate over time; mainly because of a specific adaptation to a singular type of niches. Nevertheless, at present, there is no additional information to predict the potential evolutive fate of the anammox bacteria.
5 Discussion
The anammox bacteria represent a singular bacterial group showing relative isolation within the prokaryotes. So far, their evolutive history suggests that this group potentially evolves as a result of environmental constrains presenting minimum influences from other bacterial phyla such as through HGT-type for accelerated changes. As well, to our knowledge, no phages have been reported for the anammox bacteria perhaps a consequence of their low growth rate. These points suggest the existence of genomes showing less genomic plasticity than shown for most other bacterial phyla (Johnston et al., 2014; Cuecas et al., 2017; Sheinman et al., 2021) although some genome variability within the anammox bacteria has been reported (Ding and Adrian, 2020). While this is a hypothesis that remains to be tested, present information points to this putative scenario suggesting a study case for genomic analyzes and the evolutive history of this bacterial group.
It is expected that future studies and additional microbial diversity surveys will greatly enhance the today’s relatively limited diversity within the anammox bacteria. Currently, a limiting sampling on anammox bacteria precludes to realize the actual diversity for the anammox bacteria existing on Earth (Harhangi et al., 2012; Maalcke et al., 2016; Liao et al., 2022) in a similar way that the whole prokaryotic diversity on Earth remains to be truly understood (Curtis et al., 2002). Potential issues with the slow growth rates of the anammox bacteria, potential discrimination during PCR amplification on microbial diversity surveys, added to the difficulty for culturing and the lack of monospecific cultures all sum up to make this singular bacterial group a difficult target for further research.
Diversification and speciation among prokaryotes are major research lines to be developed in the years to come. It is expected that major advances on understanding these processes will be gained during the next decade or so. Herein, we propose that the anammox bacteria could represent a singular group, both on their metabolism and evolutive history, to analyze specific phenomena related to those issues within the prokaryotes. In the anammox bacteria, different aspects converge to point this group as a singular target for evolutive studies, most importantly, a time-located LCA, their slow evolution, a common genome-wide divergence trend and the apparently scarce influence on gene exchange from other bacterial groups.
As pointed above, the environment, and specific niche requirements, might represent major evolutive factors influencing the divergence of the identified anammox clades. Currently, different niches has been reported for some of the different anammox clades which would confirm that the environment is having major influence on microbial evolution and specifically on the diversification of the anammox bacteria.
Due to the high relevance of the anammox bacteria in the environment, including their application in waste treatment plants, great interest exist on understanding the physiology, ecology, evolution and behavior of this singular bacterial group. This perspective contributes to propose the anammox bacteria as a unique case study for the analysis of evolutive trends among bacteria and specifically within the anammox-performing bacteria. Current information, mostly from WGS and MAGs, suggests slow and independent evolution which appears to be mostly forced by environmental constrains. Future research on the development of these hypotheses will confirm the role and fate of anammox bacteria, and, from those results, the extracted knowledge con assist to comprehend the evolutive history of other bacterial phyla (Goldman and Kaçar, 2022).
Data availability statement
Publicly available datasets were analyzed in this study. This data can be found at: NCBI, https://blast.ncbi.nlm.nih.gov/.
Author contributions
