 Yong Deng1Wuyuan Kong1Xiaoming Zhang1Yi Zhu1Tian Xie1Ming Chen1Li Zhu1Jingzhao Sun1Zhihua Zhang1Chaoyong Chen1Chongwen Zhu1
Yong Deng1Wuyuan Kong1Xiaoming Zhang1Yi Zhu1Tian Xie1Ming Chen1Li Zhu1Jingzhao Sun1Zhihua Zhang1Chaoyong Chen1Chongwen Zhu1 Huaqun Yin2Songqing Huang1*
Huaqun Yin2Songqing Huang1* Yabing Gu2*
Yabing Gu2*- 1Changde Tobacco Company of Hunan Province, Changde, China
- 2School of Minerals Processing and Bioengineering, Central South University, Changsha, China
Plant health states may influence the distribution of rhizosphere microorganisms, which regulate plant growth and development. In this study, the response of rhizosphere bacteria and fungi of healthy and diseased plants compared to bulk microbes was analyzed using high-throughput sequencing. Plant adaptation strategies of plants under potato virus Y (PVY) infection have been studied from a microbial perspective. The diversity and community structure of bacteria and fungi varied between bulk and rhizosphere soils, but not between healthy and diseased rhizosphere soils. A LEfSe analysis revealed the significant differences between different treatments on bacterial and fungal community compositions and identified Roseiflexaceae, Sphingomonas, and Sphingobium as the bacterial biomarkers of bulk (BCK), healthy rhizosphere (BHS), and diseased rhizosphere (BIS) soils, respectively; Rhodotorula and Ascomycota_unidentified_1_1 were identified as the fungal biomarkers of bulk (FCK) and healthy rhizosphere (FHS) soils. Bacterial networks were found to be more complex and compact than fungal networks and revealed the roles of biomarkers as network keystone taxa. PVY infection further increased the connectedness among microbial taxa to improve rhizosphere microbial community stability and resistance to environmental stress. Additionally, water content (WC) played an apparent influence on bacterial community structure and diversity, and pH showed significant effects on fungal community diversity. WC and pH greatly affected the biomarkers of bacterial rhizosphere communities, whereas the biomarkers of bulk bacterial communities were significantly affected by soil nutrients, especially for Sphingobium. Overall, the rhizosphere microbial community enrichment processes were different between healthy and diseased plants by changing the community compositions and identifying different biomarkers. These findings provide insight into the assemblage of rhizosphere microbial communities and soil physicochemical properties, which contributes to a deeper understanding of the establishment of an artificial core root microbiota to facilitate plant growth and bolstering resistance mechanisms. This knowledge contributes to a deeper understanding of the establishment of an artificial core root microbiota, thereby facilitating plant growth and bolstering resistance mechanisms.
Introduction
Potato virus Y (PVY) is a significant constituent of the Potyviridae family, exhibiting the ability to infect over 170 species across the globe. The infection by PVY results in a wide range of symptoms, such as vein necrosis and systematic mottles, with necrotic ringspots reducing crop yield and quality substantially (Chen et al., 2017). Upon infection, plants activate their defense mechanisms by producing various resistant substances, including phytoalexin chitinase, peroxidase, and plant antitoxin to enhance their tolerance (Shah et al., 2017). Furthermore, the presence of PVY in a host organism leads to the observation of numerous morphological, physiological, and histological alterations (Hinrichs-Berger et al., 1999). However, the extent to which PVY infection affects the rhizosphere, a critical component of soil-plant interaction, is constrained. Prior studies have provided evidence of the impact of plant diseases or pests on the composition of rhizosphere microbiomes (French et al., 2021; Lazcano et al., 2021; Enagbonma et al., 2023). For instance, the introduction of the downy mildew pathogen to Arabidopsis thaliana leaves induces changes in the microbial communities within the rhizosphere (Berendsen et al., 2018). Similarly, the presence of the western corn rootworm, a pest that inflicts damage on maize plants, results in the proliferation of distinct microbial taxa in the rhizosphere, such as Acinetobacter, Smaragdicoccus, and Aeromicrobium (Benitez et al., 2017).
Plants establish direct contact with numerous microorganisms in the soil through the roots (Lu et al., 2006), leading to the observable differentiation between bulk and rhizosphere soils. The rhizosphere effect, a phenomenon in which plant roots attract and accumulate certain microorganisms from the bulk soil (Hein et al., 2008), plays a vital role in governing rhizosphere microbial communities. The rhizosphere, functioning as the primary site of soil-plant interaction, typically demonstrates a higher frequency of nutrient exchange and increased microbial activities in comparison to the bulk soil (Chaparro et al., 2014). The observed dissimilarity in microbial communities between the rhizosphere and bulk soil can be ascribed to root exudates, rhizosphere metabolism, and the root system's heightened selectivity. Consequently, the disparities in microbial communities are expected to result in variations in nutrient preferences and metabolic patterns of microbial communities (Lareen et al., 2016), thereby facilitating the suppression of phytopathogens and bolstering tolerance to environmental stress. Increasing evidence demonstrated the effect of numerous factors on the shifts in microbiomes of rhizosphere and bulk soil, including abiotic factors such as disease and insect pests (Yin et al., 2021), soil types (Lopes et al., 2021), plant species (Ahmad et al., 2022), and climate factors (Zhao et al., 2021). However, root-rot disease of Zanthoxylum bungeanum trees showed significant changes in the KEGG and CAZy functional profiles of microbiomes between the rhizosphere and bulk soils, rather than microbial diversity and community composition (Liao et al., 2022). These feedback explorations about the recruitment of rhizosphere microbes from bulk soil are vital to the ecological functions of terrestrial ecosystems.
Limited investigations have been observed regarding the impacts of PVY invasion on the plant microbiome, especially for microbial interactions and community assembly.
Recent research has demonstrated that environmental factors, encompassing both biotic and abiotic factors, conventionally govern the equilibrium between stochastic and deterministic assembly processes (Aguilar and Sommaruga, 2020; He et al., 2021). It has been ascertained that a harmonized stochastic and deterministic assembly process confers benefits in upholding a diverse ecosystem. Modifications in soil physicochemical properties, such as heightened soil salinity and pH levels, may exert a deterministic impact on the composition of soil bacterial communities (Yu et al., 2022). Conversely, optimal soil pH values and diminished salinity content may contribute to the stochastic nature of soil formation. Additionally, it has been observed that disease-induced modifications in plant performance can trigger a series of consequential changes in the rhizosphere environment, thereby significantly impacting the equilibrium between deterministic and stochastic factors within the rhizosphere microbiome (Liu et al., 2022). Despite the apparent recognition of this phenomenon, comprehensive testing and examination of its intricacies have been infrequently conducted. It is possible to propose the hypothesis that the invasion of pathogens in the rhizosphere microbiome has a deterministic impact on the compositional variability of said microbiome by altering plant performance. The occurrence of pathogen invasion is frequently accompanied by alterations in the diversity of rhizosphere microorganisms (Jiang et al., 2023). As a result, it can be deduced that the invasion of pathogens affects the microbial interactions that depend on the particular types and quantities of microorganisms that exist. Moreover, the evaluation of whether the bacterial microbiome of plant roots demonstrates dynamic universality, characterized by consistent interactions between microbes and their surroundings across hosts, or if each individual's microbiota adheres to its own distinct set of principles, is yet to be determined.
This study aimed to investigate the response of rhizosphere bacteria and fungi in healthy and diseased plants, as well as the bulk microbe, in relation to microbial diversity, structure, composition, co-occurrence network, and their correlation with soil physicochemical properties. Specifically, this research examined the disparities in (1) the diversity, composition, and structure of microbial communities in the rhizosphere and bulk soil between diseased and healthy conditions, (2) the network interactions among microbial genera and subnetworks based on biomarkers and their associated taxa, and (3) the relationship between microbial communities and soil physicochemical properties to understand the adaptation strategies of rhizosphere microbes under potato virus Y infection. This study places specific emphasis on the influence of PVY infection on the assemblies of rhizosphere microbiome communities and presents a unique viewpoint on the role of the microbiome in enhancing plant resistance against diseases.
