 Pavel A. Andriyanov
Pavel A. Andriyanov Daria D. Kashina
Daria D. Kashina Alena N. Menshikova
Alena N. Menshikova- Federal Research Center for Virology and Microbiology, Branch in Nizhny Novgorod, Nizhny Novgorod, Russia
Delftia tsuruhatensis is a gram-negative, aerobic bacterium mostly known as an organic pollutant degrading and growth-promoting microorganism. However, it recently emerged as an opportunistic human pathogen. To date, the source of D. tsuruhatensis infection is not clear. The majority of studies of D. tsuruhatensis have focused on environmental or clinical strains, while investigations of D. tsuruhatensis strains isolated from food sources are limited. In the present study, we report the case of D. tsuruhatensis isolation from raw bovine milk. Classical bacteriology approaches, as well as next-generation sequencing and comparative genomics, were used to characterize the features of the D. tsuruhatensis MR-6/3H strain. The MR-6/3H strain was resistant to 19 antimicrobials among 23 tested, including all aminoglycosides, phenicol, trimethoprim-sulfamethoxazole, and almost all β-lactams. Phylogenetically, the MR-6/3H was close to clinical origin strains, including those previously isolated in Russia. Comparative genomics revealed the presence of putative antimicrobial resistance genes in the MR-6/3H isolate, mostly associated with efflux systems. Notably, genus-specific OXA-926-like β-lactamase was also detected. In all, 27 putative virulence factors were predicted, the majority of which were associated with motility, adherence, stress survival, siderophore synthesis, and immunomodulation. In the MR-6/3H genome, the five prophage regions were identified, including two with intact levels. Integrons and CRISPR-Cas systems were not detected in the MR-6/3H isolate. Thus, our findings suggest that raw milk can be the potential source of and transmission route for the dissemination of multidrug-resistant D. tsuruhatensis.
Introduction
Delftia tsuruhatensis is a gram-negative, motile, non-fermentative, aerobic bacterium. This species belongs to the Comamonadaceae family, order Burkholderiales, class Betaproteobacteria. D. tsuruhatensis was initially isolated from activated sludge collected from a domestic wastewater treatment plant in 2003 in Japan and described as a terephthalate-assimilating bacterium (Shigematsu et al., 2003). These bacteria are ubiquitous, but most frequently were isolated from polluted environments, rhizosphere soil, and activated sludge. Notably, plant growth-promoting effects of D. tsuruhatensis were observed. Strain HR4, isolated from the rhizoplane of rice (Oryza sativa) in China, was shown to suppress the growth of various plant pathogens (Han et al., 2005). Similarly, strain MTQ, isolated from the soil of the tobacco rhizosphere in China, also had antimicrobial activity against some phytopathogens (Hou et al., 2015). Interestingly, the antimicrobial and inhibitory activity of D. tsuruhatensis against such notable human ESKAPE pathogens was also reported (Malešević et al., 2019; Tejman-Yarden et al., 2019). Another remarkable feature of D. tsuruhatensis is the ability to biodegrade some organic pollutants, such as phenolic compounds, cyclopropylcarboxamides, terephthalate, etc. (Shigematsu et al., 2003; Liang et al., 2005; Zheng et al., 2007; Juárez-Jiménez et al., 2010; Juarez Jimenez et al., 2012; Ye et al., 2019). Thus, D. tsuruhatensis is interesting in terms of bioremediation, biotechnology, and new antimicrobial drug development.
Delftia tsuruhatensis was isolated from such animals as Danio rerio (Zebrafish: skin mucus) (Chaires et al., 2022) and Oreochromis niloticus (Nile tilapia: internal organs and injured eye) (Soto-Rodriguez et al., 2013; Preena et al., 2020), laboratory mice (inbred strain C57BL/6: proximal colonic tissue) (Saffarian et al., 2016), and even humans. D. tsuruhatensis was reported as an emergent opportunistic human pathogen. The first infection case was reported in 2011 by Preiswerk et al., where the catheter-related infection was observed in a 53-year-old woman from Switzerland. Moreover, isolates from collected blood had multidrug-resistant phenotype (MDR) and were resistant to ampicillin, some cephalosporins, all tested aminoglycosides, and colistin, whereas they were susceptible to amoxicillin-clavulanate, piperacillin-tazobactam, III generation cephalosporins, carbapenems, and fluoroquinolones. Ciprofloxacin-based therapy led to a positive infection outcome (Preiswerk et al., 2011). Afterward, some similar cases of D. tsuruhatensis infection were further reported, including infection in a premature infant (Tabak et al., 2013; Ranc et al., 2018; Cheng et al., 2021; Cho et al., 2021). Recent pan genomic investigations revealed some putative virulence factors in D. tsuruhatensis genomes, most of which were associated with nosocomial infections, for example, bloodstream infection, urinary tract infection, skin infection, etc. The multitude of antimicrobial resistance (AMR) genes were also predicted in some D. tsuruhatensis strains, including factors conferring resistance to aminoglycosides, fluoroquinolones, aminocyclitols, tetracyclines, phenols, and folate pathway antagonists (Yin et al., 2022). Furthermore, cases of AMR genes acquired via horizontal gene transfer (HGT) have been reported in D. tsuruhatensis. For instance, in the chromosome of the CRS1243 clinical strain, the blaIMP-1 beta-lactamase gene linked with mobile element conferring resistance to cephamycin, cephalosporin, penem, penam, and carbapenem was detected (Cho et al., 2021). Likewise, the TR1180 clinical strain was reported to harbor the unique 38-kb genomic island with the insertion of an In4-like integron containing some AMR genes (Cheng et al., 2021).
An open pan-genome with a great number of various proteins was also highlighted, which defines D. tsuruhatensis as a generalist pathogen with extensive genetic diversity and the ability to thrive in many ecological niches. Phylogenomic analysis revealed that D. tsuruhatensis strains appear to be particularly capable of animal colonizing and causing opportunistic infection (Bhat et al., 2022). Thus, it is important to investigate the possible sources of D. tsuruhatensis, as well as explore its genomic diversity and antimicrobial resistance mechanisms. In the present study, the case of D. tsuruhatensis isolation from unpasteurized milk (food source) is reported. We utilized common bacteriological methods, antimicrobial susceptibility evaluation, next-generation sequencing, phylogenomics, and comparative genomics approaches to comprehensively characterize the D. tsuruhatensis MR-6/3H strain.
Results
Bacterium isolation, cultural characteristics, and identification
Delftia tsuruhatensis MR-6/3H was isolated in February 2022 from raw bovine milk, from a retail seller in the territory of the Nizhny Novgorod region, Russia. On 5% human O-blood agar, MR-6/3H formed punctiform, circular, whitish colonies with a diameter of 0.5–2 mm and an entire margin and convex elevation (Figure 1). MR-6/3H exhibited Gamma-hemolysis on human O-group erythrocytes. On the tryptone soya agar (TSA), MR-6/3H exhibited the same colony morphology as on blood agar but had opaque colonies. The strain was positive for oxidase and catalase. Microscopic analysis revealed a typical Gram-negative, rod-shaped bacterium.
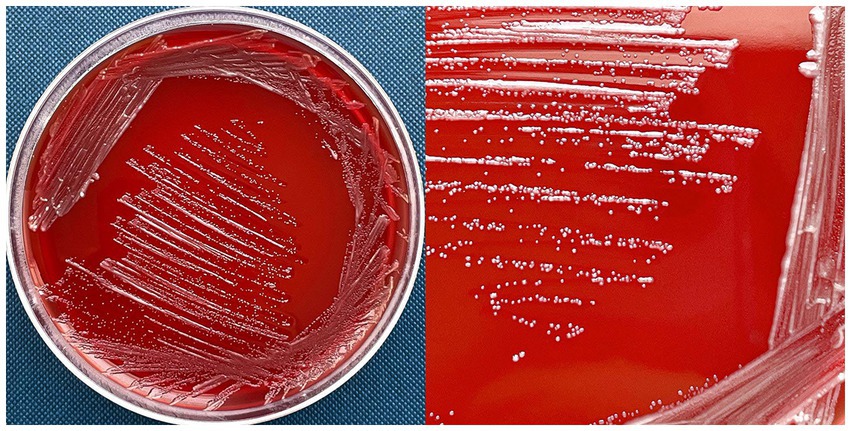
Figure 1. D. tsuruhatensis MR-6/3H strain on 5% human O-blood agar. MR-6/3H exhibited punctiform, circular, whitish colonies with a diameter of 0.5–2 mm andan entire margin and convex elevation; MR-6/3H had a gamma-hemolysis activity (absence of visible hemolysis).
We used sequencing of the 16S rRNA gene for the isolate identification. The assembled consensus of the 16S rRNA gene was 1,389 bp in length with an identity percentage of 100% with sequences of both species: D. tsuruhatensis NBRC 16741 (T) (GenBank accession no. BCTO01000107) and D. lacustris LMG 24775 (T) (GenBank accession no. jgi.1102360). MR-6/3H also had 99.5% of sequence identity with D. acidovorans 2,167 (T). Further genomic approaches were used to clarify the species identification.
Antimicrobial susceptibility profile of Delftia MR-6/3H isolate
Antimicrobial susceptibility testing was performed via the disk-diffusion method on 23 antimicrobials based on the european committee on antimicrobial susceptibility testing guidance (EUCAST v 12.02022). Delftia MR-6/3H was resistant to 19 antimicrobial drugs widely used in clinical practice including all aminoglycosides tested (gentamicin, amikacin, tobramycin, kanamycin, and neomycin), chloramphenicol, trimethoprim-sulfamethoxazole, and almost all β-lactams (ampicillin, amoxicillin-clavulanic acid, aztreonam, meropenem, imipenem, ceftobiprole, cefotaxime, cefepime, piperacillin, piperacillin-tazobactam, ticarcillin, and ticarcillin-clavulanic acid). The Delftia MR-6/3H strain was susceptible to ceftazidime, tetracycline, and both fluoroquinolones: ciprofloxacin, and levofloxacin. Such phenotype was considered multidrug-resistant (MDR). The susceptibility profile with zones of growth inhibition diameters and antibiotics tested are shown in Table 1.
