 Claire Gourin
Claire Gourin Sophie Alain
Sophie Alain Sébastien Hantz
Sébastien Hantz- 1INSERM, CHU Limoges, University of Limoges, RESINFIT, Limoges, France
- 2CHU Limoges, Laboratoire de Bactériologie-Virologie-Hygiène, National Reference Center for Herpesviruses, Limoges, France
Human cytomegalovirus (HCMV) is one of the main causes of serious complications in immunocompromised patients and after congenital infection. There are currently drugs available to treat HCMV infection, targeting viral polymerase, whose use is complicated by toxicity and the emergence of resistance. Maribavir and letermovir are the latest antivirals to have been developed with other targets. The approval of letermovir represents an important innovation for CMV prevention in hematopoietic stem cell transplant recipients, whereas maribavir allowed improving the management of refractory or resistant infections in transplant recipients. However, in case of multidrug resistance or for the prevention and treatment of congenital CMV infection, finding new antivirals or molecules able to inhibit CMV replication with the lowest toxicity remains a critical need. This review presents a range of molecules known to be effective against HCMV. Molecules with a direct action against HCMV include brincidofovir, cyclopropavir and anti-terminase benzimidazole analogs. Artemisinin derivatives, quercetin and baicalein, and anti-cyclooxygenase-2 are derived from natural molecules and are generally used for different indications. Although they have demonstrated indirect anti-CMV activity, few clinical studies were performed with these compounds. Immunomodulating molecules such as leflunomide and everolimus have also demonstrated indirect antiviral activity against HCMV and could be an interesting complement to antiviral therapy. The efficacy of anti-CMV immunoglobulins are discussed in CMV congenital infection and in association with direct antiviral therapy in heart transplanted patients. All molecules are described, with their mode of action against HCMV, preclinical tests, clinical studies and possible resistance. All these molecules have shown anti-HCMV potential as monotherapy or in combination with others. These new approaches could be interesting to validate in clinical trials.
1 Introduction
Human cytomegalovirus (CMV) is an opportunistic pathogen in the immunocompromised host. Not only in transplant recipients, but also in AIDS patients or highly immunocompromised patients with congenital immunodeficiency or immunosuppressive biotherapies. Such infections can lead to graft rejection and organ damages (Kotton et al., 2018; Ljungman et al., 2019). Due to the use of preventive strategies, either preemptive treatment or prophylaxis, CMV disease frequency has decreased. But in solid organ recipients, late disease may occur in up to 18% of patients after stopping prophylaxis (Kotton et al., 2018). In stem cell recipients, it decreased from 10–40% to 2–3% in randomized trials but 5–10% in real life cohorts despite efficient preemptive treatment (Ljungman et al., 2019). Currently, available antivirals are limited to virostatic polymerase inhibitors (ganciclovir, its oral prodrug valganciclovir, cidofovir and foscarnet). Neutropenia limits efficacy of ganciclovir or valganciclovir and this hematological toxicity prevents its use as a prophylaxis in the stem cell recipients. Cidofovir and foscarnet are highly nephrotoxic and restricted to second line treatment. The second limitation of these molecules is the emergence of resistance, favored by prolonged treatments in highly immunocompromised hosts, and use of lower doses due to renal impairment (Razonable et al., 2019).
Congenital CMV infection (cCMV) is also a leading cause of hearing loss and neurological sequelae in children. During pregnancy, the prevalence of primary CMV infection ranges from 1 to 2% in the United States and Western Europe (Hyde et al., 2010; Leruez-Ville et al., 2020), with an average cCMV birth prevalence of 0.65% (Kenneson and Cannon, 2007). If the primary maternal infection occurs during pregnancy, especially during the first trimester, more severe sequelae, including complete hearing loss, are to be feared. The risk of maternal transmission occurs in 30–40% of case with CMV primary infection. Thus, during the first trimester of pregnancy, it is essential to prevent viral transmission to the fetus to avoid neurological disability in newborns (Ornoy and Diav-Citrin, 2006; Ross et al., 2006; Chatzakis et al., 2020). Among infected neonates, 12.7% will have symptoms at birth and 40 to 58% develop permanent sequelae. As a whole, long-term sequelae from sensorineural hearing loss to neurodevelopmental disabilities may occur in 17 to 19% of infected newborns, 51 to 57% of them following maternal primary infection (Dollard et al., 2007; Leruez-Ville and Ville, 2020). Ganciclovir (GCV) and its prodrug valganciclovir (VGCV), foscarnet (FOS) and cidofovir (CDV), are proscribed during pregnancy, due to their toxicity (e.g., neutropenia, nephrotoxicity). Although a randomized study has demonstrated the efficacy of a high dose (8 g per day) of valaciclovir (VACV), a prodrug of acyclovir, in preventing transmission, only 50% of periconceptional or 1st trimester primary infection transmissions were avoided, and more efficient anti-CMV drugs are thus needed (Shahar-Nissan et al., 2020). Treatment of symptomatic newborns for 6 weeks GCV or 6 months with VGCV was shown to improve hearing skills, and is now recommended, although 49 to 63% of the treated neonates developed grade 3 or 4 neutropenia with treatment (Kimberlin et al., 2015).
The burden of long-term therapies for immunocompromised patients, and the emergence of new resistance mechanisms (Chou, 2020), the unmet need for low toxic treatments to prevent or cure cCMV, make it essential to find new antiviral targets and to develop new therapies, in order to treat CMV infections more efficiently while reducing side effects.
Recently, two antiviral drugs with new targets, high specificity and low toxicity, reached clinical development: letermovir targets the highly virus-specific terminase complex (UL56, UL98 and UL51) and maribavir inhibits the UL97 viral kinase. Letermovir (LMV) was approved in 2017 by the Food and Drug Administration (FDA) for the prophylaxis of CMV infection in hematopoietic stem cell transplant patients with high risk of CMV infections (Marty et al., 2017). This new antiviral inhibits the terminase complex, a viral component not found in human cells, thereby reducing its toxicity. Similarly, maribavir (MBV) was approved in 2021 for the treatment of adults and children presenting post-transplant CMV infections refractory or resistant to antivirals (Food and Drug Administration, 2021). It targets the viral kinase UL97 (Biron et al., 2002). Both LMV and MBV have a high oral bioavailability and a low toxicity profile. Nevertheless, resistance mutations have already been described with these new antivirals, making it crucial to continue to develop new therapies.
This is why it is necessary to find new molecules with an anti-CMV spectrum. In this context, this review summarizes the panel of molecules with antiviral activity, including direct inhibitors (brincidofovir, cyclopropavir, anti-terminase benzimidazole analogs), molecules acting through cellular pathways inhibition (artemisinin derivatives, flavonoids, leflunomide, everolimus, or anti Cox) and immunoglobulins (Figures 1, 2 and Table 1).
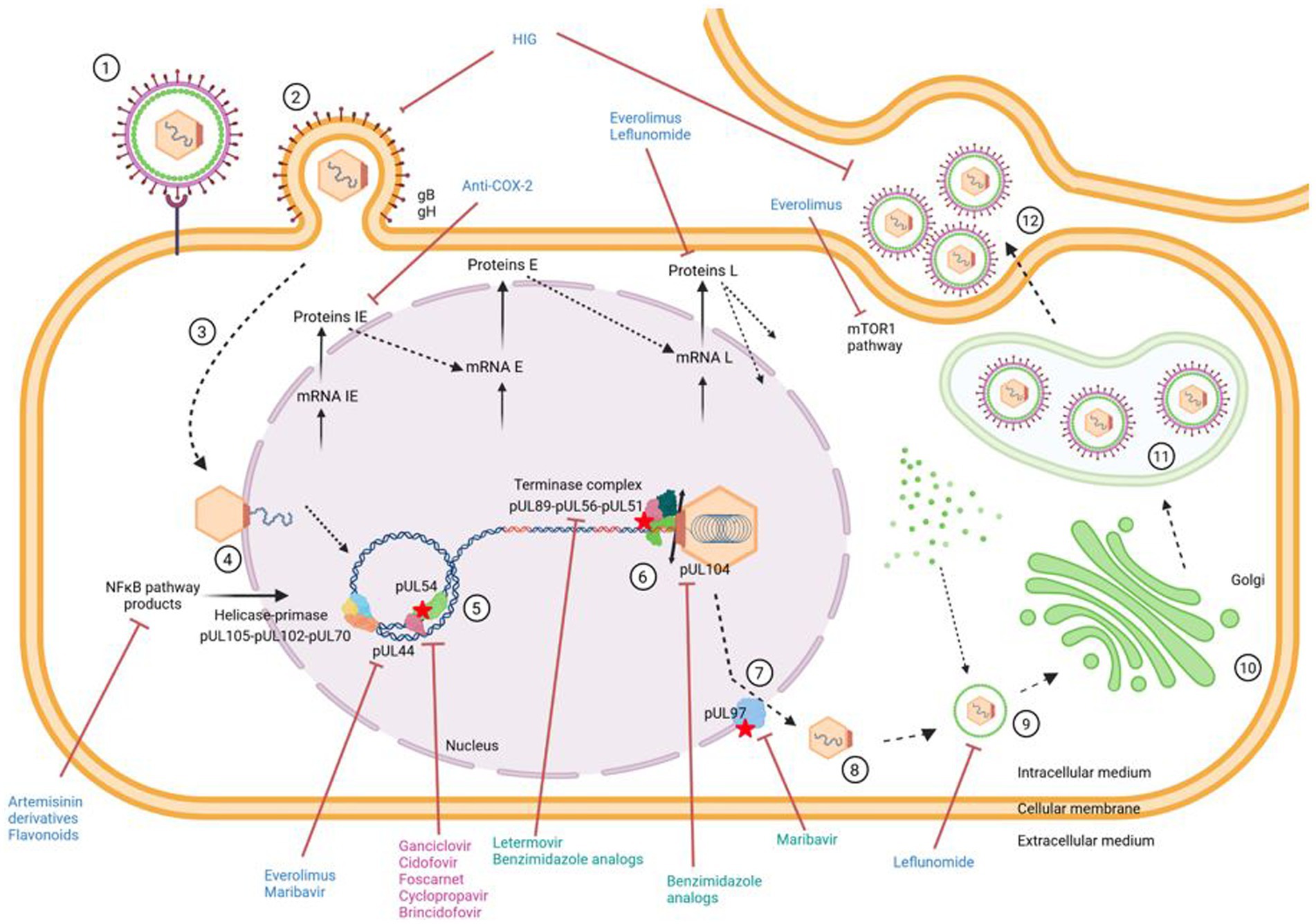
Figure 1. Antiviral targets in relication cycle of CMV. ① Virus attaches to cell. ② Entry of virus into cell and release of capsid into cytoplasm. ③ Migration of capsid to cell nucleus. ④ Release of viral genome from capsid through nuclear pore. ⑤ Replication of viral DNA by the rolling circle method using the viral polymerase pUL54. ⑥ Encapsidation of the genome into neoformed capsids via the encapsidation complex. ⑦ Nuclear exit. ⑧ Release of newly formed virions into cell cytoplasm. ⑨ Tegumentation of newly formed virions. ➉ Passage of virions through Golgi apparatus. ⑪ Acquisition of a primary envelope and transport of virions in a vesicle to the extracellular medium. ⑫ Budding, release of infectious virus particles and infection of a new cell. Indirect antivirals, direct antivirals and antivirals targeting the viral polymerase are in blue, green and pink, respectively. Created with BioRender.com
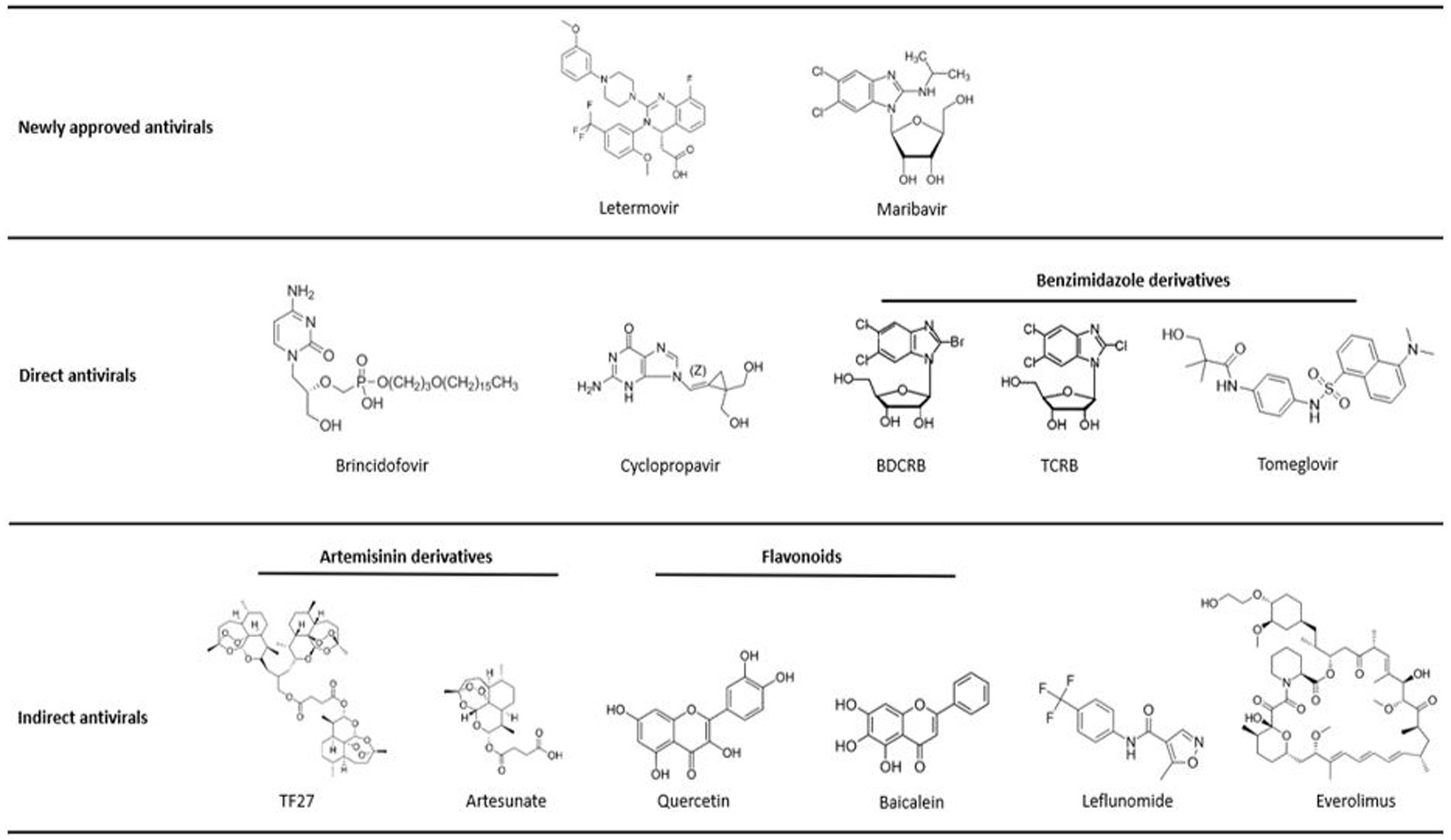
Figure 2. Molecule structures. Structures of chemical molecules with their corresponding order in the review. All molecules are classified as approved antivirals, molecules with direct antiviral activity or molecules with indirect antiviral activity.
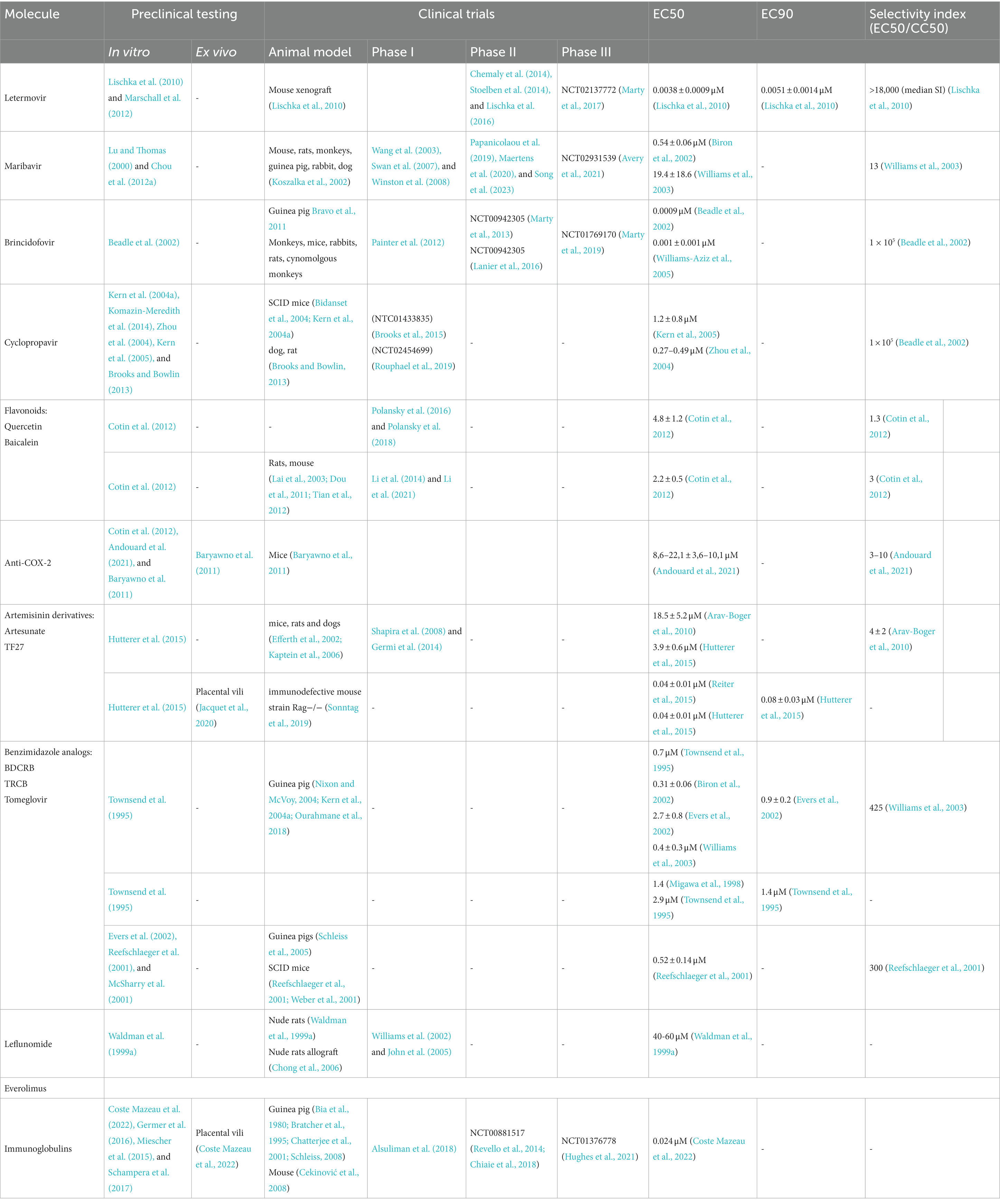
Table 1. Summary of molecules and their main characteristics.
2 Newly approved antivirals target the late stage of the viral cycle
2.1 Letermovir
Letermovir (LMV; AIC246; Prevymis™; Figure 2), an antiviral of the quinazoline class, was developed by Aicuris and further marketed by Merck. LMV acts at the late stage of the viral cycle by direct inhibition of the human CMV terminase complex (Goldner et al., 2011). This viral terminase complex has no functional equivalent in the mammalian cells and the drug is therefore highly specific.
It appears to be very specific of CMV, and has a high activity against resistant strains to DNA polymerase inhibitors (Lischka et al., 2010). In 2017, LMV has been approved by the FDA for CMV prophylaxis in stem cell transplant patients seropositive for CMV (Marty et al., 2017).
2.1.1 Mechanism of action
LMV targets pUL56, the large subunit of the CMV terminase complex that cleaves DNA prior to encapsidation of the genome in neoformed capsids. In addition, it has high specificity against CMV, even if other herpesviruses also possess a terminase complex. This could possibly be explained by a particular mode of action, as LMV probably disrupts the interaction between the subunits of the terminase complex: pUL56, pUL89 and pUL51 unlike other antivirals, which often act by blocking functional domains. LMV has been shown to inhibit primarily the viral step of genome encapsidation (Lischka et al., 2010). Moreover, it was demonstrated that LMV prevents cleavage of concatemeric DNA into units of genomes and formation of CMV mature virions (Goldner et al., 2011).
2.1.2 Preclinical studies
Preclinical studies showed its very high antiviral activity (range: 1.6–5.1 nM; 1,000-fold more potent than GCV) against clinical and laboratory strains included refractory-resistant isolates to current drugs (Marschall et al., 2012) with low toxicity levels at high doses over the EC90. LMV has a high specificity to HCMV and is well tolerated in various cell types with a mean selectivity index of 18,000. LMV has in vivo efficacy in a mouse xenograft model (Lischka et al., 2010) and shows an anti-CMV activity in histoculture of third trimester placenta (Hamilton et al., 2020). It reached concentrations above EC50 at the fetal face when perfused across a third trimester placenta (Faure Bardon et al., 2020). However, its efficacy during the first trimester is not yet validated. Drug combination assays showed additive effect and no synergistic toxicity with current CMV drugs and no effect with anti-HIV drugs (Wildum et al., 2015).
2.1.3 Clinical studies
LMV is a highly lipophilic molecule with a Cmax of between 45 min and 2.25 h and a half-life of 12 h. After administration, LMV is highly protein-bound and eliminated via the biliary tract. The efficacy, safety and pharmacokinetic parameters of oral LMV were studied in a Phase IIa trial: LMV 40 mg twice daily or 80 mg once daily was administered to patients for 14 days as a preventive treatment against CMV infection in kidney and kidney/pancreas transplant recipients (Stoelben et al., 2014). This study demonstrated that all patients responded to LMV treatment. Chemaly et al. conducted a Phase IIb variable-dose prophylaxis trial in 2014: LMV was administered daily orally at 60 mg, 120 mg or 240 mg for 12 weeks post-transplant in CMV-seropositive allogeneic hematopoietic cell recipients. The incidence of prophylaxis failure (with or without virological failure) was significantly lower in the LMV-treated groups than in the placebo group (32% for the 120 mg group, 29% for the 240 mg group vs. 64%). The incidence of virological failure was lower in the 240 mg group (6%) than in the placebo group (36%). This study demonstrated that LMV was well tolerated and that a dose of 240 mg once daily was effective in suppressing viremia (Chemaly et al., 2014).
The Phase III prophylaxis trial (NCT02137772) evaluated the efficacy of a daily oral or intravenous dose of LMV of 480 mg/day (or 240 mg/day in patients taking ciclosporin) for 14 weeks after transplantation. A significant reduction in the number of patients developing CMV infection was observed. Indeed, at week 24, 38% of patients in the LMV group developed an HCMV infection versus 61% in the placebo group. In addition, the mortality rate was higher in the placebo group (16%) compared to the LMV group (10%). This study confirms the efficacy of LMV in the prophylaxis of CMV infection after HSCT in R+ patients (Marty et al., 2017). Used as primary and secondary prophylaxis in the French Compassionate Use Program (CUP) for high-risk patients, it was well tolerated and reduced the number of CMV infections compared with historical studies (Robin et al., 2020; Beauvais et al., 2022).
LMV has also been tested in case-series for prophylaxis or treatment in organ transplanted patients. It had good virologic outcomes and was well tolerated in patient with few side effects (Aryal et al., 2019; Veit et al., 2020; Linder et al., 2021). Recently, a large study conducted in 94 centers with kidney recipients showed that LMV was non-inferior to valganciclovir for prophylaxis of CMV disease for 52 weeks, with lower rates of leukopenia or neutropenia, arguing in favor of its use in this indication (Limaye et al., 2023).
2.1.4 Resistance
In less than 5 passages, the selection of resistant strains is rapidly achieved in vitro with UL56 mutations conferring high or absolute LMV resistance (Chou, 2017). In vitro studies have also revealed mutations in the genes encoding pUL51 and pUL89. In clinical trials, the first resistant isolate appeared after a sub-optimal dose of 60 mg/day (Chemaly et al., 2014; Lischka et al., 2016). Resistant mutants can emerge rapidly under LMV if treatment is interrupted or underdosed (Alain et al., 2020). pUL56 is the main target of LMV, which explains why mutations occur more frequently in this protein than in other proteins of the terminize complex (Cherrier et al., 2018; Frietsch et al., 2019; Alain et al., 2020). A new resistance mutation A95V in pUL51 was also described in vivo after LMV treatment in combination with a L257I mutation in pUL56 (Muller et al., 2022; Figure 3).
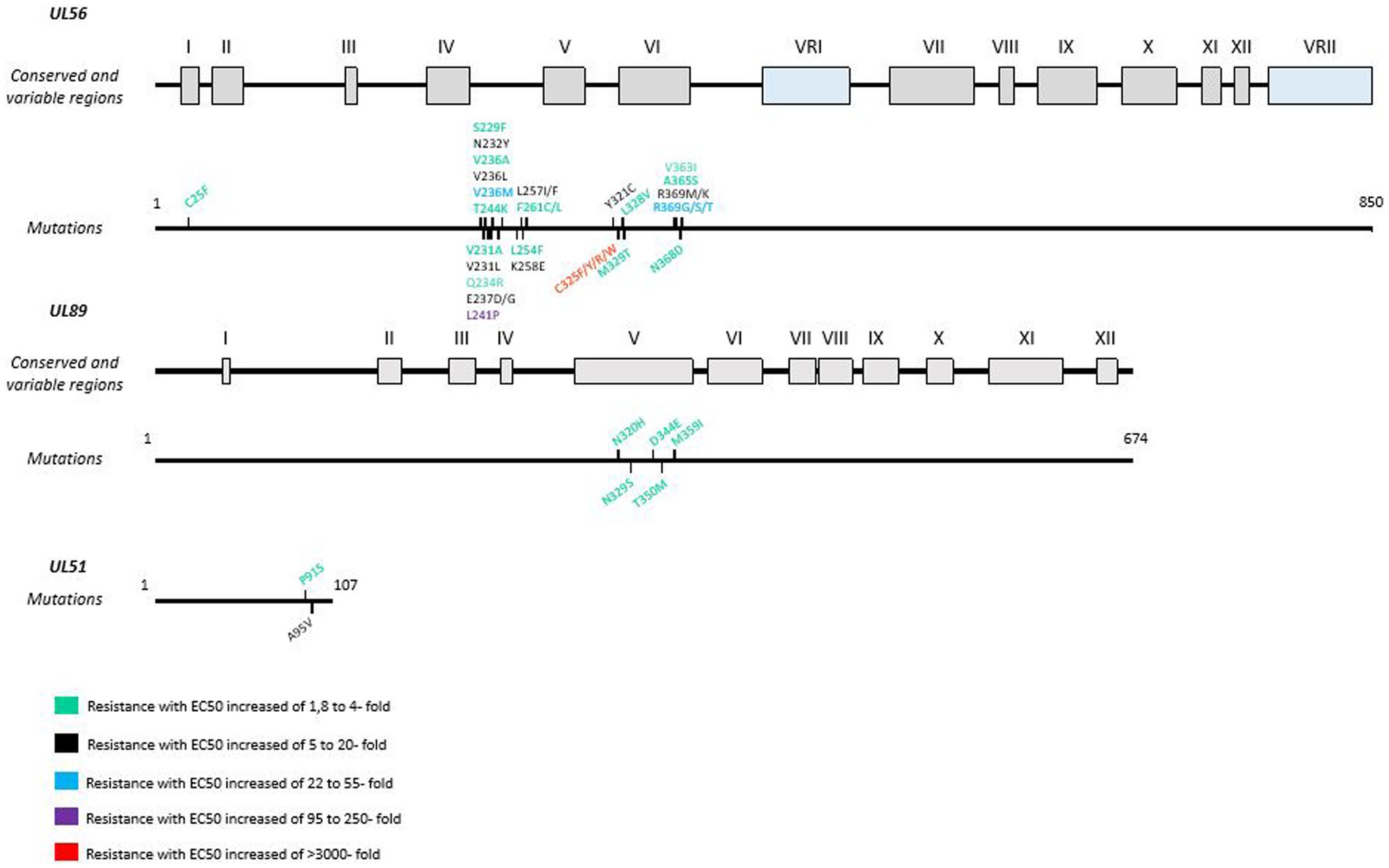
Figure 3. Resistance mutations to letermovir in UL56, UL89 and UL51 genes. LMV resistance mutations according to EC50 values. Mutations are referenced on genes under the map of conserved and variable regions. Scaled representation.
