 YangLin Cui
YangLin Cui YuMeng Guo1
YuMeng Guo1
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 27 March 2024
Sec. Microorganisms in Vertebrate Digestive Systems
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1348027
This article is part of the Research Topic The Gut-Liver Axis: the Main Role of Microbiome in Liver Diseases View all 17 articles
Background: Previous studies have suggested that the gut microbiota (GM) is closely associated with the development of autoimmune cholestatic liver disease (ACLD), but limitations, such as the presence of confounding factors, have resulted in a causal relationship between the gut microbiota and autoimmune cholestatic liver disease that remains uncertain. Thus, we used two-sample Mendelian randomization as a research method to explore the causal relationship between the two.
Methods: Pooled statistics of gut microbiota from a meta-analysis of genome-wide association studies conducted by the MiBioGen consortium were used as an instrumental variable for exposure factors. The Pooled statistics for primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC) were obtained from the R9 version of the FinnGen database (https://r9.finngen.fi/). Inverse-variance Weighted (IVW), cML-MA, MR-Egger regression, Weighted median (WME), Weighted mode (WM), and Simple mode (SM) were used to detect the association between intestinal flora and the causal relationship between intestinal flora and ACLD, in which IVW method was dominant, was assessed based on the effect indicator dominance ratio (odds ratio, OR) and 95% confidence interval (CI). Sensitivity analysis, heterogeneity test, gene pleiotropy test, MR pleiotropy residual sum and outlier test (MR-PRESSO) were combined to verify the stability and reliability of the results. Reverse Mendelian randomization analysis was performed on gut microbiota and found to be causally associated with ACLD.
Results: The IVW results showed that the relative abundance of the genus Clostridium innocuum group, genus Butyricicoccus, and genus Erysipelatoclostridium was negatively correlated with the risk of PBC, that is, increased abundance reduced the risk of PBC and was a protective, and the relative abundance of the genus Eubacterium hallii was positively correlated with the risk of PSC, which is a risk factor for PSC. Family Clostridiaceae1 and family Lachnospiraceae were negatively correlated with the risk of PSC, which is a protective factor for PSC.
Conclusion: This study found a causal relationship between gut microbiota and ACLD. This may provide valuable insights into gut microbiota-mediated pathogenesis of ACLD. It is necessary to conduct a large-sample randomized controlled trial (RCT) at a later stage to validate the associated role of the relevant gut microbiota in the risk of ACLD development and to explore the associated mechanisms.
Autoimmune cholestatic liver disease (ACLD) encompasses a cadre of immune-mediated, protracted cholestatic hepatic maladies, notably primary biliary cholangitis (PBC) and primary sclerosing cholangitis (PSC). The etiology of ACLD is the complicated interplay between genetic predispositions and environmental factors, precipitating the breakdown of immune tolerance toward biliary epithelial cells and subsequent immune activation (Liwinski et al., 2022; Richardson et al., 2022). Among these, PBC is characterized by progressive destruction of the small intrahepatic bile ducts as a pathological feature, whereas PSC manifests inflammation or fibrosis within both intrahepatic and extrahepatic conduits of medium and large calibers (Blesl and Stadlbauer, 2021). The pathophysiological mechanisms underlying ACLD remain unclear. In recent years, scholarly attention has pivoted toward the gut microbiota (GM) as a critical environmental determinant, with multiple investigations revealing a potential correlation between GM and ACLD. For instance, Furukawa et al. (2020) observed a decrease in the microbial diversity of patients with PBC, characterized by an elevated relative abundance of Lactobacilli and a diminished presence of symbiotically beneficial Clostridium compared to their normal counterparts. Moreover, evidence suggests that GM and its metabolites may be intricately linked to the progression of PBC toward hepatic fibrosis (Lammert et al., 2021). Analogously, investigations into PSC have revealed reduced GM diversity and altered abundance of specific flora. Fukui (2019), in an observational study, identified an upregulation of fecal Haemophilus, Rothia, Clostridium, Enterococcus, Streptococcus, and Veillonella in 43 Czech PSC patients compared to their healthy counterparts. Notwithstanding the aforementioned insights, observational studies encounter the challenge of controlling for confounding variables such as age, environment, dietary habits, and lifestyle, which may impinge upon the discernment of a causal relationship between GM and ACLD. Consequently, to clarify the potential causal nexus between GM and ACLD, we deployed a two-way Mendelian randomization approach.
Mendelian randomization (MR) is a methodological approach that leverages genetic variation to construct instrumental variables (IVs) for exposure, thereby facilitating the estimation of causal relationships between said exposure and the onset of a given malady (Greenland, 2000). It is a viable surrogate for Randomized Controlled Trials (RCTs), owing to the randomized assignment of genotypes from progenitors to progeny (Zhang et al., 2023). This randomization process ensures that the association between genetic variants and the resultant outcome remains impervious to common confounding variables, and the causal sequence maintains a plausible trajectory (Li et al., 2022). The methodological prowess of MR is now ubiquitously harnessed to dig deeper into the complicated tapestry of causal relationships between the intricacies of the GM and afflictions. In this study, we meticulously employed Genome-Wide Association Study (GWAS) amalgamated data from MiBioGen and the R9 version of the FinnGen Consortium to perform a two-sample Mendelian randomization (TSMR) analysis to assess the causal relationship between GM and ACLD.
In the present inquiry, we applied TSMR methodology to scrutinize the complicated nexus between GM and ACLD. This investigation involved orchestration of dual, bidirectional, two-sample Mendelian randomization analyses, employing GM as the exposure, PBC and PSC as the outcomes. Our primary analytical approach predominantly involved the Inverse Variance Weighting (IVW) method, with subsequent layers of scrutiny including sensitivity analyses, heterogeneity assessments, gene multiplicity evaluations, and application of the MR-PRESSO algorithm, all orchestrated to corroborate the steadfast reliability of the derived outcomes.
MR analysis needs to satisfy the following three assumptions: (1) IVs and exposure need to be strongly associated (association assumption), (2) IVs should not be associated with the outcome through confounders (independence assumption), and (3) IVs cannot directly affect the outcome but only through exposure (exclusivity assumption). The specific process is shown in Figure 1.
