 Xiaoliang Hu
Xiaoliang Hu Lu Yang
Lu Yang Zhige Tian
Zhige Tian
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 11 January 2024
Sec. Microbial Symbioses
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1339188
This article is part of the Research Topic Microbial Symbiosis and Infectious Disease Dynamics in Reptiles and Wildfowl View all 5 articles
Introduction: The gastrointestinal tract and oral cavity of animal species harbor complex microbial communities, the composition of which is indicative of the behavior, co-evolution, diet, and immune system of the host.
Methods: This study investigated the microbial composition in snakes from varying altitudinal ranges by assessing the fecal and oral bacterial communities in Protobothrops mucrosquamatus, Elaphe dione, and Gloydius angusticeps from Sichuan Province, China, using metagenomic sequencing.
Results and discussion: It was revealed that Bacteroidetes, Proteobacteria, Firmicutes, and Fusobacteria were the core microbial phyla in fecal samples across all three species, while Proteobacteria, Bacteroidetes, Actinobacteria, and Firmicutes were the core microbial phyla in oral samples across all three species. Notably, the dominance of Armatimonadetes was documented for the first time in the feces of all three species. Comparative analysis of the microbiomes of the three species indicated distinct microbiological profiles between snakes living at low- and high-altitude regions. Furthermore, 12 to 17 and 22 to 31 bacterial pathogens were detected in the oral and fecal samples, respectively, suggesting that snakes may serve as a novel reservoir for emerging diseases. Overall, this study provides a comparative analysis of the fecal and oral microbiomes in three snake species. Future investigations are anticipated to further elucidate the influence of age, genetics, behavior, diet, environment, ecology, and evolution on the gut and oral microbial communities of snakes.
Snakes exhibit diverse diets and live in a variety of habitats, including grasslands, wetlands, forests, agricultural fields, deserts, plateaus, and marine areas (Uetz, 2000). The distinct ecological, physiological, and behavioral characteristics of snakes markedly influence the ecology of their gastrointestinal and oral microbial communities (Costello et al., 2010; Tang et al., 2019). More than 205 snake species are distributed across mainland China (Zhou and Jiang, 2004). However, limited research has been conducted on the microbial communities within these species. Protobothrops mucrosquamatus, a medium-sized pitviper characterized by a long triangular head, slender body, and proficiency in tree climbing, is primarily found at lower altitudes in southwest and southeast China, Laos, northern Bangladesh, Vietnam, northern Myanmar, and northeastern India (Zhao, 2006). Elaphe dione lives in various habitats, including plains, hills, mountains, grasslands, fields, grass slopes, forests, and riversides, as well as around vegetable gardens, farmhouses, chicken coops, and livestock pens, and mainly feeds on lizards, rats, birds and their eggs, frogs, and insects (Schulz and Entzeroth, 1996). Gloydius angusticeps, characterized by its mostly grayish-brown body and short length, rarely exceeding 60 cm, inhabits high-altitude zones in rocky or wetland environments, with a diet primarily consisting of lizards and rats.
Microbial populations within the gastrointestinal tract and oral cavities of both vertebrates and invertebrates play an essential role in nutrient absorption (Mackie, 2002) and can influence host behavior (Archie and Theis, 2011; Ezenwa et al., 2012), immunity (Hooper et al., 2001), reproduction (Shropshire and Bordenstein, 2016), ecology, and evolution. To date, studies on oral microflora have predominantly focused on snakebites (Arroyo et al., 1980; Blaylock, 2001; Campagner et al., 2012; Barbosa et al., 2018; Panda et al., 2019). Furthermore, research on gut microbial communities has been limited to a few species, including Naja naja (Krishnankutty et al., 2018), Ophiophagus hannah (Krishnankutty et al., 2018), Python molurus (Krishnankutty et al., 2018), Rhabdophis subminiatus (Tang et al., 2019), Python bivittatus (Costello et al., 2010), Naja atra, Ptyas mucosa, Elaphe carinata, Deinagkistrodon acutus (Zhang et al., 2019), and Ptyas mucosa (Qin et al., 2019). As such, two critical areas remain to be elucidated, including the ecology of gut and oral bacterial diversity and the interactions between bacterial diversity and factors such as diet, altitude, physiology, and genetics (David et al., 2014; Moeller et al., 2014; McKenney et al., 2015).
Wildlife, noted for their vast diversity and ecological importance, are also reservoirs for a variety of bacterial, viral, and fungal organisms. These microbial communities can include zoonotic pathogens that pose threats to other animals, including rare species. For example, bats and rodents are host to more than 60 zoonotic species (Luis et al., 2013), while wild boars harbor multiple diseases that can threaten rare feline species. However, there is limited information regarding the extent of microbes and the range and nature of diseases potentially harbored by snakes.
To expand our understanding of the gut and oral microbial diversity in P. mucrosquamatus, E. dione, and G. angusticeps, we conducted metagenomic profiling to explore the diversity of bacterial species in fecal and oral samples and to examine the possibility of zoonosis in these snakes.
The Second Tibetan Plateau Scientific Expedition and Research program included a focus on gut and oral cavity bacterial diversity in reptiles. During July to September 2020, we selected several sampling sites in gullies and forests, capturing four brown-spotted pitvipers (P. mucrosquamatus) and three Dione’s rat snakes (E. dione) in the Laojun Mountains (28°43′N, 104°04′E) which was Situated west of Yibin city in Sichuan, China (Figure 1). Additionally, three G. angusticeps snakes were captured in the grasslands and along the lakeside of Ruoergai Prairie in Sichuan (average altitude of 3 300–3 600 m) (Figure 1). The snakes were captured using specialized tools and the samples were collected and processed as described previously (Tian et al., 2022). Briefly, the three kinds of snakes were individually placed in sterilized tubs and skins of which were cleaned with 75% alcohol to prevent sample contamination. All pharyngeal and anal swab samples were collected opportunistically in areas where snakes were captured. The swabs were placed in RNase-free tubes and immediately transported on dry ice to Shanghai Biozeron Biotechnology Co., Ltd. (China) the same day. The snakes were then released back into the wild. overnight to collect.
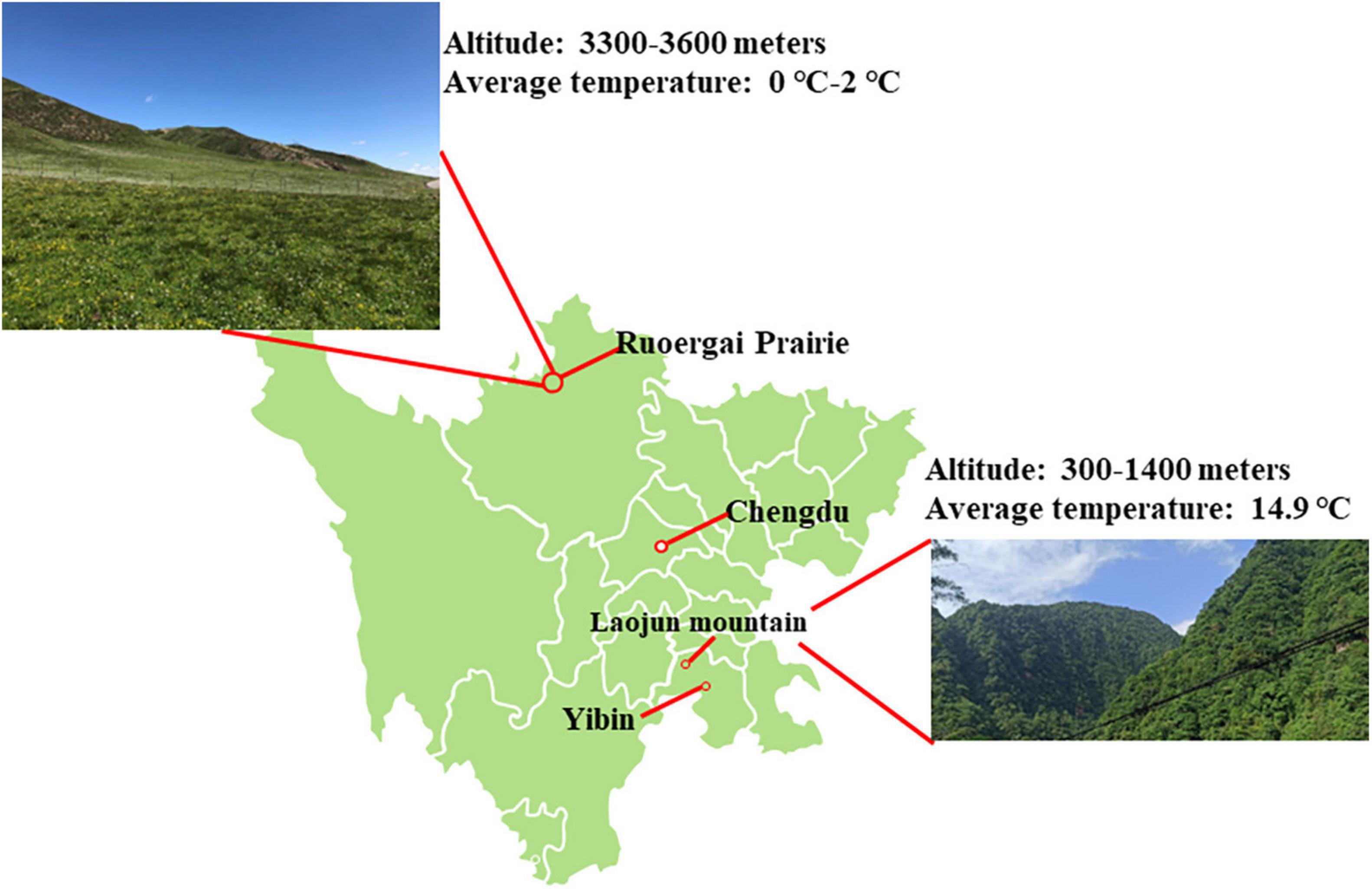
Figure 1. Map of Laojun Mountain nature reserve and Ruoergai Prairie.
