 Hanna Huuki
Hanna Huuki Johanna Vilkki2
Johanna Vilkki2 Aila Vanhatalo
Aila Vanhatalo Ilma Tapio
Ilma Tapio
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 05 March 2024
Sec. Microorganisms in Vertebrate Digestive Systems
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1353874
Early-life modulation of rumen microbiota holds promise for enhancing calf growth, health, and long-term production in ruminants. However, limited attention has been given to the impact of rumen microbiota modulation on the establishment of hindgut microbiota. In this study, fecal microbiota development was examined in identical twin calves for 12 months. The treatment group (T-group) received adult cow fresh rumen liquid inoculum during the pre-weaning period, while the control group did not (C-group). The effects of inoculum were assessed on calf gut health and as microbial seeding route into the hindgut. The early rumen modulation had no effect on age-related fecal microbiota development. The fecal bacterial community evolved gradually following dietary changes and categorized into pre-weaning and post-weaning communities. Bacterial richness increased with age and stabilized at month 9, while between-sample variation reduced in post-weaning samples. Archaeal load in fecal samples increased after month 4, while archaeal richness increased and stabilized in both groups by month 9. Between-sample similarity was higher during the pre-weaning period, with increased dissimilarity from month 4 onward. Anaerobic fungi were detected in feces at month 4, with richness peaking at month 7. Before month 6, fungal community composition distinctly differed from mature communities. When colostrum, calf rumen, and donor inoculum were evaluated as seeding sources for hindgut colonization, the calf’s own rumen was identified as the primary seeding source for fecal bacteria and fungi. Colostrum was a source for several bacteria detected in feces, but these were of temporary importance until weaning. The donor inoculum had limited impact on gut health as diarrhea rates were similar between the T-group and C-group. In conclusion, early-life microbiota modulation shows potential in ruminant development. However, a more targeted approach with bacteria adapted to the hindgut environment may be necessary to modulate hindgut effectively. This research contributes to our understanding of the complex relationship between gut microbiota and calf health and growth.
The gastrointestinal (GI) tract in young ruminants undergoes both anatomical growth and functional maturation (Baldwin et al., 2004; Guilloteau et al., 2009), which is crucial for nutrient absorption and the development of immune tolerance to symbiotic microbiota (Amin and Seifert, 2021). Upon birth, the first colostrum meal bypasses rumen and is directed toward abomasum, where it helps to create an acidic environment and triggers colostrum digestion and absorption in the small intestine (Lopez and Heinrichs, 2022). Within hours of birth, the gut of a newborn calf allows the passage of colostrum-derived immunoglobulin G, providing short-term immunity. Weaning represents a pivotal stage, transitioning the calf from milk-based digestion in the small intestine into solid feed intake and ruminal fermentation, which will also have an impact on the microbial community (Meale et al., 2016). As solid feed consumption increases, rumen capacity expands significantly, providing an enhanced surface area for the absorption of ruminal fermentation products, particularly VFAs, to meet the calf’s growth demands. Song et al. (2018) hypothesized that hindgut microbial fermentation plays an important role in providing energy to pre-weaned calves prior to the complete development of the rumen by demonstrating significant association between volatile fatty acids (VFAs) and the colonization of mucosa-attached health-related bacterial genera and carbohydrate-utilizing bacterial genera.
Functional development of the rumen is driven by solid feed consumption, while intensive microbial colonization of the GI tract starts at birth. Maternal transfer of microbes occurs during birth through contact with the dam’s vaginal and fecal microbiota and the colostrum, skin, or saliva (Yeoman et al., 2018). These initial microbial populations lay the foundation for the calf’s gut microbiota. Interaction with adult animals further contributes to microbial diversity, as the calf encounters a broader array of microorganisms through close contact and shared living spaces (Lowe et al., 1987; Bird et al., 2010). Further development of GI microbial ecosystem in young ruminants is influenced by diet and use of medication and hygiene practices in housing systems among others. Understanding complex interplay of these factors is crucial for optimizing gut health and overall well-being of young ruminants.
Although the rumen microbial composition and function have been in the center of research (Rey et al., 2014; Furman et al., 2020), the large intestine microbiome may have unexplored potential to improve the animal welfare and production performance (O’Hara et al., 2020). Hindgut microbiota has an important role in immune defense (Hooper et al., 2012), nitrogen metabolism and cycling (Hoover, 1978) or can produce a variety of compounds, e.g., vitamins (Jiang et al., 2022) and bile acid metabolites (Lin et al., 2023), which may have an impact on the host nutrient absorption, metabolism, and health. While ruminants acquire most of their energy from the ruminal fermentation, hindgut fermentation accounts for 5–10% of the total metabolizable energy (Gressley et al., 2011) with some studies suggesting association of fecal microbiota with feed efficiency (Welch et al., 2021). Compared with the rumen, much less is known about the colonization of the large intestine and the potential to modulate its microbial ecosystem through early life interventions. However, some studies have demonstrated successful impact of hindgut microbiota modulation. Yu et al. (2020) orally inoculated lambs with rumen fluid from an adult sheep and showed that inoculation during weaning had stronger effect on the colonic microbiota than the inoculation before weaning. Another study showed mild effects of fecal microbiota transplant from adult cows on microbiota of young calves and ability to alleviate weaning stress through pro-inflammatory response and increasing antioxidant potential (Rosa et al., 2021). Similarly, an oral dose of fecal material obtained from healthy calves was capable of ameliorating diarrhea in sick calves of similar age with alterations in their gut microbiota (Kim et al., 2021). In human studies, the transfer of beneficial microbiota though fecal microbiota transplantation has demonstrated high potential to ameliorate gut dysbiosis associated with Clostridium difficile infection, suggesting that colonic microbial modulation leading to health benefits for humans and animals is possible (Bakken et al., 2011). If hindgut microbial colonization in early life would have long-term effects and influence production traits later in life, this area requires further investigation.
In this experiment, we studied six monozygotic twin-calf pairs that we recruited for an early-life rumen modulation study. Starting from the 2nd week after birth to the end of the pre-weaning period, one calf from each pair was given an oral dose of adult cow rumen liquid. The results demonstrated that the inoculum stimulated earlier maturation of rumen bacterial ecosystem in treated calves before and after weaning, with additional positive effect observed on the growth in young animals (Huuki et al., 2022a,b). Later, the same animals were followed up during their first lactation period, where differences between the groups were observed in residual energy intake and milk yield at different phases of lactation among others (Huuki et al., 2022b). As the colonization routes of the rumen and hindgut are interconnected, our next objective was to elucidate if early-life rumen microbiota modulation also affected the colonization and development of the colonic microbiota or had beneficial effect on gut health in young animals. Therefore, we explored the possible colonization routes associated with the colostrum, own rumen, or donor inoculum of microbes into the large intestine. We hypothesized that the inoculum treatment could also affect the large intestine microbiota and improve the gut health.
Six pairs of identical twin calves (four female and two male pairs) were produced by embryo splitting technique at Natural Resources Institute Finland (Luke). At birth, calves were randomly assigned to either a treatment (T-group) group or a control (C-group) group and separated into individual pens (147 cm × 172 cm) for the whole pre-weaning period to avoid direct contact between the calves or with other animals. Within 2 h of birth, all the calves received 2 L of the same high-quality (Brix value 24) colostrum mix, collected in advance from 3 cows, and stored at −20°C. As a second meal, the calves were offered colostrum from their own mothers, and thereafter, bulk colostrum was offered. Management and feeding of animals during the pre-weaning period are described in detail by Huuki et al. (2022a). During weeks 2 and 3, the T-group calves were given 5 mL of fresh rumen liquid orally, which was obtained from a feed efficient adult cow (Mäntysaari et al., 2012). During weeks 4–8, the dose was increased to 10 mL. Inoculation was performed weekly on Mondays, Wednesdays, and Fridays and is described by Huuki et al. (2022a). The C-group calves remained untreated but otherwise were managed and fed as the T-group animals. All calves were weaned at the age of 8 weeks, and the animals from both the T-group and C-group were placed into a group pen and housed together first at the Natural Resources Institute Finland (Luke) research barn and later in VikingGenetics breeding facility in Hollola, Finland. The bull calves were removed from the experiment after weaning. Management and feeding of heifers during the post-weaning period are described in detail by Huuki et al. (2022b).
The general health was monitored daily, and diarrhea cases in young animals were recorded by the barn veterinarian. During the pre-weaning period, four calves had loose feces and were treated with electrolyte supplement (Benfital Plus nutrient supplement, Boehringer Ingelheim Danmark A/S), or in more severe cases, three calves were given coal paste (Lehmän HIILI-pasta, FinnCow, Finland). For the analysis of microbial community changes, a calf was recorded to have diarrhea if symptoms occurred within ±2 days around the fecal sample collection time.
Aliquots of the colostrum mix, provided as the first meal, and colostrum from each twin pair’s mother, given as the second meal, were collected into sterile 2 mL screw cap tubes and snap-frozen in dry ice. Fecal samples from calves were collected directly from rectum once a week when the calves were 1–8 weeks old. After weaning, the fecal samples were collected monthly starting from month 4 until animals reached 12 months of age. Feces, either freshly excreted or directly from the rectum, were collected before feeding at 1000–1100 h and immediately placed into sterile bags and snap-frozen in dry ice. Rumen samples from calves were collected at the age of 2, 4, 6, and 8 weeks via esophageal polyvinyl chloride tube (9/13 inner/outer diameter) with a vacuum pump (Ruminator, Germany) and from the donor animal though the fistula as described by Huuki et al. (2022a). All samples were stored at −80°C until DNA extraction.
The total DNA was extracted from 150–250 mg of feces following the protocol by Yu and Morrison (2004) for samples collected during weeks 1–4 and as described by Rius et al. (2012) for samples collected from week 5 to adulthood. Both methods included bead beating step using MP FastPrep-24 (MP Biomedicals, USA). The total DNA extraction from rumen samples was described by Huuki et al. (2022a), while the DNA from 1 mL of colostrum samples was extracted using Milk Bacterial DNA Isolation Kit (Norgen Biotek Corporation, Ontario, Canada), following the manufacturers protocol. Bacteria and archaea were determined by primers 515F and 806R (Caporaso et al., 2011) targeting the 16S rRNA gene V4 region, while for fecal anaerobic fungi identification, primers Neo 18SF and Neo5.8SR targeting the ITS1 region (Edwards et al., 2008) were used. The sequencing libraries were prepared as described by Huuki et al. (2022a) and sequenced in Finnish Functional Genomics Centre (Turku, Finland) on Illumina MiSeq platform by using 2 × 250 bp chemistry for bacteria and archaea and 2 × 300 bp for anaerobic fungi.
The demultiplexed bacterial and archaeal sequencing data were processed using QIIME 2 v2022.2 (Bolyen et al., 2019). The DADA2 (Callahan et al., 2016) workflow was applied for quality control, filtering of chimeric reads, and clustering of bacterial sequences into amplicon sequence variants (ASVs). The ASVs with less than 10 reads were removed. Silva 138 database (Quast et al., 2013) was used for bacterial taxonomy assignment and the RIM-DB database (Seedorf et al., 2014) was used for the archaeal taxonomy assignment. The anaerobic fungi sequencing data were processed with QIIME 1.9.1 (Caporaso et al., 2010) as described by Huuki et al. (2022a), and fungal taxonomy was assigned using the ITS1 (Koetschan et al., 2014) reference database. After removal of low abundance reads, the data consisted of 2,989,290 bacterial, 22,303 archaeal, and 4,569,340 fungal reads. The raw sequence reads are available in the NCBI Sequence Read Archive under BioProject PRJNA713003 with BioSample accessions SAMN36868668-36868795 and SAMN36903992-36904023. The anaerobic fungi mock communities, used as controls for sequencing, showed bias toward Piromyces sp. strain being assigned to BlackRhino clade. All BlackRhino clade OTUs and related reference sequence were compared against the NCBI database with BLASTN (Zhang et al., 2000).
The quantities of bacteria, archaea, and anaerobic fungi were estimated with qPCR by quantifying rRNA/ITS1 gene copy numbers of each taxonomic group in 1 ng of extracted DNA. The quantitation was performed as described by Huuki et al. (2022b).
Differences in microbial quantities, measured as gene amplicon copy numbers, were estimated using Linear Mixed Model GLIMMIX in SAS 9.4 (SAS Institute Inc., Cary, NC, United States). The bacteria copy numbers were log10(x) transformed for normality. Treatment, age, and the treatment × age interaction were treated as fixed effects. Pair was treated as random while age was treated as a repeated effect for each animal. Based on fit statistics, the covariance structure was set to compound symmetry for bacteria and spatial power for fungi and archaea. The difference in variance between groups was considered in bacteria and fungi.
Alpha diversity was estimated using Shannon and Simpson indexes, and the richness (number of observed ASVs) as implemented in Phyloseq (McMurdie and Holmes, 2013). Before analysis, the sequencing data were evenly subsampled to 3,187 reads for bacteria, 11,688 reads for anaerobic fungi, and 100 reads for archaea. The statistical analysis was performed using linear mixed model with fixed effects as described above and compound symmetry covariance structure. To achieve the normal distribution of residuals, the Simpson index was (x)2 transformed for bacteria and anaerobic fungi, and least square means were later reverse transformed. The effects were estimated using the Residual Maximum Likelihood (REML) method and were declared significant at p ≤ 0.05. Due to the low number of reads for archaea, the statistical analysis was performed only on samples from month 4 onward.
Changes in microbial community structure between samples were evaluated as Bray–Curtis dissimilarities following Hellinger transformation and visualized using principal coordinate analysis (PCoA) with Phyloseq. The significance of age, treatment, and treatment x age interaction was estimated using distance-based permutational multivariate analysis (Adonis) as implemented in vegan (Oksanen et al., 2022) and the pairwise comparisons between age × treatment with permutational multivariate analysis of variance with false discovery rate adjustment with RVAideMemoire (Herve, 2020).
