 Ziwei Chen*†
Ziwei Chen*† Liangzhe Zhang
Liangzhe Zhang- School of Electronic and Information Engineering, Beijing Jiaotong University, Beijing, China
Introduction: A growing body of research indicates that microorganisms play a crucial role in human health. Imbalances in microbial communities are closely linked to human diseases, and identifying potential relationships between microbes and diseases can help elucidate the pathogenesis of diseases. However, traditional methods based on biological or clinical experiments are costly, so the use of computational models to predict potential microbe-disease associations is of great importance.
Methods: In this paper, we present a novel computational model called MLFLHMDA, which is based on a Multi-View Latent Feature Learning approach to predict Human potential Microbe-Disease Associations. Specifically, we compute Gaussian interaction profile kernel similarity between diseases and microbes based on the known microbe-disease associations from the Human Microbe-Disease Association Database and perform a preprocessing step on the resulting microbe-disease association matrix, namely, weighting K nearest known neighbors (WKNKN) to reduce the sparsity of the microbe-disease association matrix. To obtain unobserved associations in the microbe and disease views, we extract different latent features based on the geometrical structure of microbes and diseases, and project multi-modal latent features into a common subspace. Next, we introduce graph regularization to preserve the local manifold structure of Gaussian interaction profile kernel similarity and add
Results: The AUC values for global leave-one-out cross-validation and 5-fold cross validation implemented by MLFLHMDA are 0.9165 and 0.8942+/−0.0041, respectively, which perform better than other existing methods. In addition, case studies of different diseases have demonstrated the superiority of the predictive power of MLFLHMDA. The source code of our model and the data are available on https://github.com/LiangzheZhang/MLFLHMDA_master.
Introduction
The interactions between biological activities and complex, diverse and dynamically changing microbial communities are intricate (Sommer and Backhed, 2013). On one hand, the relationship between humans and the microbiome is mutualistic, for example, gut microbes can synthesize essential amino acids and vitamins required by the human body and also facilitate the digestion and absorption of less easily digestible foods (Huang et al., 2017). Furthermore, there is clinical and histological evidence suggesting that the topical application of lactic acid can effectively depigment, improve skin surface roughness, and reduce mild wrinkles caused by environmental photodamage (Huang et al., 2020). On the other hand, compelling evidence suggests that disruptions in the host microbial community can increase the incidence of various complex human diseases, such as diabetes (Wen et al., 2008), asthma (Noval Rivas et al., 2016), liver diseases (Henao-Mejia et al., 2013), and even cancers (Castellarin et al., 2012; Schwabe and Jobin, 2013). Some researchers have found that dysbiosis is associated with overgrowth of microbes such as S. aureus, which employs clumping factor B (ClfB), toxins, proteases, and superantigens to colonize the skin and induce damaging inflammatory responses (Edslev et al., 2020). In addition, other studies have shown that Clostridia responds to various physiological signals and secrete Large Clostridial Toxins (LCTs), which are considered the major virulence factors for various infections.
As mentioned above, identifying the potential relationship between microbes and diseases will be beneficial in elucidating the pathogenesis of diseases and providing new medical solutions for disease prevention, diagnosis, and treatment. However, traditional approaches often require a significant expenditure of cost and time to establish novel associations between microbes and diseases through biological or clinical experiments (Chen et al., 2019). With the advancement of computer technology, it has become imperative to predict potential microbe-disease associations by constructing computational models. The HMDAD database, established by Ma et al. (2017), through manual curation from large-scale public literature, is the first human microbe-disease association database. Based on this database, excellent computational models can be developed to prioritize potential microbes for large-scale research on disease associations. Researchers have successively proposed microbe-disease prediction models based on different theories, which could be broadly categorized into the following three types (Zhao et al., 2021): (1) score function-based models, (2) network algorithm-based models, and (3) machine learning-based models.
In the score function-based models, the probability of association between diseases and microbes is calculated using score functions based on various methods. Chen et al. (2017) built a microbe-disease association network and proposed a novel computational model of the KATZ measure for Human Microbe-Disease Association prediction (KATZHMDA). In this model, the prediction of latent associations was transformed into calculating the similarity between corresponding nodes based on the lengths and quantities of paths connecting two nodes in the network. Huang et al. (2017) developed a novel computational model based on a depth-first search algorithm for predicting microbes potentially associated with diseases (PBHMDA). The authors initially established a heterogeneous network by integrating known microbe-disease associations and Gaussian interaction profile kernel similarities between microbes and diseases. Then, a specialized depth-first search algorithm was employed to traverse all connected paths between nodes in the heterogeneous network, thereby obtaining prediction scores for each microbe-disease association pair. Long and Luo (2019) developed a novel computational model for predicting disease-associated microbes (WMGHMDA) based on weighted meta-graph. In the model, the authors defined the contribution value of the association probability for a given microbe-disease pair as the product of weights of all edges included in the meta-graph. Subsequently, the sum of contribution values from all meta-graphs for a given microorganism-disease pair was used as its final prediction score. The advantages of these models are that the theory of the algorithms and computational processes involved are relatively easy to understand, and the models do not require negative samples for prediction, which is extremely difficult to obtain.
The second type of method is network algorithm-based models, Bao et al. (2017) proposed a computational method named NCPHMDA to infer latent microbes for diseases by calculating consistency projection scores. The model measured the correlation between microbes and diseases by calculating the similarity of nodes in the heterogeneous network. Wu et al. (2018) introduced a novel model for optimizing random walks and restarts on the human microbe-disease association heterogeneous network (PRWHMAD). The heterogeneous network consisted of disease networks and microbe networks from different data sources, respectively. Finally, the authors used particle swarm optimization (PSO) to optimize the parameters of the random walk and obtain the final vector of association probabilities. Niu et al. (2019) proposed the Random Walk on Hypergraph for Microbe-Disease Association (RWHMDA) model to predict potential microbe-disease associations. Specifically, Niu and colleagues constructed a novel higher-order hypergraph model and extended the well-known random walk process to hypergraphs in a modified manner. Yan et al. (2020) introduced BRWMDA, a correlation prediction method based on the similarity between microbes and diseases. The approach utilized network integration and dual random walks on disease and microbe networks. The random walks ceased when the maximum number of iterations for both networks was reached, yielding the final correlation probability matrix. Wang Y. et al. (2022) presented the MSLINE model, which constructed a Microbe Disease Heterogeneous Network (MDHN) by integrating known associations and multiple similarities. Subsequently, a random walk algorithm was implemented on the MDHN to learn its structural information. Finally, the microbe-disease associations were scored based on the structural information of each node. The main advantage of these models is that they can fully utilize the topological information in the network. In addition, these models involve fewer parameters, which greatly reduces the difficulty of parameter selection.
The third kind of approach is based on machine learning. In recent years, machine learning has been applied in bioinformatics and computational biology, such as in miRNA-disease association prediction (Liu et al., 2022; Wang C.-C. et al., 2022), metabolite-disease association prediction (Sun et al., 2022; Gao et al., 2023), miRNA-lncRNA association prediction (Wang W. et al., 2022), and lncRNA-protein association prediction (Zhao et al., 2023). To some extent, these studies have contributed to the development of computational models for predicting microbe-disease associations. For example, Peng et al. (2018) proposed a novel model called ABHMDA, which revealed microbes associated with diseases through a strong classifier composed of weak classifiers with respective weights. ABHMDA assigned different weights to multiple weak classifiers and obtained the final association. Wang et al. (2017) introduced a LRLSHMDA calculation method based on machine learning, calculated the association probability of microbe-disease pairs based on the observed microbe-disease relationship network, and used it to prioritize all candidate microbes for the diseases studied, and achieved good results. Li et al. (2021) proposed a novel calculative method called BPNNHMDA, which utilized a neural network model with a unique activation function and optimized initial connection weights based on Gaussian interaction profile kernel similarity to predict potential microbe-disease associations.
