 Xinyu Zhang
Xinyu Zhang Wei Wang
Wei Wang Yajing Wang
Yajing Wang Zhijun Cao
Zhijun Cao Hongjian Yang
Hongjian Yang Shengli Li
Shengli Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 13 May 2024
Sec. Microorganisms in Vertebrate Digestive Systems
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1336278
Introduction: The aim of this study was to investigate the effects of diets on the composition and function of rumen microbiome and metabolites in Sanhe heifers.
Methods: Metagenomic and metabolomic analyses were performed using rumen fluid samples collected from Sanhe heifers (n = 20) with similar body weights and ages from grass-fed and grain-fed systems.
Results: The grain-fed group exhibited more intensive rumen fermentation than the grass-fed group. However, the grass-fed group exhibited carbohydrate metabolism and methane production higher than that of the grain-fed group; these increases were observed as a higher abundance of various bacterial phyla (Firmicutes, Bacteroidetes, Actinobacteria, Lentisphaerae, and Verrucomicrobia), families (Lachnospiraceae, Eubacteriaceae, and Eggerthellaceae), and the archaeal family Methanobacteriaceae. A comparison of genes encoding carbohydrate-active enzymes, using Kyoto Encyclopedia of Genes and Genome profiles, revealed noteworthy differences in the functions of rumen microbiota; these differences were largely dependent on the feeding system.
Conclusion: These results could help manipulate and regulate feed efficiency in Sanhe cattle.
Sanhe cattle, a dual-purpose breed, are famous for their excellent milk (i.e., high in fat) and high meat yields (Xu et al., 2017). Sanhe cattle have rough feeding tolerance (Hu et al., 2019), strong disease resistance (Usman et al., 2017), and stable genetic performance. In previous reports, we introduced Sanhe cattle to an altitude of 3,650 m in Tibet and observed improved nutrient digestibility, indicating that Sanhe cattle exhibit strong adaptability to high-altitude environments (Zhang X. et al., 2022). However, the effects of feeding regimens on the composition and functions of gut microbiota in Sanhe heifers have not been explored.
Ruminant-derived products are important sources of meat and milk, providing high-quality protein for human consumption. Beyond being an organ, the rumen can be characterized as a complex microbial ecosystem that hosts numerous bacteria, fungi, archaea, and protozoa (Mackie and White, 1990; Russell and Rychlik, 2001); the microbial communities hosted in the rumen shape the amount of energy ruminants can extract from their food, thereby directly impacting agricultural productivity. In an anaerobic environment, the rumen microbiota digest (through fermentation) the feed consumed by cattle and produce volatile fatty acids (VFAs). VFAs, including acetate, propionate, butyrate, and valerate, affect carcass yield and meat quality traits in beef cattle. The component microbes in the rumen microbiota work together to maintain the stability of the rumen environment, associated with rumen fermentation (Morgavi et al., 2013), feed efficiency (Jewell et al., 2015; Shabat et al., 2016), and methane emissions. Various factors, including diet, age (Jami et al., 2013; Guo et al., 2020), antibiotics (Yousif et al., 2018; Holman et al., 2019), and environment, can affect the microbiota of ruminants. However, among these factors, diet has the greatest impact on the microbiota of the digestive tract. For example, cows fed a high-fiber diet expressed a higher relative abundance of Fibrobacterota in the rumen, whereas those fed a high-concentrate diet had a higher relative abundance of Bacteroidetes (Fernando et al., 2010). Similarly, increased dietary proportions of grain and starch lower bacterial richness and diversity in the rumen of cattle (Mao et al., 2013; Petri et al., 2013). Moreover, feeding regimes, such as grass- and grain-feeding, have been shown to affect the physiological health of cows, and the organoleptic and nutritional quality of the resultant beef and milk.
Metagenomics and metabolomics based on DNA levels and metabolic differences have gained increased research interest as effective tools for understanding the functional potential of the rumen microbiome (Li et al., 2018). A few studies have also used combined meta-omics analyses to explore rumen microbiome functions. Recently, two studies have explored the effects of grass- and grain-fed diets on the rumen microbiota of sheep and yaks using 16S rDNA (Belanche et al., 2019; Xu et al., 2021). We hypothesized that changes in the rumen microbiota of Sanhe heifers caused by changes in the diet might explain the changes in VFA and metabolites in the two different feeding systems.
In this study, using metagenomic and metabolomic analyses, we investigated the effect of various feeding regimens on the composition of rumen microbiota and metabolites of Sanhe heifers. These efforts are meant to bring forth insights regarding the role of the rumen microbiota in digestion and metabolism and can help assist in manipulating and regulating feeding regimens to realize maximum feed efficiency.
The Sanhe heifers utilized in our study were sourced from the Inner Mongolia Autonomous Region (119°57′E, 47°17′N; 700 m altitude). In May 2020, 150 Sanhe heifers of similar age (12 months) and body weight were chosen from a herd of over 500 Sanhe cattle that shared the same father and were housed at the Xieertala farm in Inner Mongolia. These heifers were subsequently divided into two groups, each comprising 75 individuals. One group remained at Xieertala farm (grain-fed group), while the other was transported to nearby pastures for grass feeding (grass-fed group). The grain-fed Sanhe heifers were provided with a total mixed ration (TMR) twice daily at 06:30 a.m., and 17:30 p.m. The TMR was formulated based on the National Research Council guidelines, aiming to meet the nutrient requirements of Sanhe heifers under the given conditions (NRC, 2001). The grain-fed group fed with the diet with dry matter 50.79%, organic matter 90.45%, crude protein 15.70%, ether extract 2.92%, neutral detergent 38.98%, acid detergent fiber 22.36%. The TMR was sampled weekly, dried, ground, and subjected to chemical analysis. Water was provided ad libitum. The grass-fed group had ad libitum access to pasture.
Ten healthy Sanhe heifers with similar ages (14–15 months) and mean body weights (340.71–328.65 kg) were randomly selected from each group. Rumen fluid samples were collected at 06:30 a.m. before morning feeding. The samples were divided, with one part immediately frozen in liquid nitrogen (−196°C) for bacterial analysis, and the other stored at −20°C for examining rumen fermentation parameters.
This experiment received approval from the Ethical Committee of China Agricultural University (project number AW22121202-1-2).
To assess the VFA concentrations in rumen fluid samples, each sample was thawed and subjected to centrifugation. The quantification of the supernatant was conducted, following a protocol outlined in a prior study by Erwin et al. (1961).
DNA was extracted from samples using a kit. The quantity and quality of DNA were assessed through TBS-380 and NanoDrop2000 instruments. A 1% agarose gel was used to confirm DNA integrity. Subsequently, DNA extract was fragmented to an average size of about 400 bp using Covaris M220 (Gene Company Limited, China) for paired-end library construction. Paired-end libraries were determined through the NEXTFLEX Rapid DNA-Seq kit. Adapters were attached to DNA fragments. Sequencing was detected using an Illumina NovaSeq machine.
After obtaining paired-end Illumina reads, the “fastp” tool (Chen et al., 2018) removed adapters and excluded reads (length ≤ 50 bp, quality value ≤20, or containing N bases). Metagenomic data compilation involved generating condensed de Bruijn plots using MEGAHIT (Li et al., 2015 non-redundant database, version 1.1.2). Synonyms (length ≥ 300 bp) were chosen to ensure data integrity. These synonyms were used for downstream genetic analysis and annotation.
Open reading frames (ORFs) within the compiled isoforms underwent prediction through Prodigal (Hyatt et al., 2010). Application of NCBI translation tables facilitated the identification of predicted ORFs with a length of ≤100 bp, subsequently translating them into amino acid sequences. The establishment of a non-redundant gene catalog was accomplished employing CD-HIT (Fu et al., 2012). High-quality reads were aligned to the catalog of non-redundant genes using SOAPaligner (Li et al., 2008), and gene abundance was computed with a 95% identity threshold. For taxonomic annotation, Diamond (Buchfink et al., 2015) aligned representative sequences from the non-redundant gene catalog to the non-redundant database, applying an e-value limit of 1e-5. KEGG annotation was executed through Diamond’s analysis against the KEGG database, maintaining an e-value limit of 1e-5. Carbohydrate active enzyme (CAZyme) annotations were conducted using HMMER hmmscan against the CAZy database, implementing an e-value cutoff of 1e-5.
Metabolite extraction from rumen fluid samples involved mixing with a cold solvent mixture. After vortexing and ice bath treatment, samples were centrifuged, and the supernatant was determined by liquid chromatography-mass spectrometry (LC–MS).
Rumen fluid metabolites underwent separation employing an ultra-high-performance liquid chromatography system (1,290 Infinity LC; Agilent Technologies, Santa Clara, CA, United States) coupled with a quadrupole time-of-flight mass spectrometer (TripleTOF 6,600, AB Sciex, Framingham, MA, United States). The analysis utilized a 2.1 mm × 100 mm ACQUIY UPLC BEH 1.7 μm column (Waters, Milford, MA, United States). The mobile phases facilitated positive and negative ion electrospray ionization mode. Gradient conditions were meticulously set, beginning at 85% B for 1 min, linearly decreasing to 65% over 11 min, followed by a linear decrease to 40% for 0.1 min, maintaining at 40% for 4 min, and concluding with an increase to 85% over 0.1 min. The system underwent reequilibration for 5 min before each subsequent run. The electrospray ionization source operated under specified conditions: ion source gas 1 at 60, gas 2 at 60, curtain gas at 30, source temperature at 600°C, and ionization spray voltage fluctuating ±5,500 V. During mass spectrometry acquisition, the instrument’s m/z range spanned from 60 to 1,000 Da, with a build-up time of the time-of-flight mass spectrometry scans set at 0.20 s/segment. Signal acquisition occurred in Auto-MS/MS mode within the m/z range of 25–1,000 Da, and the production scan’s accumulation time was established at 0.05 s/segment. Utilizing information-dependent acquisition, production scans operated in high-sensitivity mode with a fixed collision energy of 35 V ± 15 eV and a de-clustering potential of ±60 V.