Materials and methods
Collection of soil samples
The sampling was conducted in August 2022 at tobacco fields located in Changde city (between 29°13′30′′-29°59′19′′N and 110°28′40′′-110°58′30′′E, Hunan Province, China). Part of the plants in the sampling sites were infected by potato virus Y. Eight sample sites, naturally and randomly infected by PVY, were selected, and three soil samples were collected from each site, namely bulk soil, rhizosphere soil of a healthy plant, and rhizosphere soil of a diseased plant. A diseased plant that showed infected symptoms (the plant vein necrosis, leaf distortion, and stem necrosis) and a healthy plant were randomly selected at each sampling site. Rhizosphere soil samples were collected from both healthy (H) and diseased plants (I) by gently uprooting them and subsequently shaking off any excess soil from the roots by hand. The soil adhering to the root segment within the range of 0–4 mm from the root was identified as rhizosphere soil. The ridge soil, which was more than 20 cm away from the plant, was collected as bulk soil (C). In total, there were 24 soil samples collected in our study. A dry ice blanket was used to transport all samples to the laboratory. We divided soil samples into two parts, storing one at 80°C for microbial experiments and submitting the other to the School of Resources and Environment at Southwest University for measurement of soil properties. The pH value, water content (WC), organic matter (OM), total nitrogen (TN), alkali hydrolyzable nitrogen (AHN), total phosphorus (TP), available phosphorus (AP), total potassium (TK), and available potassium (AK) were tested following the methods outlined in previous studies (Gu et al., 2019, 2022).
DNA extraction, amplicon sequencing, and data processing
Total DNA of rhizosphere and bulk soil samples (0.5 g of fresh soil from each sample) were extracted with the FastDNA SPIN Kit for soil (MP Biomedicals, Solon, OH, United States) following the manufacturer's instructions. The total DNA concentration and quality were measured using a NanoDrop 1000 spectrophotometer (Thermo Scientific, Waltham, United States). The primers 799F (5′-AACMGGATTAGATACCCKG-3′)/1115R (5′-AGGGTTGCGCTCGTTG-3′) were used to amplify the V5-V7 region of the bacterial 16S rRNA gene, and primer fITS7 (5′-GTGARTCATCGAATCTTTG-3′)/ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) was used to amplify the fungal ITS2 region (Kembel et al., 2014; Deng et al., 2021). The polymerase chain reaction (PCR) production was used to perform paired-end sequencing using an Illumina Hiseq 2500 platform at MEGIGENE Biotechnology Co., Ltd. (Guangzhou, China). In the NCBI database, bacteria and fungi raw data were uploaded under PRJNA946037 and PRJNA946055.
The sequences were processed using QIIME2-2022.8 according to previously described methods (Zhang et al., 2022). In summary, after the elimination of adaptors and primer sequences, the raw sequences were assembled for each sample based on the distinctive barcode. The clean sequences, exhibiting a similarity of 97%, were then allocated to amplicon sequence variants (ASVs) utilizing DADA2 (Callahan et al., 2016), and their representative sequences were classified using the SILVA reference database (version 132) and the UNITE database (version 10.05) for bacteria and fungi, respectively (Kõljalg et al., 2005; Quast et al., 2012). After discarding singletons, the ASVs table was resampled for downstream analysis.
Biomarkers identified by LEfSe analysis
The linear discriminate analysis effect size (LEfSe), which was implemented in the microeco package in R (version 4.0.0), was used to obtain the biomarkers for each treatment (Qu et al., 2020). Initially, a non-factorial parametric Kruskal-Wallis (KW) sum-rank test was employed to identify significant differences in abundance, with a significance threshold of 0.05. Subsequently, LEfSe was conducted using LDA to assess the impact of each component's (species) abundance on the observed differences. A logarithmic LDA score threshold of 3.0 was set to discriminate features.
Network analysis
To examine the relationships between bacteria and fungi, a co-occurrence analysis was conducted using the SparCC method implemented in the SpiecEasi package (Friedman and Alm, 2012). The network was visualized using Gephi (version 0.9.2). Bacterial and fungal taxa at the genus level that were present at < 4 out of 8 sites were excluded from the network analysis. Correlations were calculated using SparCC, with a threshold of absolute correlation set at 0.3. The P-values were adjusted using the Benjamini–Hochberg procedure, and only those with P-values below 0.05 were retained. A range of network topological properties, including degree, modularity, betweenness centrality, and average path length, were computed in the R package igraph. The subnetworks based on biomarkers identified by LEfSe analysis and their associated nodes were selected by Cytoscape (version 3.8.1). The determination of keystone species within the networks involved the calculation of within-module connectivity (Zi) and among-module connectivity (Pi). Specifically, network hubs were identified as having Zi values ≥ to 2.5 and Pi values ≥ 0.62, module hubs had Zi values ≥ 2.5 and Pi values < 0.62, and connectors had Zi values < 2.5 and Pi values ≥ 0.62. In addition, the networks among ASVs were also constructed following the above steps, and similar changes in topological properties compared to networks at the genus level were found (Supplementary Table S1).
Statistical analysis
The community alpha diversity indices, including the Shannon index, species richness, and Pielou's evenness index, were calculated using the vegan package. The differences in alpha diversity indices and soil properties among bulk, healthy rhizosphere, and diseased rhizosphere soils were tested using ANOVA. Principal coordinate analysis (PCoA) based on Bray-Curtis distance was performed and visualized the community structure differences. Analysis of similarities (ANOSIM) with the Bray-Curtis distance and 999 permutations was carried out by “anosim” in the vegan R package. Spearman's linkages between the microbial communities and soil physicochemical properties were analyzed based on a mantel test using the “linkET” package (Sun et al., 2022). All the plots were visualized using the “ggplot” package.
Results
Soil physical and chemical properties in bulk and rhizosphere soils
Soil physical and chemical properties showed significant differences between bulk and rhizosphere soils but not between healthy and diseased rhizosphere soils (Table 1). The contents of TN, AP, TP, and AK were found to be significantly elevated in the rhizosphere soil compared to the bulk soil, as determined using ANOVA (p < 0.05). However, no significant differences were observed between the healthy and diseased rhizosphere soils in terms of these parameters. Other properties, including WC, pH, OM, AHN, and TK, showed no significant differences among bulk, healthy rhizosphere, and diseased rhizosphere soils.
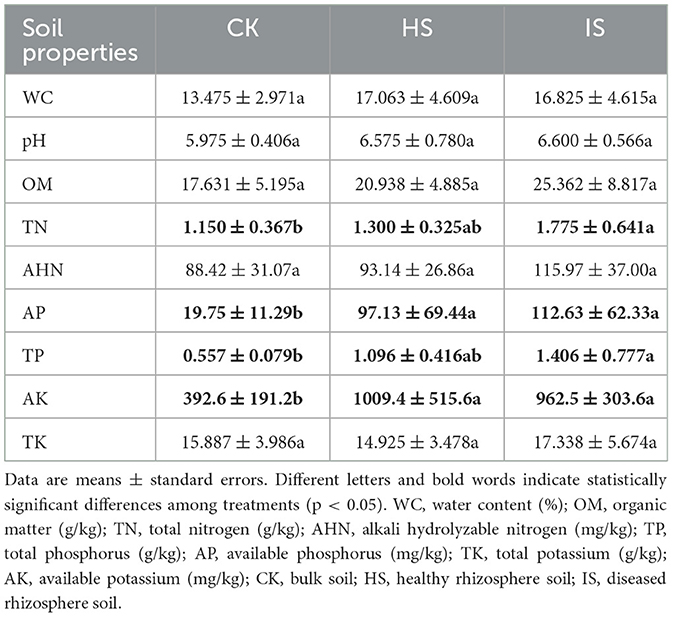
Table 1. ANOVA of bulk and rhizosphere soil physical and chemical properties among treatments based on Tukey's test.
Variations of bacterial and fungal community diversities
Both PCoA and ANOSIM analyses revealed significant differences in the beta-diversities of bacterial communities between the bulk and rhizosphere soils (ANOSIM, P < 0.05) (Figure 1A). Specifically, the beta-diversity of bulk soil bacterial community (BCK) differed significantly from that in healthy rhizosphere soil (BHS, R2 = 0.211, P = 0.04) and diseased rhizosphere soil (BIS, R2 = 0.217, P = 0.03), whereas no significant difference was observed between BHS and BIS. ANOSIM analysis results showed that the fungal community beta-diversities among bulk and rhizosphere soil could not be significantly distinguished (P > 0.05) (Figure 1B). The alpha-diversity of the microbial communities, as measured by the Shannon index, species richness, and Pielou's evenness index, was calculated. An ANOVA analysis revealed a significant difference among the CK, HS, and IS groups (Figures 1C, D). Specifically, the bacterial community alpha diversity in the BCK was significantly higher than in the BHS. However, there was no significant difference in alpha diversity between the BIS and BCK/BHS (Fisher's test, P < 0.05). In terms of the fungal communities, the species richness in FCK was significantly higher than in the FHS and FIS groups (Fisher's test, P < 0.05).