2.2 Maribavir
Maribavir (MBV; 1,263 W94; LIVTENCITY™) [formerly 1,263 W94, 5,6-dichloro-2-(isopropylamino)-1,β-l-ribofuranosyl-1-H-benzimidazole] (Figure 2) is an oral bioavailable benzimidazole riboside initially developed by Glaxo Smith Kline (Biron et al., 2002), then Viropharma, now marketed by Takeda Pharmaceuticals/a Shire company for treatment of refractory or resistant CMV infections. In November 2021, the FDA approved MBV for 400 mg twice a day-treatment of adults and children (12 years of age or older, weight > 35 kg) with post-transplant CMV infection/illness refractory/resistant to GCV, VGCV, CDV and FOS (Food and Drug Administration, 2021; Halpern-Cohen and Blumberg, 2022). In 2022, the European Commission approved MBV in the same indications.
2.2.1 Mechanism of action
Maribavir does not require activation or intracellular processing. Unlike other anti-CMV drugs, MBV targets the viral kinase UL97 and its natural substrates, which are involved in the DNA replication and viral capsid nuclear egress (Biron et al., 2002; Hamirally et al., 2009; Prichard, 2009). This mechanism of action confers MBV an in vitro and in vivo activity against GCV, FOS and CDV resistant CMV strains (Biron et al., 2002; Drew et al., 2006). The combination of MBV and GCV is therefore not recommended, as GCV activation requires three phosphorylation, the first of which being mediated by pUL97. Indeed, MBV antagonizes anti-CMV effect of GCV by increasing the 50% inhibitory concentration (IC50) of a GCV-sensitive strain by 13 fold (Chou and Marousek, 2006).
MBV competitively inhibits pUL97 (Biron et al., 2002) and blocks the phosphorylation of several downstream proteins including cellular components and the viral proteins pp65 and pUL44 the DNA polymerase accessory protein (Prichard, 2009). Like Cdc2/Cyclin-dependent kinase 1 (CDK1) in CMV-uninfected cells, the viral kinase pUL97 phosphorylates nuclear lamina components (lamin A/C), facilitating the removal of mature virions from the nucleus. Consequently, MBV treatment results in the accumulation of immature virions in the nucleus (Hamirally et al., 2009). It also inhibits CMV DNA replication through pUL44 inhibition. In vitro drug combination assays showed additive effect with foscarnet, and synergy with artesunate (Morère et al., 2015). Additive effect was also observed with cidofovir or letermovir (quinazoline), while association with BDCRB, a benzimidazole inhibitor of the terminase, or with sirolimus, a mTor inhibitor, was synergistic (Chou et al., 2019).
2.2.2 Preclinical studies
In vitro, MBV has selective activity against CMV. Its activity has been demonstrated against Epstein–Barr virus (EBV), however it is not active against herpes simplex virus, varicella-zoster virus (VZV) or human herpesviruses 6 and 8 (HHV-6 and HHV-8) (Prichard, 2009).
Preclinical studies showed that MBV has a better oral bioavailability, a better safety profile and a lower toxicity for host cells than current drugs (GCV, FOS and CDV) with theoretical benefits for the viral inhibition and cross-resistances appearing (Lu and Thomas, 2000; Koszalka et al., 2002; Chou et al., 2012a). MBV also reached concentrations above EC50 at the fetal face when perfused across a third trimester placenta (Faure Bardon et al., 2020). In addition, it inhibits CMV replication in first trimester placental villi models in histoculture, with the same EC50 as in vitro (Morère et al., 2015).
In animal models, the oral bioavailability of MBV is 90% in rats and 50% in monkeys. MBV is excreted via the biliary route and, to a lesser extent, via the metabolic and renal pathways. The minimum effect dose in rats was 100 mg/kg/day and the no-effect dose in monkeys was 180 mg/kg/day (Wang et al., 2003). It was initially distributed in the gastrointestinal tract of rats, but did not cross the blood–brain barrier. This study showed favorable results for MBV’s safety profile (Koszalka et al., 2002). In addition, Kern et al. (2004a) demonstrated that oral MBV significantly reduced HCMV replication at concentrations of 75 mg/kg twice daily in SCID-humanized mice with human fetal retinal tissue implants or thymus/liver implants. However, MBV was more effective in treating thymus/liver infection, as it was shown to be poorly absorbed by ocular tissues.
2.2.3 Clinical studies
Several Phase I clinical trials have been conducted with MBV to evaluate its safety, pharmacokinetics and efficacy against CMV infection. In fact, two Phase I clinical trials with escalating single doses of MBV (50 mg to 1,600 mg) were conducted in healthy and human immunodeficiency virus (HIV)-infected patients. 30–40% of an oral dose of MBV was absorbed, and Cmax was reached 1–3 h after administration (Wang et al., 2003). MBV was rapidly eliminated. At the 400 mg dose, no statistical difference was observed whatever the renal functions of patients (Wang et al., 2003; Swan et al., 2007). The main side effect of MBV is dysgeusia. A Phase I study with multiple oral doses of MBV was carried out to evaluate its antiviral activity. MBV was administered orally at doses of 100 mg twice daily, 400 mg once daily or 400 mg twice daily to CMV-seropositive HSCT recipients. One hundred days after transplantation, pp65 antigenemia was lower in all groups than in the placebo group (15, 19, 15% vs. 39% respectively). In addition, pharmacokinetic analysis of the 400 mg twice-daily dose showed higher Cmax and area under the curve (AUC) values than the 100 mg twice-daily dose, but with no improvement in antiviral activity and more side effects (Winston et al., 2008).
At low doses, MBV failed to meet the primary endpoints of the initial Phase III study for prophylaxis in hematopoietic stem cell allograft and liver transplant recipients. However, in a Phase II dose-ranging clinical trial, MBV ≥ 400 mg twice was active against refractory or resistant CMV infections in transplant recipients (Papanicolaou et al., 2019). This dosing also showed similar efficacy to those of valganciclovir in pre-emptive treatment of solid organ transplant and HSCT recipients (Maertens et al., 2020). MBV is mainly metabolized in the liver, and moderate hepatic impairment increased total MBV concentrations. This suggests that dose adjustment of MBV may not be necessary for individuals with mild to moderate hepatic impairment (Song et al., 2023).
A randomized Phase III trial, the Solstice study (NCT02931539), demonstrated the efficacy of MBV in SOT and HSCT patients with refractory CMV infections with or without resistance. The study was conducted on 352 patients (235 patients receiving MBV 400 mg twice daily versus 117 patients receiving investigator-assigned therapy (IAT): GCV, VGCV, FOS or CDV) for 8 weeks with a 12-week follow-up. Endpoints were CMV disappearance at the end of week 8 and MBV disappearance and symptom control at the end of week 8, maintained until week 16. Significantly, more patients in the MBV group achieved the primary endpoint (55.7% vs. 23.9%; p < 0.001) and the secondary endpoint (18.7% vs. 10.3%; p < 0.01). Side effects were less frequent in the MBV group than in the IAT group. Acute kidney injury was more frequent in patients treated with FOS (21.3% vs. 8.5%), and neutropenia was more frequent in patients treated with GCV/GCV (9.4% vs. 33.9%). In the MBV group, 13.2% of patients discontinued treatment due to drug-related adverse events, compared with 31.9% in the IAT group (Avery et al., 2021).
2.2.4 Resistance
Although MBV is a newly approved antiviral, resistance mutations (Figure 4) have already been found in viral genes UL97 and UL27. Indeed, some pUL97 mutations (V353A, L397R, L337M, T409M, H411L, H411N, H411Y, F342, C480F) confer moderate to high-level resistance to MBV, with a 3.5 to 200 fold increase of the EC50 (Chou et al., 2007, 2019; Chou and Marousek, 2008; Chou, 2020). These mutations are close to the kinase ATP-binding and catalytic domains upstream the GCV resistance mutations (Chou and Marousek, 2008). Mutations F342Y and C480F are responsible for cross-resistance to ganciclovir and may be present before MBV treatment (Chou et al., 2023). In addition, mutations in the UL27 gene confer low resistance to MBV with a 2- to 3-fold increase in EC50. These mutations (R233S, W362R, W153R, L193F, A269T, V353E, L426F, E22stop, W362stop, 218delC, and 301-311del) compensate for pUL97 inhibition by destabilizing Tip60 (histone acetyltransferase), increase p21 expression and inhibit cyclin-dependent cellular kinases (Chou et al., 2004; Chou, 2009; Kamil and Coen, 2011; Reitsma et al., 2011). In phase II and phase III clinical trials, resistance to MBV emerged in 52 and 26% of treated patients, respectively (Papanicolaou et al., 2019; Chou et al., 2023).
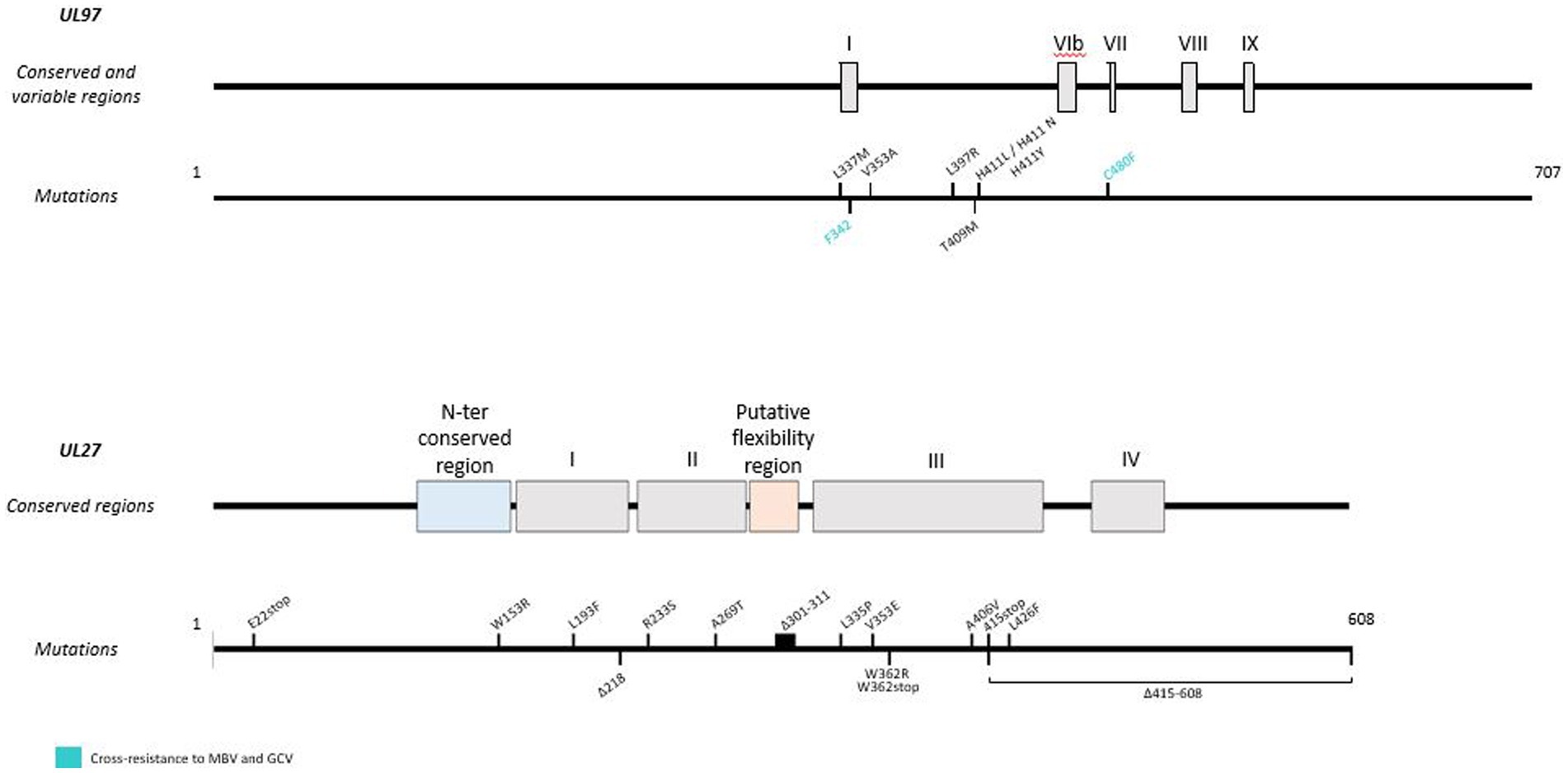
Figure 4. Resistance mutations to maribavir in UL97 and UL27 genes. Mutations responsible for cross-resistance with GCV are represented in blue. Mutations are referenced on genes under the map of conserved and variable regions. Scaled representation.
3 New molecules with activity against CMV
3.1 Direct-acting antivirals
3.1.1 Brincidofovir
Brincidofovir (BCV, CMX001; HDP-CDV) [[(S)-2-(4-amino-2-oxo-1(2H)-pyrimidinyl)-1-(hydroxymethyl)ethoxy]methyl]mono[3-(hexadecyloxy)propyl] ester (Figure 2) developed by Chimerix is a lipid antiviral conjugate (LAC) composed of a lipid [1-0-hexadecyl-oxypropyl (HDP)] covalently linked to the acyclic nucleotide analog CDV, enabling the drug to utilize the natural absorption pathways of lysophosphatidylcholine in the small intestine (i.e., passive diffusion and flipases; Lanier et al., 2016). Currently, the United-States FDA approves BCV for treatment of smallpox.
3.1.2 Mechanism of action
BCV was designed to remain intact in plasma and deliver the drug directly to target cells. It enabled enhanced cellular uptake and high intracellular levels of the converted active antiviral agent, CDV-diphosphate (CDV-PP), increasing antiviral activity against CMV by 2 to 3 orders of magnitude compared with CDV alone (Aldern et al., 2003; Williams-Aziz et al., 2005). BCV is cleaved to release CDV. Then, CDV is converted by intracellular anabolic kinases to CDV-PP, the active inhibitor of viral DNA synthesis. Unlike CDV, BCV is not a substrate for the human organic anion transporter 1, which mechanistically explains the absence of renal toxicity observed in clinical trials with BCV (Tippin et al., 2016; Figure 5).
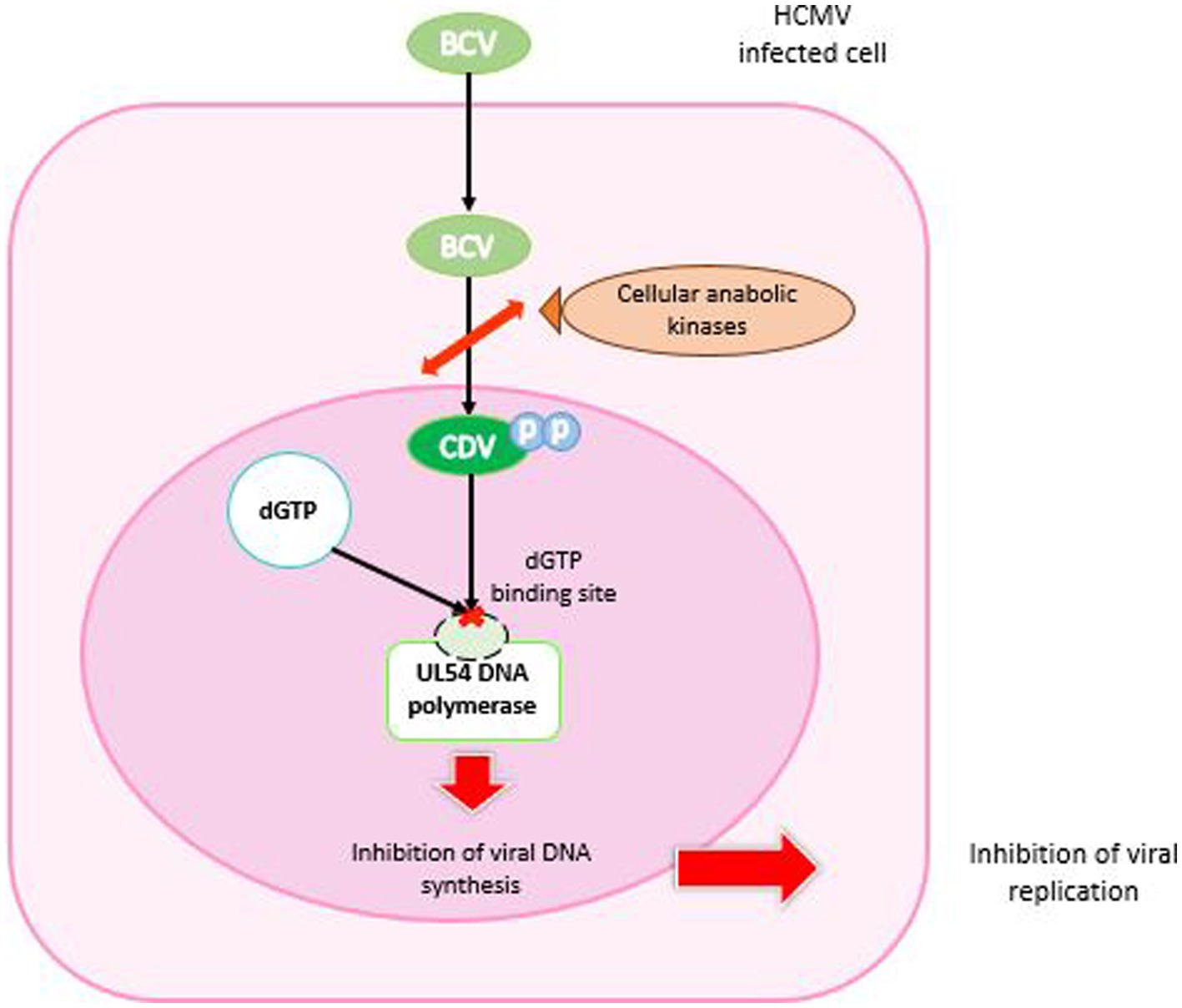
Figure 5. Mechanism of action of brincidofovir. BCV enters the HCMV-infected cell, is cleaved and phosphorylated by cellular anabolic kinases to cidofovir diphosphate. In the nucleus, CDV-PP binds competitively to the dGTP binding site of UL54 DNA polymerase. The result is inhibition of DNA synthesis and arrest of viral replication.
3.1.3 Preclinical tests
BCV has been developed for the treatment of infections by double-stranded DNA viruses. It has broad-spectrum efficacy against herpesviruses, polyomaviruses, adenoviruses, papillomaviruses and orthopoxviruses (Beadle et al., 2002; Bidanset et al., 2004; Williams-Aziz et al., 2005). BCV is effective against clinical isolates of HCMV (EC50 of 0.0009 μM against HCMV strain AD169) and HSV, including isolates resistant to GCV and ACV (Williams-Aziz et al., 2005; James et al., 2013). In addition, BCV has been shown to be 10 to 100 times more active than CDV against murine CMV (Kern et al., 2004b).
In vivo tests were carried out on animal models to evaluate its efficacy in congenital CMV infection. BCV showed antiviral activity of 0.004 μM ± 0.001 μM against guinea pig CMV (GPCMV). At the end of the second or beginning of the third trimester of gestation, guinea pigs were infected with GPCMV. Significant pup survival was observed in the BCV group (93–100% vs. 50–60%; p ≤ 0.019). Viral load was significantly reduced in the spleen and liver of pups after BCV treatment (p = 0.017 and p = 0.029 respectively). Although pup survival was improved with 4 mg/kg treatment, virus levels in fetal tissues were related to those in control tissues. This suggests that BCV could have been a good candidate for the treatment of congenital CMV infections in humans, with high tolerance (Bravo et al., 2011).
3.1.4 Clinical trials
Painter et al. evaluated the pharmacokinetics and safety of BCV in the first Phase I clinical trial in 2012. This was a randomized, double-blind, placebo-controlled, parallel-group, dose-escalation trial in healthy adults. There were no adverse events during the trial. No significant changes in pharmacokinetic parameters were reported. Gastrointestinal analyzes showed no BCV-related mucosal changes. After multiple doses, no accumulation of BCV was observed. Maximum plasma concentrations of BCV were observed 2 to 3 h after dosing. This trial showed that BCV was relatively well tolerated and had a high bioavailability with a dose of approximately 140 mg in adults (2 mg/kg) (Painter et al., 2012).
From 2009 to 2011, Marty et al. (2013) conducted a phase II clinical trial (NCT00942305) on 230 adult CMV-seropositive hematopoietic stem cell transplant recipients at 27 centers. Five sequential study cohorts were planned according to a double-blind ascending dose schedule (3:1 ratio to receive BCV or matching placebo). Drugs were administered for 9 to 11 weeks post-transplant. The primary endpoint was a CMV-related event, i.e., CMV disease or a plasma CMV DNA level above 200 copies/ml. In the BCV 100 mg twice weekly group, the incidence of CMV-related events was significantly lower than in the placebo group (10% vs. 37%; p = 0.002). The most frequent side effect of treatment was diarrhea in the BCV 200 mg weekly group. There were no reports of myelosuppression or nephrotoxicity (Marty et al., 2013).
The Phase III clinical trial (NCT01769170) was a randomized, double-blind, placebo-controlled (2:1) trial for CMV prophylaxis in 452 CMV-seropositive adults with HCT without CMV viremia. Patients received BCV or placebo until week 14 after HCT. The primary endpoint was the proportion of patients who developed clinically significant CMV infection (CS-CMVi: CMV viremia requiring preemptive treatment or CMV disease) up to week 24 after HCT. This proportion was similar in both groups (51.2% vs. 52.3%; p = 0.805). Fewer BCV-treated patients developed CMV viremia up to week 14 than placebo-treated patients (41.6%; p < 0.001). BCV resulted in more frequent adverse events (51.1% vs. 37.6%) such as acute graft-versus-host disease (32.3% vs. 6.0%) and diarrhea (6.9% vs. 2.7%). All-cause mortality at week 24 was 15.5 and 10.1% in the BCV and placebo groups, respectively. In conclusion, BCV did not reduce CMV viremia at week 24 post-transplant, and was associated with gastrointestinal toxicities (Marty et al., 2019). BCV is not available to date for the treatment of CMV infection.
3.1.5 Resistance
Since 2013, CDV resistance mutations in UL54 DNA polymerase have been shown to confer resistance to BCV. In vitro, increasing concentrations of BCV over 10 months conferred CDV and BCV resistance on a wild-type strain. Genotyping of the strain revealed a D542E mutation in pUL54, which was responsible for a more than 10-fold reduction in susceptibility to BCV and CDV in marker-transfer experiments. This mutation did not confer resistance to GCV or FOS. A smaller plaque phenotype and slower replication kinetics than the parent viruses were also demonstrated. This is the first mutation described under BCV selective pressure. This suggests that BCV may have a unique resistance profile associated with reduced viral replication and maintenance of sensitivity to FOS and GCV (James et al., 2013).
In vitro experiments under BCV pressure selected the N408K and V812L mutations in CMV DNA polymerase, which were already known to confer resistance to CDV. In addition, new substitutions in the exonuclease domain were identified: D413Y, E303D and E303G, which confer resistance to GCV and CDV, with 6- to 11-fold resistance to BCV, or 17-fold when E303G is combined with V812L. This confirmed the expected pattern of cross-resistance (Chou et al., 2016).
In a clinical study, Lanier et al. investigated CMV genotypes from a Phase II trial comparing BCV to placebo for prophylaxis of CMV infections in HCT recipients. Two mutations (M827I and R1052C) were reported in pUL54 in a small number of patients, but did not confer resistance to BCV, CDV, GCV or FOS. This study suggests that the first-line use of BCV for the prevention of CMV infection may preserve downstream options for patients (Lanier et al., 2016). Nevertheless, A987G and F412L mutations in pUL54 have been reported in other studies using BCV as rescue therapy (Kaul et al., 2011; Vial et al., 2017). These mutations were known to confer resistance to CDV, suggesting that the emergence of resistance could occur after BCV treatment.
3.2 Metylenecyclopropane analog: cyclopropavir
Cyclopropavir (CPV, Filociclovir (FCV), ZSM-I-62), (Z)-9-{[2,2-bis-(hydroxymethyl)cyclopropylidene]methyl}guanine (Figure 2) is a new analog of methylenecyclopropane (MCPN) (Zhou et al., 2004). This antiviral showed a good activity against HCMV and murine CMV in animal models (Kern et al., 2004b). In addition, CPV proved highly potent against HCMV (wild-type and GCV-resistant mutants in pUL97 and pUL54), EBV, both variants of HHV-6, HHV-7 and HHV-8 (Kern et al., 2005).
3.2.1 Mechanism of action
As with other nucleoside analogs such as GCV, activation of CPV by tri-phosphorylation is required (Littler et al., 1992). The primary phosphorylation is performed by the viral kinase pUL97 (HCMV) or pU69 (HHV-6), whereas the second and third phosphorylations are made by the guanosine monophosphate kinase (GMPK), thus resulting in tri-phosphate CPV (CPV-TP) (Kern et al., 2005; Gentry et al., 2011; Komazin-Meredith et al., 2014). Therefore, the conversion mechanism of CPV is different from that of GCV; it necessitates a single cellular enzyme to have CPV-triphosphate (CPV-TP; Figure 6).