Microbial DNA was extracted from 10 fecal and 10 oral samples in total, respectively, using an EZNA® stool DNA Kit (Omega Bio-tek, Norcross, GA, USA) in accordance with the manufacturer’s protocols. Metagenomic shotgun sequencing libraries were constructed and sequenced at Shanghai Biozeron Biological Technology Co. Ltd. (China). The sequencing reads, after undergoing quality control, were then mapped against the human genome (version: hg19) using the BWA mem algorithm (parameters: -M -k 32 -t 16).1 Reads that were filtered to remove host-genome contamination and low-quality data were identified as clean reads and utilized for subsequent analysis.
Taxonomic classification of clean reads from each sample was conducted using Kraken2 (Wood and Salzberg, 2014) with a customized Kraken database, which included all bacterial, archaeal, fungal, viral, protozoan, and algal genome sequences in the NCBI RefSeq database (release number: 90). The classification was done at seven phylogenetic levels which are domain, phylum, class, order, family, genus, species, or unclassified. Bracken2 was used to calculate the abundance of each taxonomy. The relative abundance at a specific taxonomic level represented the cumulative abundance of all species classified within that level.
The clean sequence reads were used to generate a set of contigs for each sample using MegaHit, with the parameter “–min-contig-len 500” (Li et al., 2015). Open reading frames (ORFs) of the assembled contigs were predicted using Prodigal v2.6.3 (Hyatt et al., 2010). Subsequently, the ORFs were clustered with CD-HIT (Fu et al., 2012) to create a unique gene set, with the longest sequence in each cluster serving as the representative sequence for each gene. To calculate gene abundance across all samples, Salmon (Patro et al., 2017) was used to quantify the read count for each gene. Gene abundance was then calculated using the following formulas:
Where Ab(S) represents gene abundance; Ab(U) represents single-mapping read abundance; Ab(M) represents multi-mapping read abundance; and l represents length of the gene sequence (Li et al., 2014).
Metagenomic binning was applied to the contigs of each sample. The binning process was carried out separately using metaBAT2 (Kang et al., 2015). The completeness and contamination levels of all bins were obtained using CheckM v.1.0.3 (Parks et al., 2015). Bins exhibiting a completeness greater than 50% and contamination less than 10% were classified as “filtered bins.” Assembly quality of the MAGs was enhanced using metaSPAdes (Nurk et al., 2017). All genes within the bins were transformed to protein sequences to generate the proteomes for every bin. These proteomes were used for phylogenetic tree reconstruction using Phylophlan (Segata et al., 2013).
Beta diversity was assessed using the Bray-Curtis distance metric to compare the outcomes of principal coordinates analysis (PCoA) using the community ecology package R-forge (Fouts et al., 2012). PCoA was used to explore and visualize data similarities and dissimilarities, starting with a similarity or dissimilarity matrix (= distance matrix) and mapping each item to a location in low-dimensional space to identify the main axes within the matrix. Hierarchical cluster analysis was applied to identify discrete groups with varying degrees of (dis)similarity in a dataset, as represented by a (dis)similarity matrix (like Bray-Curtis distance matrix). These groups were clustered using hierarchical clustering algorithms based on the distance matrix and presented as dendrograms using the unweighted pair-group method with arithmetic averages (UPGMA) (Hou et al., 2019). Heatmaps were generated based on the percentages of microorganisms contained in the matrix, represented as colors using the vegan package in R. Cluster trees of the microorganisms or samples were added to the heatmap analysis to show low- or high-abundant microorganisms in different modules (Fouts et al., 2012). A Venn diagram was drawn using the vegan package in R to analyze overlapping and unique gene sets affecting the bacterial communities during treatment processes (Fouts et al., 2012).
In this study, we sequenced the fecal and oral metagenomes of 10 captive snakes using the Illumina NovaSeq 6000 platform and obtained more than 200 Gb of high-quality bases, including 290 468 168 raw reads, ranging from 17 737 928 to 39 507 966 reads per sample. The assembled sequences that passed quality control filtering included 2 214 074 contigs, ranging from 50 633 to 761 127 per sample, with an average length of 464–643 bp.
Based on Venn diagram analysis, 94 263 and 20 249 contigs were common to all fecal and oral samples, respectively. In addition, 141 767, 119 903, and 139 348 contigs were detected in groups ED, GA, and PM, respectively (Figure 2A), among which 25 915 were unique to ED, 42 881 were unique to GA, and 25 535 were unique to PM (Figure 2B).
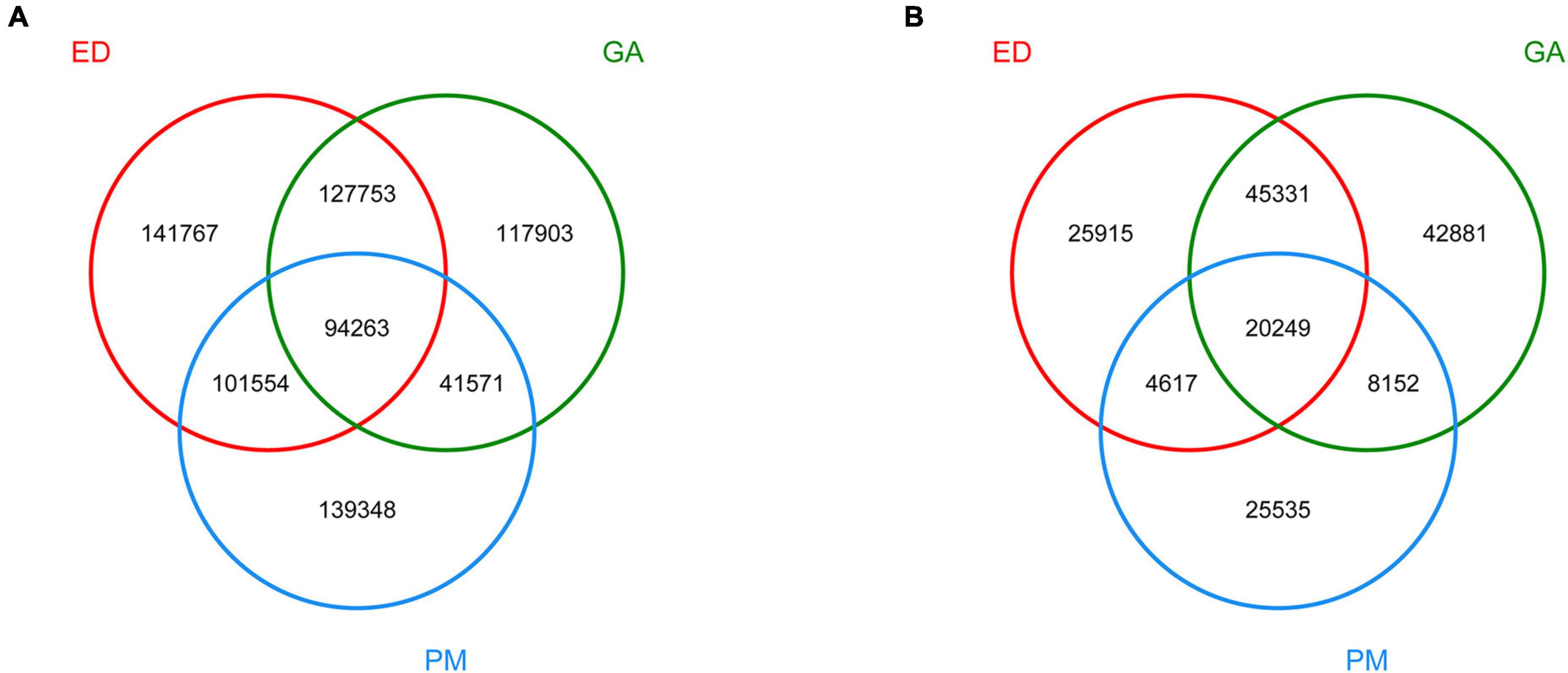
Figure 2. Venn diagram of dereplicated gene set in the fecal (A) and oral (B) samples of P. mucrosquamatus, E. dione, and G. angusticeps.
