 Tongtong Pan
Tongtong Pan Lihuang Su2†
Lihuang Su2† Yongping Chen
Yongping Chen- 1Hepatology Diagnosis and Treatment Center, The First Affiliated Hospital of Wenzhou Medical University and Zhejiang Provincial Key Laboratory for Accurate Diagnosis and Treatment of Chronic Liver Diseases, Wenzhou, China
- 2The First Affiliated Hospital of Wenzhou Medical University, Wenzhou, China
- 3Alberta Institute, Wenzhou Medical University, Wenzhou, China
Introduction: Enteric dysbacteriosis is strongly associated with nonalcoholic fatty liver disease (NAFLD). However, the underlying causal relationship remains unknown. Thus, the present study aimed to investigate the relationship between gut microbiota and NAFLD using Mendelian randomization (MR) and analyze the target genes potentially regulated by specific microbiota.
Methods: Bidirectional two-sample MR analysis was performed using inverse variance weighted (IVW) supplemented by MR-Egger, weighted median, simple mode, and weighted mode methods. Data were pooled from gut microbiota and NAFLD association studies. The least absolute shrinkage, selection operator regression, and the Support Vector Machine algorithm were used to identify genes regulated by these intestinal flora in NAFLD. The liver expression of these genes was verified in methionine choline-deficient (MCD) diet-fed mice.
Results: IVW results confirmed a causal relationship between eight specific gut microbes and NAFLD. Notably, the order Actinomycetales, NB1n, the family Actinomycetaceae, Oxalobacteraceae and the genus Ruminococcaceae UCG005 were positively correlated, whereas Lactobacillaceae, the Christensenellaceae R7 group, and Intestinibacter were negatively correlated with NAFLD onset. In NAFLD, these eight bacteria regulated four genes: colony-stimulating factor 2 receptor β, fucosyltransferase 2, 17-beta-hydroxysteroid dehydrogenase 14, and microtubule affinity regulatory kinase 3 (MAPK3). All genes, except MARK3, were differentially expressed in the liver tissues of MCD diet-fed mice.
Discussion: The abundance of eight gut microbiota species and NAFLD progression displayed a causal relationship based on the expression of the four target genes. Our findings contributed to the advancement of intestinal microecology-based diagnostic technologies and targeted therapies for NAFLD.
1 Introduction
Nonalcoholic fatty liver disease (NAFLD) is a metabolic syndrome characterized by less than 5% lipid deposits in hepatocytes, without alcohol consumption or other factors that may induce liver damage. The progressive stages of NAFLD include fatty liver (steatosis), nonalcoholic steatohepatitis (NASH), and fibrosis/cirrhosis, which eventually progress to end-stage liver failure or hepatocellular carcinoma with a low survival rate and a poor prognosis. Unfortunately, the global prevalence of NAFLD has considerably increased from 25.24% in 2016 to 32.4% in 2022 (Younossi et al., 2016; Riazi et al., 2022).
Advances in high-throughput sequencing technology have revealed strong associations between changes in the intestinal microbiome and the development of NAFLD. Several factors, including bidirectional crosstalk via the anatomical and physiological structure of the intestine–liver axis, disruption of the intestinal mucosal barrier, gut microbiota dysbiosis, harmful bacteria, and metabolite translocation, can promote the development of NAFLD lesions (Albillos et al., 2020). Currently, there are no specific drugs are approved for the treatment of NAFLD. However, interventions targeting intestinal microecology have exhibited therapeutic effects by rectifying intestinal dysbacteriosis, repairing damaged mucosal barriers, and regulating microbial metabolite production to alleviate chronic inflammation, insulin resistance, and oxidative stress (Craven et al., 2020; Juárez-Fernández et al., 2021; Zhao et al., 2021; Pan et al., 2022). However, the established associations between intestinal microbiota and NAFLD are based on cross-sectional studies that are prone to confounding factors and reverse causality. Thus, other study designs must be used to establish the causality between the intestinal microbiota and NAFLD and identify therapeutic targets.
Randomized controlled trials are the gold standard for investigating causality in epidemiology; however, their implementation is difficult owing to ethical constraints, high costs, and time constraints (Hariton and Locascio, 2018). Mendel’s law of inheritance dictates that parental alleles are randomly assigned to offspring, achieving the necessary randomization akin to randomized controlled trials without being affected by common confounding factors, including acquired or prior exposure, while excluding reverse causality. Mendelian randomization (MR) leverages genetic variation as an instrumental variable (IV), presenting an effective method for studying causal relationships between risk factors and outcomes (Hemani et al., 2018). Several genome-wide association studies (GWASs) on the intestinal microbiota and NAFLD (Bonder et al., 2016; Wang B. et al., 2016; Wang J. et al., 2016; Anstee et al., 2020; Ghodsian et al., 2021; Kurilshikov et al., 2021) have highlighted the relationships between these entities.
In the present study, we investigated the potential causality between different intestinal microbiota changes and NAFLD via a two-sample MR analysis using summary statistics from a GWAS. The least absolute shrinkage and selection operator (LASSO) regression and the Support Vector Machine (SVM) algorithm were used to screen the positively regulated genes in the intestinal flora. The expression of these genes was verified in the intestinal and liver tissues of mice fed a methionine choline-deficient (MCD) diet. This study aimed to outline a framework for developing gut microbiome-based diagnostic, preventive, and therapeutic approaches for NAFLD based on the target bacteria or genes they regulate.
2 Materials and methods
2.1 Data sources
Controls (770,180) and NAFLD cases (8,434) from a GWAS-based meta-analysis provided the summary statistics for NAFLD (Kurilshikov et al., 2021). Summary data for the 16S fecal microbiomes were obtained from a microbiome-based GWAS with 18,340 cases (Ghodsian et al., 2021). The GSE24807 Series Matrix File was downloaded from the Gene Expression Omnibus database.1 The data file (annotated as GPL2895) included the expression profile data of 17 patients, including 5 in the control group and 12 in the NAFLD group. The ethics review committees authorized each study cited in the GWAS and informed consent was obtained from all participants.