Raw MS data in wiff.scan files were converted to MzXML files via ProteoWizard MSConvert, and subsequent processing included feature detection, retention time correction, and result alignment using XCMS. Metabolite identification hinged on precise mass spectrometry analysis (precision <25 ppm) and MS/MS data corresponding to standard databases. For extracted ion characterization, variables with non-zero values >50% were retained in at least one group. Multivariate statistical analyses, including Pareto scaling, principal component analysis (PCA), and partial least squares discriminant analysis (OPLS-DA), were executed through the MetaboAnalyst web system. Model robustness underwent assessment via leave-one-out cross-validation and response replacement tests. Differences in metabolites between the low and high abundance groups were determined based on a combination of statistically significant variables’ effect on prediction (VIP) values in the OPLS-DA model and two-tailed Student’s t-tests (p-values) on raw data. Significance criteria encompassed VIp values greater than 1.0 or less than 0.05, and P values less than 0.05. Metabolite identification drew upon three databases, namely, KEGG, Human Metabolome, and Bovine Metabolome. Enrichment of KEGG metabolic pathways by distinct metabolites was evaluated using the KEGG database, and pathway significance was ascertained through Fisher’s exact test.
The t-test in SPSS software (version 22.0, SPSS, Inc., Chicago, IL, United States) was used to compare the values of rumen fermentation parameters between the two groups. Only microbial taxa with relative abundance greater than 0.01% were included in this study. Compositional profiles of the rumen microbiota were summarized at the level of phylum, family, genus, and species, whereas the archaeal community was summarized at the level of species. We analyzed the α-diversity index using the Wilcoxon rank test. Principal Coordinate Analysis (PCoA) was performed in R based on the Bray-Curtis heterogeneity matrix, and the results were visualized using the “ggplot2” package. To compare the relative abundance and microbiota function of the two groups of organisms at the phylum, family, genus, and species levels, we used the Wilcoxon method in the R software (version 4.0.5). In addition, we used the “corrplot” package (author: Taiyun Wei; publication date: 2017; version: 0.84) in the “Psych” software package (author: W Revelle; publication date: 2016; version: 1.6.9) to perform Spearman rank correlation analysis. All data are reported as means, and p < 0.05 was considered significant.
Table 1 presents the rumen fermentation parameters in Sanhe heifers under different feeding systems. The contents of acetate, butyrate, isovalerate, and total VFAs were significantly higher in the grass-fed group than those in the grain-fed group (p < 0.05). Moreover, the propionic acid content of the grain-fed group was significantly higher than that of the grass-fed group (p = 0.06).
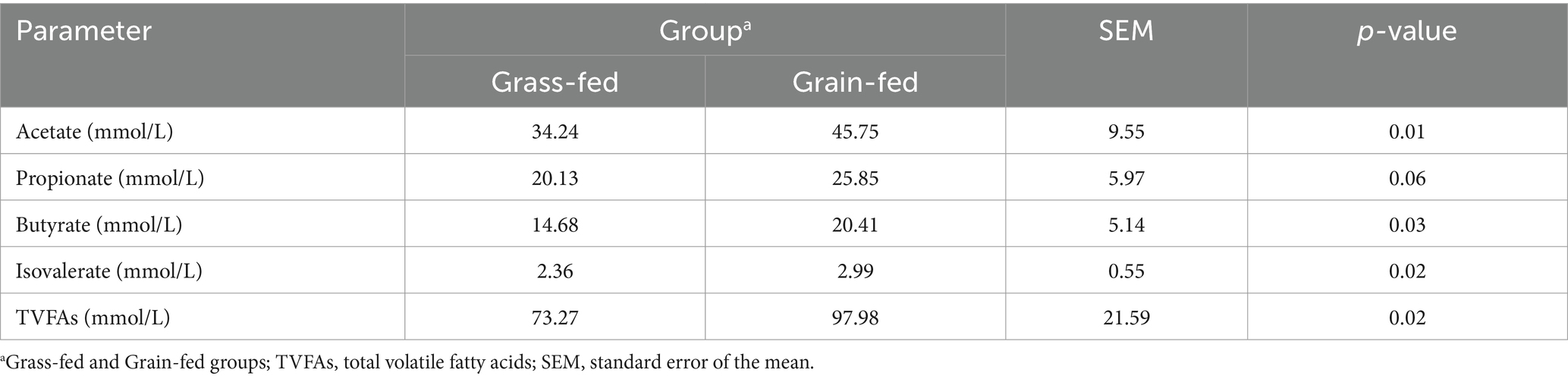
Table 1. Parameters of rumen fluid fermentation in two groups of Sanhe heifers.
Metagenome sequencing obtained 2,349,913,544 reads, with 117,495,677 ± 11,409,254 reads (mean ± standard error of the mean [SEM]) per sample (Supplementary Table S1). After quality control and removal of host genes, we obtained 2,319,213,390 ± 2,531,029 reads. Subsequently, de novo assembly yielded 36,567,343 contigs (N50 length was 609 ± 11 bp) with 1,828,367 ± 65,016 per sample.
The metagenome of Sanhe heifers identified five domains comprising 14 kingdoms; 245 phyla; 490 classes; 1,031 orders; 2,130 families; 6,159 genera; and 37,528 species. The metagenome comprised 86.83% bacteria, 8.44% Archaea, 3.53% eukaryotes, and 1.11% viruses (Supplementary Figure S1A). At the species level, the grain-fed group had 2,544 species, whereas the grass-fed group had 1,874 species (Supplementary Figure S1B). This indicates a higher diversity of microbiota in the grain-fed group, in addition to the shared species between the two groups.
Figure 1A shows archaea and eukaryotes were found to be significantly different between the two groups (adjusted p < 0.01) using the Wilcoxon rank-sum test. PCoA showed separation between the two groups based on bacterial (Figure 1B), archaeal (Figure 1C), and eukaryotic species (Figure 1D). Nevertheless, we focused on comparing bacteria and archaea in the rumen microbiota of grass- and grain-fed Sanhe heifers. Taxon abundance analysis of bacteria revealed 30,255 species belonging to 3,503 genera from 153 phyla, whereas for the archaea taxon, 1,194 species belonging to 245 genera from 23 phyla were identified.
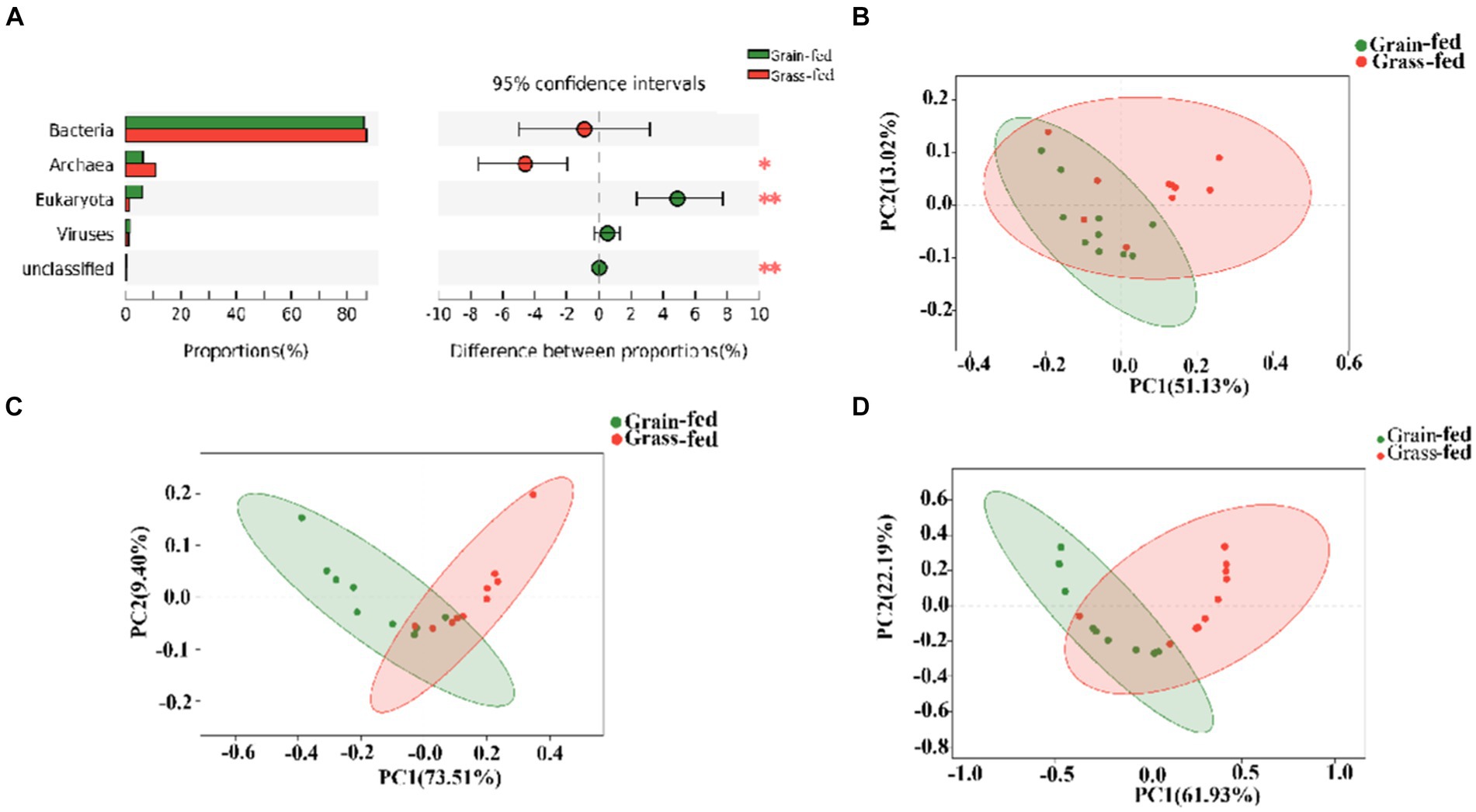
Figure 1. Microbial compositional profiles of grain-fed and grass-fed Sanhe heifers. (A) Comparison of microbial domains between grain-fed and grass-fed Sanhe heifers, with significantly different domains tested using the Wilcoxon rank-sum test and an adjusted p value of <0.05. ** p < 0.01. (B) Bacterial compositional profiles of grain-fed and grass-fed Sanhe heifer samples based on species visualized using principal coordinate analysis (PCoA). (C) Archaeal compositional profiles of grain-fed and grass-fed Sanhe heifer samples, visualized based on species using PCoA. (D) Eukaryota compositional profiles of grain-fed and grass-fed Sanhe heifer samples, visualized on the species level using PCoA.