Supplementary Table 1 presents the 10 most abundant phyla and genera found in the fecal samples of P. mucrosquamatus, E. dione, and G. angusticeps. Bacteroidetes and Proteobacteria emerged as the dominant phyla (>10% relative abundance) across all species, accounting for 16.69–62.72% and 27.34–70.65% of total contigs, respectively. Firmicutes was the sub-dominant phylum (>1% relative abundance) among the three species, accounting for 5.64–9.66%. In the fecal samples of G. angusticeps, the abundances of Fusobacteria and Actinobacteria were notably higher than in the other two species (Figure 3A). Armatimonadetes was also dominant in the guts of all three species, which is reported for the first time.
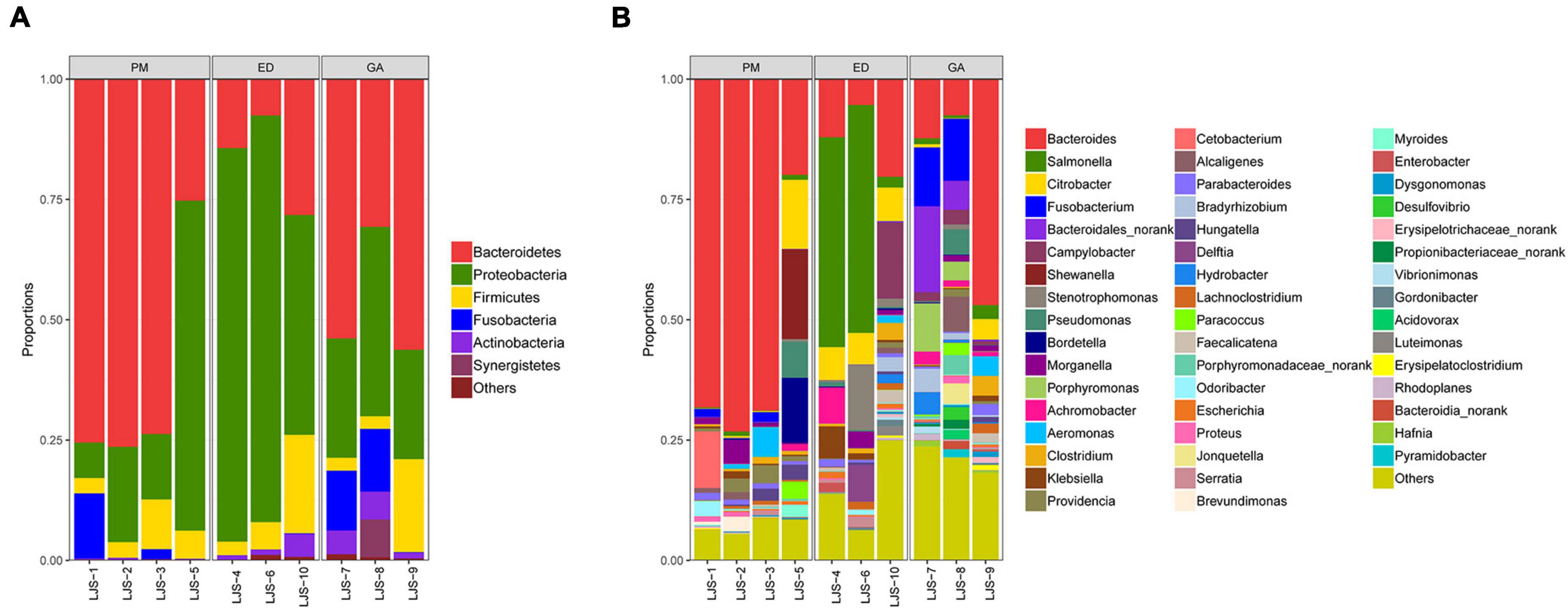
Figure 3. Relative abundance of bacterial communities at the phylum level in fecal (A) and oral samples (B) in P. mucrosquamatus, E. dione, and G. angusticeps.
Among the top 30 bacterial genera identified, 11, 12, and 16 had relative abundances higher than 1% in at least one sample. Among the abundant genera, those showing dominance (>5% relative abundance) in at least one sample included Bacteroides (P. mucrosquamatus), Bacteroides, Salmonella, Citrobacter, Campylobacter, Stenotrophomonas (E. dione), Bacteroides, and Fusobacterium (G. angusticeps). Bacteroides (>20% relative abundance) was the most dominant genus in P. mucrosquamatus and G. angusticeps, while Salmonella (>30% relative abundance) was the most dominant genus in E. dione (Figure 4A).
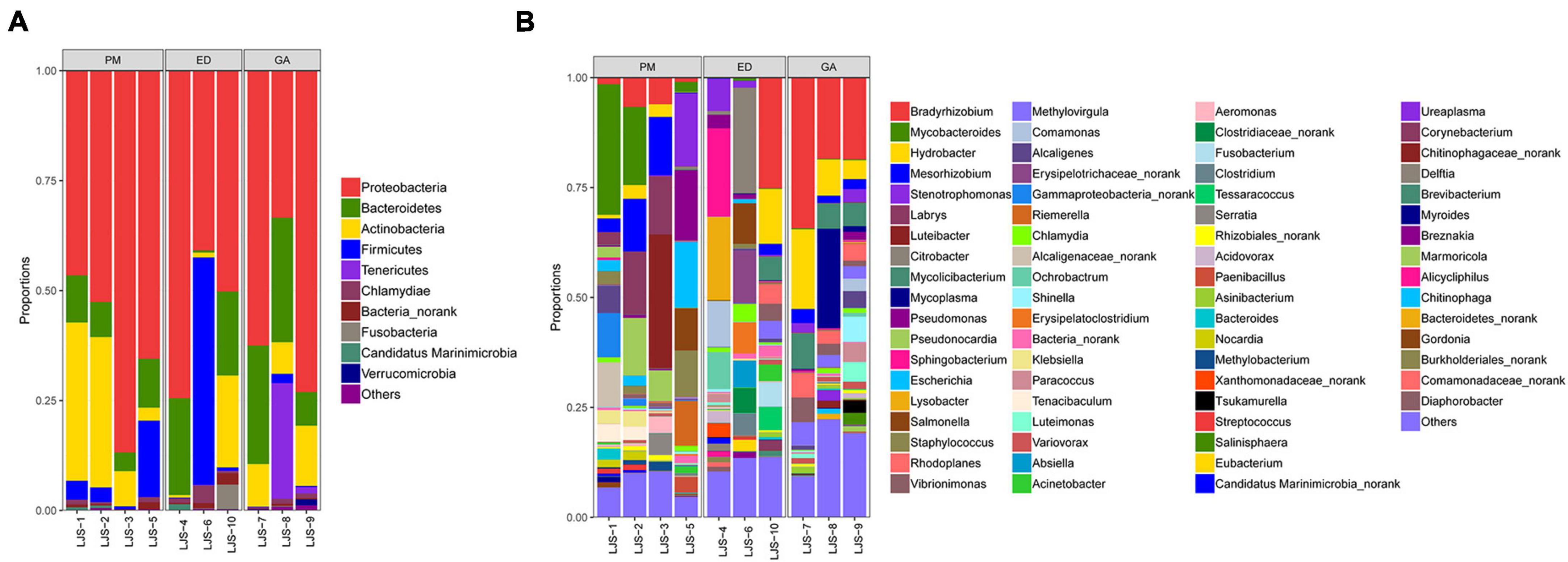
Figure 4. Relative abundance of bacterial communities at the genus level in fecal (A) and oral samples (B) in P. mucrosquamatus, E. dione, and G. angusticeps.
Proteobacteria was the dominant phylum (>55% relative abundance) in the oral samples of P. mucrosquamatus, E. dione, and G. angusticeps, accounting for 55.16, 56.32, and 62.84% of total contigs, respectively. Bacteroidetes, Actinobacteria, and Firmicutes were the sub-dominant phyla (>7% relative abundance) in the three species, accounting for 8.53–20.98%, 7.53–20.28%, and 0.82–17.54%, respectively. Chlamydiae (>0.68% relative abundance) was detected in all three species, while Tenericutes was detected in E. dione and G. angusticeps but not in P. mucrosquamatus (Figure 3B).
Among the top 30 bacterial genera identified, 16, 17, and 11 had relative abundances higher than 1% in at least one sample. Among the abundant genera, those showing dominance (>5% relative abundance) in at least one sample included Mycobacteroides, Mesorhizobium, Labrys, Luteibacter, Pseudonocardia (P. mucrosquamatus), Bradyrhizobium, Citrobacter, Sphingobacterium, Lysobacter (E. dione), Bradyrhizobium, Hydrobacter, Mycoplasma, and Mycolicibacterium (G. angusticeps). Salmonella and Chlamydia were also detected in the oral samples of all three species, while Mycoplasma was present in E. dione and G. angusticeps but absent in P. mucrosquamatus (Figure 4B).