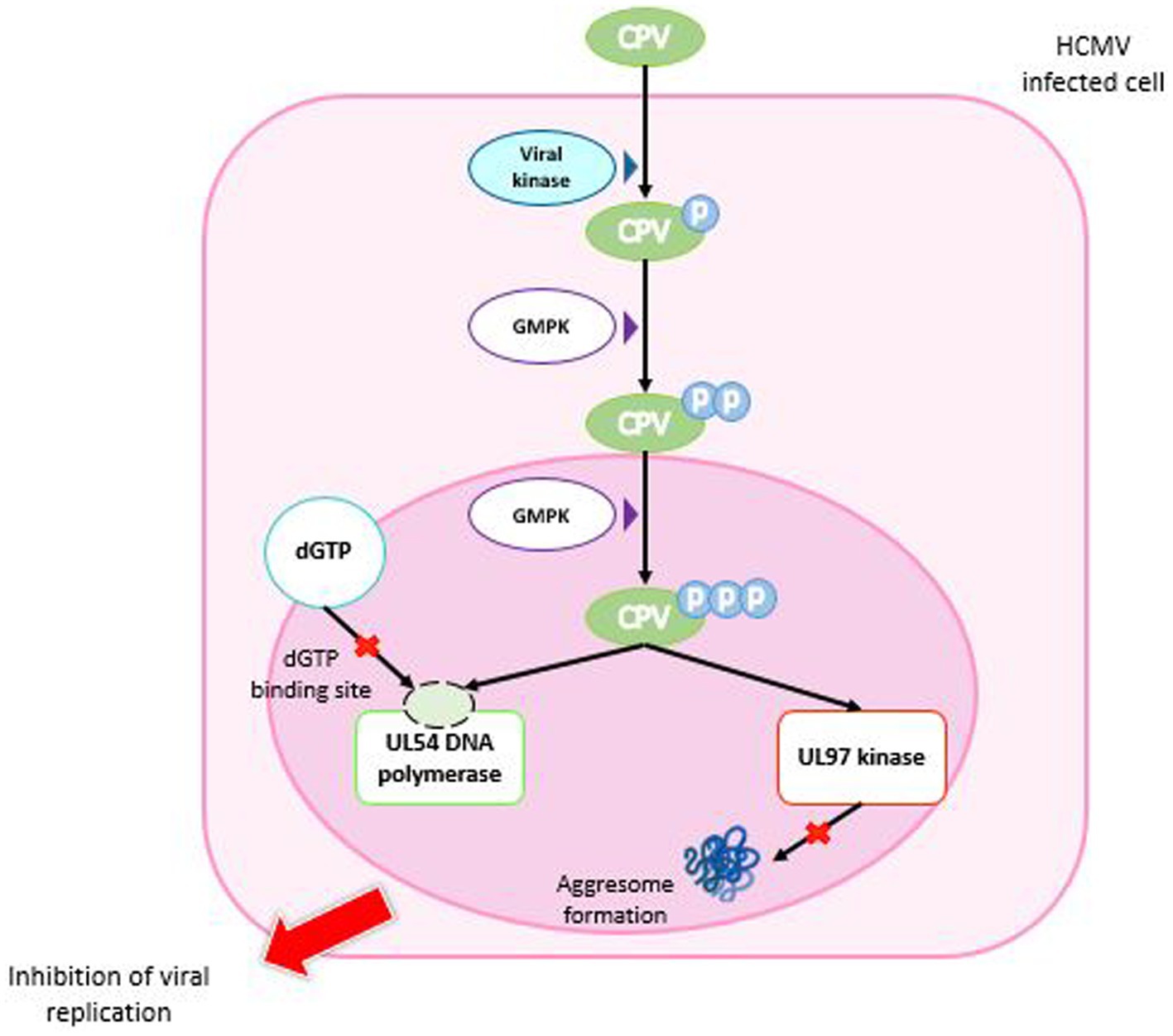
Figure 6. Mechanism of action of cyclopropavir. CPV enters the HCMV-infected cell and is phosphorylated by a viral kinase to CPV monophosphate. Two successive phosphorylations by GMPK are required to obtain CPV triphosphate. In the nucleus, CPV-triP binds competitively to the dGTP binding site of DNA polymerase UL54 and inhibits DNA synthesis. It also inhibits the ability of the viral UL97 kinase to prevent aggresome formation. Both mechanisms lead to inhibition of viral replication. GMPK: guanosine monophosphate kinase.
CPV inhibits HCMV replication by a dual mechanism, inhibiting both pUL54 DNA polymerase and UL97 kinase (James et al., 2011). Indeed, some mutations on pUL54 confer a resistance to CPV, which confirms that the viral polymerase is a target of CPV (Chou et al., 2012b). CPV-TP inhibits pUL54 by competition with dGTP and takes place as chain terminator that stops the DNA synthesis. Interestingly, of the two CPV enantiomers, (+)-CPV-TP could have a twenty-fold higher affinity with pUL54 (Chen et al., 2016). In addition, (+)-CPV is preferencially converted in (+)-CPV-TP than (−)-CPV-TP by the GMP kinase (Gentry et al., 2011). Besides this mechanism, the inhibition of normal function of pUL97 kinase by CPV was assessed by cell transfection with plasmids expressing pUL97 and a reporter plasmid expressing pp65-GFP. CPV prevented the pUL97 capacity to inhibit aggresomes formation, as MBV (James et al., 2011).
3.2.2 Preclinical tests
A study on immunocompromised SCID mice (BALB/c) infected with MCMV, orally administered CPV showed a good effectiveness compared with GCV. Mortality rates were significantly reduced with CPV. Indeed, reducing of viral replication was much more effective in CMV target organs like liver, spleen and lung (Kern et al., 2004c).
Additionally, CPV showed a greater efficacy in vitro and in vivo than GCV without any increase of toxicity (Zhou et al., 2004; Kern et al., 2005) and achieved therapeutic concentrations in vivo without prodrug modification (Wu et al., 2009). In addition, CPV was used in combination with BDCRB and produced a statistically significant synergistic effect against HCMV in vitro (O’Brien et al., 2018). On the other hand, the introduction of 1.0 or 10 nM MBV demonstrated a competitive inhibition of CPV phosphorylation with a Ki of 3.0 ± 0.3 nM (Gentry et al., 2010).
Pharmacokinetics, toxicokinetics and absorption, distribution, metabolism and excretion (ADME) datas of CPV showed good results in animals. CPV has a high oral biodisponibility. It also shown that plasma concentration were higher after the first dose of CPV than after the fourteenth daily dose. CPV did not induce or inhibit cytochrome P450 and was minimily metabolized by liver microsomes (Brooks and Bowlin, 2013).
The safety study showed that CPV did not present adverse effects on central nervous system, respiratory and cardiovasular systems. Besides, toxicology studies demonstrated that CPV did not cause haemolysis ex vivo (Brooks and Bowlin, 2013).
3.2.3 Clinical trials
The first phase 1A clinical trial (NTC01433835) included 48 healthy adults (3 males, 45 females) with a main age of 50.3 years. This randomized, placebo-controlled (3:1) trial evaluated CPV safety and pharmacokinetics after the administration of various doses ranging from 35 to 1,350 mg. No serious adverse effects were classified. Cmax were reached after 1 to 2 h after oral administration and the CPV was not detectable 24 h after last dose of treatment (Brooks et al., 2015).
A phase 1B, double blind, randomized, placebo-controlled (3:1), single center, multiple ascending doses, clinical trial (NCT02454699) was done to assess safety, tolerability and pharmacokinetics of CPV at various doses in 24 healthy adult volunteers monitored for 22 days (7 males and 17 females; main age 47.4 years). Doses of CPV were 100, 350 and 750 mg for 7 days. During this study, no serious adverse effect was highlighted. Indeed, main adverse events concerned gastrointestinal tract (17%), nervous system (11%) and skin and subcutaneous tissues (11%). The only severe adverse event appeared in the 750-mg cohort was a reversible grade 3 elevation in serum creatinine and bilirubin associated with a 1-log increase of CPV in plasma after 24 h of the initial dose. The Cmax was reached at 2 to 3 h following administration and CPV was undetectable in plasma 24 h after the last dose, as phase 1A trial. Finally, authors concluded that, in vivo, doses as low as 100 mg were sufficient to inhibit CMV (Rouphael et al., 2019).
So far, no phase II or III clinical trials are in progress (source: ClinicaTrials.gov).
3.2.4 A new antiviral against adenoviruses
Adenoviruses are responsible for a variety of infections in children, and can cause acute hepatitis with high morbidity and mortality (Kajon and St George, 2022). Recent studies have also demonstrated the antiviral activity of CPV against adenovirus (HAdV) by inhibition of the adenovirus-encoded DNA polymerase (Toth et al., 2020). Hartline et al. (2018), have shown that the strain human adenovirus type 5 (HAdV5) of the American Type Culture Collection (ATCC) was sensitive to CPV. CPV has shown a high potential to inhibit in vitro HAdV replication with a higher efficacy than CDV (5- to 10- fold higher) (Tollefson et al., 2022).
The same potential against adenoviruses has been observed with BCV. It has been demonstrated in several clinical trials, notably in the treatment of severe adenovirus infection and disease in 2022 (NTC02596997) (Alvarez-Cardona et al., 2020; Chimerix, 2022).
3.2.5 Resistance
Mutations are known to confer CPV resistance (Figure 7). Indeed, a recombinant virus with Δ498 bp mutation in the UL97 open reading frame (ORF) is resulting in a protein with a missing kinase domain (Gentry et al., 2013). The K355M mutation confers to the virus a moderate 5- to 7- fold increase in EC50 versus a 13- to 25- fold increase for FCV and GCV, respectively, (Komazin-Meredith et al., 2014). Recombinant virus lacking pUL97 kinase domain is 20 times more resistant to CPV (Kern et al., 2005), which can be explained by the need for the first phosphorylation of CPV by pUL97, essential for activation of the molecule (Gentry et al., 2013). Mutations have been identified in vitro, ranging from insignificant mutations to resistance mutation confering cross-resistance to GCV, MBV and CPV.
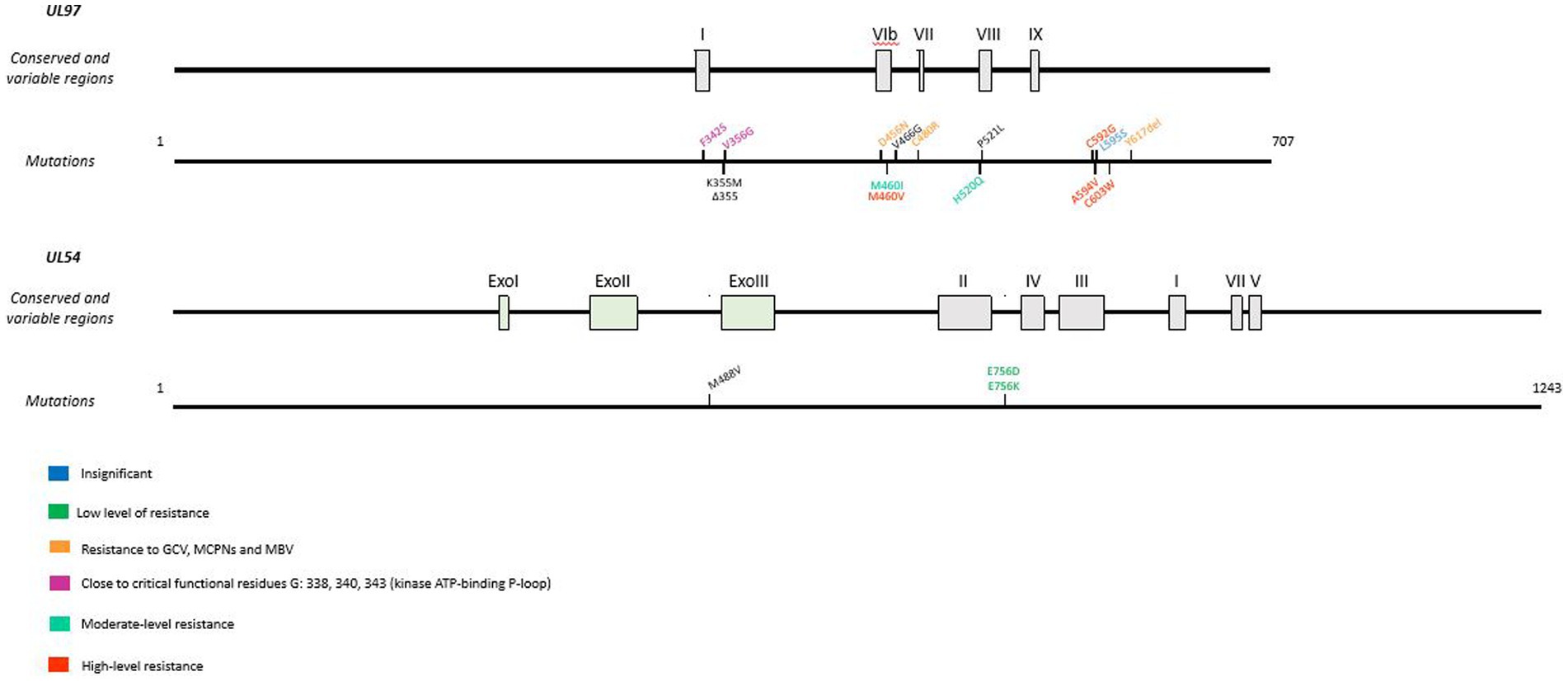
Figure 7. Resistance mutations to cyclopropavir in UL97 and UL54 genes. CPV resistance mutations according to EC50 values. In UL97, cross-resistance mutations are represented in orange and mutations next to the ATP-binding P-loop are in pink. Mutations are referenced on genes under the map of conserved and variable regions. Scaled representation.
Mutations close to critical functional residues were also identified: F342S and V356G. These mutations are close to the highly conserved residues involved in the kinase ATP-binding P-loop G338, G340 and G343 (Chou et al., 2013). The L595S mutants are resistant to GCV but remain susceptible to CPV (Chou and Bowlin, 2011). On the other hand, M460V, A594V, C592G and C603W, which confer low resistance to GCV, increase EC50 by 3 to 5-fold; M460I and H520Q induce high resistance to CPV, with a 12- to 20-fold increase in EC50 (Chou and Bowlin, 2011; James et al., 2011).
In addition, D456N, C480R and Δ617 confer resistance to GCV, MBV and all the MCPNs including CPV (Komazin-Meredith et al., 2014).
The combination H520Q-M488V (UL97-UL54) induced the highest level of resistance to CPV. Resistance mutations are close to finger and palm domains of the polymerase catalytic core (Chou et al., 2012b). Two other mutations in pUL54 were shown to confer a lower resistance to CPV: E756D and E756K. Both are involved in FOS resistance (Lurain and Chou, 2010).
3.3 Benzimidazole analogs: BDCRB, TCRB and tomeglovir
BDCRB or 2-Bromo-5,6-dichloro-1-(beta-d-ribofuranosyl)benzimidazole and TRCB or 2,5,6-Trichloro-1-(beta-D-ribofuranosyl)benzimidazole (Figure 2) are benzimidazole ribofuranoside. These two molecules are potent and selective inhibitors of HCMV replication (Townsend et al., 1995). Tomeglovir (BAY 38–4,766) or {3-hydroxy-2,2-dimethyl-N[4({[5-(dimethylamino)-1-naphthyl]sulfonyl}amino)-phenyl] propanamide} (Figure 2) is an oral, non-nucleoside compound related to the D-benzimidazole ribonucleosides. It is a potent and selective inhibitor of HCMV replication by inhibition of viral DNA concatemers processing (Reefschlaeger et al., 2001; Weber et al., 2001).
3.3.1 Mechanism of action
The mechanism of action of BDCRB and TCRB (Figure 8) does not need phosphorylation at the 5′ position and does not involve the inhibition of DNA synthesis (Krosky et al., 2002). It prevents the cleavage of high molecular weight viral DNA concatemers to monomeric genomic lengths (Underwood et al., 1998). Resistance mutations were found in HCMV genes UL56 and UL89 suggesting that BDCRB and TCRB target the encapsidation step (Krosky et al., 1998; Underwood et al., 1998). BDCRB partially inhibits the ATPase activity of pUL56 and the pUL89-associated nuclease activity at high concentrations (Scholz et al., 2003). Furthermore, it has been suggested that BDCRB causes HCMV terminase to skip the normal cleavage site and continue packing DNA until a second cleavage site is encountered 30 kb further (McVoy and Nixon, 2005). In GPCMV, monomer-length genomes are plentifully produced under BDCRB, but are slightly truncated at the left end (Nixon and McVoy, 2004). This is in accordance with the fact that BDCRB alters recognition and cleavage of DNA by the terminase complex.
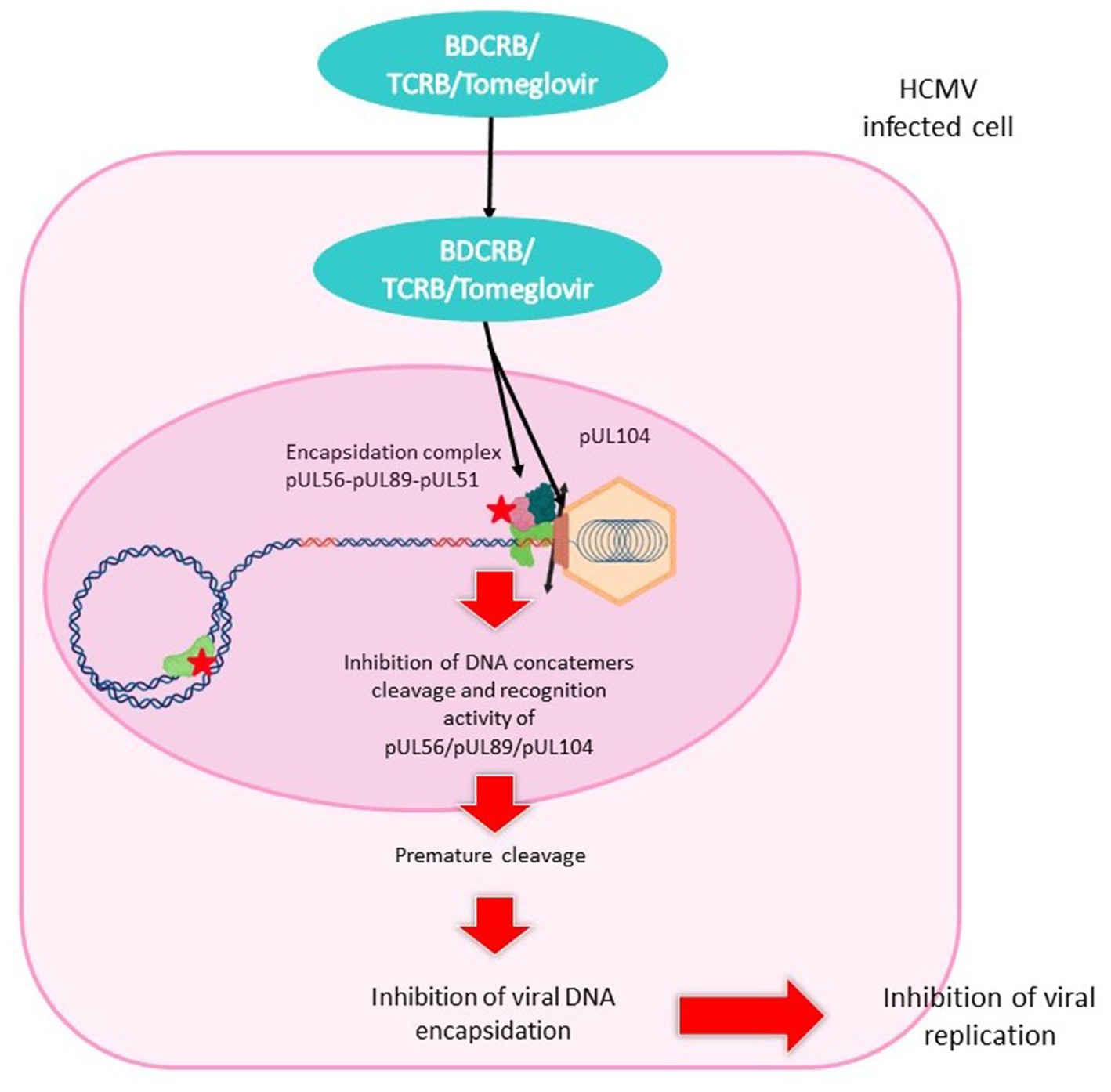
Figure 8. Mechanism of action of benzimidazol analogs. BDCRB, TCRB and tomeglovir enter the HCMV-infected cell and travel to the nucleus. They inhibit the encapsidation complex composed of pUL56, pUL89 and pUL51, and inhibit the portal protein pUL104. This mechanism leads to inhibition of DNA cleavage and concatemer recognition, or to premature cleavage. Encapsidation is interrupted and viral replication halted.
Tomeglovir mode of action (Figure 7) is the same as that of BDCRB and TCRB described above. This molecule targets HCMV-specific proteins required for cleavage and packaging of viral DNA, transforming high molecular weight viral DNA concatemers into monomeric genomes length (Reefschlaeger et al., 2001).
3.3.2 Preclinical tests
BDCRB and TCRB were effective against HCMV with EC50 of 0.7 μM and 2.9 μM in plaque assays, respectively. It was also proved that BDCRB and TCRB were inactive against HSV-1. However, the incorporation of Cl and Br into these two molecules improved dramatically their therapeutic index (Townsend et al., 1995). It was also shown that BDCRB is inactive against HHV-6 and HHV-7 (Yoshida et al., 1998). Another study demonstrated the inefficacy of BDCRB against HSV-1, HSV-2, VZV, HHV-8 but an activity against EBV (Williams et al., 2003). Furthermore, it was demonstrated that conformational changes in BDCRB structure could increase its spectrum against herpesviruses. Indeed, the L-analog of BDCRB was effective against HHV-6 (Prichard et al., 2011). Tomeglovir was effective against HCMV strains (EC50 = 0.52 ± 0.014 μM) (Reefschlaeger et al., 2001). GCV-resistant clinical isolates were also susceptible to tomeglovir (McSharry et al., 2001).
Interestingly, HCMV strain AD169 was more sensitive to benzimidazole than the strain Towne (Krosky et al., 2000). Although TCRB and BDCRB showed excellent activity in vitro, their glycosidic bonds are hydrolyzed in vivo to less active metabolites that reduce their activity (Good et al., 1994).
In addition, BDCRB showed synergy in combination with MBV, synergy at low concentrations and antagonism at higher concentrations with tomeglovir (Evers et al., 2002). Furthermore, tomeglovir had an antagonistic effect when combined with GCV (Evers et al., 2002).
The toxicity of benzimidazole analogs was tested in bone marrow cells. One hundred μM BDCRB inhibited cell proliferation by 20% over a 10-day period, while 100 μM GCV inhibited it by 52%. In other experiments on hematopoietic progenitor cell colony-forming assays, 100 μM BDCRB affected BFU-E and CFU-GM (burst forming units-erythroid and colony forming units-granulocyte/macrophage) by 31 and 47%, respectively. In contrast, GCV inhibited BFU-E by 54% and CFU-GM by 86%. However, TCRB was less effective than BDCRB. This study concludes that certain benzimidazole nucleosides are less toxic than conventional drugs (Reza Nassiri et al., 1996).
Additionally, in vitro study with BDCRB in guinea pig embryo or lung fibroblasts showed that GPCMV was sensitive to BDCRB (EC50 = 4.7 μM). BDCRB did not inhibit the formation of genome-sized GPCMV DNA, which was packaged but not protected from nuclease. Termini formed on GPCMV genome were altered by BDCRB. Then, BDCRB participated to the retention of C capsids in the nucleus (McVoy and Nixon, 2005). However, more recently, a GPCMV resistant to BDRCRB was generated and characterized. Genetic alterations were reported: an L406P substitution in GP89, the HCMV UL89 homolog; a 13.4 kb internal deletion of the GP131-GP143 non-essential ORFs; and a dramatic increase in the number of iterations of a 1 kb terminal repetitive sequence, from 0 or 1 to up to 9 at either genomic end (Ourahmane et al., 2018).
In animal model experiments, efficacy was evaluated in an ocular model of SCID-humanized mice infected with the Toledo strain of HCMV. BDCRB was administered at doses of 50 mg/kg for 28 days or 25 mg/kg twice daily for 1 week and once daily for 2 weeks. A slight but non-significant reduction in HCMV titers was observed in the 50 mg/kg group, and no reduction in mean titers was observed in the 25 mg/kg group. These results showed that BDCRB could only be active against HCMV at high concentrations. In the second experiment, HCMV-infected SCID-humanized retinal implants were treated with BDCRB or CDV for 28 days. Mice were treated with 75 mg/kg BDCRB from day one post-infection. BDCRB had no effect on reducing viral titers in retinal implant tissues. These results demonstrated the ineffectiveness of BDCRB in crossing the blood-ocular barrier in in vivo models. In addition, the same experiments were carried out with visceral organs (fetal thymus and liver) implanted in the kidney capsule. Doses of BDCRB blocked viral replication by around 2 to 3 log10 PFU/g (Kern et al., 2004a).
Tomeglovir was evaluated in MCMV-infected immunodeficient mice and reduced viral load in target organs in a manner comparable to GCV. Weight loss (consequence of viral infection) is reduced after tomeglovir administration (Reefschlaeger et al., 2001). Another study on murine model with per os administration of tomeglovir at dose ≥10 mg/kg showed similar results (Weber et al., 2001). A study in guinea pigs also demonstrated that peak plasma tomeglovir levels were 26.7 mg/mL 1 h after dosing. It reduced both viremia and DNAemia, as well as mortality following lethal GPCMV challenge in immunocompromised guinea pigs, from 83 to 17% (p < 0.0001). This study demonstrated the safety, pharmacokinetics and favorable therapeutic profiles of tomeglovir (Schleiss et al., 2005).
3.3.3 Clinical trials
Currently, no published clinical trial was performed to assess BDCRB, TCRB and tomeglovir in human (source: ClinicaTrials.gov).
3.3.4 Resistance
Resistance mutations to BDRCRB and TCRB (Table 2) are located in pUL56 (Q204R) and in pUL89 (D344E and A355T). If combined, these mutations showed a greater resistance to benzimidazole analogs than alone. Nevertheless, these mutations did not confer resistance to GCV (Krosky et al., 1998; Underwood et al., 1998; Evers et al., 2002). Additionally, mutations M360I in M89 exon II and P202A and I208N in M56 confer murine CMV resistance to tomeglovir. Mutation in M89 exon II had analogous mutations in HCMV pUL89 mentioned for BDCRB and TCRB but those in M56 did not have it in HCMV pUL56. Thus, pUL89 could be directly targeted by tomeglovir and pUL56 could compensate for restricted activities of Buerger et al. (2001). More recently, Chou (2017) highlighted new mutations in UL89 gene and UL56 responsible for tomeglovir resistance. In pUL89 N329S, T350M, H389N, N405D, D344E, C347S and V362M conferred moderate to high drug resistance. The mutation I334V did not conferred tomeglovir resistance but affected growth fitness when combined with N405D. Then, in pUL56, Q204R was shown as lower-grade resistance mutation (Chou, 2017).
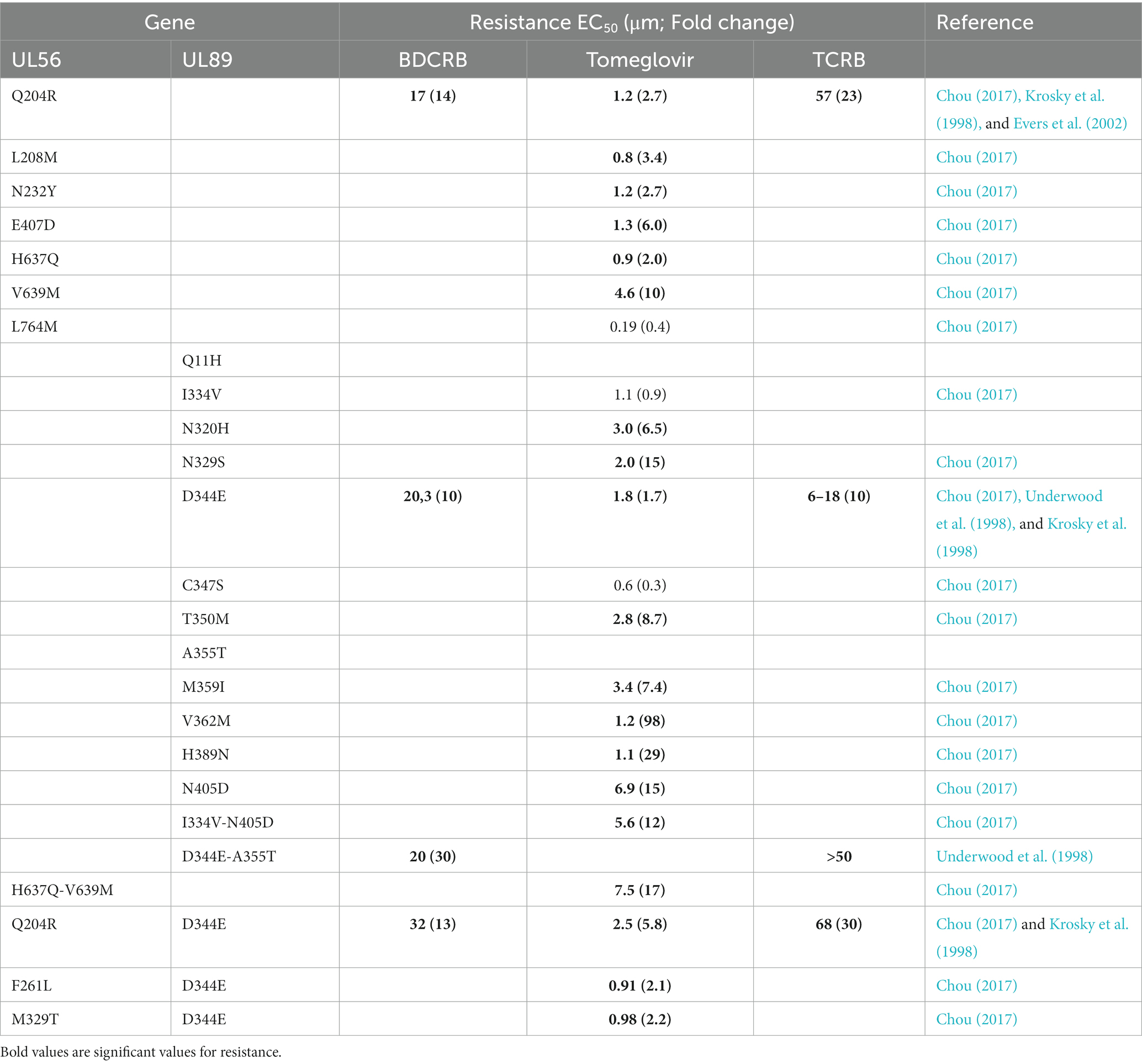
Table 2. Resistance mutations to benzimidazol analogs in UL56 and UL89 genes.