AC: Conceptualization, Formal analysis, Methodology, Writing – review & editing. MB: Formal analysis, Methodology, Writing – review & editing, Data curation. JG: Formal analysis, Methodology, Conceptualization, Funding acquisition, Supervision, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the Spanish Ministry of Science and Innovation, project PID2020-119373GB-I00 to JG funded through MCIN/AEI/10.13039/501100011033 and the European Union Next Generation (PRTR). AC and MB acknowledge support from post-doctoral program DOC_00381 (PAIDI 2020 – Regional Andalusian Government) co-funded with FEDER funds and JAEICU_23_00440, respectively.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Abe, K., Nomura, N., and Suzuki, S. (2020). Biofilms: hot spots of horizontal gene transfer (HGT) in aquatic environments, with a focus on a new HGT mechanism. FEMS Microbiol. Ecol. 96:fiaa031. doi: 10.1093/femsec/fiaa031
Akram, M., Dietl, A., Mersdorf, U., Prinz, S., Maalcke, W., Keltjens, J., et al. (2019). A 192-heme electron transfer network in the hydrazine dehydrogenase complex. Sci. Adv. 5:eaav4310. doi: 10.1126/sciadv.aav4310
Broda, E. (1977). Two kinds of lithotrophs missing in nature. J. Basic Microbiol. 17, 491–493. doi: 10.1002/jobm.19770170611
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Cuecas, A., Kanoksilapatham, W., and Gonzalez, J. M. (2017). Evidence of horizontal gene transfer by transposase gene analyses in Fervidobacterium species. PLoS One 12:e0173961. doi: 10.1371/journal.pone.0173961
Curtis, T. P., Sloan, W. T., and Scannell, J. W. (2002). Estimating prokaryotic diversity and its limits. Proc. Natl. Acad. Sci. USA 99, 10494–10499. doi: 10.1073/pnas.142680199
Dietl, A., Ferousi, C., Maalcke, W. J., Menzel, A., de Vries, S., Keltjens, J. T., et al. (2015). The inner workings of the hydrazine synthase multiprotein complex. Nature 527, 394–397. doi: 10.1038/nature15517
Ding, C., and Adrian, L. (2020). Comparative genomics in “Candidatus Kuenenia stuttgartiensis” reveal high genomic plasticity in the overall genome structure, CRISPR loci and surface proteins. BMC Genomics 21:851. doi: 10.1186/s12864-020-07242-1
Doolittle, W. F. (2000). Uprooting the tree of life. Sci. Am. 282, 90–95. doi: 10.1038/scientificamerican0200-90
Ducluzeau, A. L., van Lis, R., Duval, S., Schoepp-Cothenet, B., Russell, M. J., and Nitschke, W. (2008). Was nitric oxide the first deep electron sink? Trends Biochem. Sci. 34, 9–15. doi: 10.1016/j.tibs.2008.10.005
Folsome, C. E., Brittain, A., Smith, A., and Chang, S. (1981). Hydrazines and carbohydrazides produced from oxidized carbon in Earth’s primitive environment. Nature 294, 64–65. doi: 10.1038/294064a0
Goldman, A. D., and Kaçar, B. (2022). Very early evolution from the perspective of microbial ecology. Environ. Microbiol. 25, 5–10. doi: 10.1111/1462-2920.16144
Hammer, Ø., and Harper, D. A. (2001). Past: paleontological statistics software package for educaton and data analysis. Palaeontol. Electron. 4, 1. Available at: https://palaeo-electronica.org/2001_1/past/issue1_01.htm
Harhangi, H. R., Le Roy, M., van Alen, T., Hu, B.-L., Groen, J., Kartal, B., et al. (2012). Hydrazine synthase, a unique phylomarker with which to study the presence and diversity of anammox bacteria. Appl. Environ. Microbiol. 78, 752–758. doi: 10.1128/AEM.07113-11
Hug, L. A., Baker, B. J., Anantharaman, K., Brown, C. T., Probst, A. J., Castelle, C. J., et al. (2016). A new view of the tree of life. Nat. Microbiol. 1:16048. doi: 10.1038/nmicrobiol.2016.48
Jensen, M. M., Kuypers, M. M. M., and Lavik, G. (2008). Rates and regulation of anaerobic ammonium oxidation and denitrification in the Black Sea. Limnol. Oceanogr. 53, 23–36. doi: 10.4319/lo.2008.53.1.0023
Jia, T. Z., Caudan, M., and Mamajanov, I. (2021). Origin of species before origin of life: the role of speciation in chemical evolution. Life 11:154. doi: 10.3390/life11020154
Johnston, C., Martin, B., Fichant, G., Polard, P., and Claverys, J. P. (2014). Bacterial transformation: distribution, shared mechanisms and divergent control. Nat. Rev. Microbiol. 12, 181–196. doi: 10.1038/nrmicro3199
Kartal, B., Kuenen, J. G., and van Loosdrecht, M. C. (2010). Engineering. Sewage treatment with anammox. Science 328, 702–703. doi: 10.1126/science.1185941
Kartal, B., Maalcke, W. J., de Almeida, N., Cirpus, I., Gloerich, J., Geerts, W., et al. (2011). Molecular mechanism of anaerobic ammonium oxidation. Nature 479, 127–130. doi: 10.1038/nature10453
Kuenen, J. G. (2008). Anammox bacteria: from discovery to application. Nat. Rev. Microbiol. 6, 320–326. doi: 10.1038/nrmicro1857
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kuypers, M. M. M., Marchant, H. K., and Kartal, B. (2018). The microbial nitrogen-cycling network. Nature Rev. Microbiol. 16, 263–276. doi: 10.1038/nrmicro.2018.9