Heatmap and abundance analyses revealed distinct bacterial community structures influenced by geographic distribution and species. At the phylum level, Synergistetes, Chlamydiae, Chloroflexi, Chlorobi, Armatimonadetes, Deinococcus-Thermus, and Tenericutes were abundant in the G. angusticeps fecal samples (Figure 5A). The phylum Bacteroidetes was significantly more abundant in the fecal samples of P. mucrosquamatus than in the two species (Supplementary Table 2). The genus Salmonella was significantly more abundant in the E. dione fecal samples than in the other two species (Supplementary Table 2). In contrast, Verrucomicrobia, Tenericutes, Planctomycetes, and Chloroflexi were abundant in the G. angusticeps oral samples, while Fusobacteria was the only phylum enriched in the oral samples of E. dione (Figure 5B).
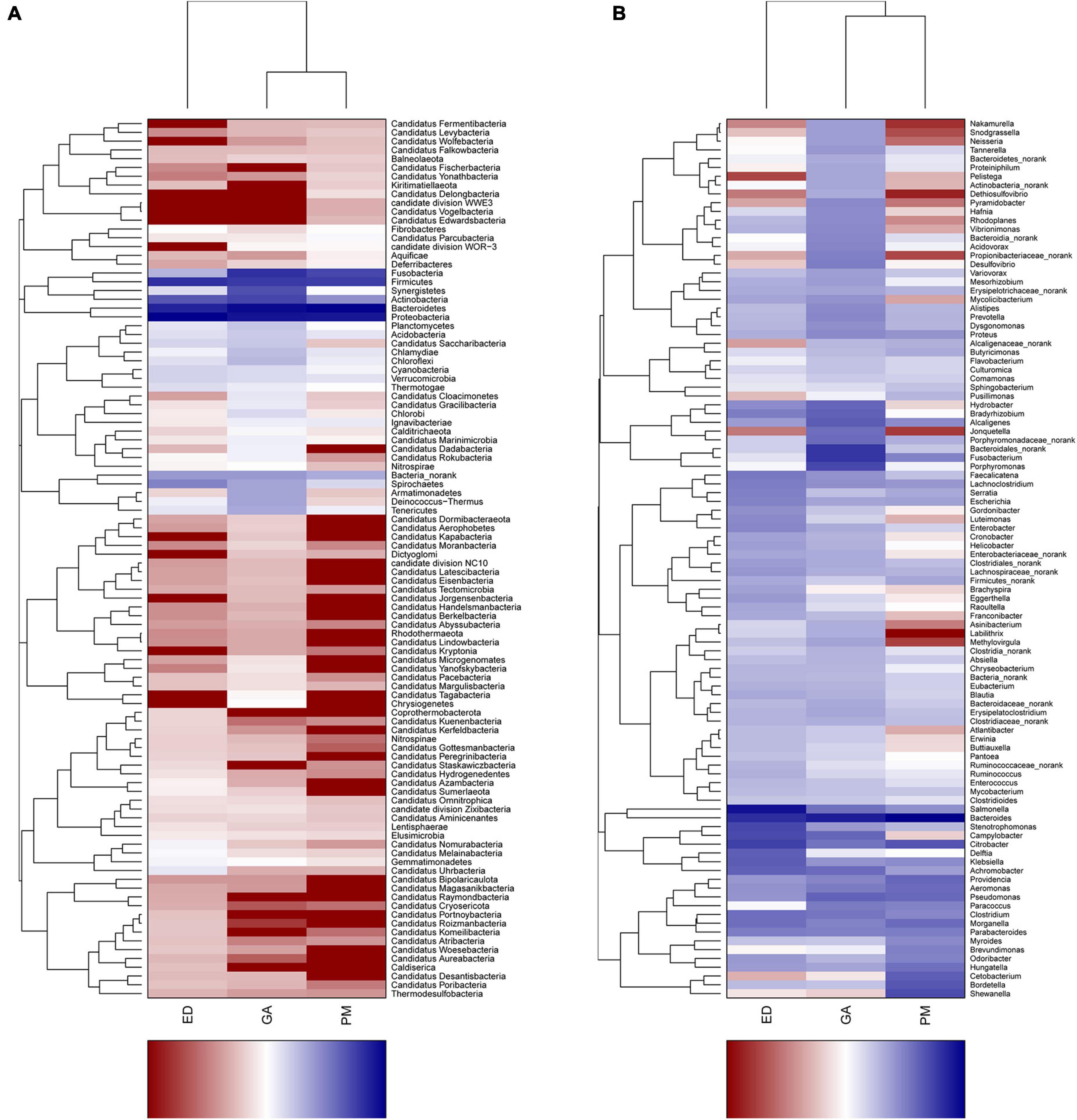
Figure 5. Heatmap showing phylum-level bacterial community composition in fecal (A) and oral samples (B) of P. mucrosquamatus, E. dione, and G. angusticeps.
At the genus level, Nakamurella, Snodgrassella, Neisseria, Pelistega, Dethiosulfovibrio, Pyramidobacter, and Porphyromonas were abundant in the fecal samples of G. angusticeps, whereas Delftia was enriched in the fecal samples of E. dione and Myroides, Brevundimonas, Cetobacterium, and Shewanella were abundant in the fecal samples of P. mucrosquamatus (Figure 6A). Pseudopropionibacterium, Aestuariimicrobium, Brevibacterium, Corynebacterium, Fusobacterium, Tessaracoccus, Acinetobacter, Citrobacter, Lysobacter, and Diaphorobacter were abundant in the oral samples of E. dione. Labrys, Serratia, Pseudonocardia, Methylobacterium, Aminobacter, Pandoraea, and Brevundimonas were enriched in the oral samples of P. mucrosquamatus. Salinisphaera, Tsukamurella, Luteimonas, Shinella, Mycoplasma, Ureaplasma, Altererythrobacter, and Marmoricola were abundant in oral samples of G. angusticeps (Figure 6B). Mycolicibacterium, Rhodoplanes, and Methylovirgula were significantly more abundant in the oral samples of G. angusticeps compared to the other two species (Supplementary Table 2).
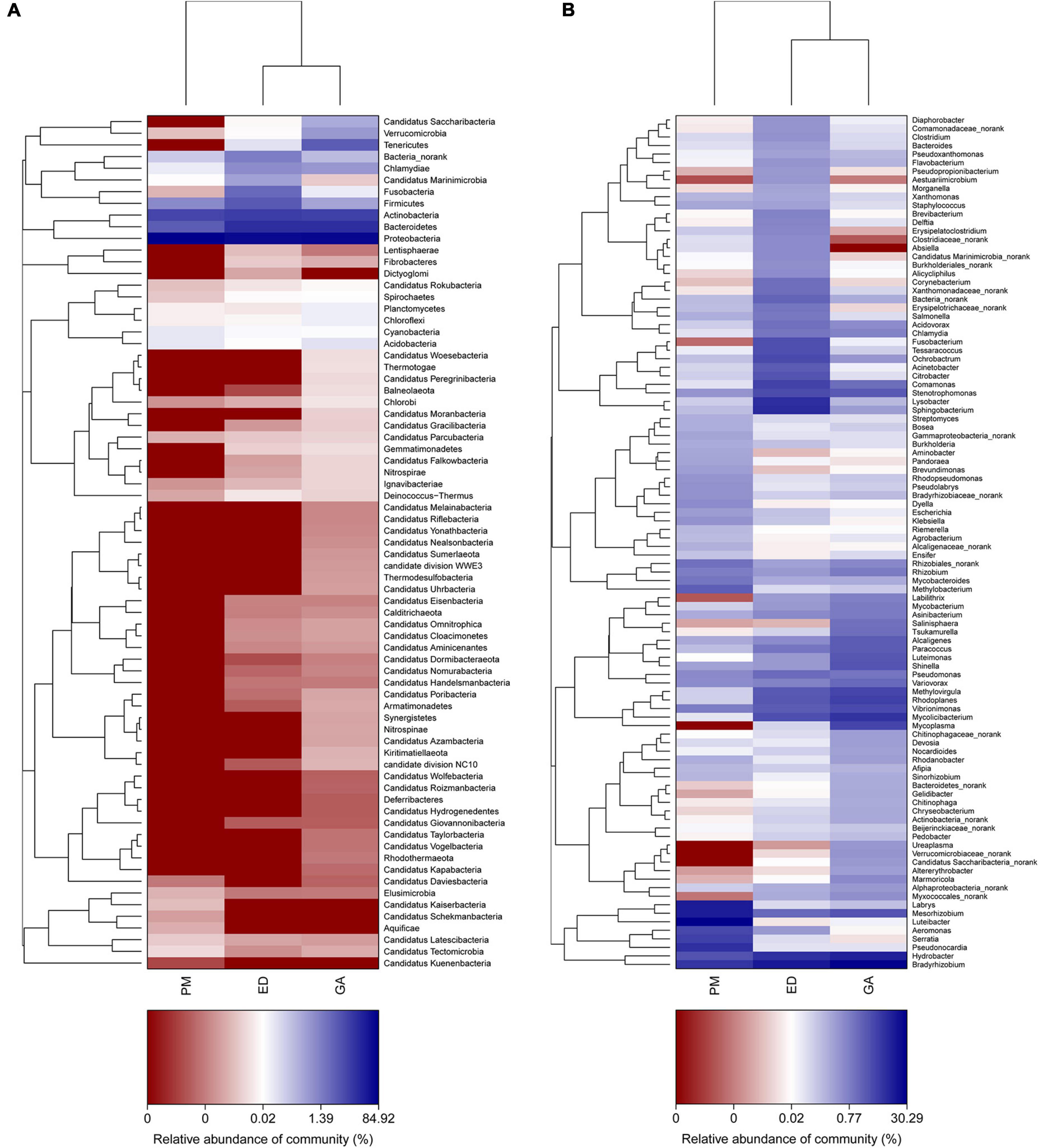
Figure 6. Heatmap showing genus-level bacterial community composition in fecal (A) and oral samples (B) of P. mucrosquamatus, E. dione, and G. angusticeps.