The most abundant bacterial phyla in the rumen microbiota were Firmicutes (64.99%), Bacteroidetes (16.91%), Actinobacteria (5.14%), Proteobacteria (1.81%), Lentisphaerae (1.06%), and Verrucomicrobia (0.70%). The dominant bacterial families were Lachnospiraceae (18.62%), Ruminococcaceae (9.19%), Clostridiaceae (6.05%), Prevotellaceae (5.98%), Eubacteriaceae (2.85%), Eggerthellaceae (2.46%), Rikenellaceae (2.12%), Erysipelotrichaceae (2.10%), Bacteroidaceae (1.79%), Atopobiaceae (1.18%), Acidaminococcaceae (0.81%), and Selenomonadaceae (0.78%, Figure 2A). Prevotella (5.33%), Butyrivibrio (3.43%), Clostridium (3.16%), Ruminococcus (2.66%), Eubacteroides (2.31%), Sarcina (1.58%), Olsenella (1.05%), and Succiniclasticum were the dominant bacterial genera (0.67%, Figure 2B). The dominant bacterial species were Clostridiales_bacterium (13.55%), Lachnospiraceae (5.48%), Bacteroidales_bacterium (2.01%), Ruminococcaceae_bacterium (1.89%), Clostridia_bacterium (1.64%), Rikenellaceae_bacterium (1.52%), Sarcina_sp._DSM_11001 (1.25%), Bacterium_F082 (1.17%), Eggerthellaceae_bacterium (1.04%), Lentisphaerae_bacterium (0.83%), and Succiniclasticum_bacterium (0.67%; Figure 2C).
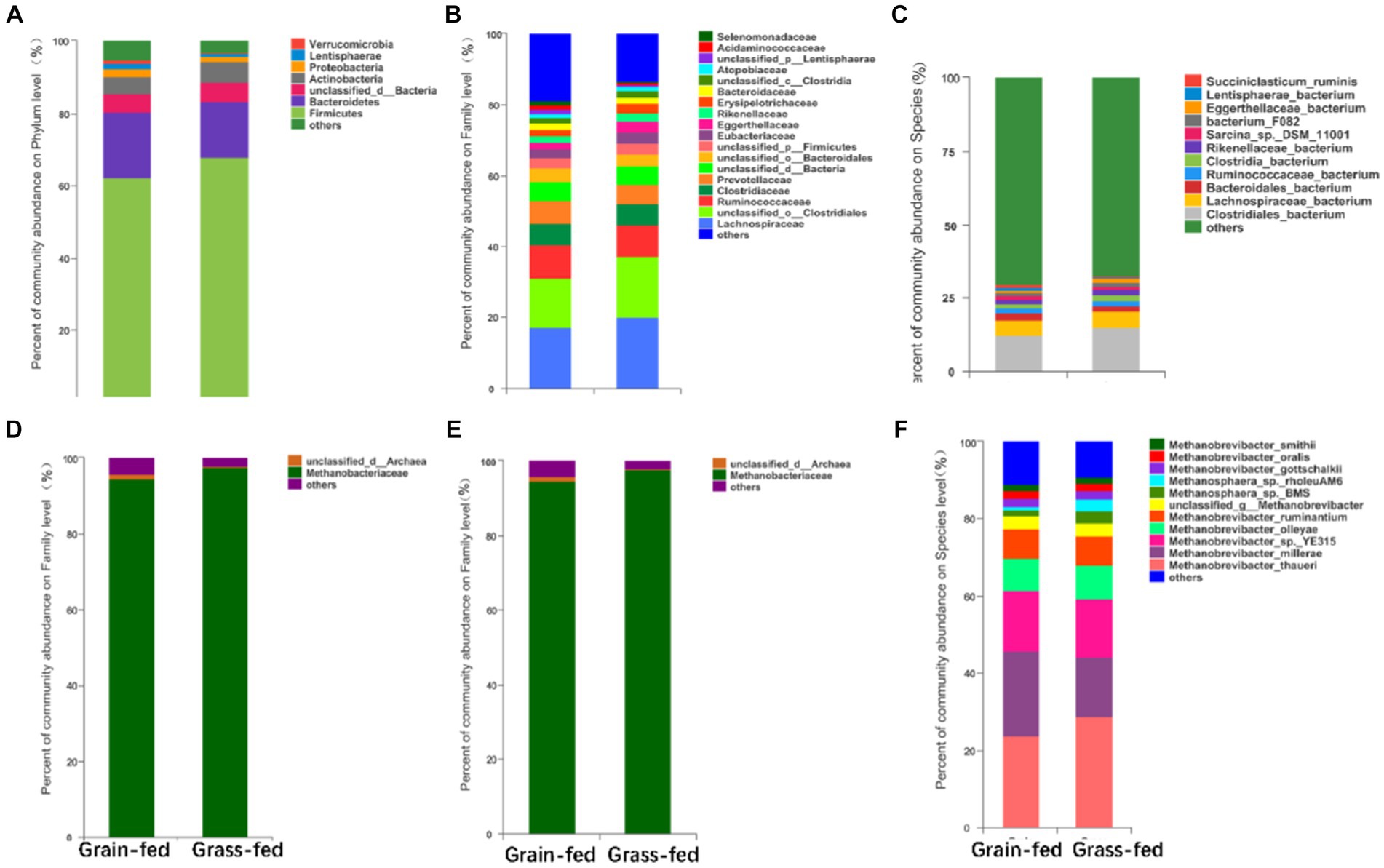
Figure 2. Compositional profiles of the rumen microbiome between grain-fed and grass-fed Sanhe heifers. Relative abundance of (A) phyla, (B) family, and (C) genus of bacterial species; Relative abundance of (D) phyla, (E) family, and (F) genus of archaeal species.
The differential abundance comparison analysis of bacteria at the phylum level revealed that the abundances of Proteobacteria, Lentisphaerae, Verrucomicrobia, Planctomycetes, Candidatus Saccharibacteria, Fibrobacteres, Fusobacteria, Cyanobacteria, Candidatus Gracilibacteria, Elusimicrobia, Candidatus Hydrogenedentes, Armatimonadetes, Kiritimatiellaeota, Candidatus Marinimicrobia, and Candidatus Nealsonbacteria were significantly higher in the rumen of the grain-fed group than those in the rumen of the grass-fed group (adjusted p < 0.05; Figure 3A). At the family level, Lachnospiraceae, Eubacteriaceae, and Eggerthellaceae were highly abundant in the grass-fed group, while the abundance of Acidaminococcaceae, Selenomonadaceae, Fibrobacteraceae, and Mycoplasmataceae were high in the grain-fed group (adjusted p < 0.05; Figure 3B). At the species level, Clostridia_bacterium, Eggerthellaceae_bacterium, Erysipelotrichaceae_bacterium, Erysipelotrichaceae_bacterium_NK3D112, Clostridiales_bacterium_NK3B98, Butyrivibrio_fibrisolvens, Butyrivibrio_sp._AE2032, and [Eubacterium]_cellulosolvens were significantly enriched in the grass-fed group. In contrast, Bacteroidales_bacterium, Lentisphaerae_bacterium, Succiniclasticum_ruminis, Ruminocoaceae_bacterium_P7, Verrucomicrobia_bacterium, and Eubacterium_sp. showed significant enrichment in the grain-fed Sanhe heifers (p < 0.05; Figure 3C).
