
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 09 April 2024
Sec. Evolutionary and Genomic Microbiology
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1335997
 Jetsi Mancilla-Rojano1,2
Jetsi Mancilla-Rojano1,2 Víctor Flores3
Víctor Flores3 Miguel A. Cevallos4
Miguel A. Cevallos4 Sara A. Ochoa2
Sara A. Ochoa2 Julio Parra-Flores5
Julio Parra-Flores5 José Arellano-Galindo6
José Arellano-Galindo6 Juan Xicohtencatl-Cortes2*
Juan Xicohtencatl-Cortes2* Ariadnna Cruz-Córdova1,2*
Ariadnna Cruz-Córdova1,2*Introduction: The Acinetobacter calcoaceticus–Acinetobacter baumannii complex, or Acb complex, consists of six species: Acinetobacter baumannii, Acinetobacter calcoaceticus, Acinetobacter nosocomialis, Acinetobacter pittii, Acinetobacter seifertii, and Acinetobacter lactucae. A. baumannii is the most clinically significant of these species and is frequently related to healthcare-associated infections (HCAIs). Clustered regularly interspaced short palindromic repeat (CRISPR) arrays and associated genes (cas) constitute bacterial adaptive immune systems and function as variable genetic elements. This study aimed to conduct a genomic analysis of Acb complex genomes available in databases to describe and characterize CRISPR systems and cas genes.
Methods: Acb complex genomes available in the NCBI and BV-BRC databases, the identification and characterization of CRISPR-Cas systems were performed using CRISPRCasFinder, CRISPRminer, and CRISPRDetect. Sequence types (STs) were determined using the Oxford scheme and ribosomal multilocus sequence typing (rMLST). Prophages were identified using PHASTER and Prophage Hunter.
Results: A total of 293 genomes representing six Acb species exhibited CRISPR-related sequences. These genomes originate from various sources, including clinical specimens, animals, medical devices, and environmental samples. Sequence typing identified 145 ribosomal multilocus sequence types (rSTs). CRISPR–Cas systems were confirmed in 26.3% of the genomes, classified as subtypes I-Fa, I-Fb and I-Fv. Probable CRISPR arrays and cas genes associated with CRISPR–Cas subtypes III-A, I-B, and III-B were also detected. Some of the CRISPR–Cas systems are associated with genomic regions related to Cap4 proteins, and toxin–antitoxin systems. Moreover, prophage sequences were prevalent in 68.9% of the genomes. Analysis revealed a connection between these prophages and CRISPR–Cas systems, indicating an ongoing arms race between the bacteria and their bacteriophages. Furthermore, proteins associated with anti-CRISPR systems, such as AcrF11 and AcrF7, were identified in the A. baumannii and A. pittii genomes.
Discussion: This study elucidates CRISPR–Cas systems and defense mechanisms within the Acb complex, highlighting their diverse distribution and interactions with prophages and other genetic elements. This study also provides valuable insights into the evolution and adaptation of these microorganisms in various environments and clinical settings.
There are more than 50 Acinetobacter species. The Acinetobacter calcoaceticus–Acinetobacter baumannii complex or Acb complex is formed by six species that are phenotypically indistinguishable: Acinetobacter baumannii, Acinetobacter calcoaceticus, Acinetobacter nosocomialis, Acinetobacter pittii, Acinetobacter seifertii, and Acinetobacter lactucae (Villalon et al., 2015).
The Acb complex includes opportunistic pathogens, mainly related to healthcare-associated infections (HCAIs), multidrug-resistant phenotypes, and resistance to desiccation and disinfectants (Manchanda et al., 2010). Other authors have described epidemiological differences among these species, although they frequently share hospital environments (Wisplinghoff et al., 2012; Calix et al., 2019). A. baumannii is the main species with the most clinical importance; it is commonly isolated in intensive care units and is related to illness types such as ventilator-associated pneumonia, meningitis, bloodstream, and urinary tract infections (Moradi et al., 2015). A. baumannii is resistant to a broad spectrum of antibiotics, limiting therapeutic options. Recently, A. baumannii has become resistant to carbapenems and has been included in the list of priority pathogens resistant to antibiotics published by the World Health Organization (World Health Organization, 2017).
The A. baumannii genome shows great plasticity, which exposes it not only to rapid changes due to mutations but also to the transfer and acquisition of exogenous material (Yakkala et al., 2019), that is, the dynamic conversion of genetic information for acquisition and removal (Bondy-Denomy and Davidson, 2014). A. baumannii acquires genetic material through bacteriophages via a transduction mechanism (Chevallereau et al., 2022).
Bacteria can develop adaptive defense mechanisms against when exposed to different exogenous genetic material. The clustered regularly interspaced short palindromic repeats (CRISPR)–CRISPR-associated enzyme (Cas) system has also been associated with the acquisition of mobile genetic material and is an adaptive mechanism of immunity and resistance used by bacteria and archaea against bacteriophages and/or plasmids (Dy et al., 2013; Makarova et al., 2020).
CRISPR systems comprise palindromic elements interspersed with sequences of constant length (Mojica and Montoliu, 2016) flanking the spacers acquired from bacteriophages, plasmids, or any genetic material. The CRISPR–Cas system comprises two main components: a guide RNA (crRNA or gRNA) and genes that encode Cas proteins, which are essential for the adaptation and incorporation of genetic material (Koonin et al., 2017).
The mechanism of action of the CRISPR–Cas system involves three stages: acquisition, expression, and interference. In the acquisition stage, elements that carry genetic material are identified and recognized (protospacer); this information is cut and integrated into the CRISPR locus at the 5′ end followed by the leader sequence. In the expression stage, the sequences that are added to the CRISPR locus are recognized as spacers, which are expressed in the form of primary transcripts or precrRNAs and are cut into smaller fragments (crRNAs) through endonucleases (Waddington et al., 2016). Finally, in the interference stage, when the bacterium receives exogenous genetic material, the crRNA accompanied by Cas proteins binds via base complementarity to the previously acquired sequence, signaling to the nucleases that the external genetic element must cleave (Bhaya et al., 2011). Moreover, the CRISPR system mediates the transfer of genetic material between genomes (Marraffini and Sontheimer, 2008; O'Meara and Nunney, 2019). Genomes displaying a CRISPR array but lacking associated cas genes, it is proposed that bacteria could capture information from bacteriophages and incorporate it into their genome as a prophage. In contrast, bacteria without a CRISPR-Cas system can be infected by bacteriophages, acquiring information that encodes genes linked to resistance and virulence, thereby enhancing their adaptive capacity (Leungtongkam et al., 2020).
CRISPR–Cas IFb has been proposed as a method for subtyping A. baumannii strains, allowing identification of the route of origin and dissemination (Touchon et al., 2014; Karah et al., 2015). The CRISPR–Cas system identified in A. baumannii is characterized by the presence of the cas1, cas2, cas3, cas5, cas7, and cas8 genes, which have been identified in chromosome and plasmid sequences (Mangas et al., 2019). A few studies have provided information about the prevalence of these systems in clinical strains of the Acb complex. This study aimed to perform genomic analysis of the Acb complex to identify and characterize CRISPR arrays and/or the cas genes, using bioinformatics programs.
A flowchart for data collection for the Acb complex genomes, detection and analysis of the CRISPR–Cas systems is shown in Figure 1.
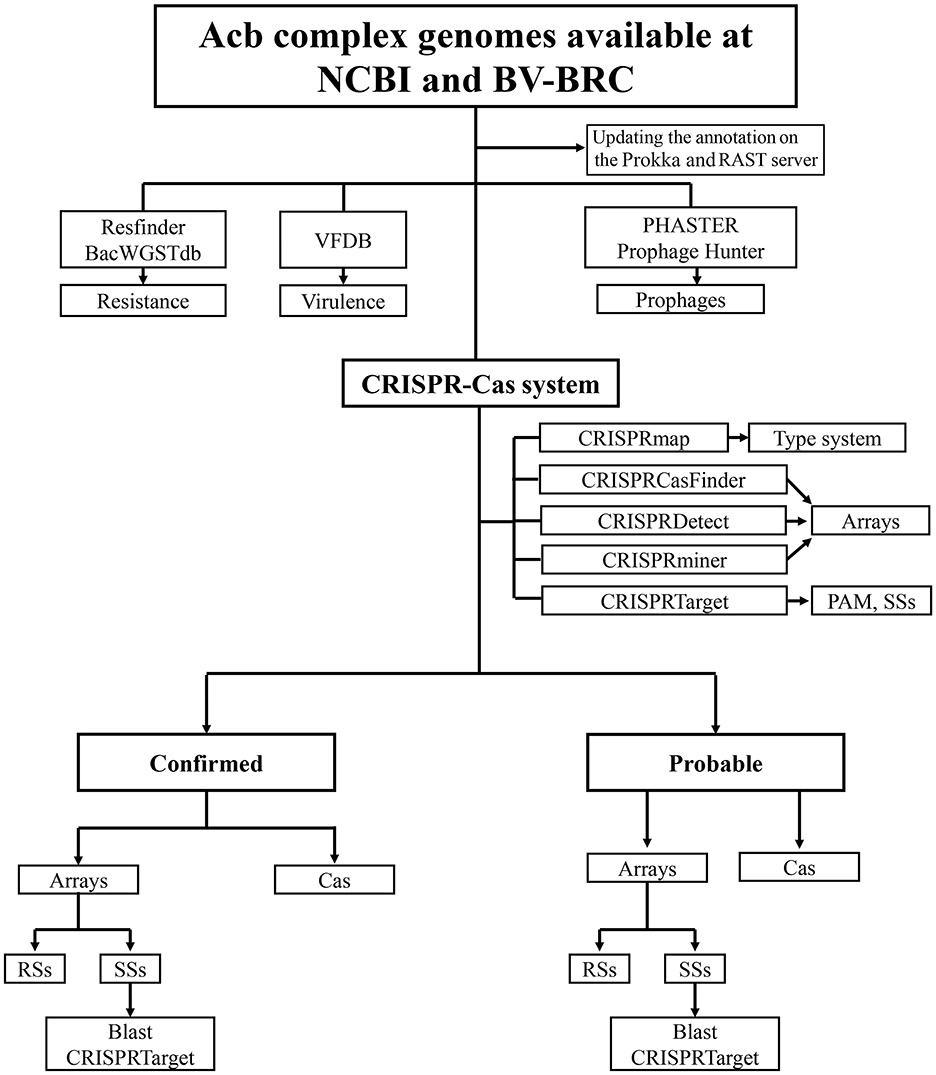
Figure 1. Flowchart for the search for CRISPR–Cas systems. Flowchart indicating data collection for Acb complex genomes, detection and analysis of the CRISPR–Cas systems, resistance and virulence genes, and prophage-associated sequences.
