 Yanyan Chen
Yanyan Chen Yijing Lin
Yijing Lin Jingyi Zhu
Jingyi Zhu Jiayin Zhou
Jiayin Zhou Haoyi Lin
Haoyi Lin Yiting Fu
Yiting Fu Yan Zhou
Yan Zhou
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 28 February 2024
Sec. Aquatic Microbiology
Volume 15 - 2024 | https://doi.org/10.3389/fmicb.2024.1323160
This article is part of the Research Topic Women in Aquatic Microbiology: 2023 View all 18 articles
The acceleration of the nitrogen cycle and the nitrogen excess observed in some coastal waters has increased interest into understanding the biochemical and molecular basis of nitrogen metabolism in various microorganisms. To investigate nitrogen metabolism of a novel heterotrophic nitrification and aerobic denitrification bacterium Klebsiella aerogenes strain (B23) under nitrogen-rich conditions, we conducted physiological and transcriptomic high-throughput sequencing analyses on strain B23 cultured on potassium nitrate–free or potassium nitrate–rich media. Overall, K. aerogenes B23 assimilated 82.47% of the nitrate present into cellular nitrogen. Further, 1,195 differentially expressed genes were observed between K. aerogenes B23 cultured on potassium nitrate–free media and those cultured on potassium nitrate-rich media. Gene annotation and metabolic pathway analysis of the transcriptome were performed using a series of bioinformatics tools, including Gene Ontology, Kyoto Encyclopedia of Genes and Genomes, and Non-Redundant Protein Database annotation. Accordingly, the nitrogen metabolism pathway of K. aerogenes B23 was analyzed; overall, 39 genes were determined to be involved in this pathway. Differential expression analysis of the genes involved in the nitrogen metabolism pathway demonstrated that, compared to the control, FNR, NarK/14945, fdx, gshA, proB, proA, gapA, argH, artQ, artJ, artM, ArgR, GAT1, prmB, pyrG, glnS, and Ca1 were significantly upregulated in the nitrogen-treated K. aerogenes B23; these genes have been established to be involved in the regulation of nitrate, arginine, glutamate, and ammonia assimilation. Further, norV, norR, and narI were also upregulated in nitrogen-treated K. aerogenes B23; these genes are involved in the regulation of NO metabolism. These differential expression results are important for understanding the regulation process of key nitrogen metabolism enzyme genes in K. aerogenes B23. Therefore, this study establishes a solid foundation for further research into the expression regulation patterns of nitrogen metabolism–associated genes in K. aerogenes B23 under nitrogen-rich conditions; moreover, this research provides essential insight into how K. aerogenes B23 utilizes nutritional elements.
Marine microorganisms are diverse, abundant, and widely distributed throughout marine ecosystems. These microorganisms act as both producers and consumers, making significant contributions to primary productivity and biomass within the ocean (Fuhrman and Hagström, 2008). However, the marine environment presents a complex and varied set of environmental characteristics across different regions, which have been attributed to both natural factors and human activities; these characteristics include high salinity (Xu et al., 2023), high and low temperatures (Cheung et al., 2021; Kang et al., 2021), and oligotrophic conditions (Duarte et al., 2013). Microorganisms can adapt and survive within complex marine environments; ultimately, this can lead to the formation of unique community structures and distribution characteristics. Microorganisms contribute to almost all biochemical reactions and biogeochemical cycles within the ocean. Therefore, understanding their community composition, structure, and distribution across different habitats, as well as elucidating their specific metabolic characteristics, becomes necessary for developing an in-depth understanding of the ecological functions of marine microorganisms.
Nitrogen is an essential element for life and a crucial component of biogeochemical cycles in aquatic ecosystems (Stüeken et al., 2016). However, the acceleration of the nitrogen cycle, owing to increased industrial activity and the extensive use of nitrogen fertilizers, has led to significant and challenging environmental issues that have a direct impact on human populations worldwide. Microbial nitrogen assimilation primarily occurs via the conversion of nitrate into ammonia by a series of enzymes such as nitrate reductase and nitrite reductase (Matassa et al., 2016). These microorganisms then use this ammonia for amino acid synthesis and transformation (Mena-Rivera et al., 2023). Amino acids are primarily used to synthesize proteins that can then be modified, sorted, transported, and stored in microbial organisms (Xu et al., 2015). This process is coordinated with microbial carbon metabolism and is a fundamental pathway for microbial life. Prior research has indicated that most oceans are currently experiencing nitrogen limitation (Buchanan et al., 2021). In recent years, with increasing marine biogeochemical research, the biochemical and molecular basis of nutrient metabolism in various microorganisms has become a key subject of interest (Kuypers et al., 2018). Studying nitrogen metabolism can, ultimately, improve our understanding regarding the utilization of nitrogen nutrients by marine microorganisms; in turn, this can contribute to the protection of the marine environment and promote the healthy development of the aquaculture industry. Studies on microbial-mediated marine nitrogen cycling have employed techniques such as isotope tracing (Glaze et al., 2022), quantitative PCR (Mosley et al., 2022), and metagenomics (Sun and Ward, 2021); these approaches have been instrumental in investigating how microorganisms absorb and utilize different nitrogen sources in different marine habitats at the physiological, genetic, and protein levels. In recent years, Archaea (Kirchman, 2012), γ-proteobacteria (Ravcheev et al., 2005) and Bacillus (Yang et al., 2021) have been isolated from seawater and have, ultimately, been determined to participate in various biological processes such as the metabolism of urea, reduction of nitrate and nitrite, and denitrification. Fungi, Sphingomonadales, and Pseudomonadales play important roles in ammonia oxidation, nitrate assimilation, and nitrification processes in the deep sea (Hawley et al., 2014; Li et al., 2018).
Overall, this study focused on Klebsiella aerogenes B23, which was newly isolated from a marine aquaculture area and has outstanding nitrogen-removing ability (Chen et al., 2023); ultimately, we aimed to determine the transcriptome of this marine microorganism in the presence or absence of nitrogen (potassium nitrate). Then a nitrogen metabolism pathway was constructed based on transcriptome data, and the differential expression of genes encoding nitrogen metabolism–related enzymes was analyzed. The primary objective of this study was to elucidate the nitrogen utilization mechanism of this K. aerogenes strain at the molecular level and lay the foundation for studying its adaptation mechanisms in nitrogen-rich seawater.
Klebsiella aerogenes B23 was isolated by Zhanjiang Yuehai Aquatic Seeding Company Limited on Donghai Island, Zhanjiang, China (Chen et al., 2023). The denitrification medium (DM1&2) used in this study was composed of 5.0 g of sucrose, 1.0 g of K2HPO4, 1.0 g of KH2PO4, 5.0 g of NaCl, and 2 mL of trace elements (containing MnSO4 1.1 g, MgSO4 1.0 g, CuSO4 1.6 g, FeSO4 1.8 g per liter of distilled water per liter of distilled water; these medium was produced either without KNO3 (DM1) or with 0.36 g of KNO3 (DM2) as the sole nitrogen source. The Luria–Bertani (LB) medium contained 10 g of peptone, 5 g of yeast extract, and 5 g of NaCl per 1 L of distilled water. Additionally, 1 × phosphate buffered saline (PBS) was purchased from BioSharp Biotech (Beijing, China). All culture media were autoclaved at 121°C for 20 min before use.
Strain B23 was activated, inoculated into LB broth, and cultured until it reached logarithmic growth phase (OD600 = 1.0). After washing with PBS three times, strain B23 samples were resuspended; this bacterial suspension was then inoculated into DM1 or DM2 with 5% (v/v) inoculum, before being sealed with breathable sealing films. The flasks were incubated in a shaking incubator at 30°C for 48 h. Finally, bacterial samples were collected by centrifugation at 24,200 × g for 10 min at 48 h before being stored at −80°C and sent to Majorbio Company (China) for transcriptome sequencing.