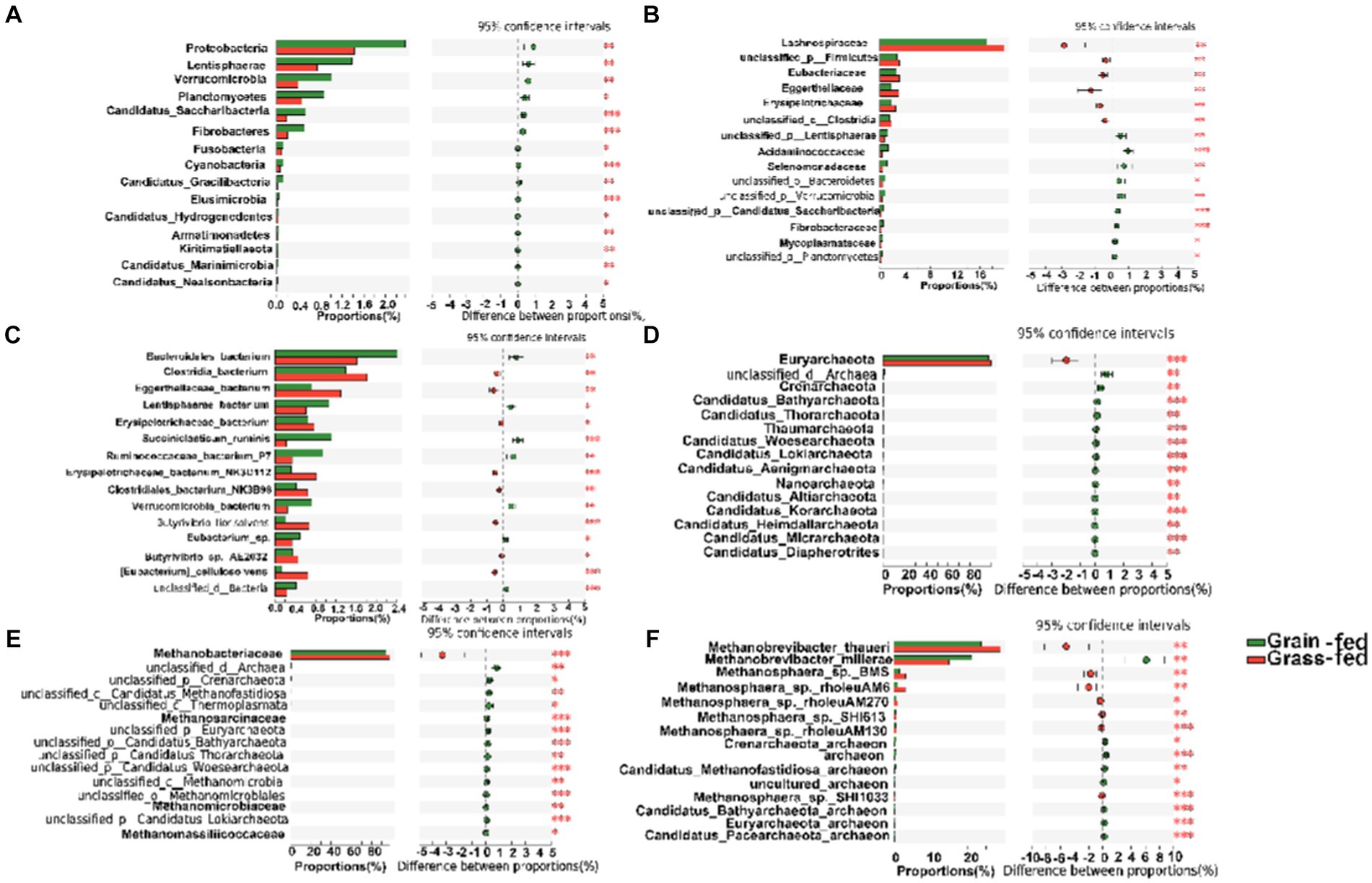
Figure 3. Comparative analysis of the composition of rumen bacteria and archaea between grain-fed and grass-fed Sanhe heifers. (A) Comparison of bacterial composition at phylum level. (B) Comparison of bacterial composition at family level. (C) Comparison of bacterial composition at species level. (D) Comparison of archaeal composition at phylum level. (E) Comparison of archaeal composition at family level. (F) Comparison of archaeal composition at species level. Statistical significance was assessed using the Wilcoxon rank-sum test, with an adjusted p value of < 0.05. ** p < 0.01.
The differential analysis of the abundance of archaea in the rumen of heifers in two feeding regimens identified Euryarchaeota (98.13%) as the most abundant archaeal phylum, which was significantly higher in the rumen of grass-fed heifers than in that of grain-fed heifers (adjusted p < 0.05; Figures 2D, 3D). At the family level, the abundance of Methanobacteriaceae (95.87%) was significantly enhanced in the grass-fed Sanhe heifers, whereas the abundances of other differential families were all significantly high in the rumen of the grain-fed groups (adjusted p < 0.05; Figures 2E, 3E). At the species level (relative abundance >1%), the abundances of Methanobrevibacter_thauer (26.18%), the most abundant archaeal species, followed by Methanosphaera_sp._BMS (2.31%) and Methano sphaera_sp._rholeuAM6 (1.98%) were significantly higher in the rumen of the grass-fed group than those in the rumen of the grain-fed group. In contrast, Methanobrevibacter_millerae (17.99%) was highly abundant in the grain-fed Sanhe heifers (adjusted p < 0.05, Figures 2F, 3F).
The comparison of the KEGG profiles and genes encoding CAZymes identified 329 endogenous third-level pathways as rumen microbial metabolic pathways (Supplementary Table S2). These pathways belonged to six first-level categories.
At the second level of KEGG pathways, 46 functional categories were identified, including “Global and overview maps,” “Carbohydrate metabolism,” “Amino acid metabolism,” “Replication and repair,” “Energy metabolism,” and “Metabolism of cofactors and vitamins.” PCoA indicated a significant separation in the functional potential between the two feed systems (p < 0.05; Supplementary Figure S2). Figure 4A displays the top 15 KEGG pathways that were significantly different at the second level. At the third level of KEGG analysis, the top 15 significantly different pathways included “Metabolic pathways,” and “Biosynthesis of secondary metabolites” (Figure 4B). These pathways were enriched in the grass-fed group and belong to three level 1 pathways, including “Environmental Information Processing,” “Genetic Information Processing,” and “Cellular Processes.”
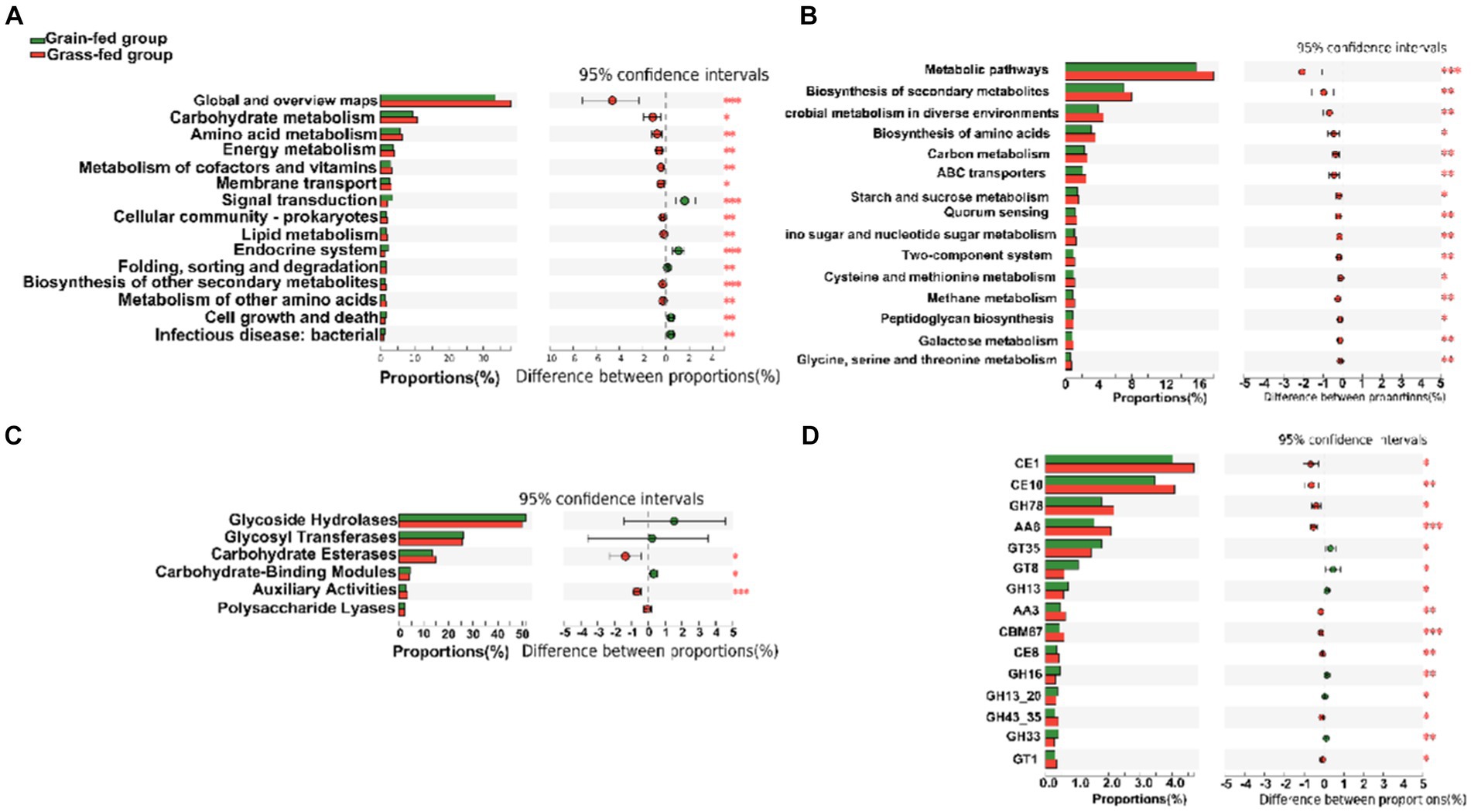
Figure 4. The comparison of KEGG pathways and carbohydrate-active enzymes (CAZy) between grain-fed and grass-fed Sanhe heifers; (A) The top 15 KEGG pathways at the second level that were found to be significantly different between the two groups; (B) The top 15 of significantly different KEGG pathways at KEGG third-level; (C) The carbohydrate-active enzyme (CAZyme) profiles at the class level; (D) The top 15 of significantly different CAZyme profiles at the family level.