A total of 7,743 genomes, described as “complete” (1,144) and “draft” (6,599), including 952 plasmids of the Acb complex from the National Center for Biotechnology Information (NCBI), and Bacterial and Viral Bioinformatics Resource Center (BV-BRC) databases (https://www.ncbi.nlm.nih.gov/and https://www.bv-brc.org/; accessed on September 2022), were included to screen for sequences associated with CRISPR–Cas systems. These were from the following species: A. baumannii (6,824), A. nosocomialis (246), A. pittii (435), A. calcoaceticus (31), A. seifertii (200), and A. lactucae (7).
The metadata of each strain were manually extracted, including the date, GenBank accession numbers, species, genetic material, strain ID, year of isolation, associated disease, sample, host, collection place, rST, ST, origin, site of isolation, and size (Supplementary Table 1).
Allele designation for sequence typing (ST) in chromosomal genomes was performed by ribosomal multilocus sequence typing (rMLST) (https://pubmlst.org/bigsdb?db=pubmlst_rmlst_seqdef_kiosk) (Jolley et al., 2018). The ST and Clonal Complexes (CC) have been determined using the Oxford scheme (Bartual et al., 2005).
The search for CRISPR systems was carried out in the chromosome and plasmid sequences using the programs CRISPRCasFinder (https://crispr.i2bc.paris-saclay.fr/; Couvin et al., 2018), CRISPRminer (http://www.microbiome-bigdata.com/CRISPRminer/; Zhang et al., 2018), and CRISPRDetect (http://crispr.otago.ac.nz/CRISPRDetect/predict_crispr_array.html; Biswas et al., 2016). The following search parameters were used: minimum length of the repeated sequence, 11 base pairs (bp); maximum length of the repeated sequence, 50 bp; minimum size of spacer sequences based on repeat size, 0.6; maximum size of spacers based on repeat size, 2.5; and maximum percentage of similarity between spacers, 60% (Cruz-López et al., 2021).
The CRISPR-Cas flanking genomic regions were manually reviewed, considering the 20 000 bp sequences upstream and downstream of the arrays. The sequences, predicted structures, and functional annotations of the proteins were analyzed using the UniProt database and I-TASSER server.
The matrices were extracted manually from the output file. The number, size, and location of RSs (repeated sequences) and SSs (spacer sequences) were determined. The RSs were analyzed with the CRISPRDetect program to establish the variations into RSs and associated with a consensus sequence. The SSs were analyzed using the CRISPRTarget program (http://crispr.otago.ac.nz/CRISPRTarget/crispr_analysis.html; Biswas et al., 2013) to identify the protospacer adjacent motif (PAM) and the genes associated with each spacer. The types and subtypes of the CRISPR systems were analyzed with the CRISPRmap program (http://rna.informatik.uni-freiburg.de/CRISPRmap/Input.jsp; Lange et al., 2013) using the consensus RS file as the input file.
The Cas protein sequences in fasta format were obtained from the RefSeq and TIGRFAM databases (ftp://ftp.ncbi.nih.gov/genomes/; http://www.jcvi.org/cgi-bin/tigrfams/index.cgi; Abby and Rocha, 2017) and were identified using the MacSyFINDER program (v 1.0.5) and makeblastdb (v2.2.28) implemented in Python (https://github.com/gem-Pasteur/macsyfinder; Abby and Rocha, 2017). According to the data obtained with this program, classification of the CRISPR–Cas systems was carried out.
The functional annotation was updated by Prokka v1.12 (Seemann, 2014) and RAST programs (Rapid Annotation using Subsystem Technology) (Aziz et al., 2008). The antibiotic resistance genes were identified using ResFinder (https://cge.cbs.dtu.dk/services/ResFinder/; Zankari et al., 2012) and BacWGSTdb (http://bacdb.cn/BacWGSTdb/analysis_single.php; Ruan and Feng, 2016). The virulence genes were identified using the Virulence Factor Database (VFDB) (http://www.mgc.ac.cn/cgi-bin/VFs/genus.cgi?Genus=Acinetobacter; Chen et al., 2005).
The prophages were identified by PHASTER (https://phaster.ca/; Arndt et al., 2016) classified as intact (score >90), incomplete (score 70–90) or related (score <70) prophages, and with Prophage Hunter (https://pro-hunter.genomics.cn/index.php/Home/Index/index.html; Song et al., 2019) classified as active (score of 0.8), ambiguous (score of 0.5–0.8), or inactive (<0.5 score) prophages. The prophage number was determined by removing overlaps of the same prophages. The sequences recognized as related or inactive prophages were eliminated.
The phylogenetic analysis was performed by VBCG (Validated Bacterial Core Genes) (https://github.com/tianrenmaogithub/vbcg, Tian and Imanian, 2023) with validated genes. The tree was inferred using an approximately-maximum-likelihood phylogenetic with FastTree.
The GenBank accession numbers of the genome sequences used in this study are listed in Supplementary Table 1. The data generated in this work are available at the following link: https://github.com/JetsiMancilla/System-CRISPR-Cas.
The data generated with the CRISPRCasFinder, CRISPRDetect, and CRISPRRminer programs allowed the detection of CRISPR arrays (confirmed and probable) and/or Cas proteins. Only in 293 Acb complex genomes described as “complete genomes (108)” or “draft genomes (105),” which included 80 plasmids, were CRISPR–Cas, CRISPR arrays or Cas proteins identified in the following species: A. baumannii (150), A. nosocomialis (42), A. pittii (76), A. calcoaceticus (12), A. seifertii (11), and A. lactucae (2) (Supplementary Table 1).
The analyzed chromosomal genomes (213) had sizes between 3.4 and 4.3 Mb, and the plasmid (80) genomes had sizes between 0.004437 and 0.33 Mb (Supplementary Table 1). The genomes exhibited a GC content of 39% (±0.1). The RAST annotation showed more than 3,690 to 4,181 coding regions in the genomes, linked to between 303 and 323 subsystems. In contrast, the Prokka annotation revealed a range of 3,427 to 5,415 coding regions (https://github.com/JetsiMancilla/System-CRISPR-Cas).
The genomes included in this study were mainly from strains isolated from patients with pneumonia, nosocomial infection, wounds, urinary tract infection, bacteremia, meningitis, or septicemia (Supplementary Table 1). The strains were mainly isolated from blood (18.8%, 55/293), sputum (9.2%, 27/293), wound (4.4%, 13/293), urine (3.7%, 11/293), and the respiratory tract (3.4%, 10/293) (Supplementary Figure 1).
According to the analysis of the 293 genomes, 145 different rSTs were established with the following prevalence values: rST8482 at 8.5% (25/293), rST8237 at 3.4% (10/293), rST8274 and rST8863 at 2.4% (7/293), rST8770 at 2.0% (6/293), and rST31297 at 1.7% (5/293). The rMLST analysis confirmed that the strains belonged to the Acb complex; however, this analysis also showed that rST8378 was present in A. baumannii and A. pittii (Supplementary Table 1). In addition to the rMLST, 18 CC (Clonal Complex) with 130 STs were defined, the most frequently were: ST208 11.5% (15/130), ST231 8.5% (11/130) and ST540 6.2% (8/130), whereas 19 ST belonging to CC92 were found (Table 1, Supplementary Table 1).