After the bacterial suspension was inoculated into DM1 and DM2, growth (OD600 value) was measured using an ultraviolet spectrophotometer (UV-3600PLUS, SHIMADZU, China); additionally, nitrogen utilization (total nitrogen, nitrate, ammonium, and nitrite) was tested at 0 and 48 h. The concentrations of total nitrogen, nitrate, ammonium, and nitrite were determined using alkaline potassium persulfate photometry, phenol disulfonic acid photometry, Nessler’s reagent spectrophotometry, and N-(1-naphthyl) ethylenediamine photometry (Gilcreas, 1966). All assays were performed in triplicate.
Total RNA was extracted from the bacterial culture using TRIzol reagent, following the manufacturer’s instructions (Invitrogen, Carlsbad, CA, USA); genomic DNA was extracted using bacterial DNA extraction kit (Takara Bio, Shiga, Japan). Then, RNA quality was assessed using an Agilent 2,100 Bioanalyzer (USA) and quantified using an ND-2000 spectrophotometer (NanoDrop Technologies, USA). High-quality RNA samples, with an OD260/280 ≥ 1.8, OD260/230 ≥ 1.0, RNA integrity number ≥ 6.5, 23S:16S ≥ 1.0, concentration ≥ 50 ng/μl, and total amount ≥ 1 μg, were selected for subsequent library construction.
RNA libraries for each sample (DM1_1, DM1_2, DM1_3, DM2_1, DM2_2, and DM2_3) were constructed using the TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA). The Ribo-Zero Magnetic Kit (Epicenter, USA) was used to remove rRNA; then, the mRNA was randomly fragmented into small pieces of approximately 200 bp. Finally, double-stranded cDNA was synthesized using mRNA as the template and random primers (Illumina); this was conducted using a SuperScript double-stranded cDNA synthesis kit (Invitrogen). During synthesis of the second strand of cDNA, dUTP was used instead of dTTP. The resulting double-stranded cDNA was then repaired to obtain blunt ends, phosphorylation at the 5′ end, and an A base addition to the 3′ end. Finally, the cDNA was ligated to a Y-shaped sequencing adapter. The second strand of the cDNA, containing dUTP, was removed using uracil-DNA glycosylase, thereby producing a library containing only the first strand of cDNA.
This cDNA library was enriched and amplified using the Phusion DNA polymerase (New England Biolabs) over 15 PCR cycles. After quantification using TBS380 (Turner Biosystems, USA), RNA-seq paired-end sequencing was performed using Illumina NovaSeq (2× 150 bp). The raw reads generated in this study have been deposited in the NCBI database (accession number: PRJNA984342).
Using the Illumina platform, sequencing image signals were converted into text signals via CASAVA base calling and stored in the FASTQ format as raw data. Adapter sequences were removed from reads. Additionally, 5′ ends that contained non-A, G, C, or T bases were trimmed. Reads of low sequencing quality (sequencing quality value < Q20) were also trimmed. Further, reads with an N content ratio of ≥10% were removed. Finally, fragments with lengths <25 bp were discarded after adapter removal and quality trimming, thereby resulting in clean data. Bowtie2 was then used to align the high-quality reads in each sample to the reference genome of K. aerogenes.
To obtain comprehensive annotation information for genes and transcripts, genomic sequences were compared using several databases, including Gene Ontology (GO), Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups of Proteins (COG), Non-Redundant Protein Database (NR), Swiss-Prot, and Pfam.
The transcripts Per Million reads (TPM) method was used to calculate the expression levels of transcripts in the DM1 and DM2 groups. After obtaining the read counts of these transcripts, differential gene expression analysis between the samples was performed using DEseq2 software (Love et al., 2014). Differentially expressed genes (DEGs) were identified between DM1 and DM2 samples with |log2FC| > 1 and q-value ≤0.05. Then the functions of these DEGs were investigated using cluster analysis, functional annotation (COG, GO, and KEGG), and functional enrichment analysis (GO and KEGG).
qRT-PCR analysis was performed according to the procedure described by Zhou et al. (2022). Primers were constructed using Primer Premier (version 5.0; Supplementary Table S1). The amplified PCR products ranged from 113 bp to 232 bp in length. To estimate the relative expression levels of the DEGs in DM1 and DM2 samples, we used the 2–ΔΔCt method (Livak and Schmittgen, 2001), with actin as a reference marker.
SPSS version 21.0 (SPSS Inc., Chicago, IL, USA) was used to perform one-way analysis of variance (ANOVA). Duncan’s new multiple-range test was used to analyze the means, with a significance level of p < 0.05. The data presented in the tables and figures represent the mean ± standard error (N = 3).
In this study, nitrogen balance was analyzed by testing and calculating the changes in different nitrogen forms (Table 1). It became clear that K. aerogenes B23 growth was significantly hindered when cultured in DM1, which was attributed to the absence of a nitrogen source. Trace ammonium and a slight decrease in intracellular N were observed after 48 h, which may be due to the presence of N in the cytoplasm of some cells. In DM2, the majority of nitrate supplied was utilized by this K. aerogenes strain. Specifically, 82.47% of the initial nitrate was transformed into intracellular nitrogen by assimilation, while 16.16% of the initial nitrate was lost through conversion into gas.
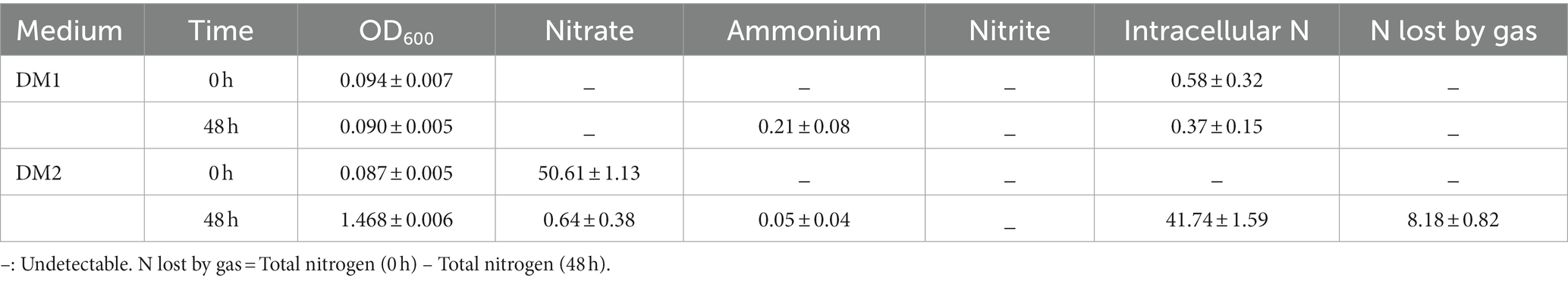
Table 1. The growth and nitrogen balance of strain B23 during denitrification process (unit:mg/L).
For the DM1_1, DM1_2, DM1_3, DM2_1, DM2_2, and DM2_3 samples, 24.77 Gb of data were analyzed. Overall, we obtained 161,397,874 clean reads with a Q30 score > 94.39% after filtering out low-quality reads and adapters (Supplementary Table S2). Parallelism among the groups was good and sequence alignment was high. All 5,028 genes were determined to match known genes in the listed databases at least once. These Genes were found to have the most hits in the NR (4,994 genes; 99.32%) and Pfam databases (4,477 genes; 89.04%); this was followed by 4,308 (85.68%) in the Swiss-Prot database, 4,258 (84.69%) in the COG database, 3,350 (66.63%) in the KEGG database, and 3,044 (60.54%) in the GO database (Table 2).