The L406P mutation described in GPCMV QP89 was more than 50 residues away from the positions of the confirmed resistance mutations in HCMV pUL89. This mutation does not confer significant resistance of GPCMV to BDCRB but may have a compensatory function in enhancing replication by making easier genomic cleavage at cleavage sites containing multiple repeats. Furthermore, deletion of the E region of HindIII is unlikely to contribute directly to resistance to BDCRB. Thus, the accumulation of terminal repeats could be a response to BDCRB pressure and the resulting increase in genome length resulted in compensatory deletion of the HindIII E region (Ourahmane et al., 2018).
BDCRB and TCRB do not show any cross-resistance with GCV because of their different mechanisms of action and their different gene targets. Surprisingly, a cross-resistance with MBV is responsible for an increase of 2-3-fold EC50 even if these molecules do not have the same mechanism of action. Indeed, BDCRB and TCRB are DNA processing inhibitors and MBV is a DNA synthesis inhibitor (Evers et al., 2002).
In addition, a mutation was described in pUL104, the portal protein of HCMV, which is colocalized with pUL56, in resistant strains to benzimidazole nucleosides. However, this L21F pUL104 mutation alone did not prove sufficient to ensure resistance of HCMV to BDCRB (Komazin et al., 2004; Table 2).
4 Host-targeting antivirals
Several molecules targeting cellular metabolism have antiviral activity by interfering with cellular components participating in the viral replication cycle. These components have various targets, efficacy, and share the absence of identified viral resistance.
4.1 Artemisinin derivatives: artesunate, artemisone, and TF27
Artemisinin is an antimalarial drug that is an active compound of A. annua (1972). Many derivatives of this drug were developed. In this context, Saokim Ltd. (Hanoi, Vietnam) synthesized artesunate (Figure 2), a semisynthetic drug of artemisinin. This compound is available as intravenous or oral formulation to treat life-threatening malaria access. In 2010, the World Health Organization (WHO) recommended this drug as quinine to treat severe malarial infections during the first trimester of pregnancy (McGready et al., 2012; Roussel et al., 2017). Besides this activity, artesunate (ART) was shown as a good inhibitor of HCMV infections in vitro (Efferth et al., 2002, 2008). The laboratory of Sensitive Biology Therapy (S.B.T) synthesized its trimeric derived compound, TF27 (Figure 2; Hutterer et al., 2015; Reiter et al., 2015; Hahn et al., 2018). Recently, LH54, the heterologous hybrid compound of artesunate was developed in the laboratory of S.B.T. and has also a good antiviral activity (Wild et al., 2020). All these artesunate derivatives have shown good efficacy.
4.1.1 Mechanism of action
Artemisin and its derivatives are primarily metabolized in dihydroartemisin (DHA) by cytochrome P-450 monooxygenase enzyme (CYP) 2B6 in human liver microsomes. This step is followed by the conversion into inactive metabolites via other enzyme systems. DHA has a half-life of about 45 min and is also an anti-malarial drug.
In HCMV infections, ART inhibits cellular transcription factors Sp1 and NF-κB (Hutterer et al., 2015). Indeed, its derivatives, TF79 and TF27, inhibit NF-κB signaling by approximatively 90% at concentration of 3 μM to 0.3 μM, with a correlation between the diminution of NF-κB pathway and the antiviral activity (Hahn et al., 2018). Previous studies have suggested that sustained NF-κB activation is necessary for viral replication (Hiscott et al., 2001; Figure 9).
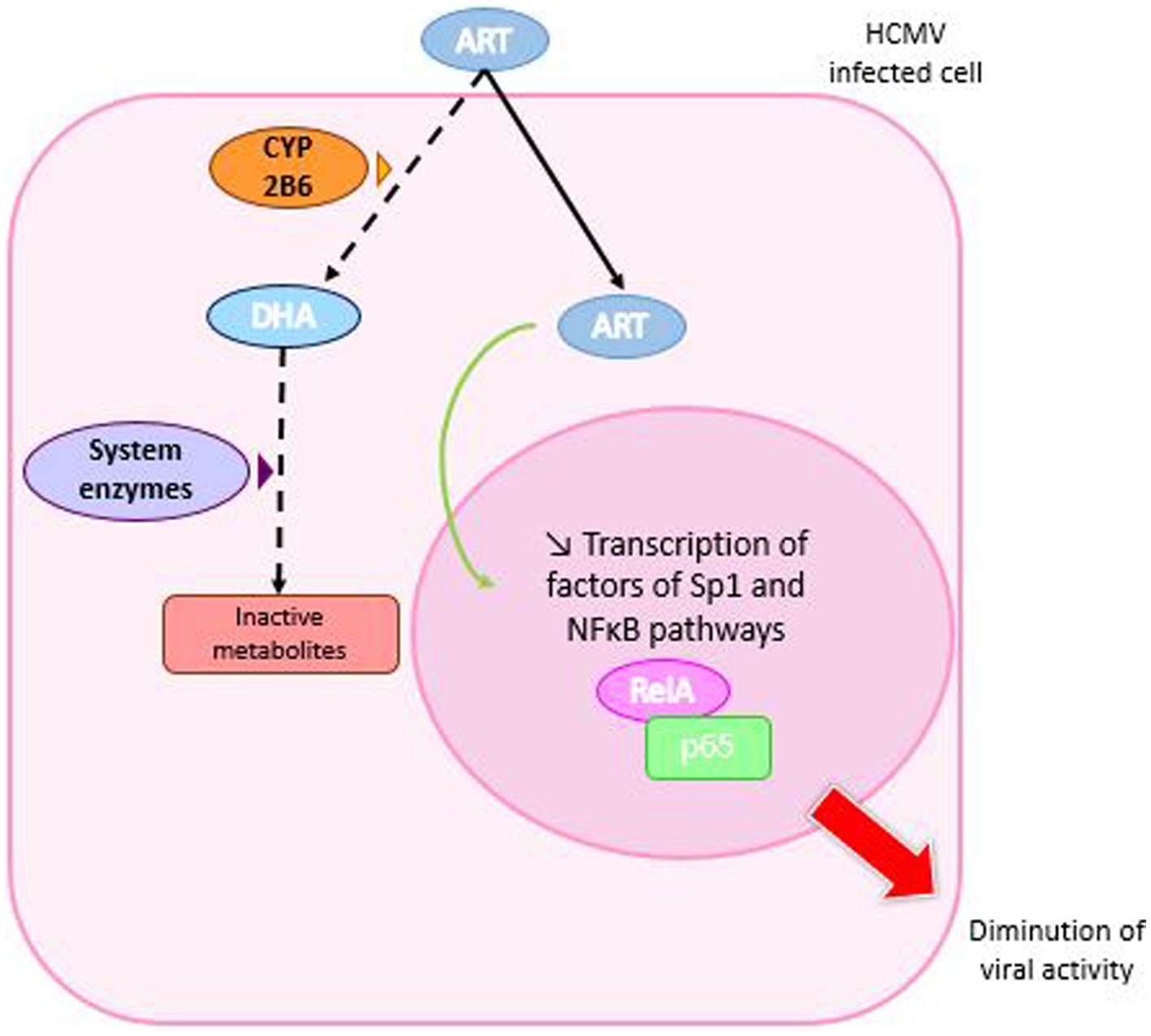
Figure 9. Mechanism of action of artemisinin derivatives. Artemisinin derivatives enter HCMV-infected cells and are metabolized to dihydroartemisin (DHA) by the cytochrome P-450 monooxygenase 2B6 (CYP 2B6) enzyme, which is then converted to inactive metabolites. In addition, ART moves to the cell nucleus where it inhibits the production of RelA and p65 involved in the NFκ-B pathway, which is necessary for viral replication.
4.1.2 Preclinical tests
Artemisinin derivatives are well tolerated in humans (Ribeiro and Olliaro, 1998; Adjuik et al., 2004). Rarely, slight and reversible adverse effects were observed such as first-degree heart block and neutropenia (Ribeiro and Olliaro, 1998; Adjuik et al., 2004). However, in animal models like mice, rats and dogs, neurotoxic effects were reported (Toovey, 2006). Studies showed that a shorter exposure to artemisinin derivatives with higher concentrations was less neurotoxic than a longer exposure with lower concentrations (Li et al., 2002).
ART was shown to be efficient against all types of herpesviruses, more than artemisinin. ART inhibits HCMV in vitro and in vivo (Efferth et al., 2002; Kaptein et al., 2006), both HHV-6 variants (Milbradt et al., 2009; Hakacova et al., 2013), Epstein–Barr virus (Auerochs et al., 2011) and Herpes simplex virus 1 (Efferth et al., 2008).
In addition, ART can be combined with other conventional anti-CMV drugs such as GCV, CDV and FOS to decrease risk of resistance mutation emergence. ART also shows a pronounced synergistic effect with MBV (Chou et al., 2011). Furthermore, its activity was confirmed in a model of 1st trimester placental villi infection (Morère et al., 2015). In combination with MBV, ART showed a synergistic effect at low concentrations, with IC50 of 0.25 and 2 μM for the two molecules, respectively. However, the combination with baicalein had an antagonistic effect (Morère et al., 2015). In the same year, Drouot et al. (2016) demonstrated that combining ART with GCV, CDV and MBV was associated with synergy, while combining it with FOS or LMV produced only moderate synergy.
Artemisin was also tested against HCMV in different ratios in combination with the anti-HCMV drugs BDCRB, LMV, GCV, CDV, BCV and MBV. This study revealed synergistic antiviral activity with no microscopically apparent cell toxicity or reduction in cell viability (Oiknine-Djian et al., 2019).
ART derivative, TF27, also showed an anti-HCMV activity at nanomolar concentrations (Hutterer et al., 2015; Reiter et al., 2015; Fröhlich et al., 2018). Besides, antiviral activity against HCMV infections was also shown in ex vivo placental villi explant model (Jacquet et al., 2020). Recently, Sonntag et al., demonstrated its antiviral efficacy in vivo by using an established model of murine CMV infection of an immunodeficient mouse strain Rag−/− (Sonntag et al., 2019). TF27 has a higher antiviral activity than ART: the EC50 of 0.04 ± 0.01 μM against HCMV strain (Hutterer et al., 2015) was 100 fold lower than the EC50 of artesunate (Hahn et al., 2018).
4.1.3 Clinical tests
ART is a good inhibitor for clinical use in the treatment of drug-resistant HCMV infection. The first report of treatment of CMV infection with ART in a HCT recipient with resistance to foscarnet and ganciclovir (DNA polymerase L776M mutation) dates back to 2008. Treatment with GCV, CDV and intravenous immunoglobulin had failed. ART was started at a dose of 100 mg daily after other treatments had been discontinued. A favorable response was observed, followed by a rapid reduction in viral load and improvement in hematopoiesis. During the 30 days of treatment, there were no adverse events and no increase in viremia. The patient received a third transplant with a recurrent episode of viremia but controlled by a new antiretroviral treatment regimen. Nevertheless, retinitis was diagnosed during treatment, reflecting the limited penetration of ART into the eye. Combining ART with GCV resolved this local infection and controlled the viral load. This study demonstrates the potential of ART to control CMV infection (Shapira et al., 2008).
ART was used in a case series of 6 SCT recipients as a preventive treatment for CMV infection to calculate its antiviral efficacy by studying viral kinetics. Two patients showed a decrease in viral load (0.8 to 2.1 log after 7 days). Antiviral efficacy was described as heterogeneous, ranging from 43 to 90%, and depended on the basic growth dynamics of the virus (Wolf et al., 2011).
In another study, ART was evaluated in five patients with resistant CMV infection. ART was unsuccessful in two cases of severe CMV disease with high CMV viral load and pulmonary involvement. However, these patients also suffered from diseases (Wegener’s granulomatosis and Hodgkin’s lymphoma) that may have accounted for some of the deaths. In conclusion, ART may be useful in the treatment of mild CMV disease due to multidrug-resistant strains. However, further data are needed on the risk factors associated with ART failure. In addition, it should be noted that ART is not sufficient to treat serious CMV disease with pulmonary involvement due to its poor diffusion in lung tissue, as has been reported in animal models (Zhao and Song, 1993; Germi et al., 2014).
4.1.4 Resistance
To date, no resistance to artemisinin derivatives has been reported in CMV.
4.2 Flavonoids
Flavonoids are metabolites found in fruits and vegetables with high biological activity and low toxicity. Flavonoids with well-classified structures and well-defined structure–function relationships include flavans, flavanones, flavones, flavanonols, flavonols, catechins, anthocyanidins, isoflavones and chalcons (Tsao, 2010). Over 5,000 flavonoids were defined as molecules with potential health benefits as antioxidative, anti-inflammatory, antitumoral, antiviral and antibacterial effects (Middleton, 1998; Beecher, 2003; Cazarolli et al., 2008). Besides, flavonoids can specially modulate activities of cellular enzymes and inhibit protein kinases (Middleton, 1998). These metabolites are therefore a real option for current antiviral therapies. Indeed, it was shown that kaempferol inhibits herpes simplex virus (Amoros et al., 1992) and that baicalein and genistein interact with the first steps of HCMV infection (Evers et al., 2005). In an exploratory in vitro study, Cotin et al. (2012) showed that baicalein and quercetin were the most potent flavonoids to inhibit HCMV in vitro. Their combination had an additive effect. In addition, the combination of these two molecules with chalcone to reduce toxicity was tested against HCMV. The result was a synergistic effect for baicalein, while an antagonistic effect was observed with quercetin (Andouard et al., 2016). Both molecules were also combined with MBV, and quercetin did not improve the efficacy of MBV alone, unlike baicalein, which reduced infection by 90% at low concentrations (2.2 μM baicalein; 1 μM MBV; Morère et al., 2015). In this review, quercetin and baicalein will be explored for their potential antiviral activity.
4.2.1 Quercetin
Quercetin or (3,3′,4′,5,7-pentahydroxy-2-phenylchromen-4-one; Figure 2) is the main representative of the flavonoid subclass, flavonols. The fruits and vegetables with the highest concentration of quercetin are apples, cherries, onions, asparagus and red leaf lettuce (Nishimuro et al., 2015). It is also found in herbs such as licorice.
In food, quercetin is present as quercetin glycosides that are hydrolyzed and released as aglycone. Then, aglycone is absorbed and metabolized into glucuronidated, methylated and sulfated derives (Kawabata et al., 2015). However, the stability of quercetin and its derivatives in the organism can be influenced by pH, temperature, metal ions and other compound as glutathione (GSH) (Boots et al., 2005; Moon et al., 2008). This could affect the efficacy of the molecule.
4.2.1.1 Mechanism of action
Quercetin was shown to act by inhibition of early viral proteins IE-1and IE-2 expression (Cotin et al., 2012). Through the downregulation of IE-2 of VZV and HCMV, it inhibits viral lytic gene expression and replication (Kim et al., 2020) and modulates NF-κB, mitochondrial and ROS pathways (Hung et al., 2015; De Oliveira et al., 2016). Quercetin also inhibits the activation of IRF3 and NF-kB induced by HSV-1 infection in a TLR3-dependent manner that results in a lower production of TNF-α (Lee et al., 2017). Quercetin was then shown to prevent EBV-induced B cell immortalization and proliferation of lymphoblastoid cell lines by interrupting the dialectic between IL-6 and STAT3, promoting autophagy and reducing ROS levels and p62 accumulation (Granato et al., 2019; Figure 10).
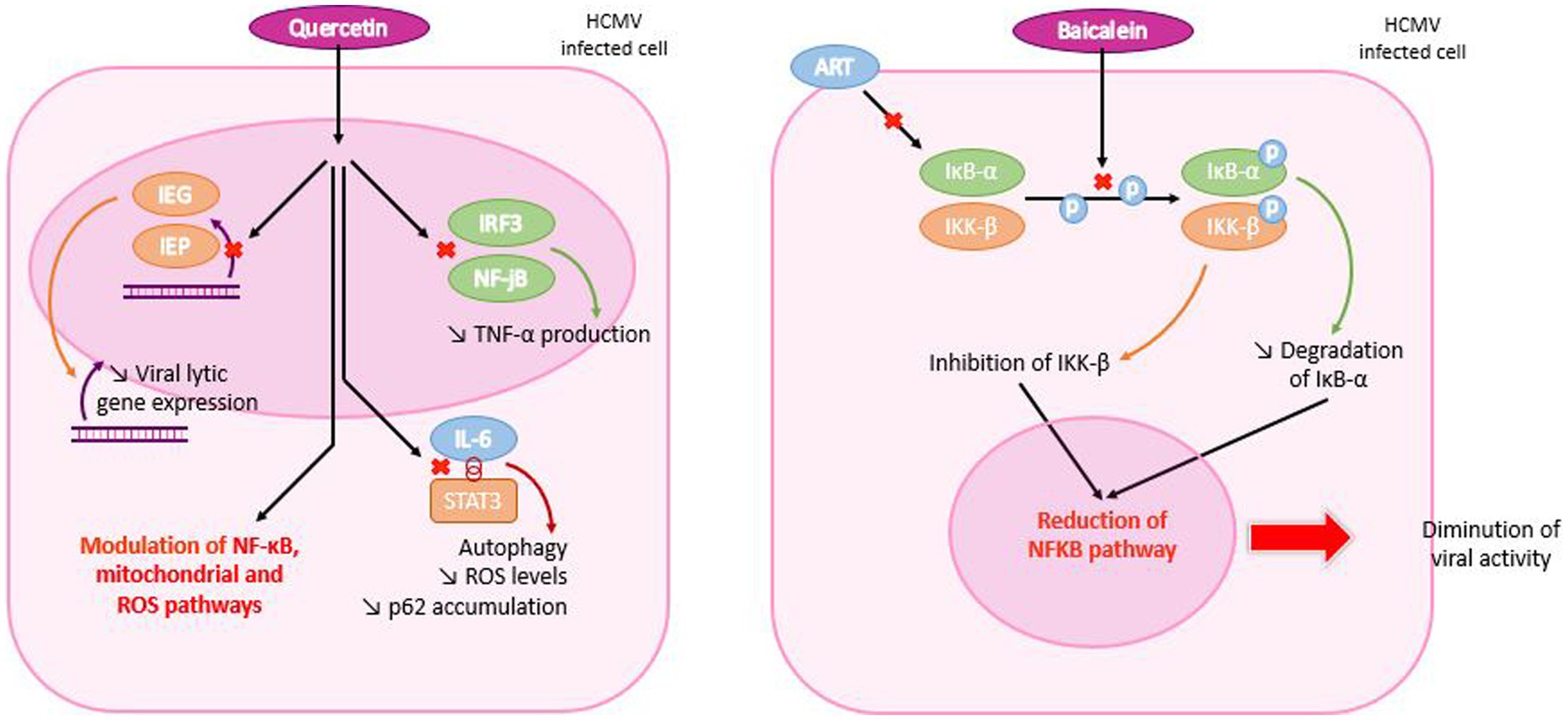
Figure 10. Mechanism of action of quercetin and baicalein in HCMV infected cells. Quercetin enters the HCMV-infected cell and travels to the nucleus, where it inhibits early viral protein and TNF-α production. It also inhibits contact between interleukin 6 (IL-6) and STAT3, resulting in reduced ROS levels and p62 accumulation. Both mechanisms are responsible for modulating NF-κB, mitochondrial and ROS pathways. In the case of baicalein, the molecule penetrates the cell and inhibits phosphorylation of IκB-α and IKK-β, which are degraded and inhibited, respectively. Each molecular mechanism leads to a reduction in the NF-κB pathway and, consequently, in viral activity.
4.2.1.2 Preclinical tests
Many studies showed the antiviral activity of quercetin. Indeed, quercetin inhibits HBsAg and HBeAg secretion in Hepatitis B virus infected cells (Wu et al., 2007). It is also an active agent against HIV-1 reverse transcriptase, protease and α-glucosidase with an EC50 value of 60 μM (Yu et al., 2007).
Quercetin was tested in vitro against herpesvirus, reducing intracellular replication of HSV-1 and HCMV. The antiviral activity against HCMV infected cells was 4.8 μM and 145 μM against HSV-1 (Cotin et al., 2012). In 2022, a formulation of polyxamer 188 and quercetin (QP188) amplified the in vitro GCV antiviral activity against HCMV. Indeed, QP188 was tunable, bioactive and rapidly internalized in NIH/3 T3 cells. This formulation had a dose-dependent activity combined synergistically with GCV. These results could be interesting for finding means to reduce GCV toxicities (Kjar et al., 2022).
4.2.1.3 Clinical study
In 2018, an herbal treatment (Gene-Eden-VIR/Novirin) composed of five ingredients including quercetin was tested in a clinical trial for the treatment of oral herpes. The study included 68 participants who took 1 to 4 capsules a day for an average duration of 10.4 months. Efficacy was assessed from symptom onset to complete resolution and also included analysis of recurrence rates. Treatment was compared with two conventional drugs: valaciclovir (VAVC) and aciclovir (ACV). Gene-Eden-VIR/Novirin was more effective in reducing the number and duration of oral herpes epidemics, and more secure than ACV and VAVC. Gene-Eden-VIR/Novirin reduced the duration of outbreaks from 5.83 days to 3.21 days in the treated group (p < 0.0001). In addition, 46.4% of patients on herbal treatment were relapse-free (p < 0.0001), and no adverse events were observed (Polansky et al., 2018). However, results of this study must be confirmed with further investigations. The same comparison was done with famciclovir (FCV) in 2016 (Polansky et al., 2016).
4.2.1.4 Resistance
Currently, no resistance mutation to quercetin was documented.
4.2.2 Baicalein
Baicalein (5,6,7-trihydroxyflavone; C15H10O5) (Figure 2) belongs to the flavone sub-family of flavonoids. This molecule is isolated from the roots of Scutellaria baicalensis with different properties: antioxidant, anti-inflammatory, anticancer, antidiabetic, antithrombotic, anxiolytic, anti-convulsive, cardioprotective, hepatoprotective and neuroprotective agent (Chang et al., 2016; Gao et al., 2016; Dinda et al., 2017; Cristelli et al., 2019; Tuli et al., 2020).
4.2.2.1 Mechanism of action
Pretreatment with baicalein failed in suppressing viral replication in cells while post-treatment was effective. These results suggest that baicalein may be effective at the post-entry stage of viral infection (Luo et al., 2020). Previous studies indicated that baicalein had an inhibitory effect on NF-κB activation induced by pathological factors (Li et al., 2016).
In vitro, baicalein has been shown to act on IκB-α and therefore to have an antagonistic effect with ART, as they share the same target, which could lead to competitive inhibition (Morère et al., 2015). In agreement, Luo et al., reported that baicalein blocks NF-κB activation by inhibiting phosphorylation of IKK-β and IκB-α. By reducing IκB-α degradation, baicalein could inhibit viral replication (Luo et al., 2020). Thus, HSV-1 infections are prevented by a dual mechanism: the suppression of IKK-β phosphorylation and the decrease of NF-κB activation. This study also demonstrated that baicalein inactivates HSV-1 particles in a direct manner (Luo et al., 2020). However, further studies are needed to explain how baicalein acts on IKK-β phosphorylation (Figure 10).
4.2.2.2 Preclinical tests
Baicalein has a poor oral bioavailability and a low aqueous solubility, which are the major disadvantage of this molecule. Studies of oral administration of baicalein have demonstrated that it is glucuronized in the intestinal wall and livers of rats and humans (Nagashima et al., 2000; Zhang et al., 2005, 2007). Additionally, baicalein is well absorbed by the small intestine and stomach (Taiming and Xuehua, 2006).
Some studies demonstrated baicalein is metabolized to baicalein and baicalein 6-O-sulfate in blood (Muto et al., 1998; Zhang et al., 2004; Dou et al., 2011). Following intravenous administration in rats, 75.7% of circulating baicalein in blood was as conjugated metabolites form (Lai et al., 2003). The bioavailability of baicalein in monkeys reached 23.0% after oral and intravenous administrations (Tian et al., 2012).
Cotin et al. (2012) demonstrated that baicalein inhibits in vitro CMV early proteins production. The inhibition of the tyrosine kinase activity of the EGF factor was already proved in a previous study (Evers et al., 2005). Additionally, combinations of quercetins and baicalein revealed additive effects particularly when baicalein was added at fixed quercetin concentrations. That reflected the probably higher efficacy of baicalein (Cotin et al., 2012).
4.2.2.3 Clinical study
A randomized, double-blind, single-dose phase I trial of baicalein (100–2,800 mg) was conducted in 72 healthy adults. Analysis of baicalein and baicalin (baicalein’s 1st metabolite) was performed by liquid chromatography–tandem mass spectrometry on various biological fluids. Urinary clearance of baicalein and baicalin was 1, and 27% of baicalein was eliminated unchanged in feces. Eleven treatment-related side effects were recorded, but these were defined as moderated and resolved without further treatment. Baicalein is well tolerated by healthy patients, and no liver or kidney toxicity was observed (Li et al., 2014).
In 2021, another trial was conducted by Li et al. using data from the 2014 Phase I clinical trial. It was a randomized, placebo-controlled, multi-dose, and escalating trial of 36 healthy subjects who received 200, 400, and 600 mg of baicalein or placebo tablets. The drug was administered once on days 1 and 10, and three times daily from day 4 to 9. To analyze the pharmacokinetics of baicalein, blood and urine samples were taken from the 600 mg group. This study showed that baicalein tablets were safe and well tolerated. Mild adverse effects were observed, but none were not resolved. Maximum plasma concentrations were observed within 2 h of baicalein administration. Urinary excretion of baicalein and its metabolites peaked in 2 h, followed by a tendency to double the peak in 12 h. These results support the launch of a Phase II clinical trial (Li et al., 2021).
4.2.2.4 Resistances
No resistance mutation to baicalein was reported in CMV.
4.3 Anti-COX-2
COX-2 inhibitors (cyclooxygenase-2) have been developed to reduce the adverse effects associated with the use of aspirin and indomethacin (gastric hemorrhage or perforation and hepatotoxicity; Nagi et al., 2015). Structural studies have highlighted the inhibitory activity of COX-2 as a heterocyclic or carbocyclic structure and substituting sulfonamide or methylsulfonyl in position para on one of the aromatic rings (Chakraborti et al., 2010). Nevertheless, some anti-COX-2 agents have been shown to cause serious adverse effects and have been withdrawn from the market (Mukherjee, 2002). Thus, certain anti-COX-2 agents have been identified in plants with fewer side effects than polyphenols. In this category, chalcones are a sub-category of the flavonoid family (Cerella et al., 2010).