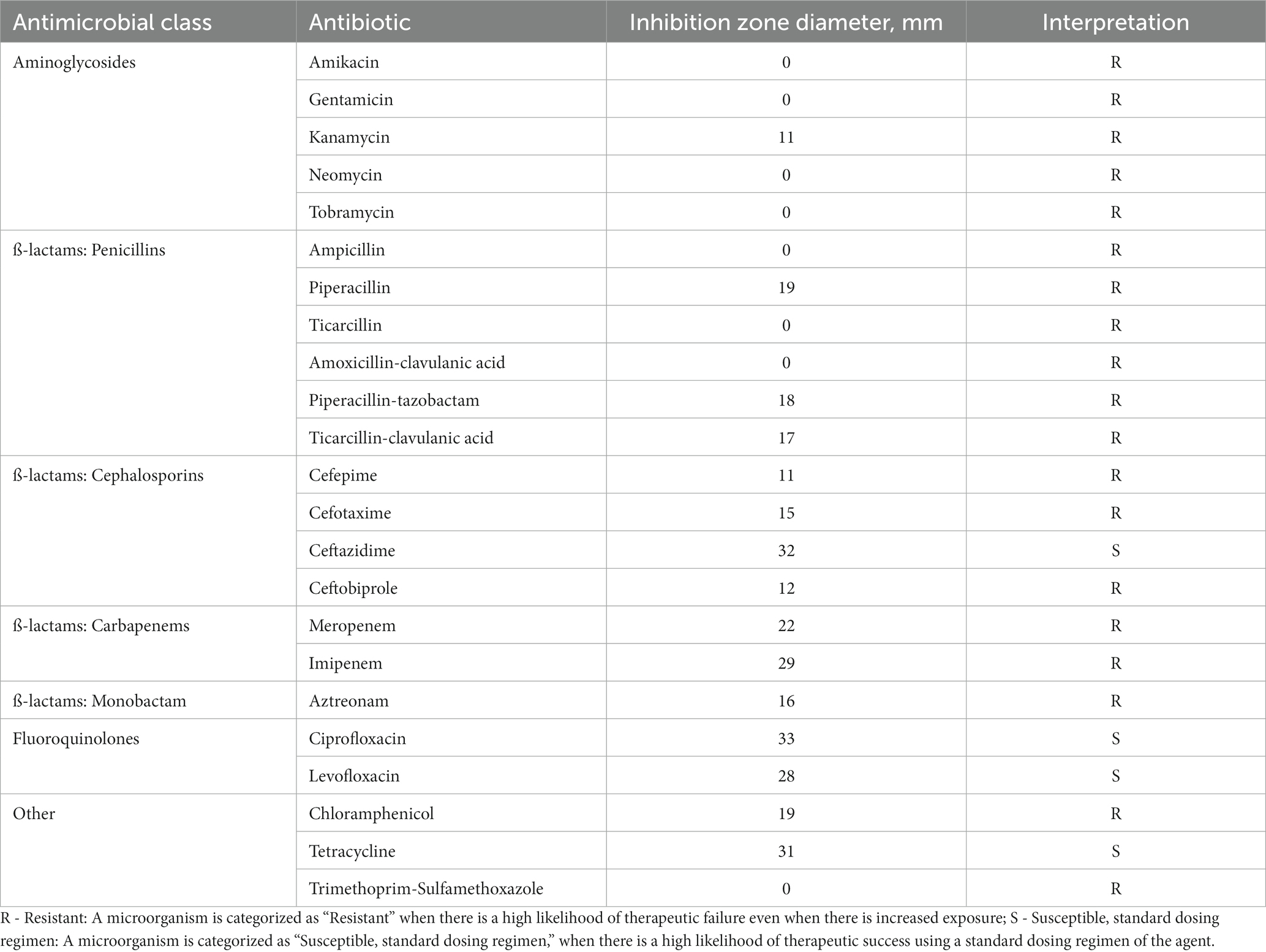
Table 1. List of antibiotics tested, inhibition zone diameter, and susceptibility profile.
Next-generation sequencing, assembly, and annotation
Assembly statistics are shown in Table 2. The assembly of the Delftia MR-6/3H genome contained 52 contigs with a total length of 6,468,367 bp (~6,4 Mb) and a GC content of 66.69%. The average read coverage was 315. In total 6,115 Open reading frames (ORFs) were detected by Rapid Annotation using Subsystem Technology (RAST), including 71 tRNA and 3 rRNA genes, 4,619 proteins with functional assignments, and 1,496 hypothetical proteins.
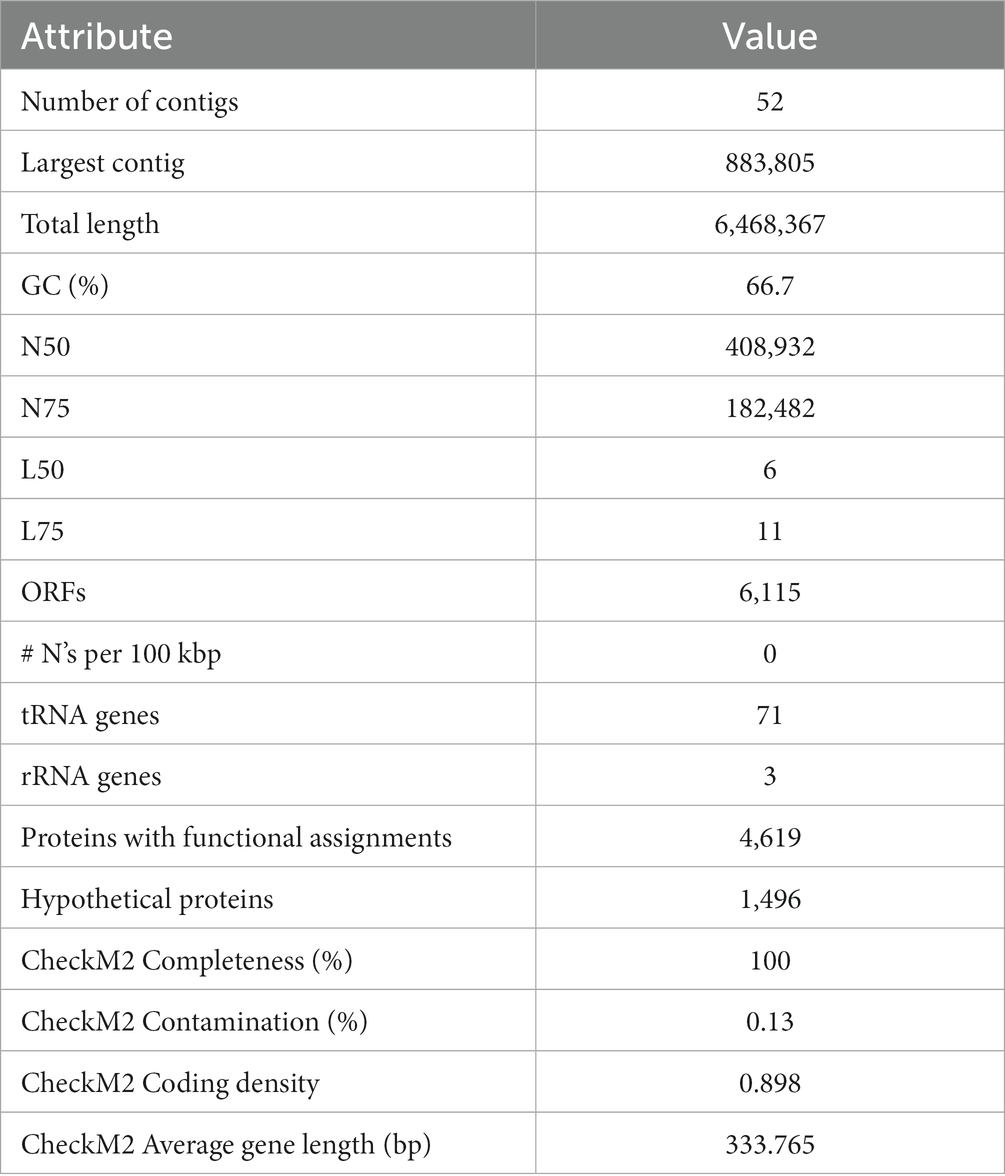
Table 2. General features and quality control of Delftia MR-6/3H genome assembly.
Phylogenomic analysis
As the 16S rRNA gene sequencing method is unable to delineate between D. tsuruhatensis and D. lacustris species, gold standard approaches based on next-generation sequencing both Average Nucleotide Identity (ANI) and Type Strain Genome Server (TYGS) were used for further identification.
The final cladograms are shown in Figure 2. The Delftia MR-6/3H strain clustered together with D. tsuruhatensis as a separate lineage (A clade) from D. acidovorans species clade (B clade). This topology was observed in both cladograms; it suggests that Delftia MR-6/3H (labeled with a pink circle) belongs to the D. tsuruhatensis species. Notably, being different species, D. lacustris and D. tsuruhatensis formed a single clade (A clade). Additionally, there was a putative subspecies clade inside the D. acidovorans cluster (C clade). These observations were supported by ANI matrix data (Figure 3) where ANI ranged between members of mentioned clades: 98–100% for the A clade and 99–100% for the C clade. The members of the latter clade had only 96% with other D. acidovorans strains and 99–100% between each other.