While researchers continue to explore potential microbe-disease relationships, current computational models still have some limitations. Firstly, most scoring function-based models are not applicable to new diseases. Some network-based methods heavily rely on experimentally validated microbe-disease associations and are unable to predict new diseases or microbes in the absence of known association information. Secondly, the microbe-disease association network is often sparse, which hinders the prediction of microbe-disease associations (MDAs). Additionally, many methods struggle to effectively extract feature matrices from the microbe-disease association matrix, resulting in a diminished model generalization ability. Lastly, some methods focus solely on a single representation of the disease or microbe space, which can negatively impact the predictive performance of the model.
In this study, we introduce a novel approach using Multi-View Latent Feature Learning for Human Microbe-Disease Association prediction (MLFLHMDA) to reveal the associations between diseases and microbes. The approach takes a disease and microbe view to infer the microbes associated with the disease. Figure 1 shows the flowchart of the MLFLHMDA. Firstly, the method constructs the microbe-disease association matrix and Gaussian interaction profile (GIP) kernel similarity from known MDAs. Considering that the sparsity of MDAs is not conducive to ensemble learning, the weighted K nearest known neighbors (WKNKN) method is used to preprocess the microbe-disease association matrix. Secondly, we employ Principal Component Analysis (PCA) on GIP kernel similarity of microbes and diseases to extract potential features of microbes and diseases. We then project multi-modal latent features into a common subspace of the microbe and disease spaces and utilize an integrated latent feature learning approach to infer potentially disease-associated microbes. Furthermore, we enhance model interpretability and performance by incorporating graph regularization and implementing -norm constraints in the learning task. Finally, to optimize the learning problem, we use an alternate iteration algorithm and score and rank each microbe and disease pair. In the global leave-one-out cross-validation (LOOCV) and 5-fold cross validation, MLFLHMDA has a good performance with the area under the receiver operating characteristic curve (AUC) of 0.9165 and 0.8942+/−0.0041, respectively. In addition, case studies of four different diseases further demonstrate that the MLFLHMDA is a useful tool to effectively identify potential MDAs.
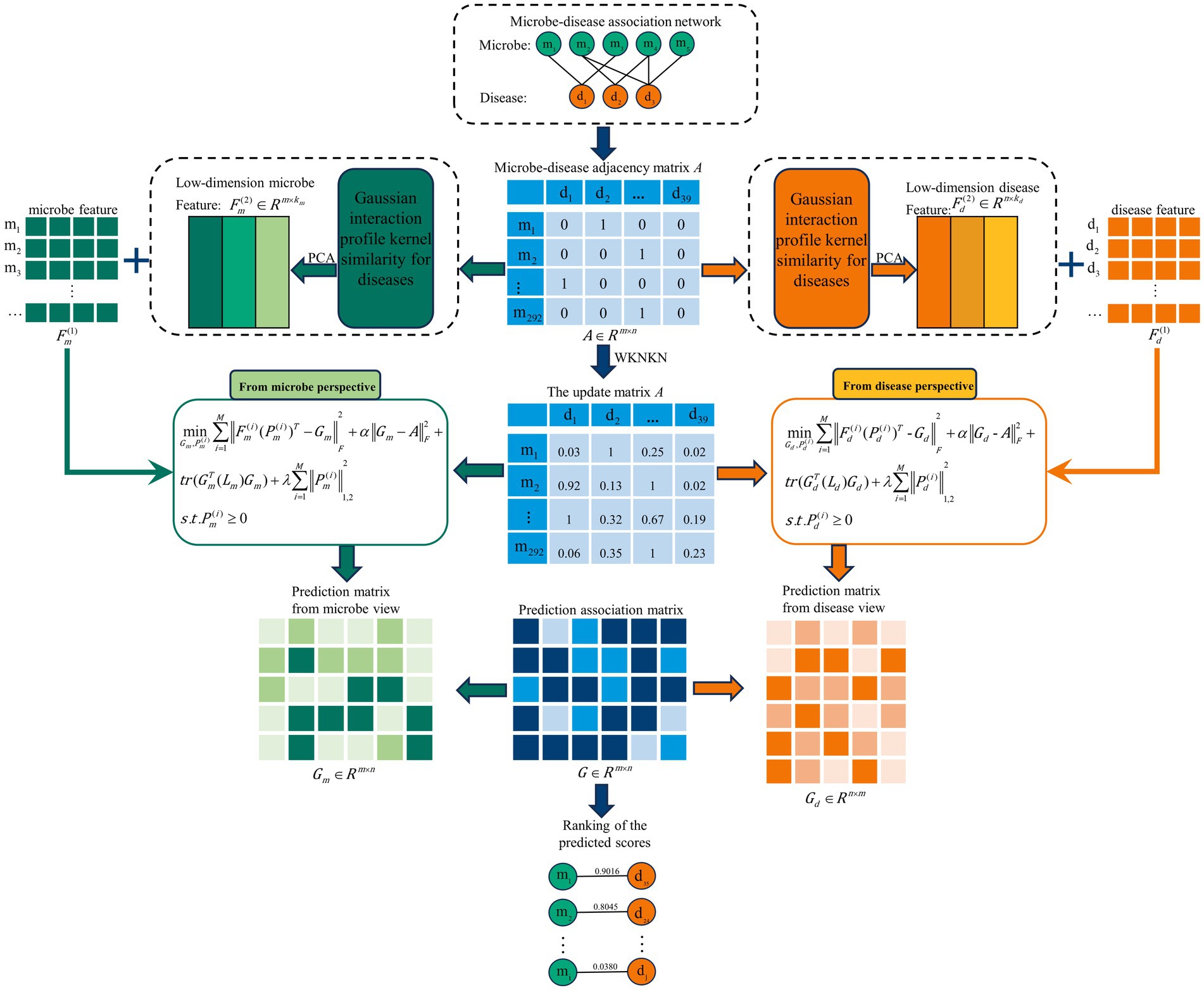
Figure 1. Flowchart of potential microbe-disease association prediction based on the computational model of MLFLHMDA.
The main contributions of this paper are as follows:
• Firstly, we provide a new approach to multi-view latent feature learning that infers disease-associated microbes from a microbe and disease view and combines similarity features, multi-modal latent features, and known association information.
• Secondly, we perform graph regularization on the similarity features to efficiently capture the graph structure information in the data space and to constrain the learning task.
• Finally, to improve the interpretability of the model and to mitigate the effect of noise inherent in the feature space of microbes and diseases, we impose -norms on the projection matrix to obtain the most representative sparse features.
Materials and methods
Human microbe-disease associations
We download the database from HMDAD1 (Ma et al., 2017), which contains 483 confirmed associations involving 292 microbes and 39 diseases. However, the dataset has some duplicate associations. After removing duplicate records, we ultimately obtain 450 unique associations. Next, we construct the adjacency matrix for the microbe-disease association network, with the variables and representing the quantities of microbes and diseases under study, respectively. is equal to 1 if there is a known association between microbe and disease , otherwise, the value is 0. In the adjacency matrix , each row binary vector corresponds to a microbe, and each column vector corresponds to a disease.
Gaussian interaction profile kernel similarity
We operate under the assumption that diseases with similar characteristics are likely to have associations with functionally similar microbes and that there are analogous patterns of interaction and non-interaction between diseases and microbes (Chen and Yan, 2013). To capture the microbe similarity, we construct the Gaussian interaction profile kernel similarity for microbes, denoted as . Let and represent the binary interaction profile vectors of microbes and , corresponding to the - and - rows in the adjacency matrix . The Gaussian interaction profile kernel similarity between microbe and is computed based on their interaction profiles as shown in Eq. (1).
here parameter is used to regulate the kernel bandwidth, and it introduces another parameter , to denote the average quantity of all microorganisms related to diseases. The calculation of is calculated as follows:
where the value of is set to 1 in Eq. (2). The definition of GIP kernel similarity for disease is similar to .