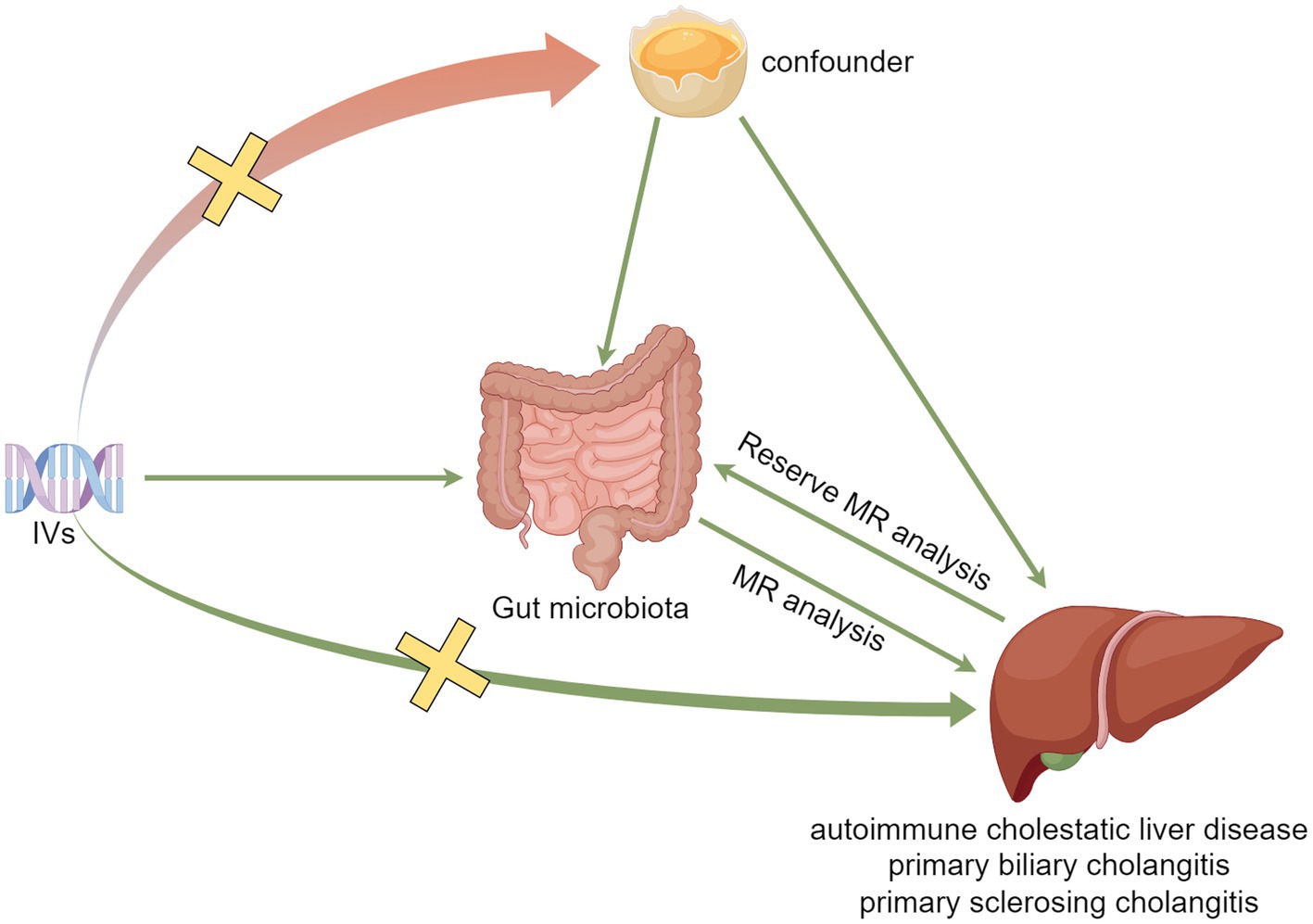
Figure 1. Model plot for MR analysis between GM and ACLD.
Single nucleotide polymorphisms (SNPs) intricately linked to the gut microbiota (GM) have been meticulously curated as instrumental variables for exposure. The consolidated dataset for these SNPs emanated from the MiBioGen consortium, an august assembly that scrutinized a comprehensive assemblage of 18,340 samples featuring 16S rRNA gene sequencing, drawn from a mosaic of 24 population-based cohorts, predominantly of European lineage. The microbial composition was meticulously analyzed, targeting three distinct variable regions of the 16S rRNA gene (V4, V3-V4, and V1-V2), with subsequent taxonomic classification achieved through direct taxonomic binning. To discern the complicated nuances of host genetic influence on gut bacterial taxa abundance, the endeavor embraced Microbiota Quantitative Trait Locus (mbQTL) positional analyses (Kurilshikov et al., 2021). Genetic datasets pertinent to both PBC and PSC—integral components of ACLD spectrum—were gleaned from the R9 iteration of the FinnGen Consortium, accessible at https://r9.finngen.fi/ (Kurki et al., 2023). The PBC cohort comprised 281,684 participants, with the case group numbered 557, and the healthy control cohort comprised 281,127 individuals. This collective was characterized by an exhaustive 20,167,312 SNPs. In parallel, the PSC dataset encompasses 332,618 participants, including 1,715 in the case group and 330,903 in the healthy control cohort, with an analogous wealth of 20,169,171 SNPs. Detailed information on ACLD data can be found in Supplementary Table 1.
A meticulous screening process was undertaken to identify IVs from a cohort of 211 gut microbes, adhering to the following exclusion criteria: (1) SNPs achieving a significance threshold (p < 1.0 × 10−5) within the designated locus range were discerningly chosen as potential IVs (Zhang et al., 2023). (2) To guarantee the independence of the included SNPs, linkage disequilibrium (LD) was meticulously expunged using a reference panel sourced from 1,000 Genome Project European samples, with a stringent criterion of r2 < 0.001 and a window size of 10,000 kb. Finally, data for the above selected SNPs were extracted from the outcome variables.
To obviate the potential sway of feeble instrumental biases in the estimation of the association effect, a rigorous evaluation of the robustness of instrumental variables was conducted. This scrutiny involved a meticulous assessment of the F-value, derived from the formula F = β2/se2 (Xie et al., 2023), where β represents the effect value and se signifies the standard error. It was posited that the absence of significant weak instrumental bias could be affirmed when the calculated F-value exceeded the threshold of 10 (Staiger and Stock, 1997).
Within the confines of this investigation, the primary modality employed to scrutinize the potential causal linkage between gut microbial abundance and the proclivity for ACLD rested on IVW analysis. This was seamlessly complemented by an ensemble of supplementary methodologies, including cML-MA, MR-Egger, Weighted median (WME), Weighted mode (WM), and Simple mode (SM). The imprimatur of a relatively steadfast causal association was imputed when the p-value fell below 0.05 for the IVW methods and concordantly for cML-MA, MR-Egger, WME, WM, and SM when their vectors were aligned (Zhang et al., 2023). Furthermore, a meticulous dissection of the MR-Egger intercept term, denoted as the Egger-intercept, served as a sentinel for horizontal pleiotropy. An Egger-intercept infinitesimally proximal to 0 or statistically insignificant signaled the dearth of genetic pleiotropy (Burgess and Thompson, 2017). Additionally, the study used the MR-PRESSO methodology, an instrument adept at identifying potential outliers. Subsequent to the excision of outliers (p-value <0.05), a recalibration of the causal analysis was performed (Verbanck et al., 2018). Sensitivity analyses, executed using the leave-one-out method, were instrumental in gauging the impact of each SNP on causality. This intricate procedure involves the sequential removal of individual SNPs to ascertain the cumulative effect value of the residual SNPs (Hemani et al., 2018). To gauge the specter of heterogeneity, the study invoked the Cochran’s Q test was used to discern potential discordance among the instrumental variables. A p-value less than 0.05 signified a noteworthy presence of heterogeneity. In a judicious effort to avert spurious positives due to the vicissitudes of multiple testing, a stringent false discovery rate (FDR) correction was implemented. Results attaining a PFDR < 0.1 were deemed statistically significant (Storey and Tibshirani, 2003; Li et al., 2022). In a bid to unravel the causal intricacies between GM and ACLD, a reciprocal MR analysis was executed for bacterial taxa pinpointed as causally entwined with ACLD in antecedent MR exploration. The genome-wide significance threshold for the exposure dataset was rigidly set at p < 5.0 × 10−6, with all other criteria and parameters aligned harmoniously with those governing anterior MR scrutiny.
Statistical analyses were diligently executed employing the Two Sample MR package (version 0.5.7) (Hemani et al., 2018), the MR-PRESSO package (version 1.0) (Verbanck et al., 2018), and MR-cML (Xue et al., 2021) within the expanse of the R software (version 4.3.1).
Eradicating 15 unclassified genera, a collective of 196 intestinal microorganisms spanning 9 phyla, 16 orders, 20 orders, 32 families, and 119 genera were judiciously included in this inquiry. Following the stringent criteria delineated in the previous section, 2,774 SNPs (Supplementary Table 2) intricately linked to the intestinal flora were identified. Notably, each of these SNPs boasted F-values surpassing the threshold of 10, thus effectually obviating the distortions introduced by feeble instrumental variables.