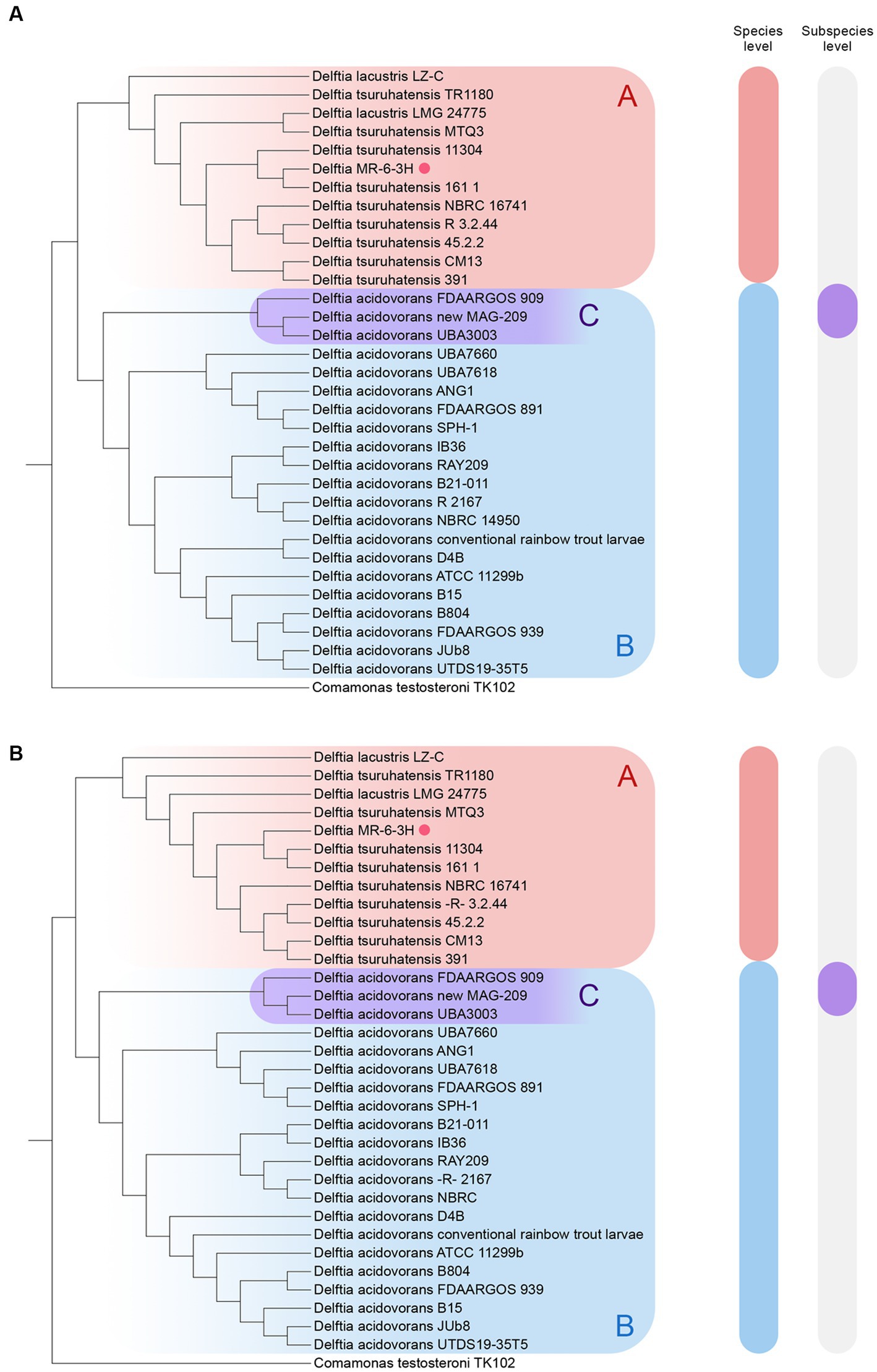
Figure 2. Reconstruction of the whole-genome cladograms of 33 Delftia spp. with Delftia MR-6/3H strain (labeled with pink circle): (A) TYGS cladogram. (B) ANI cladogram. A clade (Red-labeled) - D. tsuruhatensis/lacustris; B clade (blue-labeled) - D. acidovorans, C clade (violet-labeled) - putative D. acidovorans subspecies. The following type strains were used: D. acidovorans 2167 (T), D. lacustris LMG 24775 (T), and D. tsuruhatensis NBRC 16741 (T). Comamonas testosteroni TK102 was utilized as an outgroup.
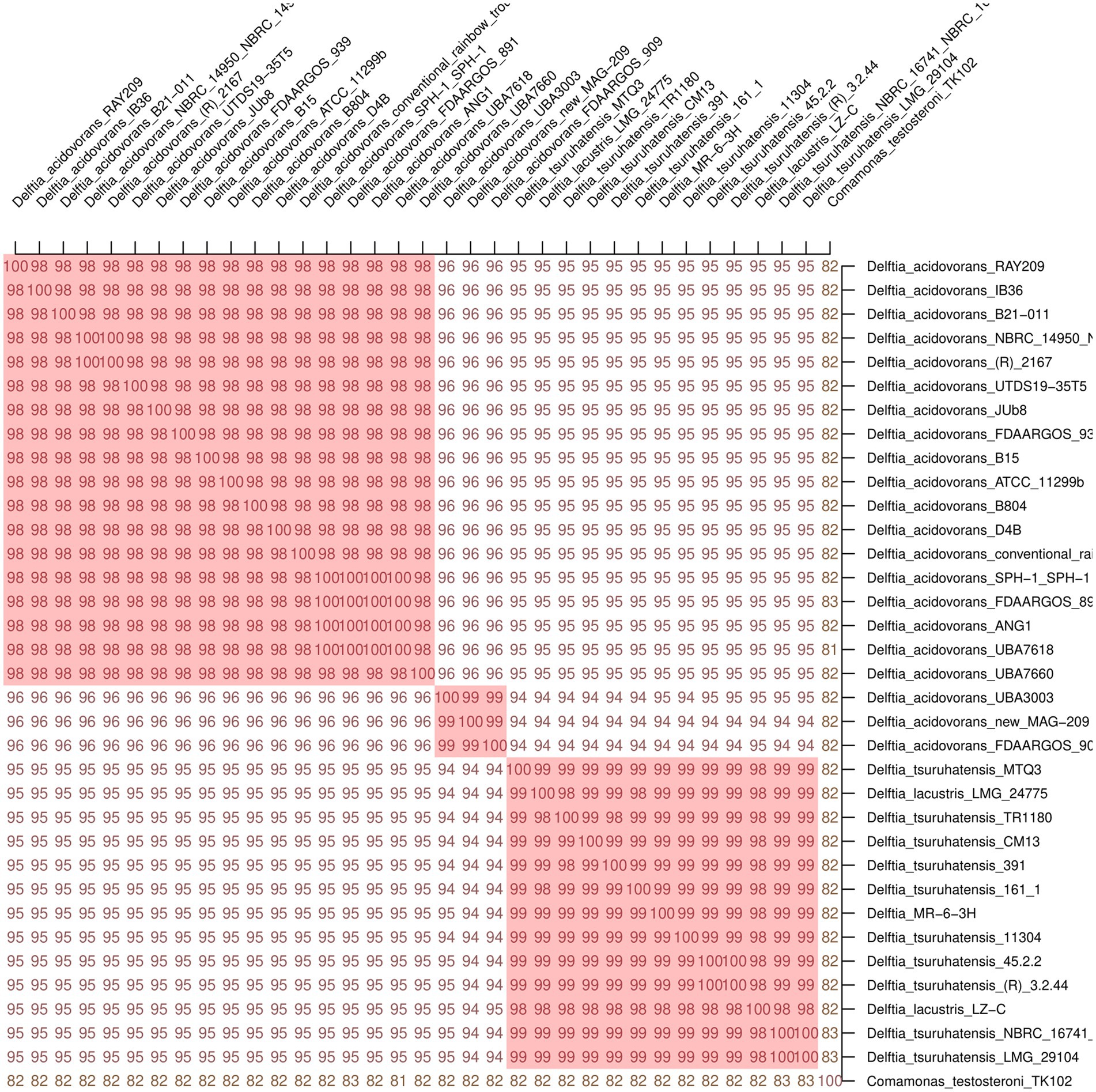
Figure 3. Average Nucleotide Identity (ANI) matrix of 33 Delftia spp. with Delftia MR-6/3H strain. As a cutoff value for species delineation >95% ANI was used. Delftia MR-6/3H strain formed a common cluster on the matrix with D. tsuruhatensis-D. lacustris species. Clusters with ANI ≥ 98 are marked in red: D. acidovorans cluster (on the top), putative D. acidovorans subspecies cluster (in the middle), and D. tsuruhatensis-D. lacustris cluster (at the bottom). Comamonas testosteroni TK102 was used as an outgroup.
The members of the A clade (D. tsuruhatensis/lacustris), to which Delftia MR-6/3H belonged, were chosen for further investigation. To clarify the sophisticated phylogenetic relationships among Delftia spp. strains and Delftia MR-6/3H strain, the REALPHY web tool was utilized (Figure 4). In general, the REALPHY cladogram was almost identical to the ANI and TYGS ones. Delftia MR-6/3H formed a separate clade with D. tsuruhatensis 161/1 and D. tsuruhatensis 11304 strains. Interestingly, both of them have a clinical origin. Strain D. tsuruhatensis 161/1 was isolated from a human host in Samara, Russia in February 2020. Strain D. tsuruhatensis 11304 has a “missing” particular source (but has a clinical origin) and was isolated in Serbia in March 2019. The mentioned clade formed a common phylogeny cluster with D. tsuruhatensis NBRC 16741 (T) type strain. Strains with animal origin (3.2.44, 45.2.2, and CM13) clustered together with D. tsuruhatensis 391, which was isolated from a human host. Both of the abovementioned clades (Delftia MR-6/3H with D. tsuruhatensis 161/1 and D. tsuruhatensis 11304, D. tsuruhatensis NBRC 16741 (T) and strains with animal origin and D. tsuruhatensis 391) formed a single sister group.
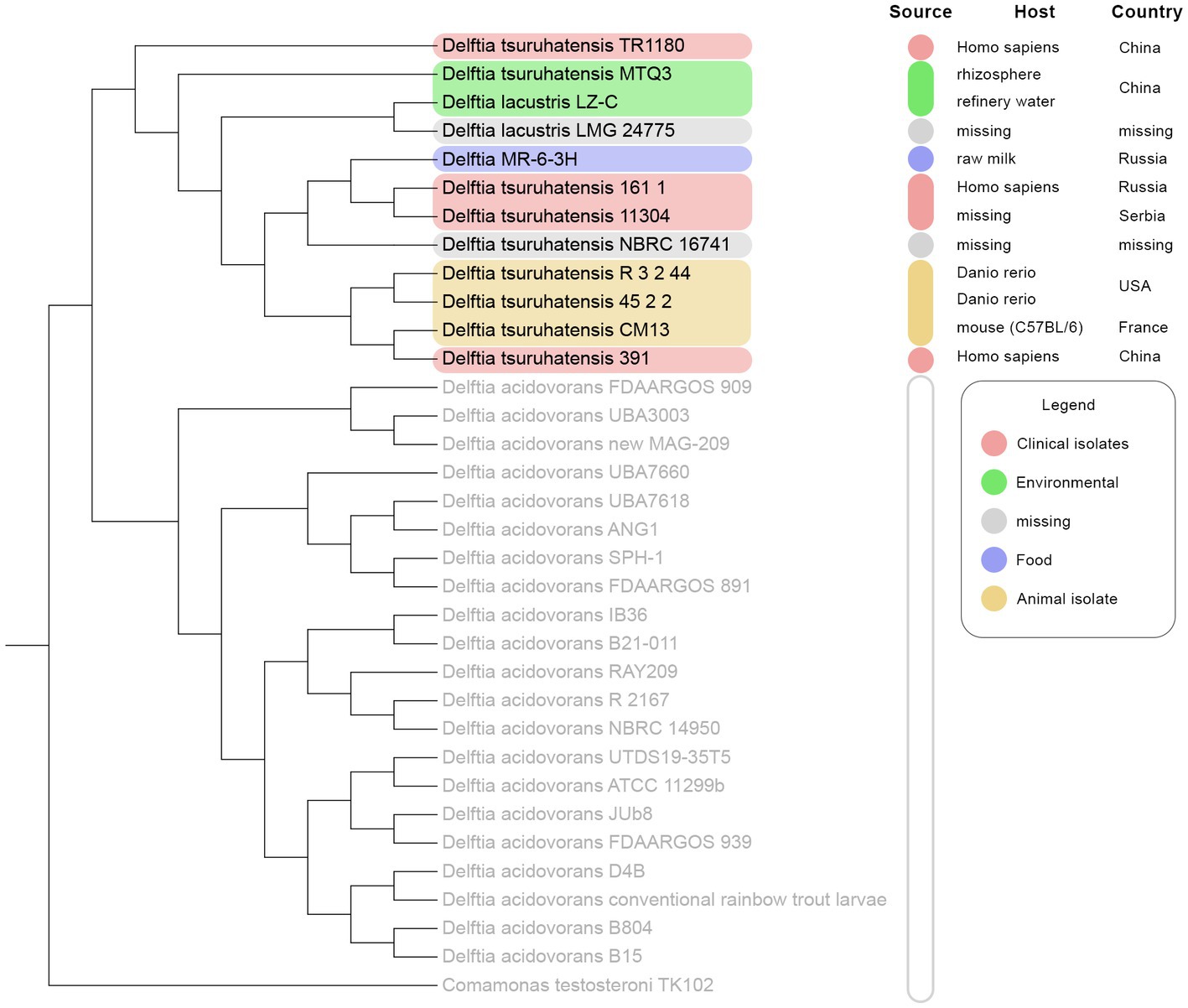
Figure 4. REALPHY-based phylogenomic analysis of 33 Delftia spp. strains: D. tsuruhatensis/D. lacustris clade is highlighted in colors depending on the source type. D. acidovorans 2167 (T), D. lacustris LMG 24775 (T), and D. tsuruhatensis NBRC 16741 (T) were used as reference genomes.