Bray-Curtis distance analysis revealed that bacterial community differences were minor within each species, with samples from the same species clustering together (Figures 7A, B). PCoA demonstrated that gut and oral microbiota from the same host species were more similar to each other than to those of other host species, indicating a higher similarity in gut microbiota within the same snake species (Figures 7C, D).
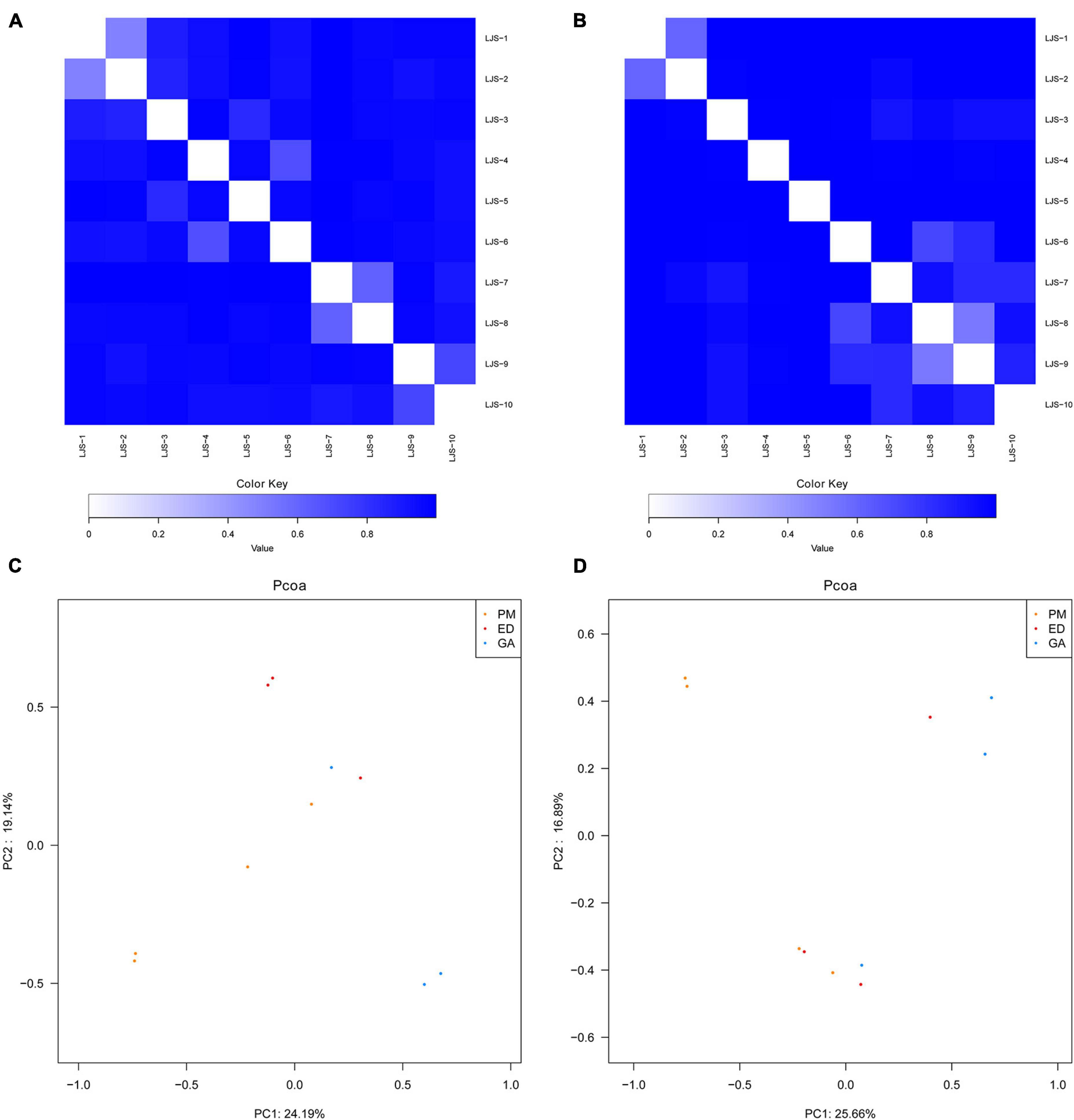
Figure 7. Bray-Curtis distance analysis and principal component analysis of bacterial communities in fecal and oral samples in P. mucrosquamatus, E. dione, and G. angusticeps. Bray-Curtis distance analysis of fecal (A) and oral samples (B). Principal component analysis of fecal (C) and oral samples (D).
A total of 34 zoonotic pathogens were identified in the three snake species studied. Specifically, in P. mucrosquamatus, 12 pathogens were found in oral samples and 22 in fecal samples; in E. dione, 19 were found in oral samples and 29 in fecal samples; and in G. angusticeps, 17 were found in oral samples and 31 in fecal samples. Interestingly, G. angusticeps harbored more pathogens than either E. dione or P. mucrosquamatus. The oral samples from all three snakes contained several pathogens commonly found in urban areas, including Streptococcus pneumoniae, Staphylococcus aureus, Mycobacterium tuberculosis, Listeria monocytogenes, Chlamydia trachomatis, Campylobacter jejuni, and Salmonella enterica. Bartonella and Mycoplasma pneumoniae were only found in the oral samples of G. angusticeps, while Clostridium perfringens, Fusobacterium russii, Fusobacterium mortiferum, Streptobacillus moniliformis, Citrobacter freundii, and Shigella dysenteriae were only found in E. dione.
Reptiles, a group of vertebrates with ancient origins, encompass over 11 000 species, with more than 30% classified within the snake clade. Despite their considerable diversity, reptiles have been largely overlooked in gut and oral microbiota studies (Uetz, 2000). Few studies have been conducted on the microbial communities of lizards (Hong et al., 2011; Ren et al., 2016; Jiang et al., 2017; Kohl et al., 2017; Zhang et al., 2018; Zhou et al., 2020; Tian et al., 2022; Zhu et al., 2022), snakes (Costello et al., 2010; Krishnankutty et al., 2018; Tang et al., 2019), turtles (Campos et al., 2018), and crocodilians (Keenan et al., 2013; Keenan and Elsey, 2015). Our team has identified the presence of various viral families, such as Adenoviridae, Iflaviridae, Circoviridae, Retroviridae, and Parvoviridae in the mouths and guts of P. mucrosquamatus, E. dione, and G. angusticeps (Liu et al., 2023).
In the present study, the most dominant phyla found in the fecal samples of P. mucrosquamatus, E. dione, and G. angusticeps were Bacteroidetes (62.7, 16.7, and 46.9%), Proteobacteria (27.3, 70.7, and 29.0%), and Firmicutes (5.6, 9.7, and 8.2%), collectively accounting for 84.1% of the sequences in the three snake species. Previous studies have also reported that presence of Firmicutes (61.8%), Bacteroidetes (20.6%), and Proteobacteria (10.1%) in Burmese pythons (Costello et al., 2010) and Proteobacteria (65.3%), Firmicutes (9.5%), and Bacteroidetes (9.03%) in Rhabdophis subminiatus (Tang et al., 2019). In our study, Bacteroidetes showed higher abundance in two of the three Chinese snakes (P. mucrosquamatus and G. angusticeps), but relatively lower abundance in Burmese pythons and R. subminiatus. The phylum Bacteroidetes, known to be abundant in many mammalian gut communities, exhibits lower abundance in insectivorous mammals (Ley et al., 2008; Shinohara et al., 2019), consistent with our previous findings in lizards (Tian et al., 2022). Further investigation is required to clarify the role of Bacteroidetes in snake evolution, behavior, and digestion. Proteobacteria, commonly present in domestic insectivorous lizards, is thought to enhance cellulose activity and promote nutrient absorption in hosts (Colston and Jackson, 2016; Tian et al., 2022). However, whether the abundance of Proteobacteria in E. dione and R. subminiatus is related to diet remains unclear. Interestingly, the dominance of Armatimonadetes in the three species, the first report of such, suggests a potential symbiotic role in degrading C5 sugars in hemicelluloses (Lee et al., 2014). Among the Chinese snakes, G. angusticeps exhibited a more diverse bacterial composition with seven abundant phyla compared to P. mucrosquamatus and E. dione. This may be attributed to G. angusticeps inhabiting high-altitude regions with minimal human contact, suggesting that unique habitats may influence gut microbial composition. However, the small sample size of snakes used in this study may introduce bias, warranting caution in interpreting these results. Overall, these findings provide new insights into the bacterial composition of reptiles from diverse habitats, particularly differences between high- and low-altitude environments, laying the groundwork for future research regarding the impact of altitude on bacterial diversity in reptiles.