Age- and group-related taxonomic differences were evaluated at phylum and genus level for bacteria and at species level for archaea and anaerobic fungi. Samples with less than 10 archaeal reads were excluded from the analysis. All data were log(1 + x) transformed for normalization. The significance of age and treatment × age interaction was evaluated using ANOVA.
The microbiota differences between the healthy calves and the calves experiencing diarrhea symptoms were identified with the linear discriminant effect size (LEfSe) analysis, with logarithmic LDA score of 2.0 for discriminative features and 0.05 alpha value for both factorial test among classes (healthy, diarrhea) and pairwise test for subclasses (T-group, C-group) (Segata et al., 2011). Before analysis, the ASVs that were observed in less than 4 samples were removed.
The SourceTracker version 1.0.1 (Knights et al., 2011) was used to assess the possible seeding sources of hindgut prokaryote ASVs (bacteria and archaea) and fungal OTUs. For prokaryote seeding, colostrum samples from twin calves’ mothers, calves’ rumen samples from weeks 2, 4, 6, and 8, and the inoculum samples from donor rumen were treated as sources. The fecal samples from week 1 to week 8 were treated as sinks. For hindgut anaerobic fungi seeding, the rumen samples from months 4–9 and the donor samples were treated as sources, with fecal samples from the same time period were treated as sinks. For the analysis, the core ASVs/OTUs for the rumen, colostrum, and donor inoculum samples were shortlisted for each time-point separately with detection rate of at least 70% and relative abundance of above 0.001% for bacteria and 0.1% for anaerobic fungi.
The quantity of fecal bacteria in the T-group between weeks 2 and 4 was numerically higher as compared with C-group, but the differences between both groups at the same time-points were not significant (Figure 1A). The fecal bacterial load changed with age (p = 0.02). The copy-numbers were low at week 1 but increased in quantity right after and reached a peak between weeks 2 and 3. After that, a decline in bacterial quantity was observed until week 8 when the calves were weaned. Post-weaning quantity in fecal bacteria increased moderately and stabilized in approximately 10 months of age.
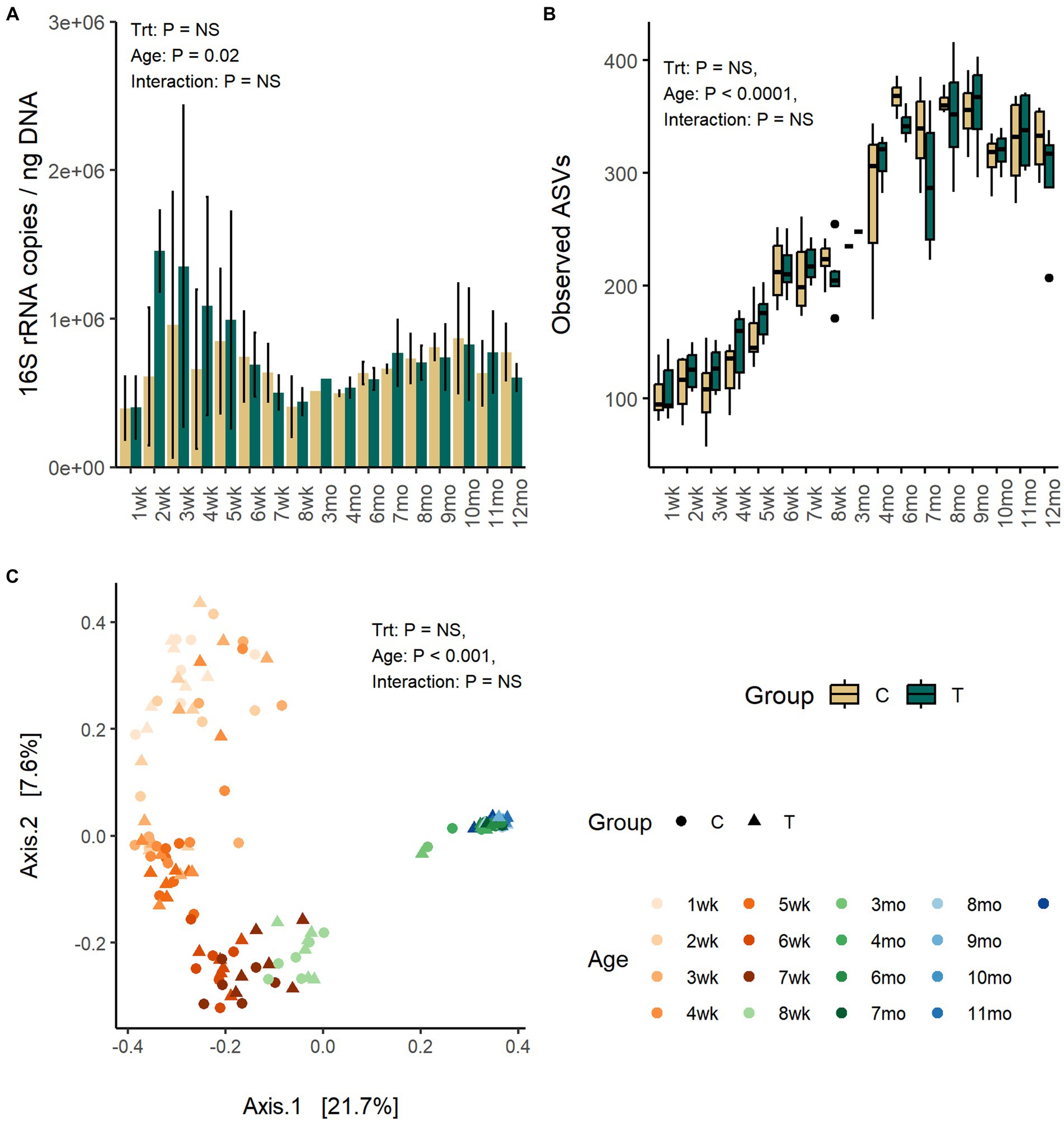
Figure 1. The number of bacteria 16S rRNA gene copies per ng of extracted DNA (A), the number of observed ASVs (B), and the PCoA visualization of Bray–Curtis distances (C) of fecal bacteria communities in rumen liquid inoculated (T) and control animals (C) from 1 weeks to 12 months age.
Fecal alpha diversity was significantly affected by age but not by inoculum treatment or interaction between treatment and age. The number of observed ASVs and Shannon diversity was increased with age (p < 0.0001) and was highest between months 6 and 9 and thereafter stabilized (Figure 1B; Supplementary Figure S1).
The PCoA visualization of Bray–Curtis distances revealed large between-animal variation in fecal bacterial community composition at weeks 1 and 2, which shifted to the first intermediate state for the weeks 3–5 and second state for weeks 6–8, finally becoming mature and uniform after month 3 (Adonis: p = 0.001) (Figure 1C). The changes in community composition likely reflected the dietary changes from milk only to increasing amount of solid feed towards increasing starter concentrate diet and later introduction of silage. The treatment (Adonis: p = 0.26) or interaction between treatment and age (Adonis: p = 0.86) had no effect on the community composition.
Firmicutes (57–87%) and Bacteroidota (9–31%) were the predominant fecal phyla at all time-points (Supplementary Figure S2). Proteobacteria, mainly dominated by Escherichia-Shigella (4–11%), and Actinobacteriota (genus Bifidobacterium) (2–12%) were present in young animals but reduced in abundance toward weaning and diminished to <1% abundance in heifers (p < 0.0001). Verrucomicrobiota (Akkermansia and WCHB1-41) (0–3%) was increased in abundance with age (p < 0.0001), in particular after month 4. Akkermansia was numerically more abundant in the C-group at approximately week 8 (Treatment: p = 0.07).
Out of the 79 genera, with relative abundance above 0.2%, 76 were affected by age and categorized fecal bacterial community into pre-weaning and post-weaning genera (Figure 2). Bacteroides, Lachnospiraceae spp., Dorea, Ruminococcaceae spp., Akkermansia, and Coprococcus were among the few genera present in feces throughout the first year of life, although their abundance varied with age. While calves were fed liquid diet (weeks 1–4), feces was dominated by a small number of higher abundant taxa, including Bifidobacterium (Actinobacteriota), Bacteroides (Bacteroidota), Lactobacillus, Streptococcus, Dorea, Blautia, Faecalibacterium, Butyricoccus, Ruminococcus gnavus group (Firmicutes), Escherichia-Shigella (Proteobacteria), and several lower abundant genera representing Firmicutes. When gradual weaning started at week 5, most of the taxa observed earlier remained present. In addition, Alistipes, Parabacteroides, Butyricimonas, Oscillospiraceae UCG-005, Christensenellaceae R-7 group, or [Eubacterium] coprostanoligenes group appeared at abundance above 1%. The biggest change was visible after weaning starting from month 3, where pre-weaning-related taxa were replaced at different time-points by 34 genera, which were associated with maturation of the fecal bacterial community. Post-weaning fecal bacterial community was composed of many more taxa, as compared with early life samples, each sharing low to moderate (0.5–5%) abundance. There was an increase in diversity of Bacteroidota, where Rikenellaceae RC9 gut group, Prevotellaceae UCG-004, and Bacteroidales RF16 group that were not detected in early life samples became abundant. Among Firmicutes, a different process was observed. Many pre-weaning-related Firmicutes genera were diminished in abundance or were not anymore detected in post-weaning samples (members of Negativicutes class or genera such as Faecalibacterium, Butyricoccus, Fournierella, Subdoligranulum, [Ruminococcus] gnavus group, and Enterococcus). Instead, several other genera belonging to Oscillospirales, Peptostreptococcales-Tissierellales, or Christensenellales orders increased in abundance when animals reached adulthood (Figure 2).
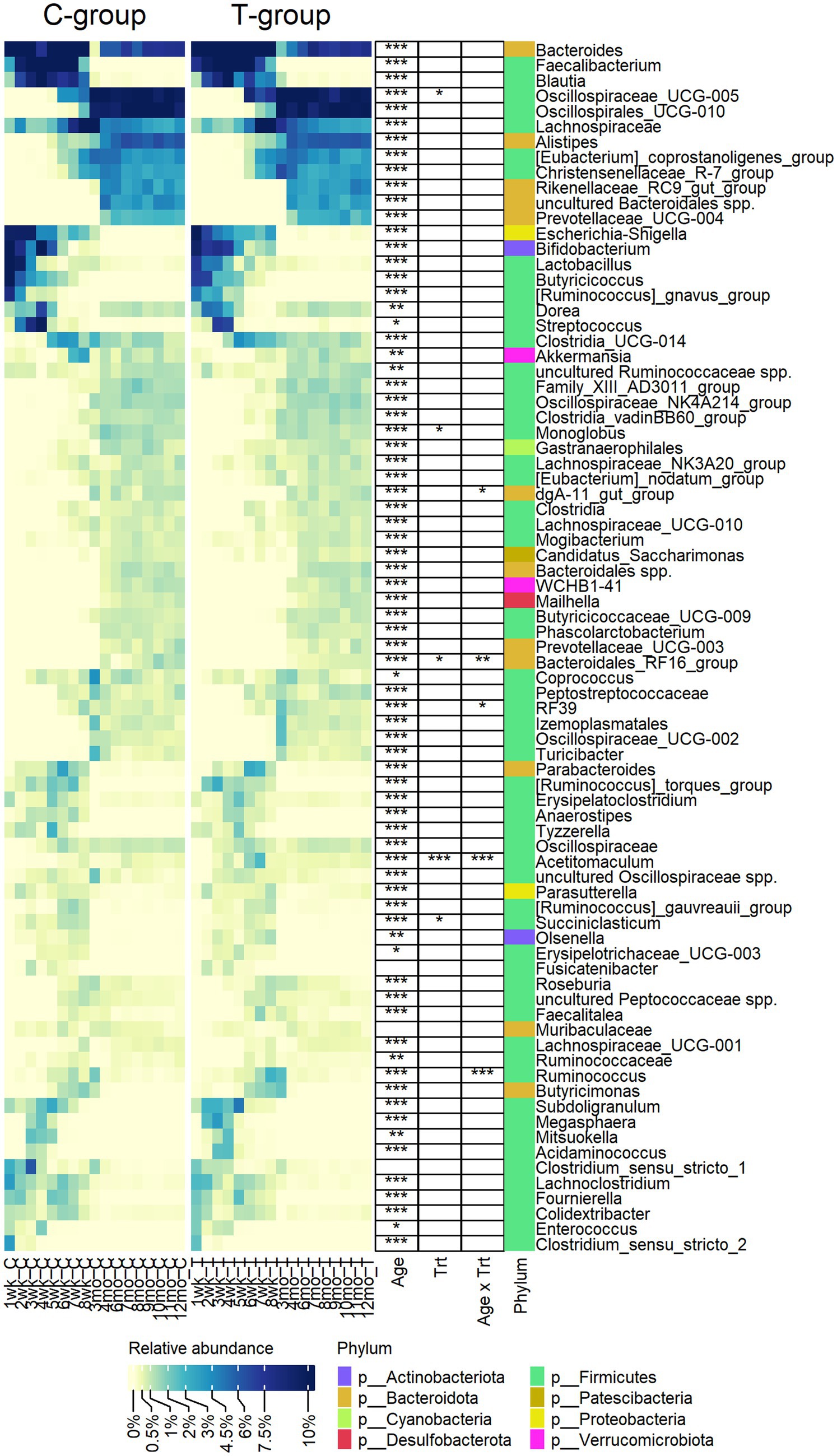
Figure 2. The relative abundance of bacterial genera observed in the feces of rumen liquid inoculated (T) and control (C) animals from week 1 to 12 months of age. The statistically significant effects of age (Age), treatment (Trt), and age x treatment interaction (Age x Trt). The colour bar indicates the affiliation to phylum.
Eight genera were significantly affected by treatment or interaction between age and treatment (Figure 2). The T-group had an overall lower abundance of Succiniclasticum (p = 0.013), Monoglobus (p = 0.05), and Bacteroidales RF16 group but significantly more abundance of Oscillospiraceae UCG-005. Other differences between the groups were related to temporary fluctuation in the abundance of several genera, e.g., Acetitomaculum was more abundant in the T-group between weeks 4 and 8 (Interaction: p < 0.0001), while Ruminococcus was significantly higher (p < 0.0001) and RF39 was lower (pairwise: p = 0.003) in T-group on week 8.