Within this investigation, a compendium of six MR methodologies was meticulously employed, with subsequent result scrutiny via the FDR correction method. In particular, Inverse-variance Weighted (IVW) outcomes unveiled six discernible gut microorganisms intricately implicated in the etiology of ACLD, each registering a noteworthy statistical significance (PFDR < 0.1). The relative abundance of three genera, including genus Clostridium innocuum group (OR = 0.54, 95% CI: 0.37–0.80, PFDR = 0.011), genus Butyricoccus (OR = 0.34, 95% CI: 0.15–0.74, PFDR = 0.034), and genus Erysipelatoclostridium (OR = 0.57, 95% CI: 0.36–0.90, PFDR = 0.063), and the relative abundance of the three types of intestinal microorganisms were negatively correlated with the risk of developing PBC, and the above three types of intestinal microorganisms were protective factors for PBC; the relative abundance of genus Eubacterium hallii group (OR = 1.45, 95% CI: 1.06–1.97, PFDR = 0.09) was positively correlated with the risk of developing PSC, i.e., it was a risk factor for PSC, family Clostridiaceae1 (OR = 0.64, 95% CI: 0.43–0.96, PFDR = 0.09) and family Lachnospiraceae (OR = 0.63, 95% CI: 0.41–0.98, PFDR = 0.09) were negatively correlated with the incidence of PSC, and were protective factors for PSC thereby manifesting as protective factors. In tandem, the complementary method cML-MA-BIC lent credence to the causal associations discerned in the aforementioned six gut microbial contributions to ACLDs. Visual representations in Figures 2–5 concurred with the directional alignment of effect values across the six Two-Sample Mendelian Randomization (TSMR) methodologies, solidifying the inference that the six identified intestinal bacterial taxa were substantively and causally interwoven with the complex etiology of ACLD, thereby accentuating the robustness of the results.
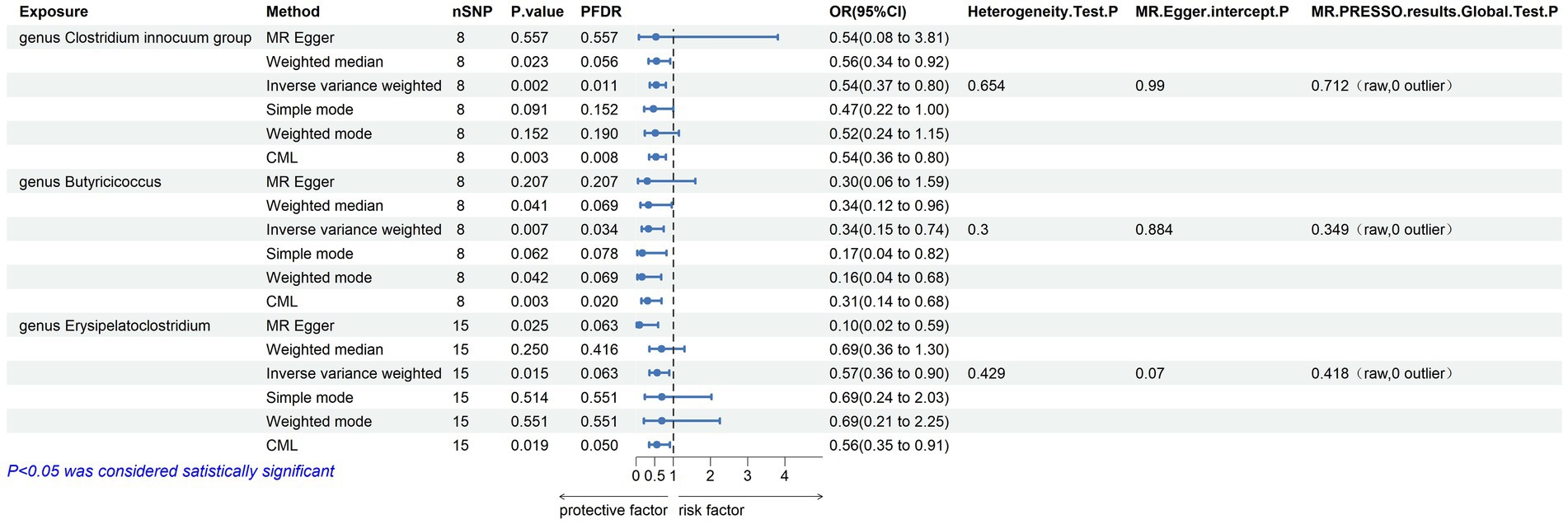
Figure 2. Results of MR analysis of 6 methods of GM and PBC and Egger-intercept, MR-PRESSO, and Cochran Q tests of IVW methods.
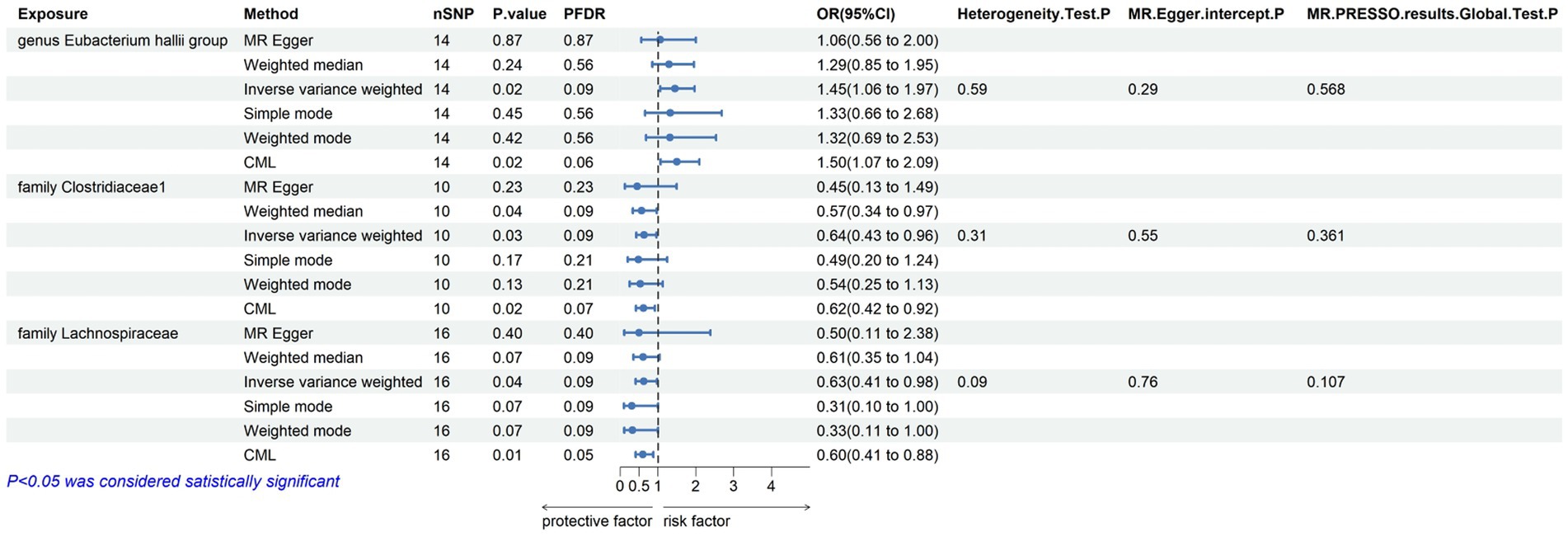
Figure 3. Results of MR analysis of 6 methods of GM and PSC and Egger-intercept, MR-PRESSO, and Cochran Q tests of IVW methods.