Network-based multi-modal feature extraction
Based on known microbe-disease associations, we construct the adjacency matrix and similarity networks to facilitate the extraction of multi-modal features for microbes and diseases. The adjacency matrix reflects the associations between each microbe. The GIP kernel similarity of microbes (or diseases) not only contains similar information but also valuable information for the construction of latent features. Therefore, we use it to extract an alternative set of latent features for both microbes and diseases. Furthermore, given that as the dimensionality increases, data sparsity becomes more pronounced, Principal Component Analysis (PCA) can effectively project the data into a lower-dimensional subspace to reduce dimensionality by finding the most dominant direction of variance in the data (Lu et al., 2018). Therefore, we perform additional latent extraction using PCA on and and utilize singular value decomposition (SVD) to decompose PCA. Since and are symmetric, they can be decomposed as , where is a unitary matrix and is a diagonal matrix with singular values arranged in descending order along the diagonal. Following the dominating energy strategy, we extract and as the dimensions, denoting them as and , which represent the low-dimensional feature vectors for microbe and disease . The dimensions and are defined as shown in Eqs. (3) and (4):
and
here and are set to 0.7 as suggested by Xiao et al. (2020).
MLFLHMDA
Inspired by this article Xiao et al. (2020), the main idea of MLFLHMDA is to integrate the views of microbes and diseases by using similarity features and multi-modal latent features in learning for disease-related microbe inference tasks. The main symbol descriptions are listed in Table 1.
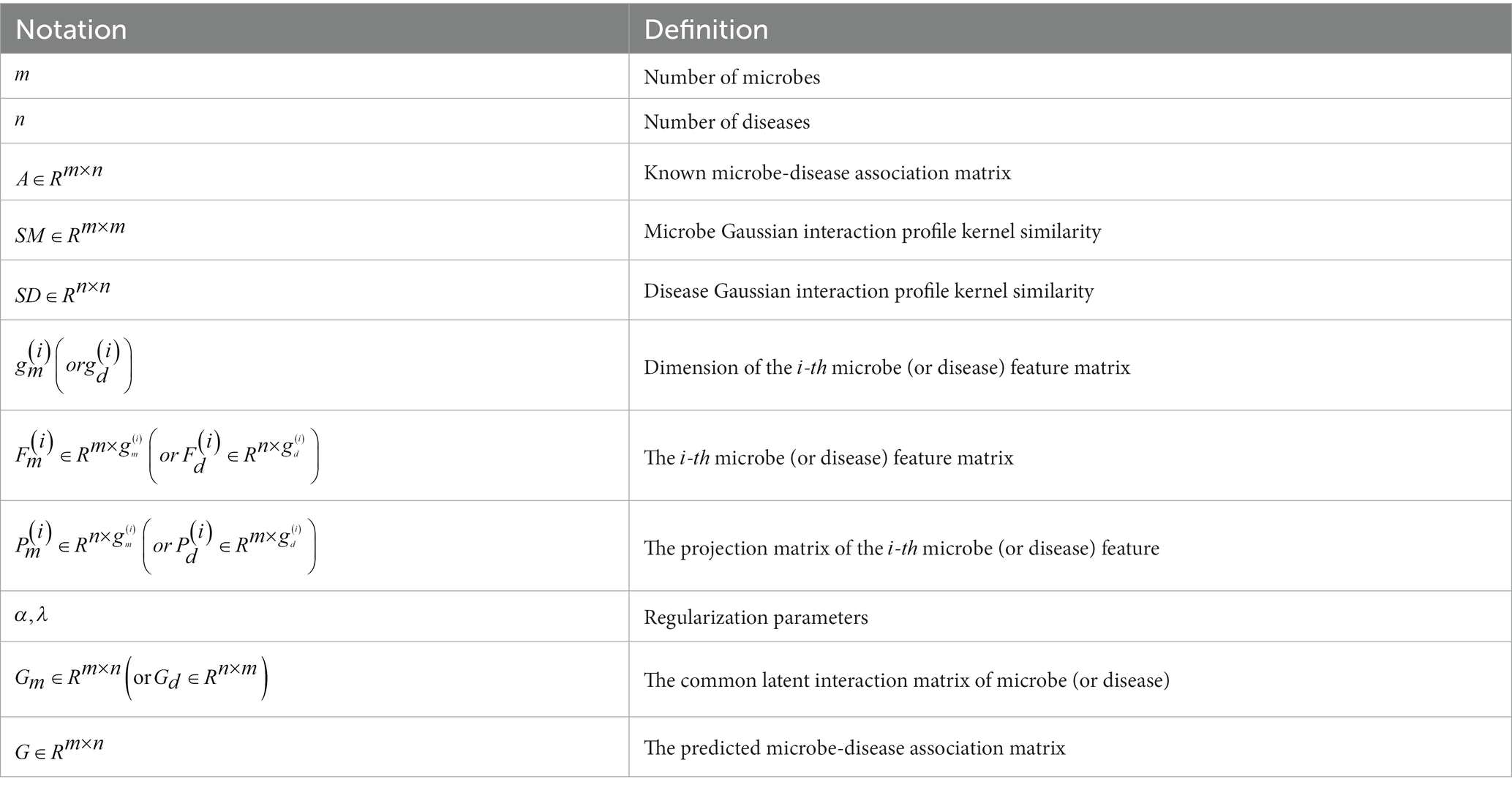
Table 1. Symbol description.
Firstly, we integrate the GIP similarity matrices for microbes and diseases . Subsequently, we employ Principal Component Analysis to extract low-dimensional latent feature matrices from these matrices. Secondly, the available information on microbe-disease associations is highly limited. The resulting adjacency matrix is sparse, and the values in the interaction profiles and for novel microbes or diseases are all zeros. Therefore, we employ a preprocessing step called Weighted K-Nearest Known Neighbors (WKNKN) to transform the microbe-disease associations matrix into values ranging from 0 to 1 (Wu et al., 2020). Based on the functional similarity between microbe and its K nearest known neighbors, we obtain the interaction profile for each microbe as follows:
where to are the K nearest known neighbors arranged in descending order based on their similarity to ; represents the weight coefficient, and . denotes a decay term, and is the normalization term. Similarly, the interaction profile for each disease is as follows:
where to are the K nearest known neighbors arranged in descending order based on their similarity to ; is the weight coefficient, and . is the normalization term. Following Xiao et al. (2018), K is set to 5 in the data space of microbes and diseases.
Then, in Eq. (7), we integrate and obtained separately from the two datasets mentioned above, replace with an associated likelihood score, then the original adjacency matrix can be updated by taking the average of the updated interaction likelihood profiles:
where is the weight coefficient and = = 1.
Mathematical formulation
In the microbe view, there are two types of feature matrices: represents the adjacency matrix and represents the low-dimensional feature matrix obtained after dimension reduction. To consider the different features of microbes, we use a linear transformation to project these two distinct feature matrices into a common latent interaction subspace. By using , where is the projection matrix of the - microbe feature matrix, = 1,2. Thus, the common latent interaction matrix of microbe view can be approximately by , which can be expressed as where represents the Frobenius norm.
Furthermore, the matrix is also designed to approximate the adjacency matrix Therefore, the following objective function can be represented mathematically as Eq. (9):
where 0 is a regularization parameter and =2.
It has been shown that the graph regularization can effectively utilize the geometric structure of data to ensure a part-based representation (Gao et al., 2020). Here, we perform the graph regularization on the microbe similarity matrix to capture graph structural information (Cai et al., 2010). The graph regularization for microbe is defined as shown in Eq. (10):
where is the graph Laplacian matrix of in Eq. (11), and is a diagonal matrix.
Research has indicated that the mixed-norm can mitigate the influence of inherent noise in data space and filter out sparse features with high correlation which can have the effect of improving the interpretability of the model (Zhang et al., 2020). The projection matrix represents the weights of microbe features. Therefore, imposing the -norm on the projection matrix served to reduce its sparsity. The definition of -norm is as shown in Eq. (12):
Finally, the objective function of MLFLHMDA is mathematically formulated as follows:
here is a regularization coefficient used to control the sparsity of . The first term of the equation is the projection of different feature matrices into a common latent interaction subspace. The second term ensures that the predicted matrix approximates the known associated matrix . The third term is a graph of Laplacian regulation. Finally, the two regularization coefficients and are set to and respectively as suggested by Xiao et al. (2020).