Comparative resistome analysis
Next-generation sequencing data were used to find putative antimicrobial resistance factors in 11 strains of Delftia spp., including the Delftia MR-6/3H strain (Supplementary Table S1). Antimicrobial resistance (AMR) gene homologs prediction was performed by BLASTp searching against the CARD database.
Among all 11 strains, 31 AMR gene homologs were predicted. Individually, each strain possessed from 11 to 17 AMR genes. All detected homologs were grouped into five subgroups regarding their resistance mechanism: efflux pumps, β-lactamases, aminoglycoside inactivation enzymes, diaminopyrimidine resistance, and others. The following factors were observed in all genomes tested, including Delftia MR-6/3H: efflux pumps (ceoB, mexD, paer_soxR, adeB, mdtC, oprJ, and abeS) and OXA-926-like β-lactamase. These genes confer resistance to a wide range of antimicrobials as well as disinfectants, for instance: penams, cephalosporins carbapenems, aminoglycosides, fluoroquinolones, phenols, diaminopyrimidine antibiotics, tetracycline, aminocoumarin, macrolide antibiotics, acriflavine antiseptic, and various disinfecting agents. In addition, the Delftia MR-6/3H strain harbored three additional efflux pump genes that were missing in some other strains: oqxB, muxB, and mexC. The latter is known to be involved in the extrusion of toxic substrates, including a wide range of antibacterials, but additionally, they can pump monobactams as well as nitrofuran compounds outside the cell.
Some Delftia spp. strains harbored unique AMR factors with high amino acid identity percentages that were not observed among other strains. For example, D. tsuruhatensis 391 possessed sul1, which confers resistance to sulfonamide antibiotics. D. tsuruhatensis TR1180 possessed also sul1, but additionally dfrA16 (trimethoprim resistance), aadA3 (aminoglycoside resistance), cmlA9 (phenicol resistance), and tet (G) (tetracycline resistance). D. lacustris LZ-C harbored cmx (phenicol resistance), and aph (3″)-Ib, aph (6)-Id genes (aminoglycoside resistance).
Comparative analysis of putative virulence genes
In total, 47 homologs of virulence factors (VF) were predicted among 11 Delftia spp. strains using BLASTp search against the Virulence Factor Database (VFDB) (Supplementary Table S2). Each strain possessed from 24 to 31 putative VF genes. All observed factors formed eight groups based on their virulence category: motility, adherence, immune modulation, stress survival, secretion system, siderophore, fitness, and others. Four motility-associated factors were detected in all 11 Delftia spp. strains (cheY, fliP, fliI, and fliN); these factors are responsible for flagellar biosynthesis, assembly, and function, which is consistent with the motility phenotype of Delftia species. The adherence group contained four genes common for all strains: pilT, pilT2, pilG (twitching motility proteins), and htpB (heat shock protein). Notably, three genes from the immune modulation category were observed among strains that had only clinical and animal (45.2.2 and CM13) origin, and were detected in the MR-6/3H isolate as well: gmd (GDP-mannose 4,6-dehydratase), wzt (O-antigen export system ATP-binding protein), and per (perosamine synthetase). Stress survival factors such as katA (catalase), sodB (superoxide dismutase), and clpP (ATP-dependent Clp protease) were also detected in all strains. Some genes related to the type IV secretion system were found among all genomes: STM0274 (EvpB family type VI secretion protein), vipA (tubule-forming protein), and clpV1 (type VI secretion system AAA+ family ATPase). Genes coding for siderophore transport were also predicted, among them two permease proteins (bauD and bauC), periplasmic siderophore-binding protein (bauB), and ferrienterobactin ABC transporter ATPase (fepC). Full sets of the abovementioned siderophore genes were found only in the following Delftia strains: MR-6-3H, 11304, 16741 (T), MTQ3, and LZ-C. Two fitness factors were identified only in the D. tsuruhatensis 391 strain, which was isolated from a human patient: mtrD (multiple transferable resistance system protein MtrD) and acrB (acriflavine resistance protein B). Additionally, two factors, namely, adeG (cation/multidrug efflux pump), which is related to biofilm formation, and pvdF (pyoverdine synthetase F), were observed among all Delftia spp. strains.
Compared to the strains with clinical origin (161/1, 11304, 391, and TR1180) the MR-6/3H strain was the closest to the 161/1 and TR1180 strains based on the VF gene set. Moreover, our strain harbored the bauB gene that was absent in the two abovementioned strains. The bauB gene codes for an additional periplasmic siderophore-binding protein (a part of a ferric siderophore ABC transporter). There were also three genes from the immune modulation category observed among strains that had only clinical and animal (45.2.2 and CM13) origin, but were present in the MR-6/3H isolate as well: gmd (GDP-mannose 4,6-dehydratase), wzt (O-antigen export system ATP-binding protein), and per (perosamine synthetase).
Integron possessing analysis
Integrons are known as mobile genetic elements that contribute greatly to antimicrobial resistance dissemination. Integrons and their elements were predicted via IntegronFinder 2.0 among 11 Delftia spp. genomes (Supplementary Table S3). A complete integron sequence was detected in D. tsuruhatensis TR1180 only, which was isolated from a human patient. It consisted of an integrase gene (int), two I class promoters: Pint_1 (integrase promoter) and Pc_1 (cassette promoter), one attI and attC sites, and carried two gene cassettes (Figure S1a). CARD search showed that the first of them (NZ_CP045291.1_3624) is dihydrofolate reductase gene dfrA16 and the second one (NZ_CP045291.1_3624) is aminoglycoside nucleotidyltransferase gene aadA3.
In the genome of D. tsuruhatensis 161/1, the CALIN (clusters of attC sites lacking integron-integrases) was detected. The latter was composed of three gene cassettes and two attC sites (Supplementary Figure S1). All of these proteins were annotated as unknown proteins (or hypothetical proteins) using BLASTp search against the GenBank protein database. We used HMMER search against reference proteomes as a more sensitive algorithm, especially in a homology search for unknown proteins, to identify their homology. Notably, HMMER search showed that one of the aforementioned proteins (NZ_JAKZJM010000052.1_2) matched with an unknown protein from the genome of the eukaryotic organism (fish) Oreochromis niloticus with an E-value of 5.5e-08. Gene Ontology (GO) annotation of this protein following the Unipro database showed that it is a putative cytoplasmic protein NLRC5 with an innate immune response function. The second protein (NZ_JAKZJM010000052.1_3) matched with an unknown protein from Microvirga aerophila with an E-value of 1.0e-16. The GO annotation for this protein was calcium-binding membrane protein with homophilic cell adhesion activity. The third one (NZ_JAKZJM010000052.1_4) matched with an unknown protein from Sunxiuqinia dokdonensis that had an e-value of 1.9e-37. The GO annotation for this protein was the ABC transporter protein.
In the chromosome of D. lacustris LZ-C, the CALIN was also found. It contained two attC and one gene cassette. This protein was matched with an unknown phage protein from Pseudoalteromonas luteoviolacea DSM 6061.
Prophage and CRISPR-Cas search in the Delftia MR-6/3H genome
In the Delftia MR-6/3H genome, five prophage regions were identified. Among them, two had an intact level (scores of 150 and 100), one had a questionable level (score of 90), and two had an incomplete level (scores of 40 and 50). Two intact prophages had 46.5 Kb genome length with GC 64.82%, which had the closest match with Burkholderia phage BcepMigl, and 38 Kb genome length with GC 64.31%, which had the closest match with Pseudomonas phage JBD18. The BcepMigl-like prophage contained 32 CDS, among them integrase, portal proteins, fiber proteins, head proteins, and terminase as well as some phage-like proteins and hypothetical proteins that were annotated. The JBD18-like prophage contained 44 CDC, among them head proteins, tail proteins, phage-like proteins, and hypothetical proteins, and the absence of integrase was observed. Other prophages also contained different virion proteins and hypothetical proteins, but integrases were absent. CRISPR-Cas systems were absent in the MR-6/3H strain.
Discussion
Delftia tsuruhatensis is known as a bacterium that can promote the growth of plants as well as biodegrade some organic pollutants in the environment, but also as an emergent opportunistic pathogen with natural resistance to some antimicrobials. In the present study, we report the case of multidrug-resistant D. tsuruhatensis isolation from unpasteurized bovine milk, collected in Russia. Classical microbiology methods, next-generation sequencing, and analysis approaches were applied to characterize our isolate.