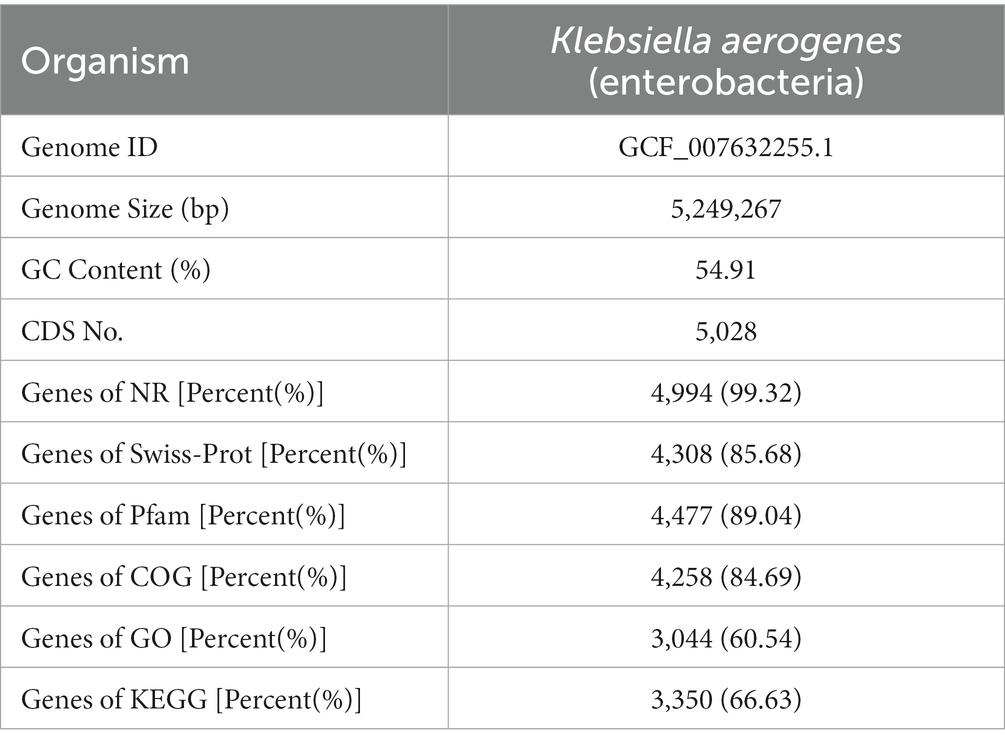
Table 2. Statistics of annotation results.
The 3,044 genes in the GO database were enriched in biological processes, cellular components, and molecular functions (Supplementary Figure S1). The top three terms in the biological process category were determined to be regulation of DNA-templated transcription (98), transmembrane transport (72), and translation (58). For cellular components, the top 3 terms were integral component of membranes (753), plasma membrane (465), and cytoplasm (417). Finally, for molecular functions, the top three terms were ATP binding (296), DNA binding (271), and metal ion binding (254).
Next, we used the COG database to annotate 4,258 genes, which were then classified into 24 distinct categories (Supplementary Figure S2). Three of these categories, each containing over 400 genes, were identified as follows: (G) carbohydrate transport and metabolism (505, 10.56%), (E) amino acid transport and metabolism (461, 9.64%), and (K) transcription (418, 8.74%). In total, 3,350 K. aerogenes genes were assigned to 40 KEGG categories (Supplementary Figure S3). Notably, the most frequently occurring pathways were carbohydrate metabolism (366), membrane transport (327), prokaryotic cellular community (152), translation (83), and antimicrobial drug resistance (70), which were categorized as metabolism, environmental information processing, cellular processes, genetic information processing, and human diseases, respectively.
Overall, RNA-seq reads from the DM1 and DM2 groups were aligned at an average mapping rate of 96.00% for K. aerogenes (Supplementary Table S2). The Pearson’s correlation coefficient between the three biological replicates of the DM1 and DM2 groups showed a strong correlation, ranging from approximately 0.982–1.0 (Supplementary Figure S5). Principal component analysis revealed distinct transcriptomic characteristics between the DM1 and DM2 groups (Supplementary Figure S6); these results suggested that DM2 KNO3 treatment significantly affected the transcriptome-wide gene expression of K. aerogenes. A total of 1,195 DEGs between the DM1 and DM2 groups were separated into 10 clusters of gene co-expression patterns (Supplementary Figure S4 and Supplementary Dataset S1, S2). Cluster 1 exhibited significantly higher gene expression in the DM2 group than in the DM1 group (control), with 616 upregulated genes in DM2 K. aerogenes (Figure 1 and Supplementary Dataset S1). GO analysis revealed that these upregulated genes were associated with the regulation of nitrogen compound metabolism, organonitrogen compound metabolism, cellular nitrogen compound metabolic process, nitrogen utilization, isoleucine metabolism, leucine metabolism, valine metabolism, glycine catabolism, and serine family amino acid catabolism (Figure 1A). Nonetheless, compared to the DM1 group, the 400 genes in cluster 2 showed significantly lower gene expression in the DM2 group (Supplementary Dataset S2). These downregulated genes were determined to be involved in nitrogen compound metabolism, organonitrogen compound metabolism, cellular nitrogen compound metabolism, cellular amino acid metabolism, glycine decarboxylation via glycine cleavage, and serine family amino acid catabolism (Figure 1B).
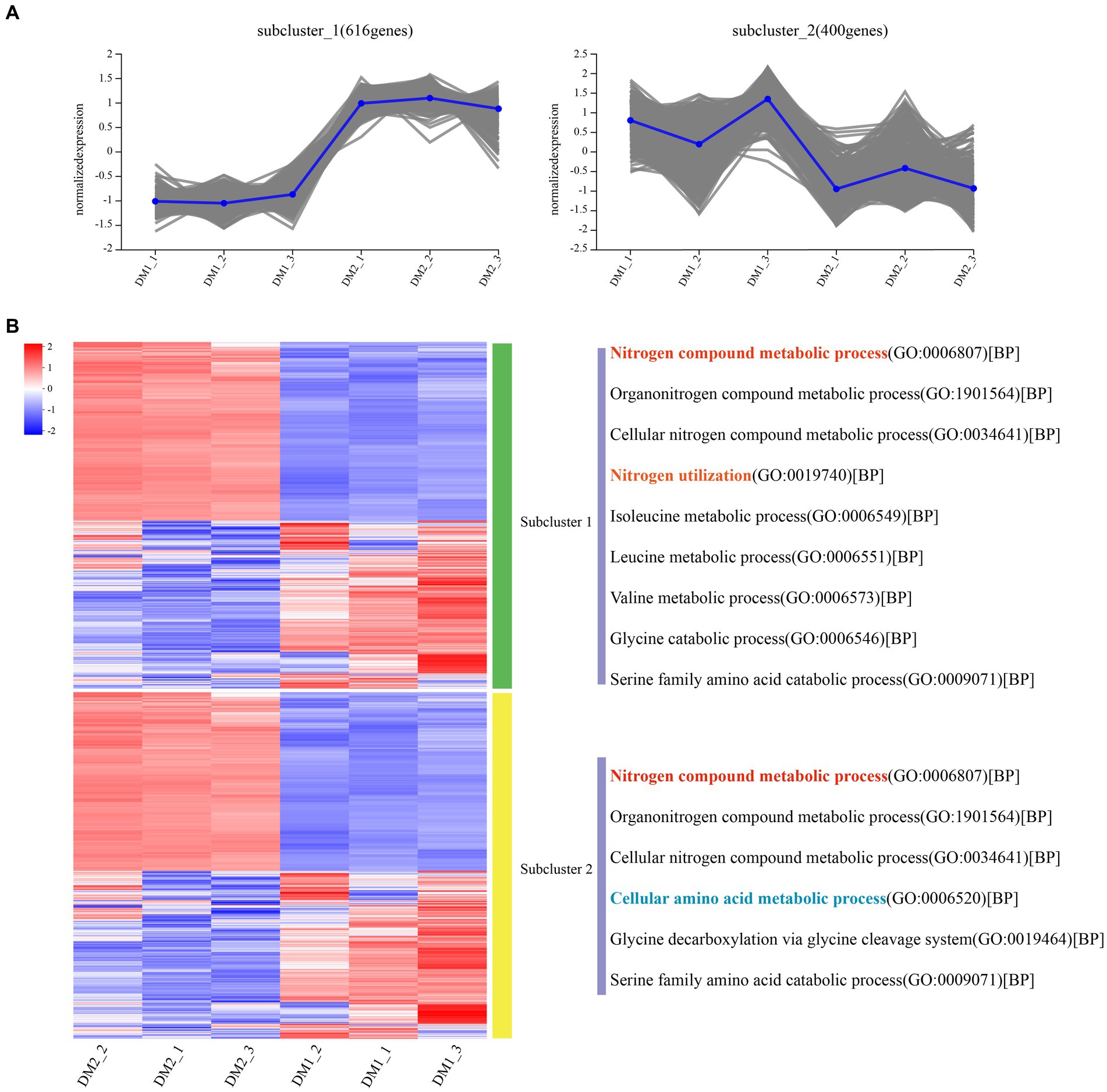
Figure 1. Gene co-expression clusters and heatmap analysis of DEGs in K. aerogenes. (A) Gene number of co-expression clusters and these gene expression patterns, (B) Heatmap and GO terms related to clusters of enriched gene co-expression. DM1_1, DM1_2, and DM1_3 indicate 3 replicates of the control group, while DM2_1, DM2_2, and DM2_3 indicate 3 replicates of the treated group.