2.2 Study design
MR analysis was performed to determine the causal relationship between the intestinal microbes and NAFLD. Quality control experiments, such as heterogeneity and gene multiplicity tests, were conducted to confirm the reliability of causality results. Three hypotheses were assumed in the MR analysis: IVs are closely related to exposure, confounding variables that affect ‘exposure–outcome’ are not linked to IVs, and IVs affect outcome only through exposure.
2.3 Selection of IVs
Single-nucleotide polymorphisms (SNPs) with genome-wide importance in the gut microbiota were selected for pooling. The following selection criteria were used to choose the IVs: (1) In the forward MR analysis, p < 1.0 × 10−5 of SNPs was selected as the potential IVs; and in the reverse MR analysis, p < 5.0 × 10−6 of SNPs was selected as the potential IVs (Burgess et al., 2017). (2) The linkage disequilibrium parameter (r2) was set at a threshold of 0.001, and a genetic distance of 10,000 kb was required to select SNPs (Cheng et al., 2020). (3) The strength of the selected SNPs was assessed after removing palindromic SNPs. The strength of genetic variation as IVs was assessed using the F-statistic, and an F-statistic ≥10 indicates a strong IV with a low likelihood of introducing significant bias in the analysis (Shang et al., 2013).
2.4 MR analysis and sensitivity analysis
Chain imbalance analysis, MR analysis, and quality control experiments were performed using the R statistical programming language and two-sample MR (Hemani et al., 2018). Five methods were used to estimate causal effects (Bowden et al., 2015; Verbanck et al., 2018; Parker et al., 2020): inverse variance weighted, MR-Egger, weighted median, simple mode, and weighted mode. IVW served as the main method. The leave-one-out approach was used for sensitivity analysis and to compute the values of the remaining cumulative effects of the SNPs (Curtis et al., 2019). Horizontal genetic pleiotropy tests were performed using the intercept terms from the MR-Egger regression to determine whether IVs affected the outcomes through pathways other than exposure. The relationship between the gut microbiome and the risk of developing NAFLD was summarized as odds ratios (ORs) and 95% confidence intervals (95% CIs). A threshold of statistical significance was set at a Bonferroni-corrected p < 0.05 to address the issue of multiple comparisons. Figure 1 presents an overview of the analysis pipeline.
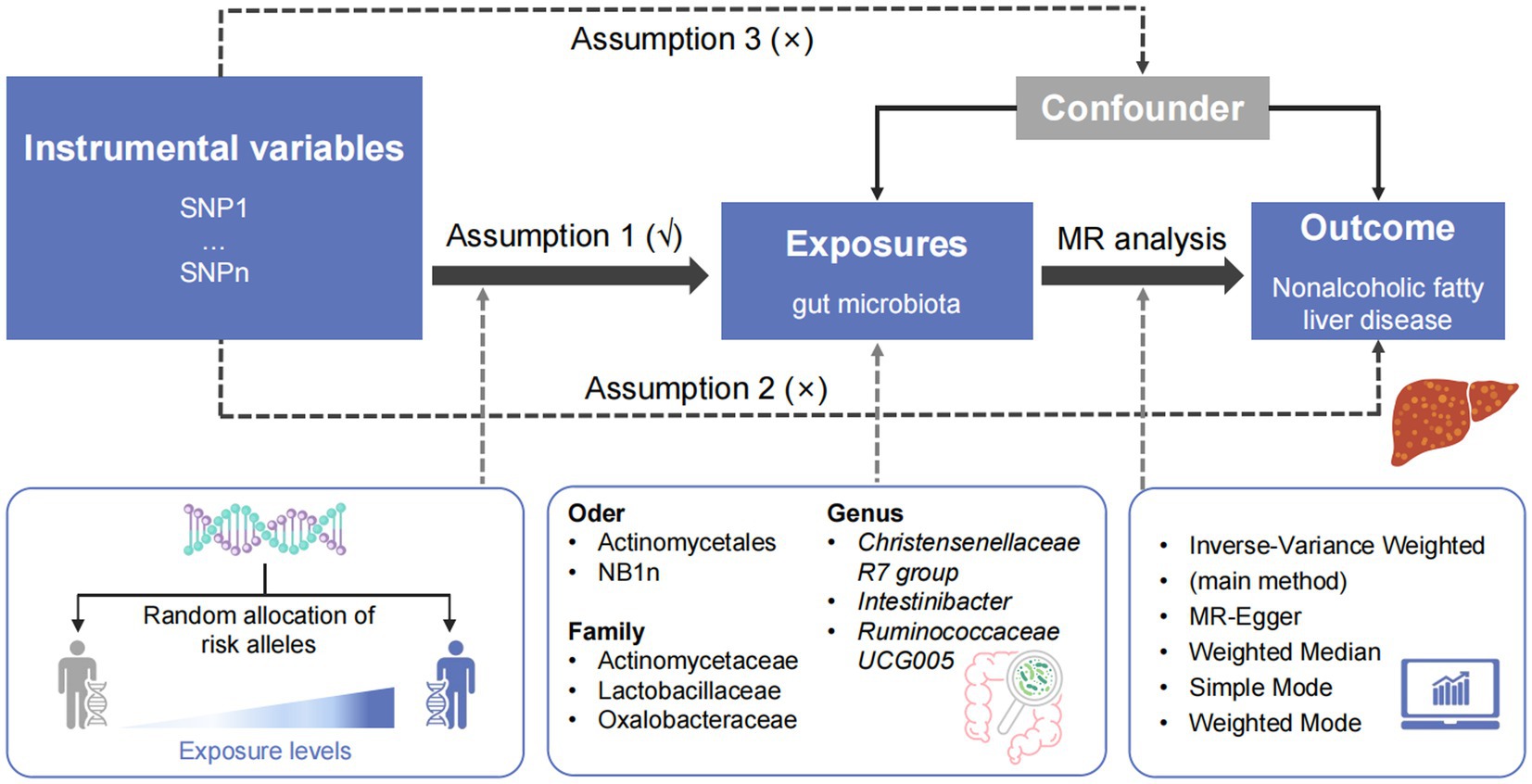
Figure 1. Overview of the study design. SNP, single-nucleotide polymorphism; MR, Mendelian randomization.