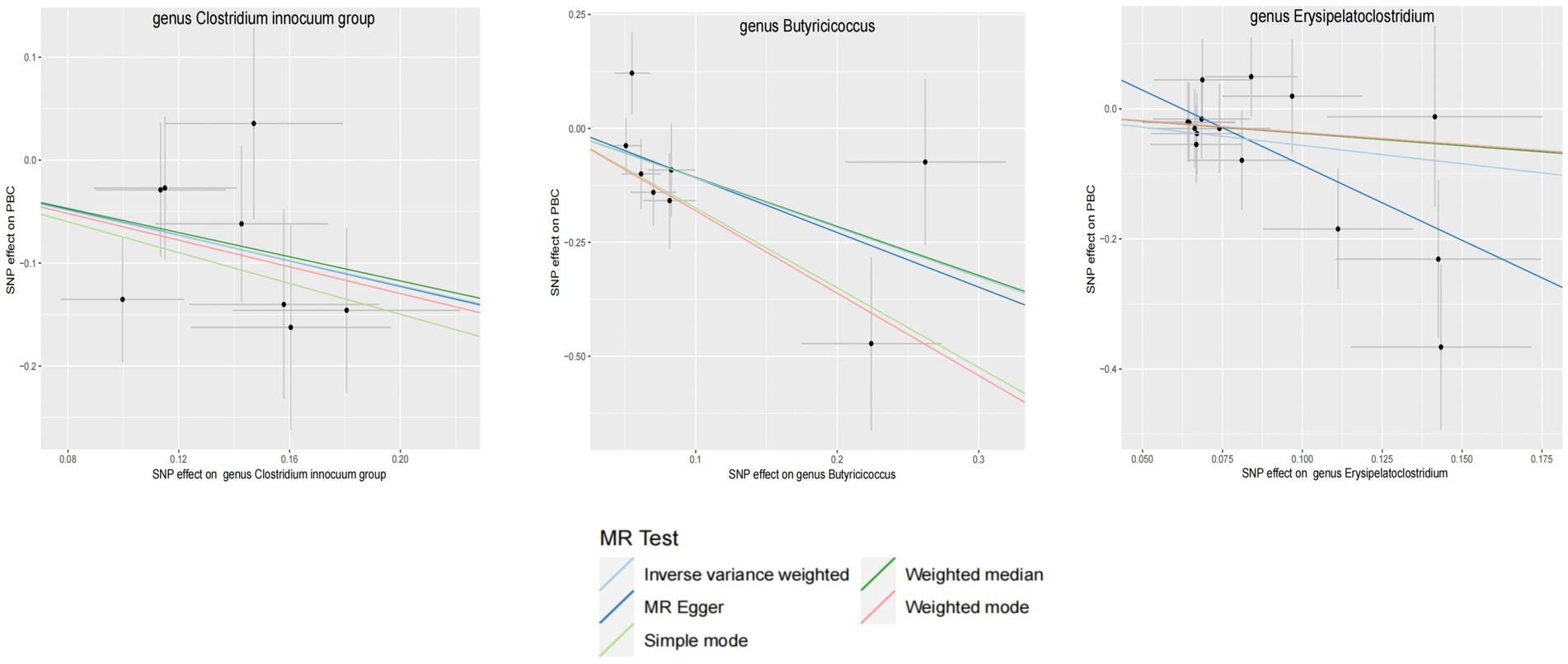
Figure 4. GM with causal relationship with PBC and the scatterplot of the 6 MR models.
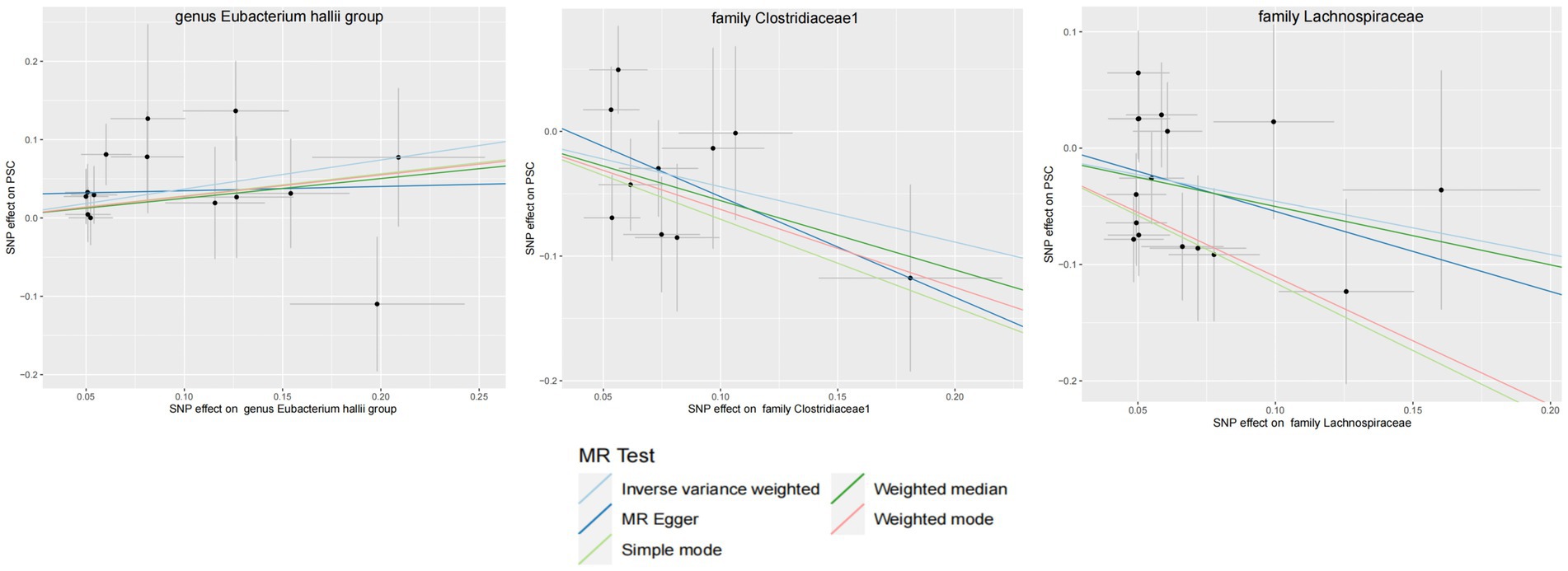
Figure 5. GM with causal relationship with PSC and the scatterplot of the 6 MR models.
The leave-one-out method analysis meticulously demonstrated that the stepwise exclusion of SNPs engendered no substantive deviation between the amalgamated effect and cumulative effect of the residual SNPs, as elegantly delineated in Figures 6, 7. Akin to this, the discerning MR-PRESSO analysis, expounded in Figures 2, 3, corroborated the absence of outliers (p > 0.05), thereby amplifying the veracity of the results. Furthermore, both the Cochran Q test and the Egger-intercept test unveiled p values surpassing the threshold of 0.05, attesting to the dearth of significant heterogeneity within the instrumental variables of this inquiry. Notably, these findings underscore the imperviousness of the results regarding the influence of genetic pleiotropy.