Firmicutes, Actinobacteria, Proteobacteria, and Bacteroidetes are commonly found in the oral bacterial compositions of various species, including humans, mice, felines, canines, chimpanzees, hawks, and lizards (Chun et al., 2010; Sturgeon et al., 2013; Gong et al., 2014; Dewhirst et al., 2015; Adler et al., 2016; Tian et al., 2022). However, research on the oral microbiota of snakes remains limited. In the present study, Proteobacteria (62.8, 55.2, and 56.3%), Bacteroidetes (8.5, 13.9, and 21.0%), Actinobacteria (20.2, 7.5, and 10.2%), and Firmicutes (6.4, 17.5, and 0.8%) were also prevalent in the oral samples of P. mucrosquamatus, E. dione, and G. angusticeps, indicating a bacterial composition similar to the abovementioned hosts. Verrucomicrobia, Tenericutes, Planctomycetes, and Chloroflexi were more abundant in G. angusticeps compared to P. mucrosquamatus and E. dione. Fusobacteria was particularly enriched in the oral samples of E. dione. Interestingly, Tenericutes, known for playing a key role in the gut communities of fish and juvenile amphibians (Colston and Jackson, 2016), was also enriched in G. angusticeps. Moreover, the genera Mycolicibacterium, Rhodoplanes, and Methylovirgula were markedly more abundant in G. angusticeps than in P. mucrosquamatus and E. dione (Table 1). Rhodoplanes is broadly distributed in aquatic habitats (Srinivas et al., 2014), while Methylovirgula is widely distributed in forest soils and acidic wetlands (pH 3–5) (Vorob’ev et al., 2009) and Mycolicibacterium is known to thrive in acid-resistant environments (Xia et al., 2022). These findings indicate that the oral environment of G. angusticeps may be more acidic than that of the other two species, consistent with its habits and wetland environment. Overall, our study suggests that oral bacterial composition varies in different snakes based on geography, diet, and habitat.
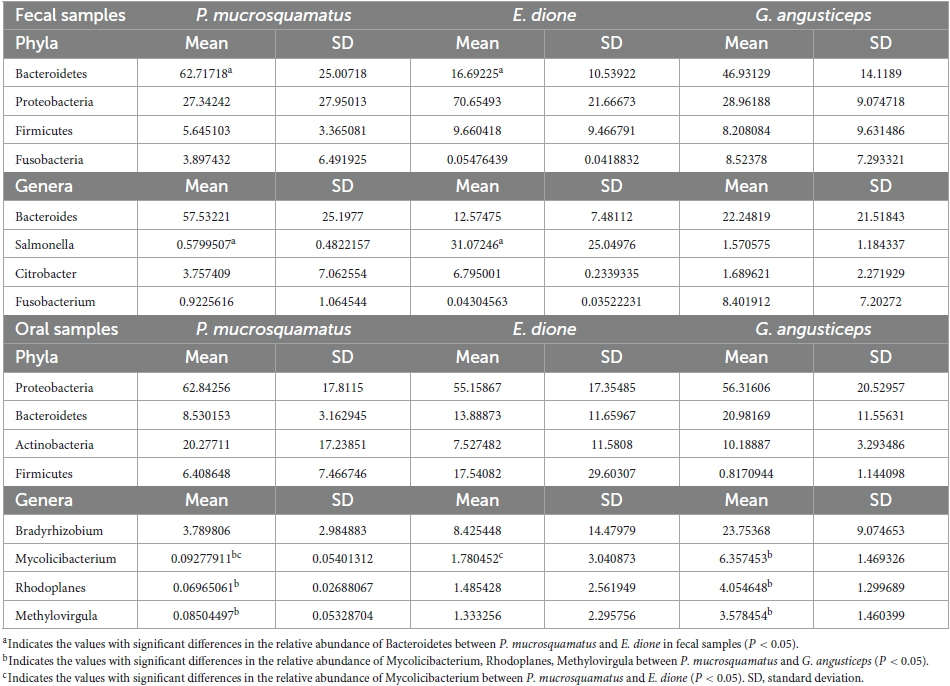
Table 1. The differences in relative abundance (% ± SD) of the abundant phylum and genera of three snake species.
Limited microbiological data exist regarding wound infections from snakebites. However, understanding the origin of bacteria in the mouth of snakes is crucial given the differences in bacterial infection severity from the bites of certain species, such as the cobra, relative to those from other snakes (Mao et al., 2016). In the current study, we identified a total of 34 bacterial and parasitic pathogens in the three Chinese snake species, some of which were zoonotic. Notably, 10 pathogenic species (Clostridium botulinum, Streptococcus pneumoniae, Pseudomonas aeruginosa, Comamonas testosteroni, Staphylococcus aureus, Mycobacterium tuberculosis, Listeria monocytogenes, Chlamydia trachomatis, Campylobacter jejuni, and Salmonella enterica) were detected in the oral samples of all three snakes, while more than 24 pathogens (Clostridium perfringens, Clostridium baratii, Clostridium botulinum, Fusobacterium russii, Fusobacterium mortiferum, Prevotella loescheii, Enterococcus durans, Streptococcus pneumoniae, Citrobacter freundii, Pseudomonas aeruginosa, Comamonas testosterone, Enterococcus cecorum, Staphylococcus aureus, Mycobacterium tuberculosis, Listeria monocytogenes, Chlamydia trachomatis, Yersinia pseudotuberculosis, Yersinia enterocolitica, Campylobacter jejuni, Campylobacter coli, Salmonella enterica, Leptospira, Bartonella, Shigella dysenteriae) were found in the gut samples of all three snakes. Various wild animals, such as rodents, boars, bats, and birds, are known carriers of many pathogens, posing considerable disease risk to both other wildlife and humans (Benskin et al., 2009; Selvin et al., 2019; Liu et al., 2022; Tapia-Ramírez et al., 2022). Therefore, the role of snakes as potential vectors of zoonosis is an important area of focus.
Bartonella, an increasing recognized vector-borne pathogen, infects a variety of hosts including humans, foxes (Kosoy and Goodrich, 2018), wolves (Greco et al., 2021), badgers (Bitam et al., 2009), hedgehogs (Bitam et al., 2009), roe deer (Dehio et al., 2001), and canids (Gerrikagoitia et al., 2012). This study marks the first identification of Bartonella in the gastrointestinal tract of the three snake species, as well as in the oral cavity of G. angusticeps. Given that Bartonella species are transmitted by arthropods, these observations underscore the need for more targeted research into the interactions between snakes and arthropod vectors.
In conclusion, metagenomic analysis of fecal and oral samples from three Chinese snake species revealed a diverse bacterial composition, dominated by Bacteroidetes, Proteobacteria, and Firmicutes in the fecal samples and by Proteobacteria, Bacteroidetes, Actinobacteria, and Firmicutes in oral samples across all three species. Notably, however, our results also showed that the microbiological data from G. angusticeps, which inhabits high-altitude regions, differed significantly from those of P. mucrosquamatus and E. dione, which reside in low-altitude regions. Furthermore, a variety of bacterial pathogens were identified in both the oral and fecal samples, which resulted in the secondary infections of bite wounds and was transmitted to the other wild field animals, suggesting that snakes may serve as a natural reservoir of zoonotic diseases. Future research should focus on (1) exploring how gut and oral microbial communities influence the ecology and evolution of snakes, and (2) investigating interactions between snakes and wildlife, including arthropods, birds, rodents, and bats, to better understand the transmission of emerging infectious diseases.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI–SRP406182.
The animal study was approved by the Committee on the Animal Ethics Procedures and Guidelines of Experiment Animals Committee affiliated with Yibin University, Yibin, China. The study was conducted in accordance with the local legislation and institutional requirements.