2.5 Feature selection process using LASSO regression and the SVM algorithm
The IVs listed in Supplementary Table S1 were converted to the corresponding SNPs using the “gwasrapidd” software package. LASSO logistic regression and the SVM algorithm were used to select the diagnostic markers for diseases using the “glmnet” and “e1071” software packages. After extracting SNPs, glmnet function of “glmnet” software package was used to fit linear model, family was set to binomial, alpha was set to 1, and then cv.glmnet was used for 10 cross-validation. When λ value reached the minimum, gene results were output.
2.6 Animals and diets
Twelve seven-week-old male wild-type C57BL/6 mice were purchased from the SLAC Laboratory Animal Co., Ltd. (Shanghai, China). All mice were housed under specific pathogen-free laboratory animal barrier environmental conditions with a 12/12 h light/dark cycle, temperature of 24 ± 2°C, and humidity of 50 ± 10%. Mice were randomly analyzed in two groups with six mice in each group. The control group was fed normal chow (NC) and had ad libitum access to water. Mouse models of NASH were established by feeding the mice an MCD diet (A02082002B; Research Diets, New Brunswick, NJ, United States) for 4 weeks. n = 6 per group. All experiments were conducted in accordance with the established guidelines and were approved by the Institutional Animal Care and Use Committee of Wenzhou Medical University (No. wydw2018-0252).
2.7 Quantitative real-time polymerase chain reaction
Total RNA was isolated from the liver samples and cultured cells using TRIzol™ reagent (Invitrogen). RNA was reverse-transcribed into cDNA using the High-Capacity cDNA Reverse Transcription Kit (Takara, Shiga, Japan). qRT-PCR was performed using SYBR Green Master Mix (Thermo Fisher Scientific) in the Real-Time PCR System (Thermo Fisher Scientific) using cDNA. β-actin was used as the invariant control. The RT-qPCR results were analyzed using the comparative Ct method (2−∆∆Ct). The primer sequences used for PCR are listed in Supplementary Table S2.
2.8 Histopathology
The liver tissues were fixed with 4% paraformaldehyde. All histological analyses were performed using paraffin-embedded tissue sections. The sections were stained with hematoxylin and eosin (Sigma, MO, United States) according to a standard procedure to visualize the lipid accumulation pattern. The Histological Activity Index (HAI) scores were quantified using ImageJ software (version 1.8.0, National Institutes of Health, Bethesda, MD, United States).
2.9 Biochemical analyses
Serum was isolated from blood via centrifugation at 1,300 rpm for 10 min and stored at −80°C until use. The levels of serum triglycerides (TG), total cholesterol (TC), non-esterified fatty acid (NEFA), aspartate transaminase (AST), and alanine transaminase (ALT) were measured according to the manufacturer’s instructions (Nanjing Jiancheng Bioengineering Institute, Jiangsu, China).
2.10 Statistical analyses
The experimental data were analyzed using GraphPad Prism 10.1.0.316 for Windows (San Diego, CA, United States). The Shapiro–Wilk test showed that the data were normally distributed, so the data were presented as the mean ± standard deviation. Then, Student’s t-test and one-or two-way analysis of variance were used to compare the data between groups. Statistical significance was set at p < 0.05.
3 Results
3.1 Selection of genetic IVs
SNPs were selected using GWAS summary statistics (Ghodsian et al., 2021). Ninety-eight SNPs were used as IVs for eight gut microbes based on the selection criteria for IVs. Supplementary Table S1 lists the details of the selected IVs.
3.2 Causal effects of gut microbiota on NAFLD
The causal relationship between the relative abundance of enterobacteria and NAFLD was analyzed at three levels (family, genus, and order) using 52-sample MR methods, primarily based on the IVW method. A total of eight intestinal microorganisms were found to be associated with NAFLD (Figure 2).