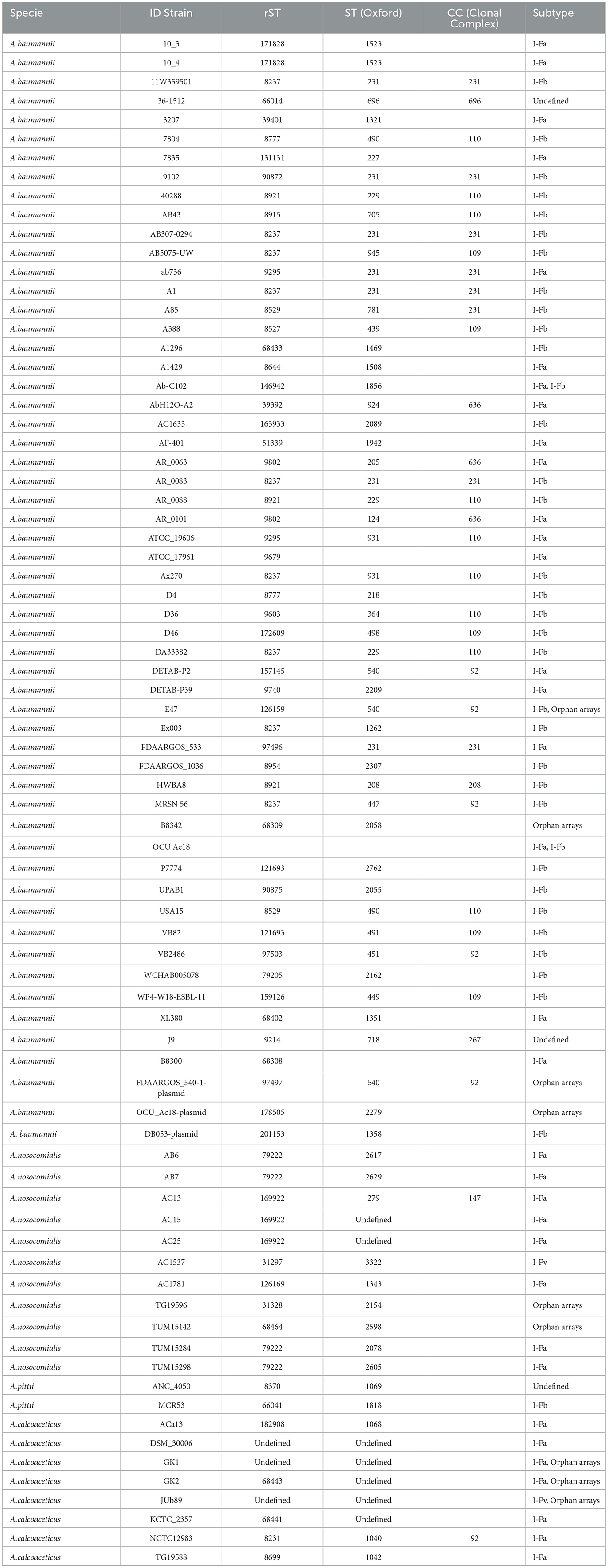
Table 1. CRISPR-Cas systems confirmed in Acb complex genomes.
The CRISPR arrays characterized by RSs (repeat sequences) and SSs (spacer sequences) were confirmed in 26.30% (77/293) of the Acb complex genomes (including three plasmids). Interestingly, more than one CRISPR array was detected in the same genome or plasmid, being a total of 85 CRISPR arrays. Briefly, 70.2% (60/85) of the CRISPR arrays corresponded to A. baumannii, 14.3% (12/85) to A. calcoaceticus, 13.1% (11/85) to A. nosocomialis, and 2.4% (2/85) to A. pittii (Supplementary Table 2).
The number of RSs ranged from three to 158, with lengths of 24 to 29 bp in the CRISPR arrays. The RSs consensus sequences obtained by CRISPRDetect were compared with the RS sequences identified in this study (Figure 2, Supplementary Table 2).
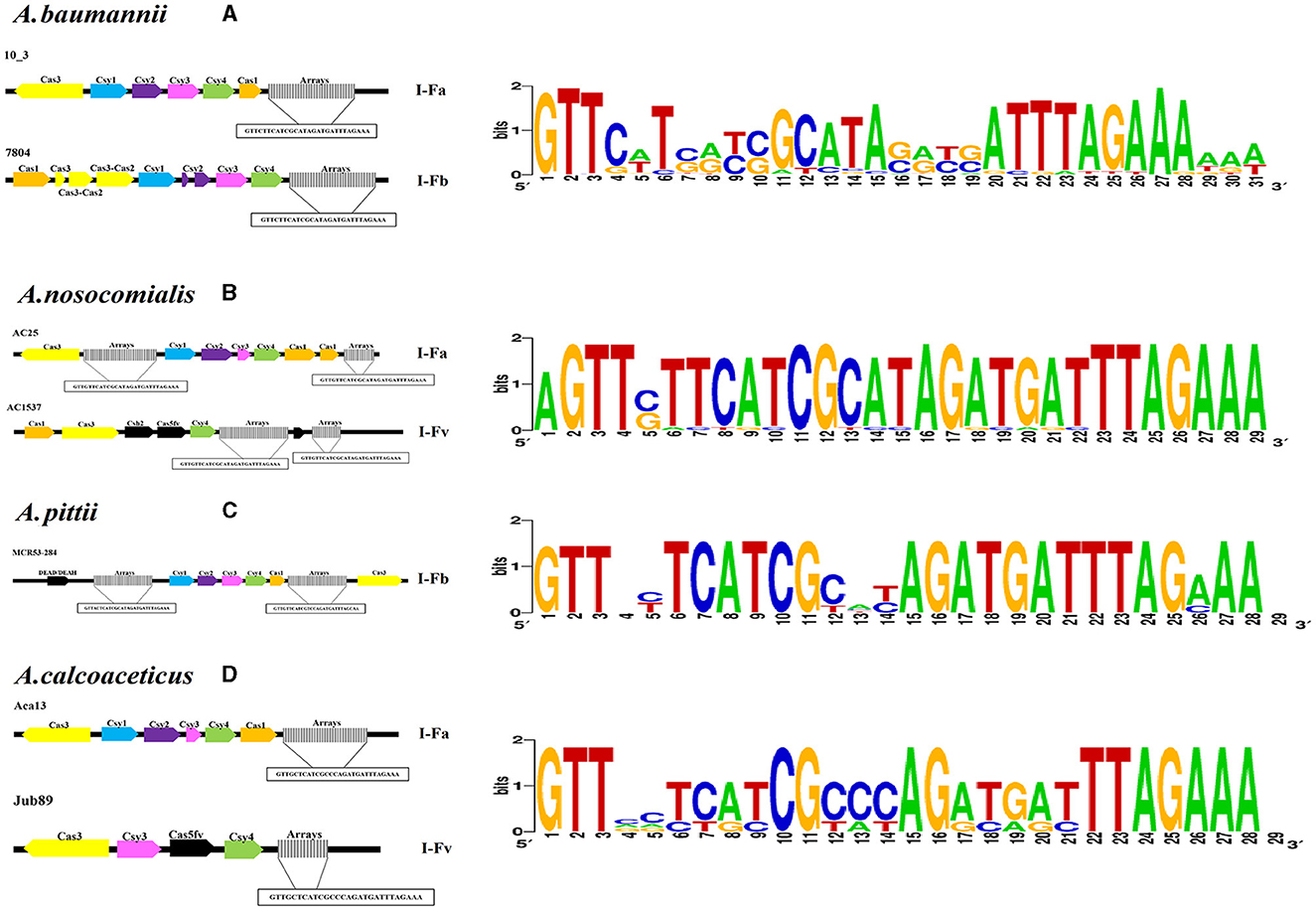
Figure 2. Identification of CRISPR–Cas systems through bioinformatics tools. Characteristics of the CRISPR–Cas systems detected in Acb complex chromosomes showing the SR consensus sequence and its variations. (A) Subtypes of CRISPR–Cas systems identified in A. baumannii chromosomes. (B) Subtypes of CRISPR–Cas systems identified in A. nosocomialis chromosomes. (C) Subtypes of CRISPR–Cas systems identified in A. pittii chromosomes. (D) Subtypes of CRISPR–Cas systems identified in A. calcoaceticus chromosomes. A graphic of the sequences was made with WebLogo (https://weblogo.berkeley.edu/., Crooks et al., 2004).
The RS consensus sequences were clustered into 14 distinct groups, with the sequence “GTTCATGGCGGCATACGCCATTTAGAAA” being the most frequent. Interestingly, the consensus RS described in this study showed a minimum of four variations and a maximum of 11 variations compared with the consensus type I-Fb RS reported previously (Karah et al., 2015). Additionally, four RSs were detected in three plasmids, only one of which was closely related (IX) to the RS identified on chromosomes (Supplementary Figure 2).
Furthermore, 2,768 SSs with lengths between 29 and 38 bp were identified in this study. Interestingly, the A. baumannii and A. calcoaceticus genomes had the greatest numbers of SSs (Supplementary Table 3).