Previously, strains of D. tsuruhatensis species were isolated from a wide range of sources such as active sludge (Shigematsu et al., 2003; Li et al., 2020), seawater (Juárez-Jiménez et al., 2010), soil (Zheng et al., 2007), rhizosphere (Guo et al., 2016), rice husk and swine manure (Chen et al., 2022), fish (Danio rerio and Oreochromis niloticus) (Preena et al., 2020; Chaires et al., 2022), mice (inbred strain C57BL/6) (Saffarian et al., 2016), and from human patients (Preiswerk et al., 2011; Tabak et al., 2013; Ranc et al., 2018; Cheng et al., 2021; Cho et al., 2021). Notably, the isolation of five D. tsuruhatensis strains from raw milk was also reported by Elionora Hantsis-Zacharov and Malka Halpern in 2007 in Israel (Accession no. EF204212 to-16) (Hantsis-Zacharov and Halpern, 2007). This indicates the ubiquitous nature of this bacterium. Recent phylogenomic investigations of the genus Delftia revealed that the members of the latter encode plenty of proteins (have open pan-genome) that allow them to succeed in many ecological niches (Bhat et al., 2022; Yin et al., 2022). However, information about microbiological and genomic analysis of D. tsuruhatensis strains isolated from food and raw milk is absent or limited. In our study, we cannot determine the certain source of the MR-6/3H isolate, because unpasteurized milk samples can be contaminated through various sources from the dairy cow itself to the hands of sellers. Yet, in the previous research work where five isolates of D. tsuruhatensis were isolated from raw milk, it was reported that milking was performed using modern automated milking facilities, and milk samples were taken under sterile conditions (Hantsis-Zacharov and Halpern, 2007). It reduces the internal contamination likelihood and may indicate that raw milk can be a source of D. tsuruhatensis. However, more investigations are needed to elucidate possible sources of this bacterium.
The 16S rRNA gene sequencing approach is recommended for the majority of bacterial species identification and delineation (Stackebrandt et al., 2002). However, we faced some ambiguities during identification via this method: strain MR-6/3H had the same identity percentage with both D. tsuruhatensis and D. lacustris. Similar observations were reported by investigators who dealt with Delftia identification, where 16S rRNA gene sequencing could not delineate between D. tsuruhatensis and D. lacustris (Preiswerk et al., 2011; Cheng et al., 2021). In our case, the abovementioned issue was resolved through ANI and TYGS, which are both gold-standard methods based on next-generation sequencing. Interestingly, in compliance with our analysis, D. tsuruhatensis and D. lacustris species formed a common clade. This observation is also supported by values of the ANI matrix as well as REALPHY phylogeny. The same findings were recently reported by Bhat et al. (2022). Our findings indicate that both D. tsuruhatensis and D. lacustris belong to the same species. This taxonomic confusion could arise because the aforementioned species were initially described based on 16S rRNA phylogeny, which cannot delineate species of the Delftia genus, and conventional DNA–DNA hybridization (DDH), which is error-prone and a significantly inaccurate method compared to in silico WGS approaches such as ANI and TYGS (Goris et al., 2007; Meier-Kolthoff and Göker, 2019). Similarly, we observed a subclade made up of three strains in the D. acidovorans species cluster in all three cladograms. Presumably, these strains can belong to the subspecies of D. acidovorans. The same observation was also reported recently (Bhat et al., 2022). We suggest that some species of the Delftia genus require taxonomic revision by the use of a polyphasic taxonomy approach. It is worth noting that the WGS approach is most effective when used in conjunction with other morphological, ecological, and molecular biology data.
Delftia tsuruhatensis is known as an emergent human opportunistic pathogen. To date, infections caused by this bacterium are known only in immunocompromised persons and newborns. D. tsuruhatensis can cause severe life-threatening infections, manifested primarily as pneumonia and catheter-related bloodstream infections (Preiswerk et al., 2011; Tabak et al., 2013; Ranc et al., 2018; Cheng et al., 2021; Cho et al., 2021). Interestingly, our D. tsuruhatensis MR-6/3H strain collected from raw milk was phylogenetically close to strains with clinical origin (D. tsuruhatensis 161/1 and D. tsuruhatensis 11304), and they formed a single sister group. Notably, D. tsuruhatensis 161/1 was isolated in Samara, Russia, in early 2020 from a human patient. D. tsuruhatensis 11304 strain was isolated in 2014 in Serbia, but a particular source was missing. Comparative analysis of putative virulence genes revealed a similar set of genes detected among the abovementioned strains. The genome of the MR-6/3H strain harbored genes that are related to flagella machinery function (cheY, tufA, flgG, fliI, and fliN), which well correlates with the motile phenotype of Delftia spp. Flagella is known as an important virulence factor, which serves for both motility and adhesion in such pathogens as Pseudomonas aeruginosa and Escherichia coli (Haiko and Westerlund-Wikström, 2013). Another group of genes (tufA, pilT, htpB, pilT2, and pilG) coding for adherence factors, including elongation factor Tu, twitching motility proteins (TMPs), and heat shock protein HtpB, were also predicted. For instance, TMPs were shown to be the major virulence factor of P. aeruginosa, which are involved not only in motility but also in invasion in a multilayer epithelial cell barrier (Alarcon et al., 2009). Lipopolysaccharide (LPS), also known as endotoxin, is a component of the outer membrane of gram-negative bacteria that contributes greatly to virulence due to its immunomodulatory and pro-inflammatory properties, including septic shock (Aurell and Wistrom, 1998). Three genes of LPS synthesis and excretion were detected in the D. tsuruhatensis MR-6/3H strain: gmd, wzt, and per. Other types of putative virulence genes were also observed in the MR-6/3H genome coding for type IV secretion system machinery, stress survival factors (catalase, superoxide dismutase, and urease), siderophores, etc. This indicates that MR-6/3H possesses a particular set of VFs that can confer virulence properties comparable to those of clinical origin strains: D. tsuruhatensis 161/1 and D. tsuruhatensis 11304. Notably, a similar set of putative virulence genes was detected in D. tsuruhatensis 11304 (Cheng et al., 2021). The genes mentioned above, coding for LPS synthesis and excretion, were observed only among strains with clinical or animal origin. Besides the immunomodulation effect that LPS has, the crucial role of this factor in animal and human colonization was also noted for such pathogens as Aeromonas hydrophila (Merino et al., 1996), Salmonella Typhimurium (Licht et al., 1996), Helicobacter pylori, and P. aeruginosa (Maldonado et al., 2016). Probably, an association of such LPS genes transports with animal and human origins of some strains is not accidental. Nonetheless, predicted virulence factors were detected using the protein homology model at the genotype level and need to be further experimentally investigated to show their contribution to the virulence of D. tsuruhatensis.
Antimicrobial resistance (AMR) is a crucial global health concern. A wide number of pathogens can acquire and exchange AMR genes, which leads to antimicrobial therapy failure and consequently to an increase in mortality (Murray et al., 2022). Antimicrobial resistance profile, particularly the multidrug-resistant (MDR) profile of clinical D. tsuruhatensis strains, was reported several times. In 2011, Preiswerk et al. reported the first case of D. tsuruhatensis infection, in which a blood-collected strain was resistant to ampicillin, I and II generation cephalosporins, all tested aminoglycosides, and colistin, but was susceptible to amoxicillin-clavulanate, piperacillin-tazobactam, III generation cephalosporins, fluoroquinolones, and carbapenems (Preiswerk et al., 2011). In another case reported in 2021, sputum specimen isolate TR1180 was resistant to β-lactams (ampicillin and cefazolin), all tested aminoglycosides, tetracycline, and trimethoprim-sulfamethoxazole based on MIC evaluation (Cheng et al., 2021). Both the abovementioned AMR profiles are quite similar. Our MR-6/3H strain had almost the same AMR phenotype, including resistance to almost all β-lactams, all aminoglycosides, chloramphenicol, and trimethoprim-sulfamethoxazole, but had susceptibility to ceftazidime, tetracycline, ciprofloxacin, and levofloxacin. In general, the AMR phenotype of the MR-6/3H strain correlated with the observed resistome, except for tetracycline and fluoroquinolones. Such discrepancy may be explained by impairments in some efflux systems encoded, misregulation of these genes, as well as their reduced expression. Alternatively, resistance to fluoroquinolones is usually related to point mutation in the gyrA gene (DNA gyrase) and with efflux to a lesser extent (Jacoby, 2005). Resistome analysis also revealed a resemblance between putative resistance factors, which were primarily made up of efflux genes, among different Delftia strains. Efflux pumps are capable of extruding a wide range of toxic compounds, including antibiotics. In 2021, Cong Cheng et al. reported similar resistance factors, including the variety of efflux pumps, when analyzing the multidrug-resistant D. tsuruhatensis TR1180 strain (mexC, mexD, oprJ, muxB, oqxB, adeB, adeS, mdtC, etc.) (Cheng et al., 2021). Apparently, the main intrinsic resistance mechanism of Delftia species is related to the regulation of membrane permeability and efflux systems, as is typical for P. aeruginosa, which uses multidrug efflux (Mex) systems and porins mainly (Pang et al., 2019). Additionally, we found the chromosome-encoded genus-specific β-lactamase, which was close to Klebsiella OXA-926 with an amino acid sequence identity of 70–73%. The latter was identified in 2021 from Klebsiella pneumoniae ST29 (Liu et al., 2021). OXA belongs to the class D β-lactamases, which have various substrate specificities against β-lactams but also can be mobilized by plasmids (Sawa et al., 2020). OXA-926 was reported to confer reduced susceptibility to piperacillin, piperacillin-tazobactam, and cephalothin as well as partial resistance to avibactam. The particular substrate specificity of Delftia-specific OXA β-lactamase is vague, but the fact of its existence itself raises concerns about its mobilization probability and further evolution in clinical D. tsuruhatensis strains. Nonetheless, the observed homologs of AMR genes were detected using genomics, and further experiments are needed to determine which specific genetic determinants are responsible for multidrug resistance in the D. tsuruhatensis strain MR-6/3H. Besides intrinsic genes, we also found acquired factors encoded by integrons in the D. tsuruhatensis TR1180 strain isolated from a human patient. TR1180 possessed dfrA16 and aadA3, conferring resistance to trimethoprim and aminoglycosides, respectively. Both of them were the cassettes inside the complete integron sequence. These findings were consistent with the previously reported genomic investigation of TR1180 (Cheng et al., 2021). Integron class 3 possession was also reported in the D. tsuruhatensis A90 strain in 2007 (Xu et al., 2007). In 2021, Sun-Mi Cho et al. detected the integron class 1 in a clinical isolate of D. tsuruhatensis, which carried a blaIMP-1 gene with IMP-1 metallo-β-Lactamase production as well as some other integron-related AMR genes (Cho et al., 2021). It indicates the horizontal gene transfer (HGT) between Delftia spp. and other bacterial taxons. Therefore, D. tsuruhatensis can acquire mobile elements with AMR genes, which can cause the selection of resistant clones and rapid evolution of this bacterium, especially in the clinical environment. Moreover, one of the cassettes inside CALIN in the D. tsuruhatensis 161/1 strain matched with Oreochromis niloticus (fish) protein with an innate immune response function. Interestingly, there were also two cases of D. tsuruhatensis isolation from the abovementioned fish species, including isolation from an injured eye (Soto-Rodriguez et al., 2013; Preena et al., 2020). Such distant homology may indicate the ancient HGT evolutionary event between Oreochromis niloticus and D. tsuruhatensis. After transfer, this protein could be further used by bacteria to establish as a pathogen or symbiont in fish, depending on selection. To date, fewer cross-domain HGT (eukaryote-to-bacteria) events have been described (Dunning Hotopp, 2011).