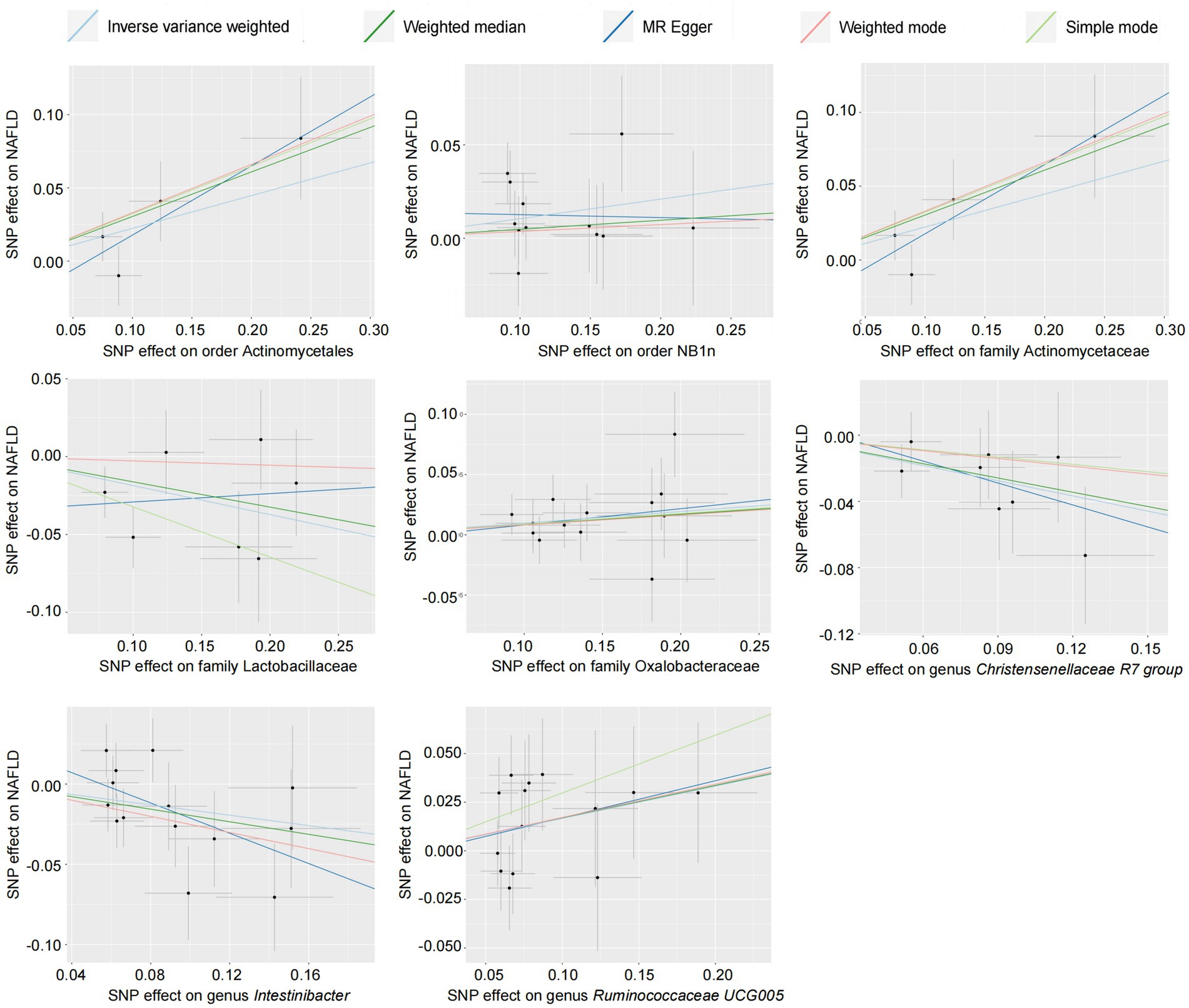
Figure 2. Scatter plot of the gut microbiota and NAFLD (forward results). SNP, single-nucleotide polymorphism; NAFLD, nonalcoholic fatty liver disease.
Forward MR analysis of the gut microbiota and NAFLD revealed that Actinomycetaceae, Oxalobacteraceae, Ruminococcaceae UCG005, Actinomycetales, and NB1n were positively associated with NAFLD. In contrast, Lactobacillaceae, Christensenellaceae R7 group, and Intestinibacter were negatively associated with NAFLD (Table 1, Figure 3, and Supplementary Table S3). Reverse MR analysis of the NAFLD-to-gut-microbiota ratio was performed to further evaluate the confounding factors and verify the relationship between enterobacteria and NAFLD. We observed that NAFLD had no effect on the eight gut microbiota mentioned above (Table 2, Figure 4, and Supplementary Table S4).
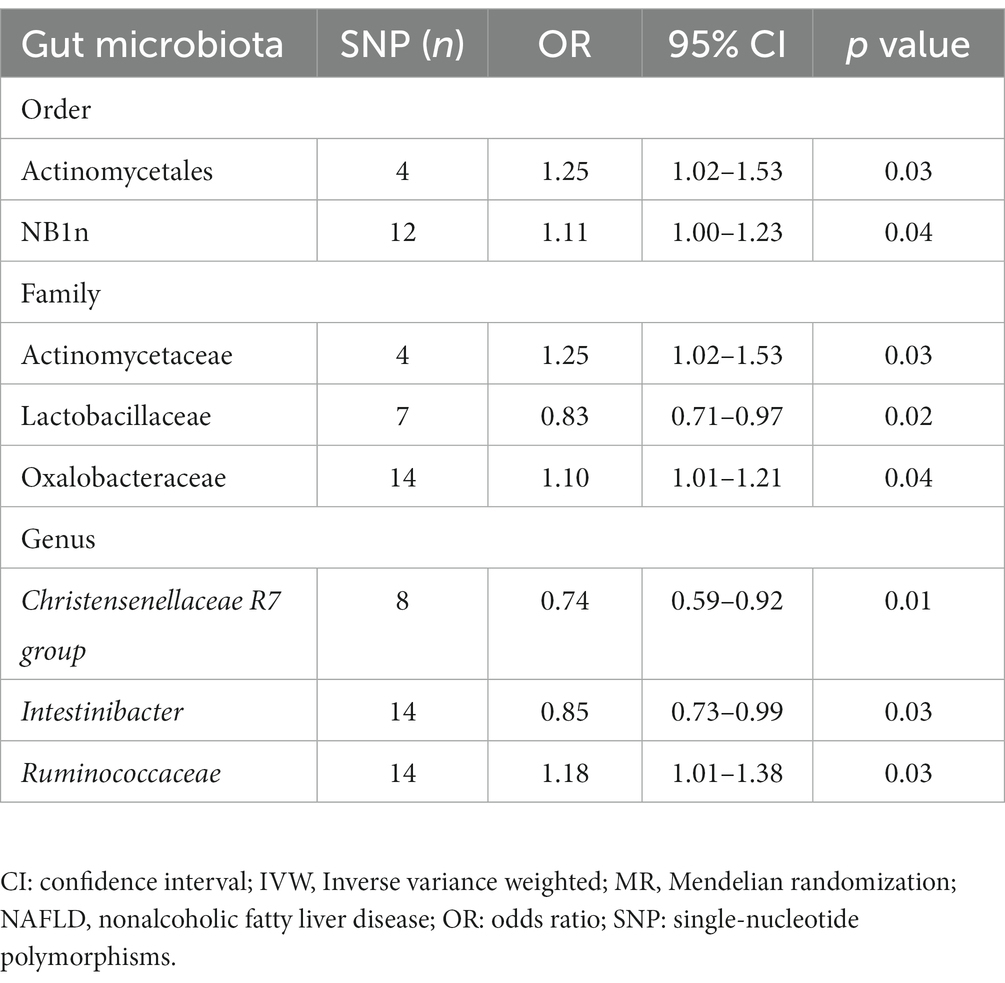
Table 1. Forward MR analysis of gut microbiota and NAFLD risk based on the IVW method.