For CAZyme profiles at the family level, 590 genes encoding CAZymes were identified (Supplementary Table S3). Among these, 22 encoded auxiliary activity (AA), 74 encoded carbohydrate-binding modules (CBM), 16 encoded carbohydrate esterases (CE), 259 encoded glycoside hydrolases (GH), 90 encoded glycosyltransferases (GT), and 81 encoded polysaccharide lyases (PL). Six types of CAZymes were identified in the two groups at the class level. Grass-fed Sanhe heifers had an enhanced abundance of GEs and AAs (p < 0.05), whereas grain-fed Sanhe heifers exhibited an increased abundance of CBMs (p < 0.05, Figure 4C). Furthermore, among the top 15 significantly different CAZyme profiles (Figure 4D), nine were higher in the rumens of the grass-fed group (p < 0.05), including three CE- (CE1, CE10, and CE8), two GH- (GH78 and GH43_35), two AA- (AA6 and AA3), one CBM- (CBM67), and one GT- (GT1) encoding CAZymes. In contrast, six CAZymes encoding GTs (GT35 and GT8) and GHs (GH13, GH16, GH13_20, and GH33) were highly abundant in the grain-fed group (p < 0.05).
Spearman’s rank correlations were computed to evaluate the relationships between the top 15 differential microbial species and metabolic pathways within the network. Notably, species affiliated with Bacteroidetes, Firmicutes, and Actinobacteria exhibited robust correlations with four primary KEGG pathways: metabolism, environmental information processing, genetic information processing, and cellular processes (|r| > 0.5, p < 0.05; Figure 5).
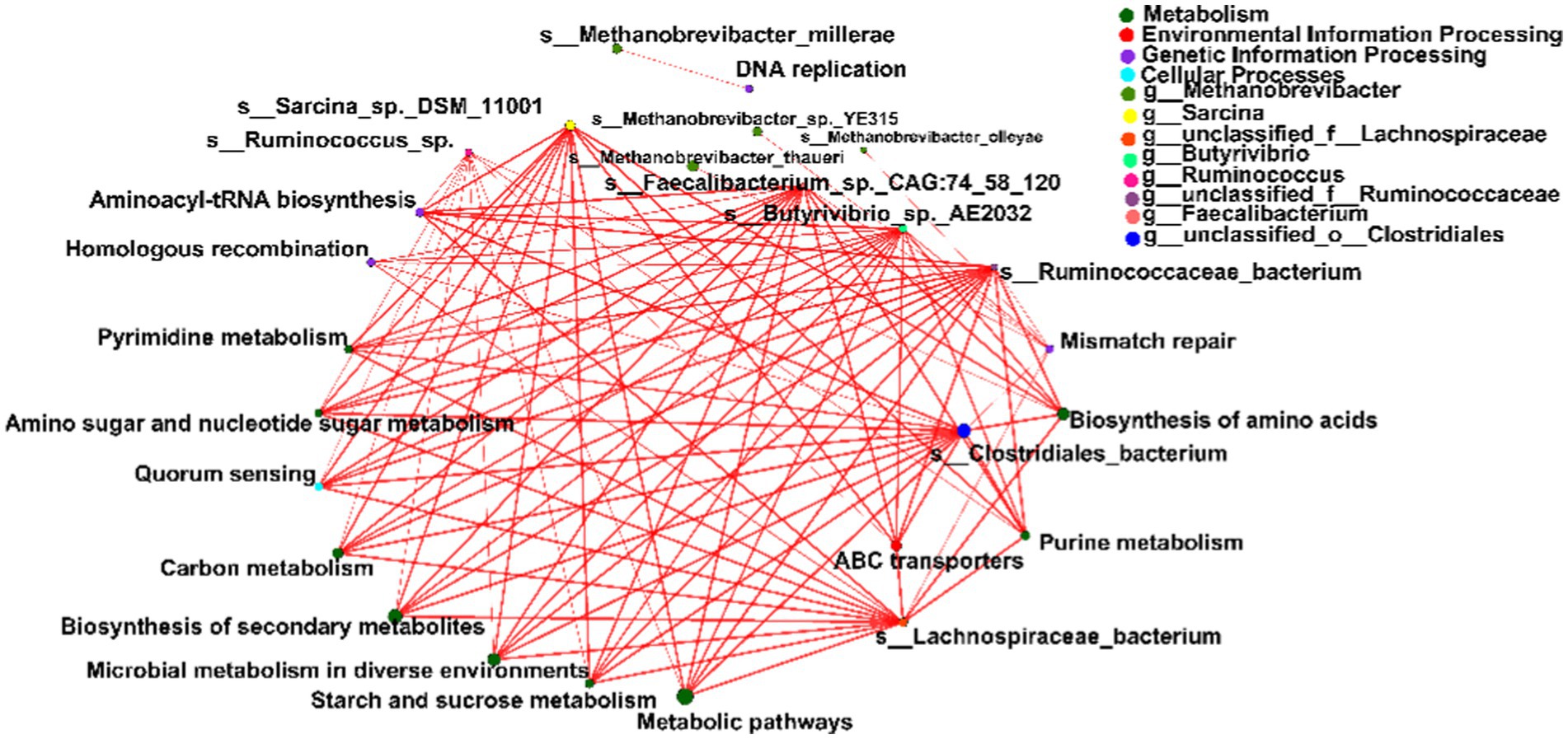
Figure 5. Spearman’s rank correlation constructed between the top 15 differential microbial species and metabolic pathways. Significant differences were determined using the Wilcoxon rank-sum test with an adjusted p value of < 0.05. ** p < 0.01.
These rumen metabolites varied significantly between the two groups (Figures 6A,B). Statistical analysis and VIP values from the OPLS-DA analysis supported this finding (Supplementary Figure S3). In total, 2,502 compounds were identified in the ruminal metabolome. After the t-test and VIP filtering for the relative concentrations of rumen metabolites, 1,130 metabolites were significantly different between the two groups of Sanhe heifers (p < 0.05, VIP > 1, FC > 2, or FC < 0.5). Among these, 256 metabolites were identified in the negative mode, of which 178 were enriched in the grass-fed group and 78 in the grain-fed group. A total of 874 different metabolites were identified in the positive mode, of which 590 and 284 were enriched in the grass- and grain-fed groups, respectively. Metabolic pathway analysis revealed the enrichment of 20 pathways (Figure 6C), Table 2 displays the differential metabolites identified in the top 20 enriched KEGG pathways. Among these, 68 metabolites were downregulated, and 25 were upregulated.