Optimization
To solve the optimization problem in Eq. (13), we employ an iterative parameter method, alternately updating and to obtain the optimal solution, thus obtaining the corresponding prediction matrix.
Fix , and solve for . The optimization problem is reduced to the following sub-problem for :
By differentiating Eq. (14) and setting it to zero, the update rule for can be derived as follows:
Fix , and solve for . Similarly, the optimization problem is reduced to the following sub-problem for :
To optimize Eq. (16), we introduce the Lagrange multiplier for the constraint of , and thus the sub-problem is formulated as shown in Eq. (17):
The partial derivative of the function
is given by Eq. (18):
Then, setting the derivative to zero and utilizing the KKT condition = 0, we have:
where is defined as the element in the - row and - column of matrix , denotes the Hadamard product in Eq. (19). is a vector and denotes the number of features in the feature matrix .
Considering that elements in may be negative, the modified iterative equation for is as follows:
where the matrices with negative and positive symbols are defined as and .
Finally, we can utilize Eq. (15) and Eq. (20) to perform alternating iterations on and until convergence.
The above optimization is conducted from a microbe view, while the model and optimization from a disease view are similar. Table 2 describes the MLFLHMDA method to predict new microbe-disease associations. The original information’s adjacency matrix is represented as , represents disease similarity profiles, and represents the disease feature matrix. Finally, the prediction matrices from the microbe and disease views are averaged and weighted to obtain the final matrix, denoted as matrix , the values of the entities in the matrix stand for the pairwise correlation scores between microbes and diseases.

Table 2. Description of algorithm MLFLHMDA.
Results
Performance evaluation
To evaluate the predictive performance of the MLFLHMDA, we conduct global LOOCV and 5-fold cross validation using the HMDAD database with validated associations. In global LOOCV, each known association sample is treated sequentially as a test sample, with the remaining associations used as training samples, while unvalidated microbe-disease associations are considered candidate samples. In each round, the test sample is transformed into an unvalidated status and used to test the model. The test sample is ranked based on its predicted score among all candidate samples, and it is considered a correct prediction only when the test sample’s rank exceeds a certain threshold. In the 5-fold cross validation, akin to global LOOCV, we randomly divide the observed microbe-disease associations into five groups. Each group serves as a test sample in turn, while the remaining four groups are used as training samples for model learning. Unvalidated associations are considered candidate samples. We perform 100 times random partitioning in the 5-fold cross validation to mitigate potential errors arising from the random allocation of sample regions. After configuring a range of thresholds, we plot the Receiver operating characteristic (ROC) curve with the true positive rate (TPR, sensitivity) on the horizontal axis and the false positive rate (FPR, 1-specificity) on the vertical axis. Ultimately, we compute the Area Under the ROC Curve (AUC) as a fundamental performance evaluation metric.
As shown in Figure 2, under global LOOCV, the AUC of MLFLHMDA is 0.9165, which is 1.25, 2.13, 2.96, 3.22, 5.21 and 1.19% higher than that of NTSHMDA (Luo and Long, 2020), BiRWHMDA (Zou et al., 2017), ABHMDA (Peng et al., 2018), LRLSHMDA (Wang et al., 2017), KATZHMDA (Chen et al., 2017), and PRWHMDA (Wu et al., 2018) respectively. Similarly, under 5-fold cross validation, the AUC of MLFLHMDA is 0.8942, which is 0.5, 1.71, 9.79, 1.93, 3.72, and 0.68% higher than that of NTSHMDA, BiRWHMDA, ABHMDA, LRLSHMDA, KATZHMDA and PRWHMDA, respectively.
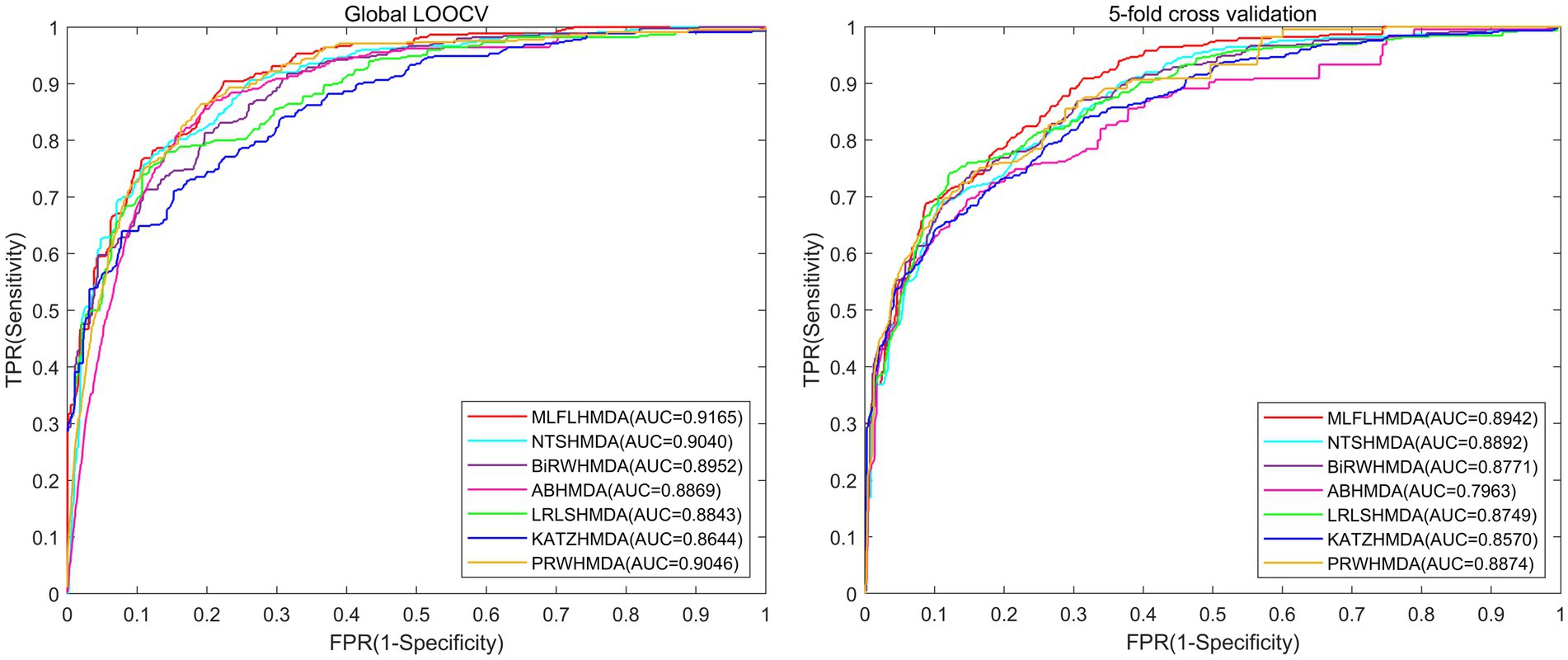
Figure 2. Performance comparison between MLFLHMDA and six classical microbe-disease association prediction models (NTSHMDA, BiRWHMDA, ABHMDA, LRLSHMDA, KATZHMDA, and PRWHMDA) in terms of ROC curves and AUCs based on global LOOCV and 5-fold cross validation. As a result, MLFLHMDA outperformed other models by achieving an AUC of 0.9165 in global LOOCV and an AUC of 0.8942 in 5-fold cross validation.
Ablation study
Ablation experiments are performed on the model under global leave-one-out cross-validation, and the effects of graph regularization and -norms are evaluated separately. Table 3 shows that both graph regularization and -norms improved the performance of the model. It is due to the ability of graph regularization to effectively capture the graph structure information of the data space and constrain the optimization iterations. The -norms also improve model performance by reducing noise in the data space and capturing the most characteristic features.

Table 3. Comparison of adding graph regularization and -norms in global leave-one-out cross-validation.