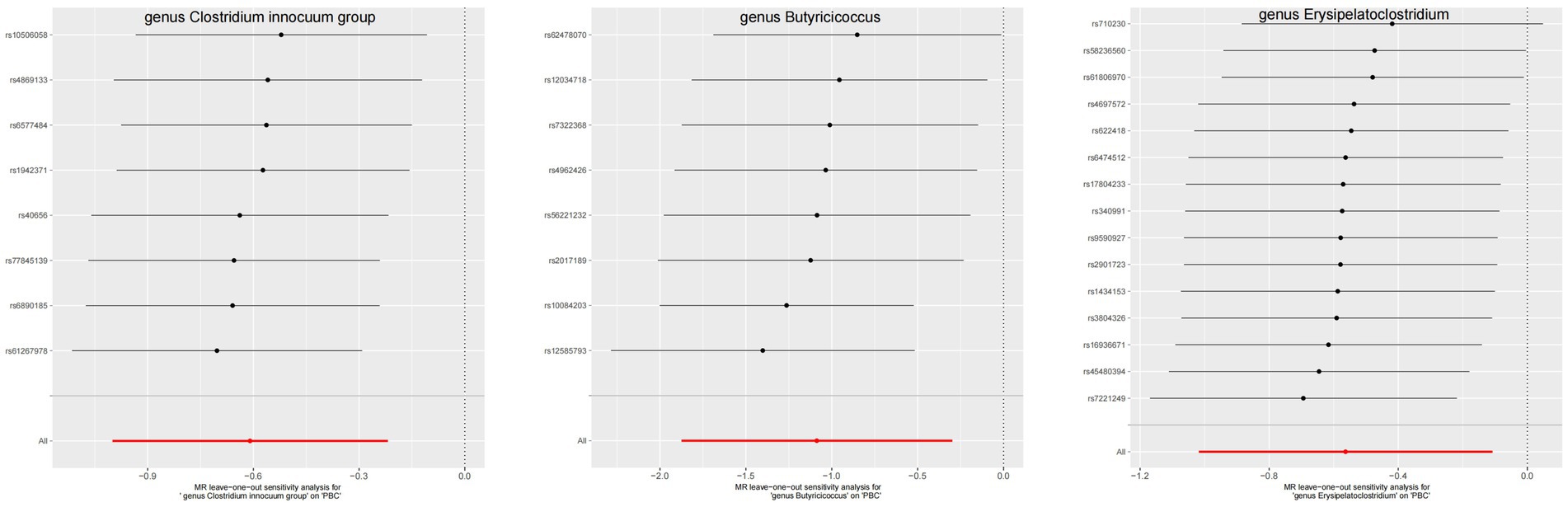
Figure 6. The leave-one-out plots of GM with causal relationship with PBC.
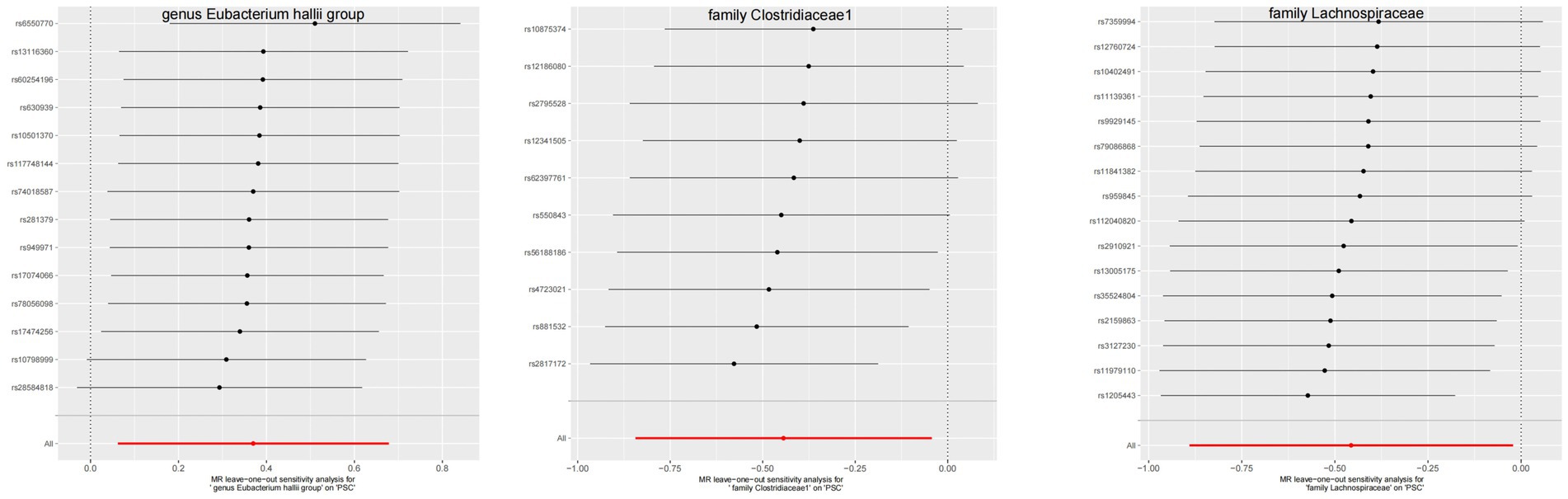
Figure 7. The leave-one-out plots of GM with causal relationship with PSC.
In the realm of intestinal microbiota, specifically comprising two familial entities and four generative cohorts, whose causal affiliations with ACLD were prominently underscored through the lens of forward MR analysis, a subsequent deconstruction was embarked upon via reverse MR scrutiny. The findings thereof failed to unveil any discernible causal nexus between ACLD and the intricate tapestry of intestinal flora, as substantiated by the statistical insignificance denoted in Table 1 (p > 0.05). The exposure data from the reverse MR analysis are detailed in Supplementary Tables 3, 4.
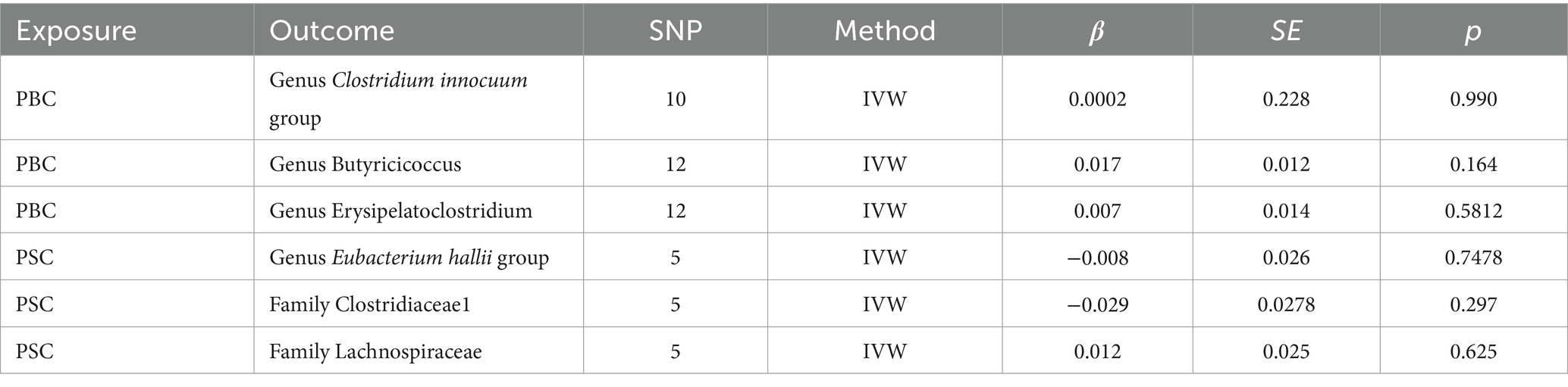
Table 1. Results of reverse MR analysis.