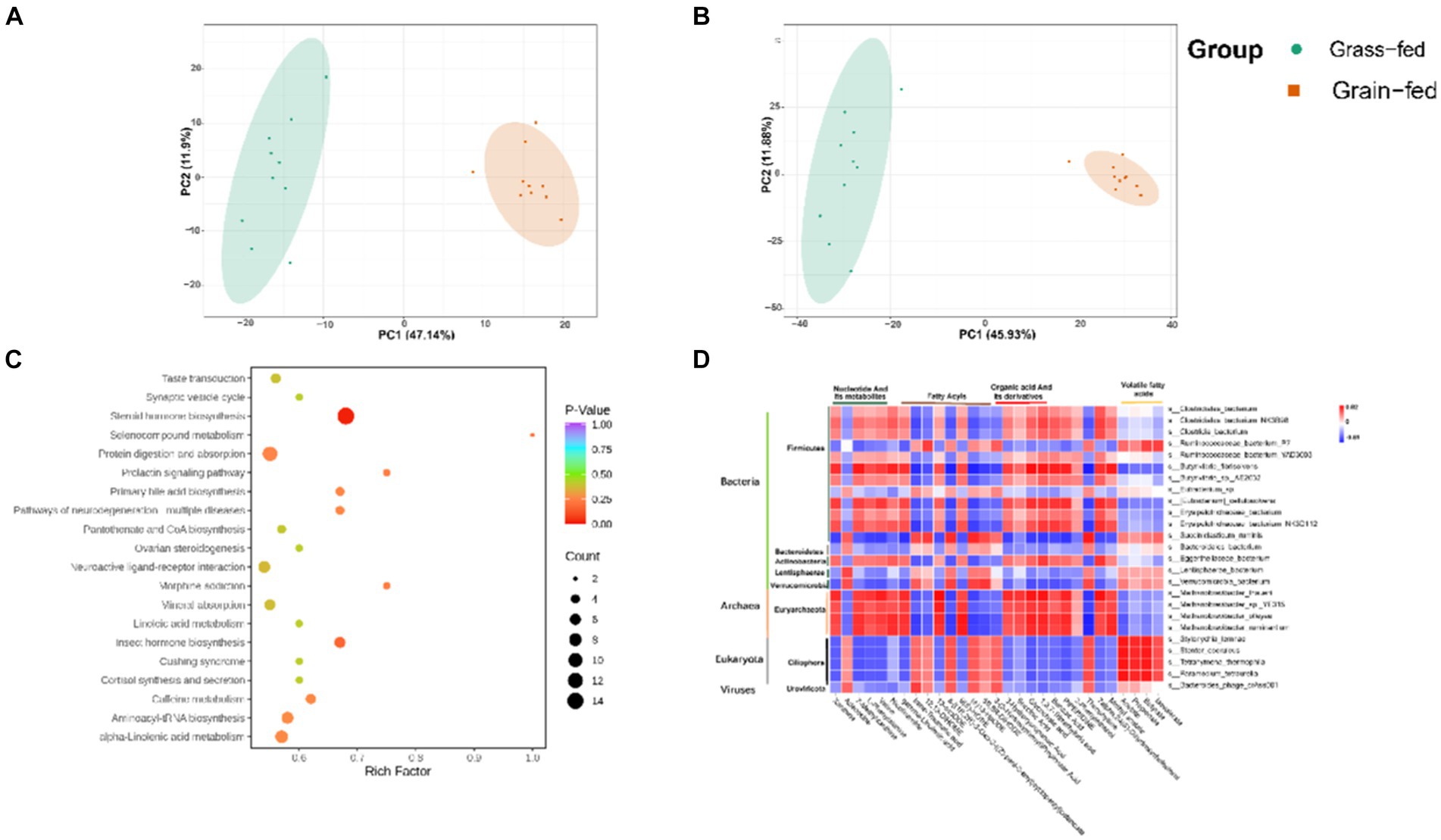
Figure 6. Rumen metabolome comparison of grass-fed and grain-fed Sanhe heifers. (A) Scatter plots of the principal component analysis (PCA) model based on all identified metabolite features of rumen samples from the two groups (negative mode); (B) Scatter plots of the PCA model based on all identified metabolite features of rumen samples from the two groups (positive mode); (C) The pathway enrichment analysis conducted using the significantly different rumen metabolites between grass-fed and grain-fed Sanhe heifers; (D) The relationship between rumen fermentation parameters, the top 25 different rumen microbiota, and the top 25 significantly different rumen metabolites using Spearman’s rank correlations.
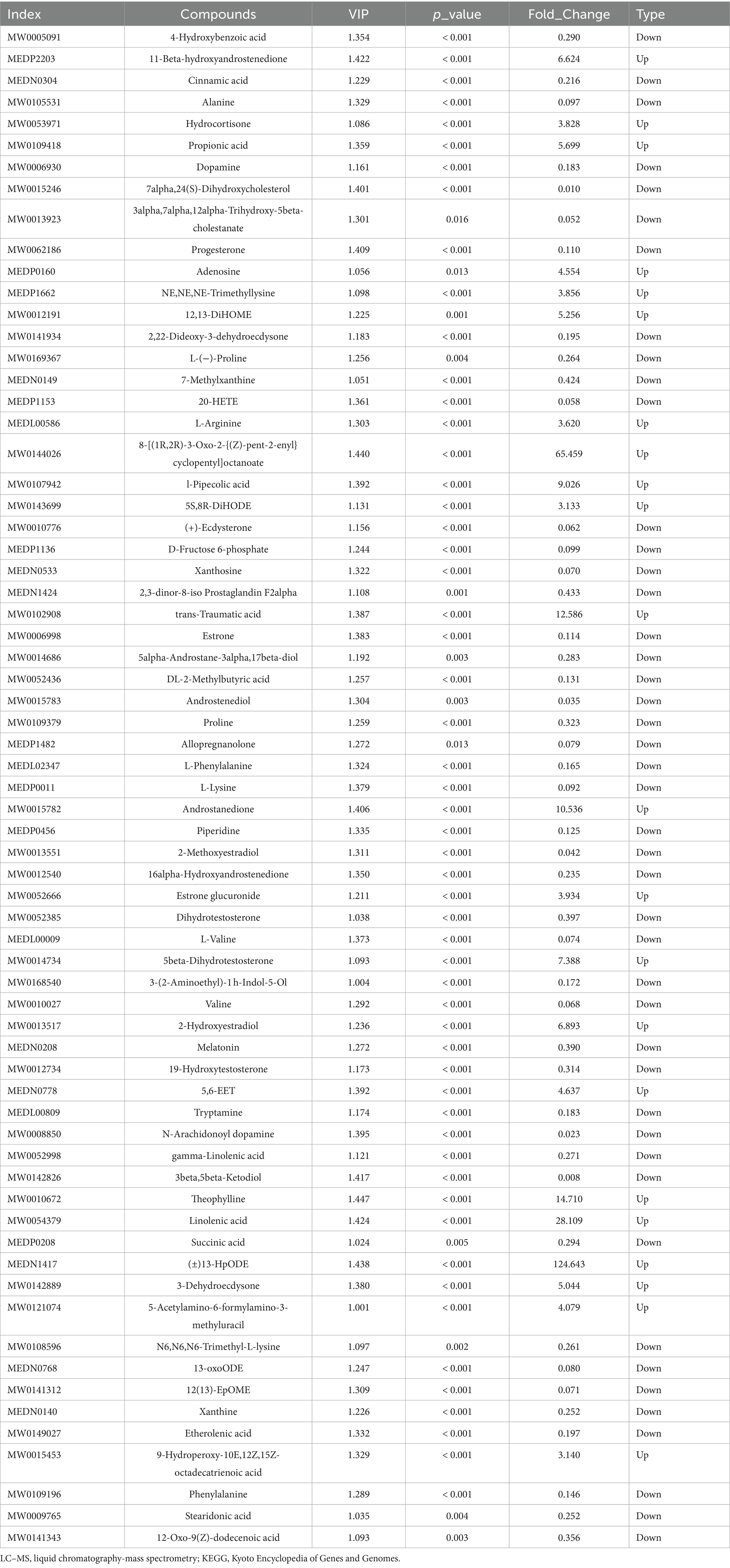
Table 2. Differential metabolites determined using hydrophilic interaction LC–MS analysis of the top 20 enriched differential KEGG pathways between two distinct groups.
Spearman’s rank correlations were determined among rumen fermentation parameters, the top 25 significant rumen metabolites, and the top 25 rumen microbiota. As shown in Figure 6D, 16 bacteria species (Clostridiales_bacterium, Clostridiales_bacterium_NK3B98, Clostridia_bacterium, Ruminococcaceae_bacterium_P7, Ruminococcaceae_bacterium_YAD3003, Butyrivibrio_fibrisolvens, Butyrivibrio_sp._AE2032, Eubacterium_sp., [Eubacterium]_cellulosolvens, Erysipelotrichaceae_bacterium, Erysipelotrichaceae_bacterium, Erysipelotrichaceae_bacterium_NK3D112, Succiniclasticum_ruminis, Bacteroidales_bacterium, Eggerthellaceae_bacterium, Lentisphaerae_bacterium, and Verrucomicrobia_bacterium); four Archaeon species (Methanobrevibacter_thaueri, Methanobrevibacter_sp._YE315, Methanobrevibacter_olleyae, and Methanobrevibacter_ruminantium); four Eukaryota species (Stylonychia_lemnae, Stentor_coeruleus, Tetrahymena_thermophila, and Paramecium_tetraurelia); and one virus species (Bacteroides_phage_crAss001) were identified to have a strong association with the top 25 different metabolites. These metabolites included five nucleotides and their metabolites, two amino acids and their metabolites, eight fatty acids, five organic acids and their derivatives, one alcohol and amine, one heterocyclic compound, one phenolic acid, one heterocyclic compound, one hormone and its related compounds, and one aldehyde, one ketone, and one ester. The microbiota in this study belonged to five phyla: Firmicutes, Bacteroidetes, Actinobacteria, Lentisphaerae, and Verrucomicrobia. These phyla are primarily associated with the degradation of carbohydrates. Additionally, the microbiota species belonging to Archaea was Euryarchaeota, which is known to be involved in methane production.
Dietary feed type compositions affect not only the available fermentation substrates, but also the ruminal microenvironment, whereas VFA profiles regulate the metabolic pathways of microbiota (Newbold and Ramos-Morales, 2020). In this study, we provide evidence supporting our hypothesis, i.e., that diet-induced changes in the rumen microbiota of Sanhe heifers explain the different effects of two feeding regimes (i.e., grain and grass) on the VFA and metabolite content of the host.