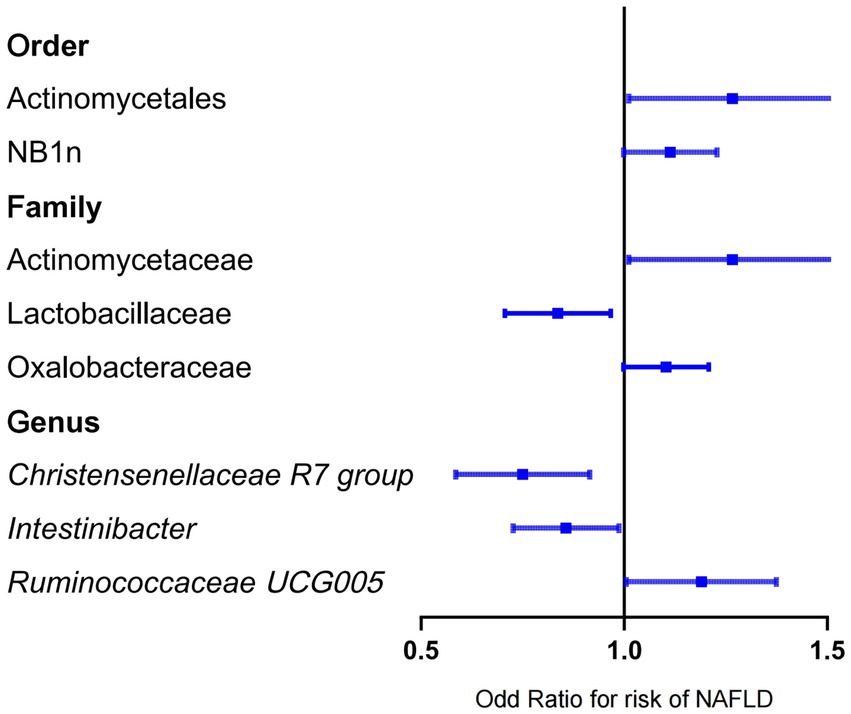
Figure 3. Mendelian randomization results of the positive association between gut microbiota and NAFLD based on the inverse variance weighted method. NAFLD, nonalcoholic fatty liver disease; OR, odds ratio.
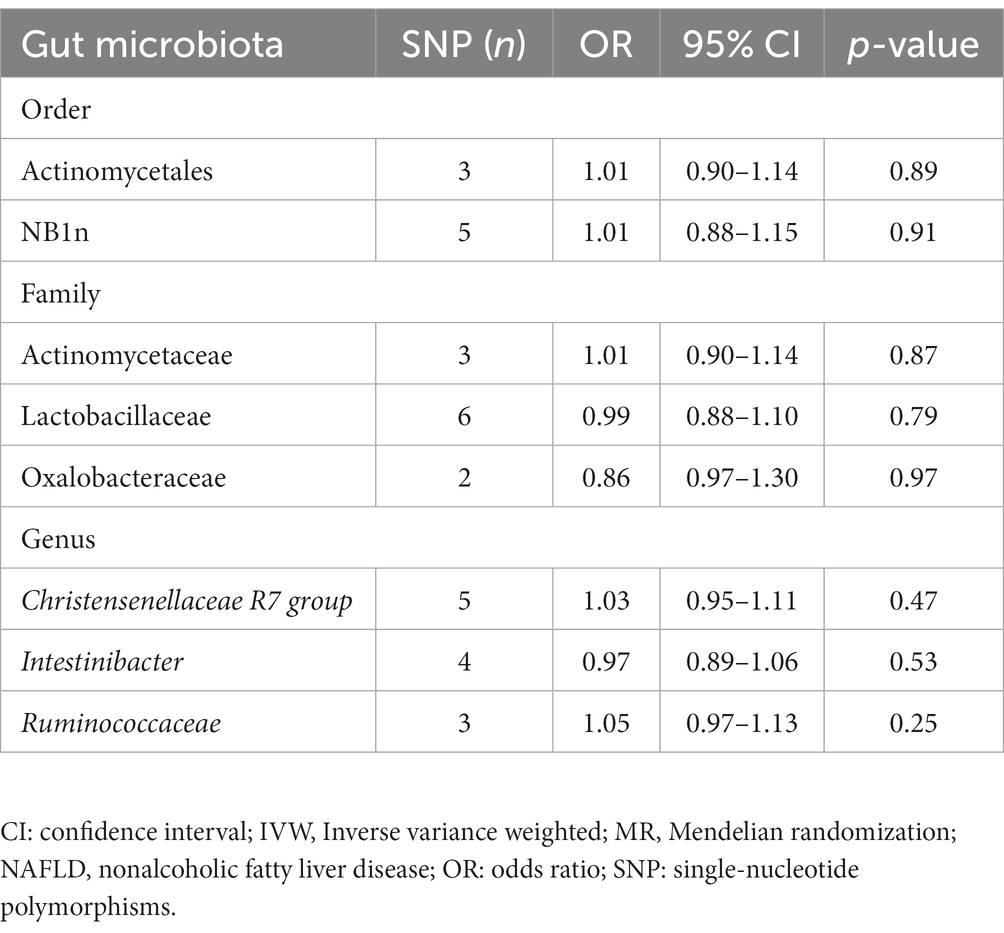
Table 2. Reverse MR results for the gut microbiota causally associated with NAFLD based on the IVW method.