In this study, we utilized GWAS-pooled data from MiBioGen and Finngen, subjected to analysis using TSMR. Our findings revealed a negative correlation between the risk of PBC and the relative abundance of three genera: Clostridium innocuum group, Butyricicoccus, and Erysipelatoclostridium. Conversely, the relative abundance of the genus Eubacterium hallii group exhibited a positive correlation with the risk of PSC. Furthermore, we observed a negative correlation between the development of PSC and the relative abundance of family Clostridiaceae1 and family Lachnospiraceae. These results contribute to our understanding of the intricate relationships between specific microbial taxa and risks associated with PBC and PSC.
An increasing body of research underscores the role of intestinal flora in the development of ACLD through the gut-liver axis. This pathway involves microbial translocation, which is a pivotal feature of ACLD pathogenesis (Blesl and Stadlbauer, 2021). Bacteria and their metabolites, entering the circulation, are recognized by hepatic immune receptors, initiating intrahepatic inflammatory responses that contribute to the development and progression of ACLD (Tripathi et al., 2018). Moreover, structural alterations in the intestinal flora result in dysregulation of hepatic and intestinal bile acid circulation. This dysregulation precipitates cholestasis and accumulation of bile acid toxicity, ultimately inducing ACLD and progressive liver injury (Mousa et al., 2021).
A study conducted by Tedesco et al. revealed that translocation of Lactobacillus gasseri triggered the production of interleukin-17 (IL-17) by hepatic γδ T-cell receptor-positive (γδ TCR) cells in mice. This translocation is associated with hepatic inflammatory injury. Additionally, γδ TCR cells were isolated from the livers of patients with PSC, implying that intestinal flora may play a role in inducing cholestatic liver disease. Specifically, the mediation of γδ TCR cells to produce IL-17 has been identified as a potential mechanism contributing to the development of cholestatic liver disease (Tedesco et al., 2018). Observational studies have further supported the link between dysbiosis of gut flora and ACLD. For instance, Sabino et al. reported a higher proportion of Enterococcus, Fusobacterium, and Lactobacillus genera in the feces of 66 Belgian patients with PSC compared to controls (Sabino et al., 2016). Furukawa et al. (2020) also found relevant associations; they identified decreased bacterial diversity, a high abundance of Lactobacillus, and a low abundance of Clostridiales in the intestines of patients with PBC compared to the healthy population. Furthermore, in the ursodeoxycholic acid (UDCA)-treated nonresponder group, there was a significant decrease in the Faecalibacterium genus. This suggests that the presence of Faecalibacterium may influence the long-term prognosis of patients with PBC.
In alignment with prior investigations, our study identified significant associations using MR analysis. Specifically, the genus Clostridium innocuum exhibited a negative correlation with the risk of developing PBC. Conversely, the genus Eubacterium hallii and the family Lachnospiraceae were positively and negatively associated with the development of PSC, respectively. Earlier research has posited the absence of beneficial Clostridiales commensals in the intestines of PBC patients (Furukawa et al., 2020). Abe et al. (2018) reported a reduction in Clostridium subcluster XIVa among PBC patients compared to controls. Furthermore, patients with PBC with abnormal hepatic function exhibited decreased levels of Clostridium cluster IV and Clostridium subcluster XIVa, particularly in those with abnormal liver function. These findings suggest a potential protective role for Clostridium in PBC and indicate its possible influence on liver function. Scholars have affirmed that Clostridium, through 7-α-Dehydroxylation, converts Primary bile acids into secondary bile acids (Staley et al., 2017). Given that ACLD patients often experience bile acid metabolism dysregulation, reduced Dehydroxylation reaction, and cytotoxicity, the concentration of hydrophobic Primary bile acids increases, leading to toxicity accumulation and bile duct damage (Schmucker et al., 1990; Li et al., 2017; Torres et al., 2018; Fiorucci et al., 2021). Therefore, Clostridium may mitigate the risk of PBC development by improving bile acid metabolism. In a study by Kummen et al. in 2020, fecal DNA sequencing of patients with PSC revealed a significant depletion of various Eubacterium species (Kummen et al., 2021). Concurrently, patient treatment with UDCA was associated with elevated levels of Eubacterium rectale. While this study suggests a protective role for Eubacterium in PSC, our findings indicate that the genus Eubacterium hallii is a risk factor for PSC, necessitating further exploration of its role in PSC development. Quraishi et al. (2020) in the same year observed a notable reduction in the family Lachnospiraceae in patients with PSC, suggesting a potential protective effect against PSC. Previous research has confirmed the ability of family Lachnospiraceae to produce butyrate (Winston et al., 2021), a short-chain fatty acid known for its anti-inflammatory, bile acid metabolism-regulating, and intestinal barrier-improving properties (Cui et al., 2018; Ye et al., 2021; Liu et al., 2022; Wang et al., 2023). This may help mitigate inflammatory damage associated with bile acid stagnation and bacterial translocation. An earlier study in 2018 demonstrated a positive correlation between Faecalibacterium enrichment and Erysipelatoclostridium in healthy infants, which was not observed in cholestatic infants (Guo et al., 2018). This suggests a potential association between dysregulation of genus Erysipelatoclostridium and cholestasis. However, protective associations for PBC necessitate further investigation. While no direct study establishes the association of genus Butyricicoccus and family Clostridiaceae1 with ACLD, but because genus Butyricicoccus is the main microbiota producing butyrate, it was found to be positively associated with the concentration of Secondary bile acids, which may contribute to the conversion of Primary bile acids to Secondary bile acids (Yang et al., 2021). Certain genera within family Clostridiaceae, such as Clostridium, have been implicated in the conversion of Primary bile acids to Secondary bile acids (Furukawa et al., 2020). Therefore, we posit that genus Butyricicoccus and family Clostridiaceae1 may exert protective effects against ACLD through the production of butyrate and regulation of bile acids. Further research is warranted to validate these hypotheses.
It is also worth noting that the occurrence and development of non-alcoholic fatty liver disease (NAFLD) is also closely related to dysregulation of gut microbiota and bile acid metabolism, for example (Sorrentino et al., 2005). Qilong Zhai et al. found in an MR study that Enterobacteriales, Enterobacteriaceae, Lachnospiraceae UCG-004, and Prevotella9 increased the risk of NAFLD, and Dorea and Veillonella increased the risk of NASH. Oscillospira and Ruminococcaceae UCG-013 were able to reduce their risk, with Veillonella, Lachnospiraceae UCG-004 being able to participate in bile acid metabolism. It has also been suggested that dysregulation of BA homeostasis and signaling may further contribute to abnormal lipid metabolism and lipotoxicity in NAFLD (Arab et al., 2017), and functional cholestasis may be related to the pathogenesis of NASH (Segovia-Miranda et al., 2019). Thus, it can be seen that the disorder of bile acid hepatic and intestinal circulation caused by dysbiosis of gut microbiota is likely to be an important cause of the development and progression of NAFLD, which has some overlap with the mechanism of ACLD triggered by gut microbiota, and thus it has been pointed out that cholestasis and NAFLD can share some of the therapeutic targets in their treatments (Trauner and Fuchs, 2022). Most of the positive gut microbiota we found in this study exerted their promotional or inhibitory effects on ACLD by affecting bile acid metabolism, and it should be noted that two protective genera, Clostridium and Butyricicoccus, were also found to be in increased abundance in patients with NAFLD regression after bariatric surgery in this study (Pérez-Rubio et al., 2023), and thus we hypothesize that they may be able to contribute to the development of common therapeutic targets to ameliorate both cholestasis and NAFLD by modulating the gut microbiota.