The SSs were clustered into 30 groups according to their sequence. A total of 30.5% (845/2,768) of the SSs were shared among the genomes, and 69.4% (1,923/2,768) of the SSs were exclusive, which means that they were not commonly found in the other analyzed genomes. Interestingly, in 45.0% (36/85) of the CRISPR arrays, the same spacer was identified two or more times (Figure 3, Supplementary Table 3).
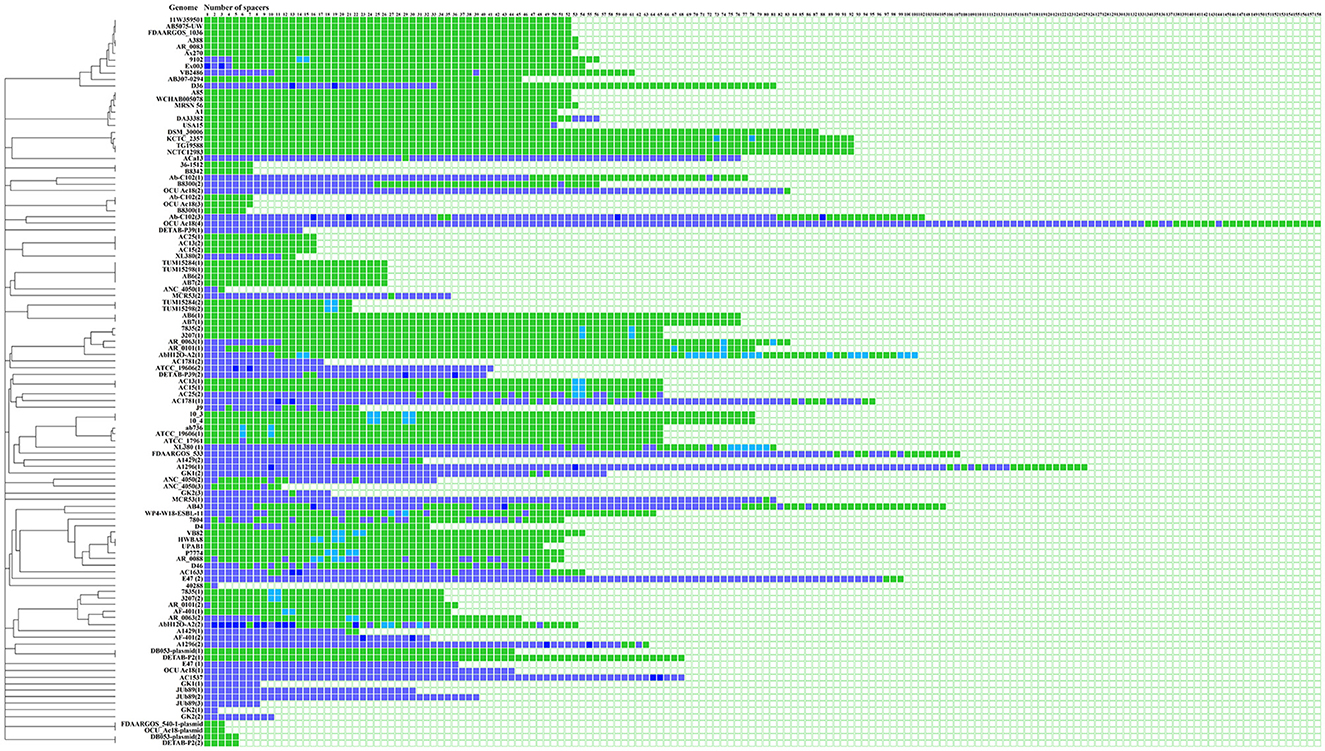
Figure 3. Identification of SSs in CRISPR–Cas systems. The number of spacers is indicated on each confirmed array. Shared SSs between genomes are highlighted in light green boxes, and unique spacers are shown in purple boxes. SSs that appear multiple times in the array and are shared between genomes are represented by light blue boxes. Unique SSs that appear multiple times in the array are shown in navy blue. Visualization was performed with the iTol program (Letunic and Bork, 2021).
The Acb complex genomes showed the presence of CRISPR arrays, and adjacent sequences corresponding to the six cas genes (cas1, csy1, csy2, csy3, csy4, and cas3) identified subtypes I-Fa, I- Fb and I-Fv (Supplementary Figures 3–12). The size of the CRISPR–Cas system in the genome ranged from 4 617 to 750 328 bp (Table 1).
The CRISPR–Cas systems identified in A. baumannii genomes were classified into the I-Fa 33.3% (20/60) and I-Fb 55% (34/60) subtypes, which included one plasmid (Supplementary Figures 3–8, Supplementary Table 4). Additionally, 3.3% (2/60) of the CRISPR systems were considered undefined and displayed CRISPR arrays associated with the DEAD/DEAH box helicase motif (36-1,512) and the cas3 gene (J9) (Supplementary Figure 10, Supplementary Table 4). Four solitary (orphan) arrays were defined among the A. baumannii genomes and plasmids (Supplementary Figure 9).
The A. nosocomialis CRISPR–Cas subtypes I-Fa and I-Fv were detected in 72.3% (8/11) and 9.1% (1/11) of the genomes, respectively. Two orphan CRISPR arrays were identified at loci other than the cas2 gene and DEAD/DEAH box helicase motif in the two genomes (Supplementary Figure 10).
Two systems were confirmed to be present in the A. pittii genomes, and only one system was found to be complete, with two arrays associated with a set of cas genes characteristic of the I-Fb subtype; however, the remaining system in the A. pittii genomes presented three small arrays and only the cas3 gene (Figure 2, Supplementary Figure 11).
Overall, 87.5% (7/8) and 12.3% (1/8) of the A. calcoaceticus genomes exhibited the CRISPR–Cas I-Fa and I-Fv subtypes, respectively (Supplementary Figure 12).
The 200 potential CRISPR arrays were determined with the CRISPRCas Finder program. These arrays were associated with 44 consensus RSs and included between two and six RSs with lengths of 18–36 bp (Supplementary Table 4). In Acb complex genomes and plasmids, putative CRISPR arrays were found in 56.8% (121/213) and 98.8% (79/80), respectively.
The putative CRISPR array genes upstream and downstream encoded 7.5% (15/200) of the A. baumannii genome to the cmr5 gene (CRISPR type III-B/BAMP) and 3% (6/200) to the DEAD/DEAH box helicase motif (Supplementary Table 4). Interestingly, the RS “GGACAAAGCGTTGCTTTGTTTATCCTC” sequence identified in two putative CRISPR arrays in A. baumannii genomes has been described in the CRISPR–Cas type I-B system.
Furthermore, the sequences predicted by CRISPR–Cas Finder were similar (>60%) to those of the cas3 gene in 65 genomes, corresponding to A. nosocomialis (36.9%, 24/65), A. pittii (60%, 39/65), and A. calcoaceticus (3.7%, 2/65). In the A. pittii genome, the csm3 and cmr5 genes were within the loci of the CRISPR–Cas III-A and III-B systems, respectively; however, CRISPR arrays were not found in these genomes (Supplementary Table 5).
Analysis of genes upstream and downstream (the flanking regions) revealed sequences that encode Cap4 proteins, which can recognize cyclic oligonucleotide-based antiphage signaling systems (CBASSs), in two A. baumannii CRISPR arrays (the 10_3 and 10_4 genomes). Similarly, CRISPR-associated primase-polymerases (CAPPs) were identified in both genomes (Figure 4, Supplementary Figure 12).