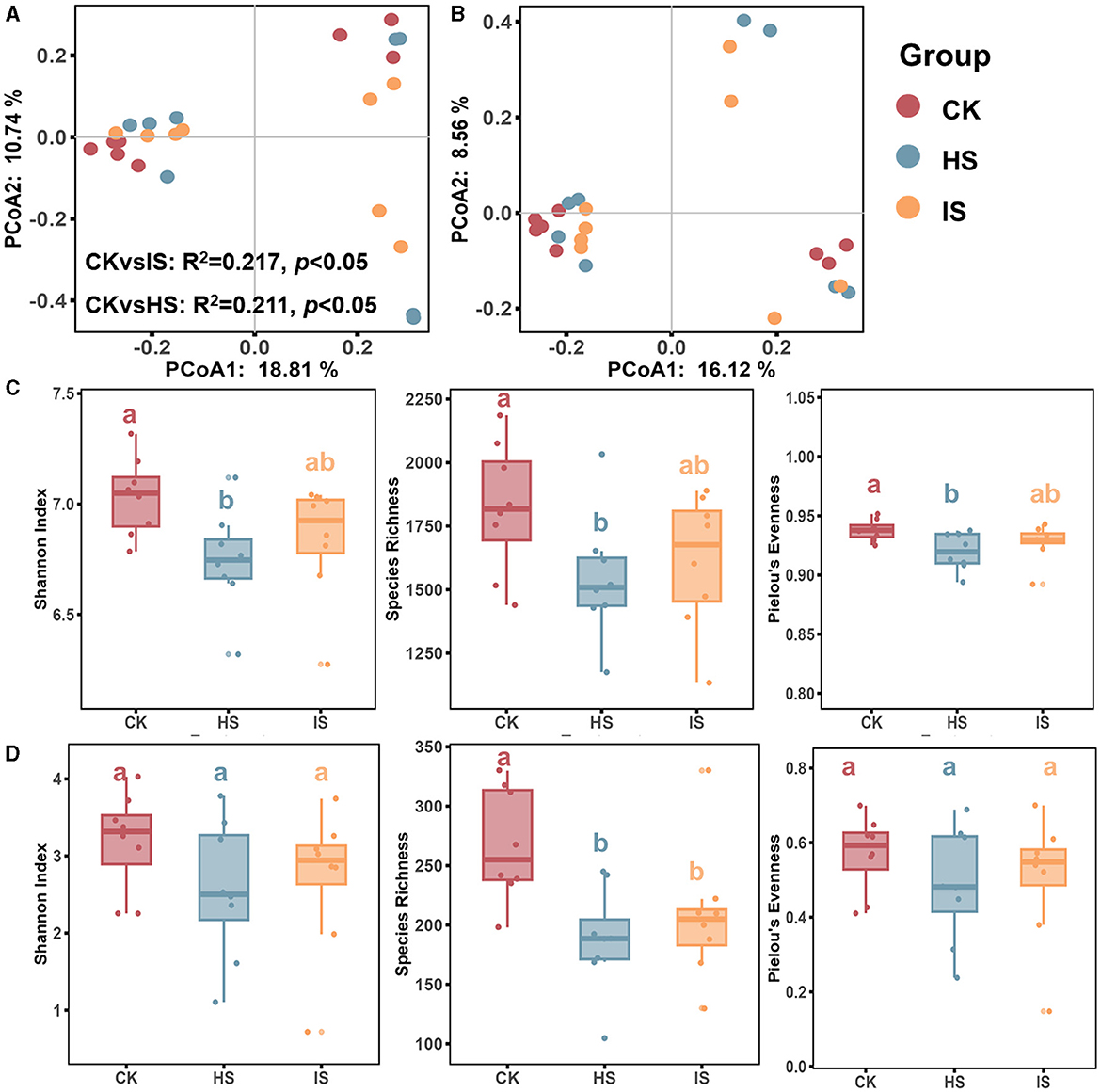
Figure 1. Microbial community diversity analysis in rhizosphere and bulk soils. PCoA of bacterial (A) and fungal (B) communities of bulk soil (CK), healthy rhizosphere soil (HS), and diseased rhizosphere soil (IS). ANOSIM analysis based on Bray-Curtis distance was conducted to reveal the community dissimilarity among CK, HS, and IS. Differences between rhizosphere and bulk soil bacterial (C) and fungal (D) community diversity indices, including the Shannon Index, species richness, and Pielou's evenness index. Different letters indicated a significant difference based on one-way ANOVA (Fisher's test) with P < 0.05.
Biomarkers differentiating bulk and rhizosphere soil samples
The LEfSe analyses were carried out to identify which taxa define the bacterial and fungal communities in CK, HS, and IS (Figure 2). There were 223 and 43 taxa found to be unique in the bacterial and fungal communities (P < 0.05), respectively. Accordingly, 11 out of 20 bacterial taxa were identified as the top bulk soil taxa with LDA of >4.0, whereas IS and HS soils had 8 and 1 taxa, respectively. The top bacterial taxa at the genus level in CK were Roseiflexaceae, which belonged to the phyla Chloroflex, and unclassified taxa belonging to the phyla Acidobacteria (LDA > 4.0). The top taxon in HS was Sphingomonas, which belonged to Proteobacteria. The top taxon in IS was Sphingobium, which belonged to Proteobacteria. For fungal communities, five out of nine fungal taxa were identified as the top bulk soil taxa, whereas four taxa were found in HS soil. The top fungal taxa associated with CK at the genus level were Rhodotorula, belonging to the phyla Basidiomycota, and Pezizaceae family, belonging to the phyla Ascomycota. The top taxon associated with HS was Ascomycota at the phylum level. For the IS soil communities, the defining taxa were Guehomyces belonging to the phyla Basidiomycota (LDA = 2.245). Therefore, Roseiflexaceae, Sphingomonas, and Sphingobium were identified as the bacterial biomarkers of CK, HS, and IS, respectively; Rhodotorula and Ascomycota_unidentified_1_1 were identified as the fungal biomarkers of CK and HS.
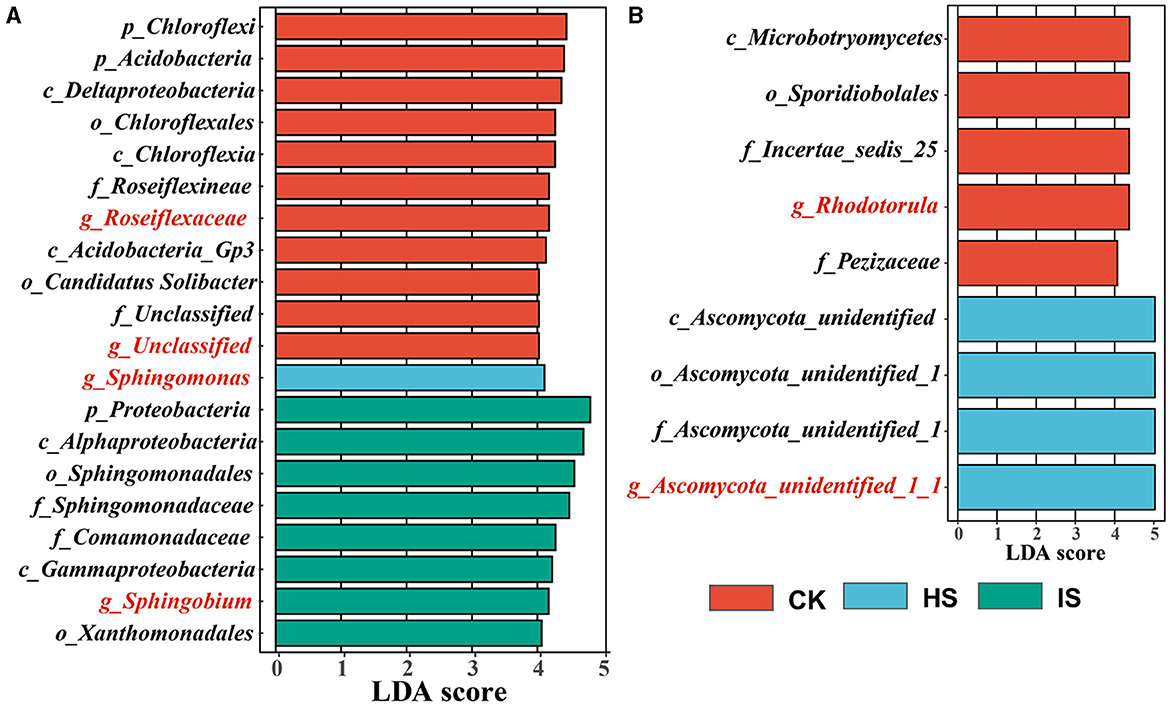
Figure 2. Taxa identified by linear discriminant effect size (LEfSe) analysis that explains the differences of bacterial (A) and fungal (B) communities among bulk soil (CK), healthy rhizosphere soil (HS), and diseased rhizosphere soil (IS). The threshold on the logarithmic LDA score for discriminative features was set to 4 (P < 0.05).