4.3.1 Mechanism of action
HCMV has been shown to increase the amount of COX-2 enzyme in infected cells, establishing an inflammatory state to promote replication (Speir et al., 1998). As part of the analysis of anti-inflammatory activity, certain chalcone derivatives and those of 2′-hydroxychalocone were determined to inhibit COX-2 and the production of PGE2 catalyzed by this enzyme. Chalcones appear to act before the early stage of viral replication, by reducing the production of IE-1 and IE-2 proteins (Andouard et al., 2021).
4.3.2 Preclinical study
Celecoxib, a COX-2 inhibitor, was evaluated against HCMV in in vitro and in vivo (mouse) models of medulloblastoma. Its efficacy was compared with that of VGC. Both drugs inhibited HCMV replication in vitro, inhibited PGE2 production and reduced growth of medulloblastoma tumor cell in vitro and in vivo (Baryawno et al., 2011).
In a study carried out in 2021, the anti-HCMV activity of new 2′-hydroxychalcone compounds was assessed. These molecules were designed to inhibit PGE2 synthesis. To achieve this, a COX-2 pharmacophore (sulfonamide motif) and other substituents (chlorine, fluorine and methyl group) were introduced into the 2′-hydroxychalcone backbone (Zarghi and Arfaei, 2011). The selection of 4 anti-COX-2 agents was based on their significant activity against PGE2 production. However, three of them proved to be toxic to cells and one had a CC50 of 1,500 μM in growing cells and 185 μM in static cells, which was related to indomethacin (a non-specific cyclooxygenase inhibitor). Toxicity was potentially increased by the presence of the SO2NH2 group in the molecules, whereas the presence of the chlorine atom reduced it. These 2’hydroxychalcones were defined as less toxic than the tri-hydroxychalcones (Cotin et al., 2012). The molecules were tested against strain AD169-GFP. EC50s were up to 16 times higher than GCV (EC50 = 19.6 ± 10.1 μM; 8.6 ± 10.1 μM; 10.5 ± 3.6 μM; 22.1 ± 7.7 μM; 15.1 ± 5.9 μM for the 4 chalcones and indomethacin respectively). EC50s were also determined on clinical isolates and proved effective against resistant strains. Three chalcones tested proved capable of inhibiting IE1-72 production (Andouard et al., 2021).
In addition, a synthesized anti-COX-2 was combined with other anti-CMV drugs such as GCV, MBV, baicalein, quercetin and ART. This resulted in a synergistic effect with MBV or baicalein. An additive effect was demonstrated with GCV or ART, and an antagonistic effect was observed with quercetin (Andouard et al., 2021).
4.3.3 Clinical study
No clinical study was done for anti-COX-2 molecules against HCMV (source: ClinicaTrials.gov).
4.3.4 Resistances
So far, no resistance mutations in HCMV genome were defined.
5 Immunomodulating molecules
5.1 Leflunomide
Leflunomide (LEF) (HWA 486; A77 1726, Arava®) or N-(4-trifluoromethylphenyl)-5-methylisoxazol-4-carboxamide (Figure 2) is an antirheumatic agent used to treat rheumatoid arthritis. It was also demonstrated as effective in HCMV infection in HCT and renal transplant.
5.1.1 Mechanism of action
After administration, LEF is converted to an active matabolite, teriflunomide (A77 1726) that blocks lymphocyte enzyme dihydroorotate dehydrogenase and the pyrimidine biosynthetic pathway (Williamson et al., 1996). This activity results in a lower T-cell proliferation and changes in immune response (Dayer and Cutolo, 2005; Chong et al., 2006). At a later stage in virion assembly, it prevents viral nucleocapsides from acquiring integument. LEF has a dose-dependent effect on the infectious production of HCMV (Waldman et al., 1999a). Unlike polymerase inhibitors, LEF did not inhibit HCMV DNA replication, but it did appear to interfere with tegument assembly by inhibiting protein phosphorylation (Xu et al., 1995; Waldman et al., 1999b; Figure 11).
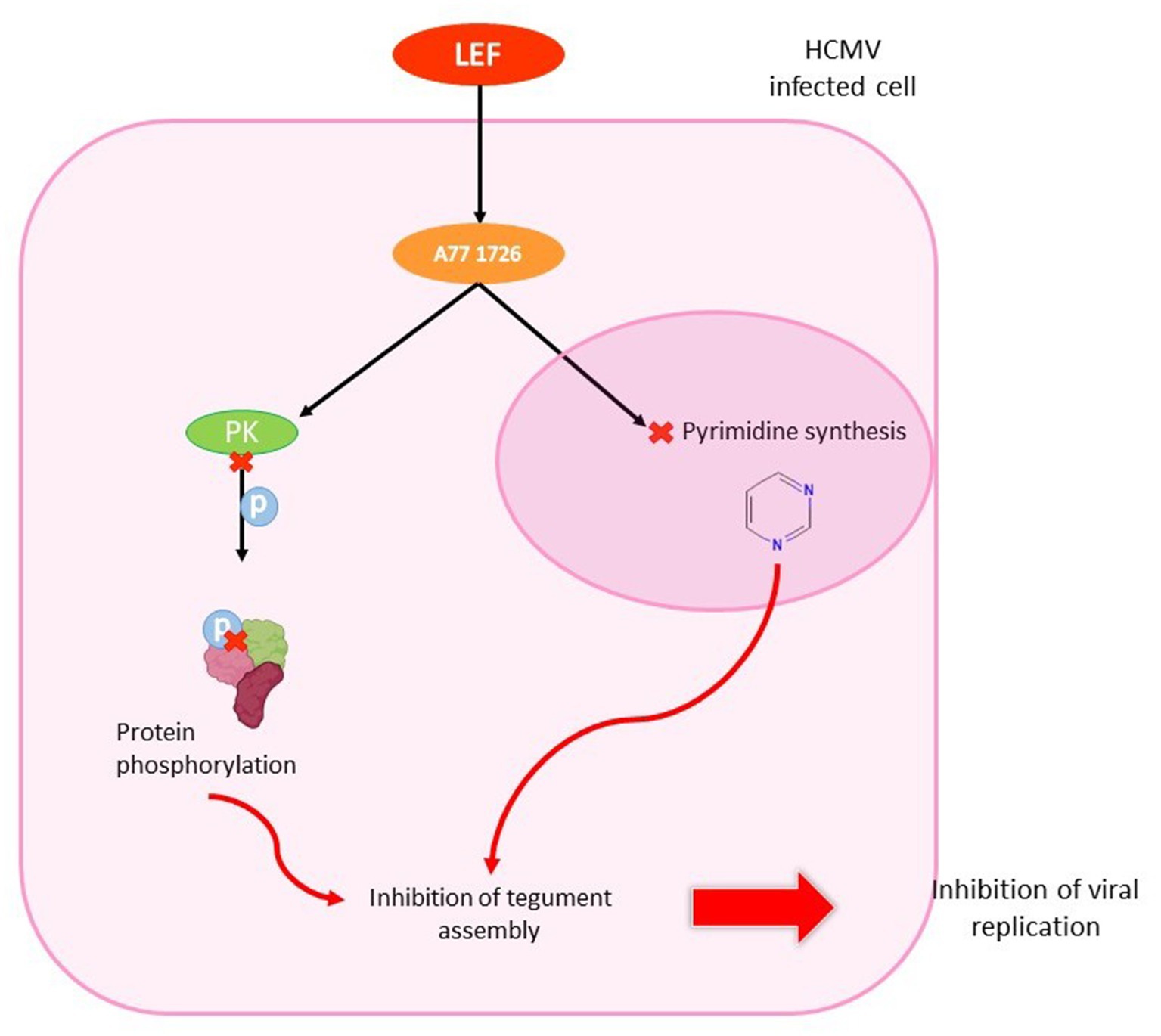
Figure 11. Antiviral effect of leflunomide in HCMV infected cell. LEF enters the HCMV-infected cell and is converted to teriflunomide (A77 1726). The metabolite inhibits protein kinase, which is unable to phosphorylate proteins. It also penetrates the nucleus to inhibit pyrimidine synthesis. These two mechanisms are involved in inhibiting tegument assembly, thereby stopping viral replication.
5.1.1.1 Preclinical tests
HCMV isolates in human fibroblats and endothelial cells, including multidrug-resistant viruses (EC50 40–60 μM), are inhibited by LEF (Waldman et al., 1999b). LEF also inhibits HSV-1 with same mechanism of action than with HCMV (Knight et al., 2001).
Furthermore, the LEF was evaluated in vivo using animal models. Immunodeficient rats were inoculated with rat CMV (Maastricht strain of RCMV) and administered 15 mg/kg/day LEF for 14 days, 10 mg/kg/day GCV for 5 days, or a drug-free vehicle. Plaque assay from tissue homogenates (salivary glands, spleen and lung) showed a decrease of 75 to 99% of viral load in the organs of animals treated with LEF, and 85 to 99% in those treated with GCV. Thus, LEF is an effective agent in decreasing viral load in vivo (Waldman et al., 1999a). After that, it was demonstrated the efficacy of LEF in an allogenic cardiac transplant model of RCMV infection with low toxicity (Chong et al., 2006).
5.1.1.2 Clinical study
Due to its synergy with calcineurin phosphatase inhibitors and its inhibitory effects on herpesvirus replication, LEF was presented as a promising drug for experimental transplantation (Williams et al., 2002). Fifty-three recipients of LEF were analyzed in a retrospective study. A single-dose pharmacokinetic study was performed in stable renal transplant recipients with a target serum concentration of 100 μg/mL to require a loading dose of 1,200 to 1,400 mg over a 7-day period. Anemia in the renal transplant patients and increase of liver enzymes in liver-transplanted patients were the major observed toxicities (Williams et al., 2002). Another study reported same side effects and diarrhea after a therapy with a mean duration of 3.5 months. The recommended dose of LEF was 100 mg/day for 5 days followed by 40 mg/day, based on the serum metabolite levels of A77 1726 teriflunomide (Avery et al., 2010).
In one study, John and colleagues analyzed 17 patients who underwent kidney transplantation and were infected with CMV. Patients were treated with monitored doses of leflunomide. Among these 17 patients, 88% responded clinically to leflunomide therapy with viral clearance in the blood and healing of the organs involved. The cost of treatment was cheaper than that of ganciclovir: 64 $ for 6 months against 721 $ for 2 weeks, respectively, (John et al., 2005).
Three cases of resistant HCMV infections were reported with LEF treatment. It was showed that LEF is not efficient enough in monotherapy and should be combined with GCV or FOS to better control CMV infection. It was also only used for CMV maintenance therapy (El Chaer et al., 2016). As an oral treatment, LEF is also a convenient alternative that does not need to stay in hospital to reach undetectable viral load (Gómez Valbuena et al., 2016). Then, after lung transplant, LEF was assessed in a case of drug resistant CMV retinitis. In spite of intravitreal FOS administration and oral VACV, HCMV disease progressed. Oral LEF helped in control of retinitis and allowed cessation of intravitreal treatment. No recurrence of infection was noticed (Rifkin et al., 2017).
A more recent study on case series assessed LEF in patients treated with GCV and FOS with adverse effects reported in 50% of cases. In 66.67% of cases, resistance mutations to polymerase inhibitors were present before LEF treatment. LEF was prescribed to treat HCMV infection in 75% of patients and as secondary prophylaxis in 25% of them. A primary reduction of HCMV viremia was observed after the beginning of LEF treatment in 77.7% of recipients but was transient in 22.2%. In 58.3% of recipients, LEF suppressed HCMV infection for long-term. Adverse effects were responsible for treatment discontinuation in 25% of cases. This study showed that LEF can be an effective treatment for transplant recipients with GCV-resistant infections, whether alone or combined with other drugs, even though the small number of subjects was a limitation. It can also be used as a secondary prophylaxis (Silva et al., 2018). LEF was also proposed in combination with hyperimmune globulins in cardiothoracic grafts and was associated with decreasing viremia (Santhanakrishnan et al., 2019).
In other hand, there is few numbers of studies with LEF in allogeneic HCT. One reported that LEF had efficacy in HCMV clearance in 38% of cases. Nevertheless, treatment significantly succeeded (53%; p = 0.022) only when LEF was used in patients with HCMV viral load <2.103copies/mL. Furthermore, it was demonstrated as ineffective in patients with terminal organ disease. Thus, LEF could be used in prophylaxis in stem cell transplants (Gokarn et al., 2019).
5.1.1.3 Resistance
Currently, there is no reported resistance mutation to leflunomide in HCMV.
5.2 mTOR inhibitor: everolimus
Everolimus (SDZ RAD; Certican®, ZORTRESS®; EVR) or 40-O-(2-hydroxyethyl)-rapamycin (Figure 2) is an oral mammalian target of a sirolimus-derived rapamycin inhibitor. It is used in immunosuppressive therapy after SOT. FDA approved it for the prevention of rejection in kidney transplant recipients at low to moderate risk (Gabardi and Baroletti, 2010).
5.2.1 Mechanism of action
Intracellular immunophilin (FKBP12) is bound by EVR, but it does not inhibit calcineurin, it binds to the mechanistic target of Rapamycin (mTOR). It is at the origin of the inhibition of a multifunctional serine–threonine kinase, preventing both the synthesis of DNA and proteins that leads to a cell cycle shutdown. More precisely, after stimulation of the IL-2 receptor on the activated T-cell, EVR inhibits p70S6 kinase which acts at a later stage in the T-cell mediated response (Roy and Arav-Boger, 2014). EVR inhibits HCMV through improved CD8+/CD4+ T cell responses specific to HCMV (Havenith et al., 2013; Roy and Arav-Boger, 2014; Figure 12).
Figure 12. Indirect antiviral action of everolimus in HCMV infected T-cell. EVR binds to the interleukin-2 receptor (IL-2R) on HCMV-infected T cells. The molecule enters the cell and binds to the immunophilin FKBP12. The complex binds to mTOR, which is unable to phosphorylate the p70S6 kinase. The kinase does not phosphorylate S6, which is involved in DNA and protein synthesis. HCMV replication is inhibited by cell cycle arrest.
5.2.1.1 Preclinical tests
So far, no preclinical test of the in vitro anti-HCMV efficacy of EVR was done because it is not the primary function of this molecule.
5.2.1.2 Clinical study
The effectiveness of EVR against HCMV has been demonstrated for several years. A multicenter Phase III trial included 634 heart transplant patients receiving immunosuppressive therapies (1.5 mg/day EVR; 3 mg/day EVR or azathioprine). The EVR groups had significant lower HCMV incidence (p < 0.001). Thus, the use of EVR suggests an additional benefit in cardiac transplant recipients as it is linked to lower rates of HCMV infection, syndrome or organ damage (Hill et al., 2007). In accordance with these results, same doses of EVR were assessed in heart and renal transplants recipients compared to mycophenolic acid (MPA). A significantly lower HCMV incidence was observed in the EVR group at 3.0 mg (3%) than in the MPA group (7%) (p < 0.04). The same observation was made for organ involvement in the EVR 1.5 mg group (0.9%) than in the MPA group (3%) (p < 0.04). Therefore, the EVR was associated with a decrease in EVR events compared to the MPA (Brennan et al., 2011). In a study of data from 3 randomized trials of de novo cardiac transplant recipients, Kobashigawa et al. reported that EVR was linked to a significantly lower incidence of HCMV infections compared to azathioprine and mycophenolate mofetil by combining its immunosuppressive efficacy with antiproliferative effects that may have a positive impact on long-term results (Kobashigawa et al., 2013).
In another study, EVR was compared with valganciclovir (VGC) in heart transplant recipients. EVR was introduced at 1.5 mg/day to discontinue the use of mycophenolate mophetil in combination with VGC that caused neutropenia. HCMV antigenemia was negative even after discontinuation of VGC and both renal function and neutrophil counts were normalized. No major side effects or rejection due to EVR were observed. Thus, EVR was described as an alternative or additive option in immunosuppressive therapy for heart transplant recipients because of maintenance of immune tolerance, prophylactic potency against HCMV, reduced myelosuppression and potential sparing of renal function (Imamura et al., 2012).
The impact of EVR on HCMV infection at both systemic or pulmonary level was also evaluated in lung transplant recipients (n = 32 patients). Eighteen patients were on EVR-immunosuppressive regimens. No differences were described in HCMV viremia occurrence between EVR-based and EVR-free immunosuppressive regimens. However, patients with EVR treatment experienced fewer high-load HCMV episodes defined as ≥105 copies/mL during EVR administration. It validate the reduction of HCMV events in EVR-based regimens transplanted patients, as lung transplant recipients (Rittà et al., 2015).
The international randomized phase IV TRANSFORM trial (NCT01950819) was conducted in de novo renal transplant patients randomized to RVE with reduced exposure to CNI or MPA with standard exposure to CNI, treated with induction and corticosteroids. EVR caused more adverse reactions than MPA, such as hyperlipidemia, interstitial lung disease, peripheral edema, proteinuria, stomatitis/mouth ulceration, thrombocytopenia and wound healing complications. However, EVR has been associated with viral infections less frequently than MPA. Indeed, HCMV infections and HCMV syndrome were lower (8.1% vs. 20.1%; p < 0.001 and 13.6% vs. 23.0%; p < 0.044 respectively). The same result was observed for BKV infections. EVR was more often stopped due to rejection or delayed healing (Tedesco-Silva et al., 2019). Another phase IV trial (NCT02096107) with EVR in kidney transplants was also conducted and reported that EVR with low tacrolimus exposure produced related efficacy to tacrolimus and MPA with significantly lower BK and HCMV levels (Taber et al., 2019). A more recent Phase IV trial was conducted with 186 seropositive HCMV kidney recipients randomized (1:1) to receive EVR or MPA in combination with basiliximab, cyclosporine and steroids. In seropositive recipients, HCMV DNAemia is prevented by EVR treatment until it is no longer tolerated or stopped (Kaminski et al., 2022a). In addition, the same authors reported that the T-cell phenotype may offer a new biomarker for predicting post-transplant infection and classifying patients who should be eligible for EVR treatment (Kaminski et al., 2022b).
5.2.1.3 Resistances
Currently, there is no resistance mutation to EVR that was reported in CMV.
6 Immunoglobulins
Two types of immunoglobulins are available in therapy: intravenous administered immunoglobulins (IVIG) and hyperimmune immunoglobulins (CMV-HIG). We will focus on HCMV-specific immunoglobulins which are able to neutralize viral infectivity (Aiba et al., 2017). Immunoglobulins may be used in preventing congenital infections or in association with other antivirals to cure HCMV infection in transplantation (El-Qushayri et al., 2021).
CMV-HIG are obtained by purification of adult human plasma-derived immunoglobulin products in pools selected for high levels of anti-HCMV antibodies. IVIG are acquired from the plasmas of healthy blood donors on the basis of a high level of antibodies against HCMV (Maniez-Montreuil et al., 1984). Using a correlation test between HCMV antibody titer and viral neutralization titer, it was shown that HIG has a higher level of anti-HCMV IgG than IVIG (Germer et al., 2016; Schampera et al., 2017). Currently, there is two options of HCMV-HIG which are, respectively, authorized in United States and Europe: Cytogam® (CMVIG CG; CSL Behring, Berne, Switzerland) and Cytotect CP® (Biotest AG).
Cytogam® is an HCMV-HIG derived from human plasma with high titers of anti-HCMV antibodies [112,5 PEIU/mL (Paul Ehrlich Institute Units)] (Germer et al., 2016). It contains a standardized amount of Ig (5 ± 0.1%). In the United States, it is approved for the prophylaxis of HCMV disease in heart, liver, lung, kidney and pancreas transplant recipients.
Cytotect CP® contains 5% Ig (50 mg/mL), 96% of which is IgG (110.1 PEIU/mL) (Germer et al., 2016). Its maximum IgA level is 2 mg/mL, and its anti-HCMV antibody level is 100 U/mL. It is approved in Europe for the prophylaxis of HCMV infection in patients treated with immunosuppressants and in solid organ transplant patients. In France, this solution is available as part of a patient-nominated program for the prevention or treatment of HCMV infection.
Cytotect CP® and Cytogam® preparations have high avidity indexes (90.5 and 91.0% respectively) and both have been tested in immunoblot assays against antigenic HCMV glycoproteins with similar results (Germer et al., 2016).
6.1 Mechanism of action
HCMV-HIG involve the neutralization of viral particles in extracellular environment, opsonization for phagocytosis (ADP), activation of the cellular immune system (ADCC) and immune adaptation with complement activation (Carbone, 2016; Figure 13).
Figure 13. The four modes of action of anti-CMV immunoglobulins. In the first mechanism, HIGs neutralize the viral particle by binding its epitope to viral envelope glycoproteins. Virus adsorption and cellular penetration are inhibited. In the second mechanism, HIGs induce complement pathways leading to the formation of the membrane attack complex (MAC), which creates a hole in the cell membrane leading to cytolysis. In the third mechanism (ADCC), the Fc fraction of the HIG-neutralizing viral particle is recognized by the cytotoxic cell’s Fc receptor (FcR). This leads to the release of cytotoxic molecules that create a cytolysis signal. In the final mechanism, after neutralization of the viral particle by HIGs, macrophage FcRs recognize HIG Fc, resulting in a phagocytosis signal. The viral particle-HIG complex is phagocytosed. The phagosome fuses with the lysosome, forming a phagolysosome and destroying the complex. Created with BioRender.com
6.2 Preclinical tests
Efficacy of HCMV-HIG was proved in vitro with a higher HCMV neutralizing activity than IVIG (Miescher et al., 2015; Germer et al., 2016; Schampera et al., 2017).
In 2022 and 2023, an in vitro and ex vivo (in placental villi of the first trimester) study analyzed the efficacy and mode of action of Cytotect CP® and showed good efficacy and low toxicity in different routes of HCMV infection (Coste Mazeau et al., 2022, 2023). The development of infection foci was blocked by Cytotect CP® with a DN50 of 0.011 to 0.033 U/mL on endothelial strains (TB40E/VHLE) in in vitro neutralization assays. Samely, on day 7, Cytotect CP® prevents CMV infection with an EC50 of 0.024 U/mL in placental villi. Although, once infected, viral growth was not inhibited in explants. The viability of villi has not been affected. An additional study shows the need for renewing Cytotect CP® every 7 days in the medium to maintain efficacy at day 14 in the explants. This was coherent with the recent pharmacokinetics study from Kagan et al. showing the decrease of plasma concentrations within 2 weeks (Kagan et al., 2021).
Potency of CMV-HIGs during pregnacy was assessed in animal models. Guinea pigs were used as model to study HCMV-HIG because congenital infection has similarities between HCMV and GPCMV. The placental barrier can be crossed by both viruses that can create an in utero infection. In guinea pig experiments, pup survival is the endpoint as GPCMV causes their death (Schleiss, 2008). This model evaluated passive immunization of the fetus. Indeed, studies have used pregnant guinea pigs with GPCMV infection prior to HIG administration or after to assess the inhibition of viral particle and gB. In two studies, fetal survival was increased after administration of CMV-HIG but the viral load was not affected (Bia et al., 1980; Chatterjee et al., 2001). Thus, anti-gB CMV-HIG were used in another study and allowed a reduction in fetal infection, inflmmation of placenta, death of the fetus and increased fetal development. Results were independent of CMV-HIG administration time. Infection rate of fetuses was significantly reduced with the administration of CMV-HIG from 39 to 0%. Anti-gB HIG also reduced inflammation of placenta and increased fetal development (Bratcher et al., 1995).
The mouse model was used to assess passive immunization in the fetal brain because the mouse CMV (mCMV) does not pass through the placental barrier. Peritoneal cavity of new-born mice was infected with mCMV. The viral infection was disseminated in mice brains with associated inflammatory lesions such as infiltrations of mononuclear cells and prominent glial nodules. Treatment with HIG or monoclonal antibody specific of gB glycoprotein led to decrease of viral load in the brains and a 5-fold reduction of inflammatory lesions. This study showed that CMV-HIG are responsible for a limitation of viral replication and its associated lesions in the brain (Cekinović et al., 2008).
6.3 Clinical trials
CMV-HIG treatments were shown to be effective against severe HCMV infections in immunocompromised patients and congenital infections. Whereas, retrospective studies assessed the potency of the HCMV-HIG for the prevention of cCMV infection. To date, the only two published trials have reported that anti-HCMV IgG has not been effective (Revello et al., 2014; Hughes et al., 2021). The effectiveness of treatment is determined according to the frequency of treatment, the concentration of HCMV-HIG and the time of seroconversion of the patient (Kagan et al., 2019, 2021). A new administration protocol with higher concentrations at the first stage of pregnancy will begin as a third Phase III trial (NCT 05170269).
Concerning adverse events, in the phase II study, Cytotect® CP was associated with a tendancy to a low birth weight for treated newborns (Revello et al., 2014). Nevertheless, these results were disclaimed by a newer investigation of Chiaie et al. that was conducted in 50 women with a dose of 200 unit/kg taken twice during pregnancy. No side effects caused by HCMV-HIG were reported in this study (Chiaie et al., 2018).
Recently, an observational study was undertaken in 149 pregnant women to assess the effectiveness of Cytotect CP® in pregnant women with HCMV primary infection during the first trimester or periconceptional period. This study was based on pharmacokinetics results showing the half-life of HCMV-HIGs, which is about 10 days. Every 2 weeks, a dose of 200 IU/kg body weight of Cytotect CP® was injected to women with primary infection before 14 weeks of amenorrhea. Intravenous injection should begin within 3 weeks of discovery of the primary infection. HCMV-HIG injections were made until the 18th week of amenorrhea. A significantly lower rate of maternal infection transmitted to the fetus was observed with 7.5% in the intervention group and 35.2% in the control group (Kagan et al., 2021).
In comparison with the historical cohort of Feldman, Seidel et al. conducted an observational survey using the same doses as Kagan et al. and found a significant reduction in the rate of mother-to-fetus transmission, regardless of the term of pregnancy (23.9% versus 39.9% for the Feldman control group; p = 0.026; Feldman et al., 2011; Seidel et al., 2020).
Some observational studies suggest that HCMV-HIG may have a protecting effect on the fetus after maternal primary infection (Buxmann et al., 2012). Cytotect® CP was evaluated on 592 cases of maternal primary infection before the 19th week of amenorrhea. After an administration of 200 unit/kg, HCMV-linked symptoms in the newborn decreased (Visentin et al., 2012).
Nevertheless, Cytotect CP® has also been evaluated in rescue therapy for hematopoietic stem cell recipients with resistant refractory infections. One study determined the safety and efficacy profiles of Cytotect CP® in 23 patients with refractory HCMV and GVHD (74%) and/or steroid treatment (64%). After 15 days, a response was observed in 18 patients. Of the 18, four had CMV reactivation, one died of CMV infection, and 4 died of CMV-related causes within 100 days of starting treatment. Total 100-day survival was 69.6% after Cytotect CP®. However, no statistical difference between respondents and non-respondents was reported. Thus, it showed that Cytotect CP® was well tolerated and effective as a recovery treatment (Alsuliman et al., 2018).
Another rescue therapy study was conducted in cardiothoracic transplant recipients. This was a 6-year retrospective single-center experiment in 35 patients. The rescue therapy consisted of Cytotect CP® supplemented by antiviral treatment (GCV/VGCV/LEF). HCMV-HIGs were well tolerated by patients and safe; only two patients had adverse events, but their symptoms were resolved after reducing HCMV-HIG doses to 1.5 mg/kg. CMV DNA was reduced in all patients and, after 4 weeks, undetectable in 73% of them. HCMV-HIG were shown as effective to control viral replication in cardiothoracic transplant recipients (Santhanakrishnan et al., 2019).
6.4 Resistances
So far, there is no reported case of resistant CMV to HIG.
7 Discussion
The CMV DNA polymerase inhibitors (GCV, FOS, and CDV) are essential molecules for decreasing the morbidity and mortality rate associated with CMV infection in transplant recipients (SOT or HSCT). However, they are often responsible for toxicity (hematologic or renal) and emergence of resistance that may limit their use. Nevertheless, they are always the most used in clinical practice. The approval of letermovir represents an important innovation for CMV prevention in HSCT. A decisive step forward in the management of refractory and/or resistant infections has been achieved with the validation of maribavir in transplant recipients. However, in case of multidrug resistance, or in non-transplanted patient or also in the prevention and treatment of cCMV infection, finding new antivirals or molecules able to inhibit CMV replication with the lowest toxicity remains a critical need.