Thus, the D. tsuruhatensis MR-6/3H strain isolated from raw milk and investigated in the present study was phylogenetically close to strains of clinical origin and possessed a common set of putative virulence factors with the latter ones. Raw milk can be the possible source of as well as a potential transmission route for the dissemination of MDR D. tsuruhatensis. However, the particular source of this isolate remains unclear. MR-6/3H also exhibited a multidrug-resistant phenotype, which was also consistent with performed resistome analysis. The carriage of a variety of genes encoding for efflux pumps indicates that the antimicrobial resistance mechanisms of D. tsuruhatensis are based mainly on the efflux of antibiotic molecules. However, considering the restrictions of the NGS approach, further experiments are needed to investigate which specific genes are responsible for antimicrobial resistance as well as for the pathogenicity of D. tsuruhatensis. In general, the Delftia genus requires a taxonomic revision, which was noted in the present as well as in some other studies. Additionally, considering the fact of ubiquity, high ecological plasticity, possession of some putative virulence factors, and intrinsic resistance determinants, D. tsuruhatensis has the great potency to succeed as an emergent nosocomial pathogen. Furthermore, it seems that D. tsuruhatensis readily acquires new genetic material via HGT, which can cause HGT-mediated rapid evolution, again, especially in the clinical environment. Further epidemiological investigation, particularly infection source detection and the development of clinical recommendations for antimicrobial therapy, are urgently required for the appropriate surveillance of D. tsuruhatensis.
Materials and methods
Bacterium isolation and cultural characteristics investigation
MR-6/3H was isolated from an unpasteurized bovine milk sample in the course of studies related to the screening for antibiotic-resistant bacteria in raw bovine milk. The milk sample was obtained from a retail seller in the Nizhny Novgorod district located in the European part of Russia in February 2022. Initially, 50 mL of milk sample was centrifuged at 8,000 rpm for 15 min to concentrate the bacterial biomass. The sediment was diluted in 2.5 mL of sterile phosphate buffer solution pH 7.2 (PBS, Himedia, Mumbai, India) and then aseptically added to 2.5 mL of tryptic soy broth (TSB, Himedia, Mumbai, India) supplemented with antibiotics. The following antibiotics were used in the final concentration: 50 μg/mL ampicillin, 25 μg/mL gentamicin, and 25 μg/mL kanamycin. Ampicillin was chosen as a beta-lactam representative with a comparatively wide antimicrobial spectrum (increased activity against gram-positive and some gram-negative species) (Wade and Benjamin, 2011). Gentamicin (II-aminoglycoside generation) and kanamycin (I-aminoglycoside generation) were used as aminoglycoside representatives to broader the spectrum due to the in vitro synergistic effect with ampicillin (Thieme et al., 2018). Additionally, 50 μg/mL fluconazole in the media was used to inhibit fungi growth. TSB was incubated for 48 h at 37°C in glass tubes without shaking. A loopful of broth was streaked onto tryptic soy agar (TSA, Himedia, Mumbai, India) media supplemented with the concentrations of antimicrobials noted above. The inoculated agar plate was incubated for 24 h at 37°C. The growth culture was further checked for purity through gram staining. Additionally, the OXITEST (Erba Lachema, Brno, Czechia) for oxidase activity detection and the 3% hydrogen peroxide test to identify catalase activity were carried out. To investigate hemolytic activity and cultural characteristics the 5% human O-blood agar was used (a blood sample was aseptically collected from a healthy adult volunteer with his documented consent; all procedures were performed in accordance with relevant guidelines and regulations). The stock culture was stored at-80°C in TSA (Himedia, Mumbai, India) with 15% glycerol.
Isolate identification based on the 16S rRNA gene sequencing
Sequencing of a nearly full-length 16S rRNA gene was implemented for identification. Bacterial DNA was isolated via thermal lysis of cell suspension at 95°C for 20 min (a single colony from an agar plate was used). We performed a polymerase chain reaction with the following universal primers: 27F 5’-AGAGTTTGATCMTGGCTCAG-3′ and 1492R 5’-TACGGYTACCTTGTTACGACTT-3′ (Weisburg et al., 1991). PCR products of ~1,500 bp were detected in 1% agarose gel. Amplicons were extracted from the gel and purified using a DNA extraction kit (Dia-m, Moscow, Russia). Further DNA sequencing was performed using the ABI PRISM BigDye Terminator v. 3.1 reagent kit (PE Applied Biosystems, Foster City, CA) according to the manufacturer’s protocols, followed by an analysis of the reaction products on an automatic sequencer Applied Biosystems 3,730 DNA Analyzer. Obtained forward and reverse sequences were assembled into consensus using UGENE (v41.0) software (Okonechnikov et al., 2012). The EzTaxon server database was utilized to compare the resulting consensus to the sequences of other bacterial 16S rRNA genes (Yoon et al., 2017). To further clarify the strain identity, the average nucleotide identity (ANI) value, as well as DDH in silico, were calculated.
Antimicrobial susceptibility via the Kirby-Bauer disk-diffusion method
The Kirby-Bauer disk-diffusion method (DDM) on Mueller-Hinton agar plates was performed according to the guidelines of EUCAST 2022 (the european committee on antimicrobial susceptibility testing). Breakpoint tables for interpretation of MICs and zone diameters, version 12.0, 2022,1 to assess the susceptibility profile of the MR-6/3H isolate were used. The following antibiotic disks were used: ampicillin (10 μg), amoxicillin-clavulanic acid (20–10 μg), aztreonam (30 μg), meropenem (10 μg), imipenem (10 μg), ceftobiprole (5 μg), cefotaxime (5 μg), cefepime (30 μg), ceftazidime (10 μg), piperacillin (30), piperacillin-tazobactam (100–10 μg), ticarcillin (75 μg), ticarcillin-clavulanic acid (75–10 μg), gentamicin (10 μg), amikacin (30 μg), tobramycin (10 μg), kanamycin (30 μg), neomycin (10 μg), ciprofloxacin (5 μg), levofloxacin (5 μg), chloramphenicol (30 μg), tetracycline (30 μg), and trimethoprim-sulfamethoxazole (1.25–23.75 μg). In total, the susceptibility to 23 antibacterials was tested. Mueller-Hinton media, as well as most antibiotic-containing disks, were obtained from HiMedia (Mumbai, India). Amoxicillin-clavulanate, gentamicin, polymyxin, and trimethoprim-sulfamethoxazole were obtained from NICF (Saint Petersburg, Russia). Aztreonam, piperacillin-tazobactam, ticarcillin, piperacillin, ticarcillin-clavulanate, tobramycin, and amikacin were obtained from Bioanalyse (Ankara, Turkey). Cefotaxime, cefepime, and ceftazidime were obtained from Erba Lachema (Brno, Czech Republic). As members of the Delftia genus are uncommon for clinical practice, EUCAST 2022 has no breakpoints for interpreting the results of DDM. In such cases, EUCAST recommends using the breakpoints of phylogenetically related bacteria. Therefore, we used Burkholderia pseudomallei and Enterobacteriales breakpoints to extend the number of tested antibiotics. Escherichia coli ATCC 25922 was used as a reference strain for quality control.
Next-generation sequencing, assembly, and annotation
Genomic DNA was extracted from overnight MR-6/3H pure culture via the QIAamp DNA Kit (Qiagen, Hilden, Germany). The concentration of DNA was evaluated with the Qubit dsDNA BR Assay Kit on a Qubit 3.0 fluorometer (Fisher Scientific Inc., Waltham, MA, USA). Further Illumina Hiseq 1,500 (Illumina, CA, USA) paired-end (2×150) sequencing of D. tsuruhatensis MR-6/3H genome was performed by Geneanalytics LLC (Moscow, Russia). Raw read libraries were deposited at SRA (accession PRJNA903655).
Read library quality was evaluated using FastQC v0.11.9. Obtained reads were trimmed with Trimmomatic software (v0.4.8) using the automatic mode of adapter removal (Bolger et al., 2014). Unicycler v0.4.8. on the PATRIC resource center2 was utilized to assemble the genome (Brettin et al., 2015; Davis et al., 2020). Contigs less than 1,000 bp in length were removed from the resulting assembly. To assess the assembly quality, QUAST v5.0.2 and CheckM2 v1.0.2 tools were used (Gurevich et al., 2013; Chklovski et al., 2023). Genome annotation was performed at the Rapid Annotations using the Subsystems Technology (RAST) server (Aziz et al., 2008; Overbeek et al., 2014; Brettin et al., 2015) and the Prokaryote Genome Annotation Pipeline. The draft genome sequence has been deposited at GenBank (accession JAPMID000000000).
Phylogenomic analysis
Phylogenomic analysis or phylogenomics in contrast to phylogenetic analysis (based on one or a small number of genes) implies the use the massive genomic data and demonstrates higher accuracy. Phylogenomics can be used to investigate evolutionary relationships between different taxons, accurate species identification, investigation of population structure, and pathogens epidemiology (Kapli et al., 2020).
Phylogenomic analysis of the MR-6/3H strain was performed for accurate species identification and population structure investigation. The list of strains used for phylogenomic analysis is shown in Supplementary Table S4. Strains were selected based on their origin, and members with missing origins were mostly excluded (except for type strains). Comamonas testosteroni TK102 was utilized as an outgroup. Genome-based species identification was carried out using the Average Nucleotide Identity Matrix calculator (ANI-matrix with ≥95% cutoff for species level) developed by Kostas lab, which estimates all-vs-all distances in a collection of genomes and Type (Strain) Genome Server (TYGS) (Rodriguez-R and Konstantinidis, 2016; Meier-Kolthoff and Göker, 2019). Phylogeny investigation was carried out with the Reference sequence Alignment based Phylogeny builder (REALPHY v1.13) with read length parameter 150 (RL = 150), which allows comparison between different species. D. acidovorans 2,167 (T), D. lacustris LMG 24775 (T), and D. tsuruhatensis NBRC 16741 (T) were used as reference genomes. The accuracy of REALPHY is comparable with the single-nucleotide polymorphism (SNP) approach. Moreover, REALPHY can consider more than one reference genome, which improves phylogeny reconstruction (Bertels et al., 2014). All cladograms were visualized and edited in iTOL v5 (Letunic and Bork, 2021).