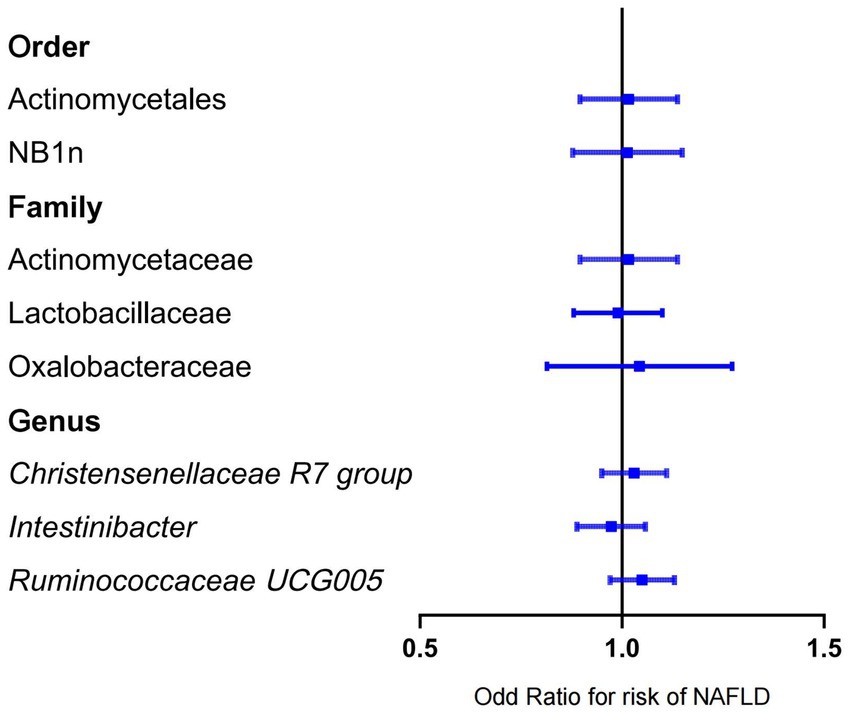
Figure 4. Mendelian randomization results of the negative association between gut microbiota and NAFLD based on inverse variance weighted method. NAFLD, nonalcoholic fatty liver disease; OR, odds ratio.
3.3 Sensitivity analysis of the reliability of the conclusion
According to the results of the MR analysis (Figure 2) and leave-one-out analysis (Figure 5) of all clusters based on different SNPs for Actinomycetaceae, Actinomycetales, Christensenellaceae R7 group, Intestinibacter, and Ruminococcaceae UCG005, all MR analyses were consistent with the primary analysis and the MR-Egger regression intercept term test. The Egger regression intercept test indicated that bacterial pleiotropy did not introduce bias to the results (p > 0.05). The results of the primary and reliability analyses using leave-one-out cross-validation supported these findings (Figure 5). Oxalobacteraceae exhibited consistent results in the multiple MR tests (Figure 2), and reliability analysis from the leave-one-out method (Figure 5) confirmed the plausibility of the outcomes. The MR-Egger regression intercept term test indicated that multipotency did not affect the abundance of Lactobacillaceae. The findings for NB1n were consistent with the main results except for those of the MR-Egger test, indicating that multipotency did not introduce bias to the results (p > 0.05). As expected, the results of the reliability analysis using the leave-one-out method were consistent with those of the primary analysis (Figure 5).