XH: Funding acquisition, Writing – original draft, Writing – review and editing. LY: Software, Writing – original draft. YZ: Software, Writing – original draft. MY: Investigation, Writing – original draft. JL: Formal analysis, Writing – original draft. YF: Methodology, Writing – original draft. PG: Methodology, Writing – review and editing. ZT: Data curation, Funding acquisition, Writing – review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Doctor Launch Project of Yibin University (Nos. 2019QD09 and 2019QD10).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1339188/full#supplementary-material
Adler, C. J., Malik, R., Browne, G. V., and Norris, J. M. (2016). Diet may influence the oral microbiome composition in cats. Microbiome 4:23. doi: 10.1186/s40168-016-0169-y
Archie, E. A., and Theis, K. R. (2011). Animal behaviour meets microbial ecology. Anim. Behav. 82, 425–436. doi: 10.1016/j.anbehav.2011.05.029
Arroyo, O., Bolaños, R., and Muñoz, G. (1980). The bacterial flora of venoms and mouth cavities of Costa Rican snakes. Bull. Pan Am. Health Organ. 14, 280–285.
Barbosa, L. N., Ferreira, R. S. Jr., Luiza Mello, P., Garcia Garces, H., Luana Chechi, J., Frachin, T., et al. (2018). Molecular identification and phylogenetic analysis of Bothrops insularis bacterial and fungal microbiota. J. Toxicol. Environ. Health. Part A 81, 142–153. doi: 10.1080/15287394.2017.1395581
Benskin, C. M., Wilson, K., Jones, K., and Hartley, I. R. (2009). Bacterial pathogens in wild birds: A review of the frequency and effects of infection. Biol. Rev. Camb. Philosoph. Soc. 84, 349–373. doi: 10.1111/j.1469-185X.2008.00076.x
Bitam, I., Rolain, J. M., Kernif, T., Baziz, B., Parola, P., and Raoult, D. (2009). Bartonella species detected in rodents and hedgehogs from Algeria. Clin. Microbiol. Infect. 15 Suppl. 2, 102–103. doi: 10.1111/j.1469-0691.2008.02180.x
Blaylock, R. S. (2001). Normal oral bacterial flora from some southern African snakes. Onderstepoort J. Vet. Res. 68, 175–182.
Campagner, M. V., Bosco, S. M., Bagagli, E., Cunha, M. L., Jeronimo, B. C., Saad, E., et al. (2012). Microbiological evaluation of different strategies for management of snakes in captivity. J. Toxicol. Environ. Health. Part A 75, 1070–1080. doi: 10.1080/15287394.2012.697837
Campos, P., Guivernau, M., Prenafeta-Boldú, F. X., and Cardona, L. (2018). Fast acquisition of a polysaccharide fermenting gut microbiome by juvenile green turtles Chelonia mydas after settlement in coastal habitats. Microbiome 6:69. doi: 10.1186/s40168-018-0454-z
Chun, J., Kim, K. Y., Lee, J. H., and Choi, Y. (2010). The analysis of oral microbial communities of wild-type and toll-like receptor 2-deficient mice using a 454 GS FLX Titanium pyrosequencer. BMC Microbiol. 10:101. doi: 10.1186/1471-2180-10-101
Colston, T. J., and Jackson, C. R. (2016). Microbiome evolution along divergent branches of the vertebrate tree of life: What is known and unknown. Mol. Ecol. 25, 3776–3800. doi: 10.1111/mec.13730
Costello, E. K., Gordon, J. I., Secor, S. M., and Knight, R. (2010). Postprandial remodeling of the gut microbiota in Burmese pythons. ISME J. 4, 1375–1385. doi: 10.1038/ismej.2010.71
David, L. A., Maurice, C. F., Carmody, R. N., Gootenberg, D. B., Button, J. E., Wolfe, B. E., et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. doi: 10.1038/nature12820
Dehio, C., Lanz, C., Pohl, R., Behrens, P., Bermond, D., Piémont, Y., et al. (2001). Bartonella schoenbuchii sp. nov., isolated from the blood of wild roe deer. Int. J. Syst. Evol. Microbiol. 51, 1557–1565. doi: 10.1099/00207713-51-4-1557
Dewhirst, F. E., Klein, E. A., Bennett, M. L., Croft, J. M., Harris, S. J., and Marshall-Jones, Z. V. (2015). The feline oral microbiome: A provisional 16S rRNA gene based taxonomy with full-length reference sequences. Vet. Microbiol. 175, 294–303. doi: 10.1016/j.vetmic.2014.11.019
Ezenwa, V. O., Gerardo, N. M., Inouye, D. W., Medina, M., and Xavier, J. B. (2012). Microbiology. Animal behavior and the microbiome. Science 338, 198–199. doi: 10.1126/science.1227412
Fouts, D. E., Szpakowski, S., Purushe, J., Torralba, M., Waterman, R. C., MacNeil, M. D., et al. (2012). Next generation sequencing to define prokaryotic and fungal diversity in the bovine rumen. PLoS One 7:e48289. doi: 10.1371/journal.pone.0048289
Fu, L., Niu, B., Zhu, Z., Wu, S., and Li, W. (2012). CHIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gerrikagoitia, X., Gil, H., García-Esteban, C., Anda, P., Juste, R. A., and Barral, M. (2012). Presence of Bartonella species in wild carnivores of northern Spain. Appl. Environ. Microbiol. 78, 885–888. doi: 10.1128/AEM.05938-11
Gong, H., Shi, Y., Zhou, X., Wu, C., Cao, P., Xu, C., et al. (2014). Microbiota in the throat and risk factors for laryngeal carcinoma. Appl. Environ. Microbiol. 80, 7356–7363. doi: 10.1128/AEM.02329-14
Greco, G., Zarea, A. A. K., Sgroi, G., Tempesta, M., D’Alessio, N., Lanave, G., et al. (2021). Zoonotic Bartonella species in Eurasian wolves and other free-ranging wild mammals from Italy. Zoonoses Public Health 68, 316–326. doi: 10.1111/zph.12827
Hong, P. Y., Wheeler, E., Cann, I. K., and Mackie, R. I. (2011). Phylogenetic analysis of the fecal microbial community in herbivorous land and marine iguanas of the Galápagos Islands using 16S rRNA-based Pyrosequencing. ISME J. 5, 1461–1470. doi: 10.1038/ismej.2011.33
Hooper, L. V., Wong, M. H., Thelin, A., Hansson, L., Falk, P. G., and Gordon, J. I. (2001). Molecular analysis of commensal host-microbial relationships in the intestine. Science 291, 881–884. doi: 10.1126/science.291.5505.881
Hou, X., Zhu, L., Zhang, X., Zhang, L., Bao, H., Tang, M., et al. (2019). Testosterone disruptor effect and gut microbiome perturbation in mice: Early life exposure to doxycycline. Chemosphere 222, 722–731. doi: 10.1016/j.chemosphere.2019.01.101
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinformat. 11:119. doi: 10.1186/1471-2105-11-119
Jiang, H. Y., Ma, J. E., Li, J., Zhang, X. J., Li, L. M., He, N., et al. (2017). Diets alter the gut microbiome of crocodile lizards. Front. Microbiol. 8:2073. doi: 10.3389/fmicb.2017.02073
Kang, D. D., Froula, J., Egan, R., Wang, Z., and Meta, B. A. T. (2015). MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ 3:e1165. doi: 10.7717/peerj.1165
Keenan, S. W., and Elsey, R. M. (2015). The good, the bad, and the unknown: Microbial symbioses of the American alligator. Integrat. Comp. Biol. 55, 972–985. doi: 10.1093/icb/icv006
Keenan, S. W., Engel, A. S., and Elsey, R. M. (2013). The alligator gut microbiome and implications for archosaur symbioses. Sci. Rep. 3:2877. doi: 10.1038/srep02877
Kohl, K. D., Brun, A., Magallanes, M., Brinkerhoff, J., Laspiur, A., Acosta, J. C., et al. (2017). Gut microbial ecology of lizards: Insights into diversity in the wild, effects of captivity, variation across gut regions and transmission. Mol. Ecol. 26, 1175–1189. doi: 10.1111/mec.13921
Kosoy, M., and Goodrich, I. (2018). Comparative ecology of Bartonella and Brucella infections in wild carnivores. Front. Vet. Sci. 5:322. doi: 10.3389/fvets.2018.00322
Krishnankutty, S. P., Muraleedharan, M., Perumal, R. C., Michael, S., Benny, J., Balan, B., et al. (2018). Next-generation sequencing analysis reveals high bacterial diversity in wild venomous and non-venomous snakes from India. J. Venomous Anim. Toxins Incl. Trop. Dis. 24:41. doi: 10.1186/s40409-018-0181-8
Lee, K. C., Morgan, X. C., Dunfield, P. F., Tamas, I., McDonald, I. R., and Stott, M. B. (2014). Genomic analysis of Chthonomonas calidirosea, the first sequenced isolate of the phylum Armatimonadetes. ISME J. 8, 1522–1533. doi: 10.1038/ismej.2013.251