Case study
To further validate the predictive capabilities of the MLFLHMDA, we conduct two independent case studies involving significant human health conditions. In the first case study, we sort all unknown samples under the same disease and verify whether the association between the top 10 microbes and the disease under study is validated by the relevant literature. In the second case study, the aim is to assess the model’s capability to predict associations between unknown microbes and diseases in the absence of any known relevant microbe. Specifically, we reset all microbe associations for a particular disease in the adjacency matrix to zero. After model predictions, we verify the number of microbe samples within the top 10 rankings for diseases that are confirmed by relevant literature. In this context, we conduct the first case studies for asthma, colon cancer, and inflammatory bowel disease, and the second case study for Type 1 diabetes. The number of confirmed results from the literature for these four diseases is 10, 9, 9, and 8, respectively.
Asthma is a chronic disease that affects the airways of the lungs and is one of the most common respiratory disorders (Wu et al., 2019). According to statistics, asthma affects over 300 million people worldwide, and it is estimated to increase to 400 million people by the year 2025 (Barcik et al., 2020). Although the exact mechanisms underlying asthma remain unclear, the disease is associated with a variety of genetic, environmental, infectious, and nutritional factors. For instance, probiotics like Lactobacillus (First in the prediction list) can effectively treat allergic diseases or gastrointestinal inflammation. Research indicates that asthma patients have a disproportionately high proportion of Proteobacteria compared to non-asthmatic individuals, including certain pathogenic bacteria that may cause acute respiratory illnesses, such as Burkholderia species (Second in the prediction list) and Pseudomonas (Third in the prediction list) (Huang et al., 2011; Beigelman et al., 2014). In this study, there are 9 out of the top 10 microbes predicted by MLFLHMDA have been experimentally validated to be associated with asthma, as shown in Table 4.
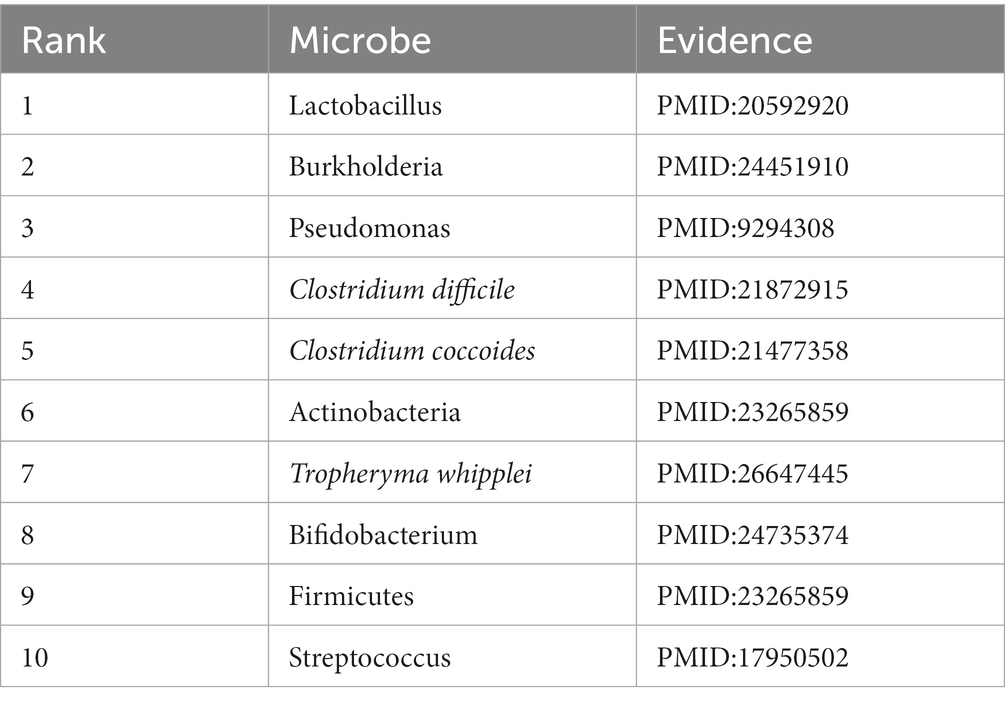
Table 4. Prediction results for the top 10 asthma-associated microbes.
Colorectal cancer (CRC) ranks as one of the most prevalent cancer types, occupying the third position in terms of the incidence of malignant tumors. Furthermore, it stood as the second leading cause of cancer-related mortality in the year 2020 (Ou et al., 2023). Studies have indicated a close association between gut microbe and the onset of colon cancer (Marmol et al., 2017). For example, Proteobacteria (First in the prediction list) is a significant bacterial taxonomic unit, and in colorectal carcinoma tissues, there is a higher abundance of Corynebacterium (Zhu et al., 2014). In addition, a strong association of spontaneous C. septicum (Third in the prediction list) infection with hematological and colorectal malignancies has been reported (Larson et al., 1995; Ramkissoon et al., 2000; Kennedy et al., 2005; Powell et al., 2008). In this study, there are 9 out of the top 10 microbes predicted by the model have been experimentally validated to be associated with colon cancer, as shown in Table 5.
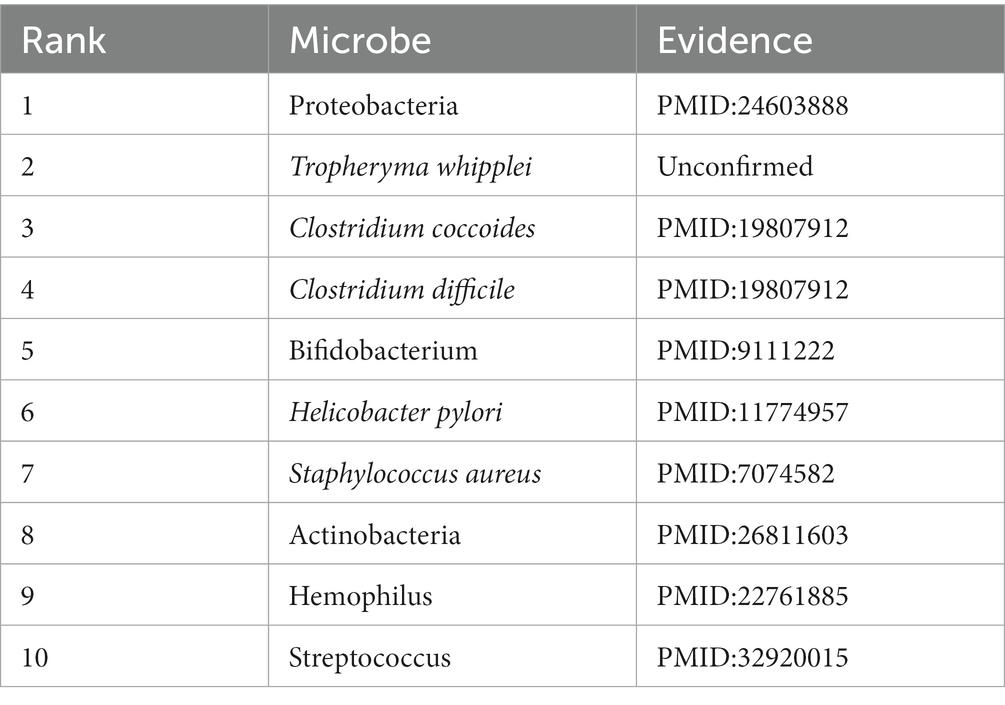
Table 5. Prediction results for the top 10 colorectal cancer-associated microbes.
Inflammatory Bowel Disease (IBD) is a group of chronic inflammatory gastrointestinal disorders, including Crohn’s disease and ulcerative colitis (Baumgart and Carding, 2007). While the exact causes of IBD are not fully understood, it is generally believed to involve genetic, environmental, and immune factors. There is already evidence suggesting a close connection between the gut microbe and IBD. For example, in IBD, especially in different variants, a reduction in Bacteroidetes (First in the prediction list) and Firmicutes (Eighth in the prediction list) occurs (Walters et al., 2014). Furthermore, studies propose that in IBD patients, especially during active phases, bacteria from the Clostridium coccoides group (Second in the prediction list), such as Faecalibacterium prausnitzii, as well as Firmicutes and Bifidobacteria, are less abundant in the gut microbe, this may be associated with disease onset and the protection of the intestinal mucosa (Sokol et al., 2009). In this study, there are 9 out of the top 10 microbes predicted by the model have been experimentally validated to be associated with Inflammatory Bowel Disease, as shown in Table 6.