The quantity of fecal archaea was low until month 4, indicating that the community started to properly develop only after weaning (Figure 3A). The copy numbers increased until month 8, after which they stabilized (p < 0.001). Treatment or interaction between treatment and age did not affect the quantity of archaea (Treatment: p = 0.99, Interaction: p = 0.27).
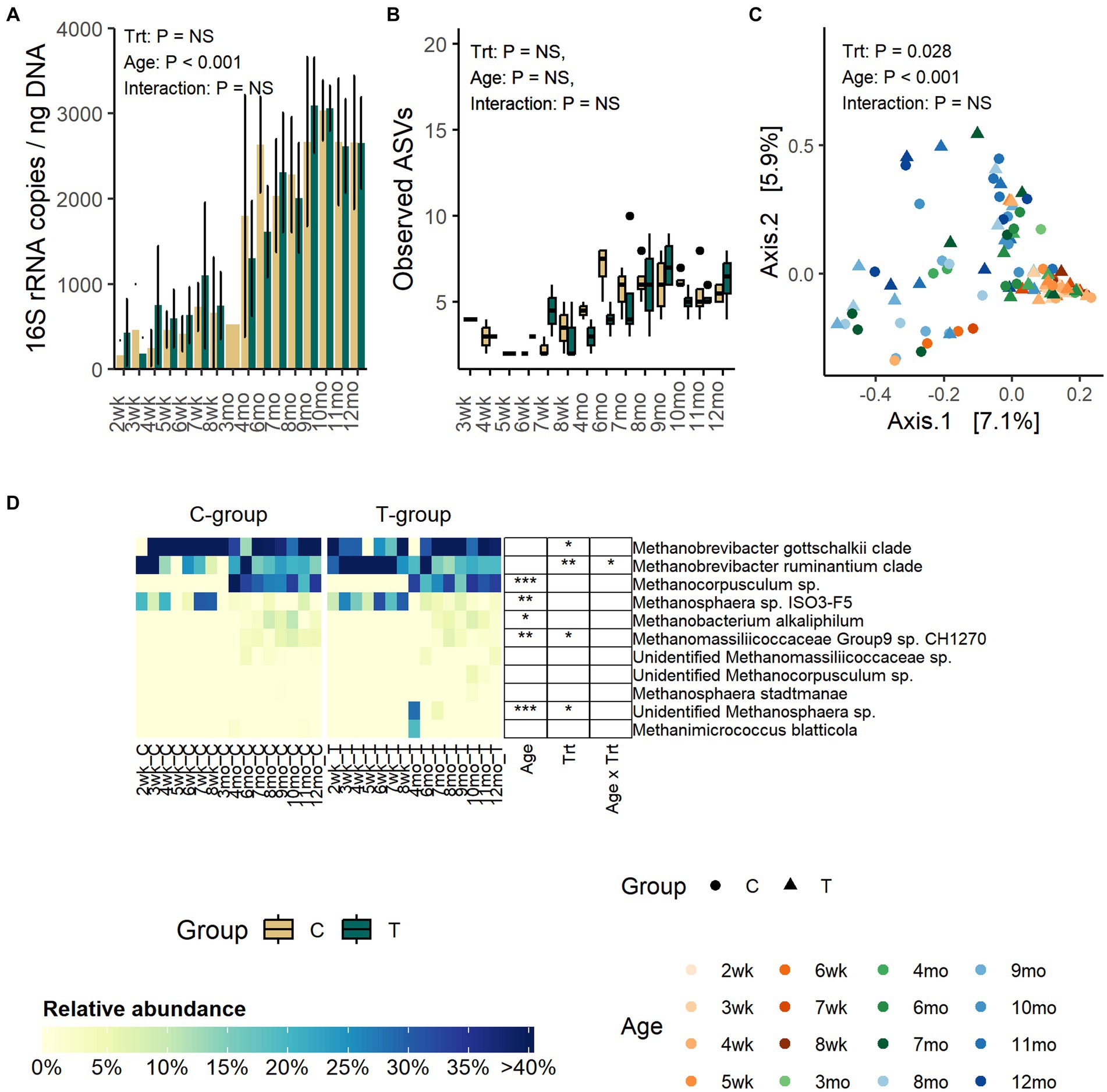
Figure 3. The number of archaea 16S rRNA gene copies per ng of extracted DNA (A), the number of observed ASVs (B), the PCoA visualization of Bray–Curtis distances of archaea communities (C) and the relative abundance of fecal archaea species (D) in rumen liquid inoculated (T) and control animals (C) from week 1 to 12 months of age. In panel (B) statistical analysis was performed for months 4–12 samples only due to low number of reads and missing observations in pre-weaning samples. For alpha diversity calculation, archaea data were evenly subsampled, and samples with less than 100 reads were removed from the analysis.
Alpha diversity was not affected by treatment or interaction between treatment and age (Figure 3B; Supplementary Figure S3). Numerically, richness remained low until month 4 after which the number of observed ASVs started to increase and reached the peak in the C-group on month 6 and in the T-group on month 9. After month 9, the observed ASVs decreased and plateaued.
The PCoA visualization of Bray–Curtis distances showed that the pre-weaning archaeal community composition was very similar between individuals, but after weaning the variation greatly increased (Adonis: Age: p = 0.001) (Figure 3C).
Before month 4, the fecal archaeal community was dominated by Methanobrevibacter gottschalkii clade (17–100%), Mbb. ruminantium clade (7–92%), and Methanosphaera sp. ISO3-F5 (4–32%) (Figure 3D). From month 4 onwards, the community experienced changes. Mbb. gottschalkii and Mbb. ruminantium clades continued as the most abundant species, while Methanosphaera sp. ISO3-F5 abundance diminished. Instead, Methanocorpusculum sp., which was not detected in pre-weaning samples, increased in abundance (16 – 41%) (Age: p < 0.001). Methanobacterium alkaliphilum and Methanomassiliicoccaceae Group 9 sp. were detected in fecal samples in month 6, and they remained in the community thereafter.
In comparison to the C-group, the T-group had a higher overall abundance of Mbb. ruminantium clade (Treatment: p = 0.007) and lower abundance of Mbb. gottschalkii clade, with differences apparent between months 1 and 3. During the post-weaning period, the T-group had less of Methanomassiliicoccaceae Group9 sp. CH1270 (Treatment: p = 0.045). Two calves in the T-group were observed to have Methanosphaera sp., which was not observed in any of the C-group calves (Treatment: p = 0.02; Interaction: p < 0.0001).
Anaerobic fungi could be reliably detected in fecal samples by the qPCR method only after weaning. Between months 4 and 12, the copy numbers were significantly increased (Age: p = 0.01) but were not affected by treatment (Treatment: p = 0.47, Interaction: p = 0. 68) (Figure 4A).
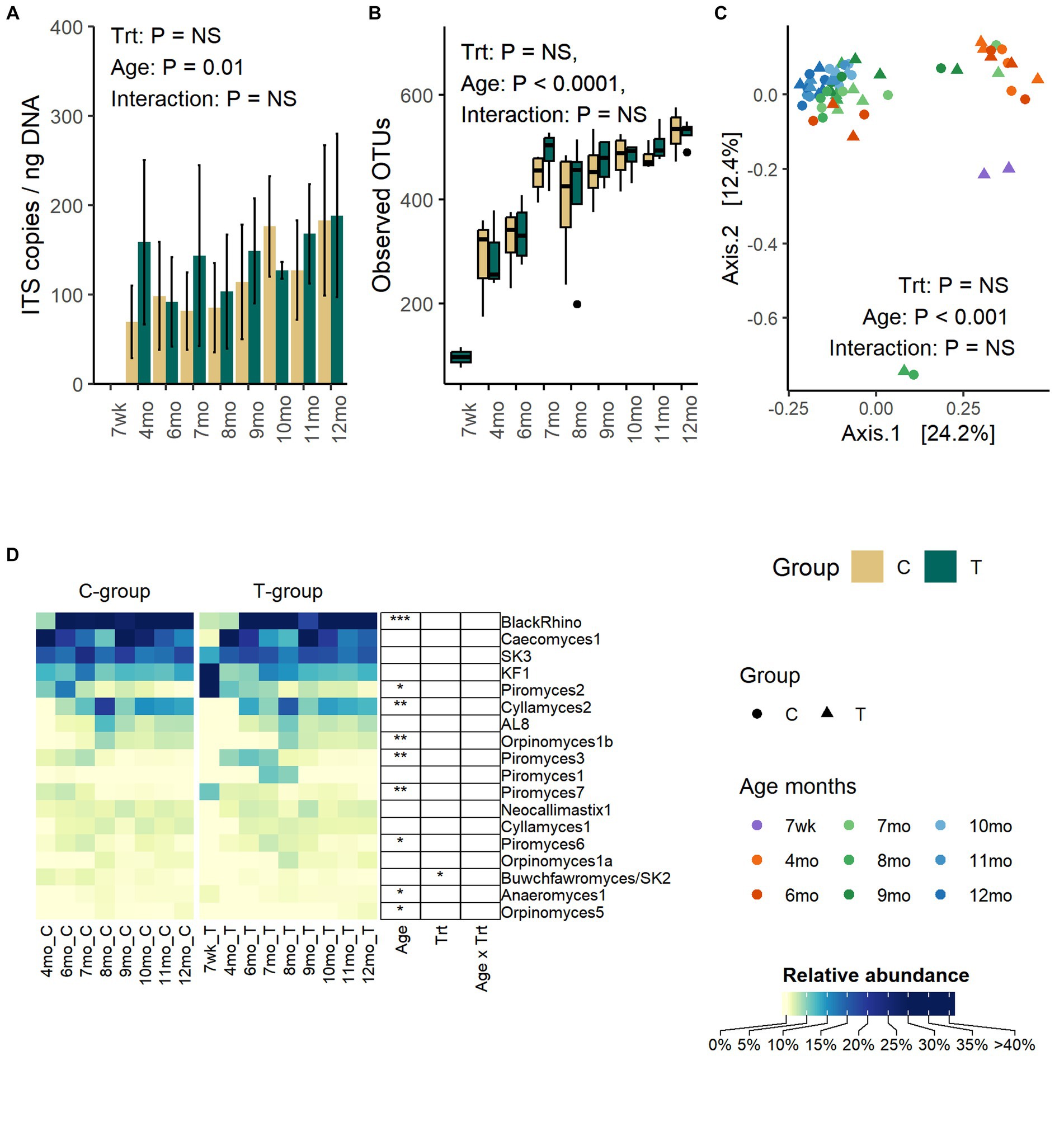
Figure 4. The number of anaerobic fungi ITS1 gene copies per ng of extracted DNA (A), the number of observed OTUs (B), the PCoA visualization of Bray – Curtis distances (C), and the average relative abundance of anaerobic fungi in the feces of rumen liquid inoculated (T-group) and control (C-group) animals between week 7 to month 12 of age (D). The qPCR method could only reliably detect anaerobic fungi after month 4, although some sequencing data could be obtained from 2 T-group samples at week 7. The data for samples at week 7 are visualized in the figure but excluded from the statistical analysis.
The number of observed OTUs was increased significantly with age until month 8 after which the richness stabilized (Age: p < 0.0001) (Figure 4B). Similarly, Shannon diversity (Age: p < 0.0001) and Simpsons evenness (Age: p < 0.0001) increased until month 7 and then stabilized (Supplementary Figure S4). Treatment or interaction between treatment and age had no significant effect on any of the alpha diversity metrices.
The age, but not treatment, affected the fecal fungal community composition (Adonis: Treatment: p = 0.65, Age: p = 0.001, Interaction: p = 1.0) (Figure 4C). The PCoA visualization of Bray–Curtis distance categorized communities into clusters based on age, where samples between week 7 and month 4 grouped together. Between months 6 and 9, communities were transitioned into a more mature state, but the composition varied between individual heifers. At months 9–12, the fungal communities of all heifers were similar and grouped together.
Although pre-weaning samples exhibited low amplification success with other PCR methods, the sequencing of ITS1 libraries from two T-group calf samples proved successful on week 7. Because of the low observation rate and low number of reads, these samples were excluded from the statistical analyses, but the taxonomic composition is visualized (Figure 4D). The week 7 samples differed from later samples, predominantly being composed of Piromyces 2 (44%) and KF1 (38%). By month 4, KF1 and Piromyces 2 groups had decreased by more than six folds, and the fecal fungal community was dominated by Caecomyces 1 (63%), SK3 (16%), and five other less abundant species (0.4–3.5%). On month 6, the BlackRhino became the most abundant clade (33%), which was accompanied by five other abundant (5.5–20%) species-level clades: Caecomyces 1, SK3, Piromyces 2, Cyllamyces 2, and KF1. All BlackRhino OTUs in our data set were associated with one reference ITS1 sequence (MH039104) that showed over 99% identity to the same Piromyces sp. SFH682 strain, which has been previously shown to be identical to BlackRhino (Fliegerova et al., 2021). On month 7, the community maintained BlackRhino, Caecomyces 1, SK3, Cyllamyces 2, and KF1 as the top five most abundant species, while Piromyces 2 started to decline, and this trend remained the same until month 12. The highest species-level richness (18 species level taxa) was observed by month 8. Between months 8 and 9, Cyllamyces 2. Orpinomyces spp., and AL8 experienced fluctuation in relative abundance, but between months 10 and 12, the community composition stabilized. On month 12, all Piromyces spp. had reduced below 1% relative abundance. Taking into consideration the relatedness of BlackRhino to Piromyces species, the results indicate that different Piromyces spp. were important at early and late post-weaning stages. The T-group had an overall higher abundance of Buwchfawromyces/SK2 species (p = 0.023), but the abundance was not affected by age or interaction between treatment and age.