Network analysis of bacterial and fungal communities at the genus level
To explore the co-associated interactions of microbes in bulk soil, healthy rhizosphere soil, and diseased rhizosphere soil, a Spiec-Easi network analysis at the genus level was conducted (Figure 3), and the network topological properties of these networks in each soil compartment were calculated. For bacterial networks, the node numbers, link numbers, degree, and clustering coefficient of BHS were lower than those of BCK and BIS (Table 2), and the BHS network had a higher positive link proportion. In addition, these topological properties among BCK and BIS were similar. We also analyzed the sub-networks of biomarkers (Roseiflexaceae, Sphingobium, and Sphingomonas) and their associated nodes. Roseiflexaceae showed 11 links with other nodes, including six negative links and five positive links in the BCK network; Sphingobium had two positive links; and Sphingomonas had three positive links within four links. In the BHS network, Roseiflexaceae had two positive links within three links with other nodes, Sphingobium had one positive link, and Sphingomonas had five negative links within six positive links. In the BIS network, Roseiflexaceae had three negative links with other nodes, Sphingobium had six negative links within 13 links, and Sphingomonas had eight negative links within nine links. Furthermore, Roseiflexaceae and Sphingomonas worked as connectors in the overall networks of the BIS and BHS networks, whereas Sphingomonas worked as connectors in the BCK networks. Fungal networks were smaller, with fewer nodes and links compared to bacterial networks. Similar to bacterial networks, nodes and links in the FHS network were the smallest. The topological properties of the FIS network, including degree and clustering coefficient, were the highest compared to those of the FCK and FHS networks, suggesting that FIS was the most compact. For subnetworks based on biomarkers and their associated nodes (Ascomycota_unidentified_1_1 and Rhodotorula), the nodes and links were the smallest in FIS and the largest in FHS. Interestingly, more than half of the links associated with keystone taxa (Ascomycota_unidentified_1_1 in FHS and Rhodotorula in FCK) in the FCK and FHS subnetworks were positive, whereas almost all the links associated with Ascomycota_unidentified_1_1 and Rhodotorula in the FIS network were negative. Furthermore, Ascomycota_unidentified_1_1 and Rhodotorula both played the role of connectors in the FCK network, and Ascomycota_unidentified_1_1 also worked as connectors in the FHS and FIS networks.
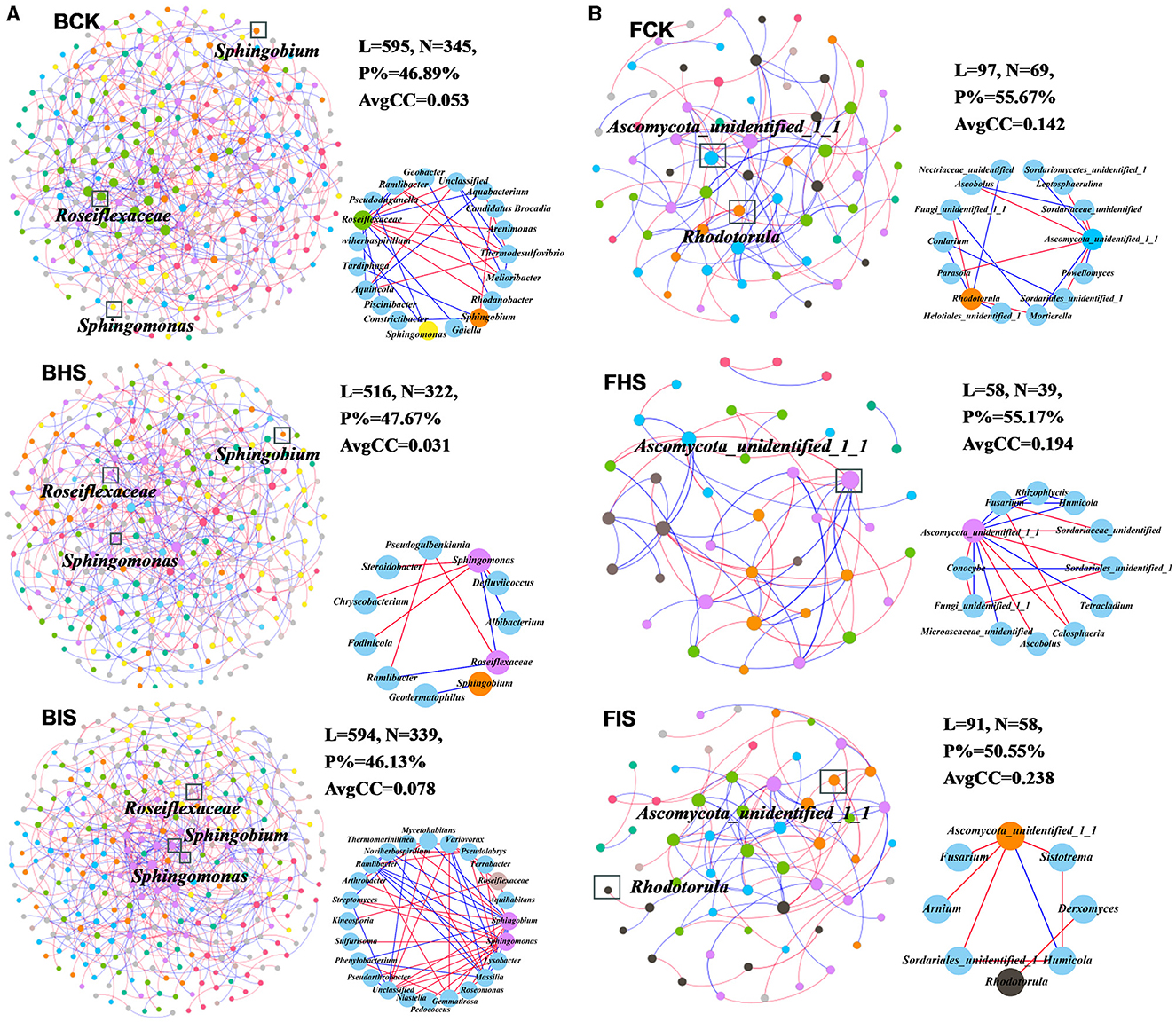
Figure 3. Overall networks and sub-networks of microbial communities at the genus taxonomic classification level. (A) Bacterial networks of bulk soil (BCK), healthy rhizosphere soil (BHS), and diseased rhizosphere soil (BIS). (B) Fungal networks of FCK, FHS, and FIS. Sub-networks were conducted based on biomarkers (Roseiflexaceae, Sphingobium, and Sphingomonas in bacterial communities and Ascomycota_unidentified_1_1 and Rhodotorula in fungal communities) and their associated nodes. Nodes were colored by module assignment. The red line represented negative links between nodes, and the blue line represented positive links.
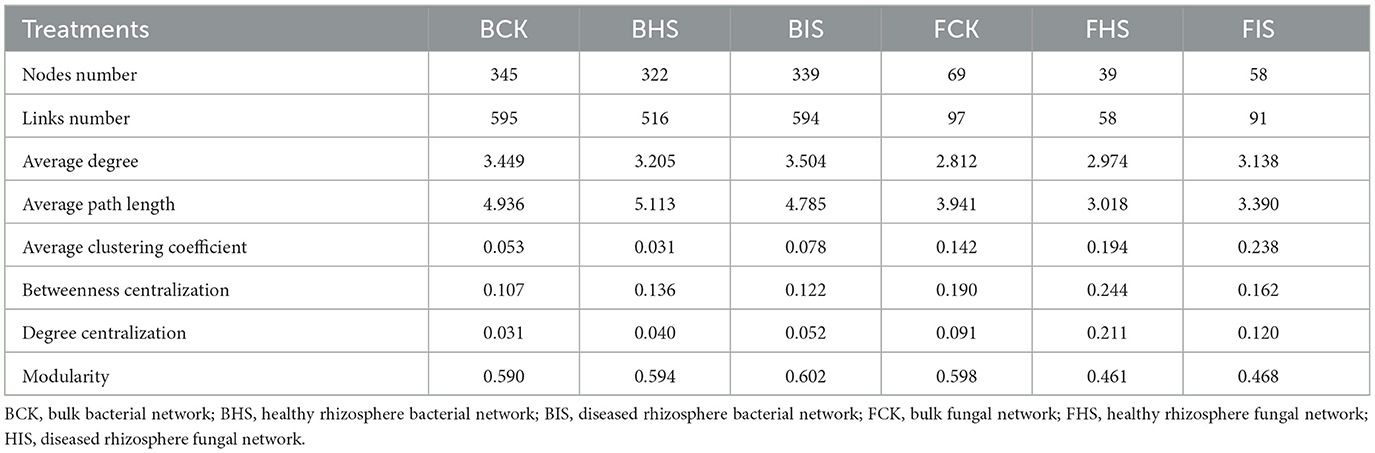
Table 2. Topological properties of bacterial and fungal networks in bulk, healthy rhizosphere, and diseased rhizosphere soils.