Ley, R. E., Hamady, M., Lozupone, C., Turnbaugh, P. J., Ramey, R. R., Bircher, J. S., et al. (2008). Evolution of mammals and their gut microbes. Science 320, 1647–1651. doi: 10.1126/science.1155725
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, J., Jia, H., Cai, X., Zhong, H., Feng, Q., Sunagawa, S., et al. (2014). An integrated catalog of reference genes in the human gut microbiome. Nat. Biotechnol. 32, 834–841. doi: 10.1038/nbt.2942
Liu, A., Tian, Z., Yin, C., Zou, J., Wu, S., Luo, Y., et al. (2023). The analysis of oral and fecal virome detects multiple novel emerging viruses in snakes. Transbound. Emerg. Dis. 2023, 1–13. doi: 10.1155/2023/4214812
Liu, A., Xue, T., Zhao, X., Zou, J., Pu, H., Hu, X., et al. (2022). Pseudorabies virus associations in wild animals: Review of potential reservoirs for cross-host transmission. Viruses 14:2254. doi: 10.3390/v14102254
Luis, A. D., Hayman, D. T., O’Shea, T. J., Cryan, P. M., Gilbert, A. T., Pulliam, J. R., et al. (2013). A comparison of bats and rodents as reservoirs of zoonotic viruses: Are bats special? Proc. Biol. Sci. 280:20122753. doi: 10.1098/rspb.2012.2753
Mackie, R. I. (2002). Mutualistic fermentative digestion in the gastrointestinal tract: Diversity and evolution. Integr. Comp. Biol. 42, 319–326. doi: 10.1093/icb/42.2.319
Mao, Y. C., Liu, P. Y., Hung, D. Z., Lai, W. C., Huang, S. T., Hung, Y. M., et al. (2016). Bacteriology of Naja atra snakebite wound and its implications for antibiotic therapy. Am. J. Trop. Med. Hygiene 94, 1129–1135. doi: 10.4269/ajtmh.15-0667
McKenney, E. A., Williamson, L., Yoder, A. D., Rawls, J. F., Bilbo, S. D., and Parker, W. (2015). Alteration of the rat cecal microbiome during colonization with the helminth Hymenolepis diminuta. Gut Microbes 6, 182–193. doi: 10.1080/19490976.2015.1047128
Moeller, A. H., Li, Y., Mpoudi Ngole, E., Ahuka-Mundeke, S., Lonsdorf, E. V., Pusey, A. E., et al. (2014). Rapid changes in the gut microbiome during human evolution. Proc. Natl. Acad. Sci. U.S.A. 111, 16431–16435. doi: 10.1073/pnas.1419136111
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: A new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Panda, S. K., Padhi, L., and Sahoo, G. (2019). Evaluation of cultivable aerobic bacterial flora from Russell’s viper (Daboia russelii) oral cavity. Microb. Pathog. 134:103573. doi: 10.1016/j.micpath.2019.103573
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Qin, Z., Wang, S., Guo, D., Zhu, J., Chen, H., Bai, L., et al. (2019). Comparative analysis of intestinal bacteria among venom secretion and non-secrection snakes. Sci. Rep. 9:6335. doi: 10.1038/s41598-019-42787-6
Ren, T., Kahrl, A. F., Wu, M., and Cox, R. M. (2016). Does adaptive radiation of a host lineage promote ecological diversity of its bacterial communities? A test using gut microbiota of Anolis lizards. Mol. Ecol. 25, 4793–4804. doi: 10.1111/mec.13796
Schulz, K. D., and Entzeroth, A. (1996). A monograph of the colubrid snakes of the genus Elaphe Fitzinger. Seattle, WA: Koeltz Scientific Books.
Segata, N., Börnigen, D., Morgan, X. C., and Huttenhower, C. (2013). PhyloPhlAn is a new method for improved phylogenetic and taxonomic placement of microbes. Nat. Commun. 4:2304. doi: 10.1038/ncomms3304
Selvin, J., Lanong, S., Syiem, D., De Mandal, S., Kayang, H., Kumar, N. S., et al. (2019). Culture-dependent and metagenomic analysis of lesser horseshoe bats’ gut microbiome revealing unique bacterial diversity and signatures of potential human pathogens. Microb. Pathog. 137:103675. doi: 10.1016/j.micpath.2019.103675
Shinohara, A., Nohara, M., Kondo, Y., Jogahara, T., Nagura-Kato, G. A., Izawa, M., et al. (2019). Comparison of the gut microbiotas of laboratory and wild Asian house shrews (Suncus murinus) based on cloned 16S rRNA sequences. Exp. Anim. 68, 531–539. doi: 10.1538/expanim.19-0021
Shropshire, J. D., and Bordenstein, S. R. (2016). Speciation by symbiosis: The microbiome and behavior. mBio 7:e01785. doi: 10.1128/mBio.01785-15
Srinivas, A., Sasikala, C., and Ramana, C. V. (2014). Rhodoplanes oryzae sp. nov., a phototrophic alphaproteobacterium isolated from the rhizosphere soil of paddy. Int. J. Syst. Evol. Microbiol. 64, 2198–2203. doi: 10.1099/ijs.0.063347-0
Sturgeon, A., Stull, J. W., Costa, M. C., and Weese, J. S. (2013). Metagenomic analysis of the canine oral cavity as revealed by high-throughput Pyrosequencing of the 16S rRNA gene. Vet. Microbiol. 162, 891–898. doi: 10.1016/j.vetmic.2012.11.018
Tang, W., Zhu, G., Shi, Q., Yang, S., Ma, T., Mishra, S. K., et al. (2019). Characterizing the microbiota in gastrointestinal tract segments of Rhabdophis subminiatus: Dynamic changes and functional predictions. MicrobiologyOpen 8:e00789. doi: 10.1002/mbo3.789
Tapia-Ramírez, G., Lorenzo, C., Navarrete, D., Carrillo-Reyes, A., and Retana, Ó, and Carrasco-Hernández, R. (2022). A review of mammarenaviruses and rodent reservoirs in the Americas. EcoHealth 19, 22–39. doi: 10.1007/s10393-022-01580-0
Tian, Z., Pu, H., Cai, D., Luo, G., Zhao, L., Li, K., et al. (2022). Characterization of the bacterial microbiota in different gut and oral compartments of splendid japalure (Japalura sensu lato). BMC Vet. Res. 18:205. doi: 10.1186/s12917-022-03300-w
Vorob’ev, A. V., de Boer, W., Folman, L. B., Bodelier, P. L., Doronina, N. V., Suzina, N. E., et al. (2009). Methylovirgula ligni gen. nov., sp. nov., an obligately acidophilic, facultatively methylotrophic bacterium with a highly divergent mxaF gene. Int. J. Syst. Evol. Microbiol. 59, 2538–2545. doi: 10.1099/ijs.0.010074-0
Wood, D. E., and Salzberg, S. L. (2014). Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 15:R46. doi: 10.1186/gb-2014-15-3-r46
Xia, J., Ni, G., Wang, Y., Zheng, M., and Hu, S. (2022). Mycolicibacter acidiphilus sp. nov., an extremely acid-tolerant member of the genus Mycolicibacter. Int. J. Syst. Evol. Microbiology 72. doi: 10.1099/ijsem.0.005419
Zhang, B., Ren, J., Yang, D., Liu, S., and Gong, X. (2019). Comparative analysis and characterization of the gut microbiota of four farmed snakes from southern China. PeerJ 7:e6658. doi: 10.7717/peerj.6658
Zhang, W., Li, N., Tang, X., Liu, N., and Zhao, W. (2018). Changes in intestinal microbiota across an altitudinal gradient in the lizard Phrynocephalus vlangalii. Ecol. Evol. 8, 4695–4703. doi: 10.1002/ece3.4029
Zhou, J., Zhao, Y. T., Dai, Y. Y., Jiang, Y. J., Lin, L. H., Li, H., et al. (2020). Captivity affects diversity, abundance, and functional pathways of gut microbiota in the northern grass lizard Takydromus septentrionalis. MicrobiologyOpen 9:e1095. doi: 10.1002/mbo3.1095
Zhou, Z., and Jiang, Z. (2004). International trade status and crisis for snake species in China. Conserv. Biol. 18, 1386–1394. doi: 10.1111/j.1523-1739.2004.00251.x
Keywords: Protobothrops mucrosquamatus, Elaphe dione, Gloydius angusticeps, microbiome, bacterial pathogens
Citation: Hu X, Yang L, Zhang Y, Yang M, Li J, Fan Y, Guo P and Tian Z (2024) Fecal and oral microbiome analysis of snakes from China reveals a novel natural emerging disease reservoir. Front. Microbiol. 14:1339188. doi: 10.3389/fmicb.2023.1339188
Received: 15 November 2023; Accepted: 22 December 2023;
Published: 11 January 2024.
Edited by:
Wei Zhu, Chinese Academy of Sciences (CAS), ChinaReviewed by:
Jin Tian, Chinese Academy of Agricultural Sciences, ChinaCopyright © 2024 Hu, Yang, Zhang, Yang, Li, Fan, Guo and Tian. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhige Tian, YmVpc2h1Z2VnZUAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.