To assess the potential effect of inoculum treatment on gut health in pre-weaning animals, we examined microbiota differences between healthy calves and calves with reported diarrhea symptoms. Five of the C-group calves and four of the T-group calves were observed to have diarrhea between 2 and 4 weeks of age. The diarrhea did not affect the bacteria quantity (Figure 5A) but significantly reduced the alpha diversity (Figure 5B). The LEfSe analysis identified 34 differentially abundant ASVs between healthy and diarrhea samples, which were affiliated with 10 genus- or species-level taxa (Supplementary Figure S5; Figure 6). Fecal microbiota in healthy calves between weeks 2 and 4 exhibited higher abundance of ASVs associated with Ruminococcus gnavus group sp., Bifidobacterium bifidum, Butyricicoccus pullicaecorum, and Faecalibacterium sp., while fecal microbiota of diarrhea samples was characterized by higher abundance of ASVs associated with Escherichia-Shigella spp., Streptococcus spp., Clostridium sensu stricto 1 sp., and Lactobacillus spp. None of the differentially abundant ASVs were significantly associated with the donor inoculum treatment. In all individuals, taxa that differentiated between healthy and diarrhea cases in early life disappeared from the fecal community by month 6 (Figure 6).
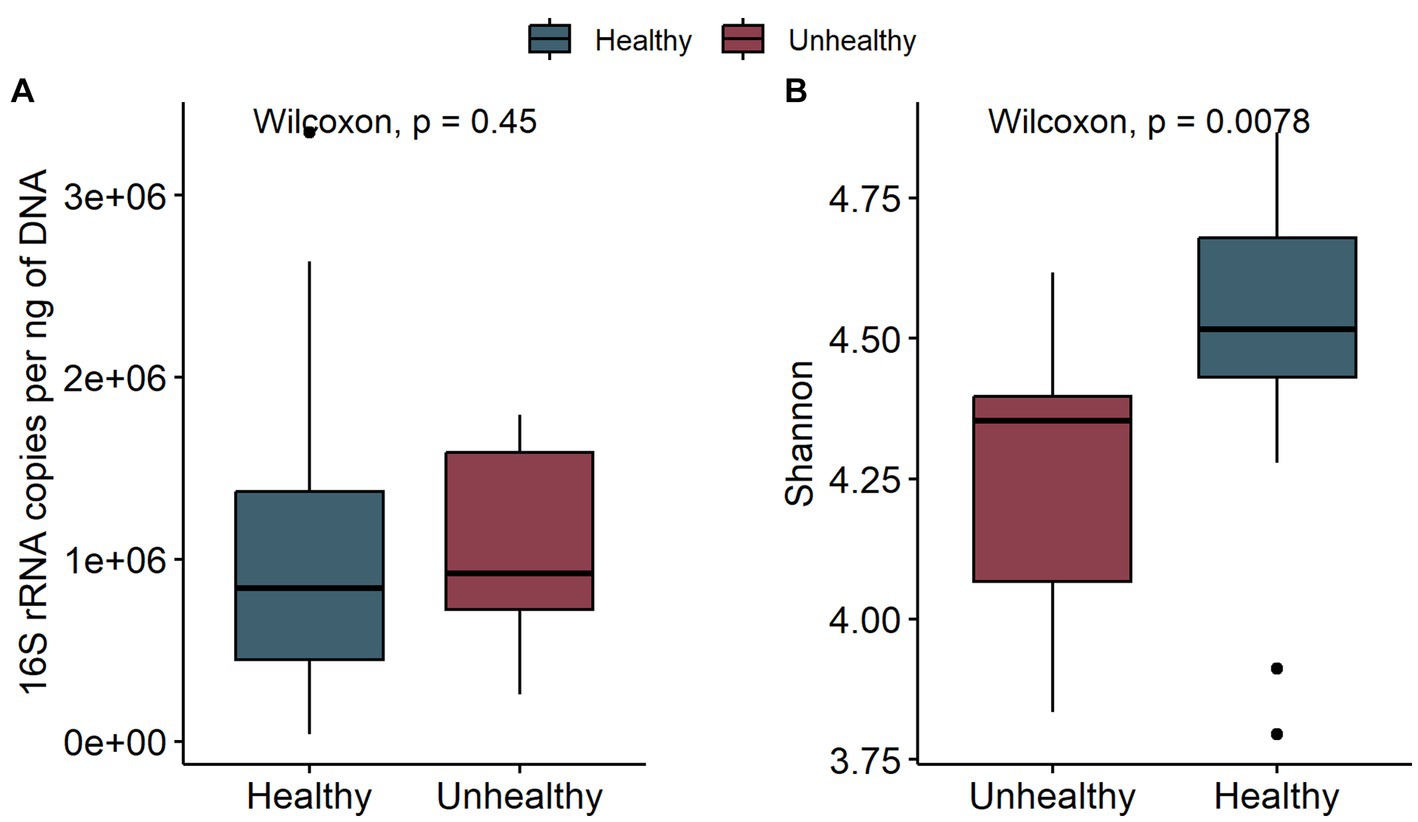
Figure 5. The comparison of bacteria 16S rRNA gene copies per ng DNA (A) and the Shannon diversity (B) of fecal samples in healthy calves (healthy) and calves with observed diarrhea (unhealthy) at 2 to 4 weeks of age.
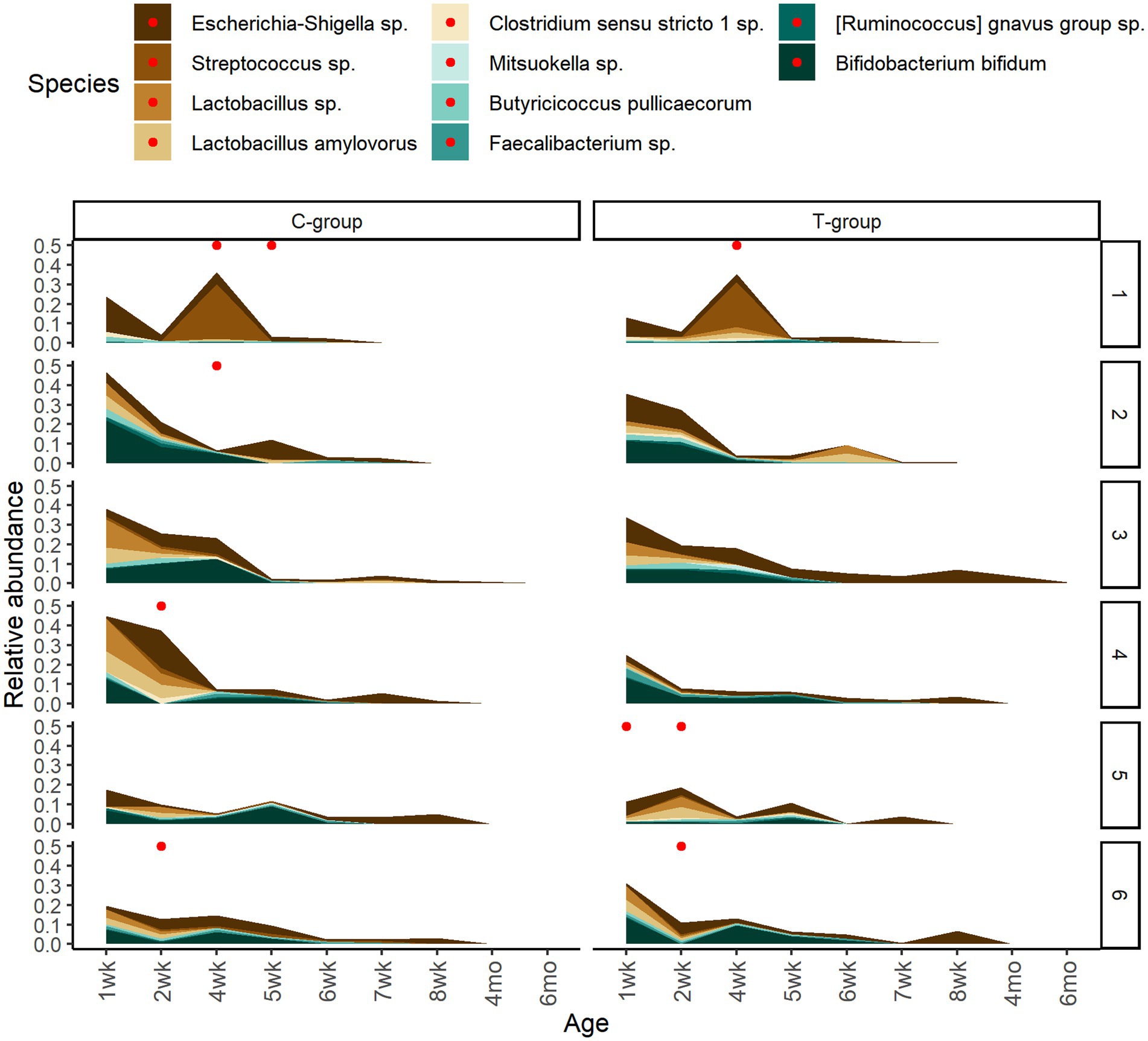
Figure 6. The temporary changes in the relative abundances of differently abundant bacterial ASVs between healthy calves and calves with observed diarrhea cases grouped at species level. The reported diarrhea occurrences (within +/− 2 days from sampling day) are indicated with red dots.
The cow colostrum demonstrated large between-sample variation in bacterial composition. Therefore, for seeding analyses, we shortlisted colostrum bacteria to 22 ASVs, present in 70% of samples (Figure 7). From them, only one Enterococcus sp. ASV was present in feces between weeks 1 and 8, Kocuria between weeks 1 and 7, and Prevotella between weeks 2 and 8. Acinetobacter was detected in feces shortly between weeks 3 and 6, while Methanobrevibacter was present in feces between weeks 5 and 8. All colostrum-derived ASVs were transient in feces, and none of them remained during the post-weaning period.
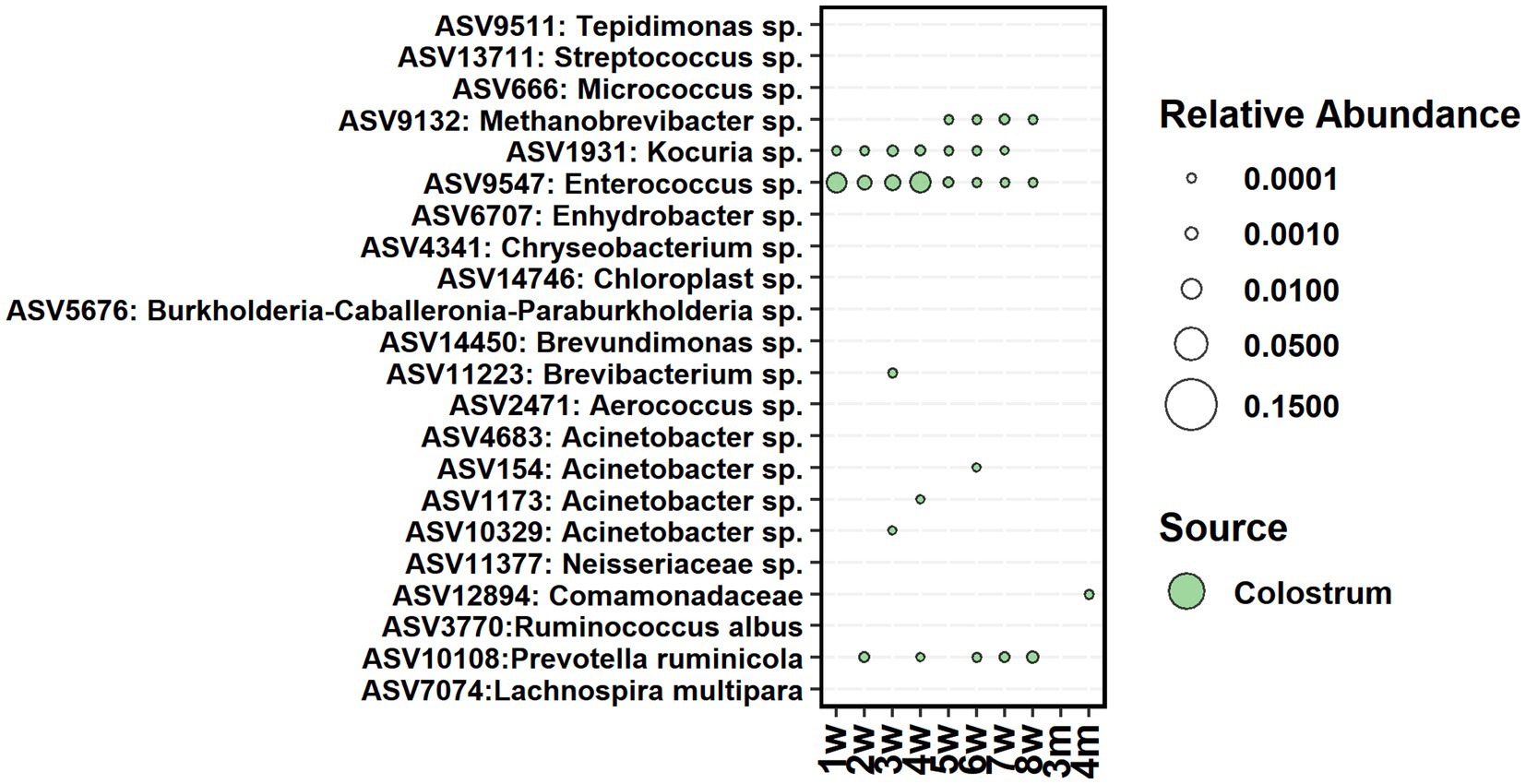
Figure 7. The average relative abundance of colostrum core ASVs detected in fecal samples of calves between week 1 and 4 months of age.