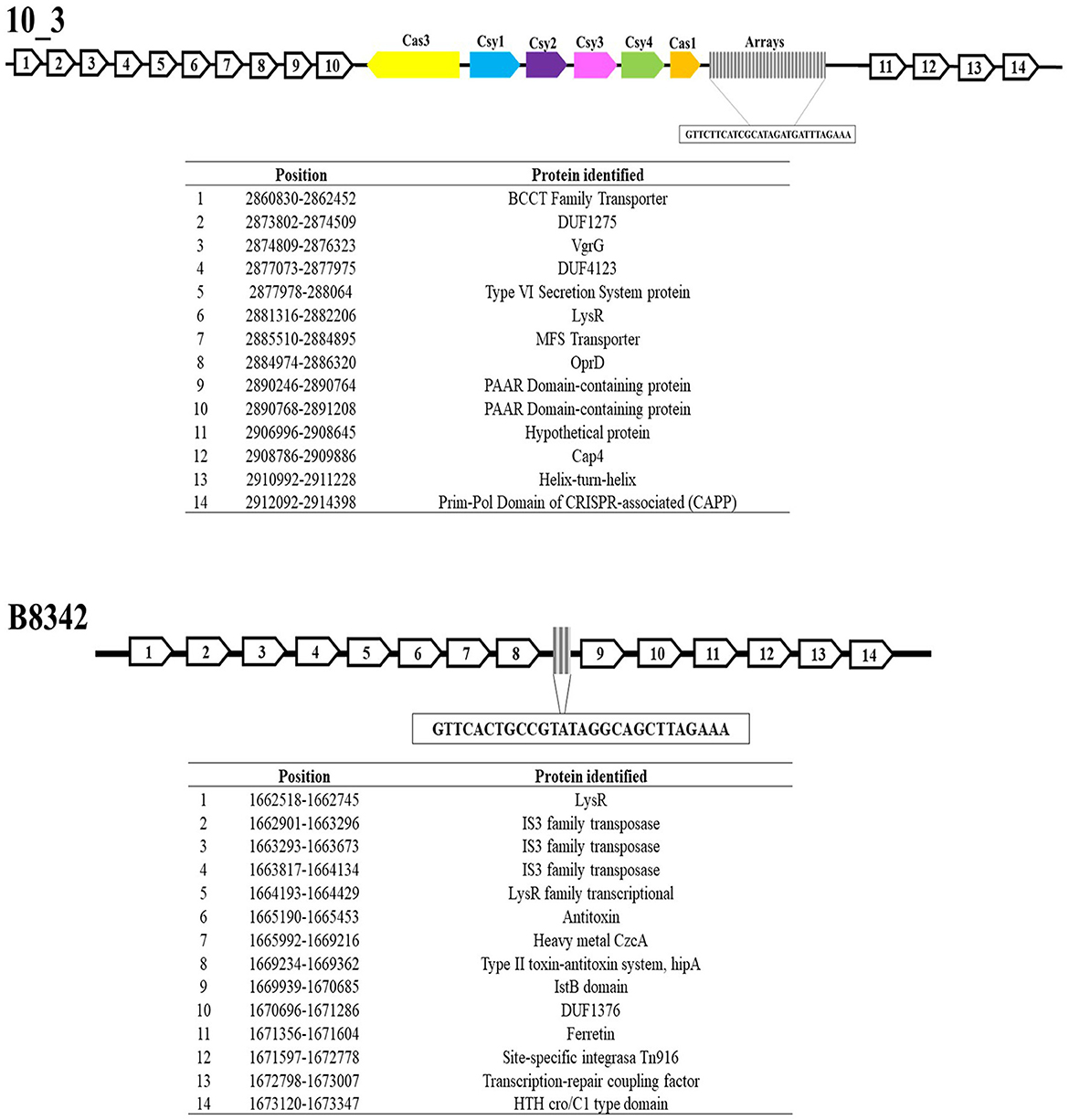
Figure 4. CRISPR-flanking genomic regions of the Acb complex. This figure shows the regions associated with the arrays, highlighting the regions encoding Cap4, CAPPSs, transposons, and toxin–antitoxin systems.
On the other hand, 9.4% (8/85) of the CRISPR–Cas systems exhibited sequences associated with the TIGR03984 protein family, and 4.7% (4/85) exhibited sequences associated with toxin–antitoxin systems. Sixty percent (6/10) of the orphan arrays displayed sequences encoding IS3, IS5, and IS6 transposases, ammonium transporters and glutamate genes (Figure 4).
Prophage genomes were identified in 68.94% (202/293) of the Acb complex genomes. Among these genomes, 79.8% (161/202) exhibited 1 to 8 complete prophages, and 20.2% (41/202) exhibited incomplete prophages. The prophage genomes found in this study (complete and incomplete) have been reported for several bacterial genera, such as Acinetobacter, Enterobacter, Salmonella, Aeromonas, Moraxella, Pseudomonas, Escherichia, and Ralstonia (Supplementary Table 6, Supplementary Figure 14). The 67.5% (52/77) of Acb complex genomes with confirmed CRISPR-Cas systems displayed at least one intact prophage; the 48.07% (25/52) were associated with the prophage PHAGE_Acinet_YMC11/11/R3177, and 46.15% (24/52) were linked to the prophage PHAGE_Acinet_Bphi_B1251 (Supplementary Figure 14).
Forty-two percent (1180/2760) of the SSs were associated with regions of prophages and plasmids belonging to the Acinetobacter genus, as well as 22 other genera, including mainly Staphylococcus, Aeromonas, Bacillus, Pseudoalteromonas, Salmonella, and Escherichia. The spacer target was a specific sequence within the prophages; however, different SSs matched the same bacteriophage (Supplementary Table 7). While some genomes possess SS that provides immunity against specific prophages, the detection of the same SS in different CRISPR arrays has been identified (Figure 5).

Figure 5. Correlation between spacers and intact prophages in the genomes. Briefly, the correlations and interactions between intact prophages and spacers targeting them are described. The purple circles represent the genomes with CRISPR–Cas systems, and the colored circles represent the intact prophages identified among the studied genomes. The networks show the interactions among the genomes that carry spacers associated with the identified prophages. The analysis and visualization of the networks were carried out with Gephi 0.10.1 (https://gephi.org/, Bastian et al., 2009).
PHAGE_Acinet_YMC11/11/R3177 was identified in three genomes of A. baumannii (7,835, 3,207, and 9,201). The AcrF11 anti-CRISPR sequence showed 100% identity with Aba7835_06455 (QFY68330.1), Aba3207_05070 (ANC36029.1), and Aba9201_17950 (AXX42741.1). In the LAC4 genome, 95% identity was confirmed for the AcrF11 sequence, but this sequence was not found in an intact phage (Figure 6, Supplementary Table 8). The search for anti-CRISPR systems led to the identification of 15 A. pittii genomes and sequences related to DUF2829 domains associated with AcrIIA7 (https://github.com/JetsiMancilla/System-CRISPR-Cas).
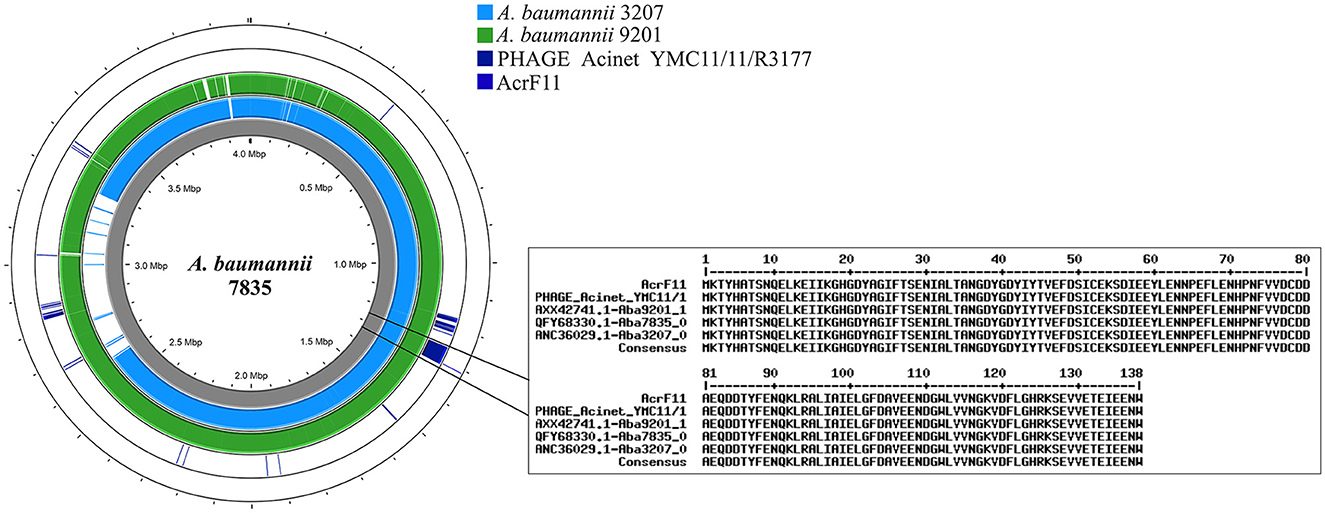
Figure 6. Anti-CRISPR proteins encoded in the genomes of the Acb complex. Identification of the AcrF11 protein in the LAC4, 7835, 3207, and 9201 genomes. The position of the protein coincides with the position of the PHAGE_Acinet_YMC11/11/R3177 in the genomes of the Acb complex.
Analysis of the Acb complex genomes revealed genes involved in colonization and virulence, including genes associated with adherence, biofilm formation, immune evasion, iron absorption, regulation, quorum sensing, and serum resistance. The genes encoded on the chromosome, including the OmpA protein involved in adhesion, invasion, persistence, and dissemination, were detected in 96.7% (206/213) of the genomes, and the genes encoding efflux systems (AdeFGH pumps), 82.6% (176/213) of the Csu fimbriae and 97.2% (207/213) of the PNAG polysaccharides were associated with the formation of biofilms.
Only 68.1% (145/213) of the genes encoded the Bap protein, which also participates in biofilm formation. On the other hand, 97.9% (208/213) of the Acb complex genomes encoded genes related to two-component systems and quorum sensing proteins involved in biofilm formation and cell adhesion (BfmRS). In the Acb complex genome, operons related to capsule Lpx and LPS were identified in 84.0% (179/213). Interestingly, 72.8% (155/213) of the genomes of A. baumannii, A. pittii, and A. nosocomialis contained genes associated with iron acquisition and acinetobactin biosynthesis (Supplementary Table 9).