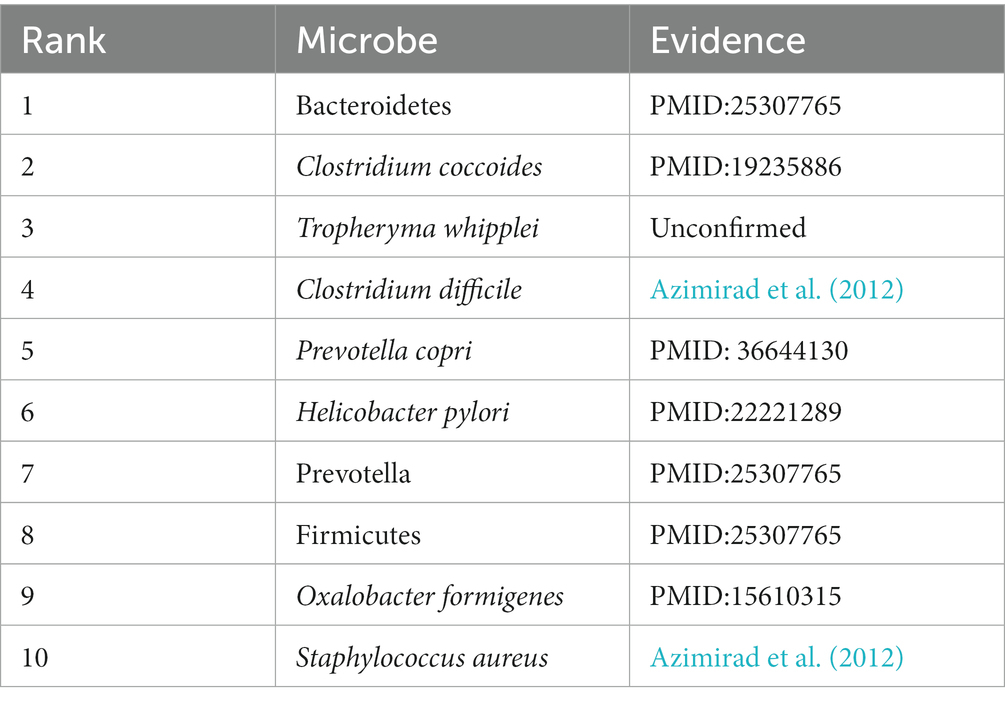
Table 6. Prediction results for the top 10 inflammatory bowel disease-associated microbes.
Type 1 Diabetes mellitus (T1DM) is an autoimmune-mediated chronic disease, accounting for 5–10% of diabetes cases. It is characterized by the destruction of the pancreatic beta cells that produce insulin, driven by the autoimmune system (Liu et al., 2023). Studies have suggested that the gut microbe may play a role in regulating glucose levels, potentially impacting energy balance and nutrient absorption (Liu et al., 2023). Some researchers find that, in T1D patients, the analysis of next-generation sequencing (NGS) of the microbiota reveals an increased abundance of Prevotella copri (Third in the prediction list) at the time of disease onset (Traversi et al., 2022). In our second case study on Type 1 diabetes, we assess the predictive capability of MLFLHMDA for potential microbe-related diseases. The results reveal that among the top 10 predicted microbiota associated with the disease, eight show varying degrees of validation. Six of these associations are confirmed using the HMDAD database. The specific ranking is shown in Table 7.
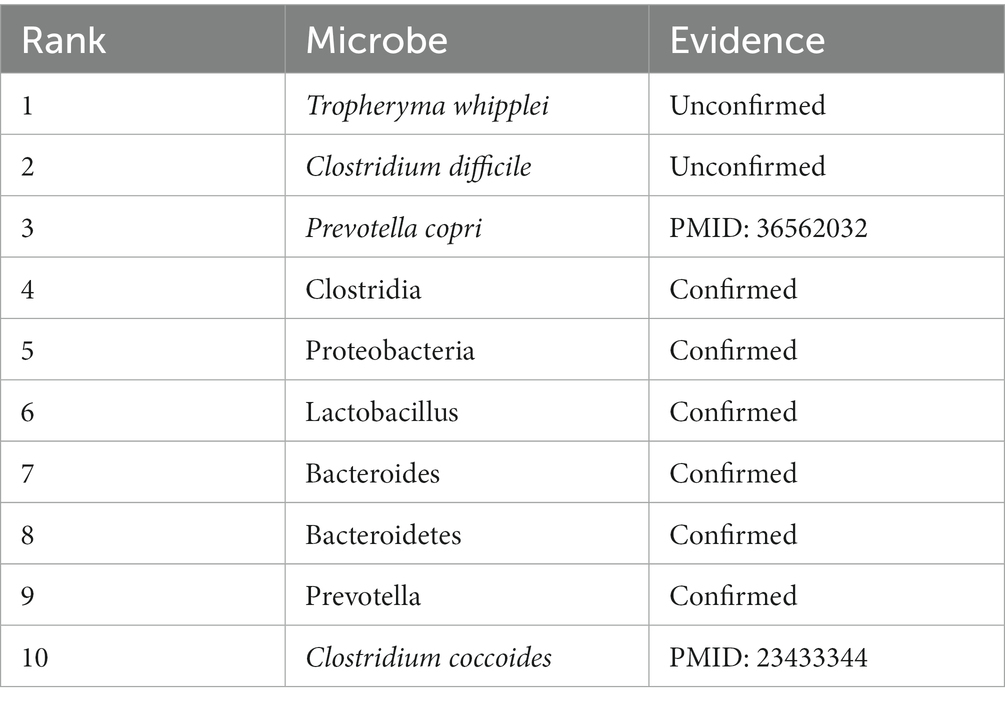
Table 7. Prediction results for the top 10 type 1 diabetes mellitus-associated microbes.
The case studies on the four complex human diseases have confirmed the outstanding predictive capabilities of MLFLHMDA. To facilitate further research and validation, we provide the probability rankings for all unconfirmed disease-microbe associations (See Supplementary Table 1). It is expected that the highly ranked candidate microbe-disease pairs will offer valuable leads and will be experimentally verified shortly.
Conclusion
Microbes play a significant role in human diseases and physiological processes. They are numerous, diverse, and interconnected within ecosystems. Exploring the potential associations between microbes and diseases is beneficial for maintaining human health and understanding disease mechanisms.
In this study, we develop a prediction model called MLFLHMDA based on multi-view latent feature learning. This method infers microbes associated with diseases from both microbe and disease views. Since the lack of known microbe-disease associations leads to sparsity in the microbe-disease association matrix, we preprocess the association matrix using WKNKN to add more interaction information. Additionally, we construct Gaussian interaction profile kernel similarity between microbes and diseases and use PCA to extract potential features in GIP kernel similarity. To obtain more potential information, multi-modal potential features are projected into the common subspace. We impose graph regularization and -norms in the integrated latent feature learning to enhance the model’s generalization performance, and score and rank each microbe and disease pair in the alternating iteration algorithm. In global LOOCV and 5-fold cross validation, the AUC of MLFLHMDA is 0.9165 and 0.8942, respectively, which outperforms the other six methods. What’s more, case studies of four diseases further validate the good predictive performance of the model.
However, MLFLHMDA still has certain limitations that need to be addressed in future work. The model solely relies on the HMDAD dataset, and collecting more experimentally validated disease-microbe relationships could improve the predictive ability of MLFLHMDA. In future work, we intend to integrate more association information as well as similarity information, and combine the advantages of existing models to construct a more superior predictive model. We believe that this method could guide medical experiments to get the potential associations and inspire in other bioinformatics fields such as circRNA-disease association prediction (Wang et al., 2021), MiRNA-disease association prediction (Chen et al., 2018), and so on. In addition, considering the high application value of genetic information, the introduction of the genetics information of the host in the future microbe-disease prediction field will inevitably benefit the association prediction. What is more, after more and more potential microbe-disease associations are predicted and confirmed, we could further predict the association between microbes and drugs based on the microbe-disease association information and other related data, which is favorable to provide new strategy design for drugs and human disease treatment (Han et al., 2023).