The core bacteria from calf rumen shortlisted for seeding analysis was comprised of 23, 22, 21, and 25 ASVs on weeks 2, 4, 6, and 8, respectively. Most of the ASVs in the rumen core community of 2 weeks old calves were also detected in 2-week-old fecal samples (Figure 8) and showed 5–41% contribution to fecal bacterial colonization (Supplementary Figure S6). The majority of them were transient: Actinomyces and Blautia were present in feces until week 5, while Prevotella, Streptococcus, Lachnoclostridium, and Succiniclasticum disappeared from feces after week 8. Bacteroides was still detected in feces at month 3, and Butyricimonas was detected at month 4. On the other hand, Escherichia-Shigella, Methanobrevibacter, Methanosphaera, and Mogibacterium colonized early and remained in feces permanently. After week 2, the proportion of calf rumen core ASVs shared with feces decreased, suggesting that the rumen and large intestine started developing more specialized microbial ecosystems. Acetitomaculum and Lachnospiraceae NK3A20 groups were transient in feces between weeks 3 and 8, while Atopobium, Family XIII AD3011 group, and Christensenellaceae R-7 group populated at weeks 3–4 and remained in feces permanently, suggesting that these taxa might be needed for the initiation of solid feed digestion. Interestingly, ASV affiliated with Lactobacillus fructivorans that was not detected in the rumen before week 6 and in feces before week 7 remained in both ecosystems until month 12 and indicated an association with the transition to solid diets. Based on the SourceTracker analysis, donor inoculum was not identified as the major seeding source for fecal bacteria in calves (Supplementary Figure S6).
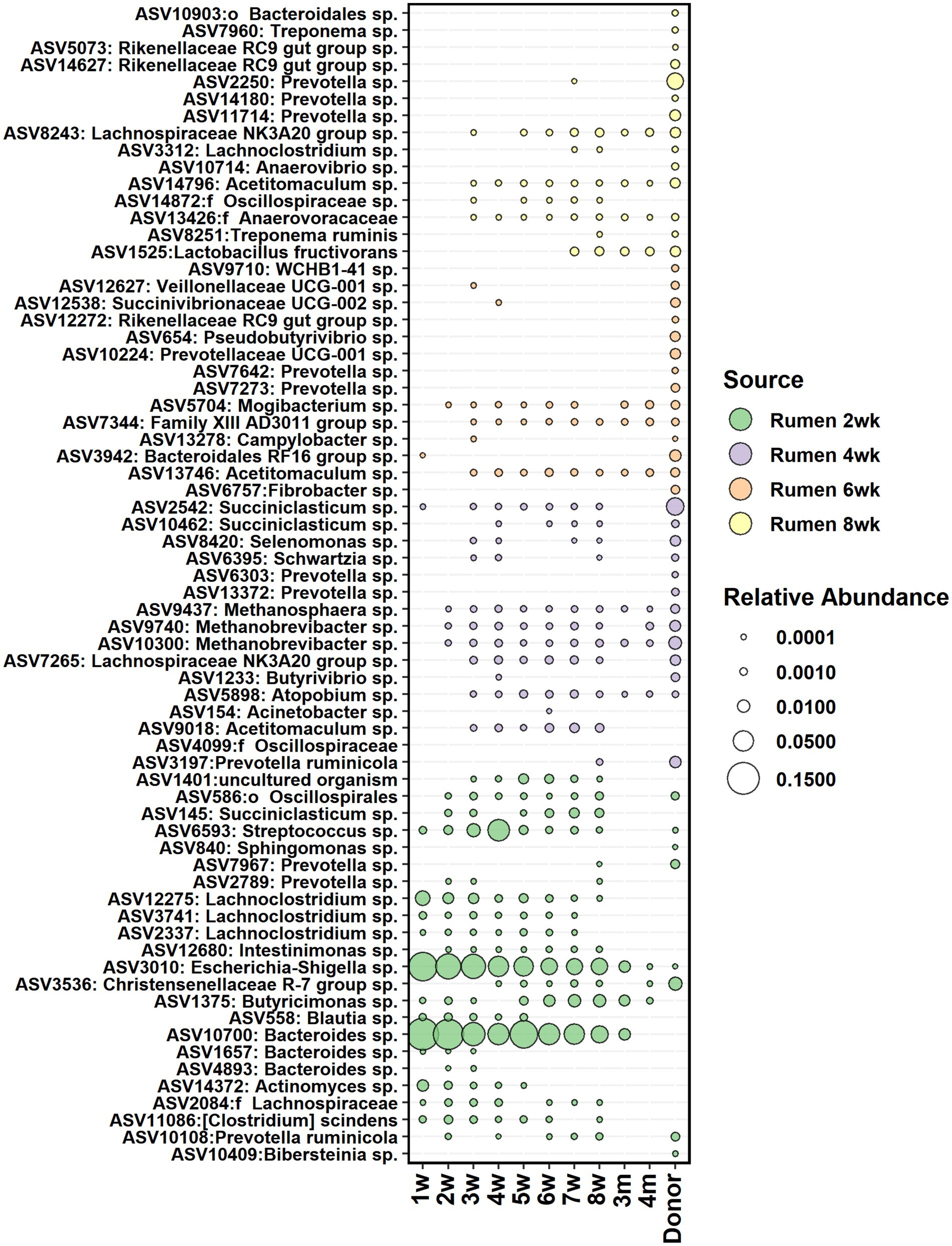
Figure 8. The average relative abundance of rumen core bacteria ASVs from week 2, 4, 6, and 8 detected in fecal samples of calves between week 1 and 4 months of age and in donor inoculum. The different colors indicate new ASVs introduced to the core community during each time-point.
According to the SourceTracker analysis, the calf’s own rumen was identified as the primary seeding source of anaerobic fungi in feces, accounting for 98.7, 68.5, 79.2, 94.0, and 95.6% of the fecal community composition between months 4 and 9, respectively (Supplementary Figure S7). The heifer rumen core anaerobic fungi OTUs shortlisted for seeding analysis comprised of 33, 32, 52, 36, and 59 OTUs on months 4–9, respectively. On months 4 and 6, all rumen OTUs of anaerobic fungi were consistently detected in feces and persisted in the community (Figure 9).
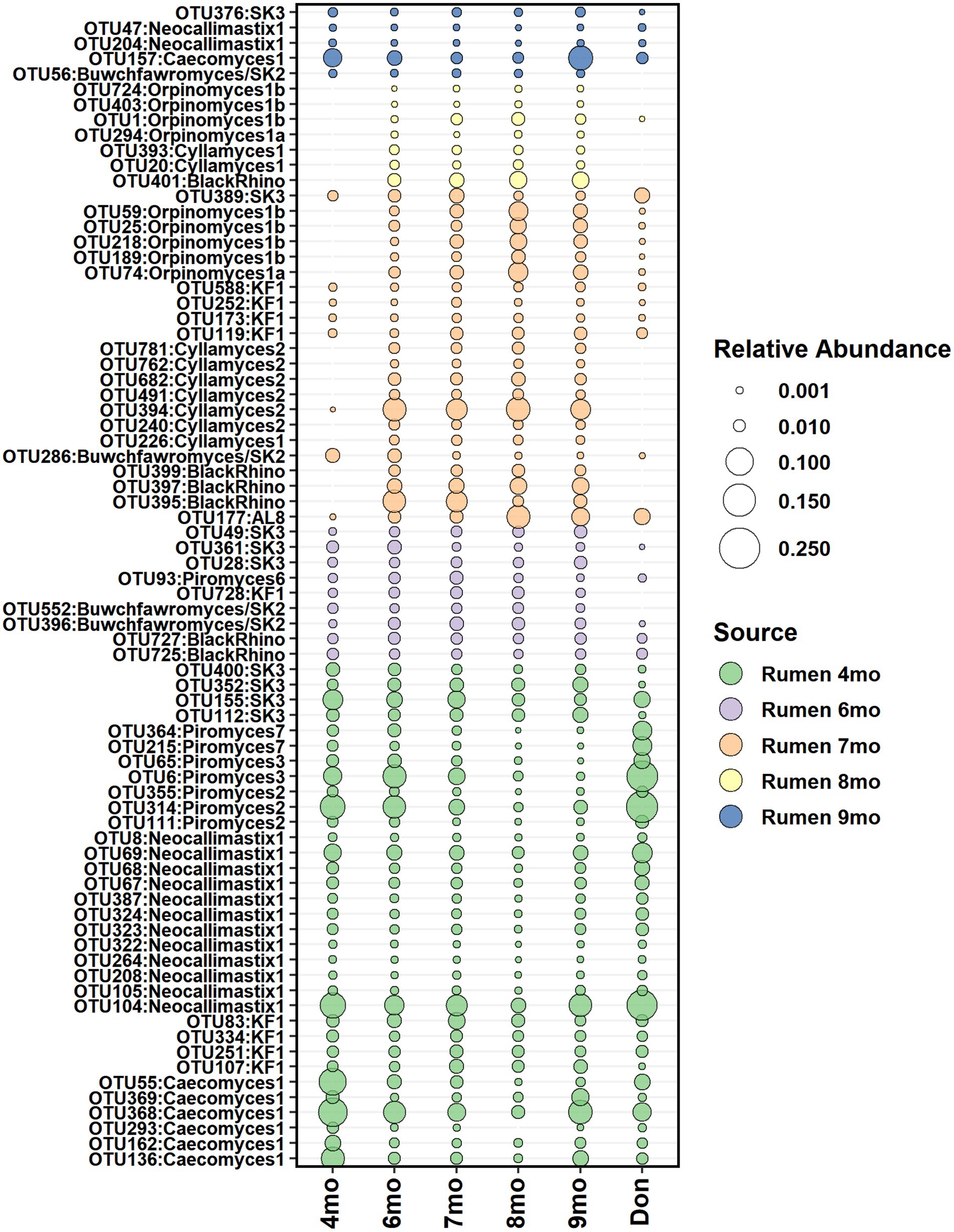
Figure 9. The average relative abundance of rumen core anaerobic fungi OTUs detected in fecal samples of heifers between month 4 and month 9 of age and in donor inoculum. The different colors indicate new OTUs introduced to the core community during each time-point.
Donor inoculum did not show contribution as seeding source at month 4, as all rumen anaerobic fungi detected in heifers at month 4 were also observed in the donor. However, starting from month 6, several new rumen anaerobic fungi OTUs were observed in feces, and these were distinct from the donor community. The timing coincided with the transfer of calves to a different barn, where exposure and acquisition of new fungi from new barn environment was possible.
The commercial milk replacer offered to calves contained Bacillus licheniformis (DSM 5749), Bacillus subtilis (DSM 5750), and Enterococcus faecium (NCIMB 11181). We tested if these species could be detected in fecal microbiota of calves. BLAST analysis of fecal Enterococcus ASVs against the NCBI database did not return 100% similarity match to given species. The closest 99.7% similarity was observed in E. faecalis and other uncultured Enterococcus sp., suggesting that the 16S rRNA V4 region is not able to separate between different species of Enterococcus. The one fecal ASV from the genus Bacillus did not match with either B. subtilis or B. licheniformis.
In this study, we examined the influence of early-life rumen microbiota modulation on the colonization and maturation of the fecal microbiota in dairy heifers over a 12-month period. We also explored modulation’s potential benefits for gut health in young animals. This research is an extension of our previous study, which demonstrated that dosing calves with fresh rumen liquid from an adult donor cow accelerated the maturation of rumen bacterial and archaeal communities, promoted the growth of calves (Huuki et al., 2022a,b), and impacted animal production performance during their 1st lactation (Huuki et al., 2022b). In the current study, we demonstrated that colostrum and the young animal rumen serve as sources for seeding the fecal bacterial and archaeal communities, while the donor rumen inoculum treatment had no impact on the composition of fecal microbiota. In addition, we provided new evidence that rumen also greatly contributes to hindgut anaerobic fungi colonization as a seeding source.
Our results showed very distinct bacterial communities between pre-weaning and post-weaning samples, which coincided with the changes in the diet. After the first week, the bacterial load in feces quickly reached a stable state, but the community richness continued to increase until month 4. During the first 2 weeks, while the calves were fed milk diet, the fecal bacteria consisted mainly of Bacteroides, Bifidobacterium, Faecalibacterium, Blautia, Lactobacillus, and Ruminococcus gnavus group, well-known commensal bacteria. Many of them are known to have important roles in the developing gastrointestinal tract of calves (Liu et al., 2021; Gomez et al., 2022) and have been previously associated with large intestine microbiota in pre-weaning animals (Dias et al., 2018; Song et al., 2018). After the second week, when the milk diet was changed to milk replacer, Lactobacillus were replaced mainly by another facultative anaerobe Streptococcus. Lactobacillus is associated with milk digestion and is able to utilize lactose and starch for lactate production (Hammes and Hertel, 2015). In addition, Streptococci are able to utilize a variety of carbohydrates, e.g., lactose and starch, in fermentation (Whiley and Hardie, 2015). However, in vitro co-culturing of oral Streptococcus sp. and Lactobacillus sp. strains has shown pH-dependent competition among the genera (Bowden and Hamilton, 1989). The change in diet composition may have altered the available substrates and may have caused changes in hindgut fermentation, leading to altered pH, and caused the decrease in Lactobacillus.
As a seeding source, the colostrum’s contribution to the bacterial colonization in feces was temporary. Only Enterococcus sp., Kocuria sp., and Prevotella ruminicola were shared between colostrum and feces of calves during the pre-weaning period, suggesting the association of these taxa with the digestion of liquid diet. Enterococci are commonly found in the feces of pre-ruminant calves (Devriese et al., 1992) and are facultatively anaerobic, lactic acid-producing bacteria that are capable of utilizing various sugars (Švec and Devriese, 2015). Kocuria are typically associated with the skin microbial community and have been found among udder skin microbiota (Braem et al., 2013) and in milk of cows (Quigley et al., 2013; Shinozuka et al., 2021). The colostrum-related ASVs disappeared from the fecal community after weaning, which indicates that they had a functional role only during the pre-weaning period. Facultative anaerobes such as Enterococcus play a crucial role in creating and maintaining the anaerobic environment, which enable the colonization and function of obligate anaerobes. Therefore, we propose that colostrum-related taxa in fecal samples may serve a similar role in maintaining an anaerobic environment and digesting milk or liquid feed. Our results are in line with previous studies (Yeoman et al., 2018; Klein-Jöbstl et al., 2019) that colostrum acts as a seeding source for some important hindgut bacteria, but it also plays an important role in providing immunoglobulins and nutrients to host and microbes and thereby indirectly affects the community structure.