Toxin-antitoxin systems were identified in only 12.5% (10/80) of the plasmids. Twenty percent (16/80) of the genes were encoded to the stress response proteins, and 8.7% (7/80) were associated with resistance to toxic compounds.
Among the chromosomally encoded resistance genes, 28.6% (61/213) were related with aminoglycoside resistance, 69.0% (147/213) with beta-lactam resistance, and only 20.2% (43/213) with genes linked to tetracycline resistance.
The Acb complex plasmid analysis revealed genes associated with resistance to aminoglycosides, antagonists of the folate pathway, beta-lactams, tetracyclines, phenicols, macrolides, and polymyxins. Briefly, 12.5% (10/80) of the genes were related to macrolide resistance [msr(E) and mph(E)] and encoded efflux pumps. Moreover, 3.8% (3/80) of the genes conferred resistance to streptomycin (strA and strB), gentamicin [ant (3”) Ia], and tobramycin. Regarding the genes encoding resistance to beta-lactams, 18.8% (15/80) of the plasmids contained blaOXA − 23, blaOXA − 24, blaOXA − 58, blaOXA − 72, and blaIMP − 1 carbapenem resistance genes, such as those conferring resistance to imipenem, meropenem and doripenem.
The cephalosporin resistance genes blaADC − 25, blaPER − 1, and blaNDM − 1 were identified in one plasmid of A. baumannii and A. pittii (Supplementary Table 9).
Several regions encoding copper ATPases, copper chaperones, regulatory proteins, and copper oxidases identified in plasmids have also been associated with the transport and oxidation of copper. The data suggested that these elements interfere with the processes of colonization and immune evasion in A. baumannii; however, these elements were located only in plasmids from A. seiferttii and A. pittii. Our data indicate that A. seiferttii and A. pittii plasmids encode more resistance mechanisms than A. baumannii plasmids.
A. baumannii has been implicated in healthcare-associated infections (HCAIs), such as ventilator-associated pneumonia, meningitis, bloodstream infections, and urinary tract infections. A. baumannii together with A. pitti, A. nosocomialis, A. seiferttii, and A. lactucae belongs to the Acb complex. A. pitti, A. nosocomialis, A. seiferttii, and A. lactucae also cause infections and have been associated with resistance to multiple drugs (Fitzpatrick et al., 2015; Li et al., 2021; Alonso et al., 2023; Bajaj et al., 2023; Kang et al., 2023). A. calcoaceticus is mainly found in environmental samples and may also carry antibiotic resistance determinants (Al Atrouni et al., 2016).
The use of bioinformatics tools and the availability of genome sequences have facilitated the analysis and characterization of A. baumannii genomes. These studies have contributed to understanding A. baumannii genome dynamics and the functions of its different genetic determinants, including its response to selective environmental pressure during the evolutionary process. The analysis of A. baumannii genomes has allowed us to explore the presence of repeated sequences associated with CRISPR–Cas systems. However, these systems have yet to be investigated in other members of the Acb complex. The matrices that make up the repeated sequences associated with CRISPR Cas systems belong to types I-F, characterized by the presence of the cas1, cas2, cas3, cas5, cas6, and cas7 genes (Karah et al., 2015; Mangas et al., 2019; Yadav and Singh, 2022).
The CRISPR–Cas system was found in one plasmid because it has been established that CRISPR–Cas systems in plasmids provide bacteria with similar efficacy in protecting against bacteriophage infections compared to those encoded in the chromosome (Siedentop et al., 2024). Additionally, we identified other plasmids characterized by a series of orphan arrays. This phenomenon has been observed in plasmids from other bacteria and archaea, where systems on plasmids are often incomplete or composed of small orphan arrays (Pinilla-Redondo et al., 2022). The prevalence of CRISPR–Cas systems in these genomes may also be associated with the evolution and adaptation of these species in diverse environments (Westra et al., 2019).
The availability of several bioinformatic tools (online and for command-line use) has facilitated the identification of CRISPR–Cas systems. This study identified and confirmed 85 CRISPR arrays and Cas genes according to the CRISPRCasFinder, CRISPRDetect, and CRISPRminer programs.
In this study identified CRISPR arrays or cas gene harbored subtypes I-B and III-B, as previously described by other authors (Yadav and Singh, 2022). Also, CRISPR arrays or genes associated with subtypes III-A and III-B in A. pittii were described for the first time. These systems are considered non-functional. However, there are reports suggesting that these small arrays could be evolutionarily preserved (Shmakov et al., 2023). Various phenomena could explain the isolation of these arrays; one possibility is the emergence of de novo arrays, or it could be that their acquisition is linked to transfer through mobile elements (Westra et al., 2019; Shmakov et al., 2023), which could explain the presence of transposases adjacent to these arrays in our study.
An interesting aspect with CRISPR-arrays was their diversity. The RSs exhibited a clear modularity; even if it's not preserved in all the identified systems. Although these sequences are not directly associated with the exogenous material that makes up the system, it is important to demonstrate their modularity, since they play a crucial role in the marking and organization of the spacer sequences, as well as in the expression and interference processes (Nethery et al., 2021).
RSs also plays a crucial role in marking and organizing spacer sequences in genomes (Nethery et al., 2021). RSs sequences can exhibit modularity; that is, they are uniform and repeated throughout the CRISPR array (Yair and Gophna, 2019). The diversity of the RSs may also be related to the diversity of spacer sequences identified.
Spacer sequences in CRISPR–Cas systems provide information on the prior exposure of bacteria to various elements (Garrett, 2021). Our data showed that the analysis of these sequences corresponded to spacers and regions of interest, thereby ensuring potential immunity against prophages and plasmids, as described by other authors (Maniv et al., 2016).
Analysis of SS in the CRISPR-Cas system revealed a correlation between the identified bacteriophages in the genomes and those for which the bacteria store information within the spacers of the CRISPR-Cas system. Furthermore, the existence of genome groups is observed, which, although not sharing identical spacer sequences, may contain information related to the same prophage.
The variability of intact prophages in Acb complex genomes analyzed in this study suggest diversity. When the environmental bacteriophage diversity is high, CRISPR–Cas systems confer an advantage to bacteria harboring these gene elements. As already described, spacers can be effective against a specific bacteriophage fraction. In this context, the low diversity of prophages among genomes suggests that those carrying CRISPR–Cas systems confer greater fitness than those lacking them (Zaayman and Wheatley, 2022).
The identification of CRISPR–Cas systems involves exploring the flanking regions of the genome. Importantly, these flanking regions in all genomes carried by CRISPR–Cas systems were not always conserved. Our study revealed that the flanking regions encoded to Cap4 proteins associated with clustered bacterial immune systems (CBASSs) (Lowey et al., 2020), which interfere with the replication of prophages, providing bacteria with a protective mechanism against bacteriophage infections. Our data suggested that there could be genomes that harbor more than two defense mechanisms against bacteriophages in parallel, leading to a decrease in the fitness of the bacteria. In contrast, other authors have proposed that a CRISPR–Cas system in association with another mechanism enhances the fitness of bacteria; therefore, two systems can coexist, i.e., CRISPR–Cas systems, restriction-modification (RM) systems, and Argonaute systems (Makarova et al., 2009; Oliveira et al., 2014; Lisitskaya et al., 2018).
Interestingly, bacteria carrying CRISPR–Cas systems have the ability to defend themselves against infection by bacteriophages. However, the bacteriophages have mechanisms that allow them to evade the action of CRISPR–Cas systems. Recent data indicate that these elements act at the DNA level or interfere with the binding of DNA to Cas proteins (Parsaeimehr et al., 2022) and in the presence of anti-CRISPR proteins in the genomes of A. baumannii (Yadav and Singh, 2022). However, more specific data detailing the characterization of A. baumannii and A. pitti, where anti-CRISPR Cas proteins have also been found, are still needed. These anti-CRISPR proteins seem to have different modes of action; in A. baumannii, they have been shown to act via recognition of the PAM sequence, and in A. pitti, they act by inhibiting the activity of the Cas protein (Niu et al., 2020; Forsberg, 2023). Interestingly, this study determined how bacteriophages carry anti-CRISPR–Cas information; in contrast, other bacteria do not contain anti-CRISPR–Cas information to act against the CRISPR–Cas system.
This study elucidates the diversity and complexity of CRISPR–Cas systems and other defense mechanisms in strains of the Acb complex. These systems play a crucial role in the adaptation and fitness of these microorganisms in various environments. The confirmed arrays displayed size and sequence variations, with consensus sequences proving difficult to link to specific Acb complex species.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
JM-R: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft. VF: Supervision, Validation, Writing – review & editing. MAC: Supervision, Validation, Writing – review & editing. SAO: Investigation, Resources, Writing – review & editing. JP-F: Investigation, Resources, Writing – review & editing. JA-G: Investigation, Resources, Writing – review & editing. JX-C: Funding acquisition, Project administration, Resources, Supervision, Validation, Writing – review & editing. AC-C: Conceptualization, Formal analysis, Funding acquisition, Investigation, Project administration, Resources, Supervision, Validation, Visualization, Writing – review & editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by Federal Funds (HIM/2017/003 SSA.1299 and HIM/2021/027 SSA.1719) at the HIMFG. This work was also supported by the “Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCYT)” with number grant 319563 at the “Announcement for Basic Science and/or Frontier Science, Modality: Paradigms and Controversies of Science 2022.” JM-R also received support from the “Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCYT),” México, PDCPN 605309.