Finally, GO enrichment analysis revealed that 4,587 genes were significantly upregulated in the DM2 group compared to the DM1 group. These genes were determined to be associated with ribosomal subunits (GO:0044391), the ribonucleoprotein complex (GO:1990904), rRNA binding (GO:0019843), structural constituent of ribosome (GO:0003735), and structural molecule activity (GO:0005198) (Figure 2A). Alternatively, 1857 genes were determined to be downregulated in the DM2 group compared to The DM1 group; these genes were associated with phenylacetate catabolism (GO:0010124), branched-chain amino acid metabolism (GO:0009081), branched-chain amino acid biosynthesis (GO:0009082), isoleucine biosynthesis (GO:0009097), and isoleucine metabolism (GO:0006549) (Figure 2B).
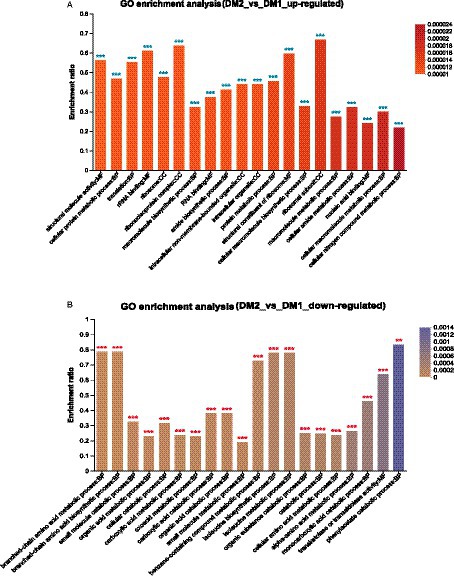
Figure 2. GO enrichment analysis of the DEGs in K. aerogenes between the DM1 and DM2 groups. (A) Upregulated genes, (B) downregulated genes. The label “**” indicates p < 0.01, and “***” indicates p < 0.001.
Nine candidate DEGs (eight upregulated and one downregulated) were randomly selected; the expression levels of these DEGs were measured using qRT-PCR with specific primers to validate the reliability of the RNA-seq data for K. aerogenes under nitrogen-rich conditions (Supplementary Table S1). The expression patterns of the nitrogen treatment–associated DEGs, as observed by qRT-PCR, closely aligned with those from previously established RNA-seq data of K. aerogenes (Figure 3); this highlighted the reliability of the RNA-seq data obtained in this study. Additionally, this indicated that the identified DEGs are suitable candidates for further investigation of K. aerogenes under nitrogen-rich conditions.
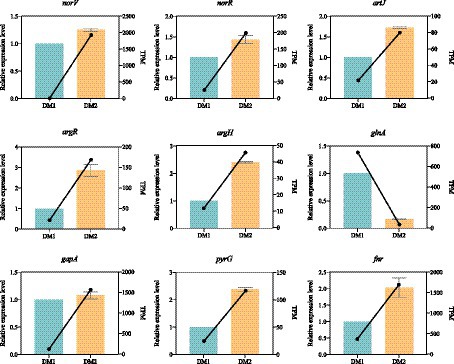
Figure 3. Expression pattern validation of 9 selected DEGs in K. aerogenes determined by RNA-seq and qRT-PCR.
Overall, eight genes were determined to be involved in nitrate assimilation under nitrogen-dependent conditions in K. aerogenes (Table 3). Specifically, five of these genes (norV/05380, norR/05390, narI/14925, FNR/RS13560 and NarK/14945) were upregulated, and three (narG/11865, NarK/11870 and ntrB/14980) were downregulated under nitrogen-rich conditions compared to the control. Additionally, in the nitrogen-treated group, five genes involved in arginine metabolism (artQ/16875, artJ/16870, artM/16880, argR/02445 and argH/23940) were upregulated, while two (tatE/18075 and argT/07020) were downregulated. Further, under nitrogen-rich conditions, five glutamate metabolism–associated genes (gshA/05525, proB/19730, proA/19735, gapA/09905 and fdx/06275) were upregulated, while seven (glnA/24240, putA/15685, gltB/02550, astD/10040, trpD/10200, gltS/00465 and ndoA/03960) were downregulated. Finally, in the nitrogen-treated group, five ammonia assimilation–associated genes (GAT1/06645, prmB/06915, pyrG/05015, glnS/17865 and Ca1/novel0138) were upregulated, while seven (glnH/17190, glnP/17195, glnQ/17200, GAT1/02860, FmdA_AmdA/21860, arcC/14025 and CA/22225) were downregulated.