When calves’ own rumen microbiota was evaluated as a seeding source, an especially strong influx from the rumen into feces was observed at weeks 2–4, and these taxa mostly remained in feces only until animals shifted to solid diets. To be able to colonize the large intestine, bacteria have to have suitable nutrients for growth, and they have to be able to withstand the acidic conditions in the abomasum and the bile acids and digestive enzymes. Our observations suggest that at an early age, when the rumen environment still resembles more than that of the hindgut, rumen serves as a reservoir for certain bacteria, which can transfer to and establish in the hindgut. However, after week 4, only a few new bacteria were introduced to the fecal community from the rumen likely due to functional specialization of both the rumen and hindgut microbial communities. The bacteria in the large intestine (like Bacteroides) were benefited by relying more on the host mucosa-derived glycans and xylans and cross-feeding of their breakdown products (Lin et al., 2023). Silage was introduced to the diet on 7th week, and by week 8, a small seeding from the more mature rumen community was observed. This was likely caused by the introduction of new substrates that provided new niches into the hindgut environment.
After week 4, when the gradual increase in solid feed intake started, Bacteroides, Faecalibacterium, and Blautia remained abundant, while Bifidobacterium decreased in abundance. Bifidobacterium are specialized in utilizing complex milk and mucus oligosaccharides (Marcobal et al., 2010; Zhang et al., 2022), while Bacteroides and Blautia are capable of not only utilizing milk and host-derived oligosaccharides and glycoconjugates (Song et al., 2015; Liu et al., 2021) but also can utilize a wider range of plant carbohydrates (Singh, 2019). The dietary transition was also accompanied by an increase in the abundance of many fiber-degrading Firmicutes (e.g., Oscillospiraceae UCG-005, Lachnospiraceae, and Christensenellaceae R-7 group). The rumen enzymatic activity reaches a stable state only after 1 month of age (Rey et al., 2012), and the starch digestion of the small intestine is not yet very efficient in calves (Gilbert et al., 2015). Increased flow of solid feed digesta into the large intestine may have benefitted these bacteria.
In comparison to the rumen bacterial community that matured at approximately 6 months of age (Huuki et al., 2022b), we found that the maturation of fecal bacterial community occurred earlier at approximately month 4. The community richness greatly increased, and the remaining pre-weaning related taxa (e.g., Faecalibacterium and Blautia) reduced and were replaced by several Firmicutes and Bacteroidetes genera, like Rikenellaceae spp. and Prevotellaceae spp., with broader capability to utilize plant carbohydrates. The earlier maturation of fecal bacteria community may be due to more stable digesta consistency and environment in the large intestine. When animals shift to solid diets and rumen fermentation becomes the major source of energy, the composition and flow of digesta into the hindgut becomes fairly constant (Constable et al., 2005, 2006). On the contrary, in the rumen, the change in volume size and development of muscle mass still continues while animals grow during the post-weaning period. The diurnal variation between ingestion, rumination, and resting bouts or adjustment to changing passage rate may require more time for the rumen bacterial community to mature. Moreover, the digesta retention time is shorter, and digesta substrate consistency in the large intestine is less complex in comparison to the ingested diet in the rumen, as the complex dietary carbohydrates have undergone digestion by ruminal microbial enzymes and gastric enzymes in the abomasum and small intestine. Therefore, the hindgut bacteria community can probably cope with a more limited functional capacity, and the community is therefore able to mature earlier.
Significantly less information is available regarding methanogenesis and archaea community composition in the hindgut of ruminants compared with the rumen. The archaeal load in feces was lower before weaning than after weaning, indicating that the proper community establishment occurred after weaning. Archaea richness is increased at least until month 6 and is consistent with previous observations according to Dill-McFarland et al. (2017, 2019), who documented proper archaea establishment between weaning and 1 year of age. While archaea community composition was more similar between calves during the pre-weaning period, the variation increased over time. In contrast, some previous studies have reported individual variation to decrease with age (Dill-McFarland et al., 2017, 2019). However, due to the restricted number of reads per sample, particularly in the pre-weaning phase, it is essential to exercise caution when interpreting the results.
Methanobrevibacter dominated the pre-weaning community and persisted after weaning, gradually accompanied by other archaea. Methanocorpusculum appeared only after weaning, which suggests that they are associated with solid diet. These findings align with previous studies (Dill-McFarland et al., 2017, 2019; Li et al., 2022). Methanocorpusculum is more commonly found in feces than in the rumen (Tapio et al., 2016; Holman and Gzyl, 2019). They exhibit flexibility in energy precursors, utilizing CO2, H2, and formate, and in some species, secondary alcohols were used as electron donors (Chong and Boone, 2015). These archaea also possess gene-encoding bile salt hydrolases (Lin et al., 2023), potentially aiding in their adaptation to the hindgut. Methanobrevibacter gottschalkii was generally more abundant in feces compared with Mbb. ruminantium. Unlike Mbb. ruminantium, Mbb. gottschalkii is known to be bile tolerant (Miller, 2002). Previous studies have suggested a negative interaction between Mbb. gottchalkii and Mbb. rumimantium clades in the rumen environment, possibly due to different expression levels of methyl-coenzyme reductase I (MCRI) and methyl-coenzyme reductase II (MCRII) isozymes (Kittelmann et al., 2013). These enzymes, crucial for the final step in methane production, are expressed differently in response to varying H2 and CO2 concentrations; MCRI is expressed in low gas concentration, and MCRII is expressed in high gas concentration (Bonacker et al., 1992). Some Mbb. ruminantium strains (i.e., M1) possess only MCRI operon, while many other Methanobrevibacter species have both MCRI and MCRII operons (Leahy et al., 2010; Pitta et al., 2022).
In this study, Methanobrevibacter sp. was a part of the colostrum core community, which seeded in feces from 5th week onwards. Compared with milk bacteria, the milk archaea have been less studied. However, recent evidence suggests that there is a rich community of archaea in milk (Hoque et al., 2019; Shinozuka et al., 2021), and archaea have also been found in the uterus (Yeoman et al., 2018) and GI tract of calves right after birth (Guzman et al., 2015). Our results suggest that colostrum could serve as a seeding source for some archaea, but the majority were seeded from young animal rumen early in life and remained in the large intestine thereafter. However, further studies are needed to elucidate the seeding sources and succession of archaea in the hindgut.
Anaerobic fungi establishment in the hindgut of ruminants is less explored compared with the rumen. Consequently, we investigated how the community of anaerobic fungi matures in the hindgut of growing animals and whether early-life rumen modulation has an impact on fecal fungal establishment. We were unable to detect anaerobic fungi in feces before weaning. This implies that the anaerobic fungi colonization in the large intestine requires transition into solid feeds, particularly intake of silage, with fungal richness increasing until month 10. Our results agree with a previous study, indicating that fecal anaerobic fungi establish sometime after weaning but before 1 year of age (Dill-McFarland et al., 2019). In a study using culturing techniques, anaerobic fungi were detected in feces of 4-week-old calf, with all studied animals acquiring fecal fungi by the age of 15 weeks (Theodorou et al., 1993). In contrast to our results and the earlier observations, a recent study suggested the presence of fecal anaerobic fungi as early as day 1 after birth, with quantities increasing until month 4 (Jones et al., 2023).
The anaerobic fungi colonization in feces is age related. The two pre-weaning samples from the T-group were mainly comprised of Piromyces 2, uncultured KF1, and uncultured SK3 clades. Certain Piromyces strains have demonstrated robust growth on various substrates, including lactose and starch (Teunissen et al., 1992). It is possible that the pre-weaning diet, consisting of milk replacer along with concentrate and silage, provided favorable substrates for the growth of these fungi. Month 4 appeared as a transition time-point with strong predominance of Caecomyces, while starting from month 6, dominance was more evenly distributed among five fungal genera, with BlackRhino replacing Caecomyces as the most abundant group. Although originally isolated from Black rhinoceros, the BlackRhino clade was shown to be identical to the cultured Piromyces sp. SFH682 (Fliegerova et al., 2021). Considering the similarity of BlackRhino to Piromyces, our results agree with Jones et al. (2023) who showed that the post-weaning fungal communities had higher abundance of Caecomyces, Piromyces, and Cyllamyces in comparison to the pre-weaning communities. Our results also indicated that different Piromyces species might be accustomed to the hindgut environment at different age stages in growing animals. From month 8 onwards, while BlackRhino (Piromyces) continued to have high relative abundance, other Piromyces species began to decrease, disappearing almost entirely from the feces by month 12. This time-point coincided with a shift in the maintenance diet to a lower-energy diet with higher roughage content (Huuki et al., 2022b). Because easily fermented carbohydrates are, to a large extent, consumed in the rumen, and in some cases Piromyces have been observed to degrade straw less efficiently than Neocallimastix and Caecomyces (Gordon and Phillips, 1989), it is possible that some Piromyces species were not able to compete on the remaining available substrates.
Previously, we demonstrated that a proper establishment of anaerobic fungi in the rumen of dairy calves took place only after weaning and reached the mature-stable state at approximately 10 months age (Huuki et al., 2022a,b). When young animals’ own rumen was evaluated as a seeding source, we demonstrated that from month 4 onwards, both rumen and feces shared the same fungal taxonomical groups; only different fungal species showed predominance in feces as compared with the rumen. For example, Neocallimastix 1 was the most abundant clade after month 4 in rumen samples (Huuki et al., 2022b), while BlackRhino (Piromyces) group and Caecomyces 1 group were among the most abundant taxa in fecal samples. Recently, Meili et al. (2023) have shown evidence of the phylogenetic co-evolution of anaerobic fungal taxa, where Piromyces show preference for hindgut fermenters, while more recently evolved Neocallimastix may have co-evolved with the rumen. Caecomyces are found in both foregut and hindgut fermenters (Meili et al., 2023) and have been found to be more abundant in fecal samples of cows in comparison to the rumen (Tapio et al., 2016).
The anaerobic fungi transfer in the developing rumen might be also facilitated by the shared environment, and to a large extent, the fungal community is shared by weaned young and adult animals within the barn, while the inter-animal differences in abundances of different taxa are caused by host effects (Huuki et al., 2022a, 2022b). Indeed, anaerobic fungi are found in the mouth (Lowe et al., 1987; Tapio et al., 2016), and due to resistant structures, they can be viable months after defecation (McGranaghan et al., 1999) and can therefore be easily transmitted through contact and cross-contamination of bedding and feed with feces. In this study, when the heifers were moved into a different facility after month 4, we observed an increase in the rumen and fecal fungal diversity. The changes in the community composition were likely affected by dietary and host-related factors, such as functional development and the growth of the rumen and intestine. Nevertheless, the exposure to the new fungal taxa in the new barn environment may have contributed to the increase in taxonomical diversity. Therefore, we hypothesize that the rumen is the direct seeding source for fungi in feces, and the colonization of both the rumen and feces takes place around the same time, and that the fungi are being continuously seeded from the environment to the rumen, and when entering the hindgut, the hindgut conditions, diet, and interaction with other microbes finally determine the community structure.
During the pre-weaning period, some of the calves in both groups experienced diarrhea. The diarrhea can be caused by bovine rotavirus, coronavirus, norovirus, and bacteria such as Salmonella, Escherichia coli, Clostridium perfringens, or protozoa Cryptosporidium parvum, which cause dysbiosis of microbial community (Cho and Yoon, 2014). In the present study, diarrhea reduced the fecal bacterial diversity in calves, which is in line with previous observations (Gomez et al., 2022). Our results showed that the reduction of diversity in diarrhea cases was associated with the lower relative abundance of beneficial genera, e.g., Bifidobacterium and Faecalibacterium and with simultaneously increased potential pathogenic genera such as Escherichia-Shigella, Streptococcus, Clostridium sensu stricto 1, and Lactobacillus. Faecalibacterium and Bifidobacterium are butyrate-producing bacteria that, in humans, promote a balanced microbial community, reinforce the gut barrier, produce anti-inflammatory substances, competitively exclude pathogens, and modulate immune responses (O’Callaghan and Van Sinderen, 2016; Martín et al., 2023). The decrease in butyrate-producing taxa (Faecalibacterium and Bifidobacterium) and the increase in facultative anaerobic lactic acid-producing taxa (e.g., Escherichia-Shigella, Streptococcus) in diarrhea cases could be related to increased oxygen, leaking into the hindgut lumen, which inhibits the growth of oxygen-sensitive beneficial obligate anaerobes, and decreased pH and increased lactate production in the gut, which promotes the growth of facultative anaerobes such as Escherichia-Shigella and Streptococcus (Gomez et al., 2022). However, our results showed that the beneficial taxa associated with healthy samples disappeared from the feces of all calves before weaning, and the potential pathogenic taxa by month 6, suggesting that their negative impact and role in the gut health is restricted to early life period only.
We hypothesized that early-life rumen microbiota modulation could also modulate hindgut microbiota and be beneficial to gut health, as the donor inoculum promoted microbiota development in the rumen (Huuki et al., 2022a,b). However, despite effects to the rumen, the treatment neither reduced diarrhea cases nor showed differences between the groups in fecal bacterial community composition before and after diarrhea. Treatment did not have a marked impact on early-life fecal microbiota colonization either and did not serve as a major seeding source of fecal microbiota. In the present study, the inoculum originated from adult cows’ rumen which is well adapted to plant fiber fermentation, while some special characteristics (such as gastric acid and bile tolerance and/or alternate substrates for energy source) are needed for the microbes to pass the abomasum and survive in the lower intestine. Previous studies have noted that inoculation with rumen liquid (Bu et al., 2020) or fecal microbiome transplantation (FMT) (Kim et al., 2021; Rosa et al., 2021) may reduce diarrhea in young ruminants, and FMT has an effect on the fecal microbiota composition. The microbial communities in fecal transplant have already adapted to the hindgut environment, which may have contributed to the slightly better success in treating diarrhea with FMT treatment. Therefore, the target environment and the age of the animal should be taken into account when designing microbial inoculations to improve gut health, gut development, or production performance.