This paper is part of the requirements for obtaining a Doctoral degree at the Posgrado en Ciencias Biológicas, UNAM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1335997/full#supplementary-material
HCAIs, healthcare-associated infections; CRISPR, clustered regularly interspaced short palindromic; gRNA, guide RNA; NCBI, National Center for Biotechnology Information; BV-BRC, Bacterial and Viral Bioinformatics Resource Center; rMLST, ribosomal multilocus sequence typing; bp, base pairs; RS, repeat sequences; SSs, spacer sequences; PAM, protospacer adjacent motif; VFDB, Virulence Factor Database; UPGMA, unweighted pair group method with arithmetic; CBASSs, cyclic oligonucleotide-based antiphage signaling systems; CAPPs, CRISPR-associated primase-polymerases.
Abby, S. S., and Rocha, E. P. C. (2017). Identification of protein secretion systems in bacterial genomes using MacSyFinder. Methods Mol. Biol. 1615, 1–21. doi: 10.1007/978-1-4939-7033-9_1
Al Atrouni, A., Kempf, M., Eveillard, M., Rafei, R., Hamze, M., and Joly-Guillou, M. L. (2016). First report of Oxa-72-producing Acinetobacter calcoaceticus in Lebanon. New Microbes New Infect. 9, 11–12. doi: 10.1016/j.nmni.2015.11.010
Alonso, C. A., Choque-Matos, J., Guibert, F., Rojo-Bezares, B., López, M., Egoávil-Espejo, R., et al. (2023). First description of an infection by Acinetobacter pitti/lactucae subcomplex in Peru. Rev. Peru. Med. Exp. Salud Pública 40, 377–378. doi: 10.17843/rpmesp.2023.403.12721
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST Server: rapid annotations using subsystems technology. BMC Gen. 9:75. doi: 10.1186/1471-2164-9-75
Bajaj, T., Ismail, N., Trivedi, A., and Sarav, M. (2023). Peritoneal dialysis-related peritonitis with Acinetobacter pittii: a case report. J. Investig. Med. High Impact Case Rep. 11:23247096221148264. doi: 10.1177/23247096221148264
Bartual, S. G., Seifert, H., Hippler, C., Luzon, M. A., Wisplinghoff, H., and Rodríguez-Valera, F. (2005). Development of a multilocus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J. Clin. Microbiol. 43, 4382–4390. doi: 10.1128/JCM.43.9.4382-4390.2005
Bastian, M., Heymann, S., and Jacomy, M. (2009). “Gephi: an open source software for exploring and manipulating networks,” in International AAAI Conference on Weblogs and Social Media. doi: 10.1609/icwsm.v3i1.13937
Bhaya, D., Davison, M., and Barrangou, R. (2011). CRISPR-Cas systems in bacteria and archaea: versatile small RNAs for adaptive defense and regulation. Annu. Rev. Genet. 45, 273–297. doi: 10.1146/annurev-genet-110410-132430
Biswas, A., Gagnon, J. N., Brouns, S. J., Fineran, P. C., and Brown, C. M. (2013). CRISPRTarget: bioinformatic prediction and analysis of crRNA targets. RNA Biol. 10, 817–827. doi: 10.4161/rna.24046
Biswas, A., Staals, R. H. J., Morales, S. E., Fineran, P. C., and Brown, C. M. (2016). CRISPRDetect: a flexible algorithm to define CRISPR arrays. BMC Gen. 17:356. doi: 10.1186/s12864-016-2627-0
Bondy-Denomy, J., and Davidson, A. R. (2014). To acquire or resist: the complex biological effects of CRISPR-Cas systems. Trends Microbiol. 22, 218–225. doi: 10.1016/j.tim.2014.01.007
Calix, J. J., Burnham, J. P., and Feldman, M. F. (2019). Comparison of the clinical characteristics of hospital-acquired and non-hospital-acquired Acinetobacter calcoaceticus-baumannii complex in a large midwest US health care system. Open Forum. Infect. Dis. 6:ofz423. doi: 10.1093/ofid/ofz423
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328. doi: 10.1093/nar/gki008
Chevallereau, A., Pons, B. J., van Houte, S., and Westra, E. R. (2022). Interactions between bacterial and phage communities in natural environments. Nat. Rev. Microbiol. 20, 49–62. doi: 10.1038/s41579-021-00602-y
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/nar/gky425
Crooks, G. E., Hon, G., Chandonia, J. M., and Brenner, S. E. (2004). WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190. doi: 10.1101/gr.849004
Cruz-López, E. A., Rivera, G., Cruz-Hernández, M. A., Martínez-Vázquez, A. V., Castro-Escarpulli, G., Flores-Magallón, R., et al. (2021). Identification and characterization of the CRISPR/Cas system in Staphylococcus aureus strains from diverse sources. Front. Microbiol. 12:656996. doi: 10.3389/fmicb.2021.656996
Dy, R. L., Pitman, A. R., and Fineran, P. C. (2013). Chromosomal targeting by CRISPR-Cas systems can contribute to genome plasticity in bacteria. Mob. Genet. Elements 3:e26831. doi: 10.4161/mge.26831
Fitzpatrick, M. A., Ozer, E., Bolon, M. K., and Hauser, A. R. (2015). Influence of ACB complex genospecies on clinical outcomes in a U.S. hospital with high rates of multidrug resistance. J. Infect. 70, 144–152. doi: 10.1016/j.jinf.2014.09.004
Forsberg, K. J. (2023). Anti-CRISPR discovery: using magnets to find needles in haystacks. J. Mol. Biol. 435, 167952. doi: 10.1016/j.jmb.2023.167952
Garrett, S. C. (2021). Pruning and tending immune memories: spacer dynamics in the CRISPR array. Front. Microbiol. 12:664299. doi: 10.3389/fmicb.2021.664299
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications. Wellcome Open Res. 3:124. doi: 10.12688/wellcomeopenres.14826.1
Kang, H. M., Yun, K. W., and Choi, E. H. (2023). Molecular epidemiology of Acinetobacter baumannii complex causing invasive infections in Korean children during 2001–2020. Ann. Clin. Microbiol. Antimicrob. 22:32. doi: 10.1186/s12941-023-00581-3
Karah, N., Samuelsen, Ø., Zarrilli, R., Sahl, J. W., Wai, S. N., and Uhlin, B. E. (2015). CRISPR-cas subtype I-Fb in Acinetobacter baumannii: evolution and utilization for strain subtyping. PLoS ONE 10:e0118205. doi: 10.1371/journal.pone.0118205
Koonin, E. V., Makarova, K. S., and Zhang, F. (2017). Diversity, classification and evolution of CRISPR-Cas systems. Curr. Opin. Microbiol. 37, 67–78. doi: 10.1016/j.mib.2017.05.008
Lange, S. J., Alkhnbashi, O. S., Rose, D., Will, S., and Backofen, R. (2013). CRISPRmap: an automated classification of repeat conservation in prokaryotic adaptive immune systems. Nucleic Acids Res. 41, 8034–8044. doi: 10.1093/nar/gkt606
Letunic, I., and Bork, P. (2021). Interactive Tree Of Life (Itol) v5: an online tool for phylogenetic tree display and annotation. Nucl. Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Leungtongkam, U., Thummeepak, R., Kitti, T., Tasanapak, K., Wongwigkarn, J., Styles, K. M., et al. (2020). Genomic analysis reveals high virulence and antibiotic resistance amongst phage susceptible Acinetobacter baumannii. Sci. Rep. 10:16154. doi: 10.1038/s41598-020-73123-y
Li, L. H., Yang, Y. S., Sun, J. R., Huang, T. W., Huang, W. C., Chen, F. J., et al. (2021). Clinical and molecular characterization of Acinetobacter seifertii in Taiwan. J. Antimicrob. Chemother. 76, 312–321. doi: 10.1093/jac/dkaa432
Lisitskaya, L., Aravin, A. A., and Kulbachinskiy, A. (2018). DNA interference and beyond: structure and functions of prokaryotic Argonaute proteins. Nat. Commun. 9:5165. doi: 10.1038/s41467-018-07449-7
Lowey, B., Whiteley, A. T., Keszei, A. F. A., Morehouse, B. R., Mathews, I. T., Antine, S. P., et al. (2020). CBASS immunity uses CARF-related effectors to Sense 3′-5′- and 2′-5′-linked cyclic oligonucleotide signals and protect bacteria from phage infection. Cell 182, 38–49.e17. doi: 10.1016/j.cell.2020.05.019
Makarova, K. S., Wolf, Y. I., Iranzo, J., Shmakov, S. A., Alkhnbashi, O. S., Brouns, S. J. J., et al. (2020). Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat. Rev. Microbiol. 18, 67–83. doi: 10.1038/s41579-019-0299-x