In this review, we have listed the main compounds with potential activity against CMV. They belong to different families of molecules, some with specific antiviral activity, others known for their antimicrobial or immunosuppressive activities but with anti-CMV efficacy, and still others with a completely different mode of action, such as immunoglobulins.
Some direct antivirals like brincidofovir or cyclopropavir have an interesting profile for CMV treatment, but the development of the first was stopped after emergence of toxicity in a phase II clinical trial whereas the second did not enter in phase II trials until now. The search of cidofovir derivatives or pro-drugs is not yet stopped: recently, a new family of antiviral acyclonucleoside analogs with high bioavailability and potential activity against HCMV was patented (Roy and Agrofoglio, 2022) and are under evaluation (personal data). Concerning anti-terminase benzimidazole analogs, their poor biodisponibility limited their clinical use. As the effectiveness of letermovir validates terminase inhibitors as a clinically relevant class of antiviral agents, the development of other terminase inhibitors may be considered and research on these inhibitors should be encouraged (Ligat et al., 2018; Gentry et al., 2019). Indirect antivirals are also an interesting area to explore because of their cellular targets, which do not select for resistance to direct antivirals. Artemisin derivatives have shown their efficacy to control HCMV replication in some transplant patients but with varying degrees of effectiveness. A degree of uncertainty therefore remains when these treatments are used as monotherapy but artesunate is still an alternative alone or in association in multidrug resistant infections. Other indirect antivirals like flavonoids or anti-cox derivatives have demonstrated good efficacy in vitro but few or no study were performed until now with these molecules. Expanding these families of chemical compounds could be a complementary approach. Concerning immunomodulating agents, leflunomide, mTOR inhibitors and immunoglobulins could be used in combination with other antiviral drugs, as their use as monotherapy is not sufficiently effective to be recommended for the control of high level HCMV replication.
With agents acting by new modes of action as LTV and MBV available in clinical use, association therapy for the treatment of CMV infection and disease can move from concept to reality. In vitro studies support at least one additive (and sometimes synergistic) effect of association of LTV or MBV with DNA polymerase inhibitors. MBV targeting the kinase pUL97, his mode of action differs from the DNA polymerase or terminase inhibitors, but pUL97 being the kinase essential for GCV activation, association of both antivirals are antagonist. Moreover, we and others have already demonstrated in vitro that combination of indirect antiviral with DNA polymerase inhibitors have additive or synergistic activity (Morère et al., 2015; Wildum et al., 2015; Drouot et al., 2016; Chou et al., 2019). So far, clinical studies are needed to assess which combination therapy for HCMV is superior to monotherapy and which combination regimens are most effective. Combination therapy has already proved its relevance to treat other viral infections such as human immunodeficiency virus and hepatitis C virus (Hepatitis, 2018; Saag et al., 2020). Use of immunoglobulins in addition with antiviral therapy should also be considered in immunosuppressed patients, especially those with weak or null cellular response against CMV. Nevertheless, the effectiveness of this approach must be confirmed in clinical trials to better define the indications according to the patient profile, the history of CMV infection and the antivirals already used. Since few agents are currently being studied in humans, a combination therapy with existing agents and possibly with indirect acting anti-HCMV molecules approved for other indications not suitable for use in monotherapy should be considered.
Author contributions
CG: Writing – original draft. SA: Writing – review & editing. SH: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was granted by the University of Limoges and Santé Publique France (National Reference Center for Herpesviruses).
Conflict of interest
SA is an expert for Takeda, MSD, Merck and Biotest with no personal link.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Adjuik, M., Babiker, A., Garner, P., Olliaro, P., Taylor, W., and White, N. (2004). Artesunate combinations for treatment of malaria: meta-analysis. Lancet 363, 9–17. doi: 10.1016/S0140-6736(03)15162-8
Aiba, N., Shiraki, A., Yajima, M., Oyama, Y., Yoshida, Y., Ohno, A., et al. (2017). Interaction of immunoglobulin with cytomegalovirus-infected cells. Viral Immunol. 30, 500–507. doi: 10.1089/vim.2016.0151
Alain, S., Feghoul, L., Girault, S., Lepiller, Q., Frobert, E., Michonneau, D., et al. (2020). Letermovir breakthroughs during the French named patient Programme: interest of monitoring blood concentration in clinical practice. J. Antimicrob. Chemother. 75, 2253–2257. doi: 10.1093/jac/dkaa135
Aldern, K. A., Ciesla, S. L., Winegarden, K. L., and Hostetler, K. Y. (2003). Increased antiviral activity of 1-O-hexadecyloxypropyl-[2-(14)C]cidofovir in MRC-5 human lung fibroblasts is explained by unique cellular uptake and metabolism. Mol. Pharmacol. 63, 678–681. doi: 10.1124/mol.63.3.678
Alsuliman, T., Kitel, C., Dulery, R., Guillaume, T., Larosa, F., Cornillon, J., et al. (2018). Cytotect®CP as salvage therapy in patients with CMV infection following allogeneic hematopoietic cell transplantation: a multicenter retrospective study. Bone Marrow Transplant. 53, 1328–1335. doi: 10.1038/s41409-018-0166-9
Alvarez-Cardona, J. J., Whited, L. K., and Chemaly, R. F. (2020). Brincidofovir: understanding its unique profile and potential role against adenovirus and other viral infections. Future Microbiol. 15, 389–400. doi: 10.2217/fmb-2019-0288
Amoros, M., Simões, C. M., Girre, L., Sauvager, F., and Cormier, M. (1992). Synergistic effect of flavones and flavonols against herpes simplex virus type 1 in cell culture. Comparison with the antiviral activity of propolis. J. Nat. Prod. 55, 1732–1740. doi: 10.1021/np50090a003
Andouard, D., Gueye, R., Hantz, S., Fagnère, C., Liagre, B., Bernardaud, L., et al. (2021). Impact of new cyclooxygenase 2 inhibitors on human cytomegalovirus replication in vitro. Antivir. Ther. 26, 117–125. doi: 10.1177/13596535211064078
Andouard, D., Mazeron, M. C., Ligat, G., Couvreux, A., Pouteil-Noble, C., Cahen, R., et al. (2016). Contrasting effect of new HCMV pUL54 mutations on antiviral drug susceptibility: benefits and limits of 3D analysis. Antivir. Res. 129, 115–119. doi: 10.1016/j.antiviral.2016.02.004
Arav-Boger, R., He, R., Chiou, C. J., Liu, J., Woodard, L., Rosenthal, A., et al. (2010). Artemisinin-derived dimers have greatly improved anti-cytomegalovirus activity compared to artemisinin monomers. PLoS One 5:e10370. doi: 10.1371/journal.pone.0010370
Aryal, S., Katugaha, S. B., Cochrane, A., Brown, A. W., Nathan, S. D., Shlobin, O. A., et al. (2019). Single-center experience with use of letermovir for CMV prophylaxis or treatment in thoracic organ transplant recipients. Transpl. Infect. Dis. 21:e13166. doi: 10.1111/tid.13166
Auerochs, S., Korn, K., and Marschall, M. (2011). A reporter system for Epstein-Barr virus (EBV) lytic replication: anti-EBV activity of the broad anti-herpesviral drug artesunate. J. Virol. Methods 173, 334–339. doi: 10.1016/j.jviromet.2011.03.005
Avery, R. K., Alain, S., Alexander, B. D., Blumberg, E. A., Chemaly, R. F., Cordonnier, C., et al. (2021). Maribavir for refractory cytomegalovirus infections with or without resistance post-transplant: results from a phase 3 randomized clinical trial. Clin. Infect. Dis. 75, 690–701. doi: 10.1093/cid/ciab988
Avery, R. K., Mossad, S. B., Poggio, E., Lard, M., Budev, M., Bolwell, B., et al. (2010). Utility of Leflunomide in the treatment of complex cytomegalovirus syndromes. Transplantation 90, 419–426. doi: 10.1097/TP.0b013e3181e94106
Baryawno, N., Rahbar, A., Wolmer-Solberg, N., Taher, C., Odeberg, J., Darabi, A., et al. (2011). Detection of human cytomegalovirus in medulloblastomas reveals a potential therapeutic target. J. Clin. Invest. 121, 4043–4055. doi: 10.1172/JCI57147
Beadle, J. R., Hartline, C., Aldern, K. A., Rodriguez, N., Harden, E., Kern, E. R., et al. (2002). Alkoxyalkyl esters of Cidofovir and cyclic Cidofovir exhibit multiple-log enhancement of antiviral activity against cytomegalovirus and herpesvirus replication in vitro. Antimicrob. Agents Chemother. 46, 2381–2386. doi: 10.1128/AAC.46.8.2381-2386.2002
Beauvais, D., Robin, C., Thiebaut, A., Alain, S., Coiteux, V., Ducastelle-Lepretre, S., et al. (2022). Effective Letermovir prophylaxis of CMV infection post allogeneic hematopoietic cell transplantation: results from the French temporary authorization of use compassionate program. J. Clin. Virol. 148:105106. doi: 10.1016/j.jcv.2022.105106
Beecher, G. R. (2003). Overview of dietary flavonoids: nomenclature, occurrence and intake. J. Nutr. 133, 3248S–3254S. doi: 10.1093/jn/133.10.3248S
Bia, F. J., Griffith, B. P., Tarsio, M., and Hsiung, G. D. (1980). Vaccination for the prevention of maternal and fetal infection with Guinea pig cytomegalovirus. J. Infect. Dis. 142, 732–738. doi: 10.1093/infdis/142.5.732
Bidanset, D. J., Beadle, J. R., Wan, W. B., Hostetler, K. Y., and Kern, E. R. (2004). Oral activity of ether lipid Ester prodrugs of Cidofovir against experimental human cytomegalovirus infection. J. Infect. Dis. 190, 499–503. doi: 10.1086/421912
Biron, K. K., Harvey, R. J., Chamberlain, S. C., Good, S. S., Smith, A. A. III, Davis, M. G., et al. (2002). Potent and selective inhibition of human cytomegalovirus replication by 1263W94, a Benzimidazole l-riboside with a unique mode of action. Antimicrob. Agents Chemother. 46, 2365–2372. doi: 10.1128/AAC.46.8.2365-2372.2002
Boots, A. W., Balk, J. M., Bast, A., and Haenen, G. R. M. M. (2005). The reversibility of the glutathionyl-quercetin adduct spreads oxidized quercetin-induced toxicity. Biochem. Biophys. Res. Commun. 338, 923–929. doi: 10.1016/j.bbrc.2005.10.031
Bratcher, D. F., Bourne, N., Bravo, F. J., Schleiss, M. R., Slaoui, M., Myers, M. G., et al. (1995). Effect of passive antibody on congenital cytomegalovirus infection in Guinea pigs. J. Infect. Dis. 172, 944–950. doi: 10.1093/infdis/172.4.944
Bravo, F. J., Bernstein, D. I., Beadle, J. R., Hostetler, K. Y., and Cardin, R. D. (2011). Oral Hexadecyloxypropyl-Cidofovir therapy in pregnant Guinea pigs improves outcome in the congenital model of cytomegalovirus infection. Antimicrob. Agents Chemother. 55, 35–41. doi: 10.1128/AAC.00971-10
Brennan, D. C., Legendre, C., Patel, D., Mange, K., Wiland, A., McCague, K., et al. (2011). Cytomegalovirus incidence between Everolimus versus mycophenolate in De novo renal transplants: pooled analysis of three clinical trials. Am. J. Transplant. 11, 2453–2462. doi: 10.1111/j.1600-6143.2011.03674.x
Brooks, J. L., and Bowlin, T. L. (2013). Preclinical pharmacokinetic, safety pharmacology and in vitro and Oral toxicology studies of MBX-400, a potent inhibitor of human cytomegalovirus.
Brooks, J., Bowlin, T., and Usa, M.. (2015). Phase 1A clinical trial results of MBX-400; a potent antiviral in development for CMV and HHV-6.
Buerger, I., Reefschlaeger, J., Bender, W., Eckenberg, P., Popp, A., Weber, O., et al. (2001). A novel nonnucleoside inhibitor specifically targets cytomegalovirus DNA maturation via the UL89 and UL56 gene products. J. Virol. 75, 9077–9086. doi: 10.1128/JVI.75.19.9077-9086.2001
Buxmann, H., Stackelberg, O. M., Schlößer, R. L., Enders, G., Gonser, M., Meyer-Wittkopf, M., et al. (2012). Use of cytomegalovirus hyperimmunoglobulin for prevention of congenital cytomegalovirus disease: a retrospective analysis. J. Perinat. Med. 40, 439–446. doi: 10.1515/jpm-2011-0257
Carbone, J. (2016). The immunology of Posttransplant CMV infection: potential effect of CMV immunoglobulins on distinct components of the immune response to CMV. Transplantation 100, S11–S18. doi: 10.1097/TP.0000000000001095
Cazarolli, L. H., Zanatta, L., Alberton, E. H., Bonorino Figueiredo, M. S., Folador, P., Damazio, R. G., et al. (2008). Flavonoids: prospective drug candidates. Mini-Rev. Med. Chem. 8, 1429–1440. doi: 10.2174/138955708786369564
Cekinović, D., Golemac, M., Pugel, E. P., Tomac, J., Čičin-Šain, L., Slavuljica, I., et al. (2008). Passive immunization reduces murine cytomegalovirus-induced brain pathology in newborn mice. J. Virol. 82, 12172–12180. doi: 10.1128/JVI.01214-08
Cerella, C., Sobolewski, C., Dicato, M., and Diederich, M. (2010). Targeting COX-2 expression by natural compounds: a promising alternative strategy to synthetic COX-2 inhibitors for cancer chemoprevention and therapy. Biochem. Pharmacol. 80, 1801–1815. doi: 10.1016/j.bcp.2010.06.050
Chakraborti, A. K., Garg, S. K., Kumar, R., Motiwala, H. F., and Jadhavar, P. S. (2010). Progress in COX-2 inhibitors: a journey so far. Curr. Med. Chem. 17, 1563–1593. doi: 10.2174/092986710790979980
Chang, Y., Lu, C. W., Lin, T. Y., Huang, S. K., and Wang, S. J. (2016). Baicalein, a constituent of Scutellaria baicalensis, reduces glutamate release and protects neuronal cell against Kainic acid-induced excitotoxicity in rats. Am. J. Chin. Med. 44, 943–962. doi: 10.1142/S0192415X1650052X
Chatterjee, A., Harrison, C. J., Britt, W. J., and Bewtra, C. (2001). Modification of maternal and congenital cytomegalovirus infection by anti–glycoprotein B antibody transfer in Guinea pigs. J. Infect. Dis. 183, 1547–1553. doi: 10.1086/320714
Chatzakis, C., Ville, Y., Makrydimas, G., Dinas, K., Zavlanos, A., and Sotiriadis, A. (2020). Timing of primary maternal cytomegalovirus infection and rates of vertical transmission and fetal consequences. Am. J. Obstet. Gynecol. 223, 870–883.e11. doi: 10.1016/j.ajog.2020.05.038
Chemaly, R. F., Ullmann, A. J., Stoelben, S., Richard, M. P., Bornhäuser, M., Groth, C., et al. (2014). Letermovir for cytomegalovirus prophylaxis in hematopoietic-cell transplantation. N. Engl. J. Med. 370, 1781–1789. doi: 10.1056/NEJMoa1309533
Chen, H., Li, C., Zemlicka, J., Gentry, B. G., Bowlin, T. L., and Coen, D. M. (2016). Potency and Stereoselectivity of Cyclopropavir triphosphate action on human cytomegalovirus DNA polymerase. Antimicrob. Agents Chemother. 60, 4176–4182. doi: 10.1128/AAC.00449-16
Cherrier, L., Nasar, A., Goodlet, K. J., Nailor, M. D., Tokman, S., and Chou, S. (2018). Emergence of letermovir resistance in a lung transplant recipient with ganciclovir-resistant cytomegalovirus infection. Am. J. Transplant. 18, 3060–3064. doi: 10.1111/ajt.15135
Chiaie, L. D., Neuberger, P., Vochem, M., Lihs, A., Karck, U., and Enders, M. (2018). No evidence of obstetrical adverse events after hyperimmune globulin application for primary cytomegalovirus infection in pregnancy: experience from a single Centre. Arch. Gynecol. Obstet. 297, 1389–1395. doi: 10.1007/s00404-018-4703-y
Chimerix. (2022) An intermediate-size, expanded access protocol to provide Brincidofovir for the treatment of serious adenovirus infection or disease. Report No.: NCT02596997. Available at: https://clinicaltrials.gov/study/NCT02596997. (Accessed Jan 1, 2023).
Chong, A. S., Zeng, H., Knight, D. A., Shen, J., Meister, G. T., Williams, J. W., et al. (2006). Concurrent antiviral and immunosuppressive activities of Leflunomide in vivo. Am. J. Transplant. 6, 69–75. doi: 10.1111/j.1600-6143.2005.01152.x
Chou, S. (2009). Diverse cytomegalovirus UL27 mutations adapt to loss of viral UL97 kinase activity under Maribavir. Antimicrob. Agents Chemother. 53, 81–85. doi: 10.1128/AAC.01177-08
Chou, S. (2017). Comparison of cytomegalovirus Terminase gene mutations selected after exposure to three distinct inhibitor compounds. Antimicrob. Agents Chemother. 61:e01325. doi: 10.1128/AAC.01325-17
Chou, S. (2020). Advances in the genotypic diagnosis of cytomegalovirus antiviral drug resistance. Antivir. Res. 176:104711. doi: 10.1016/j.antiviral.2020.104711
Chou, S., Alain, S., Cervera, C., Chemaly, R. F., Kotton, C. N., Lundgren, J., et al. (2023). Drug resistance assessed in a phase 3 clinical trial of Maribavir therapy for refractory or resistant cytomegalovirus infection in transplant recipients. J. Infect. Dis. :jiad293. doi: 10.1093/infdis/jiad293
Chou, S., and Bowlin, T. L. (2011). Cytomegalovirus UL97 mutations affecting Cyclopropavir and ganciclovir susceptibility. Antimicrob. Agents Chemother. 55, 382–384. doi: 10.1128/AAC.01259-10
Chou, S., Ercolani, R. J., and Lanier, E. R. (2016). Novel cytomegalovirus UL54 DNA polymerase gene mutations selected in vitro that confer Brincidofovir resistance. Antimicrob. Agents Chemother. 60, 3845–3848. doi: 10.1128/AAC.00214-16
Chou, S., Ercolani, R. J., Marousek, G., and Bowlin, T. L. (2013). Cytomegalovirus UL97 kinase catalytic domain mutations that confer multidrug resistance. Antimicrob. Agents Chemother. 57, 3375–3379. doi: 10.1128/AAC.00511-13
Chou, S., Hakki, M., and Villano, S. (2012a). Effects on maribavir susceptibility of cytomegalovirus UL97 kinase ATP binding region mutations detected after drug exposure in vitro and in vivo. Antivir. Res. 95, 88–92. doi: 10.1016/j.antiviral.2012.05.013
Chou, S., and Marousek, G. I. (2006). Maribavir antagonizes the antiviral action of ganciclovir on human cytomegalovirus. Antimicrob. Agents Chemother. 50, 3470–3472. doi: 10.1128/AAC.00577-06
Chou, S., and Marousek, G. I. (2008). Accelerated evolution of Maribavir resistance in a cytomegalovirus exonuclease domain II mutant. J. Virol. 82, 246–253. doi: 10.1128/JVI.01787-07
Chou, S., Marousek, G., Auerochs, S., Stamminger, T., Milbradt, J., and Marschall, M. (2011). The unique antiviral activity of artesunate is broadly effective against human cytomegaloviruses including therapy-resistant mutants. Antivir. Res. 92, 364–368. doi: 10.1016/j.antiviral.2011.07.018
Chou, S., Marousek, G., and Bowlin, T. L. (2012b). Cyclopropavir susceptibility of cytomegalovirus DNA polymerase mutants selected after antiviral drug exposure. Antimicrob. Agents Chemother. 56, 197–201. doi: 10.1128/AAC.05559-11
Chou, S., Marousek, G. I., Senters, A. E., Davis, M. G., and Biron, K. K. (2004). Mutations in the human cytomegalovirus UL27 gene that confer resistance to Maribavir. J. Virol. 78, 7124–7130. doi: 10.1128/JVI.78.13.7124-7130.2004
Chou, S., Van Wechel, L. C., and Marousek, G. I. (2007). Cytomegalovirus UL97 kinase mutations that confer Maribavir resistance. J. Infect. Dis. 196, 91–94. doi: 10.1086/518514
Chou, S., Wu, J., Song, K., and Bo, T. (2019). Novel UL97 drug resistance mutations identified at baseline in a clinical trial of maribavir for resistant or refractory cytomegalovirus infection. Antivir. Res. 172:104616. doi: 10.1016/j.antiviral.2019.104616
Coste Mazeau, P., Berto, L., Andouard, D., El Hamel, C., Chianea, T., Hantz, S., et al. (2023). New therapeutic perspective in the prevention of congenital cytomegalovirus infection. Antivir. Res. 216:105661. doi: 10.1016/j.antiviral.2023.105661
Coste Mazeau, P., Jacquet, C., Muller, C., Courant, M., El Hamel, C., Chianea, T., et al. (2022). Potential of anti-CMV immunoglobulin Cytotect CP® in vitro and ex vivo in a first-trimester placenta model. Microorganisms 10:694. doi: 10.3390/microorganisms10040694
Cotin, S., Calliste, C. A., Mazeron, M. C., Hantz, S., Duroux, J. L., Rawlinson, W. D., et al. (2012). Eight flavonoids and their potential as inhibitors of human cytomegalovirus replication. Antivir. Res. 96, 181–186. doi: 10.1016/j.antiviral.2012.09.010
Cristelli, M. P., Felipe, C. R., Prizmic, P. S. S., De Azevedo, V. F. D., Viana, L. A., Tavares, M. G., et al. (2019). Use of mTOR inhibitor as prophylaxis for cytomegalovirus disease after kidney transplantation: a natural experiment. Clin. Transpl. 33:e13689. doi: 10.1111/ctr.13689
Dayer, J. M., and Cutolo, M. (2005). Is there a rationale to using leflunomide in early rheumatoid arthritis? Clin. Exp. Rheumatol. 23, 404–412.
De Oliveira, M. R., Nabavi, S. M., Braidy, N., Setzer, W. N., Ahmed, T., and Nabavi, S. F. (2016). Quercetin and the mitochondria: a mechanistic view. Biotechnol. Adv. 34, 532–549. doi: 10.1016/j.biotechadv.2015.12.014
Dinda, B., Dinda, S., DasSharma, S., Banik, R., Chakraborty, A., and Dinda, M. (2017). Therapeutic potentials of baicalin and its aglycone, baicalein against inflammatory disorders. Eur. J. Med. Chem. 131, 68–80. doi: 10.1016/j.ejmech.2017.03.004
Dollard, S. C., Grosse, S. D., and Ross, D. S. (2007). New estimates of the prevalence of neurological and sensory sequelae and mortality associated with congenital cytomegalovirus infection. Rev. Med. Virol. 17, 355–363. doi: 10.1002/rmv.544
Dou, J., Chen, L., Xu, G., Zhang, L., Zhou, H., Wang, H., et al. (2011). Effects of baicalein on Sendai virus in vivo are linked to serum baicalin and its inhibition of hemagglutinin-neuraminidase. Arch. Virol. 156, 793–801. doi: 10.1007/s00705-011-0917-z
Drew, W. L., Miner, R. C., Marousek, G. I., and Chou, S. (2006). Maribavir sensitivity of cytomegalovirus isolates resistant to ganciclovir, cidofovir or foscarnet. J. Clin. Virol. 37, 124–127. doi: 10.1016/j.jcv.2006.07.010
Drouot, E., Piret, J., and Boivin, G. (2016). Artesunate demonstrates in vitro synergism with several antiviral agents against human cytomegalovirus. Antivir. Ther. 21, 535–539. doi: 10.3851/IMP3028
Efferth, T., Marschall, M., Wang, X., Huong, S. M., Hauber, I., Olbrich, A., et al. (2002). Antiviral activity of artesunate towards wild-type, recombinant, and ganciclovir-resistant human cytomegaloviruses. J. Mol. Med. 80, 233–242. doi: 10.1007/s00109-001-0300-8
Efferth, T., Romero, M. R., Wolf, D. G., Stamminger, T., Marin, J. J. G., and Marschall, M. (2008). The antiviral activities of artemisinin and Artesunate. Clin. Infect. Dis. 47, 804–811. doi: 10.1086/591195
El Chaer, F., Mori, N., Shah, D., Oliver, N., Wang, E., Jan, A., et al. (2016). Adjuvant and salvage therapy with leflunomide for recalcitrant cytomegalovirus infections in hematopoietic cell transplantation recipients: a case series. Antivir. Res. 135, 91–96. doi: 10.1016/j.antiviral.2016.08.027
El-Qushayri, A. E., Ghozy, S., Abbas, A. S., Dibas, M., Dahy, A., Mahmoud, A. R., et al. (2021). Hyperimmunoglobulin therapy for the prevention and treatment of congenital cytomegalovirus: a systematic review and meta-analysis. Expert Rev. Anti-Infect. Ther. 19, 661–669. doi: 10.1080/14787210.2021.1846521
Evers, D. L., Chao, C. F., Wang, X., Zhang, Z., Huong, S. M., and Huang, E. S. (2005). Human cytomegalovirus-inhibitory flavonoids: studies on antiviral activity and mechanism of action. Antivir. Res. 68, 124–134. doi: 10.1016/j.antiviral.2005.08.002
Evers, D. L., Komazin, G., Shin, D., Hwang, D. D., Townsend, L. B., and Drach, J. C. (2002). Interactions among antiviral drugs acting late in the replication cycle of human cytomegalovirus. Antivir. Res. 56, 61–72. doi: 10.1016/S0166-3542(02)00094-3
Faure Bardon, V., Peytavin, G., Lê, M. P., Guilleminot, T., Elefant, E., Stirnemann, J., et al. (2020). Placental transfer of Letermovir and Maribavir in the ex vivo human cotyledon perfusion model. New perspectives for in utero treatment of congenital cytomegalovirus infection. PLoS One 15:e0232140. doi: 10.1371/journal.pone.0232140
Feldman, B., Yinon, Y., Tepperberg Oikawa, M., Yoeli, R., Schiff, E., and Lipitz, S. (2011). Pregestational, periconceptional, and gestational primary maternal cytomegalovirus infection: prenatal diagnosis in 508 pregnancies. Am. J. Obstet. Gynecol. 205, 342.e1–342.e6. doi: 10.1016/j.ajog.2011.05.030
Food and Drug Administration. (2021). FDA approves first treatment for common type of post-transplant infection that is resistant to other drugs. US Food Drug Administration. Available at: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-common-type-post-transplant-infection-resistant-other-drugs.