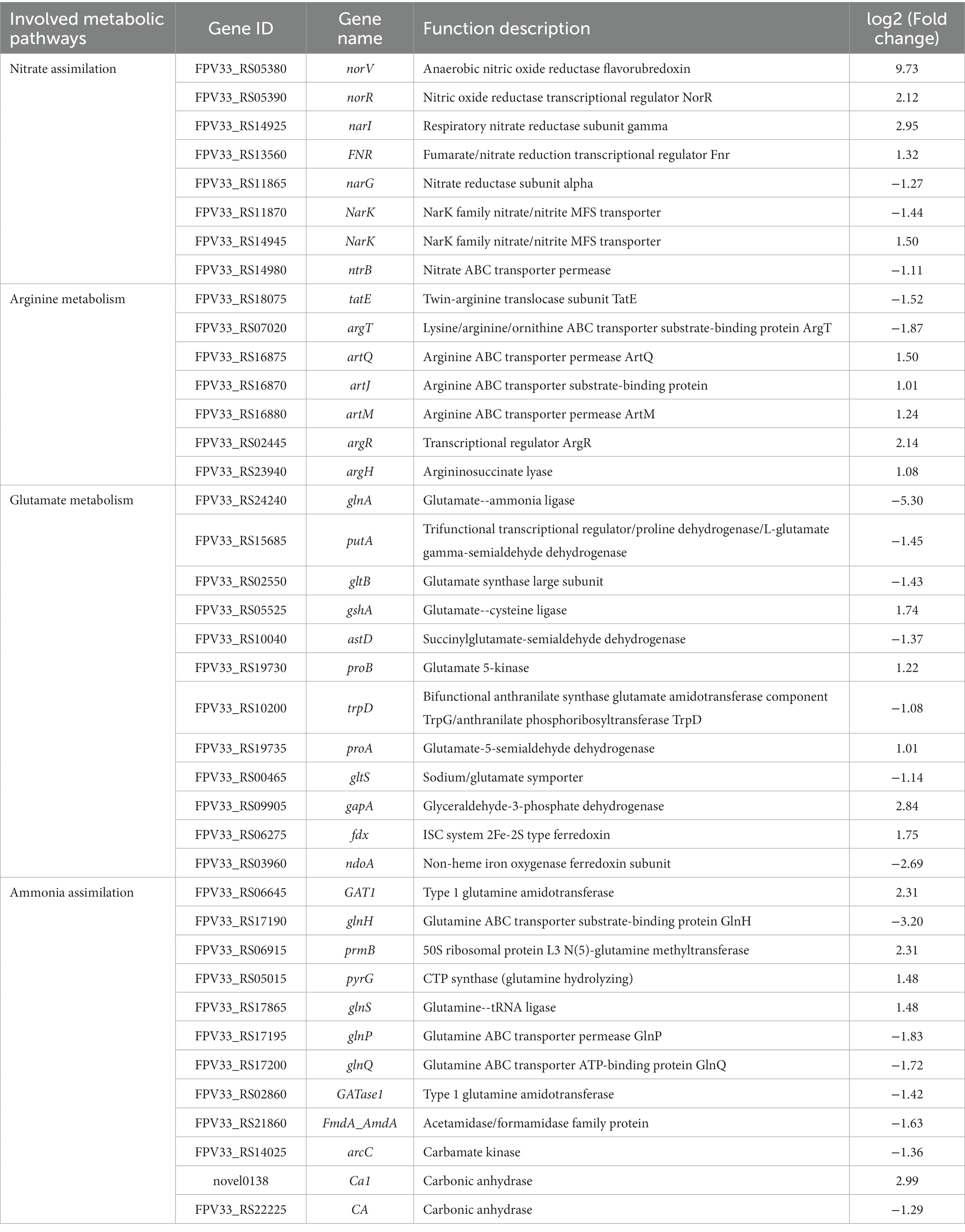
Table 3. Genes involved in nitrogen metabolism pathways in K. aerogenes between the DM1 and DM2 groups.
Microorganism play a vital role in driving the nitrogen cycle, a crucial component of the biogeochemical cycle. Nitrogen fixation is the process by which nitrogen-fixing microorganisms convert atmospheric N2 directly into biologically available ammonia; ultimately, this ammonia acts as the primary source of newly supplied nitrogen to the ocean (Fripiat et al., 2021). In the present study, K. aerogenes B23 exhibited effective growth in nitrogen-containing medium; additionally, this K. aerogenes strain utilized the nitrate provided, with 82.47% of this nitrate being assimilated into intracellular nitrogen. Previous studies have demonstrated that the formation of various enzymes in K. aerogenes is dependent on the quality and quantity of the nitrogen source provided in the growth substrate. It is understood that this nitrogen-dependent regulation of enzymes requires the action of the nitrogen regulatory system (Schwacha and Bender, 1993). The Klebsiella K312 has been established to convert nitrate to ammonia under nitrate-limited conditions; additionally, this process has been linked to the synthesis of nitrate reductase and nitrite reductase (Dunn et al., 1979). Based on the RNA-seq data of this study, we established how nitrate, from nitrate-containing medium, is utilized and assimilated by K. aerogenes B23. Specifically, this K. aerogenes strain grown in nitrate-containing medium exhibited unique and distinct expression patterns.
In the present study, 1,195 DEGs were found in K. aerogenes B23 grown in nitrate-containing medium. Cluster 1 contained 616 highly overexpressed genes in nitrate-treated K. aerogenes B23. Then, GO analysis indicated that these overexpressed genes were closely related to nitrogen compound metabolism, organonitrogen compound metabolism, cellular nitrogen compound metabolism, nitrogen utilization, isoleucine metabolism, leucine metabolism, valine metabolism, glycine catabolism, and serine family amino acid catabolism (Figure 1). To better explain the molecular mechanism behind the transport and assimilation of potassium nitrate in B23, we analyzed the associated KEGG metabolic pathways. We constructed the nitrogen metabolism pathway of K. aerogenes B23, which included nitrate assimilation, urea cycle (arginine and glutamate metabolism), and ammonia assimilation (Table 3 and Figure 4). Nitrogen metabolism is initiated by the absorption of inorganic nutrients from the environment. Nitrate/nitrite transporter proteins are the components responsible for this transport of nitrate/nitrite into cells for further assimilation (Senior, 1975). Nitrate assimilation is the process by which bacteria convert externally absorbed nitrate into ammonia, which is then used to form organic matter and store energy (Bothe et al., 2007). Nitrate reductase, the initiating enzyme for nitrogen utilization, is widely present in bacterial cells and is a key limiting and regulating enzyme in nitrate assimilation. Specifically, it converts the oxidized forms of nitrogen compounds absorbed by bacteria into their reduced forms, thereby completing the nitrogen metabolism cycle (Jiang and Jiao, 2016). In the K. aerogenes B23 transcriptome, we identified the transcriptional isoforms of genes that encode enzymes involved in the nitrate assimilation pathway (Table 3). Additionally, the expression levels of FNR, NarK/14945, and fdx were significantly upregulated under nitrogen-rich conditions. During the transition from aerobic to anaerobic growth in Escherichia coli, the transcription factor FNR plays a crucial role in regulating the reduction of succinate and nitrate (Constantinidou et al., 2006). In denitrifying bacteria, the transport of nitrate and nitrite is typically mediated by transmembrane transport proteins that are part of the major facilitator superfamily, which belongs to the NarK subfamily (Alvarez et al., 2019). In this study, NarK genes were found to be upregulated and involved in nitrate transport within K. aerogenes B23. The first step in the nitrate assimilation pathway is catalyzed by nitrate reductase, which converts nitrate to nitrite. However, owing to the toxicity of nitrite to cells, a second step involving nitrite reduction is necessary; this process utilizes ferredoxin and NADH as coenzymes to convert nitrite into ammonia (Ding et al., 2022). In this study, the gene encoding ferredoxin (fdx) of K. aerogenes B23 exhibited high expression in the presence of nitrate.