Correlation between bulk and rhizosphere soil properties and microbial communities
Mantel tests were employed to ascertain the biomarkers that exert an influence on the bacterial and fungal communities in both bulk and rhizosphere soils (Figure 4). It was observed that the bacterial communities exhibited a greater susceptibility to soil properties when compared to the fungal communities. Specifically, WC, AP, TP, and TK were identified as significant factors impacting the structure and diversity of bacterial communities (p < 0.05). In fungal communities, pH and TP had significant effects on community diversity (p < 0.01), and there was no factor found to significantly affect community structure. Furthermore, the biomarkers of both bacterial and fungal communities were discovered to be influenced by the fluctuations in soil properties. The variations of Roseiflexaceae among bulk soil, healthy rhizosphere soil, and diseased rhizosphere soil were significantly influenced by TP and TK (p < 0.05), WC, pH, and AK were the key factors affecting the relative abundance of Sphingobium (p < 0.01), and WC was the key factor affecting Sphingomonas (p < 0.05). The fungal biomarkers Ascomycota_unidentified_1_1 were significantly affected by WC, AP, TP, and TK, whereas no significant key factor was found for Rhodotorula genera (p < 0.05). Overall, bacterial and fungal communities had different key drivers, and bacterial communities had more driving factors than fungal communities.
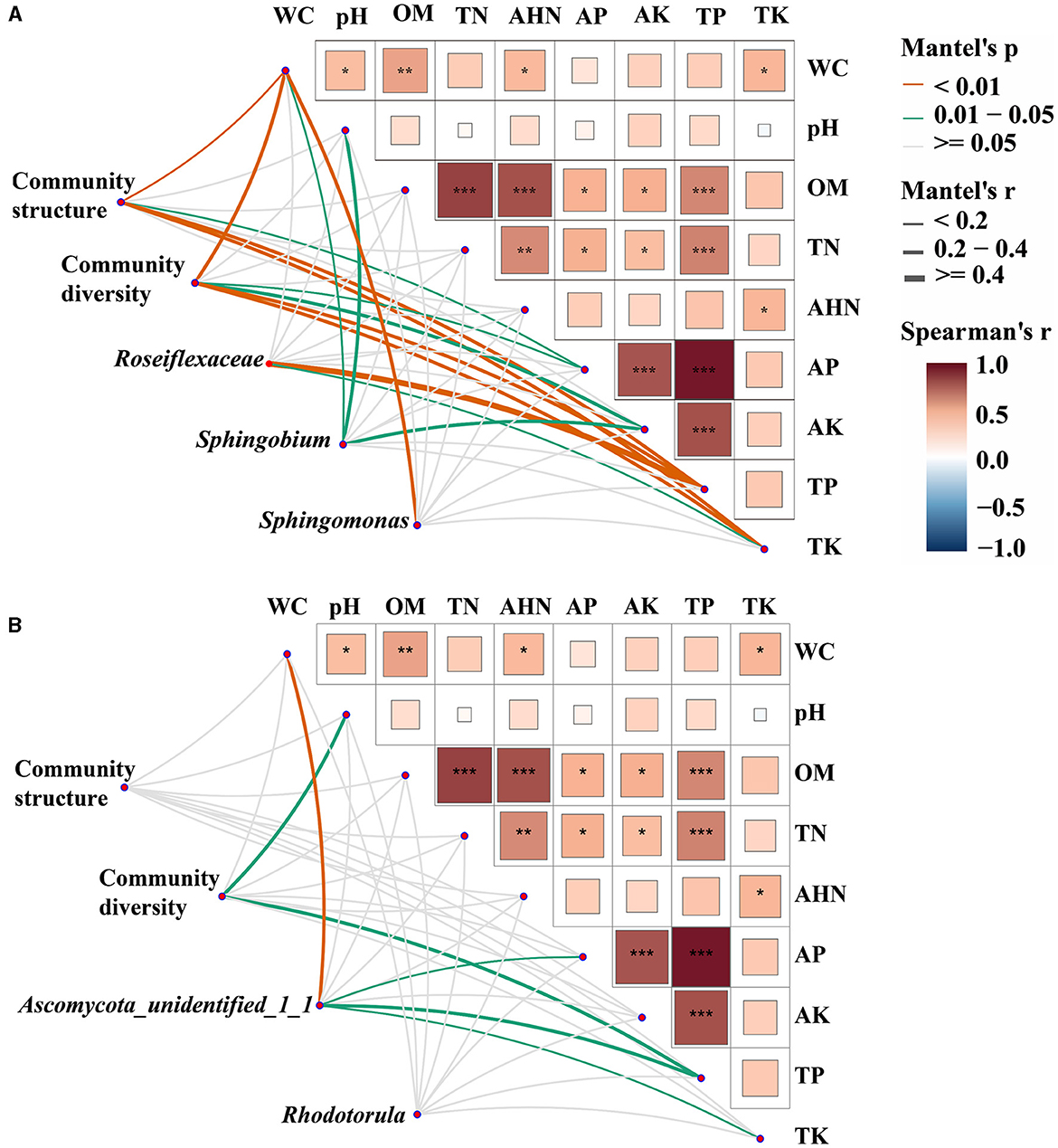
Figure 4. Environmental drivers of bacterial (A) and fungal (B) communities. Pairwise comparisons of soil physicochemical properties were shown, with a color gradient denoting Spearman's correlation coefficients. Microbial community structure (PCoA based on Bray-Curtis distance), community diversity indices (Shannon index, species richness, and Pielou's evenness index), and the relative abundance of biomarkers (Roseiflexaceae, Sphingobium, and Sphingomonas in bacterial communities and Ascomycota_unidentified_1_1 and Rhodotorula in fungal communities) were related to each soil physicochemical property through partial Mantel tests. Edge width corresponds to Mantel's r statistic for the corresponding distance correlations, and edge color denotes the statistical significance based on 9,999 permutations. *p < 0. 05; **p < 0.01; ***p < 0.001.
Discussion
Differences in the diversity, composition, and community between bacteria and fungi in bulk soil, healthy rhizosphere soil, and diseased rhizosphere soil
The diversity of microbial communities in the rhizosphere is crucial to soil nutrient cycling and many plant processes (Lebeis, 2015). In agreement with most previous studies, rhizosphere soil microbial community diversities were significantly decreased compared with bulk soil (Bakker et al., 2015). Significant differences in community structure and a decline in community diversity were observed in this study. However, the microbial community diversity and structure of rhizosphere soils showed no significant correlation with the plant's healthy state in this study. The presence of plant diseases, including root-rot disease (Liao et al., 2022), Verticillium wilt (Wei et al., 2019), and ginseng rusty root (Bian et al., 2020), has been observed to induce alterations in the microbiomes of the root endosphere, rhizosphere, and soil. Different types of pathogens might be possible reasons for the different results of this study compared to previous studies. Previous studies usually focused on bacterial and fungal pathogens, whereas viruses are the pathogen for PVY disease. The transmission modes and infection types were different among bacterial, fungal, and virus-pathogen diseases. The infection of viruses into plants typically does not result in immediate mortality but rather induces modifications in their growth and developmental mechanisms, leading to alterations in plant pigmentation or morphology, commonly referred to as discoloration and deformity (Koziel et al., 2021). Thus, the effects of PVY infection on the rhizosphere might be relatively delayed compared with bacterial and fungal diseases. Differences among bacterial and fungal communities under PVY infection were also observed and can be attributed to different responses to soil physicochemical properties. However, there were no significant changes in community structures between bulk soil and rhizosphere soil. Several previous studies have shown similar results (Ai et al., 2018; Xue et al., 2022).