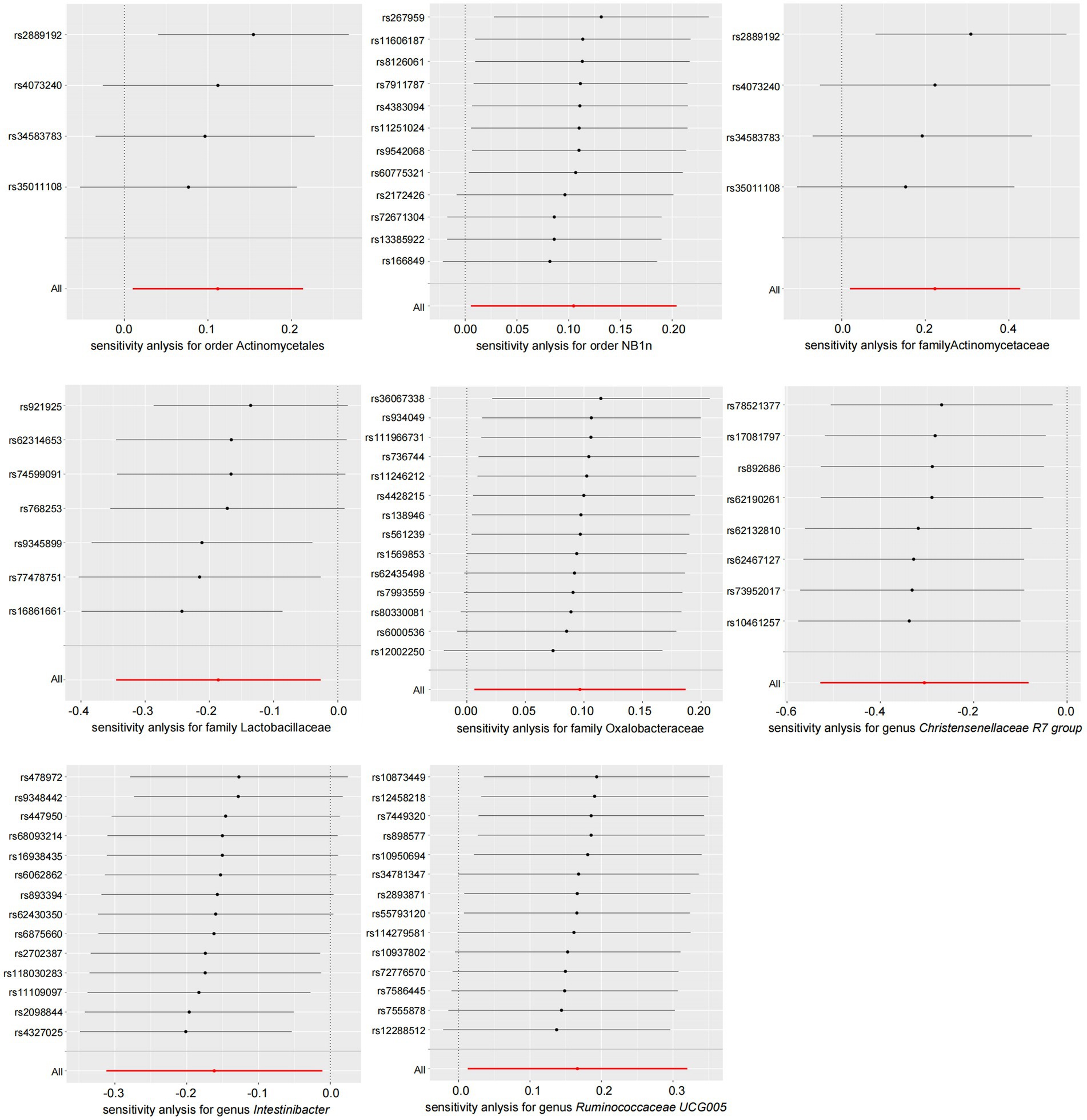
Figure 5. Leave-one-out plot of gut microbiota and NAFLD (forward results). NAFLD, nonalcoholic fatty liver disease.
3.4 Identification of the key SNP-harboring genes regulated by positive gut microbiota and verification of their expression in mice with NASH
The IVs in Supplementary Table S1 were converted into 106 genes using the “gwasrapidd” pack-age, the principal component analysis (PCA) diagram shows a significant difference between our control group and the NAFLD disease group (Figure 6A). Further, the corresponding characteristic genes in NAFLD were screened using LASSO regression and the SVM algorithm. After ten cross-validations, LASSO regression identified five NAFLD characteristic genes (Figures 6B,C). However, the top five feature genes with the highest accuracy were screened using the SVM algorithm and intersected with the feature genes screened using LASSO regression (Figure 6D). Four intersection genes were accordingly identified as key genes involved in the regulation of the differential intestinal flora in NAFLD: colony-stimulating factor 2 receptor β (CSF2RB), fucosyltransferase 2 (FUT2), 17-beta-hydroxysteroid dehydrogenase 14 (HSD17B14), and microtubule affinity-regulated kinase 3 (MARK3) (Figure 6E).
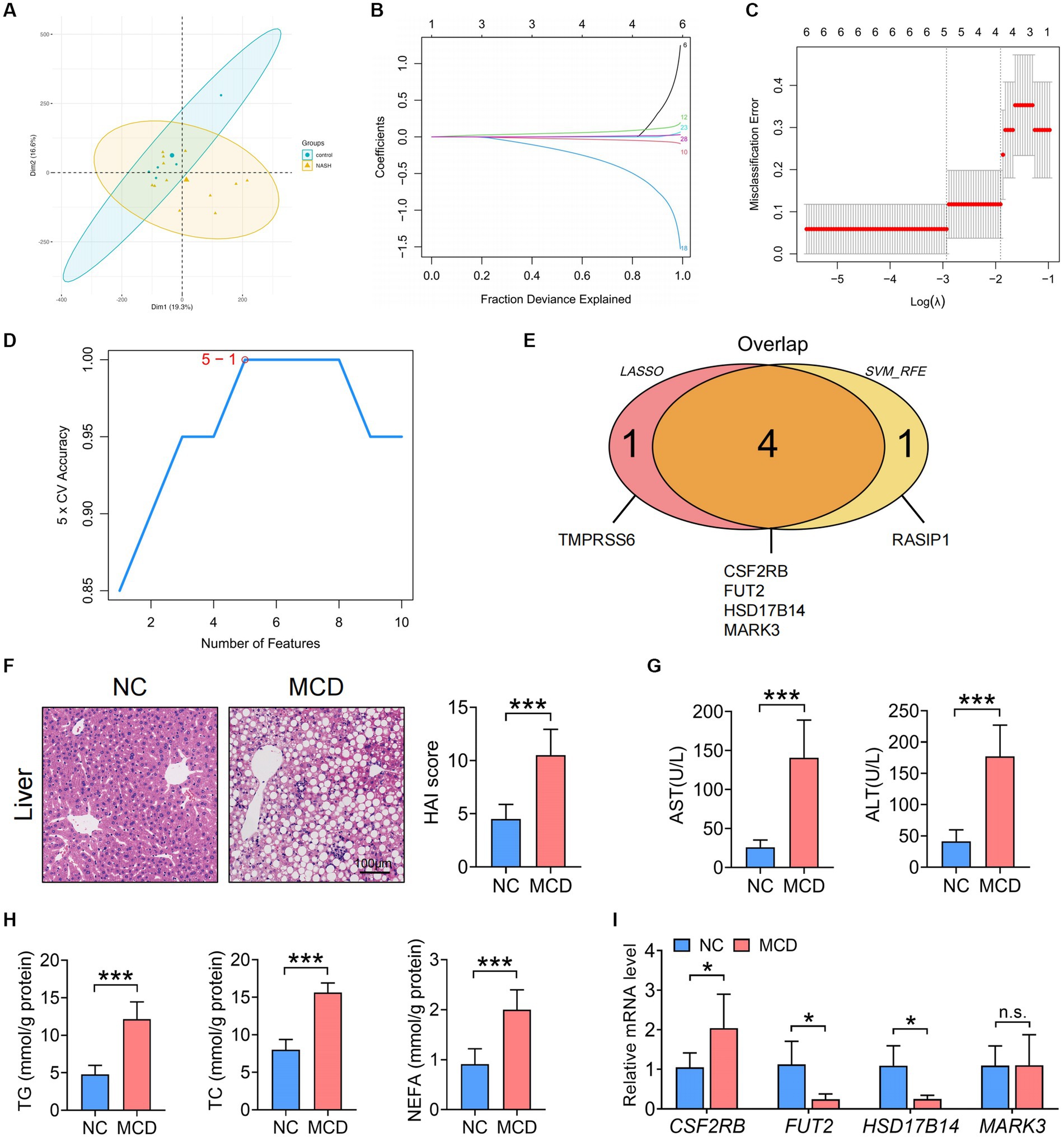
Figure 6. Identification of the key SNP-harboring genes corresponding to the gut microbiota and verification of their expression in the liver and colon of MCD-fed mice with NASH. (A) PCA results. (B) LASSO coefficient distribution of the differential genes. (C) Ten cross-validations of tuning parameter selection in the LASSO model. (D) The highest accuracy of the SVM model. (E) Wayne graphs for the LASSO and SVM algorithms. (F) Representative images of liver hematoxylin and eosin staining and statistical results of the HAI scores of NC or MCD diet-fed mice. Scale =100 μm. (G) Serum ALT and AST levels in MCD diet-fed mice. (H) Serum TG, TC, and NEFA levels in NC or MCD diet-fed mice. (I) qRT-PCR analysis of the key SNP-harbouring genes (CSF2RB, FUT2, HSD17B14, and MARK3) in the liver of NC-or MCD diet-fed mice. (F,I) n = 6 per group. Data are presented as mean ± standard deviation. (F−H) Two-tailed unpaired Student’s t-test; (I) Two-way analysis of variance (ANOVA) followsed by Sidak. MCD, methionine choline-deficient; NASH, nonalcoholic steatohepatitis; PCA, Principal component analysis; SNP, single-nucleotide polymorphism; LASSO, least absolute shrinkage and selection operator; SVM, support vector machine; alanine transaminase, ALT; aspartate transaminase, AST; HAI. Histological Activity Index; TG, triglyceride; TC, total cholesterol; NEFA, non-esterified fatty acid; NC, normal chow.