Intensive feeding improves the performance of cattle and provides economic benefits. In this study, grain-fed Sanhe heifers produced more VFA than grass-fed Sanhe heifers, which is consistent with the results of previous studies (Wang et al., 2020). The rumen microbiota plays a crucial role in converting feed into VFAs and provides energy to the ruminants. Consistent with previous studies (Liu et al., 2019), our study demonstrated that rumen volatile acid production in Sanhe heifers differed significantly under different feeding regimes; this differentiation could be attributed to taxonomic and functional differences among the microbes that are supported by the two feeding regimes.
We found a higher diversity of microbiota species in grain-fed Sanhe heifers than in grass-fed Sanhe heifers; however, this result is inconsistent with a previous study (Xu et al., 2021). At the family level, we observed greater abundances of Lachnospiraceae, Eubacteriaceae, Eggerthellaceae, and Erysipelotrichaceae in the grass-fed group. This finding is expected as the diet exclusively consisted of various grasses, making it conducive to bacterial members associated with fiber degradation. Additionally, it has been suggested that Lachnospiraceae strains present in the rumen might be capable of bio-hydrogenating fatty acids (Boeckaert et al., 2009). Lachnospiraceae (Vacca et al., 2020) and Eubacteriaceae (Zeng et al., 2021) contain butyrate producers. Moreover, Eggerthellaceae are involved in the transformation of dietary polyphenols (Selma et al., 2014) and are potential probiotics (Langella et al., 2019). We also showed higher abundances of Acidaminococcaceae, Selenomonadaceae, Fibrobacteraceae, and Mycoplasmataceae in the grain-fed group. Acidaminococcaceae, an important glutamate-fermenting family of bacteria, produces ammonia as the principal end-product through glutamate fermentation (Zhang L. et al., 2022). Selenomonadaceae are common gut inhabitants that are abundant in animals on a high-starch diet and utilize saccharides and lactate (Korpela et al., 2014). Mycoplasmataceae are related to the host immune system (Wood et al., 2021). All known methanogens are the members of the Euryarchaeota (Mizrahi et al., 2021). In this study, we revealed that the abundance of Methanobacteriaceae, the most prominent family of methanogens, was higher in the grass-fed group than in the grain-fed group. This result explains why grass-fed cattle emit more methane from the perspective of microbial microbiota (Capper, 2012). Methane emissions result in a large loss of energy and decrease animal efficiency (Zhou et al., 2009), which is consistent with our finding that grass-fed Sanhe heifers produce fewer VFAs and more methane than grain-fed Sanhe heifers. However, there are reports that grass-fed systems can decrease carbon footprints (Lynch, 2019).
Metagenomic analyses have been successfully used to determine the composition and role of various microbiome in the rumen (Li and Guan, 2017; Shen et al., 2020). Mu et al. (2022) found that the feeding structure of a high-concentrate diet affected ruminant microbiome composition. We revealed a higher number of KEGG pathways in the grass-fed group compared with the grain-fed group. Most of these pathways are associated with carbohydrate degradation, which may be caused by the different carbohydrate compositions of diets under different feeding systems. Additionally, we found that methane metabolism was higher in the grass-fed group, which is consistent with our previous results of an increased abundance of methane-producing microbiota in the grass-fed group. Regarding the carbohydrate-active enzymes in the rumen microbiome, the GH-encoding genes had the widest distribution in the genomes (Stewart et al., 2018), mainly comprising enzymes involved in polysaccharide metabolism, including cellulose and starch. GTs are enzymes involved in the production of oligosaccharides, polysaccharides, and glycoconjugates and can catalyze the formation of glycosidic bonds to generate glycosides (Breton et al., 2006). Although no significant differences were observed, the abundances of GHs and GTs in the grain-fed group were higher in our study, which is consistent with the results of a previous study (Wang et al., 2019). The abundance of CBMs was higher in the grain-fed group. CBMs are non-enzymatic entities that can improve enzyme catalytic efficiency by preferentially binding to polysaccharides and increasing enzyme concentration (Jones et al., 2018). In contrast, a higher abundance of CEs, AAs, and PLs was identified in the grass-fed group than that in the grain-fed group, which is consistent with the results of Wang et al. (2019). Together, these results indicate that the rumen microbiome of the grain-fed group exhibited a higher prevalence of enzymes such as GHs, GTs, and CBMs, which are involved in starch degradation and binding. This allows them to efficiently digest a relatively larger proportion of concentrate than the grass-fed group. In contrast, a higher abundance of CEs and PLs as cell wall-degrading enzymes than that in the grain-fed group contributed to the digestion of fiber in the grass group.
The metabolomic analyses also revealed the enrichment of pathways related to carbohydrate metabolism, including “Propanoate metabolism,” the grain-fed group supporting our metagenomic results. Moreover, the enriched pathways are associated with lipid metabolism, including “Steroid hormone biosynthesis,” “Primary bile acid biosynthesis,” “Linoleic acid metabolism,” “Arachidonic acid metabolism,” and “Alpha-linolenic acid metabolism.”
In conclusion, we analyzed the dynamics of the rumen microbiota of Sanhe heifers under two different feeding regimes (i.e., grass and grain) using metagenomic and metabolomic analyses. We found that grain intensified rumen fermentation, and that the composition and function of the rumen microbiota varies depending on the feeding system, with notable differences in metabolic pathways, including carbohydrate digestion, amino acid metabolism, and methane production. Other studies employing non-targeted metabolomics have also reported similar outcomes. In summary, omics analysis has revealed noteworthy distinctions in both the composition of the rumen microbiota and their resultant metabolites under different feeding regimes.
The original contributions presented in the study are publicly available. This data can be found at: https://www.ncbi.nlm.nih.gov, BioProject: PRJNA942501.
The study protocol was approved by the Ethical Committee of China Agricultural University (project number AW22121202-1-2). The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
XZ: Writing – original draft. WW: Writing – review & editing. YW: Writing – review & editing. ZC: Writing – review & editing. HY: Writing – review & editing. SL: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The Nation Key R&D Program of China (No. 2022YFD 1301001) is supported by the earmarked fund for the China Agriculture Research System of MOF and MARA (CARS36).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1336278/full#supplementary-material
Belanche, A., Kingston-Smith, A. H., Griffith, G. W., and Newbold, C. J. A. (2019). A multi-kingdom study reveals the plasticity of the rumen microbiota in response to a shift from non-grazing to grazing diets in sheep. Front. Microbiol. 10:122. doi: 10.3389/fmicb.2019.00122
Boeckaert, C., Morgavi, D. P., Jouany, J. P., Maignien, L., Boon, N., and Fievez, V. (2009). Role of the protozoan Isotricha prostoma, liquid-, and solid-associated bacteria in rumen biohydrogenation of linoleic acid. Animal 3, 961–971. doi: 10.1017/S1751731109004285
Breton, C., Snajdrová, L., Jeanneau, C., Koca, J., and Imberty, A. (2006). Structures and mechanisms of glycosyltransferases. Glycobiology 16, 29R–37R. doi: 10.1093/glycob/cwj016
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Capper, J. L. (2012). Is the grass always greener? Comparing the environmental impact of conventional, natural and grass-fed beef production systems. Animals (Basel) 2, 127–143. doi: 10.3390/ani2020127
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Erwin, E. S., Marco, G. J., and Emery, E. M. (1961). Volatile fatty acid analyses of blood and rumen fluid by gas chromatography. J. Dairy Sci. 44, 1768–1771. doi: 10.3168/jds.S0022-0302(61)89956-6
Fernando, S. C., Purvis, H. T., Najar, F. Z., Sukharnikov, L. O., Krehbiel, C. R., Nagaraja, T. G., et al. (2010). Rumen microbial population dynamics during adaptation to a high-grain diet. Appl. Environ. Microbiol. 76, 7482–7490. doi: 10.1128/AEM.00388-10
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Guo, C. Y., Ji, S. K., Yan, H., Wang, Y. J., Liu, J. J., Cao, Z. J., et al. (2020). Dynamic change of the gastrointestinal bacterial ecology in cows from birth to adulthood. Microbiology 9:e1119. doi: 10.1002/mbo3.1119
Holman, D. B., Yang, W., and Alexander, T. W. (2019). Antibiotic treatment in feedlot cattle: a longitudinal study of the effect of oxytetracycline and tulathromycin on the fecal and nasopharyngeal microbiota. Microbiome 7:86. doi: 10.1186/s40168-019-0696-4