This study highlights the plasticity of the GI tract microbial colonization, where crosstalk between the colostrum, rumen, and hindgut allows the GI tract to adjust in time and space to changing dietary and developmental needs. We showed that the colostrum may be a source for some of the hindgut bacteria and archaea, but their role in the gut mostly is restricted to the pre-weaning period. In comparison to the rumen, the fecal bacterial community matures earlier, potentially due to a more stable environment. Although present in the feces of pre-weaning calves, archaea increased in quantity and diversity after weaning when animals shift to utilizing the solid feeds only. Anaerobic fungi in feces occurred after weaning, and the diversity increased until reaching adulthood. The fecal anaerobic fungi are seeded through the rumen, and the communities of the rumen and large intestine mature at the same time. Despite early-life rumen microbiota modulation having a positive effect on rumen microbiota development, no effect was observed either on the development of fecal microbiota or on the pre-weaning gut health. The future applications of gut microbiota modulation should be designed taking into account the intended trait, gut region, and the age of the animal, e.g., rumen and hindgut communities have adapted to different environments with different niches.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/, BioProject PRJNA713003.
The animal study was approved by Project Authorisation Board (Regional Administrative Agency for Southern Finland, Hämeenlinna, Finland; ESAVI/17310/2021). The study was approved by the National Ethics Committee (ESAVI/5687/04.10.07/2017, Hämeenlinna, Finland) The study was conducted in accordance with the local legislation and institutional requirements.
HH: Data curation, Formal analysis, Investigation, Methodology, Visualization, Writing – original draft. JV: Conceptualization, Writing – review & editing. AV: Supervision, Writing – review & editing. IT: Conceptualization, Funding acquisition, Investigation, Project administration, Resources, Supervision, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the Natural Resources Institute Finland (Luke) strategic project (41007–00132300) and the European Union’s Horizon 2020 Research and Innovation program MASTER project (under grant agreement no. 818368). Author HH was supported by the doctoral program of Sustainable Use of Renewable Resources, Department of Agricultural sciences, Faculty of Agriculture and Forestry, University of Helsinki, and personal grants “Suomi kasvaa ruoasta” (no. 20210072) funded by Oiva Kuusisto foundation, and August Johannes and Aino Tiura research foundation (no. 635).
The authors thank Tuula-Marjatta Hamama (Luke) for embryo work in creating twin calves, Luke research barn staff and Viking Genetics and Tuula Nyström for animal care and co-operation in sample collection. The Finnish Functional Genomics Centre supported by the University of Turku, Åbo Akademi University, and Biocenter Finland is acknowledged for sequencing. The authors wish to acknowledge CSC – IT Center for Science, Finland, for computational resources.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1353874/full#supplementary-material
SUPPLEMENTARY FIGURE S1 | The Shannon diversity (A) and the Simpsons diversity index (B) of fecal becteria communities in rumen liquid inoculated (T) and control animals (C) from week 1 to month 12 of age.
Supplementary FIGURE S2 | The relative abundances of bacterial phyla in fecal samples of rumen liquid inoculated (T) and control heifers (C) from week 1 to month 12 of age.
Supplementary FIGURE S3 | The Shannon diversity (A) and the Simpsons diversity index (B) of fecal archaea communities in rumen liquid inoculated (T) and control animals (C) from week 1 to month 12 of age. Statistical analysis was performed for month 4-12 samples only, due to low number of reads and missing observations in pre-weaning samples.
Supplementary FIGURE S4 | The Shannon diversity (A) and the Simpsons diversity index (B) of fecal anaerobic fungi in rumen liquid inoculated (T) and control animals (C) from month 4 to month 12 of age.
Supplementary FIGURE S5 | The average relative abundance of differently abundant ASVs between healthy samples (green) and samples with diarrhea symptoms (red) in samples from weeks 2 to 4.
Supplementary FIGURE S6 | The average contribution of own mothers’ colostrum, shared colostrum mix, donor inoculum and calves’ own rumen community at 2, 4, 6, and 8 weeks, and at 4 months age as a seeding source to fecal bacteria community in calves at the age from 1 week to 4 months.
Supplementary FIGURE S7 | The average contribution of donor inoculum and heifers’ own rumen community between ages from 4 to 9 months as a seeding source to the fecal fungi community of heifers the age from 1 week to 4 months.
Amin, N., and Seifert, J. (2021). Dynamic progression of the calf’s microbiome and its influence on host health. Comput. Struct. Biotechnol. J. 19, 989–1001. doi: 10.1016/j.csbj.2021.01.035
Bakken, J. S., Borody, T., Brandt, L. J., Brill, J. V., Demarco, D. C., Franzos, M. A., et al. (2011). Treating Clostridium difficile infection with fecal microbiota transplantation. Clin. Gastroenterol. Hepatol. 9, 1044–1049. doi: 10.1016/j.cgh.2011.08.014
Baldwin, R. L., McLeod, K. R., Klotz, J. L., and Heitmann, R. N. (2004). Rumen development, intestinal growth and hepatic metabolism in the pre- and postweaning ruminant. J. Dairy Sci. 87, E55–E65. doi: 10.3168/jds.S0022-0302(04)70061-2
Bird, S. H., Hegarty, R. S., and Woodgate, R. (2010). Modes of transmission of rumen protozoa between mature sheep. Anim. Prod. Sci. 50, 414–417. doi: 10.1071/AN09216
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., Al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Bonacker, L. G., Baudner, S., and Thauer, R. K. (1992). Differential expression of the two methyl-coenzyme M reductases in Methanobacterium thermoautotrophicum as determined immunochemically via isoenzyme-specific antisera. Eur. J. Biochem. 206, 87–92. doi: 10.1111/j.1432-1033.1992.tb16904.x
Bowden, G. H. W., and Hamilton, I. R. (1989). Competition between Streptococcus mutans and Lactobacillus casei in mixed continuous culture. Oral Microbiol. Immunol. 4, 57–64. doi: 10.1111/j.1399-302X.1989.tb00100.x
Braem, G., De Vliegher, S., Verbist, B., Piessens, V., Van Coillie, E., De Vuyst, L., et al. (2013). Unraveling the microbiota of teat apices of clinically healthy lactating dairy cows, with special emphasis on coagulase-negative staphylococci. J. Dairy Sci. 96, 1499–1510. doi: 10.3168/jds.2012-5493
Bu, D., Zhang, X., Ma, L., Park, T., Wang, L., Wang, M., et al. (2020). Repeated inoculation of Young calves with rumen microbiota does not significantly modulate the rumen prokaryotic microbiota consistently but decreases diarrhea. Front. Microbiol. 11:1403. doi: 10.3389/fmicb.2020.01403
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., and Holmes, S. P. (2016). DADA2: high-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Caporaso, J. G., Lauber, C. L., Walters, W. A., Berg-lyons, D., Lozupone, C. A., Turnbaugh, P. J., et al. (2011). Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. PNAS 108, 4516–4522. doi: 10.1073/pnas.1000080107
Cho, Y., and Yoon, K.-J. (2014). An overview of calf diarrhea - infectious etiology, diagnosis, and intervention. J. Vet. Sci. 15, 1–17. doi: 10.4142/jvs.2014.15.1.1
Chong, S. C., and Boone, D. R. (2015). “Methanocorpusculum” in Bergey’s manual of systematics of Archaea and Bacteria. ed. W. B. Whitman (Hoboken, NJ: John Wiley & Sons, Ltd.), 1–4.
Constable, P. D., Ahmed, A. F., and Misk, N. A. (2005). Effect of suckling Cow’s Milk or Milk replacer on Abomasal luminal pH in dairy calves. J. Vet. Intern. Med. 19, 97–102. doi: 10.1111/j.1939-1676.2005.tb02665.x
Constable, P. D., Wittek, T., Ahmed, A. F., Marshall, T., Sen, I., and Nouri, M. (2006). Abomasal pH and emptying rate in the calf and dairy cow and the effect of commonly administered therapeutic agents. in Proceedings of XXVth World Buiatrics Congress, October 15th 2006, Nice, France (Nice: World Association for Buiatrics). Available at: https://www.researchgate.net/publication/228631194
Devriese, L. A., Laurier, L., Herdt, P. D., and Haesebrouck, F. (1992). Enterococcal and streptococcal species isolated from faeces of calves, young cattle and dairy cows. J. Appl. Bacteriol. 72, 29–31. doi: 10.1111/j.1365-2672.1992.tb04877.x
Dias, J., Marcondes, M. I., Motta de Souza, S., Cardoso da Mata e Silva, B., Fontes Noronha, M., Tassinari Resende, R., et al. (2018). Bacterial community dynamics across the gastrointestinal tracts of dairy calves during preweaning development. Appl. Environ. Microbiol. 84:e02675-17. doi: 10.1128/AEM.02675-17
Dill-McFarland, K. A., Breaker, J. D., and Suen, G. (2017). Microbial succession in the gastrointestinal tract of dairy cows from 2 weeks to first lactation. Sci. Rep. 7:40864. doi: 10.1038/srep40864
Dill-McFarland, K. A., Weimer, P. J., Breaker, J. D., and Suen, G. (2019). Diet influences early microbiota development in dairy calves without long-term impacts on Milk production. Appl. Environ. Microbiol. 85, 1–12. doi: 10.1128/AEM.02141-18
Edwards, J. E., Kingston-Smith, A. H., Jimenez, H. R., Huws, S. A., SkØt, K. P., Griffith, G. W., et al. (2008). Dynamics of initial colonization of nonconserved perennial ryegrass by anaerobic fungi in the bovine rumen. FEMS Microbiol. Ecol. 66, 537–545. doi: 10.1111/j.1574-6941.2008.00563.x
Fliegerova, K. O., Podmirseg, S. M., Vinzelj, J., Grilli, D. J., Kvasnov, S., Schierov, D., et al. (2021). The effect of a high-grain diet on the rumen microbiome of goats with a special focus on anaerobic Fungi. Microorganisms 9:157. doi: 10.3390/microorganisms9010157
Furman, O., Shenhav, L., Sasson, G., Kokou, F., Honig, H., Jacoby, S., et al. (2020). Stochasticity constrained by deterministic effects of diet and age drive rumen microbiome assembly dynamics. Nat. Commun. 11:1904. doi: 10.1038/s41467-020-15652-8
Gilbert, M. S., Pantophlet, A. J., Berends, H., Pluschke, A. M., Van den Borne, J. J. G. C., Hendriks, W. H., et al. (2015). Fermentation in the small intestine contributes substantially to intestinal starch disappearance in calves. J. Nutr. 145, 1147–1155. doi: 10.3945/jn.114.208595
Gomez, D. E., Li, L., Goetz, H., Macnicol, J., Gamsjaeger, L., and Renaud, D. L. (2022). Calf diarrhea is associated with a shift from obligated to facultative anaerobes and expansion of lactate-producing Bacteria. Front. Vet. Sci. 9:846383. doi: 10.3389/fvets.2022.846383
Gordon, G. L., and Phillips, M. W. (1989). Degradation and utilization of cellulose and straw by three different anaerobic fungi from the ovine rumen. Appl. Environ. Microbiol. 55, 1703–1710. doi: 10.1128/aem.55.7.1703-1710.1989
Gressley, T. F., Hall, M. B., and Armentano, L. E. (2011). Ruminant nutrition symposium: productivity, digestion, and health responses to hindgut acidosis in ruminants. J. Anim. Sci. 89, 1120–1130. doi: 10.2527/jas.2010-3460
Guilloteau, P., Zabielski, R., and Blum, J. W. (2009). Gastrointestinal tract and digestion in the young ruminant: ontogenesis, adaptations, consequences and manipulations. J. Physiol. Pharmacol. 60 Suppl 3, 37–46. Available at: https://www.jpp.krakow.pl/journal/archive/10_09_s3/pdf/37_10_09_s3_article.pdf.
Guzman, C. E., Bereza-Malcolm, L. T., De Groef, B., and Franks, A. E. (2015). Presence of selected methanogens, fibrolytic bacteria, and proteobacteria in the gastrointestinal tract of neonatal dairy calves from birth to 72 hours. PLoS One 10:e0133048. doi: 10.1371/journal.pone.0133048
Hammes, W. P., and Hertel, C. (2015). “Lactobacillus” in Bergey’s manual of systematics of Archaea and Bacteria. Ed. W. B. Whitman (Hoboken, NJ: John Wiley & Sons, Ltd.), 1–76.