In comparison with previous literature, we found some similarities in the design of the two studies, but there are obvious differences in data sources, findings and research methods, etc. The study of Yang et al. (2024) found that order Bacillales, family Peptostreptococcaceae, family Ruminococcaceae, genus Anaerotruncu was associated with a reduced risk of developing PBC, order Selenomonadales, family Bifidobacteriaceae may be a factor that increases the risk of PBC, order Selenomonadales, family Rhodospirillaceae, and genus RuminococcaceaeUCG013 was positively associated with PSC, order Actinomycetales, family Actinomycetaceae, genus Actinomyces, genus Alloprevotella, genus Barnesiella, and genus Peptococcus were negatively correlated with PSC risk. Zhang et al. (2023) similarly found that Order Selenomonadales, Order Bifidobacteriales and Genus Lachnospiraceae UCG_004 were associated with higher risk of PBC, and Family Peptostreptococcaceae and Family Ruminococcaceae were protective against PBC. The above two studies and the content of the present study are from different databases, so the results produce a large difference, which also indicates that there may be some differences in the gut microorganisms affecting the pathogenesis of PBC and PSC in different sample populations. This study can further add candidate gut microorganisms affecting the pathogenesis of cholestatic liver disease, providing more complete basic research content for future studies.
The strengths and limitations of this study are delineated as follows. Notably, the utilization of the MR method stands out as a strength, enabling the analysis of the causal relationship between GM and ACLD, and can be complemented by the results of previous studies. This approach offers potential candidate gut microorganisms for subsequent investigations. Additionally, the study leveraged genetic data derived from a substantial population sample, mitigating the impact of confounding factors more effectively than small observational studies. Consequently, the results were deemed more reliable. However, certain limitations merit consideration. Firstly, the GWAS data primarily originated from European populations, raising the possibility of variation in results when extrapolated to other ethnic groups. Furthermore, the reliance on 16S rRNA gene sequencing limits taxonomic resolution to the genus level, precluding an exploration of the causal relationship between GM and ACLD at the species level. In specific instances, such as the prominence of the genus Eubacterium hallii in this study, discrepancies in its direction of action compared to prior research on its correlation with PSC were observed. Additionally, the absence of direct evidence confirming a causal relationship between the genus Butyricicoccus and the family Clostridiaceae1 with ACLD pathogenesis is acknowledged. This underscores the necessity for future studies to validate and substantiate these potential associations. Consequently, the study calls for further investigation to solidify the understanding of these relationships and their implications for ACLD development.
In summary, this study identified six intestinal microbial taxa with causal associations to PBC and PSC through Mendelian randomization analysis. This finding establishes a foundational basis for investigating the link between GM and ACLD. Nevertheless, to comprehensively illustrate the specific mechanisms underlying the protective or triggering effects of these relevant flora on ACLD, further investigations employing large sample clinical Randomized Controlled Trial (RCT), pathway analyses, and laboratory studies are imperative.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
No animal/human studies are presented in the manuscript.
YC: Data curation, Methodology, Writing – original draft, Investigation. YG: Methodology, Software, Writing – original draft. YK: Data curation, Software, Writing – original draft. GZ: Supervision, Writing – review & editing, Project administration.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We acknowledge the participants and investigators of the FinnGen study and the Mibiogen consortium. We thank www.figdraw.com/ for their help in MR modeling and process graph construction.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1348027/full#supplementary-material
Abe, K., Takahashi, A., Fujita, M., Imaizumi, H., Hayashi, M., Okai, K., et al. (2018). Dysbiosis of oral microbiota and its association with salivary immunological biomarkers in autoimmune liver disease. PLoS One 13:e0198757. doi: 10.1371/journal.pone.0198757
Arab, J. P., Karpen, S. J., Dawson, P. A., Arrese, M., and Trauner, M. (2017). Bile acids and nonalcoholic fatty liver disease: molecular insights and therapeutic perspectives. Hepatology 65, 350–362. doi: 10.1002/hep.28709.27358174
Blesl, A., and Stadlbauer, V. (2021). The gut-liver axis in cholestatic liver diseases. Nutrients 13:1018. doi: 10.3390/nu13031018
Burgess, S., and Thompson, S. G. (2017). Interpreting findings from Mendelian randomization using the MR-egger method. Eur. J. Epidemiol. 32, 377–389. doi: 10.1007/s10654-017-0255-x
Cui, H., Cai, Y., Wang, L., Jia, B., Li, J., Zhao, S., et al. (2018). Berberine regulates Treg/Th17 balance to treat ulcerative colitis through modulating the gut microbiota in the Colon. Front. Pharmacol. 9:571. doi: 10.3389/fphar.2018.00571
Fiorucci, S., Distrutti, E., Carino, A., Zampella, A., and Biagioli, M. (2021). Bile acids and their receptors in metabolic disorders. Prog. Lipid Res. 82:101094. doi: 10.1016/j.plipres.2021.101094
Fukui, H. (2019). Role of gut dysbiosis in liver diseases: what have we learned so far? Diseases 7:58. doi: 10.3390/diseases7040058
Furukawa, M., Moriya, K., Nakayama, J., Inoue, T., Momoda, R., Kawaratani, H., et al. (2020). Gut dysbiosis associated with clinical prognosis of patients with primary biliary cholangitis. Hepatol. Res. 50, 840–852. doi: 10.1111/hepr.13509
Greenland, S. (2000). An introduction to instrumental variables for epidemiologists. Int. J. Epidemiol. 29, 722–729. doi: 10.1093/ije/29.4.722
Guo, C., Li, Y., Wang, P., Li, Y., Qiu, C., Li, M., et al. (2018). Alterations of gut microbiota in cholestatic infants and their correlation with hepatic function. Front. Microbiol. 9:2682. doi: 10.3389/fmicb.2018.02682
Hemani, G., Zheng, J., Elsworth, B., Wade, K. H., Haberland, V., Baird, D., et al. (2018). The MR – base platformsupports systematic causal inference across the humanphenome. eLife 7:e34408. doi: 10.7554/eLife.34408
Kummen, M., Thingholm, L. B., Rühlemann, M. C., Holm, K., Hansen, S. H., Moitinho-Silva, L., et al. (2021). Altered gut microbial metabolism of essential nutrients in primary sclerosing cholangitis. Gastroenterology 160, 1784–1798.e0. doi: 10.1053/j.gastro.2020.12.058
Kurilshikov, A., Medina-Gomez, C., Bacigalupe, R., Radjabzadeh, D., Wang, J., Demirkan, A., et al. (2021). Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165. doi: 10.1038/s41588-020-00763-1
Kurki, M. I., Karjalainen, J., Palta, P., Sipilä, T. P., Kristiansson, K., Donner, K. M., et al. (2023). FinnGen provides genetic insights from a well-phenotyped isolated population. Nature 613, 508–518. doi: 10.1038/s41586-022-05473-8.36653562
Lammert, C., Shin, A., Xu, H., Hemmerich, C., O'Connell, T. M., and Chalasani, N. (2021). Short-chain fatty acid and fecal microbiota profiles are linked to fibrosis in primary biliary cholangitis. FEMS Microbiol. Lett. 368:fnab038. doi: 10.1093/femsle/fnab038