Lam, P., and Kuypers, M. M. M. (2011). Microbial nitrogen cycling processes in oxygen minimum zones. Annu. Rev. Mar. Sci. 3, 317–345. doi: 10.1146/annurev-marine-120709-142814
Li, L., Ling, Y., Wang, H. Y., Chu, Z. S., Yan, G. K., Li, Z. W., et al. (2020). N2O emission in partial nitritation-anammox process. Chin. Chem. Lett. 31, 28–38. doi: 10.1016/j.cclet.2019.06.035
Liao, T., Wang, S., Stüeken, E. E., and Luo, H. (2022). Phylogenomic evidence for the origin of obligate anaerobic anammox bacteria around the great oxidation event. Mol. Biol. Evol. 39:msac170. doi: 10.1093/molbev/msac170
Lodha, T., Narvekar, S., and Karodi, P. (2021). Classification of uncultivated anammox bacteria and “Candidatus Uabimicrobium” into new classes and provisional nomenclature as “Candidatus Brocadiia” classis nov. and “Candidatus Uabimicrobiia” classis nov. of the phylum Planctomycetes and novel family “Candidatus Scalinduaceae” fam. Nov to accommodate the genus “Candidatus Scalindua”. Syst. Appl. Microbiol. 44:126272. doi: 10.1016/j.syapm.2021.126272
Maalcke, W. J., Reimann, J., de Vries, S., Butt, J. N., Dietl, A., Kip, N., et al. (2016). Characterization of anammox hydrazine dehydrogenase, a key N2-producing enzyme in the global nitrogen cycle. J. Biol. Chem. 291, 17077–17092. doi: 10.1074/jbc.M116.735530
Oshiki, M., Satoh, H., and Okabe, S. (2016). Ecology and physiology of anaerobic ammonium oxidizing bacteria. Environ. Microbiol. 18, 2784–2796. doi: 10.1111/1462-2920.13134
Pitcher, A., Villanueva, L., Hopmans, E. C., Schouten, S., Reichart, G. J., and Sinninghe Damsté, J. S. (2011). Niche segregation of ammonia-oxidizing archaea and anammox bacteria in the Arabian Sea oxygen minimum zone. ISME J. 5, 1896–1904. doi: 10.1038/ismej.2011.60
Santana, M. M., Gonzalez, J. M., and Cruz, C. (2017). Nitric oxide accumulation: the evolutionary trigger for phytopathogenesis. Front. Microbiol. 8:1947. doi: 10.3389/fmicb.2017.01947
Sengupta, S., and Azad, R. K. (2022). Reconstructing horizontal gene flow network to understand prokaryotic evolution. Open Biol. 12:220169. doi: 10.1098/rsob.220169
Seuntjens, D., Carvajal-Arroyo, J. M., Ruopp, M., Bunse, P., De Mulder, C. P., Lochmatter, S., et al. (2018). High-resolution mapping and modeling of anammox recovery from recurrent oxygen exposure. Water Res. 144, 522–531. doi: 10.1016/j.watres.2018.07.024
Sheinman, M., Arkhipova, K., Arndt, P. F., Dutilh, B. E., Hermsen, R., and Massip, F. (2021). Identical sequences found in distant genomes reveal frequent horizontal transfer across the bacterial domain. eLife 10:e62719. doi: 10.7554/eLife.62719
Sonthiphand, P., Hall, M. W., and Neufeld, J. D. (2014). Biogeography of anaerobic ammonia-oxidizing (anammox) bacteria. Front. Microbiol. 5:399. doi: 10.3389/fmicb.2014.00399
Stein, L. Y., and Klotz, M. G. (2016). The nitrogen cycle. Curr. Biol. 26, R94–R98. doi: 10.1016/j.cub.2015.12.021
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix- choice. Nucl. Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
van der Star, W. R. L., Abma, W. R., Blommers, D., Mulder, J. W., Tokutomi, T., Strous, M., et al. (2007). Startup of reactors for anoxic ammonium oxidation: experiences from the first full-scale anammox reactor in Rotterdam. Water Res. 41, 4149–4163. doi: 10.1016/j.watres.2007.03.044
Wang, Y., Xu, L., Wang, S., Ye, F., and Zhu, G. (2019). Global distribution of anaerobic Ammonia oxidation (Anammox) Bacteria - field surveys in wetland, dryland, Groundwater Aquifer and Snow. Front. Microbiol. 10:2583. doi: 10.3389/fmicb.2019.02583
Zhang, L., and Okabe, S. (2020). Ecological niche differentiation among anammox bacteria. Water Res. 171:115468. doi: 10.1016/j.watres.2020.115468
Zhao, R., Babbin, A. R., Roerdink, D. L., Thorseth, I. H., and Jørgensen, S. L. (2023). Nitrite accumulation and anammox bacterial niche partitioning in Arctic Mid-Ocean ridge sediments. ISME Commun. 3:26. doi: 10.1038/s43705-023-00230-y
Keywords: anammox, hydrazine dehydrogenase, phylogeny, microbial evolution, genome divergence, diversification
Citation: Cuecas A, Barrau MJ and Gonzalez JM (2024) Microbial divergence and evolution. The case of anammox bacteria. Front. Microbiol. 15:1355780. doi: 10.3389/fmicb.2024.1355780
Edited by:
Rekha Seshadri, Joint Genome Institute, Berkeley Lab (DOE), United StatesReviewed by:
Rosa María Martínez-Espinosa, University of Alicante, SpainCopyright © 2024 Cuecas, Barrau and Gonzalez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan M. Gonzalez, am1ncmF1QGlybmFzZS5jc2ljLmVz