In this study, we found the different community compositions of bulk, healthy rhizosphere, and diseased rhizosphere soils. Roseiflexaceae, Sphingomonas, and Sphingobium were found to be the biomarkers of the BCK, BHS, and BIS communities, respectively. The genus Roseiflexaceae has been identified as a biomarker for the soil microbial community's yield, functioning as chemoautotrophic bacteria involved in carbon dioxide assimilation and significantly influencing the growth of Eichhornia crassipes (Wang et al., 2022). The genera Sphingomonas and Sphingobium are commonly found in rhizosphere soil and have been recognized as beneficial microbes that positively impact soil nutrient cycling. Sphingobium has been associated with carbon and nitrogen cycling in soil (Videira et al., 2009), while Sphingomonas has been reported to adapt to neutral pH and possess nitrogen-fixing capabilities. An increased abundance of Sphingomonas and Sphingobium in the rhizosphere could result in improved conditions for crop growth. More importantly, Sphingomonas has the biocontrol potential of plant disease (Innerebner et al., 2011). Thus, enrichment of Sphingomonas could be regarded as a potential defensive mechanism against plant diseases. An unclassified genus belonging to the Ascomycota phyla and the genus Rhodotorula were also identified as the biomarkers in the fungal communities of FHS and FCK, respectively. Rhodotorula was a fungal genus with a higher relative abundance in field soil (Tan et al., 2021). Ascomycota phyla was reported to be the dominant root-associated fungus (Peng et al., 2023), which was highly adaptable and actively involved in nutrient cycling (Egidi et al., 2019). Furthermore, Ascomycota possesses essential genes responsible for encoding cellulolytic enzymes and facilitating the carbon conversion process (Hannula et al., 2012). Therefore, bacterial and fungal community compositions manifest the adaptive traits of plants in diverse environments and contribute to the facilitation of crucial ecological processes through the regulation of the abundance of these prevailing microorganisms.
PVY infection affected the stability of the rhizosphere microbial community network
A co-occurrence network is a representation of microbial flora and their interactions. Interrelationships among these organisms play an essential role in maintaining microbial communities' structure and functionality across various environments (Zhou et al., 2021). The presence of a significant number of links between microbial taxa enhances network cohesion, while increased connectivity and network interactions play a crucial role in promoting stability within the microbial communities (Morrien et al., 2017). We found that the relationships between taxa in bacterial communities were closer and more complex than those in fungi communities. However, there were very slight differences between bulk soil and rhizosphere soil. In detail, the network complexity and size showed larger changes in healthy rhizosphere soil compared to diseased rhizosphere soil. Therefore, PVY infection weakened the selection of plant roots for microbial communities and increased their network complexity and size. Therefore, PVY infection resulted in a higher degree of connectivity among microbial taxa in the rhizosphere, enhancing rhizosphere microbial community stability. The network degree of taxa within the fungal community exhibited lower values compared to those within bacterial communities, possibly attributable to dissimilarities in the anticipated metabolic activities of these organisms. In response to alterations in environmental conditions and the abundance of other taxa, archaea and bacteria generally demonstrate more prompt reactions than eukaryotes, including fungi (Paul, 2007). Fungi possess the ability to enzymatically degrade intricate organic matter extracellularly, thereby diminishing their obligatory reliance on bacterial and archaeal taxa (Pivato et al., 2008). This characteristic may elucidate the relatively reduced number of correlative interactions observed among fungi.
Microbial taxa were shown to have cooperative and competitive relationships in the interaction network via positive and negative edges (Price et al., 2021). Our results showed that fungal networks were dominated by positive links, whereas bacterial networks showed more negative links. Similar to other studies, the fungal community may adapt to soil physicochemical properties through cooperative symbiosis or mutualism (Fan et al., 2018; Peng et al., 2023). Topologically, network hubs and connectors can be seen as regulators, mediators, or adaptors, while module hubs can be considered integral components within specific modules (Han et al., 2004). In this study, we have classified these hubs and connectors as key species, acknowledging their crucial roles in network structure and their potential as targets for microbial modulation aimed at enhancing crop productivity (Olesen et al., 2007). Bacterial biomarkers, Roseiflexaceae and Sphingomonas, were found to be connectors in bacterial networks. Fungal biomarkers Ascomycota_unidentified_1_1 and Rhodotorula were also identified as connectors in fungal networks. In addition, subnetworks of biomarkers further indicated their changes among bulk, healthy rhizosphere, and diseased rhizosphere soils through greater competitive ability and more complex interaction. The proportion of negative links and the link numbers of Roseiflexaceae, Sphingomonas, and Sphingobium with other taxa were the highest in the BCK, BHS, and BIS networks, respectively. In the fungal network FIS, the links between biomarkers and other taxa were dominated by negative links.
The bacterial communities of bulk and rhizosphere soils were more sensitive to soil physicochemical properties than fungal communities
The composition and structure of rhizosphere microorganisms are influenced by intricate soil properties (Reinhold-Hurek et al., 2015). Mantel analysis revealed that the factors influencing bulk and rhizosphere soils differed between bacterial and fungal communities. Compared to fungal communities, we found more key underlying factors in bacterial communities. In detail, WC, AP, TP, and TK were all key factors affecting bacterial community structure and diversity. However, fungal community diversity was affected by pH and TP, and there was no factor found to significantly affect community structure. This discovery aligns with previous research findings (Ai et al., 2018; Xue et al., 2022). This may be because bacterial and fungal communities have different influence mechanisms for their different characteristics, such as strategies and ecological niches. The impact of WC on the structure and diversity of bacterial communities was evident; however, it had no significant effect on fungal communities. A significant influence of pH was observed in the diversity of the fungal community but not in the bacterial community. A comparable outcome was also documented, indicating that alterations in soil water content had a notable impact on the diversity of rhizosphere bacterial communities, while no significant impact was observed on the composition of the soil, rhizosphere, and root endosphere fungal communities (Naylor et al., 2017). Additionally, the contents of TP, TK, AP, and AK were identified as crucial factors influencing bacterial community diversity, while AP was found to influence fungal community diversity. Mantel analysis also indicated that the key factors affecting the biomarkers of microbial communities were different between bulk, healthy rhizosphere, and diseased rhizosphere soils. WC and pH were key factors affecting the biomarkers of bacterial rhizosphere communities, whereas the biomarkers of bulk bacteria were significantly affected by soil nutrients (e.g., TP and TK). This observation can be attributed to the rhizosphere environment's elevated nutrient conditions compared to the bulk soil, which offers more favorable resources for microbial activities and plant growth (Peng et al., 2022). In addition, the rhizosphere soil of the diseased plant provided higher restricting factors for the bacterial community, thus the biomarkers of Sphingobium in BIS showed closer relationships with soil properties. Therefore, our research indicates that soil physicochemical properties exerted a greater influence on bacterial communities compared to fungal communities, and the factors influencing bulk soil and rhizosphere soil differed.
Conclusion
Our findings demonstrate that soil environment properties significantly impacted soil microbiomes, particularly the bacterial community, while PVY infection did not significantly affect the structure and diversity of rhizosphere bacterial and fungal communities. LEfSe analysis further indicated the significant differences in community compositions and identified the biomarkers among bulk, healthy rhizosphere, and diseased rhizosphere soils. Bacterial networks were more complex and compact than fungal networks. PVY infection increased rhizosphere microbial community stability and resistance to environmental stress through greater connectedness among microbial taxa. Mantel analyses further revealed that WC played an apparent influence on bacterial community structure and diversity, and pH showed significant effects on fungal community diversity. WC and pH were key factors affecting the biomarkers of bacterial rhizosphere communities, whereas the biomarkers of bulk bacteria were significantly affected by soil nutrients, especially for the biomarkers of Sphingobium. In summary, this study explored the bacterial and fungal adaptation strategies from bulk soil to rhizosphere soil in healthy and diseased plants, and regulation of community composition and interaction networks were the main adaptation strategies to Hs and IS.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
YD: Conceptualization, Writing—original draft. WK: Writing—review & editing. XZ: Writing—review & editing. YZ: Writing—review & editing. TX: Writing—review & editing. MC: Writing—review & editing. LZ: Writing—review & editing. JS: Writing—review & editing. ZZ: Writing—review & editing. CC: Writing—review & editing. CZ: Writing—review & editing. HY: Writing—review & editing. SH: Writing—original draft. YG: Writing—original draft.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This project was supported by the Changde Tobacco Company of Hunan Province (CD2022KJ01).