Frietsch, J. J., Michel, D., Stamminger, T., Hunstig, F., Birndt, S., Schnetzke, U., et al. (2019). In vivo emergence of UL56 C325Y cytomegalovirus resistance to Letermovir in a patient with acute myeloid leukemia after hematopoietic cell transplantation. Mediterr. J. Hematol. Infect. Dis. 11:e2019001. doi: 10.4084/MJHID.2019.001
Fröhlich, T., Hahn, F., Belmudes, L., Leidenberger, M., Friedrich, O., Kappes, B., et al. (2018). Synthesis of artemisinin-derived dimers, trimers and dendrimers: investigation of their antimalarial and antiviral activities including putative mechanisms of action. Chemistry 24, 8103–8113. doi: 10.1002/chem.201800729
Gabardi, S., and Baroletti, S. A. (2010). Everolimus: a proliferation signal inhibitor with clinical applications in organ transplantation, oncology, and cardiology. Pharmacotherapy 30, 1044–1056. doi: 10.1592/phco.30.10.1044
Gao, Y., Snyder, S. A., Smith, J. N., and Chen, Y. C. (2016). Anticancer properties of baicalein: a review. Med. Chem. Res. 25, 1515–1523. doi: 10.1007/s00044-016-1607-x
Gentry, B. G., Bogner, E., and Drach, J. C. (2019). Targeting the terminase: an important step forward in the treatment and prophylaxis of human cytomegalovirus infections. Antivir. Res. 161, 116–124. doi: 10.1016/j.antiviral.2018.11.005
Gentry, B. G., Gentry, S. N., Jackson, T. L., Zemlicka, J., and Drach, J. C. (2011). Phosphorylation of antiviral and endogenous nucleotides to di- and triphosphates by guanosine monophosphate kinase. Biochem. Pharmacol. 81, 43–49. doi: 10.1016/j.bcp.2010.09.005
Gentry, B. G., Kamil, J. P., Coen, D. M., Zemlicka, J., and Drach, J. C. (2010). Stereoselective phosphorylation of Cyclopropavir by pUL97 and competitive inhibition by Maribavir. Antimicrob. Agents Chemother. 54, 3093–3098. doi: 10.1128/AAC.00468-10
Gentry, B. G., Vollmer, L. E., Hall, E. D., Borysko, K. Z., Zemlicka, J., Kamil, J. P., et al. (2013). Resistance of human cytomegalovirus to Cyclopropavir maps to a base pair deletion in the open Reading frame of UL97. Antimicrob. Agents Chemother. 57, 4343–4348. doi: 10.1128/AAC.00214-13
Germer, M., Herbener, P., and Schüttrumpf, J. (2016). Functional properties of human cytomegalovirus Hyperimmunoglobulin and standard immunoglobulin preparations. Ann. Transplant. 21, 558–564. doi: 10.12659/AOT.898050
Germi, R., Mariette, C., Alain, S., Lupo, J., Thiebaut, A., Brion, J. P., et al. (2014). Success and failure of artesunate treatment in five transplant recipients with disease caused by drug-resistant cytomegalovirus. Antivir. Res. 101, 57–61. doi: 10.1016/j.antiviral.2013.10.014
Gokarn, A., Toshniwal, A., Pathak, A., Arora, S., Bonda, A., Punatar, S., et al. (2019). Use of Leflunomide for treatment of cytomegalovirus infection in recipients of allogeneic stem cell transplant. Biol. Blood Marrow Transplant. 25, 1832–1836. doi: 10.1016/j.bbmt.2019.04.028
Goldner, T., Hewlett, G., Ettischer, N., Ruebsamen-Schaeff, H., Zimmermann, H., and Lischka, P. (2011). The novel Anticytomegalovirus compound AIC246 (Letermovir) inhibits human cytomegalovirus replication through a specific antiviral mechanism that involves the viral Terminase. J. Virol. 85, 10884–10893. doi: 10.1128/JVI.05265-11
Gómez Valbuena, I., Alioto, D., Serrano Garrote, O., and Ferrari Piquero, J. M. (2016). Use of leflunomide in a cytomegalovirus infection resistant: a report of a case. Farm Hosp. 40, 52–54. doi: 10.7399/fh.2016.40.1.10161
Good, S. S., Owens, T. L. B., and Drach, J. C. (1994). The disposition in rats and monkeys of 2-bromo-5,6-dichloro-1-(β-D-ribofuranosyl)-benzimidazole (BDCRB) and its 4,5,6,-trichloro-cohner (TCRB). Antivir. Res. 23:103. doi: 10.1016/0166-3542(94)90251-8
Granato, M., Gilardini Montani, M. S., Zompetta, C., Santarelli, R., Gonnella, R., Romeo, M. A., et al. (2019). Quercetin interrupts the positive feedback loop between STAT3 and IL-6, promotes autophagy, and reduces ROS, preventing EBV-driven B cell immortalization. Biomol. Ther. 9:482. doi: 10.3390/biom9090482
Hahn, F., Fröhlich, T., Frank, T., Bertzbach, L. D., Kohrt, S., Kaufer, B. B., et al. (2018). Artesunate-derived monomeric, dimeric and trimeric experimental drugs – their unique mechanistic basis and pronounced antiherpesviral activity. Antivir. Res. 152, 104–110. doi: 10.1016/j.antiviral.2018.02.013
Hakacova, N., Klingel, K., Kandolf, R., Engdahl, E., Fogdell-Hahn, A., and Higgins, T. (2013). First therapeutic use of Artesunate in treatment of human herpesvirus 6B myocarditis in a child. J. Clin. Virol. 57, 157–160. doi: 10.1016/j.jcv.2013.02.005
Halpern-Cohen, V., and Blumberg, E. A. (2022). New perspectives on antimicrobial agents: Maribavir. Antimicrob. Agents Chemother. 66, e02405–e02421. doi: 10.1128/aac.02405-21
Hamilton, S. T., Marschall, M., and Rawlinson, W. D. (2020). Investigational antiviral therapy models for the prevention and treatment of congenital cytomegalovirus infection during pregnancy. Antimicrob. Agents Chemother. 65, e01627–e01620. doi: 10.1128/AAC.01627-20
Hamirally, S., Kamil, J. P., Ndassa-Colday, Y. M., Lin, A. J., Jahng, W. J., Baek, M. C., et al. (2009). Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear Lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 5:e1000275. doi: 10.1371/journal.ppat.1000275
Hartline, C. B., Keith, K. A., Eagar, J., Harden, E. A., Bowlin, T. L., and Prichard, M. N. (2018). A standardized approach to the evaluation of antivirals against DNA viruses: Orthopox-, adeno-, and herpesviruses. Antivir. Res. 159, 104–112. doi: 10.1016/j.antiviral.2018.09.015
Havenith, S. H. C., Yong, S. L., van Donselaar-van der Pant, K. A. M. I., van Lier, R. A. W., ten Berge, I. J. M., and Bemelman, F. J. (2013). Everolimus-treated renal transplant recipients have a more robust CMV-specific CD8+ T-cell response compared with cyclosporine- or mycophenolate-treated patients. Transplantation 95, 184–191. doi: 10.1097/TP.0b013e318276a1ef
Hepatitis, C. (2018). Guidance 2018 update: AASLD-IDSA recommendations for testing, managing, and treating Hepatitis C virus infection. Clin. Infect. Dis. 67, 1477–1492. doi: 10.1093/cid/ciy585
Hill, J. A., Hummel, M., Starling, R. C., Kobashigawa, J. A., Perrone, S. V., Arizón, J. M., et al. (2007). A lower incidence of cytomegalovirus infection in De novo heart transplant recipients randomized to Everolimus. Transplantation 84, 1436–1442. doi: 10.1097/01.tp.0000290686.68910.bd
Hiscott, J., Kwon, H., and Génin, P. (2001). Hostile takeovers: viral appropriation of the NF-kB pathway. J. Clin. Invest. 107, 143–151. doi: 10.1172/JCI11918
Hughes, B. L., Clifton, R. G., Rouse, D. J., Saade, G. R., Dinsmoor, M. J., Reddy, U. M., et al. (2021). A trial of Hyperimmune globulin to prevent congenital cytomegalovirus infection. N. Engl. J. Med. 385, 436–444. doi: 10.1056/NEJMoa1913569
Hung, C. H., Chan, S. H., Chu, P. M., and Tsai, K. L. (2015). Quercetin is a potent anti-atherosclerotic compound by activation of SIRT1 signaling under oxLDL stimulation. Mol. Nutr. Food Res. 59, 1905–1917. doi: 10.1002/mnfr.201500144
Hutterer, C., Niemann, I., Milbradt, J., Fröhlich, T., Reiter, C., Kadioglu, O., et al. (2015). The broad-spectrum antiinfective drug artesunate interferes with the canonical nuclear factor kappa B (NF-κB) pathway by targeting RelA/p65. Antivir. Res. 124, 101–109. doi: 10.1016/j.antiviral.2015.10.003
Hyde, T. B., Schmid, D. S., and Cannon, M. J. (2010). Cytomegalovirus seroconversion rates and risk factors: implications for congenital CMV. Rev. Med. Virol. 20, 311–326. doi: 10.1002/rmv.659
Imamura, T., Shiga, T., Kinugawa, K., Kato, N., Endo, M., Inaba, T., et al. (2012). Successful conversion to Everolimus after cytomegalovirus infection in a heart transplant recipient. Int. Heart J. 53, 199–201. doi: 10.1536/ihj.53.199
Jacquet, C., Marschall, M., Andouard, D., El Hamel, C., Chianea, T., Tsogoeva, S. B., et al. (2020). A highly potent trimeric derivative of artesunate shows promising treatment profiles in experimental models for congenital HCMV infection in vitro and ex vivo. Antivir. Res. 175:104700. doi: 10.1016/j.antiviral.2019.104700
James, S. H., Hartline, C. B., Harden, E. A., Driebe, E. M., Schupp, J. M., Engelthaler, D. M., et al. (2011). Cyclopropavir inhibits the Normal function of the human cytomegalovirus UL97 kinase▿. Antimicrob. Agents Chemother. 55, 4682–4691. doi: 10.1128/AAC.00571-11
James, S. H., Price, N. B., Hartline, C. B., Lanier, E. R., and Prichard, M. N. (2013). Selection and recombinant phenotyping of a novel CMX001 and Cidofovir resistance mutation in human cytomegalovirus. Antimicrob. Agents Chemother. 57, 3321–3325. doi: 10.1128/AAC.00062-13
John, G. T., Manivannan, J., Chandy, S., Peter, S., Fleming, D. H., Chandy, S. J., et al. (2005). A prospective evaluation of Leflunomide therapy for cytomegalovirus disease in renal transplant recipients. Transplant. Proc. 37, 4303–4305. doi: 10.1016/j.transproceed.2005.10.116
Kagan, K. O., Enders, M., Hoopmann, M., Geipel, A., Simonini, C., Berg, C., et al. (2021). Outcome of pregnancies with recent primary cytomegalovirus infection in first trimester treated with hyperimmunoglobulin: observational study. Ultrasound Obstet. Gynecol. 57, 560–567. doi: 10.1002/uog.23596
Kagan, K. O., Enders, M., Schampera, M. S., Baeumel, E., Hoopmann, M., Geipel, A., et al. (2019). Prevention of maternal–fetal transmission of cytomegalovirus after primary maternal infection in the first trimester by biweekly hyperimmunoglobulin administration. Ultrasound Obstet. Gynecol. 53, 383–389. doi: 10.1002/uog.19164
Kajon, A. E., and St George, K. (2022). Mysterious cases of acute hepatitis in children: is adenovirus still a lead suspect? Emerg. Microbes Infect. 11, 1787–1789. doi: 10.1080/22221751.2022.2095933
Kamil, J. P., and Coen, D. M. (2011). HATs On for drug resistance. Cell Host Microbe 9, 85–87. doi: 10.1016/j.chom.2011.02.001
Kaminski, H., Kamar, N., Thaunat, O., Bouvier, N., Caillard, S., Garrigue, I., et al. (2022a). Incidence of cytomegalovirus infection in seropositive kidney transplant recipients treated with everolimus: a randomized, open-label, multicenter phase 4 trial. Am. J. Transplant. 22, 1430–1441. doi: 10.1111/ajt.16946
Kaminski, H., Marseres, G., Yared, N., Nokin, M. J., Pitard, V., Zouine, A., et al. (2022b). mTOR inhibitors prevent CMV infection through the restoration of functional αβ and γδ T cells in kidney transplantation. J. Am. Soc. Nephrol. 33, 121–137. doi: 10.1681/ASN.2020121753
Kaptein, S. J. F., Efferth, T., Leis, M., Rechter, S., Auerochs, S., Kalmer, M., et al. (2006). The anti-malaria drug artesunate inhibits replication of cytomegalovirus in vitro and in vivo. Antivir. Res. 69, 60–69. doi: 10.1016/j.antiviral.2005.10.003
Kaul, D. R., Stoelben, S., Cober, E., Ojo, T., Sandusky, E., Lischka, P., et al. (2011). First report of successful treatment of multidrug-resistant cytomegalovirus disease with the novel anti-CMV compound AIC246. Am. J. Transplant. 11, 1079–1084. doi: 10.1111/j.1600-6143.2011.03530.x
Kawabata, K., Mukai, R., and Ishisaka, A. (2015). Quercetin and related polyphenols: new insights and implications for their bioactivity and bioavailability. Food Funct. 6, 1399–1417. doi: 10.1039/C4FO01178C
Kenneson, A., and Cannon, M. J. (2007). Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev. Med. Virol. 17, 253–276. doi: 10.1002/rmv.535
Kern, E. R., Bidanset, D. J., Hartline, C. B., Yan, Z., Zemlicka, J., and Quenelle, D. C. (2004c). Oral activity of a methylenecyclopropane analog, Cyclopropavir, in animal models for cytomegalovirus infections. Antimicrob. Agents Chemother. 48, 4745–4753. doi: 10.1128/AAC.48.12.4745-4753.2004
Kern, E. R., Collins, D. J., Wan, W. B., Beadle, J. R., Hostetler, K. Y., and Quenelle, D. C. (2004b). Oral treatment of murine cytomegalovirus infections with ether lipid esters of Cidofovir. Antimicrob. Agents Chemother. 48, 3516–3522. doi: 10.1128/AAC.48.9.3516-3522.2004
Kern, E. R., Hartline, C. B., Rybak, R. J., Drach, J. C., Townsend, L. B., Biron, K. K., et al. (2004a). Activities of Benzimidazole d- and l-Ribonucleosides in animal models of cytomegalovirus infections. Antimicrob. Agents Chemother. 48, 1749–1755. doi: 10.1128/AAC.48.5.1749-1755.2004
Kern, E. R., Kushner, N. L., Hartline, C. B., Williams-Aziz, S. L., Harden, E. A., Zhou, S., et al. (2005). In vitro activity and mechanism of action of methylenecyclopropane analogs of nucleosides against herpesvirus replication. Antimicrob. Agents Chemother. 49, 1039–1045. doi: 10.1128/AAC.49.3.1039-1045.2005
Kim, C. H., Kim, J. E., and Song, Y. J. (2020). Antiviral activities of quercetin and Isoquercitrin against human herpesviruses. Molecules 25:2379. doi: 10.3390/molecules25102379
Kimberlin, D. W., Jester, P. M., Sánchez, P. J., Ahmed, A., Arav-Boger, R., Michaels, M. G., et al. (2015). Valganciclovir for symptomatic congenital cytomegalovirus disease. N. Engl. J. Med. 372, 933–943. doi: 10.1056/NEJMoa1404599
Kjar, A., Wadsworth, I., Vargis, E., and Britt, D. W. (2022). Poloxamer 188 – quercetin formulations amplify in vitro ganciclovir antiviral activity against cytomegalovirus. Antivir. Res. 204:105362. doi: 10.1016/j.antiviral.2022.105362
Knight, D. A., Hejmanowski, A. Q., Dierksheide, J. E., Williams, J. W., Chong, A. S. F., and Waldman, W. J. (2001). Inhibition of herpes simplex virus type 1 by the experimental immunosuppressive agent leflunomide. Transplantation 71, 170–174. doi: 10.1097/00007890-200101150-00031
Kobashigawa, J., Ross, H., Bara, C., Delgado, J. F., Dengler, T., Lehmkuhl, H. B., et al. (2013). Everolimus is associated with a reduced incidence of cytomegalovirus infection following de novo cardiac transplantation. Transpl. Infect. Dis. 15, 150–162. doi: 10.1111/tid.12007
Komazin, G., Townsend, L. B., and Drach, J. C. (2004). Role of a mutation in human cytomegalovirus gene UL104 in resistance to Benzimidazole Ribonucleosides. J. Virol. 78, 710–715. doi: 10.1128/JVI.78.2.710-715.2004
Komazin-Meredith, G., Chou, S., Prichard, M. N., Hartline, C. B., Cardinale, S. C., Comeau, K., et al. (2014). Human cytomegalovirus UL97 kinase is involved in the mechanism of action of methylenecyclopropane analogs with 6-ether and -thioether substitutions. Antimicrob. Agents Chemother. 58, 274–278. doi: 10.1128/AAC.01726-13
Koszalka, G. W., Johnson, N. W., Good, S. S., Boyd, L., Chamberlain, S. C., Townsend, L. B., et al. (2002). Preclinical and toxicology studies of 1263W94, a potent and selective inhibitor of human cytomegalovirus replication. Antimicrob. Agents Chemother. 46, 2373–2380. doi: 10.1128/AAC.46.8.2373-2380.2002
Kotton, C. N., Kumar, D., Caliendo, A. M., Huprikar, S., Chou, S., Danziger-Isakov, L., et al. (2018). The third international consensus guidelines on the Management of Cytomegalovirus in solid-organ transplantation. Transplantation 102, 900–931. doi: 10.1097/TP.0000000000002191
Krosky, P. M., Borysko, K. Z., Nassiri, M. R., Devivar, R. V., Ptak, R. G., Davis, M. G., et al. (2002). Phosphorylation of β-d-Ribosylbenzimidazoles is not required for activity against human cytomegalovirus. Antimicrob. Agents Chemother. 46, 478–486. doi: 10.1128/AAC.46.2.478-486.2002
Krosky, P. M., Ptak, R. G., Underwood, M. R., Biron, K. K., Townsend, L. B., and Drach, J. C. (2000). Differences in DNA packaging genes and sensitivity to Benzimidazole Ribonucleosides between human cytomegalovirus strains AD169 and Towne. Antivir. Chem. Chemother. 11, 349–352. doi: 10.1177/095632020001100506
Krosky, P. M., Underwood, M. R., Turk, S. R., Feng, K. W. H., Jain, R. K., Ptak, R. G., et al. (1998). Resistance of human cytomegalovirus to Benzimidazole Ribonucleosides maps to two open Reading frames: UL89 and UL56. J. Virol. 72, 4721–4728. doi: 10.1128/JVI.72.6.4721-4728.1998
Lai, M. Y., Hsiu, S. L., Tsai, S. Y., Hou, Y. C., and Chao, P. D. L. (2003). Comparison of metabolic pharmacokinetics of baicalin and baicalein in rats. J. Pharm. Pharmacol. 55, 205–209. doi: 10.1211/002235702522
Lanier, E. R., Foster, S., Brundage, T., Chou, S., Prichard, M. N., Kleiboeker, S., et al. (2016). Analysis of mutations in the gene encoding cytomegalovirus DNA polymerase in a phase 2 clinical trial of Brincidofovir prophylaxis. J. Infect. Dis. 214, 32–35. doi: 10.1093/infdis/jiw073
Lee, S., Lee, H. H., Shin, Y. S., Kang, H., and Cho, H. (2017). The anti-HSV-1 effect of quercetin is dependent on the suppression of TLR-3 in raw 264.7 cells. Arch. Pharm. Res. 40, 623–630. doi: 10.1007/s12272-017-0898-x
Leruez-Ville, M., Foulon, I., Pass, R., and Ville, Y. (2020). Cytomegalovirus infection during pregnancy: state of the science. Am. J. Obstet. Gynecol. 223, 330–349. doi: 10.1016/j.ajog.2020.02.018
Leruez-Ville, M., and Ville, Y. (2020). Is it time for routine prenatal serological screening for congenital cytomegalovirus? Prenat. Diagn. 40, 1671–1680. doi: 10.1002/pd.5757
Li, L., Gao, H., Lou, K., Luo, H., Hao, S., Yuan, J., et al. (2021). Safety, tolerability, and pharmacokinetics of oral baicalein tablets in healthy Chinese subjects: a single-center, randomized, double-blind, placebo-controlled multiple-ascending-dose study. Clin. Transl. Sci. 14, 2017–2024. doi: 10.1111/cts.13063
Li, J., Ma, J., Wang, K. S., Mi, C., Wang, Z., Piao, L. X., et al. (2016). Baicalein inhibits TNF-α-induced NF-κB activation and expression of NF-κB-regulated target gene products. Oncol. Rep. 36, 2771–2776. doi: 10.3892/or.2016.5108
Li, Q. G., Mog, S. R., Si, Y. Z., Kyle, D. E., Gettayacamin, M., and Milhous, W. K. (2002). Neurotoxicity and efficacy of arteether related to its exposure times and exposure levels in rodents. Am. J. Trop. Med. Hyg. 66, 516–525. doi: 10.4269/ajtmh.2002.66.516
Li, M., Shi, A., Pang, H., Xue, W., Li, Y., Cao, G., et al. (2014). Safety, tolerability, and pharmacokinetics of a single ascending dose of baicalein chewable tablets in healthy subjects. J. Ethnopharmacol. 156, 210–215. doi: 10.1016/j.jep.2014.08.031
Ligat, G., Cazal, R., Hantz, S., and Alain, S. (2018). The human cytomegalovirus terminase complex as an antiviral target: a close-up view. FEMS Microbiol. Rev. 42, 137–145. doi: 10.1093/femsre/fuy004
Limaye, A. P., Budde, K., Humar, A., Vincenti, F., Kuypers, D. R. J., Carroll, R. P., et al. (2023). Letermovir vs Valganciclovir for prophylaxis of cytomegalovirus in high-risk kidney transplant recipients: a randomized clinical trial. JAMA 330, 33–42. doi: 10.1001/jama.2023.9106
Linder, K. A., Kovacs, C., Mullane, K. M., Wolfe, C., Clark, N. M., La Hoz, R. M., et al. (2021). Letermovir treatment of cytomegalovirus infection or disease in solid organ and hematopoietic cell transplant recipients. Transpl. Infect. Dis. 23:e13687. doi: 10.1111/tid.13687
Lischka, P., Hewlett, G., Wunberg, T., Baumeister, J., Paulsen, D., Goldner, T., et al. (2010). In vitro and in vivo activities of the novel Anticytomegalovirus compound AIC246. Antimicrob. Agents Chemother. 54, 1290–1297. doi: 10.1128/AAC.01596-09
Lischka, P., Zhang, D., Holder, D., and Zimmermann, H. (2016). Impact of glycoprotein B genotype and naturally occurring ORF UL56 polymorphisms upon susceptibility of clinical human cytomegalovirus isolates to letermovir. Antivir. Res. 132, 204–209. doi: 10.1016/j.antiviral.2016.06.008
Littler, E., Stuart, A. D., and Chee, M. S. (1992). Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature 358, 160–162. doi: 10.1038/358160a0
Ljungman, P., De la Camara, R., Robin, C., Crocchiolo, R., Einsele, H., Hill, J. A., et al. (2019). Guidelines for the management of cytomegalovirus infection in patients with haematological malignancies and after stem cell transplantation from the 2017 European conference on infections in Leukaemia (ECIL 7). Lancet Infect. Dis. 19, e260–e272. doi: 10.1016/S1473-3099(19)30107-0
Luo, Z., Kuang, X. P., Zhou, Q. Q., Yan, C. Y., Li, W., Gong, H. B., et al. (2020). Inhibitory effects of baicalein against herpes simplex virus type 1. Acta Pharm. Sin. B 10, 2323–2338. doi: 10.1016/j.apsb.2020.06.008
Lurain, N. S., and Chou, S. (2010). Antiviral drug resistance of human cytomegalovirus. Clin. Microbiol. Rev. 23, 689–712. doi: 10.1128/CMR.00009-10
Maertens, J., Logan, A. C., Jang, J., Long, G., Tang, J. L., Hwang, W. Y. K., et al. (2020). Phase 2 study of anti-human cytomegalovirus monoclonal antibodies for prophylaxis in hematopoietic cell transplantation. Antimicrob. Agents Chemother. 64, e02467–e02419. doi: 10.1128/AAC.02467-19
Maniez-Montreuil, M., Dupressoir-Burlet, M. V., Martinache, L., and Goudemand, M. (1984). Preparation of a specific anti-cytomegalovirus immunoglobulin for intravenous injection. Rev. Fr. Transfus. Immunohematol. 27, 375–382. doi: 10.1016/S0338-4535(84)80178-6
Marschall, M., Stamminger, T., Urban, A., Wildum, S., Ruebsamen-Schaeff, H., Zimmermann, H., et al. (2012). In vitro evaluation of the activities of the novel Anticytomegalovirus compound AIC246 (Letermovir) against herpesviruses and other human pathogenic viruses. Antimicrob. Agents Chemother. 56, 1135–1137. doi: 10.1128/AAC.05908-11
Marty, F. M., Ljungman, P., Chemaly, R. F., Maertens, J., Dadwal, S. S., Duarte, R. F., et al. (2017). Letermovir prophylaxis for cytomegalovirus in hematopoietic-cell transplantation. N. Engl. J. Med. 377, 2433–2444. doi: 10.1056/NEJMoa1706640
Marty, F. M., Winston, D. J., Chemaly, R. F., Mullane, K. M., Shore, T. B., Papanicolaou, G. A., et al. (2019). A randomized, double-blind, placebo-controlled phase 3 trial of Oral Brincidofovir for cytomegalovirus prophylaxis in allogeneic hematopoietic cell transplantation. Biol. Blood Marrow Transplant. 25, 369–381. doi: 10.1016/j.bbmt.2018.09.038
Marty, F. M., Winston, D. J., Rowley, S. D., Vance, E., Papanicolaou, G. A., Mullane, K. M., et al. (2013). CMX001 to prevent cytomegalovirus disease in hematopoietic-cell transplantation. N. Engl. J. Med. 369, 1227–1236. doi: 10.1056/NEJMoa1303688
McGready, R., Phyo, A. P., Rijken, M. J., Tarning, J., Lindegardh, N., Hanpithakpon, W., et al. (2012). Artesunate/dihydroartemisinin pharmacokinetics in acute falciparum malaria in pregnancy: absorption, bioavailability, disposition and disease effects. Br. J. Clin. Pharmacol. 73, 467–477. doi: 10.1111/j.1365-2125.2011.04103.x
McSharry, J. J., McDonough, A., Olson, B., Hallenberger, S., Reefschlaeger, J., Bender, W., et al. (2001). Susceptibilities of human cytomegalovirus clinical isolates to BAY38-4766, BAY43-9695, and ganciclovir. Antimicrob. Agents Chemother. 45, 2925–2927. doi: 10.1128/AAC.45.10.2925-2927.2001
McVoy, M. A., and Nixon, D. E. (2005). Impact of 2-Bromo-5,6-Dichloro-1-β-d-Ribofuranosyl Benzimidazole riboside and inhibitors of DNA, RNA, and protein synthesis on human cytomegalovirus genome maturation. J. Virol. 79, 11115–11127. doi: 10.1128/JVI.79.17.11115-11127.2005
Middleton, E. (1998). Effect of plant flavonoids on immune and inflammatory cell function. Adv. Exp. Med. Biol. 439, 175–182. doi: 10.1007/978-1-4615-5335-9_13
Miescher, S. M., Huber, T. M., Kühne, M., Lieby, P., Snydman, D. R., Vensak, J. L., et al. (2015). In vitro evaluation of cytomegalovirus-specific hyperimmune globulins vs. standard intravenous immunoglobulins. Vox Sang. 109, 71–78. doi: 10.1111/vox.12246
Migawa, M. T., Girardet, J. L., Walker, J. A., Koszalka, G. W., Chamberlain, S. D., Drach, J. C., et al. (1998). Design, synthesis, and antiviral activity of α-nucleosides: d- and l-isomers of Lyxofuranosyl- and (5-Deoxylyxofuranosyl)benzimidazoles. J. Med. Chem. 41, 1242–1251. doi: 10.1021/jm970545c
Milbradt, J., Auerochs, S., Korn, K., and Marschall, M. (2009). Sensitivity of human herpesvirus 6 and other human herpesviruses to the broad-spectrum antiinfective drug artesunate. J. Clin. Virol. 46, 24–28. doi: 10.1016/j.jcv.2009.05.017
Moon, Y., Wang, L., DiCenzo, R., and Morris, M. E. (2008). Quercetin pharmacokinetics in humans. Biopharm. Drug Dispos. 29, 205–217. doi: 10.1002/bdd.605
Morère, L., Andouard, D., Labrousse, F., Saade, F., Calliste, C. A., Cotin, S., et al. (2015). Ex vivo model of congenital cytomegalovirus infection and new combination therapies. Placenta 36, 41–47. doi: 10.1016/j.placenta.2014.11.003
Mukherjee, D. (2002). Selective cyclooxygenase-2 (COX-2) inhibitors and potential risk of cardiovascular events. Biochem. Pharmacol. 63, 817–821. doi: 10.1016/S0006-2952(02)00842-0
Muller, C., Tilloy, V., Frobert, E., Feghoul, L., Garrigue, I., Lepiller, Q., et al. (2022). First clinical description of letermovir resistance mutation in cytomegalovirus UL51 gene and potential impact on the terminase complex structure. Antivir. Res. 204:105361. doi: 10.1016/j.antiviral.2022.105361
Muto, R., Motozuka, T., Nakano, M., Tatsumi, Y., Sakamoto, F., and Kosaka, N. (1998). The chemical structure of new substance as the metabolite of baicalin and time profiles for the plasma concentration after oral administration of sho-saiko-to in human. Yakugaku Zasshi 118, 79–87. doi: 10.1248/yakushi1947.118.3_79
Nagashima, S., Hirotani, M., and Yoshikawa, T. (2000). Purification and characterization of UDP-glucuronate: baicalein 7-O-glucuronosyltransferase from Scutellaria baicalensis Georgi. Cell suspension cultures. Phytochemistry 53, 533–538. doi: 10.1016/S0031-9422(99)00593-2
Nagi, R., Yashoda Devi, B. K., Rakesh, N., Reddy, S. S., and Patil, D. J. (2015). Clinical implications of prescribing nonsteroidal anti-inflammatory drugs in oral health care—a review. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. 119, 264–271. doi: 10.1016/j.oooo.2014.12.002
Nishimuro, H., Ohnishi, H., Sato, M., Ohnishi-Kameyama, M., Matsunaga, I., Naito, S., et al. (2015). Estimated Daily intake and seasonal food sources of quercetin in Japan. Nutrients 7, 2345–2358. doi: 10.3390/nu7042345
Nixon, D. E., and McVoy, M. A. (2004). Dramatic effects of 2-Bromo-5,6-Dichloro-1-β-d-Ribofuranosyl Benzimidazole riboside on the genome structure, packaging, and egress of Guinea pig cytomegalovirus. J. Virol. 78, 1623–1635. doi: 10.1128/JVI.78.4.1623-1635.2004
O’Brien, M. S., Markovich, K. C., Selleseth, D., DeVita, A. V., Sethna, P., and Gentry, B. G. (2018). In vitro evaluation of current and novel antivirals in combination against human cytomegalovirus. Antivir. Res. 158, 255–263. doi: 10.1016/j.antiviral.2018.08.015
Oiknine-Djian, E., Bar-On, S., Laskov, I., Lantsberg, D., Haynes, R. K., Panet, A., et al. (2019). Artemisone demonstrates synergistic antiviral activity in combination with approved and experimental drugs active against human cytomegalovirus. Antivir. Res. 172:104639. doi: 10.1016/j.antiviral.2019.104639
Ornoy, A., and Diav-Citrin, O. (2006). Fetal effects of primary and secondary cytomegalovirus infection in pregnancy. Reprod. Toxicol. 21, 399–409. doi: 10.1016/j.reprotox.2005.02.002
Ourahmane, A., Sauer, A., Nixon, D. E., Murphy, C., Mondello, M., Douglass, E., et al. (2018). A Guinea pig cytomegalovirus resistant to the DNA maturation inhibitor BDCRB. Antivir. Res. 154, 44–50. doi: 10.1016/j.antiviral.2018.04.006
Painter, W., Robertson, A., Trost, L. C., Godkin, S., Lampert, B., and Painter, G. (2012). First pharmacokinetic and safety study in humans of the novel lipid antiviral conjugate CMX001, a broad-Spectrum Oral drug active against double-stranded DNA viruses. Antimicrob. Agents Chemother. 56, 2726–2734. doi: 10.1128/AAC.05983-11
Papanicolaou, G. A., Silveira, F. P., Langston, A. A., Pereira, M. R., Avery, R. K., Uknis, M., et al. (2019). Maribavir for refractory or resistant cytomegalovirus infections in hematopoietic-cell or solid-organ transplant recipients: a randomized, dose-ranging, double-blind, phase 2 study. Clin. Infect. Dis. 68, 1255–1264. doi: 10.1093/cid/ciy706
Polansky, H., Javaherian, A., and Itzkovitz, E. (2016). Clinical study in genital herpes: natural Gene-Eden-VIR/Novirin versus acyclovir, valacyclovir, and famciclovir. Drug Des. Devel. Ther. 10, 2713–2722. doi: 10.2147/DDDT.S112852
Polansky, H., Javaherian, A., and Itzkovitz, E. (2018). Clinical trial of herbal treatment Gene-Eden-VIR/Novirin in Oral herpes. J. Evid. Based Integr. Med. 23:2515690X1880626. doi: 10.1177/2515690X18806269
Prichard, M. N. (2009). Function of human cytomegalovirus UL97 kinase in viral infection and its inhibition by maribavir. Rev. Med. Virol. 19, 215–229. doi: 10.1002/rmv.615
Prichard, M. N., Frederick, S. L., Daily, S., Borysko, K. Z., Townsend, L. B., Drach, J. C., et al. (2011). Benzimidazole Analogs Inhibit Human Herpesvirus 6▿. Antimicrob. Agents Chemother. 55, 2442–2445. doi: 10.1128/AAC.01523-10
Razonable, R. R., Amer, H., and Mardini, S. (2019). Application of a new paradigm for cytomegalovirus disease prevention in Mayo Clinic’s first face transplant. Mayo Clin. Proc. 94, 166–170. doi: 10.1016/j.mayocp.2018.09.017
Reefschlaeger, J., Bender, W., Hallenberger, S., Weber, O., Eckenberg, P., Goldmann, S., et al. (2001). Novel non-nucleoside inhibitors of cytomegaloviruses (BAY 38-4766): in vitro and in vivo antiviral activity and mechanism of action. J. Antimicrob. Chemother. 48, 757–767. doi: 10.1093/jac/48.6.757
Reiter, C., Fröhlich, T., Gruber, L., Hutterer, C., Marschall, M., Voigtländer, C., et al. (2015). Highly potent artemisinin-derived dimers and trimers: synthesis and evaluation of their antimalarial, antileukemia and antiviral activities. Bioorg. Med. Chem. 23, 5452–5458. doi: 10.1016/j.bmc.2015.07.048
Reitsma, J. M., Savaryn, J. P., Faust, K., Sato, H., Halligan, B. D., and Terhune, S. S. (2011). Antiviral inhibition targeting the HCMV kinase pUL97 requires pUL27-dependent degradation of Tip60 acetyltransferase and cell-cycle arrest. Cell Host Microbe 9, 103–114. doi: 10.1016/j.chom.2011.01.006
Revello, M. G., Lazzarotto, T., Guerra, B., Spinillo, A., Ferrazzi, E., Kustermann, A., et al. (2014). A randomized trial of hyperimmune globulin to prevent congenital cytomegalovirus. N. Engl. J. Med. 370, 1316–1326. doi: 10.1056/NEJMoa1310214
Reza Nassiri, M., Emerson, S. G., Devivar, R. V., Townsend, L. B., Drach, J. C., and Taichman, R. S. (1996). Comparison of benzimidazole nucleosides and ganciclovir on the in vitro proliferation and colony formation of human bone marrow progenitor cells. Available at: http://deepblue.lib.umich.edu/handle/2027.42/72204. (Accessed April 4, 2023).