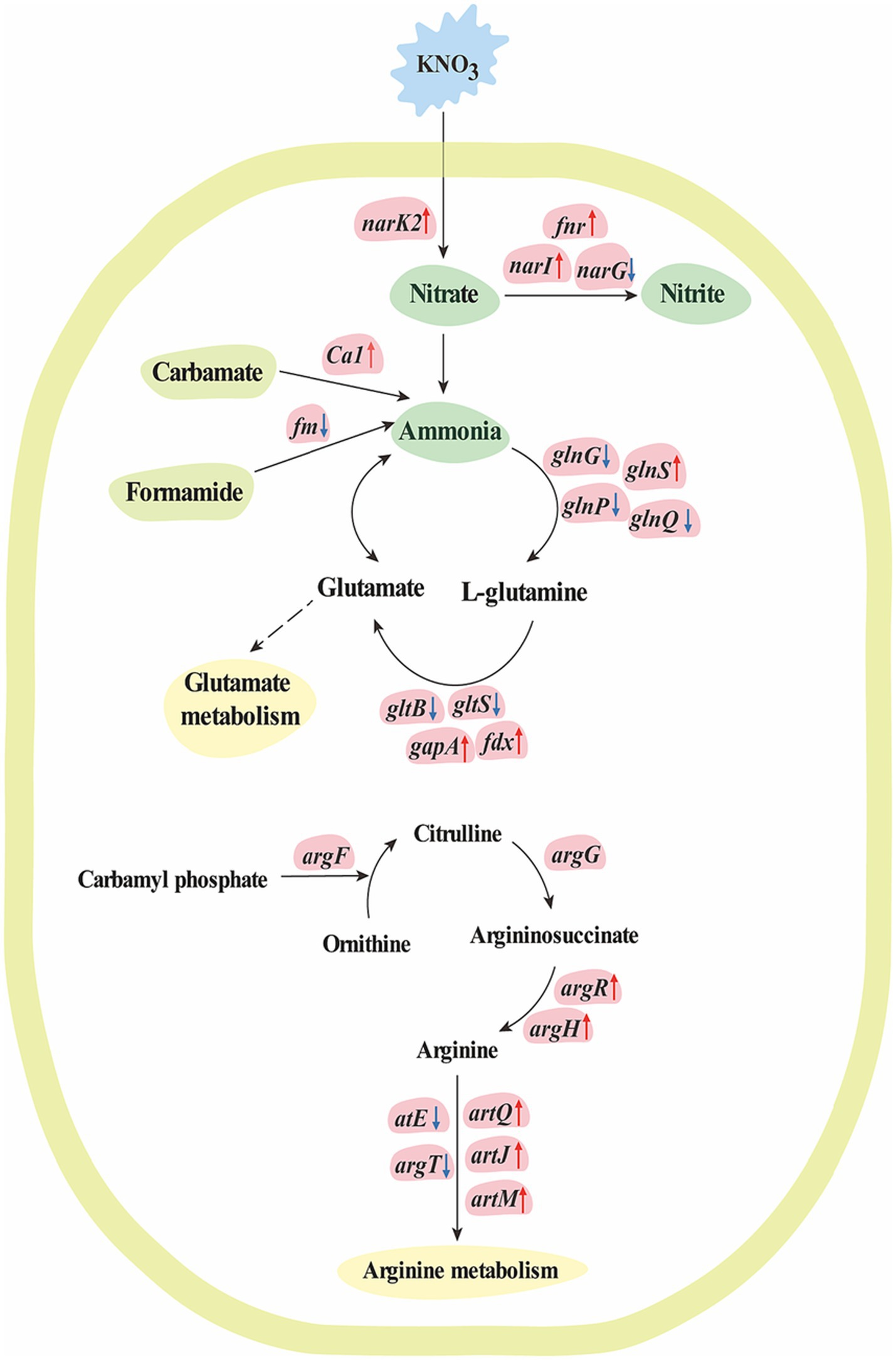
Figure 4. The molecular mechanism for nitrogen metabolism of K. aerogenes under nitrogen conditions. The text in blue block indicates K. aerogenes was cultured with KNO3, while the text in green block indicates nitrogen compound, the text in yellow-green block indicates organic compounds that involve in ammonia metabolism, and the text in yellow blocks indicates metabolism pathways. Up-regulated genes are indicated in pink blocks with up arrow, while down-regulated genes are indicated in pink blocks with down arrow, and non-significant regulated genes are indicated in pink blocks without arrow. Solid arrows indicate direct pathways for transcriptional regulation, while dashed arrows indicate indirect or unclear mechanisms.
Transcripts encoding enzymes involved in ammonia metabolism were found in the K. aerogenes B23 transcriptome. Among these, gshA, proB, proA, and gapA were significantly upregulated. Glutamine synthetase and glutamate synthase use ammonia to catalyze the production of L-glutamate and L-glutamine, respectively. These amino acids can then participate in glutamate metabolism (Zhou et al., 2020). In the present study, the large subunit of glutamate synthase, gltB, was significantly downregulated in nitrogen-treated K. aerogenes B23. These enzymes participate in the conversion of ammonia into organic peptides, contribute to the urea cycle, convert ammonia to ornithine to generate arginine, and subsequently catalyze the condensation of the guanidine group on ornithine with the amino group of aspartic acid, thereby forming argininosuccinate (Cunin et al., 1986; Charlier and Bervoets, 2019). Argininosuccinate synthase catalyzes the cleavage of argininosuccinate to produce arginine. Arginase then hydrolyzes arginine to produce urea and regenerate ornithine, thereby completing the urea cycle. Additionally, nitric oxide synthase catalyzes the interconversion of arginine and citrulline to supplement the needs of different amino acids during specific periods of the urea cycle (Mori, 2007; Lorin et al., 2014). In the present study, the gene encoding arginine succinyltransferase, argH, was significantly upregulated in nitrogen-treated K. aerogenes, resulting in the accelerated breakdown of arginine succinate. Additionally, genes encoding the arginine ABC transporters, artQ, artJ, and artM, were upregulated, which accelerated arginine transportation. The transcriptional regulator ArgR, was also upregulated in the nitrogen-treated group; specifically, this protein plays a key role in positive regulation of these arginine ABC transporters. The urea cycle is highly important for bacterial metabolism, with its intermediate products playing important roles in various biological processes. Ornithine and arginine are involved in biosynthesis and metabolism of arginine and citrulline, respectively. Urea in the internal and external environments of bacteria has important physiological significance, especially in facilitating the absorption of external nitrogen sources by plants for growth and development. Further, urea also helps maintain the balance of nitrogen metabolism and recycling within bacterial cells.
In addition to nitrate assimilation and urea decomposition, the glutamate–glutamine cycle can also supply the required ammonia for nitrogen metabolism (Esteves-Ferreira et al., 2018). Specifically, glutamine synthetase converts ammonia into glutamine. Additionally, glutamine synthetase receives the carbon skeleton from alpha-ketoglutarate via glutamate synthase (GLT1/GLTD), thereby allowing the production of glutamate. Finally, glutamate dehydrogenase (GDH1/GDH2) converts glutamate back to ammonia, thereby completing the glutamine synthetase–glutamate synthase cycle. Glutamate synthase is the rate-limiting enzyme in this cycle (Herrero et al., 2001). Nonetheless, GAT1_2.1 may function as a glutaminase, working in conjunction with glutamate dehydrogenase 2 to break down glutamine and direct 2-oxoglutarate towards the TCA cycle in the presence of excess nitrogen (Kambhampati et al., 2021). In the present study, GAT1 in K. aerogenes B23 was determined to be upregulated under nitrogen-rich conditions and was found to participate in glutamine hydrolysis. Similarly, the upregulation of prmB and pyrG in nitrate-treated K. aerogenes B23 was found to be involved in the hydrolysis of glutamine, whereas glnS upregulation was necessary for maintaining glutamine synthesis. Carbonic anhydrase (CA) can also catalyze the generation of ammonia from carbamoyl phosphate, thereby supplementing the consumption of ammonia and generating ATP. In this study, Ca1 was significantly upregulated in K. aerogenes B23 grown in nitrate-supplemented medium. CA catalyzes the decomposition of cyanates into ammonia; moreover, this enzyme can catalyze the reversible hydration reaction of carbon dioxide, ultimately providing CO2/HCO3− for further enzymatic reactions. Additionally, CA can facilitate energy production by removing CO2/HCO3−.