Hu, L., Ma, Y., Liu, L., Kang, L., Brito, L. F., Wang, D., et al. (2019). Detection of functional polymorphisms in the hsp70 gene and association with cold stress response in inner-Mongolia Sanhe cattle. Cell Stress Chaperones 24, 409–418. doi: 10.1007/s12192-019-00973-5
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Jami, E., Israel, A., Kotser, A., and Mizrahi, I. (2013). Exploring the bovine rumen bacterial community from birth to adulthood. ISME J. 7, 1069–1079. doi: 10.1038/ismej.2013.2
Jewell, K. A., McCormick, C. A., Odt, C. L., Weimer, P. J., and Suen, G. (2015). Ruminal bacterial community composition in dairy cows is dynamic over the course of two lactations and correlates with feed efficiency. Appl. Environ. Microbiol. 81, 4697–4710. doi: 10.1128/AEM.00720-15
Jones, D. R., Thomas, D., Alger, N., Ghavidel, A., Inglis, G. D., and Abbott, D. W. (2018). SACCHARIS: an automated pipeline to streamline discovery of carbohydrate active enzyme activities within polyspecific families and de novo sequence datasets. Biotechnol. Biofuels 11:27. doi: 10.1186/s13068-018-1027-x
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. doi: 10.1093/nar/gkr988
Korpela, K., Flint, H. J., Johnstone, A. M., Lappi, J., Poutanen, K., Dewulf, E., et al. (2014). Gut microbiota signatures predict host and microbiota responses to dietary interventions in obese individuals. PLoS One 9:e90702. doi: 10.1371/journal.pone.0090702
Langella, P., Guarner, F., and Martín, R. (2019). Editorial: next-generation probiotics: from commensal bacteria to novel drugs and food supplements. Front. Microbiol. 10:1973. doi: 10.3389/fmicb.2019.01973
Li, F., and Guan, L. L. (2017). Metatranscriptomic profiling reveals linkages between the active rumen microbiome and feed efficiency in beef cattle. Appl. Environ. Microbiol. 83, e00061–e00017. doi: 10.1128/AEM.00061-17
Li, R., Li, Y., Kristiansen, K., and Wang, J. (2008). SOAP: short oligonucleotide alignment program. Bioinformatics 24, 713–714. doi: 10.1093/bioinformatics/btn025
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, F., Neves, A. L. A., Ghoshal, B., and Guan, L. L. (2018). Symposium review: mining metagenomic and metatranscriptomic data for clues about microbial metabolic functions in ruminants. J. Dairy Sci. 101, 5605–5618. doi: 10.3168/jds.2017-13356
Liu, C., Wu, H., Liu, S., Chai, S., Meng, Q., and Zhou, Z. (2019). Dynamic alterations in yak rumen bacteria community and metabolome characteristics in response to feed type. Front. Microbiol. 10:1116. doi: 10.3389/fmicb.2019.01116
Lynch, J. (2019). Availability of disaggregated greenhouse gas emissions from beef cattle production: a systematic review. Environ. Impact Assess. Rev. 76, 69–78. doi: 10.1016/j.eiar.2019.02.003
Mackie, R. I., and White, B. A. (1990). Recent advances in rumen microbial ecology and metabolism: potential impact on nutrient output. J. Dairy Sci. 73, 2971–2995. doi: 10.3168/jds.S0022-0302(90)78986-2
Mao, S. Y., Zhang, R. Y., Wang, D. S., and Zhu, W. Y. (2013). Impact of subacute ruminal acidosis (SARA) adaptation on rumen microbiota in dairy cattle using pyrosequencing. Anaerobe 24, 12–19. doi: 10.1016/j.anaerobe.2013.08.003
Mizrahi, I., Wallace, R. J., and Moraïs, S. (2021). The rumen microbiome: balancing food security and environmental impacts. Nat. Rev. Microbiol. 19, 553–566. doi: 10.1038/s41579-021-00543-6
Morgavi, D. P., Kelly, W. J., Janssen, P. H., and Attwood, G. T. (2013). Rumen microbial (meta)genomics and its application to ruminant production. Animal 7, 184–201. doi: 10.1017/S1751731112000419
Mu, Y., Qi, W., Zhang, T., Zhang, J., and Mao, S. (2022). Multi-omics analysis revealed coordinated responses of rumen microbiome and epithelium to high-grain-induced subacute rumen acidosis in lactating dairy cows. mSystems 7:e0149021. doi: 10.1128/msystems.01490-21
Newbold, C. J., and Ramos-Morales, E. (2020). Review: ruminal microbiome and microbial metabolome: effects of diet and ruminant host. Animal 14, s78–s86. doi: 10.1017/S1751731119003252
Noguchi, H., Park, J., and Takagi, T. (2006). MetaGene: prokaryotic gene finding from environmental genome shotgun sequences. Nucleic Acids Res. 34, 5623–5630. doi: 10.1093/nar/gkl723
Petri, R. M., Schwaiger, T., Penner, G. B., Beauchemin, K. A., Forster, R. J., McKinnon, J. J., et al. (2013). Changes in the rumen epimural bacterial diversity of beef cattle as affected by diet and induced ruminal acidosis. Appl. Environ. Microbiol. 79, 3744–3755. doi: 10.1128/AEM.03983-12
Russell, J. B., and Rychlik, J. L. (2001). Factors that alter rumen microbial ecology. Science 292, 1119–1122. doi: 10.1126/science.1058830
Selma, M. V., Beltrán, D., García-Villalba, R., Espín, J. C., and Tomás-Barberán, F. A. (2014). Description of urolithin production capacity from ellagic acid of two human intestinal Gordonibacter species. Food Funct. 5, 1779–1784. doi: 10.1039/C4FO00092G
Shabat, S. K., Sasson, G., Doron-Faigenboim, A., Durman, T., Yaacoby, S., Berg Miller, M. E., et al. (2016). Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 10, 2958–2972. doi: 10.1038/ismej.2016.62
Shen, J., Zheng, L., Chen, X., Han, X., Cao, Y., and Yao, J. (2020). Metagenomic analyses of microbial and carbohydrate-active enzymes in the rumen of dairy goats fed different rumen degradable starch. Front. Microbiol. 11:1003. doi: 10.3389/fmicb.2020.01003
Stewart, R. D., Auffret, M. D., Warr, A., Wiser, A. H., Press, M. O., Langford, K. W., et al. (2018). Assembly of 913 microbial genomes from metagenomic sequencing of the cow rumen. Nat. Commun. 9:870. doi: 10.1038/s41467-018-03317-6
Usman, T., Wang, Y., Liu, C., He, Y., Wang, X., Dong, Y., et al. (2017). Novel SNPs in IL-17F and IL-17A genes associated with somatic cell count in Chinese Holstein and inner-Mongolia Sanhe cattle. J. Anim. Sci. Biotechnol. 8:5. doi: 10.1186/s40104-016-0137-1
Vacca, M., Celano, G., Calabrese, F. M., Portincasa, P., Gobbetti, M., and De Angelis, M. (2020). The controversial role of human gut Lachnospiraceae. Microorganisms 8:573. doi: 10.3390/microorganisms8040573
Wang, L., Zhang, G., Li, Y., and Zhang, Y. (2020). Effects of high forage/concentrate diet on volatile fatty acid production and the microorganisms involved in VFA production in cow rumen. Animals (Basel) 10:223. doi: 10.3390/ani10020223
Wang, L., Zhang, G., Xu, H., Xin, H., and Zhang, Y. (2019). Metagenomic analyses of microbial and carbohydrate-active enzymes in the rumen of Holstein cows fed different forage-to-concentrate ratios. Front. Microbiol. 10:649. doi: 10.3389/fmicb.2019.00649
Wood, A. M., Tang, M., Truong, T., Feldman, C., Pieper, C., and Murtha, A. P. (2021). Vaginal Mycoplasmataceae colonization and association with immune mediators in pregnancy. J. Matern. Fetal Neonatal Med. 34, 2295–2302. doi: 10.1080/14767058.2019.1663820
Xu, C., Liu, W., Sun, B., Zhang, S., Zhang, S., Yang, Y., et al. (2021). Multi-omics analysis reveals a dependent relationship between rumen bacteria and diet of grass- and grain-fed yaks. Front. Microbiol. 12:642959. doi: 10.3389/fmicb.2021.642959
Xu, Q., Wang, Y. C., Liu, R., Brito, L. F., Kang, L., Yu, Y., et al. (2017). Differential gene expression in the peripheral blood of Chinese Sanhe cattle exposed to severe cold stress. Genet. Mol. Res. 16:10. doi: 10.4238/gmr16029593
Yousif, M. H., Li, J. H., Li, Z. Q., Maswayi Alugongo, G. M., Ji, S. K., Li, Y. X., et al. (2018). Low concentration of antibiotics modulates gut microbiota at different levels in pre-weaning dairy calves. Microorganisms 6:118. doi: 10.3390/microorganisms6040118
Zeng, W., He, D., Xing, Y., Liu, J., Su, N., Zhang, C., et al. (2021). Internal connections between dietary intake and gut microbiota homeostasis in disease progression of ulcerative colitis: a review. Food Sci. Human Wellness 10, 119–130. doi: 10.1016/j.fshw.2021.02.016
Zhang, X., Huang, S., Li, S., and Wang, W. (2022). Effects of altitude on the digestion performance, serum antioxidative characteristics, rumen fermentation parameters, and rumen bacteria of Sanhe heifers. Front. Microbiol. 13:875323. doi: 10.3389/fmicb.2022.875323
Zhang, L., Zhuang, Z., Zhang, G., Huang, T., and Fan, D. (2022). Assessment of bidirectional relationships between 98 genera of the human gut microbiota and amyotrophic lateral sclerosis: a 2-sample Mendelian randomization study. BMC Neurol. 22:8. doi: 10.1186/s12883-021-02522-z
Keywords: metabolites, microbiota, grass-fed, grain-fed, Sanhe heifers
Citation: Zhang X, Wang W, Wang Y, Cao Z, Yang H and Li S (2024) Metagenomic and metabolomic analyses reveal differences in rumen microbiota between grass- and grain-fed Sanhe heifers. Front. Microbiol. 15:1336278. doi: 10.3389/fmicb.2024.1336278
Edited by:
Edoardo Pasolli, University of Naples Federico II, ItalyReviewed by:
Shaopu Wang, Sichuan University, ChinaCopyright © 2024 Zhang, Wang, Wang, Cao, Yang and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shengli Li, bGlzaGVuZzA2NzdAMTYzLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.