Ribeiro, I. R., and Olliaro, P. (1998). Safety of artemisinin and its derivatives. A review of published and unpublished clinical trials. Med. Trop. 58, 50–53.
Rifkin, L. M., Minkus, C. L., Pursell, K., Jumroendararasame, C., and Goldstein, D. A. (2017). Utility of Leflunomide in the treatment of drug resistant cytomegalovirus retinitis. Ocul. Immunol. Inflamm. 25, 93–96. doi: 10.3109/09273948.2015.1071406
Rittà, M., Costa, C., Solidoro, P., Sidoti, F., Libertucci, D., Boffini, M., et al. (2015). Everolimus-based immunosuppressive regimens in lung transplant recipients: impact on CMV infection. Antivir. Res. 113, 19–26. doi: 10.1016/j.antiviral.2014.10.016
Robin, C., Thiebaut, A., Alain, S., Sicre de Fontbrune, F., Berceanu, A., D’Aveni, M., et al. (2020). Letermovir for secondary prophylaxis of cytomegalovirus infection and disease after allogeneic hematopoietic cell transplantation: results from the French compassionate program. Biol. Blood Marrow Transplant. 26, 978–984. doi: 10.1016/j.bbmt.2020.01.027
Ross, S. A., Fowler, K. B., Ashrith, G., Stagno, S., Britt, W. J., Pass, R. F., et al. (2006). Hearing loss in children with congenital cytomegalovirus infection born to mothers with preexisting immunity. J. Pediatr. 148, 332–336. doi: 10.1016/j.jpeds.2005.09.003
Rouphael, N. G., Hurwitz, S. J., Hart, M., Beck, A., Anderson, E. J., Deye, G., et al. (2019). Phase Ib trial to evaluate the safety and pharmacokinetics of multiple ascending doses of Filociclovir (MBX-400, Cyclopropavir) in healthy volunteers. Antimicrob. Agents Chemother. 63, e00717–e00719. doi: 10.1128/AAC.00717-19
Roussel, C., Caumes, E., Thellier, M., Ndour, P. A., Buffet, P. A., and Jauréguiberry, S. (2017). Artesunate to treat severe malaria in travellers: review of efficacy and safety and practical implications. J. Travel Med. 24:93. doi: 10.1093/jtm/taw093
Roy, V., and Agrofoglio, L. A. (2022). Nucleosides and emerging viruses: a new story. Drug Discov. Today 27, 1945–1953. doi: 10.1016/j.drudis.2022.02.013
Roy, S., and Arav-Boger, R. (2014). New cell-signaling pathways for controlling cytomegalovirus replication. Am. J. Transplant. 14, 1249–1258. doi: 10.1111/ajt.12725
Saag, M. S., Gandhi, R. T., Hoy, J. F., Landovitz, R. J., Thompson, M. A., Sax, P. E., et al. (2020). Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2020 recommendations of the international antiviral society–USA panel. JAMA 324, 1651–1669. doi: 10.1001/jama.2020.17025
Santhanakrishnan, K., Yonan, N., Callan, P., Karimi, E., Al-Aloul, M., and Venkateswaran, R. (2019). The use of CMVIg rescue therapy in cardiothoracic transplantation: a single-center experience over 6 years (2011-2017). Clin. Transpl. 33:e13655. doi: 10.1111/ctr.13655
Schampera, M. S., Schweinzer, K., Abele, H., Kagan, K. O., Klein, R., Rettig, I., et al. (2017). Comparison of cytomegalovirus (CMV)-specific neutralization capacity of hyperimmunoglobulin (HIG) versus standard intravenous immunoglobulin (IVIG) preparations: impact of CMV IgG normalization. J. Clin. Virol. 90, 40–45. doi: 10.1016/j.jcv.2017.03.005
Schleiss, M. R. (2008). Comparison of vaccine strategies against congenital CMV infection in the guinea pig model. J. Clin. Virol. 41, 224–230. doi: 10.1016/j.jcv.2007.10.008
Schleiss, M. R., Bernstein, D. I., McVoy, M. A., Stroup, G., Bravo, F., Creasy, B., et al. (2005). The non-nucleoside antiviral, BAY 38-4766, protects against cytomegalovirus (CMV) disease and mortality in immunocompromised guinea pigs. Antivir. Res. 65, 35–43. doi: 10.1016/j.antiviral.2004.09.004
Scholz, B., Rechter, S., Drach, J. C., Townsend, L. B., and Bogner, E. (2003). Identification of the ATP-binding site in the terminase subunit pUL56 of human cytomegalovirus. Nucleic Acids Res. 31, 1426–1433. doi: 10.1093/nar/gkg229
Seidel, V., Hackelöer, M., Rancourt, R. C., Henrich, W., and Siedentopf, J. P. (2020). Fetal and maternal outcome after hyperimmunoglobulin administration for prevention of maternal–fetal transmission of cytomegalovirus during pregnancy: retrospective cohort analysis. Arch. Gynecol. Obstet. 302, 1353–1359. doi: 10.1007/s00404-020-05728-7
Shahar-Nissan, K., Pardo, J., Peled, O., Krause, I., Bilavsky, E., Wiznitzer, A., et al. (2020). Valaciclovir to prevent vertical transmission of cytomegalovirus after maternal primary infection during pregnancy: a randomised, double-blind, placebo-controlled trial. Lancet 396, 779–785. doi: 10.1016/S0140-6736(20)31868-7
Shapira, M. Y., Resnick, I. B., Chou, S., Neumann, A. U., Lurain, N. S., Stamminger, T., et al. (2008). Artesunate as a potent antiviral agent in a patient with late drug-resistant cytomegalovirus infection after hematopoietic stem cell transplantation. Clin. Infect. Dis. 46, 1455–1457. doi: 10.1086/587106
Silva, J. T., Pérez-González, V., Lopez-Medrano, F., Alonso-Moralejo, R., Fernández-Ruiz, M., San-Juan, R., et al. (2018). Experience with leflunomide as treatment and as secondary prophylaxis for cytomegalovirus infection in lung transplant recipients: a case series and review of the literature. Clin. Transpl. 32:e13176. doi: 10.1111/ctr.13176
Song, I., Chen, G., Wu, J., and Ilic, K. (2023). Maribavir pharmacokinetics and safety in participants with moderate hepatic impairment: a phase 1, open-label, single-dose, parallel group study. J. Clin. Pharmacol. 63, 250–258. doi: 10.1002/jcph.2155
Sonntag, E., Hahn, F., Bertzbach, L. D., Seyler, L., Wangen, C., Müller, R., et al. (2019). In vivo proof-of-concept for two experimental antiviral drugs, both directed to cellular targets, using a murine cytomegalovirus model. Antivir. Res. 161, 63–69. doi: 10.1016/j.antiviral.2018.11.008
Speir, E., Yu, Z. X., Ferrans, V. J., Huang, E. S., and Epstein, S. E. (1998). Aspirin attenuates cytomegalovirus infectivity and gene expression mediated by Cyclooxygenase-2 in coronary artery smooth muscle cells. Circ. Res. 83, 210–216. doi: 10.1161/01.RES.83.2.210
Stoelben, S., Arns, W., Renders, L., Hummel, J., Mühlfeld, A., Stangl, M., et al. (2014). Preemptive treatment of cytomegalovirus infection in kidney transplant recipients with letermovir: results of a phase 2a study. Transpl. Int. 27, 77–86. doi: 10.1111/tri.12225
Swan, S. K., Smith, W. B., Marbury, T. C., Schumacher, M., Dougherty, C., Mico, B. A., et al. (2007). Pharmacokinetics of Maribavir, a novel Oral Anticytomegalovirus agent, in subjects with varying degrees of renal impairment. J. Clin. Pharmacol. 47, 209–217. doi: 10.1177/0091270006296765
Taber, D. J., Chokkalingam, A., Su, Z., Self, S., Miller, D., and Srinivas, T. (2019). Randomized controlled trial assessing the impact of everolimus and low-exposure tacrolimus on graft outcomes in kidney transplant recipients. Clin. Transpl. 33:e13679. doi: 10.1111/ctr.13679
Taiming, L., and Xuehua, J. (2006). Investigation of the absorption mechanisms of baicalin and baicalein in rats. J. Pharm. Sci. 95, 1326–1333. doi: 10.1002/jps.20593
Tedesco-Silva, H., Pascual, J., Viklicky, O., Basic-Jukic, N., Cassuto, E., Kim, D. Y., et al. (2019). Safety of Everolimus with reduced Calcineurin inhibitor exposure in De novo kidney transplants: an analysis from the randomized TRANSFORM study. Transplantation 103, 1953–1963. doi: 10.1097/TP.0000000000002626
Tian, S., He, G., Song, J., Wang, S., Xin, W., Zhang, D., et al. (2012). Pharmacokinetic study of baicalein after oral administration in monkeys. Fitoterapia 83, 532–540. doi: 10.1016/j.fitote.2011.12.019
Tippin, T. K., Morrison, M. E., Brundage, T. M., and Momméja-Marin, H. (2016). Brincidofovir is not a substrate for the human organic anion transporter 1: a mechanistic explanation for the lack of nephrotoxicity observed in clinical studies. Ther. Drug Monit. 38, 777–786. doi: 10.1097/FTD.0000000000000353
Tollefson, A. E., Hussein, I. T. M., Toth, K., and Bowlin, T. L. (2022). Filociclovir is a potent inhibitor of human adenovirus F41. Antivir. Res. 208:105431. doi: 10.1016/j.antiviral.2022.105431
Toovey, S. (2006). Are currently deployed artemisinins neurotoxic? Toxicol. Lett. 166, 95–104. doi: 10.1016/j.toxlet.2006.06.001
Toth, K., Hussein, I. T. M., Tollefson, A. E., Ying, B., Spencer, J. F., Eagar, J., et al. (2020). Filociclovir is a potent in vitro and in vivo inhibitor of human adenoviruses. Antimicrob. Agents Chemother. 64, e01299–e01220. doi: 10.1128/AAC.01299-20
Townsend, L. B., Devivar, R. V., Turk, S. R., Nassiri, M. R., and Drach, J. C. (1995). Design, synthesis, and antiviral activity of certain 2,5,6-trihalo-1-(beta-D-ribofuranosyl)benzimidazoles. J. Med. Chem. 38, 4098–4105. doi: 10.1021/jm00020a025
Tsao, R. (2010). Chemistry and biochemistry of dietary polyphenols. Nutrients 2, 1231–1246. doi: 10.3390/nu2121231
Tuli, H. S., Aggarwal, V., Kaur, J., Aggarwal, D., Parashar, G., Parashar, N. C., et al. (2020). Baicalein: a metabolite with promising antineoplastic activity. Life Sci. 259:118183. doi: 10.1016/j.lfs.2020.118183
Underwood, M. R., Harvey, R. J., Stanat, S. C., Hemphill, M. L., Miller, T., Drach, J. C., et al. (1998). Inhibition of human cytomegalovirus DNA maturation by a Benzimidazole Ribonucleoside is mediated through the UL89 gene product. J. Virol. 72, 717–725. doi: 10.1128/JVI.72.1.717-725.1998
Veit, T., Munker, D., Kauke, T., Zoller, M., Michel, S., Ceelen, F., et al. (2020). Letermovir for difficult to treat cytomegalovirus infection in lung transplant recipients. Transplantation 104, 410–414. doi: 10.1097/TP.0000000000002886
Vial, R., Zandotti, C., Alain, S., Decourt, A., Jourde-Chiche, N., Purgus, R., et al. (2017). Brincidofovir use after Foscarnet crystal nephropathy in a kidney transplant recipient with multiresistant cytomegalovirus infection. Case Rep. Transplant. 2017, 1–7. doi: 10.1155/2017/3624146
Visentin, S., Manara, R., Milanese, L., Da Roit, A., Forner, G., Salviato, E., et al. (2012). Early primary cytomegalovirus infection in pregnancy: maternal Hyperimmunoglobulin therapy improves outcomes among infants at 1 year of age. Clin. Infect. Dis. 55, 497–503. doi: 10.1093/cid/cis423
Waldman, W. J., Knight, D. A., Blinder, L., Shen, J., Lurain, N. S., Miller, D. M., et al. (1999a). Inhibition of cytomegalovirus in vitro and in vivo by the experimental immunosuppressive agent Leflunomide. Intervirology 42, 412–418. doi: 10.1159/000053979
Waldman, W. J., Knight, D. A., Lurain, N. S., Miller, D. M., Sedmak, D. D., Williams, J. W., et al. (1999b). Novel mechanism of inhibition of cytomegalovirus by the experimental immunosuppressive agent leflunomide. Transplantation 68, 814–825. doi: 10.1097/00007890-199909270-00014
Wang, L. H., Peck, R. W., Yin, Y., Allanson, J., Wiggs, R., and Wire, M. B. (2003). Phase I safety and pharmacokinetic trials of 1263W94, a novel Oral anti-human cytomegalovirus agent, in healthy and human immunodeficiency virus-infected subjects. Antimicrob. Agents Chemother. 47, 1334–1342. doi: 10.1128/AAC.47.4.1334-1342.2003
Weber, O., Bender, W., Eckenberg, P., Goldmann, S., Haerter, M., Hallenberger, S., et al. (2001). Inhibition of murine cytomegalovirus and human cytomegalovirus by a novel non-nucleosidic compound in vivo. Antivir. Res. 49, 179–189. doi: 10.1016/S0166-3542(01)00127-9
Wild, M., Bertzbach, L. D., Tannig, P., Wangen, C., Müller, R., Herrmann, L., et al. (2020). The trimeric artesunate derivative TF27 exerts strong anti-cytomegaloviral efficacy: focus on prophylactic efficacy and oral treatment of immunocompetent mice. Antivir. Res. 178:104788. doi: 10.1016/j.antiviral.2020.104788
Wildum, S., Zimmermann, H., and Lischka, P. (2015). In vitro drug combination studies of Letermovir (AIC246, MK-8228) with approved anti-human cytomegalovirus (HCMV) and anti-HIV compounds in inhibition of HCMV and HIV replication. Antimicrob. Agents Chemother. 59, 3140–3148. doi: 10.1128/AAC.00114-15
Williams, S. L., Hartline, C. B., Kushner, N. L., Harden, E. A., Bidanset, D. J., Drach, J. C., et al. (2003). In vitro activities of Benzimidazole d- and l-Ribonucleosides against herpesviruses. Antimicrob. Agents Chemother. 47, 2186–2192. doi: 10.1128/AAC.47.7.2186-2192.2003
Williams, J. W., Mital, D., Chong, A., Kottayil, A., Millis, M., Longstreth, J., et al. (2002). Experiences with LEFLUNOMIDE in solid organ transplantation. Transplantation 73, 358–366. doi: 10.1097/00007890-200202150-00008
Williams-Aziz, S. L., Hartline, C. B., Harden, E. A., Daily, S. L., Prichard, M. N., Kushner, N. L., et al. (2005). Comparative activities of lipid esters of Cidofovir and cyclic Cidofovir against replication of herpesviruses in vitro. Antimicrob. Agents Chemother. 49, 3724–3733. doi: 10.1128/AAC.49.9.3724-3733.2005
Williamson, R. A., Yea, C. M., Robson, P. A., Curnock, A. P., Gadher, S., Hambleton, A. B., et al. (1996). Dihydroorotate dehydrogenase is a target for the biological effects of leflunomide. Transplant. Proc. 28, 3088–3091.
Winston, D. J., Young, J. A. H., Pullarkat, V., Papanicolaou, G. A., Vij, R., Vance, E., et al. (2008). Maribavir prophylaxis for prevention of cytomegalovirus infection in allogeneic stem cell transplant recipients: a multicenter, randomized, double-blind, placebo-controlled, dose-ranging study. Blood 111, 5403–5410. doi: 10.1182/blood-2007-11-121558
Wolf, D. G., Shimoni, A., Resnick, I. B., Stamminger, T., Neumann, A. U., Chou, S., et al. (2011). Human cytomegalovirus kinetics following institution of Artesunate after hematopoietic stem cell transplantation. Antivir. Res. 90, 183–186. doi: 10.1016/j.antiviral.2011.03.184
Wu, Z., Drach, J. C., Prichard, M. N., Yanachkova, M., Yanachkov, I., Bowlin, T. L., et al. (2009). L-valine Ester of Cyclopropavir - a new antiviral prodrug. Antivir. Chem. Chemother. 20, 37–46. doi: 10.3851/IMP782
Wu, L. L., Yang, X. B., Huang, Z. M., Liu, H. Z., and Wu, G. X. (2007). In vivo and in vitro antiviral activity of hyperoside extracted from Abelmoschus manihot (L) medik. Acta Pharmacol. Sin. 28, 404–409. doi: 10.1111/j.1745-7254.2007.00510.x
Xu, X., Williams, J. W., Bremer, E. G., Finnegan, A., and Chong, A. S. F. (1995). Inhibition of protein tyrosine phosphorylation in T cells by a novel immunosuppressive agent, Leflunomide (∗). J. Biol. Chem. 270, 12398–12403. doi: 10.1074/jbc.270.21.12398
Yoshida, M., Yamada, M., Tsukazaki, T., Chatterjee, S., Lakeman, F. D., Nii, S., et al. (1998). Comparison of antiviral compounds against human herpesvirus 6 and 7. Antivir. Res. 40, 73–84. doi: 10.1016/S0166-3542(98)00049-7
Yu, Y. B., Miyashiro, H., Nakamura, N., Hattori, M., and Park, J. C. (2007). Effects of triterpenoids and flavonoids isolated from Alnus firma on HIV-1 viral enzymes. Arch. Pharm. Res. 30, 820–826. doi: 10.1007/BF02978831
Zarghi, A., and Arfaei, S. (2011). Selective COX-2 inhibitors: a review of their structure-activity relationships. Iran J. Pharm. Res. 10, 655–683. doi: 10.1016/j.bmc.2005.11.041
Zhang, L., Lin, G., Chang, Q., and Zuo, Z. (2005). Role of intestinal first-pass metabolism of baicalein in its absorption process. Pharm. Res. 22, 1050–1058. doi: 10.1007/s11095-005-5303-7
Zhang, L., Lin, G., and Zuo, Z. (2004). High-performance liquid chromatographic method for simultaneous determination of baicalein and baicalein 7-glucuronide in rat plasma. J. Pharm. Biomed. Anal. 36, 637–641. doi: 10.1016/j.jpba.2004.07.024
Zhang, L., Lin, G., and Zuo, Z. (2007). Involvement of UDP-glucuronosyltransferases in the extensive liver and intestinal first-Pass metabolism of flavonoid Baicalein. Pharm. Res. 24, 81–89. doi: 10.1007/s11095-006-9126-y
Zhao, K. C., and Song, Z. Y. (1993). Pharmacokinetics of dihydroqinghaosu in human volunteers and comparison with qinghaosu. Yao Xue Xue Bao 28, 342–346.
Zhou, S., Breitenbach, J. M., Borysko, K. Z., Drach, J. C., Kern, E. R., Gullen, E., et al. (2004). Synthesis and antiviral activity of (Z)- and (E)-2,2-[Bis(hydroxymethyl)cyclopropylidene]methylpurines and -pyrimidines: second-generation methylenecyclopropane analogues of nucleosides. J. Med. Chem. 47, 566–575. doi: 10.1021/jm030316s
Keywords: cytomegalovirus, letermovir, maribavir, direct antivirals, indirect antivirals, immunomodulatory molecules, immunoglobulins
Citation: Gourin C, Alain S and Hantz S (2023) Anti-CMV therapy, what next? A systematic review. Front. Microbiol. 14:1321116. doi: 10.3389/fmicb.2023.1321116
Edited by:
Cécile E. Malnou, Université Toulouse III Paul Sabatier, FranceReviewed by:
Hang Su, Albert Einstein College of Medicine, United StatesBeatrice Mercorelli, University of Padua, Italy
Copyright © 2023 Gourin, Alain and Hantz. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claire Gourin, claire.gourin@unilim.fr; Sophie Alain, sophie.alain@unilim.fr; Sébastien Hantz, sebastien.hantz@unilim.fr