Comparative genomics
A comparative genomics approach was used to identify and compare putative antimicrobial resistance genes (AMR) and putative virulence factors (VFs). Comparative analysis was performed using the Galaxy Europe web server3 (Afgan et al., 2022). In all, 11 genomes of Delftia spp. strains with different origins, including Delftia MR-6/3H (Supplementary Table S1), were chosen to identify putative antimicrobial resistance genes and putative virulence factors. The strains mentioned above formed the common clade of D. tsuruhatensis and D. lacustris species and, therefore, were chosen for comparative analysis. AMR and VFs were predicted with BLASTp (v2.12.0) search against the Comprehensive Antibiotic Resistance Database (CARD v3.2.5) and virulence factor database set A (VFDB v2022.11.04) (Liu et al., 2019; Alcock et al., 2020). For both searches, the following cutoff parameters were used: e-value ≤1e-30, bitscore >100, query coverage ≥80%, and percentage of identity ≥60. The aforementioned parameters were chosen as recommended by W. Pearson for homology prediction but strengthened to avoid false-positive matching (Pearson, 2013).
Integrons were identified via IntegronFinder 2.0 with automatic parameters (Néron et al., 2022). Version 2.0 is upgraded and adapted to draft genome assemblies. IntegronFinder output consists of three types of integron: complete (include all int elements), In0 (integron-integrases lacking cassettes - include integrase only), and CALIN (clusters of attC sites lacking integron-integrases). Integron regions were visualized in Ugene v41.0 (Okonechnikov et al., 2012). A homology search of hypothetical proteins was performed via BLASTp (Madden et al., 1996) and HMMER web server (based on hidden Markov models (HMMs)) against the Reference Proteomes dataset (Potter et al., 2018).
Prophage and CRISPR-Cas analysis
Bacteriophages or phages are known to be one of the major players contributing to the evolution of bacteria. Analysis of prophage genomes carriage allows us to investigate the evolution history of certain strains and analyze possible adaptive traits introduced by phage incorporation (Fortier and Sekulovic, 2013; Touchon et al., 2016). Prophage genomes and elements were detected using the PHASTER web tool with default parameters (Arndt et al., 2016). PHASTER uses a fasta assembly file as input and performs a BLAST search against a custom prophage/phage database. Detected phage elements were then clustered into prophage regions into three subgroups based on score: intact (score > 90), questionable (score from 70 to 90), and incomplete (score < 70). CRISPR-Cas elements were detected using the CRISPRCasFinder web tool with default parameters (Couvin et al., 2018).
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Ethics statement
The studies involving humans were approved by Ethics committee of Federal Research Center for Virology and Microbiology. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
PA: Conceptualization, Formal analysis, Methodology, Project administration, Supervision, Visualization, Writing – original draft, Writing – review & editing. DK: Investigation, Validation, Writing – review & editing. AM: Investigation, Validation, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1321122/full#supplementary-material
Footnotes
References
Afgan, E., Nekrutenko, A., Grüning, B. A., Blankenberg, D., Goecks, J., Schatz, M. C., et al. (2022). The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2022 update. Nucleic Acids Res. 50, W345–W351. doi: 10.1093/NAR/GKAC247
Alarcon, I., Evans, D. J., and Fleiszig, S. M. J. (2009). The role of twitching motility in Pseudomonas aeruginosa exit from and translocation of corneal epithelial cells. Invest. Ophthalmol. Vis. Sci. 50, 2237–2244. doi: 10.1167/IOVS.08-2785
Alcock, B. P., Raphenya, A. R., Lau, T. T. Y., Tsang, K. K., Bouchard, M., Edalatmand, A., et al. (2020). CARD 2020: antibiotic resistome surveillance with the comprehensive antibiotic resistance database. Nucleic Acids Res. 48, D517–D525. doi: 10.1093/NAR/GKZ935
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/NAR/GKW387
Aurell, C. A., and Wistrom, A. O. (1998). Critical aggregation concentrations of gram-negative bacterial lipopolysaccharides (LPS). Biochem. Biophys. Res. Commun. 253, 119–123. doi: 10.1006/BBRC.1998.9773
Aziz, R. K., Bartels, D., Best, A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bertels, F., Silander, O. K., Pachkov, M., Rainey, P. B., and Van Nimwegen, E. (2014). Automated reconstruction of whole-genome phylogenies from short-sequence reads. Mol. Biol. Evol. 31, 1077–1088. doi: 10.1093/MOLBEV/MSU088
Bhat, S. V., Maughan, H., Cameron, A. D. S., and Yost, C. K. (2022). Phylogenomic analysis of the genus Delftia reveals distinct major lineages with ecological specializations. Microb. Genom. 8:mgen000864. doi: 10.1099/MGEN.0.000864
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/BIOINFORMATICS/BTU170
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/SREP08365
Chaires, A., Thomas, K., Estes, A. M., and Jozwick, A. K. S. (2022). Draft genome sequence of Delftia tsuruhatensis strain 45.2.2, colonizer of zebrafish, Danio rerio, skin mucus. Microbiol. Res. Announc. 11:e0066622. doi: 10.1128/MRA.00666-22
Chen, L. F., Chen, L. X., Pan, D., Ren, Y. L., Zhang, J., Zhou, B., et al. (2022). Ammonium removal characteristics of Delftia tsuruhatensis SDU2 with potential application in ammonium-rich wastewater treatment. Int. J. Environ. Sci. Technol. 20, 3911–3926. doi: 10.1007/S13762-022-04219-3
Cheng, C., Zhou, W., Dong, X., Zhang, P., Zhou, K., Zhou, D., et al. (2021). Genomic analysis of Delftia tsuruhatensis strain TR1180 isolated from a patient from China with In4-like Integron-associated antimicrobial resistance. Front. Cell. Infect. Microbiol. 11:663933. doi: 10.3389/FCIMB.2021.663933
Chklovski, A., Parks, D. H., Woodcroft, B. J., and Tyson, G. W. (2023). CheckM2: a rapid, scalable and accurate tool for assessing microbial genome quality using machine learning. Nat. Methods 20, 1203–1212. doi: 10.1038/s41592-023-01940-w
Cho, S. M., Hong, S. G., Lee, Y., Song, W., Yong, D., Jeong, S. H., et al. (2021). First identification of IMP-1 Metallo-β-lactamase in Delftia tsuruhatensis strain CRS1243 isolated from a clinical specimen. Ann. Lab. Med. 41, 436–438. doi: 10.3343/ALM.2021.41.4.436
Couvin, D., Bernheim, A., Toffano-Nioche, C., Touchon, M., Michalik, J., Néron, B., et al. (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for cas proteins. Nucleic Acids Res. 46, W246–W251. doi: 10.1093/NAR/GKY425
Davis, J. J., Wattam, A. R., Aziz, R. K., Brettin, T., Butler, R., Butler, R. M., et al. (2020). The PATRIC bioinformatics resource center: expanding data and analysis capabilities. Nucleic Acids Res. 48, D606–D612. doi: 10.1093/NAR/GKZ943
Dunning Hotopp, J. C. (2011). Horizontal gene transfer between bacteria and animals. Trends Genet. 27, 157–163. doi: 10.1016/j.tig.2011.01.005
Fortier, L. C., and Sekulovic, O. (2013). Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365. doi: 10.4161/VIRU.24498
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/IJS.0.64483-0
Guo, H., Yang, Y., Liu, K., Xu, W., Gao, J., Duan, H., et al. (2016). Comparative genomic analysis of Delftia tsuruhatensis MTQ3 and the identification of functional NRPS genes for Siderophore production. Biomed. Res. Int. 2016, 1–8. doi: 10.1155/2016/3687619
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/BIOINFORMATICS/BTT086
Haiko, J., and Westerlund-Wikström, B. (2013). The role of the bacterial flagellum in adhesion and virulence. Biology 2, 1242–1267. doi: 10.3390/BIOLOGY2041242
Han, J., Sun, L., Dong, X., Cai, Z., Sun, X., Yang, H., et al. (2005). Characterization of a novel plant growth-promoting bacteria strain Delftia tsuruhatensis HR4 both as a diazotroph and a potential biocontrol agent against various plant pathogens. Syst. Appl. Microbiol. 28, 66–76. doi: 10.1016/J.SYAPM.2004.09.003
Hantsis-Zacharov, E., and Halpern, M. (2007). Culturable psychrotrophic bacterial communities in raw milk and their proteolytic and lipolytic traits. Appl. Environ. Microbiol. 73, 7162–7168. doi: 10.1128/AEM.00866-07
Hou, Q., Wang, C., Guo, H., Xia, Z., Ye, J., Liu, K., et al. (2015). Draft genome sequence of Delftia tsuruhatensis MTQ3, a strain of plant growth-promoting rhizobacterium with antimicrobial activity. Genome Announc. 3:e00822. doi: 10.1128/GENOMEA.00822-15
Jacoby, G. A. (2005). Mechanisms of resistance to quinolones. Clin. Infect. Dis. 41, S120–S126. doi: 10.1086/428052
Juarez Jimenez, B., Reboleiro Rivas, P., Gonzalez Lopez, J., Pesciaroli, C., Barghini, P., and Fenice, M. (2012). Immobilization of Delftia tsuruhatensis in macro-porous cellulose and biodegradation of phenolic compounds in repeated batch process. J. Biotechnol. 157, 148–153. doi: 10.1016/J.JBIOTEC.2011.09.026
Juárez-Jiménez, B., Manzanera, M., Rodelas, B., Martínez-Toledo, M. V., Gonzalez-López, J., Crognale, S., et al. (2010). Metabolic characterization of a strain (BM90) of Delftia tsuruhatensis showing highly diversified capacity to degrade low molecular weight phenols. Biodegradation 21, 475–489. doi: 10.1007/S10532-009-9317-4