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
ZC: Conceptualization, Supervision, Writing – review & editing. LZ: Data curation, Formal analysis, Investigation, Methodology, Software, Validation, Writing – original draft. JL: Formal analysis, Software, Validation, Writing – original draft. MF: Data curation, Investigation, Validation, Writing – original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by funding from the Fundamental Research Funds for the Central Universities (2021JBM001).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1353278/full#supplementary-material
Footnotes
References
Azimirad, M., Bahreiny, R., and Hasani, Z. (2012). “Prevalence of superantigenic Staphylococcus aureus and toxigenic Clostridium difficile in patients with IBD,” in Conferences portal university of medical sciences.
Bao, W., Jiang, Z., and Huang, D. S. (2017). Novel human microbe-disease association prediction using network consistency projection. BMC Bioinform. 18:543. doi: 10.1186/s12859-017-1968-2
Barcik, W., Boutin, R. C. T., Sokolowska, M., and Finlay, B. B. (2020). The role of lung and gut microbiota in the pathology of asthma. Immunity 52, 241–255. doi: 10.1016/j.immuni.2020.01.007
Baumgart, D. C., and Carding, S. R. (2007). Inflammatory bowel disease: cause and immunobiology. Lancet 369, 1627–1640. doi: 10.1016/S0140-6736(07)60750-8
Beigelman, A., Weinstock, G. M., and Bacharier, L. B. (2014). The relationships between environmental bacterial exposure, airway bacterial colonization, and asthma. Curr. Opin. Allergy Clin. Immunol. 14, 137–142. doi: 10.1097/ACI.0000000000000036
Cai, D., He, X., Han, J., Huang, T., and Intelligence, M. (2010). Graph regularized nonnegative matrix factorization for data representation. IEEE Trans. Pattern Anal. Mach. Intell. 33, 1548–1560. doi: 10.1109/TPAMI.2010.231
Castellarin, M., Warren, R. L., Freeman, J. D., Dreolini, L., Krzywinski, M., Strauss, J., et al. (2012). Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22, 299–306. doi: 10.1101/gr.126516.111
Chen, X., Huang, Y. A., You, Z. H., Yan, G. Y., and Wang, X. S. (2017). A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 33, 733–739. doi: 10.1093/bioinformatics/btw715
Chen, X., Xie, D., Wang, L., Zhao, Q., You, Z.-H., and Liu, H. (2018). BNPMDA: Bipartite Network Projection for MiRNA-Disease Association prediction. Bioinformatics 34, 3178–3186. doi: 10.1093/bioinformatics/bty333
Chen, X., Xie, D., Zhao, Q., and You, Z.-H. (2019). MicroRNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 20, 515–539. doi: 10.1093/bib/bbx130
Chen, X., and Yan, G. Y. (2013). Novel human lncRNA-disease association inference based on lncRNA expression profiles. Bioinformatics 29, 2617–2624. doi: 10.1093/bioinformatics/btt426
Edslev, S. M., Agner, T., and Andersen, P. S. (2020). Skin microbiome in atopic dermatitis. Acta Derm. Venereol. 100:adv00164. doi: 10.2340/00015555-3514
Gao, H., Sun, J., Wang, Y., Lu, Y., Liu, L., Zhao, Q., et al. (2023). Predicting metabolite–disease associations based on auto-encoder and non-negative matrix factorization. Brief. Bioinform. 24:bbad259. doi: 10.1093/bib/bbad259
Gao, Z., Wang, Y. T., Wu, Q. W., Ni, J. C., and Zheng, C. H. (2020). Graph regularized L(2,1)-nonnegative matrix factorization for miRNA-disease association prediction. BMC Bioinformatics 21:61. doi: 10.1186/s12859-020-3409-x
Han, C.-D., Wang, C.-C., Huang, L., and Chen, X. (2023). MCFF-MTDDI: multi-channel feature fusion for multi-typed drug–drug interaction prediction. Brief. Bioinform. 24:bbad215. doi: 10.1093/bib/bbad215
Henao-Mejia, J., Elinav, E., Thaiss, C. A., Licona-Limon, P., and Flavell, R. A. (2013). Role of the intestinal microbiome in liver disease. J. Autoimmun. 46, 66–73. doi: 10.1016/j.jaut.2013.07.001
Huang, Z. A., Chen, X., Zhu, Z., Liu, H., Yan, G. Y., You, Z. H., et al. (2017). PBHMDA: path-based human microbe-disease association prediction. Front. Microbiol. 8:233. doi: 10.3389/fmicb.2017.00233
Huang, H. C., Lee, I. J., Huang, C., and Chang, T. M. (2020). Lactic acid bacteria and lactic acid for skin health and melanogenesis inhibition. Curr. Pharm. Biotechnol. 21, 566–577. doi: 10.2174/1389201021666200109104701
Huang, Y. J., Nelson, C. E., Brodie, E. L., Desantis, T. Z., Baek, M. S., Liu, J., et al. (2011). Airway microbiota and bronchial hyperresponsiveness in patients with suboptimally controlled asthma. J. Allergy Clin. Immunol. 127, 372–381.e1-3. doi: 10.1016/j.jaci.2010.10.048
Kennedy, C. L., Krejany, E. O., Young, L. F., O'connor, J. R., Awad, M. M., Boyd, R. L., et al. (2005). The alpha-toxin of Clostridium septicum is essential for virulence. Mol. Microbiol. 57, 1357–1366. doi: 10.1111/j.1365-2958.2005.04774.x
Larson, C. M., Bubrick, M. P., Jacobs, D. M., and West, M. A. (1995). Malignancy, mortality, and medicosurgical management of Clostridium septicum infection. Surgery 118, 592–597; discussion 597-8.