Herve, M. (2020). Package “RVAideMemoire” Version 0.9–75. Available at: https://CRAN.R-project.org/package=RVAideMemoire
Holman, D. B., and Gzyl, K. E. (2019). A meta-analysis of the bovine gastrointestinal tract microbiota. FEMS Microbiol. Ecol. 95:fiz072. doi: 10.1093/femsec/fiz072
Hooper, L. V., Littman, D. R., and Macpherson, A. J. (2012). Interactions between the microbiota and the immune system. Science 336, 1268–1273. doi: 10.1126/science.1223490
Hoover, W. H. (1978). Digestion and absorption in the hindgut of ruminants. J. Anim. Sci. 46, 1789–1799. doi: 10.2527/jas1978.4661789x
Hoque, M. N., Istiaq, A., Clement, R. A., Sultana, M., Crandall, K. A., Siddiki, A. Z., et al. (2019). Metagenomic deep sequencing reveals association of microbiome signature with functional biases in bovine mastitis. Sci. Rep. 9:13536. doi: 10.1038/s41598-019-49468-4
Huuki, H., Ahvenjärvi, S., Lidauer, P., Popova, M., Vilkki, J., Vanhatalo, A., et al. (2022a). Fresh rumen liquid inoculant enhances the rumen microbial community establishment in pre-weaned dairy calves. Front. Microbiol. 12:758395. doi: 10.3389/fmicb.2021.758395
Huuki, H., Tapio, M., Mäntysaari, P., Negussie, E., Ahvenjärvi, S., Vilkki, J., et al. (2022b). Long-term effects of early-life rumen microbiota modulation on dairy cow production performance and methane emissions. Front. Microbiol. 13:983823. doi: 10.3389/fmicb.2022.983823
Jiang, Q., Lin, L., Xie, F., Jin, W., Zhu, W., Wang, M., et al. (2022). Metagenomic insights into the microbe-mediated B and K2 vitamin biosynthesis in the gastrointestinal microbiome of ruminants. Microbiome 10:109. doi: 10.1186/s40168-022-01298-9
Jones, A. L., Clayborn, J., Pribil, E., Foote, A. P., Montogomery, D., Elshahed, M. S., et al. (2023). Temporal progression of anaerobic fungal communities in dairy calves from birth to maturity. Environ. Microbiol. 25, 2088–2101. doi: 10.1111/1462-2920.16443
Kim, H. S., Whon, T. W., Sung, H., Jeong, Y. S., Jung, E. S., Shin, N. R., et al. (2021). Longitudinal evaluation of fecal microbiota transplantation for ameliorating calf diarrhea and improving growth performance. Nat. Commun. 12, 161–116. doi: 10.1038/s41467-020-20389-5
Kittelmann, S., Seedorf, H., Walters, W. A., Clemente, J. C., Knight, R., Gordon, J. I., et al. (2013). Simultaneous amplicon sequencing to explore co-occurrence patterns of bacterial, archaeal and eukaryotic microorganisms in rumen microbial communities. PLoS One 8:e47879. doi: 10.1371/journal.pone.0047879
Klein-Jöbstl, D., Quijada, N. M., Dzieciol, M., Feldbacher, B., Wagner, M., Drillich, M., et al. (2019). Microbiota of newborn calves and their mothers reveals possible transfer routes for newborn calves’ gastrointestinal microbiota. PLoS One 14, 1–18. doi: 10.1371/journal.pone.0220554
Knights, D., Kuczynski, J., Charlson, E. S., Zaneveld, J., Mozer, M. C., Collman, R. G., et al. (2011). Bayesian community-wide culture-independent microbial source tracking. Nat. Methods 8, 761–763. doi: 10.1038/nmeth.1650
Koetschan, C., Kittelmann, S., Lu, J., Al-Halbouni, D., Jarvis, G. N., Müller, T., et al. (2014). Internal transcribed spacer 1 secondary structure analysis reveals a common Core throughout the anaerobic Fungi (Neocallimastigomycota). PLoS One 9:e91928. doi: 10.1371/journal.pone.0091928
Leahy, S. C., Kelly, W. J., Altermann, E., Ronimus, R. S., Yeoman, C. J., Pacheco, D. M., et al. (2010). The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS One 5:e8926. doi: 10.1371/journal.pone.0008926
Li, F., He, Z., and Guan, L. L. (2022). Metagenomic analysis provides bases on individualized shift of colon microbiome affected by delaying colostrum feeding in neonatal calves. Front. Microbiol. 13:1035331. doi: 10.3389/fmicb.2022.1035331
Lin, L., Lai, Z., Yang, H., Zhang, J., Qi, W., Xie, F., et al. (2023). Genome-centric investigation of bile acid metabolizing microbiota of dairy cows and associated diet-induced functional implications. ISME J. 17, 172–184. doi: 10.1038/s41396-022-01333-5
Liu, X., Mao, B., Gu, J., Wu, J., Cui, S., Wang, G., et al. (2021). Blautia —a new functional genus with potential probiotic properties? Gut Microbes 13, 1–21. doi: 10.1080/19490976.2021.1875796
Lopez, A. J., and Heinrichs, A. J. (2022). Invited review: the importance of colostrum in the newborn dairy calf. J. Dairy Sci. 105, 2733–2749. doi: 10.3168/jds.2020-20114
Lowe, S. E., Theodorou, M. K., and Trinci, A. P. J. (1987). Isolation of anaerobic fungi from saliva and faeces of sheep. J. Gen. Microbiol. 133, 1829–1834. doi: 10.1099/00221287-133-7-1829
Mäntysaari, P., Liinamo, A. E., and Mäntysaari, E. A. (2012). Energy efficiency and its relationship with milk, body, and intake traits and energy status among primiparous Nordic red dairy cattle. J. Dairy Sci. 95, 3200–3211. doi: 10.3168/jds.2011-4685
Marcobal, A., Barboza, M., Froehlich, J. W., Block, D. E., German, J. B., Lebrilla, C. B., et al. (2010). Consumption of human milk oligosaccharides by gut-related microbes. J. Agric. Food Chem. 58, 5334–5340. doi: 10.1021/jf9044205
Martín, R., Rios-Covian, D., Huillet, E., Auger, S., Khazaal, S., Bermúdez-Humarán, L. G., et al. (2023). Faecalibacterium: a bacterial genus with promising human health applications. FEMS Microbiol. Rev. 47:fuad039. doi: 10.1093/femsre/fuad039
McGranaghan, P., Davies, J. C., Griffith, G. W., Davies, D. R., and Theodorou, M. K. (1999). The survival of anaerobic fungi in cattle faeces. FEMS Microbiol. Ecol. 29, 293–300. doi: 10.1111/j.1574-6941.1999.tb00620.x
McMurdie, P. J., and Holmes, S. (2013). Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meale, S. J., Li, S., Azevedo, P., Derakhshani, H., Plaizier, J. C., Khafipour, E., et al. (2016). Development of ruminal and fecal microbiomes are affected by weaning but not weaning strategy in dairy calves. Front. Microbiol. 7, 1–16. doi: 10.3389/fmicb.2016.00582
Meili, C. H., Jones, A. L., Arreola, A. X., Habel, J., Pratt, C. J., Hanafy, R. A., et al. (2023). Patterns and determinants of the global herbivorous mycobiome. Nat. Commun. 14:3798. doi: 10.1038/s41467-023-39508-z
Miller, T. L. (2002). Description of Methanobrevibacter gottschalkii sp. nov., Methanobrevibacter thaueri sp. nov., Methanobrevibacter woesei sp. nov. and Methanobrevibacter wolinii sp. nov. Int. J. Syst. Evol. Microbiol. 52, 819–822. doi: 10.1099/ijs.0.02022-0
O’Callaghan, A., and Van Sinderen, D. (2016). Bifidobacteria and their role as members of the human gut microbiota. Front. Microbiol. 7:925. doi: 10.3389/fmicb.2016.00925
O’Hara, E., Neves, A. L. A., Song, Y., and Guan, L. L. (2020). The role of the gut microbiome in cattle production and health: driver or passenger? Ann. Rev. Anim. Biosci. 8, 199–220. doi: 10.1146/annurcv-animal-021419-083952
Oksanen, J., Simpson, G. L., Blanchet, F. G., Kindt, R. L., Legendre, P., Minchin, P. R., et al. (2022). Package “vegan” Community Ecology Package. Available at: https://github.com/vegandevs/vegan
Pitta, D., Indugu, N., Narayan, K., and Hennessy, M. (2022). Symposium review: understanding the role of the rumen microbiome in enteric methane mitigation and productivity in dairy cows. J. Dairy Sci. 105, 8569–8585. doi: 10.3168/jds.2021-21466
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Quigley, L., O’Sullivan, O., Stanton, C., Beresford, T. P., Ross, R. P., Fitzgerald, G. F., et al. (2013). The complex microbiota of raw milk. FEMS Microbiol. Rev. 37, 664–698. doi: 10.1111/1574-6976.12030
Rey, M., Enjalbert, F., Combes, S., Cauquil, L., Bouchez, O., and Monteils, V. (2014). Establishment of ruminal bacterial community in dairy calves from birth to weaning is sequential. J. Appl. Microbiol. 116, 245–257. doi: 10.1111/jam.12405
Rey, M., Enjalbert, F., and Monteils, V. (2012). Establishment of ruminal enzyme activities and fermentation capacity in dairy calves from birth through weaning. J. Dairy Sci. 95, 1500–1512. doi: 10.3168/jds.2011-4902
Rius, A. G., Kittelmann, S., Macdonald, K. A., Waghorn, G. C., Janssen, P. H., and Sikkema, E. (2012). Nitrogen metabolism and rumen microbial enumeration in lactating cows with divergent residual feed intake fed high-digestibility pasture. J. Dairy Sci. 95, 5024–5034. doi: 10.3168/jds.2012-5392
Rosa, F., Michelotti, T. C., St-Pierre, B., Trevisi, E., and Osorio, J. S. (2021). Early life fecal microbiota transplantation in neonatal dairy calves promotes growth performance and alleviates inflammation and oxidative stress during weaning. Animals 11:2704. doi: 10.3390/ani11092704
Seedorf, H., Kittelmann, S., Henderson, G., and Janssen, P. H. (2014). RIM-DB: a taxonomic framework for community structure analysis of methanogenic archaea fromthe rumen and other intestinal environments. PeerJ 2:e494. doi: 10.7717/peerj.494
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Shinozuka, Y., Kawai, K., Kurumisawa, T., Shimizu, Y., Imanishi, T., Ohno, A., et al. (2021). Examination of the microbiota of normal cow milk using miniontm nanopore sequencing. J. Vet. Med. Sci. 83, 1620–1627. doi: 10.1292/jvms.21-0353
Singh, R. P. (2019). Glycan utilisation system in Bacteroides and Bifidobacteria and their roles in gut stability and health. Appl. Microbiol. Biotechnol. 103, 7287–7315. doi: 10.1007/s00253-019-10012-z
Song, Y., Liu, C., and Finegold, S. M. (2015). ‘‘Bacteroides”, in: Bergey’s manual of systematics of Archaea and Bacteria. ed. W. B. Whitman (Hoboken, NJ: John Wiley & Sons, Ltd.) doi: 10.1002/9781118960608.gbm00238
Song, Y., Malmuthuge, N., Steele, M. A., and Guan, L. L. (2018). Shift of hindgut microbiota and microbial short chain fatty acids profiles in dairy calves from birth to pre-weaning. FEMS Microbiol. Ecol. 94:fix179. doi: 10.1093/femsec/fix179
Švec, P., and Devriese, L. A. (2015). “Enterococcus,” in Bergey’s manual of systematics of Archaea and Bacteria, ed. W. B. Whitman. Hoboken, NJ: John Wiley & Sons, Inc.
Tapio, I., Shingfield, K. J., McKain, N., Bonin, A., Fischer, D., Baya, A. R., et al. (2016). Oral samples as non-invasive proxies for assessing the composition of the rumen microbial community. PLoS One 11:e0151220. doi: 10.1371/journal.pone.0151220
Teunissen, M. J., De Kort, G. V. M., Op Den Camp, H. J. M., Huis In, T., and Veld, J. H. J. (1992). Production of cellulolytic and xylanolytic enzymes during growth of the anaerobic fungus Piromyces sp. on different substrates. J. Gen. Microbiol. 138, 1657–1664. doi: 10.1099/00221287-138-8-1657
Theodorou, M. K., Davies, D. R., Jordanz, M. G. C., Trinci, A. P. J., and Orpin, C. G. (1993). Comparison of anaerobic fungi in faeces and rumen digesta of newly born and adult ruminants. Mycol. Res. 97, 1245–1252. doi: 10.1016/S0953-7562(09)81293-8
Welch, C. B., Lourenco, J. M., Krause, T. R., Seidel, D. S., Fluharty, F. L., Pringle, T. D., et al. (2021). Evaluation of the fecal bacterial communities of Angus steers with divergent feed efficiencies across the lifespan from weaning to slaughter. Front. Vet. Sci. 8:597405. doi: 10.3389/fvets.2021.597405
Whiley, R. A., and Hardie, J. M. (2015). “Streptococcus” in: Bergey’s manual of systematics of Archaea and Bacteria. Ed. W. B. Whitman (Hoboken, NJ: John Wiley & Sons, Ltd.), 1–86.
Yeoman, C. J., Ishaq, S. L., Bichi, E., Olivo, S. K., Lowe, J., and Aldridge, B. M. (2018). Biogeographical differences in the influence of maternal microbial sources on the early successional development of the bovine neonatal gastrointestinal tract. Sci. Rep. 8:3197. doi: 10.1038/s41598-018-21440-8
Yu, Z., and Morrison, M. (2004). Improved extraction of PCR-quality community DNA from digesta and fecal samples. BioTechniques 36, 808–812. doi: 10.2144/04365ST04
Yu, S., Shi, W., Yang, B., Gao, G., Chen, H., Cao, L., et al. (2020). Effects of repeated oral inoculation of artificially fed lambs with lyophilized rumen fluid on growth performance, rumen fermentation, microbial population and organ development. Anim. Feed Sci. Technol. 264:114465. doi: 10.1016/j.anifeedsci.2020.114465
Zhang, B., Li, L. Q., Liu, F., and Wu, J. Y. (2022). Human milk oligosaccharides and infant gut microbiota: molecular structures, utilization strategies and immune function. Carbohydr. Polym. 276:118738. doi: 10.1016/j.carbpol.2021.118738
Keywords: fecal bacteria, archaea, anaerobic fungi, microbiome modulation, microbiome establishment, heifer, calf, colostrum
Citation: Huuki H, Vilkki J, Vanhatalo A and Tapio I (2024) Fecal microbiota colonization dynamics in dairy heifers associated with early-life rumen microbiota modulation and gut health. Front. Microbiol. 15:1353874. doi: 10.3389/fmicb.2024.1353874
Edited by:
Franck Carbonero, Washington State University Health Sciences Spokane, United StatesReviewed by:
Kristie Keeney, Lumen Bioscience, Inc., United StatesCopyright © 2024 Huuki, Vilkki, Vanhatalo and Tapio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ilma Tapio, aWxtYS50YXBpb0BsdWtlLmZp
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.