Kapli, P., Yang, Z., and Telford, M. J. (2020). Phylogenetic tree building in the genomic age. Nat. Rev. Genet. 21, 428–444. doi: 10.1038/s41576-020-0233-0
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/NAR/GKAB301
Li, H., Liu, H., Zeng, Q., Xu, M., Li, Y., Wang, W., et al. (2020). Isolation and appraisal of a non-fermentative bacterium, Delftia tsuruhatensis, as denitrifying phosphate-accumulating organism and optimal growth conditions. J. Water Process. Eng. 36:101296. doi: 10.1016/J.JWPE.2020.101296
Liang, Q., Takeo, M., Chen, M., Zhang, W., Xu, Y., and Lin, M. (2005). Chromosome-encoded gene cluster for the metabolic pathway that converts aniline to TCA-cycle intermediates in Delftia tsuruhatensis AD9. Microbiology 151, 3435–3446. doi: 10.1099/MIC.0.28137-0
Licht, T. R., Krogfelt, K. A., Cohen, P. S., Poulsen, L. K., Urbance, J., and Molin, S. (1996). Role of lipopolysaccharide in colonization of the mouse intestine by Salmonella typhimurium studied by in situ hybridization. Infect. Immun. 64, 3811–3817. doi: 10.1128/IAI.64.9.3811-3817.1996
Liu, L., Feng, Y., Wei, L., Xiao, Y., and Zong, Z. (2021). KPC-2-producing Carbapenem-resistant Klebsiella pneumoniae of the uncommon ST29 type carrying OXA-926, a novel narrow-Spectrum OXA β-lactamase. Front. Microbiol. 12:701513. doi: 10.3389/FMICB.2021.701513
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2019). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/NAR/GKY1080
Madden, T. L., Tatusov, R. L., and Zhang, J. (1996). Applications of network BLAST server. Methods Enzymol. 266, 131–137. doi: 10.1016/S0076-6879(96)66011-X
Maldonado, R. F., Sá-Correia, I., and Valvano, M. A. (2016). Lipopolysaccharide modification in gram-negative bacteria during chronic infection. FEMS Microbiol. Rev. 40, 480–493. doi: 10.1093/FEMSRE/FUW007
Malešević, M., Di Lorenzo, F., Filipić, B., Stanisavljević, N., Novović, K., Senerovic, L., et al. (2019). Pseudomonas aeruginosa quorum sensing inhibition by clinical isolate Delftia tsuruhatensis 11304: involvement of N-octadecanoylhomoserine lactones. Sci. Rep. 9, 16465–16413. doi: 10.1038/s41598-019-52955-3
Meier-Kolthoff, J. P., and Göker, M. (2019). TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 10, 2182–2110. doi: 10.1038/s41467-019-10210-3
Merino, S., Rubires, X., Aguillar, A., Guillot, J. F., and Tomás, J. M. (1996). The role of the O-antigen lipopolysaccharide on the colonization in vivo of the germfree chicken gut by Aeromonas hydrophila serogroup O:34. Microb. Pathog. 20, 325–333. doi: 10.1006/MPAT.1996.0031
Murray, C. J., Ikuta, K. S., Sharara, F., Swetschinski, L., Robles Aguilar, G., Gray, A., et al. (2022). Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis. Lancet 399, 629–655. doi: 10.1016/S0140-6736(21)02724-0
Néron, B., Littner, E., Haudiquet, M., Perrin, A., Cury, J., and Rocha, E. P. C. (2022). IntegronFinder 2.0: identification and analysis of integrons across bacteria, with a focus on antibiotic resistance in Klebsiella. Microorganisms 10:482270. doi: 10.1101/2022.02.28.482270
Okonechnikov, K., Golosova, O., Fursov, M., Varlamov, A., Vaskin, Y., Efremov, I., et al. (2012). Unipro UGENE: a unified bioinformatics toolkit. Bioinformatics 28, 1166–1167. doi: 10.1093/BIOINFORMATICS/BTS091
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/NAR/GKT1226
Pang, Z., Raudonis, R., Glick, B. R., Lin, T. J., and Cheng, Z. (2019). Antibiotic resistance in Pseudomonas aeruginosa: mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 37, 177–192. doi: 10.1016/J.BIOTECHADV.2018.11.013
Pearson, W. R. (2013). An introduction to sequence similarity (“homology”) searching. Curr. Protoc. Bioinformatics 42, 3.1.1–3.1.8. doi: 10.1002/0471250953.BI0301S42
Potter, S. C., Luciani, A., Eddy, S. R., Park, Y., Lopez, R., and Finn, R. D. (2018). HMMER web server: 2018 update. Nucleic Acids Res. 46, W200–W204. doi: 10.1093/NAR/GKY448
Preena, P. G., Dharmaratnam, A., and Swaminathan, T. R. (2020). Antimicrobial resistance analysis of pathogenic Bacteria isolated from freshwater Nile Tilapia (Oreochromis niloticus) cultured in Kerala, India. Curr. Microbiol. 77, 3278–3287. doi: 10.1007/S00284-020-02158-1
Preiswerk, B., Ullrich, S., Speich, R., Bloemberg, G. V., and Hombach, M. (2011). Human infection with Delftia tsuruhatensis isolated from a central venous catheter. J. Med. Microbiol. 60, 246–248. doi: 10.1099/JMM.0.021238-0
Ranc, A., Dubourg, G., Fournier, P. E., Raoult, D., and Fenollar, F. (2018). Delftia tsuruhatensis, an emergent opportunistic healthcare-associated pathogen. Emerg. Infect. Dis. 24, 594–596. doi: 10.3201/EID2403.160939
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 4:e1900v1. doi: 10.7287/PEERJ.PREPRINTS.1900V1
Saffarian, A., Mulet, C., Tournebize, R., Naito, T., Sansonetti, P. J., and Pédron, T. (2016). Complete genome sequence of Delftia tsuruhatensis CM13 isolated from murine proximal colonic tissue. Genome Announc. 4:e01398-16. doi: 10.1128/GENOMEA.01398-16
Sawa, T., Kooguchi, K., and Moriyama, K. (2020). Molecular diversity of extended-spectrum β-lactamases and carbapenemases, and antimicrobial resistance. J. Intensive Care 8:13. doi: 10.1186/S40560-020-0429-6
Shigematsu, T., Yumihara, K., Ueda, Y., Numaguchi, M., Morimura, S., and Kida, K. (2003). Delftia tsuruhatensis sp. nov., a terephthalate-assimilating bacterium isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 53, 1479–1483. doi: 10.1099/IJS.0.02285-0
Soto-Rodriguez, S. A., Cabanillas-Ramos, J., Alcaraz, U., Gomez-Gil, B., and Romalde, J. L. (2013). Identification and virulence of Aeromonas dhakensis, Pseudomonas mosselii and Microbacterium paraoxydans isolated from Nile tilapia, Oreochromis niloticus, cultivated in Mexico. J. Appl. Microbiol. 115, 654–662. doi: 10.1111/JAM.12280
Stackebrandt, E., Frederiksen, W., Garrity, G. M., Grimont, P. A. D., Kämpfer, P., Maiden, M. C. J., et al. (2002). Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Int. J. Syst. Evol. Microbiol. 52, 1043–1047. doi: 10.1099/00207713-52-3-1043
Tabak, O., Mete, B., Aydin, S., Mandel, N. M., Otlu, B., Ozaras, R., et al. (2013). Port-related Delftia tsuruhatensis bacteremia in a patient with breast cancer. New Microbiol. 36, 199–201.
Tejman-Yarden, N., Robinson, A., Davidov, Y., Shulman, A., Varvak, A., Reyes, F., et al. (2019). Delftibactin-a, a non-ribosomal peptide with broad antimicrobial activity. Front. Microbiol. 10:2377. doi: 10.3389/FMICB.2019.02377
Thieme, L., Klinger-Strobel, M., Hartung, A., Stein, C., Makarewicz, O., and Pletz, M. W. (2018). In vitro synergism and anti-biofilm activity of ampicillin, gentamicin, ceftaroline and ceftriaxone against Enterococcus faecalis. J. Antimicrob. Chemother. 73, 1553–1561. doi: 10.1093/JAC/DKY051
Touchon, M., Bernheim, A., and Rocha, E. P. C. (2016). Genetic and life-history traits associated with the distribution of prophages in bacteria. ISME J. 10, 2744–2754. doi: 10.1038/ismej.2016.47
Wade, K. C., and Benjamin, D. K. (2011). “Clinical pharmacology of anti-infective drugs” in Infectious diseases of the fetus and newborn infant. eds. J. S. Remington, J. O. Klein, C. B. Wilson, V. Nizet, and Y. A. Maldonado (Amsterdam: Elsevier). Available at: https://www.sciencedirect.com/book/9781416064008/infectious-diseases-of-the-fetus-and-newborn
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703. doi: 10.1128/JB.173.2.697-703.1991
Xu, H., Davies, J., and Miao, V. (2007). Molecular characterization of class 3 integrons from Delftia spp. J. Bacteriol. 189, 6276–6283. doi: 10.1128/JB.00348-07
Ye, J. X., Lin, T. H., Hu, J. T., Poudel, R., Cheng, Z. W., Zhang, S. H., et al. (2019). Enhancing chlorobenzene biodegradation by Delftia tsuruhatensis using a water-silicone oil biphasic system. Int. J. Environ. Res. Public Health 16:1629. doi: 10.3390/IJERPH16091629
Yin, Z., Liu, X., Qian, C., Sun, L., Pang, S., Liu, J., et al. (2022). Pan-genome analysis of Delftia tsuruhatensis reveals important traits concerning the genetic diversity, pathogenicity, and biotechnological properties of the species. Microbiol. Spectr. 10:e0207221. doi: 10.1128/SPECTRUM.02072-21
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/IJSEM.0.001755
Zheng, R. C., Wang, Y. S., Liu, Z. Q., Xing, L. Y., Zheng, Y. G., and Shen, Y. C. (2007). Isolation and characterization of Delftia tsuruhatensis ZJB-05174, capable of R-enantioselective degradation of 2,2-dimethylcyclopropanecarboxamide. Res. Microbiol. 158, 258–264. doi: 10.1016/J.RESMIC.2006.12.007
Keywords: Delftia tsuruhatensis, antimicrobial resistance (AMR), genomics, raw milk, phylogenomic analysis
Citation: Andriyanov PA, Kashina DD and Menshikova AN (2024) Genomic analysis of multidrug-resistant Delftia tsuruhatensis isolated from raw bovine milk. Front. Microbiol. 14:1321122. doi: 10.3389/fmicb.2023.1321122
Edited by:
Min Yue, Zhejiang University, ChinaReviewed by:
Imchang Lee, Hallym University, Republic of KoreaKotsoana Peter Montso, North-West University, South Africa
Copyright © 2024 Andriyanov, Kashina and Menshikova. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pavel A. Andriyanov, YW5kcml5YW5vdnB2bEBnbWFpbC5jb20=
†These authors have contributed equally to this work