Li, Y., Tang, R., Leung, P. S. C., Gershwin, M. E., and Ma, X. (2017). Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 16, 885–896. doi: 10.1016/j.autrev.2017.07.002
Li, P., Wang, H., Guo, L., Gou, X., Chen, G., Lin, D., et al. (2022). Association between gut microbiota and preeclampsia-eclampsia: a two-sample Mendelian randomization study. BMC Med. 20:443. doi: 10.1186/s12916-022-02657-x
Liu, X., Wang, L., Tan, S., Chen, Z., Wu, B., and Wu, X. (2022). Therapeutic effects of Berberine on liver fibrosis are associated with lipid metabolism and intestinal flora. Front. Pharmacol. 13:814871. doi: 10.3389/fphar.2022.814871
Liwinski, T., Heinemann, M., and Schramm, C. (2022). The intestinal and biliary microbiome in autoimmune liver disease-current evidence and concepts. Semin. Immunopathol. 44, 485–507. doi: 10.1007/s00281-022-00936-6
Mousa, O. Y., Juran, B. D., McCauley, B. M., Vesterhus, M. N., Folseraas, T., Turgeon, C. T., et al. (2021). Bile acid profiles in primary sclerosing cholangitis and their ability to predict hepatic decompensation. Hepatology 74, 281–295. doi: 10.1002/hep.31652
Pérez-Rubio, Á., Soluyanova, P., Moro, E., Quintás, G., Rienda, I., Periañez, M. D., et al. (2023). Gut microbiota and plasma bile acids associated with non-alcoholic fatty liver disease resolution in bariatric surgery patients. Nutrients 15:3187. doi: 10.3390/nu15143187.37513605
Quraishi, M. N., Acharjee, A., Beggs, A. D., Horniblow, R., Tselepis, C., Gkoutos, G., et al. (2020). A pilot integrative analysis of colonic gene expression, gut microbiota, and immune infiltration in primary sclerosing cholangitis-inflammatory bowel disease: association of disease with bile acid pathways. J. Crohns Colitis 14, 935–947. doi: 10.1093/ecco-jcc/jjaa021
Richardson, N., Wootton, G. E., Bozward, A. G., and Oo, Y. H. (2022). Challenges and opportunities in achieving effective regulatory T cell therapy in autoimmune liver disease. Semin. Immunopathol. 44, 461–474. doi: 10.1007/s00281-022-00940-w
Sabino, J., Vieira-Silva, S., Machiels, K., Joossens, M., Falony, G., Ballet, V., et al. (2016). Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 65, 1681–1689. doi: 10.1136/gutjnl-2015-311004
Schmucker, D. L., Ohta, M., Kanai, S., Sato, Y., and Kitani, K. (1990). Hepatic injury induced by bile salts: correlation between biochemical and morphological events. Hepatology 12, 1216–1221. doi: 10.1002/hep.1840120523
Segovia-Miranda, F., Morales-Navarrete, H., Kücken, M., Moser, V., Seifert, S., Repnik, U., et al. (2019). Three-dimensional spatially resolved geometrical and functional models of human liver tissue reveal new aspects of NAFLD progression. Nat. Med. 25, 1885–1893. doi: 10.1038/s41591-019-0660-7
Sorrentino, P., Tarantino, G., Perrella, A., Micheli, P., Perrella, O., and Conca, P. (2005). A clinical-morphological study on cholestatic presentation of nonalcoholic fatty liver disease. Dig. Dis. Sci. 50, 1130–1135. doi: 10.1007/s10620-005-2719-1.15986869
Staiger, D., and Stock, J. H. (1997). Instrumental variables regression with weak instruments. Econometrica 65, 557–586. doi: 10.2307/2171753
Staley, C., Weingarden, A. R., Khoruts, A., and Sadowsky, M. J. (2017). Interaction of gut microbiota with bile acid metabolism and its influence on disease states. Appl. Microbiol. Biotechnol. 101, 47–64. doi: 10.1007/s00253-016-8006-6
Storey, J. D., and Tibshirani, R. (2003). Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. U. S. A. 100, 9440–9445. doi: 10.1073/pnas.1530509100
Tedesco, D., Thapa, M., Chin, C. Y., Ge, Y., Gong, M., Li, J., et al. (2018). Alterations in intestinal microbiota lead to production of interleukin 17 by intrahepatic γδ T-cell receptor-positive cells and pathogenesis of cholestatic liver disease. Gastroenterology 154, 2178–2193. doi: 10.1053/j.gastro.2018.02.019
Torres, J., Palmela, C., Brito, H., Bao, X., Ruiqi, H., Moura-Santos, P., et al. (2018). The gut microbiota, bile acids and their correlation in primary sclerosing cholangitis associated with inflammatory bowel disease. United European Gastroenterol J 6, 112–122. doi: 10.1177/2050640617708953
Trauner, M., and Fuchs, C. D. (2022). Novel therapeutic targets for cholestatic and fatty liver disease. Gut 71, 194–209. doi: 10.1136/gutjnl-2021-324305.34615727
Tripathi, A., Debelius, J., Brenner, D. A., Karin, M., Loomba, R., Schnabl, B., et al. (2018). The gut-liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 15, 397–411. doi: 10.1038/s41575-018-0011-z
Verbanck, M., Chen, C. Y., Neale, B., and do, R. (2018). Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698. doi: 10.1038/s41588-018-0099-7
Wang, S., Xiang, L., li, F., Deng, W., lv, P., and Chen, Y. (2023). Butyrate protects against Clostridium difficile infection by regulating bile acid metabolism. Microbiol. Spectr. 11:e0447922. doi: 10.1128/spectrum.04479-22
Winston, J. A., Rivera, A., Cai, J., Patterson, A. D., and Theriot, C. M. (2021). Secondary bile acid ursodeoxycholic acid alters weight, the gut microbiota, and the bile acid pool in conventional mice. PLoS One 16:e0246161. doi: 10.1371/journal.pone.0246161
Xie, J., Huang, H., Liu, Z., Li, Y., Yu, C., Xu, L., et al. (2023). The associations between modifiable risk factors and nonalcoholic fatty liver disease: a comprehensive mendelian randomization study. Hepatology 77, 949–964. doi: 10.1002/hep.32728
Xue, H., Shen, X., and Pan, W. (2021). Constrained maximum likelihood-based Mendelian randomization robust to both correlated and uncorrelated pleiotropic effects. Am. J. Hum. Genet. 108, 1251–1269. doi: 10.1016/j.ajhg.2021.05.014
Yang, Z. H., Liu, F., Zhu, X. R., Suo, F. Y., Jia, Z. J., and Yao, S. K. (2021). Altered profiles of fecal bile acids correlate with gut microbiota and inflammatory responses in patients with ulcerative colitis. World J. Gastroenterol. 27, 3609–3629. doi: 10.3748/wjg.v27.i24.3609
Yang, J., Ma, G., Wang, K., Yang, H., Jiang, S., Fan, Q., et al. (2024). Causal associations between gut microbiota and Cholestatic liver diseases: a Mendelian randomization study. Front. Med. 11:1342119. doi: 10.3389/fmed.2024.1342119.38327703
Ye, X., Shen, S., Xu, Z., Zhuang, Q., Xu, J., Wang, J., et al. (2021). Sodium butyrate alleviates cholesterol gallstones by regulating bile acid metabolism. Eur. J. Pharmacol. 908:174341. doi: 10.1016/j.ejphar.2021.174341
Zhang, J., Wu, G., Tang, Y., Liu, H., Ge, X., Peng, R., et al. (2023). Causal associations between gut microbiota and primary biliary cholangitis: a bidirectional two-sample Mendelian randomization study. Front. Microbiol. 14:1273024. doi: 10.3389/fmicb.2023.1273024
Keywords: gut microbiota, autoimmune cholestatic liver disease, Mendelian randomization, primary biliary cholangitis, primary sclerosing cholangitis
Citation: Cui Y, Guo Y, Kong Y and Zhang G (2024) Association between gut microbiota and autoimmune cholestatic liver disease, a Mendelian randomization study. Front. Microbiol. 15:1348027. doi: 10.3389/fmicb.2024.1348027
Edited by:
Mauro Cataldi, University of Naples Federico II, ItalyReviewed by:
Giovanni Tarantino, University of Naples Federico II, ItalyCopyright © 2024 Cui, Guo, Kong and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: GuangYe Zhang, eGp4emZtQDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.