Makarova, K. S., Wolf, Y. I., van der Oost, J., and Koonin, E. V. (2009). Prokaryotic homologs of Argonaute proteins are predicted to function as key components of a novel system of defense against mobile genetic elements. Biol. Direct 4:29. doi: 10.1186/1745-6150-4-29
Manchanda, V., Sanchaita, S., and Singh, N. (2010). Multidrug resistant Acinetobacter. J. Glob. Infect. Dis. 2, 291–304. doi: 10.4103/0974-777X.68538
Mangas, E. L., Rubio, A., Álvarez-Marín, R., Labrador-Herrera, G., Pachón, J., Pachón-Ibáñez, M. E., et al. (2019). Pangenome of Acinetobacter baumannii uncovers two groups of genomes, one of them with genes involved in CRISPR/Cas defence systems associated with the absence of plasmids and exclusive genes for biofilm formation. Microb. Genom. 5:e000309. doi: 10.1099/mgen.0.000309
Maniv, I., Jiang, W., Bikard, D., and Marraffini, L. A. (2016). Impact of different target sequences on type III CRISPR-Cas immunity. J. Bacteriol. 198, 941–950. doi: 10.1128/JB.00897-15
Marraffini, L. A., and Sontheimer, E. J. (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. doi: 10.1126/science.1165771
Mojica, F. J. M., and Montoliu, L. (2016). On the origin of CRISPR-Cas technology: from prokaryotes to mammals. Trends Microbiol. 24, 811–820. doi: 10.1016/j.tim.2016.06.005
Moradi, J., Hashemi, F. B., and Bahador, A. (2015). Antibiotic resistance of Acinetobacter baumannii in Iran: a systemic review of the published literature. Osong Public Health Res. Perspect. 6, 79–86. doi: 10.1016/j.phrp.2014.12.006
Nethery, M. A., Korvink, M., Makarova, K. S., Wolf, Y. I., Koonin, E. V., and Barrangou, R. (2021). CRISPRclassify: repeat-based classification of CRISPR Loci. CRISPR J. 4, 558–574. doi: 10.1089/crispr.2021.0021
Niu, Y., Yang, L., Gao, T., Dong, C., Zhang, B., Yin, P., et al. (2020). A type I-F anti-CRISPR protein Inhibits the CRISPR-Cas surveillance complex by ADP-ribosylation. Mol. Cell 80, 512–524.e5. doi: 10.1016/j.molcel.2020.09.015
Oliveira, P. H., Touchon, M., and Rocha, E. P. (2014). The interplay of restriction-modification systems with mobile genetic elements and their prokaryotic hosts. Nucleic Acids Res. 42, 10618–10631. doi: 10.1093/nar/gku734
O'Meara, D., and Nunney, L. (2019). A phylogenetic test of the role of CRISPR-Cas in limiting plasmid acquisition and prophage integration in bacteria. Plasmid 104:102418. doi: 10.1016/j.plasmid.2019.102418
Parsaeimehr, A., Ebirim, R. I., and Ozbay, G. (2022). CRISPR-Cas technology a new era in genomic engineering. Biotechnol. Rep. 34:e00731. doi: 10.1016/j.btre.2022.e00731
Pinilla-Redondo, R., Russel, J., Mayo-Muñoz, D., Shah, S. A., Garrett, R. A., Nesme, J., et al. (2022). CRISPR-Cas systems are widespread accessory elements across bacterial and archaeal plasmids. Nucl. Acids Res. 50, 4315–4328. doi: 10.1093/nar/gkab859
Ruan, Z., and Feng, Y. (2016). BacWGSTdb, a database for genotyping and source tracking bacterial pathogens. Nucleic Acids Res. 44, D682–D687. doi: 10.1093/nar/gkv1004
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shmakov, S. A., Barth, Z. K., Makarova, K. S., Wolf, Y. I., Brover, V., Peters, J. E., et al. (2023). Widespread CRISPR-derived RNA regulatory elements in CRISPR-Cas systems. Nucl. Acids Res. 51, 8150–8168. doi: 10.1093/nar/gkad495
Siedentop, B., Rüegg, D., Bonhoeffer, S., and Chabas, H. (2024). My host's enemy is my enemy: plasmids carrying CRISPR-Cas as a defence against phages. Proc. Biol. Sci. 291:20232449. doi: 10.1098/rspb.2023.2449
Song, W., Sun, H. X., Zhang, C., Cheng, L., Peng, Y., Deng, Z., et al. (2019). Prophage hunter: an integrative hunting tool for active prophages. Nucl. Acids Res. 47, W74–W80. doi: 10.1093/nar/gkz380
Tian, R., and Imanian, B. (2023). VBCG: 20 validated bacterial core genes for phylogenomic analysis with high fidelity and resolution. Microbiome 11:247. doi: 10.1186/s40168-023-01705-9
Touchon, M., Cury, J., Yoon, E. J., Krizova, L., Cerqueira, G. C., Murphy, C., et al. (2014). The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences. Genome Biol. Evol. 6, 2866–2882. doi: 10.1093/gbe/evu225
Villalon, P., Valdezate, S., Cabezas, T., Ortega, M., Garrido, N., Vindel, A., et al. (2015). Endemic and epidemic Acinetobacter baumannii clones: a twelve-year study in a tertiary care hospital. BMC Microbiol. 15:47. doi: 10.1186/s12866-015-0383-y
Waddington, S. N., Privolizzi, R., Karda, R., and O'Neill, H. C. (2016). A broad overview and review of CRISPR-Cas technology and stem cells. Curr. Stem Cell Rep. 2, 9–20. doi: 10.1007/s40778-016-0037-5
Westra, E. R., van Houte, S., Gandon, S., and Whitaker, R. (2019). The ecology and evolution of microbial CRISPR-Cas adaptive immune systems. Philos. Trans. R. Soc. Lond. B Biol. Sci. 374:20190101. doi: 10.1098/rstb.2019.0101
Wisplinghoff, H., Paulus, T., Lugenheim, M., Stefanik, D., Higgins, P. G., Edmond, M. B., et al. (2012). Nosocomial bloodstream infections due to Acinetobacter baumannii, Acinetobacter pittii and Acinetobacter nosocomialis in the United States. J. Infect. 64, 282–290. doi: 10.1016/j.jinf.2011.12.008
World Health Organization (2017). Global priority list of antibiotic-resistant bacteria to guide research, discovery, and development of new antibiotics. Available online at: https://www.who.int/news/item/27-02-2017-who-publishes-list-of-bacteria-for-which-new-antibiotics-are-urgently-needed (accessed September, 2023).
Yadav, G., and Singh, R. (2022). In silico analysis reveals the co-existence of CRISPR-Cas type I-F1 and type I-F2 systems and its association with restricted phage invasion in Acinetobacter baumannii. Front. Microbiol. 13:909886. doi: 10.3389/fmicb.2022.909886
Yair, Y., and Gophna, U. (2019). Repeat modularity as a beneficial property of multiple CRISPR-Cas systems. RNA Biol. 16, 585–587. doi: 10.1080/15476286.2018.1474073
Yakkala, H., Samantarrai, D., Gribskov, M., and Siddavattam, D. (2019). Comparative genome analysis reveals niche-specific genome expansion in Acinetobacter baumannii strains. PLoS ONE 14:e0218204. doi: 10.1371/journal.pone.0218204
Zaayman, M., and Wheatley, R. M. (2022). Fitness costs of CRISPR-Cas systems in bacteria. Microbiology (Reading) 168:001209. doi: 10.1099/mic.0.001209
Zankari, E., Hasman, H., Cosentino, S., Vestergaard, M., Rasmussen, S., Lund, O., et al. (2012). Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 67, 2640–2644. doi: 10.1093/jac/dks261
Keywords: Acinetobacter baumannii, Acinetobacter calcoaceticus–Acinetobacter baumannii complex, cas genes, CRISPR systems, prophages
Citation: Mancilla-Rojano J, Flores V, Cevallos MA, Ochoa SA, Parra-Flores J, Arellano-Galindo J, Xicohtencatl-Cortes J and Cruz-Córdova A (2024) A bioinformatic approach to identify confirmed and probable CRISPR–Cas systems in the Acinetobacter calcoaceticus–Acinetobacter baumannii complex genomes. Front. Microbiol. 15:1335997. doi: 10.3389/fmicb.2024.1335997
Received: 09 November 2023; Accepted: 26 March 2024;
Published: 09 April 2024.
Edited by:
Alicja Wegrzyn, University of Gdansk, PolandReviewed by:
Andres Felipe Opazo-Capurro, University of Concepcion, ChileCopyright © 2024 Mancilla-Rojano, Flores, Cevallos, Ochoa, Parra-Flores, Arellano-Galindo, Xicohtencatl-Cortes and Cruz-Córdova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ariadnna Cruz-Córdova, YXJpYWRubmFjcnV6QHlhaG9vLmNvbS5teA==; Juan Xicohtencatl-Cortes, anVhbnhpY29AeWFob28uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.