Conflict of interest
YD, WK, XZ, YZ, TX, MC, LZ, JS, ZZ, CC, CZ, and SH were employed by Changde Tobacco Company of Hunan Province.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1333076/full#supplementary-material
References
Aguilar, P., and Sommaruga, R. (2020). The balance between deterministic and stochastic processes in structuring lake bacterioplankton community over time. Mol. Ecol. 29, 3117–3130. doi: 10.1111/mec.15538
Ahmad, J., Zervas, A., Ellegaard-Jensen, L., Hennessy, R. C., Carbone, I., Cornish, V., et al. (2022). Microbial diversity in four rhizocompartments (bulk soil, rhizosphere, rhizoplane, and endosphere). of four winter wheat varieties at the fully emerged flag leaf growth stage. Microbiol. Res. Announc. 11, e00663–00622. doi: 10.1128/mra.00663-22
Ai, C., Zhang, S., Zhang, X., Guo, D., Zhou, W., Huang, S., et al. (2018). Distinct responses of soil bacterial and fungal communities to changes in fertilization regime and crop rotation. Geoderma 319, 156–166. doi: 10.1016/j.geoderma.2018.01.010
Bakker, M. G., Chaparro, J. M., Manter, D. K., and Vivanco, J. M. (2015). Impacts of bulk soil microbial community structure on rhizosphere microbiomes of Zea mays. Plant Soil 392, 115–126. doi: 10.1007/s11104-015-2446-0
Benitez, M-. S., Osborne, S. L., and Lehman, R. M. (2017). Previous crop and rotation history effects on maize seedling health and associated rhizosphere microbiome. Sci. Rep. 7, 15709. doi: 10.1038/s41598-017-15955-9
Berendsen, R. L., Vismans, G., Yu, K., Song, Y., De Jonge, D., Burgman, R., et al. (2018). Disease-induced assemblage of a plant-beneficial bacterial consortium. The ISME J. 12, 1496–1507. doi: 10.1038/s41396-018-0093-1
Bian, X., Xiao, S., Zhao, Y., Xu, Y., Yang, H., Zhang, L., et al. (2020). Comparative analysis of rhizosphere soil physiochemical characteristics and microbial communities between rusty and healthy ginseng root. Sci. Rep. 10, 15756. doi: 10.1038/s41598-020-71024-8
Callahan, B. J., Mcmurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J. A., Holmes, S. P., et al. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Chaparro, J. M., Badri, D. V., and Vivanco, J. M. (2014). Rhizosphere microbiome assemblage is affected by plant development. The ISME J. 8, 790–803. doi: 10.1038/ismej.2013.196
Chen, S., Li, F., Liu, D., Jiang, C., Cui, L., Shen, L., et al. (2017). Dynamic expression analysis of early response genes induced by potato virus Y in PVY-resistant Nicotiana tabacum. Plant Cell Rep. 36, 297–311. doi: 10.1007/s00299-016-2080-1
Deng, Y., Huang, H., Lei, F., Fu, S., Zou, K., Zhang, S., et al. (2021). Endophytic bacterial communities of ginkgo biloba leaves during leaf developmental period. Front. Microbiol. 12, 698703. doi: 10.3389/fmicb.2021.698703
Egidi, E., Delgado-Baquerizo, M., Plett, J. M., Wang, J., Eldridge, D. J., Bardgett, R. D., et al. (2019). A few Ascomycota taxa dominate soil fungal communities worldwide. Nat. Commun. 10, 2369. doi: 10.1038/s41467-019-10373-z
Enagbonma, B. J., Fadiji, A. E., Ayangbenro, A. S., and Babalola, O. O. (2023). Communication between plants and rhizosphere microbiome: exploring the root microbiome for sustainable agriculture. Microorganisms 11, 2003. doi: 10.3390/microorganisms11082003
Fan, K., Weisenhorn, P., Gilbert, J. A., and Chu, H. (2018). Wheat rhizosphere harbors a less complex and more stable microbial co-occurrence pattern than bulk soil. Soil Biol. Biochem. 125, 251–260. doi: 10.1016/j.soilbio.2018.07.022
French, E., Kaplan, I., and Enders, L. (2021). Foliar aphid herbivory alters the tomato rhizosphere microbiome, but initial soil community determines the legacy effects. Front. Sust. Food Syst. 5, 629684. doi: 10.3389/fsufs.2021.629684
Friedman, J., and Alm, E. J. (2012). Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8, e1002687. doi: 10.1371/journal.pcbi.1002687
Gu, Y., Liu, Y., Li, J., Cao, M., Wang, Z., Li, J., et al. (2022). Mechanism of intermittent deep tillage and different depths improving crop growth from the perspective of rhizosphere soil nutrients, root system architectures, bacterial communities, and functional profiles. Front. Microbiol. 12, 759374. doi: 10.3389/fmicb.2021.759374
Gu, Y., Meng, D., Yang, S., Xiao, N., Li, Z., Liu, Z., et al. (2019). Invader-resident community similarity contribute to the invasion process and regulate biofertilizer effectiveness. J. Clean. Prod. 241, 118278. doi: 10.1016/j.jclepro.2019.118278
Han, J-. D. J., Bertin, N., Hao, T., Goldberg, D. S., Berriz, G. F., Zhang, L. V., et al. (2004). Evidence for dynamically organized modularity in the yeast protein–protein interaction network. Nature 430, 88–93. doi: 10.1038/nature02555
Hannula, S. E., Boschker, H. T. S., De Boer, D., and Van Veen, W. J.A. (2012). 13C pulse-labeling assessment of the community structure of active fungi in the rhizosphere of a genetically starch-modified potato (Solanum tuberosum). cultivar and its parental isoline. New Phytol. 194, 784–799. doi: 10.1111/j.1469-8137.2012.04089.x
He, Q., Wang, S., Hou, W., Feng, K., Li, F., Hai, W., et al. (2021). Temperature and microbial interactions drive the deterministic assembly processes in sediments of hot springs. Sci. Total Environ. 772, 145465. doi: 10.1016/j.scitotenv.2021.145465
Hein, J. W., Wolfe, G. V., and Blee, K. A. (2008). Comparison of rhizosphere bacterial communities in Arabidopsis thaliana mutants for systemic acquired resistance. Microb. Ecol. 55, 333–343. doi: 10.1007/s00248-007-9279-1
Hinrichs-Berger, J., Harfold, M., Berger, S., and Buchenauer, H. (1999). Cytological responses of susceptible and extremely resistant potato plants to inoculation with potato virus Y. Physiol. Mol. Plant Pathol. 55, 143–150. doi: 10.1006/pmpp.1999.0216
Innerebner, G., Knief, C., and Vorholt, J. A. (2011). Protection of Arabidopsis thaliana against leaf-pathogenic Pseudomonas syringae by Sphingomonas strains in a controlled model system. Appl. Environ. Microbiol. 77, 3202–3210. doi: 10.1128/AEM.00133-11
Jiang, H., Luo, J., Liu, Q., Ogunyemi, S. O., Ahmed, T., Li, B., et al. (2023). Rice bacterial leaf blight drives rhizosphere microbial assembly and function adaptation. Microbiol. Spectr. 12, e01059-23. doi: 10.1128/spectrum.01059-23
Kembel, S. W., O'Connor, T. K., Arnold, H. K., Hubbell, S. P., Wright, S. J., and Green, J. L. (2014). Relationships between phyllosphere bacterial communities and plant functional traits in a neotropical forest. Proc. Nat. Acad. Sci. 111, 13715–13720. doi: 10.1073/pnas.1216057111
Kõljalg, U., Larsson, K. H., Abarenkov, K., Nilsson, R. H., Alexander, I. J., Eberhardt, U. E., et al. (2005). UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 166, 1063–1068. doi: 10.1111/j.1469-8137.2005.01376.x
Koziel, E., Otulak-Koziel, K., and Bujarski, J. J. (2021). Plant cell wall as a key player during resistant and susceptible plant-virus interactions. Front. Microbiol. 12, 656809. doi: 10.3389/fmicb.2021.656809
Lareen, A., Burton, F., and Schäfer, P. (2016). Plant root-microbe communication in shaping root microbiomes. Plant Mol. Biol. 90, 575–587. doi: 10.1007/s11103-015-0417-8
Lazcano, C., Boyd, E., Holmes, G., Hewavitharana, S., Pasulka, A., Ivors, K., et al. (2021). The rhizosphere microbiome plays a role in the resistance to soil-borne pathogens and nutrient uptake of strawberry cultivars under field conditions. Sci. Rep. 11, 3188. doi: 10.1038/s41598-021-82768-2
Lebeis, S. L. (2015). Greater than the sum of their parts: characterizing plant microbiomes at the community-level. Curr. Opin. Plant Biol. 24, 82–86. doi: 10.1016/j.pbi.2015.02.004
Liao, L. B., Chen, X. X., Xiang, J., Zhang, N. N., Wang, E. T., Shi, F. S., et al. (2022). Zanthoxylum bungeanum root-rot associated shifts in microbiomes of root endosphere, rhizosphere, and soil. PeerJ 10, e13808. doi: 10.7717/peerj.13808
Liu, H., Sun, F., Peng, J., Shen, M., Li, J., Dong, Y., et al. (2022). Deterministic process dominated belowground community assembly when suffering tomato bacterial wilt disease. Agronomy 12, 1024. doi: 10.3390/agronomy12051024
Lopes, L. D., Hao, J., and Schachtman, D. P. (2021). Alkaline soil pH affects bulk soil, rhizosphere and root endosphere microbiomes of plants growing in a Sandhills ecosystem. FEMS Microbiol. Ecol. 97, fiab028. doi: 10.1093/femsec/fiab028
Lu, Y., Rosencrantz, D., Liesack, W., and Conrad, R. (2006). Structure and activity of bacterial community inhabiting rice roots and the rhizosphere. Environ. Microbiol. 8, 1351–1360. doi: 10.1111/j.1462-2920.2006.01028.x
Morrien, E., Hannula, S. E., Snoek, L. B., Helmsing, N. R., Zweers, H., De Hollander, D., et al. (2017). Soil networks become more connected and take up more carbon as nature restoration progresses. Nat. Commun. 8, 14349. doi: 10.1038/ncomms14349
Naylor, D., Degraaf, S., Purdom, E., and Coleman-Derr, D. (2017). Drought and host selection influence bacterial community dynamics in the grass root microbiome. The ISME J. 11, 2691–2704. doi: 10.1038/ismej.2017.118
Olesen, J. M., Bascompte, J., Dupont, Y. L., and Jordano, P. (2007). The modularity of pollination networks. Proc. Nat. Acad. Sci. 104, 19891–19896. doi: 10.1073/pnas.0706375104
Paul, E. (2007). Soil Microbiology, Ecology, and Biochemistry in Perspective. Amsterdam: Elsevier, 3–24.