Denitrification is the process in which microorganisms decompose nitrate or nitrite into N2, N2O, or NO under anaerobic conditions. It is the main biological process by which reactive nitrogen can return to the atmosphere in gaseous form. NO is a toxic metabolite in bacteria that readily reacts with [Fe-S], ferroheme cofactors, and other transition metal centers (Galloway et al., 2008; Canfield et al., 2010). Bacteria that produce NO typically possess nitric oxide reductase (NOR), which converts NO to N2O (Cole, 2018). NOR consists of a small subunit (NorC) and large subunit (NorB) (Harland et al., 2023). Additionally, NorV acts as the primary defense mechanism that aids bacteria in resisting oxidation and nitrite-based sterilization. Further, the norV gene has been determined to provide a protective advantage to Aeromonas hydrophila against Tetrahymena predation, improve bacterial survival within macrophages, and contribute significantly to bacterial virulence in zebrafish (Liu et al., 2019). The NorR regulatory protein detects the presence of NO in E. coli and triggers the activation of the genes necessary for NO detoxification under anaerobic and microaerobic conditions (Tucker et al., 2008). In the present study, norV, norR, and narI were determined to be significantly upregulated in nitrate-treated K. aerogenes B23; these genes were specifically associated with NO metabolism and, thereby, preventing the toxic effects of NO in this K. aerogenes strain.
Overall, K. aerogenes B23 can effectively grow in nitrate-rich culture media and can perform nitrogen fixation, ammonification, nitrification, denitrification, and deamination by upregulating the expression of FNR, NarK/14945, fdx, gshA, proB, proA, gapA, argH, artQ, artJ, artM, ArgR, GAT1, prmB, pyrG, glnS, and Ca1. Additionally, it can mitigate the toxic effects of NO by upregulating norV, norR, and narI. Ultimately, this study reveals a new microorganism that is involved in the marine nitrogen cycle and provides a scientific basis for studying its corresponding nitrogen metabolism mechanisms.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI Database (https://www.ncbi.nlm.nih.gov/), PRJNA984342.
YC: Formal analysis, Writing – original draft, Methodology. YL: Formal analysis, Writing – original draft. JZhu: Investigation, Writing – review & editing. JZho: Investigation, Writing – review & editing. HL: Conceptualization, Writing – review & editing. YF: Conceptualization, Writing – review & editing. YZ: Writing – review & editing, Supervision.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research was financially supported by School-level Talents Project of Lingnan Normal University (ZL2033).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2024.1323160/full#supplementary-material
Alvarez, L., Sanchez-Hevia, D., Sánchez, M., and Berenguer, J. (2019). A new family of nitrate/nitrite transporters involved in denitrification. Int. Microbiol. 22, 19–28. doi: 10.1007/s10123-018-0023-0
Bothe, H., Ferguson, S., and Newton, W. E. (2007). Biology of the nitrogen cycle. Amsterdam: Elsevier Science
Buchanan, P. J., Aumont, O., Bopp, L., Mahaffey, C., and Tagliabue, A. (2021). Impact of intensifying nitrogen limitation on ocean net primary production is fingerprinted by nitrogen isotopes. Nat. Commun. 12:6214. doi: 10.1038/s41467-021-26552-w
Canfield, D. E., Glazer, A. N., and Falkowski, P. G. (2010). The evolution and future of Earth’s nitrogen cycle. Science 330, 192–196. doi: 10.1126/science.1186120
Charlier, D., and Bervoets, I. (2019). Regulation of arginine biosynthesis, catabolism and transport in Escherichia coli. Amino Acids 51, 1103–1127. doi: 10.1007/s00726-019-02757-8
Chen, Y., Zhong, J., Li, B., Dai, W., Yang, Z., Huang, C., et al. (2023). Exploring the nitrogen removal capacity of Klebsiella aerogenes B23 isolated from shrimp farm wastewater: heterotrophic nitrification and aerobic denitrification. Aquac. Int. doi: 10.1007/s10499-023-01224-2
Cheung, W. W. L., Frölicher, T. L., Lam, V. W. Y., Oyinlola, M. A., Reygondeau, G., Sumaila, U. R., et al. (2021). Marine high temperature extremes amplify the impacts of climate change on fish and fisheries. Sci. Adv. 7:eabh0895. doi: 10.1126/sciadv.abh0895
Cole, J. A. (2018). Anaerobic bacterial response to nitrosative stress. Adv. Microb. Physiol. 72, 193–237. doi: 10.1016/bs.ampbs.2018.01.001
Constantinidou, C., Hobman, J. L., Griffiths, L., Patel, M. D., Penn, C. W., Cole, J. A., et al. (2006). A reassessment of the fnr regulon and transcriptomic analysis of the effects of nitrate, nitrite, NarXL, and NarQP as Escherichia coli K12 adapts from aerobic to anaerobic growth. J. Biol. Chem. 281, 4802–4815. doi: 10.1074/jbc.M512312200
Cunin, R., Glansdorff, N., Piérard, A., and Stalon, V. (1986). Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev. 50, 314–352. doi: 10.1128/mr.50.3.314-352.1986
Ding, L., Han, B., and Zhou, J. (2022). Characterization of the facultative anaerobic Pseudomonas stutzeri strain HK13 to achieve efficient nitrate and nitrite removal. Process Biochem. 118, 236–242. doi: 10.1016/j.procbio.2022.04.021
Duarte, C. M., Regaudie-de-Gioux, A., Arrieta, J. M., Delgado-Huertas, A., and Agustí, S. (2013). The oligotrophic ocean is heterotrophic. Annu. Rev. Mar. Sci. 5, 551–569. doi: 10.1146/annurev-marine-121211-172337
Dunn, G. M., Herbert, R. A., and Brown, C. M. (1979). Influence of oxygen tension on nitrate reduction by a Klebsiella sp. growing in chemostat culture. J. Gen. Microbiol. 112, 379–383. doi: 10.1099/00221287-112-2-379
Esteves-Ferreira, A. A., Inaba, M., Fort, A., Araújo, W. L., and Sulpice, R. (2018). Nitrogen metabolism in cyanobacteria: metabolic and molecular control, growth consequences and biotechnological applications. Crit. Rev. Microbiol. 44, 541–560. doi: 10.1080/1040841X.2018.1446902
Fripiat, F., Martínez-García, A., Marconi, D., Fawcett, S. E., Kopf, S. H., Luu, V. H., et al. (2021). Nitrogen isotopic constraints on nutrient transport to the upper ocean. Nat. Geosci. 14, 855–861. doi: 10.1038/s41561-021-00836-8
Fuhrman, J. A., and Hagström, Å. (2008). Microbial ecology of the oceans. New York: John Wiley & Sons, Inc.