Li, H., Wang, Y., Zhang, Z., Tan, Y., Chen, Z., Wang, X., et al. (2021). Identifying microbe-disease association based on a novel Back-propagation neural network model. IEEE/ACM Trans. Comput. Biol. Bioinform. 18, 2502–2513. doi: 10.1109/TCBB.2020.2986459
Liu, W., Lin, H., Huang, L., Peng, L., Tang, T., Zhao, Q., et al. (2022). Identification of miRNA-disease associations via deep forest ensemble learning based on autoencoder. Brief. Bioinform. 23:bbac104. doi: 10.1093/bib/bbac104
Liu, J., Zhou, L., Sun, L., Ye, X., Ma, M., Dou, M., et al. (2023). Association between intestinal Prevotella copri abundance and glycemic fluctuation in patients with brittle diabetes. Diabetes Metab. Syndr. Obes. 16, 1613–1621. doi: 10.2147/DMSO.S412872
Long, Y., and Luo, J. (2019). WMGHMDA: a novel weighted meta-graph-based model for predicting human microbe-disease association on heterogeneous information network. BMC Bioinformatics 20:541. doi: 10.1186/s12859-019-3066-0
Lu, C., Yang, M., Luo, F., Wu, F. X., Li, M., Pan, Y., et al. (2018). Prediction of lncRNA-disease associations based on inductive matrix completion. Bioinformatics 34, 3357–3364. doi: 10.1093/bioinformatics/bty327
Luo, J., and Long, Y. (2020). NTSHMDA: prediction of human microbe-disease association based on random walk by integrating network topological similarity. IEEE/ACM Trans. Comput. Biol. Bioinform. 17, 1341–1351. doi: 10.1109/TCBB.2018.2883041
Ma, W., Zhang, L., Zeng, P., Huang, C., Li, J., Geng, B., et al. (2017). An analysis of human microbe-disease associations. Brief. Bioinform. 18, 85–97. doi: 10.1093/bib/bbw005
Marmol, I., Sanchez-De-Diego, C., Pradilla Dieste, A., Cerrada, E., and Rodriguez Yoldi, M. J. (2017). Colorectal carcinoma: a general overview and future perspectives in colorectal cancer. Int. J. Mol. Sci. 18:197. doi: 10.3390/ijms18010197
Niu, Y.-W., Qu, C.-Q., Wang, G.-H., and Yan, G.-Y. (2019). RWHMDA: random walk on hypergraph for microbe-disease association prediction. Front. Microbiol. 10:1578. doi: 10.3389/fmicb.2019.01578
Noval Rivas, M., Crother, T. R., and Arditi, M. (2016). The microbiome in asthma. Curr. Opin. Pediatr. 28, 764–771. doi: 10.1097/MOP.0000000000000419
Ou, S., Chen, H., Wang, H., Ye, J., Liu, H., Tao, Y., et al. (2023). Fusobacterium nucleatum upregulates MMP7 to promote metastasis-related characteristics of colorectal cancer cell via activating MAPK(JNK)-AP1 axis. J. Transl. Med. 21:704. doi: 10.1186/s12967-023-04527-3
Peng, L.-H., Yin, J., Zhou, L., Liu, M.-X., and Zhao, Y. (2018). Human microbe-disease association prediction based on adaptive boosting. Front. Microbiol. 9:2440. doi: 10.3389/fmicb.2018.02440
Powell, M. J., Sasapu, K. K., and Macklin, C. (2008). Metastatic gas gangrene and colonic perforation: a case report. World J. Emerg. Surg. 3:15. doi: 10.1186/1749-7922-3-15
Ramkissoon, Y., Ghoorahoo, H., Haydock, S. F., and O'shaughnessy, K. M. (2000). An unusual complication of carcinoma of the caecum. Postgrad. Med. J. 76, 451–452. doi: 10.1136/pmj.76.897.438
Schwabe, R. F., and Jobin, C. (2013). The microbiome and cancer. Nat. Rev. Cancer 13, 800–812. doi: 10.1038/nrc3610
Sokol, H., Seksik, P., Furet, J. P., Firmesse, O., Nion-Larmurier, I., Beaugerie, L., et al. (2009). Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 15, 1183–1189. doi: 10.1002/ibd.20903
Sommer, F., and Backhed, F. (2013). The gut microbiota--masters of host development and physiology. Nat. Rev. Microbiol. 11, 227–238. doi: 10.1038/nrmicro2974
Sun, F., Sun, J., and Zhao, Q. J. (2022). A deep learning method for predicting metabolite-disease associations via graph neural network. Brief. Bioinform. 23:bbac266. doi: 10.1093/bib/bbac266
Traversi, D., Scaioli, G., Rabbone, I., Carletto, G., Ferro, A., Franchitti, E., et al. (2022). Gut microbiota, behavior, and nutrition after type 1 diabetes diagnosis: a longitudinal study for supporting data in the metabolic control. Front. Nutr. 9:968068. doi: 10.3389/fnut.2022.968068
Walters, W. A., Xu, Z., and Knight, R. (2014). Meta-analyses of human gut microbes associated with obesity and IBD. FEBS Lett. 588, 4223–4233. doi: 10.1016/j.febslet.2014.09.039
Wang, C.-C., Han, C.-D., Zhao, Q., and Chen, X. (2021). Circular RNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 22:bbab286. doi: 10.1093/bib/bbab286
Wang, F., Huang, Z. A., Chen, X., Zhu, Z., Wen, Z., Zhao, J., et al. (2017). LRLSHMDA: laplacian regularized least squares for human microbe-disease association prediction. Sci. Rep. 7:7601. doi: 10.1038/s41598-017-08127-2
Wang, Y., Lei, X., Lu, C., and Pan, Y. (2022). Predicting microbe-disease association based on multiple similarities and LINE algorithm. IEEE/ACM Trans. Comput. Biol. Bioinform. 19, 2399–2408. doi: 10.1109/TCBB.2021.3082183
Wang, C.-C., Li, T.-H., Huang, L., and Chen, X. (2022). Prediction of potential miRNA–disease associations based on stacked autoencoder. Brief. Bioinform. 23:bbac021. doi: 10.1093/bib/bbac021
Wang, W., Zhang, L., Sun, J., Zhao, Q., and Shuai, J. (2022). Predicting the potential human lncRNA-miRNA interactions based on graph convolution network with conditional random field. Brief. Bioinform. 23:bbac463. doi: 10.1093/bib/bbac463
Wen, L., Ley, R. E., Volchkov, P. Y., Stranges, P. B., Avanesyan, L., Stonebraker, A. C., et al. (2008). Innate immunity and intestinal microbiota in the development of type 1 diabetes. Nature 455, 1109–1113. doi: 10.1038/nature07336
Wu, T. D., Brigham, E. P., and Mccormack, M. C. (2019). Asthma in the primary care setting. Med. Clin. North Am. 103, 435–452. doi: 10.1016/j.mcna.2018.12.004
Wu, C., Gao, R., Zhang, D., Han, S., and Zhang, Y. (2018). PRWHMDA: human microbe-disease association prediction by random walk on the heterogeneous network with PSO. Int. J. Biol. Sci. 14, 849–857. doi: 10.7150/ijbs.24539
Wu, T. R., Yin, M. M., Jiao, C. N., Gao, Y. L., Kong, X. Z., and Liu, J. X. (2020). MCCMF: collaborative matrix factorization based on matrix completion for predicting miRNA-disease associations. BMC Bioinformatics 21:454. doi: 10.1186/s12859-020-03799-6
Xiao, Q., Luo, J., Liang, C., Cai, J., and Ding, P. (2018). A graph regularized non-negative matrix factorization method for identifying microRNA-disease associations. Bioinformatics 34, 239–248. doi: 10.1093/bioinformatics/btx545
Xiao, Q., Zhang, N., Luo, J., Dai, J., and Tang, X. (2020). Adaptive multi-source multi-view latent feature learning for inferring potential disease-associated miRNAs. Brief. Bioinform. 22, 2043–2057. doi: 10.1093/bib/bbaa028
Yan, C., Duan, G., Wu, F. X., Pan, Y., and Wang, J. (2020). BRWMDA: predicting microbe-disease associations based on similarities and bi-random walk on disease and microbe networks. IEEE/ACM Trans. Comput. Biol. Bioinform. 17, 1595–1604. doi: 10.1109/TCBB.2019.2907626
Zhang, J., Jing, L., and Tan, J. J. K.-B. S. (2020). Cross-regression for multi-view feature extraction. Knowl. Based Syst. 200:105997. doi: 10.1016/j.knosys.2020.105997
Zhao, J., Sun, J., Shuai, S. C., Zhao, Q., and Shuai, J. J. (2023). Predicting potential interactions between lncRNAs and proteins via combined graph auto-encoder methods. Brief. Bioinform. 24:bbac527. doi: 10.1093/bib/bbac527
Zhao, Y., Wang, C. C., and Chen, X. (2021). Microbes and complex diseases: from experimental results to computational models. Brief. Bioinform. 22:bbaa158. doi: 10.1093/bib/bbaa158
Zhu, Q., Jin, Z., Wu, W., Gao, R., Guo, B., Gao, Z., et al. (2014). Analysis of the intestinal lumen microbiota in an animal model of colorectal cancer. PLoS One 9:e90849. doi: 10.1371/journal.pone.0090849
Keywords: microbe, disease, microbe-disease association, multi-view, latent feature learning
Citation: Chen Z, Zhang L, Li J and Fu M (2024) MLFLHMDA: predicting human microbe-disease association based on multi-view latent feature learning. Front. Microbiol. 15:1353278. doi: 10.3389/fmicb.2024.1353278
Edited by:
Qi Zhao, University of Science and Technology Liaoning, ChinaReviewed by:
Xing Chen, Jiangnan University, ChinaLihong Peng, Chinese PLA General Hospital, China
Wei Liu, Xiangtan University, China
Copyright © 2024 Chen, Zhang, Li and Fu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ziwei Chen, endjaGVuQGJqdHUuZWR1LmNu
†These authors share first authorship