Peng, M., He, H., Wang, X., Wang, Z., and Zhuang, L. (2023). Comparison of network connectivity and environmental driving factors of root-associated fungal communities of desert ephemeral plants in two habitat soils. J. Environ. Manage. 332, 117375. doi: 10.1016/j.jenvman.2023.117375
Peng, M., He, H., Wang, Z., Li, G., Lv, X., Pu, X., et al. (2022). Responses and comprehensive evaluation of growth characteristics of ephemeral plants in the desert-oasis ecotone to soil types. J. Environ. Manage. 316, 115288. doi: 10.1016/j.jenvman.2022.115288
Pivato, B., Gamalero, E., Lemanceau, P., and Berta, G. (2008). Colonization of adventitious roots of Medicago truncatula by Pseudomonas fluorescens C7R12 as affected by arbuscular mycorrhiza. FEMS Microbiol. Lett. 289, 173–180. doi: 10.1111/j.1574-6968.2008.01391.x
Price, G. W., Langille, M. G. I., and Yurgel, S. N. (2021). Microbial co-occurrence network analysis of soils receiving short- and long-term applications of alkaline treated biosolids. Sci. Total Environ. 751, 141687. doi: 10.1016/j.scitotenv.2020.141687
Qu, Q., Zhang, Z., Peijnenburg, W. J. G. M., Liu, W., Lu, T., Hu, B., et al. (2020). Rhizosphere microbiome assembly and its impact on plant growth. J. Agric. Food Chem. 68, 5024–5038. doi: 10.1021/acs.jafc.0c00073
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Reinhold-Hurek, B., Bünger, W., Burbano, C. S., Sabale, M., and Hurek, T. (2015). Roots shaping their microbiome: global hotspots for microbial activity. Ann. Rev. Phytopathol. 53, 403–424. doi: 10.1146/annurev-phyto-082712-102342
Shah, A., Hassan, Q. P., Mushtaq, S., Shah, A. M., and Hussain, A. (2017). Chemoprofile and functional diversity of fungal and bacterial endophytes and role of ecofactors–A review. J. Basic Microbiol. 57, 814–826. doi: 10.1002/jobm.201700275
Sun, X., Zhang, X., Zhang, G., Miao, Y., Zeng, T., Zhang, M., et al. (2022). Environmental response to root secondary metabolite accumulation in Paeonia lactiflora: insights from rhizosphere metabolism and root-associated microbial communities. Microbiol. Spectr. 10, e0280022. doi: 10.1128/spectrum.02800-22
Tan, L., Zeng, W. A., Xiao, Y., Li, P., Gu, S., Wu, S., et al. (2021). Fungi-bacteria associations in wilt diseased rhizosphere and endosphere by interdomain ecological network analysis. Front. Microbiol. 12, 722626. doi: 10.3389/fmicb.2021.722626
Videira, S. S., De Araujo, D., Rodrigues Lda, J. L., Baldani, S., and Baldani, V. L. J.I. (2009). Occurrence and diversity of nitrogen-fixing Sphingomonas bacteria associated with rice plants grown in Brazil. FEMS Microbiol. Lett. 293, 11–19. doi: 10.1111/j.1574-6968.2008.01475.x
Wang, J. L., Liu, K. L., Zhao, X. Q., Gao, G. F., Wu, Y. H., Shen, R. F., et al. (2022). Microbial keystone taxa drive crop productivity through shifting aboveground-belowground mineral element flows. Sci. Total Environ. 811, 152342. doi: 10.1016/j.scitotenv.2021.152342
Wei, F., Zhao, L., Xu, X., Feng, H., Shi, Y., Deakin, G., et al. (2019). Cultivar-dependent variation of the cotton rhizosphere and endosphere microbiome under field conditions. Front. Plant Sci. 10, 1659. doi: 10.3389/fpls.2019.01659
Xue, R., Wang, C., Zhao, L., Sun, B., and Wang, B. (2022). Agricultural intensification weakens the soil health index and stability of microbial networks. Agric. Ecosyst. Environ. 339, 108118. doi: 10.1016/j.agee.2022.108118
Yin, C., Casa Vargas, J. M., Schlatter, D. C., Hagerty, C. H., Hulbert, S. H., Paulitz, T. C., et al. (2021). Rhizosphere community selection reveals bacteria associated with reduced root disease. Microbiome 9, 1–18. doi: 10.1186/s40168-020-00997-5
Yu, H., Zhong, Q., Peng, Y., Zheng, X., Xiao, F., Wu, B., et al. (2022). Environmental filtering by pH and salinity jointly drives prokaryotic community assembly in coastal wetland sediments. Front. Marine Sci. 8, 792294. doi: 10.3389/fmars.2021.792294
Zhang, M., Zhang, T., Zhou, L., Lou, W., Zeng, W., Liu, T., et al. (2022). Soil microbial community assembly model in response to heavy metal pollution. Environ. Res. 213, 113576. doi: 10.1016/j.envres.2022.113576
Zhao, K., Ma, B., Xu, Y., Stirling, E., and Xu, J. (2021). Light exposure mediates circadian rhythms of rhizosphere microbial communities. The ISME J. 15, 2655–2664. doi: 10.1038/s41396-021-00957-3
Keywords: soil-plant-microbe interactions, plant virus disease, rhizosphere microbes, biomarkers, co-occurrence network, soil properties
Citation: Deng Y, Kong W, Zhang X, Zhu Y, Xie T, Chen M, Zhu L, Sun J, Zhang Z, Chen C, Zhu C, Yin H, Huang S and Gu Y (2024) Rhizosphere microbial community enrichment processes in healthy and diseased plants: implications of soil properties on biomarkers. Front. Microbiol. 15:1333076. doi: 10.3389/fmicb.2024.1333076
Received: 04 November 2023; Accepted: 01 February 2024;
Published: 29 February 2024.
Edited by:
Rui Zhuo, Hunan University, ChinaReviewed by:
Daniela Costa, Teagasc - Irish Agriculture and Food Development Authority, IrelandShulan Shi, China University of Mining and Technology, China
Copyright © 2024 Deng, Kong, Zhang, Zhu, Xie, Chen, Zhu, Sun, Zhang, Chen, Zhu, Yin, Huang and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Songqing Huang, MjU4NDQzMDE2QHFxLmNvbQ==; Yabing Gu, Z3V5YWJpbmcwMjA3QDE2My5jb20=