Galloway, J. N., Townsend, A. R., Erisman, J. W., Bekunda, M., Cai, Z., Freney, J. R., et al. (2008). Transformation of the nitrogen cycle: recent trends, questions, and potential solutions. Science 320, 889–892. doi: 10.1126/science.1136674
Gilcreas, F. W. (1966). Standard methods for the examination of water and waste water. Am. J. Public Health Nations Health 56, 387–388. doi: 10.2105/AJPH.56.3.387
Glaze, T. D., Erler, D. V., and Siljanen, H. (2022). Microbially facilitated nitrogen cycling in tropical corals. ISME J. 16, 68–77. doi: 10.1038/s41396-021-01038-1
Harland, J. B., Samanta, S., and Lehnert, N. (2023). Bacterial nitric oxide reductase (NorBC) models employing click chemistry. J. Inorg. Biochem. 246:112280. doi: 10.1016/j.jinorgbio.2023.112280
Hawley, A. K., Brewer, H. M., Norbeck, A. D., Paša-Tolić, L., and Hallam, S. J. (2014). Metaproteomics reveals differential modes of metabolic coupling among ubiquitous oxygen minimum zone microbes. Proc. Natl. Acad. Sci. USA 111, 11395–11400. doi: 10.1073/pnas.1322132111
Herrero, A., Muro-Pastor, A. M., and Flores, E. (2001). Nitrogen control in cyanobacteria. J. Bacteriol. 183, 411–425. doi: 10.1128/JB.183.2.411-425.2001
Jiang, X., and Jiao, N. (2016). Nitrate assimilation by marine heterotrophic bacteria. Sci. China Earth Sci. 59, 477–483. doi: 10.1007/s11430-015-5212-5
Kambhampati, S., Pajak, A., and Marsolais, F. (2021). Evidence that class I glutamine amidotransferase, GAT1_2.1, acts as a glutaminase in roots of Arabidopsis thaliana. Plant Sci. 312:111033. doi: 10.1016/j.plantsci.2021.111033
Kang, B., Pecl, G. T., Lin, L., Sun, P., Zhang, P., Li, Y., et al. (2021). Climate change impacts on China’s marine ecosystems. Rev. Fish. Biol. Fisheries. 31, 599–629. doi: 10.1007/s11160-021-09668-6
Kirchman, D. L. (2012). Marine archaea take a short cut in the nitrogen cycle. Proc. Natl. Acad. Sci. U. S. A. 109, 17732–17733. doi: 10.1073/pnas.1215654109
Kuypers, M. M. M., Marchant, H. K., and Kartal, B. (2018). The microbial nitrogen-cycling network. Nat. Rev. Microbiol. 16, 263–276. doi: 10.1038/nrmicro.2018.9
Li, Y., Jing, H., Xia, X., Cheung, S., Suzuki, K., and Liu, H. (2018). Metagenomic insights into the microbial community and nutrient cycling in the western subarctic Pacific Ocean. Front. Microbiol. 9:623. doi: 10.3389/fmicb.2018.00623
Liu, J., Dong, Y., Wang, N., Ma, S., Lu, C., and Liu, Y. (2019). Diverse effects of nitric oxide reductase NorV on Aeromonas hydrophila virulence-associated traits under aerobic and anaerobic conditions. Vet. Res. 50:67. doi: 10.1186/s13567-019-0683-6
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lorin, J., Zeller, M., Guilland, J.-C., Cottin, Y., Vergely, C., and Rochette, L. (2014). Arginine and nitric oxide synthase: regulatory mechanisms and cardiovascular aspects. Mol. Nutr. Food Res. 58, 101–116. doi: 10.1002/mnfr.201300033
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Bioinformatics. 15:550. doi: 10.1101/002832
Matassa, S., Verstraete, W., Pikaar, I., and Boon, N. (2016). Autotrophic nitrogen assimilation and carbon capture for microbial protein production by a novel enrichment of hydrogen-oxidizing bacteria. Water Res. 101, 137–146. doi: 10.1016/j.watres.2016.05.077
Mena-Rivera, L., Lloyd, C. E. M., Reay, M. K., Goodall, T., Read, D. S., Johnes, P. J., et al. (2023). Tracing carbon and nitrogen microbial assimilation in suspended particles in freshwaters. Biogeochemistry 164, 277–293. doi: 10.1007/s10533-022-00915-x
Mori, M. (2007). Regulation of nitric oxide synthesis and apoptosis by arginase and arginine recycling. J. Nutr. 137, 1616S–1620S. doi: 10.1093/jn/137.6.1616S
Mosley, O. E., Gios, E., Close, M., Weaver, L., Daughney, C., and Handley, K. M. (2022). Nitrogen cycling and microbial cooperation in the terrestrial subsurface. ISME J. 16, 2561–2573. doi: 10.1038/s41396-022-01300-0
Ravcheev, D. A., Rakhmaninova, A. B., Mironov, A. A., and Gelfand, M. S. (2005). Regulation of nitrate and nitrite respiration in γ-proteobacteria: a comparative genomics study. Mol. Biol. 39, 727–740. doi: 10.1007/s11008-005-0088-7
Schwacha, A., and Bender, R. A. (1993). The product of the Klebsiella aerogenes nac (nitrogen assimilation control) gene is sufficient for activation of the hut operons and repression of the gdh operon. J. Bacteriol. 175, 2116–2124. doi: 10.1128/jb.175.7.2116-2124.1993
Senior, P. J. (1975). Regulation of nitrogen metabolism in Escherichia coli and Klebsiella aerogenes: studies with the continuous-culture technique. J. Bacteriol. 123, 407–418. doi: 10.1128/jb.123.2.407-418.1975
Stüeken, E. E., Kipp, M. A., Koehler, M. C., and Buick, R. (2016). The evolution of Earth’s biogeochemical nitrogen cycle. Earth Sci. Rev. 160, 220–239. doi: 10.1016/j.earscirev.2016.07.007
Sun, X., and Ward, B. B. (2021). Novel metagenome-assembled genomes involved in the nitrogen cycle from a Pacific oxygen minimum zone. ISME. Commun. 1:26. doi: 10.1038/s43705-021-00030-2
Tucker, N. P., D’Autréaux, B., Yousafzai, F. K., Fairhurst, S. A., Spiro, S., and Dixon, R. (2008). Analysis of the nitric oxide-sensing non-heme iron center in the NorR regulatory protein. J. Biol. Chem. 283, 908–918. doi: 10.1074/jbc.M705850200
Xu, X., Hui, D., King, A. W., Song, X., Thornton, P. E., and Zhang, L. (2015). Convergence of microbial assimilations of soil carbon, nitrogen, phosphorus and sulfur in terrestrial ecosystems. Sci. Rep. 5:17445. doi: 10.1038/srep17445
Xu, G., Zhao, X., Zhao, S., Rogers, M. J., and He, J. (2023). Salinity determines performance, functional populations, and microbial ecology in consortia attenuating organohalide pollutants. ISME J. 17, 660–670. doi: 10.1038/s41396-023-01377-1
Yang, T., Yang, Q., Shi, Y., Xin, Y., Zhang, L., Gu, Z., et al. (2021). Insight into the denitrification mechanism of Bacillus subtilis JD-014 and its application potential in bioremediation of nitrogen wastewater. Process Biochem. 103, 78–86. doi: 10.1016/j.procbio.2021.02.007
Zhou, Y., Eid, T., Hassel, B., and Danbolt, N. C. (2020). Novel aspects of glutamine synthetase in ammonia homeostasis. Neurochem. Int. 140:104809. doi: 10.1016/j.neuint.2020.104809
Keywords: Klebsiella aerogenes, nitrogen, nitrate assimilation, metabolism, differentially expressed genes
Citation: Chen Y, Lin Y, Zhu J, Zhou J, Lin H, Fu Y and Zhou Y (2024) Transcriptomic analysis of nitrogen metabolism pathways in Klebsiella aerogenes under nitrogen-rich conditions. Front. Microbiol. 15:1323160. doi: 10.3389/fmicb.2024.1323160
Edited by:
Danny Ionescu, Leibniz-Institute of Freshwater Ecology and Inland Fisheries (IGB), GermanyReviewed by:
Zhanfei He, Zhejiang University of Technology, ChinaCopyright © 2024 Chen, Lin, Zhu, Zhou, Lin, Fu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanyan Chen, MTI1NTQ5NDg4N0BhbGl5dW4uY29t; Yan Zhou, Mjg2MTM4ODI2QHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.