To investigate the expression of the aforementioned SNP-harboring genes in the liver and colon of model mice with NASH, C57BL/6 mice were fed an NC or MCD diet for 4 weeks. The MCD diet-fed model mice with NASH exhibited a more notable infiltration of liver inflammatory cells and adipose vacuoles and increased HAI scores than those fed with NC (Figure 6F). Serum ALT, AST, TG, TC, and NEFA levels exhibited a consistent upward trend (Figures 6G,H). Furthermore, the hepatic mRNA expression of CSF2RB increased, whereas that of FUT2 and HSD17B14 decreased in mice with NASH compared to that in healthy controls (Figure 6I).
4 Discussion
NAFLD has emerged as the most common factor contributing to chronic liver disease worldwide, with an increasing global prevalence and an onset at younger ages. Intestinal inflammation, intestinal mucosal barrier destruction, bacterial flora structure disorder, and bacterial metabolite translocation are strongly associated with liver inflammation and fibrosis in NAFLD (Canfora et al., 2019; Tilg et al., 2021). The present study, using bidirectional two-sample MR analysis, revealed a causal relationship between eight intestinal flora and NAFLD and analyzed the target genes regulated by them.
Several previous studies have utilized MR analysis to analyze the causal relationship between gut bacteria and NAFLD. Consistent with these studies, we demonstrated that Lactobacillaceae, Christensenellaceae R7 group, and Intestinibacter play a protective role in NAFLD, whereas Actinomycetaceae, Oxalobacteraceae, Actinomycetales and Ruminococcaceae UCG005 are harmful bacteria in NAFLD (Li et al., 2023; Zhang R. et al., 2023). These results are consistent with previously reported intestinal microecological changes in NAFLD, with an increase in the abundance of Actinomycetales detected in obese mice fed a high-fat diet (Zhao et al., 2017). The abundances of probiotics, such as Lactobacillaceae (Yu et al., 2021; González-Lozano et al., 2022), Christensenellaceae R7 group (Tavella et al., 2021), and Intestinibacter (Zhang et al., 2022) were decreased and inversely correlated with the body weight ratio, NAFLD activity score, and visceral and subcutaneous adipose tissue content. In addition, Boursier et al. (2016) have reported that patients with NAFLD and fibrosis have a higher abundance of Ruminococcaceae members than those without fibrosis. However, some studies on the abundance and role of Rumenococcaceae in NAFLD present divergent findings form our conclusions. For example, the abundance of Ruminococcaceae members was low in patients with NAFLD (Tsai et al., 2020). Two studies compared the obesity status of the gut microecology in Latin American NASH and Asian NAFLD cohorts and revealed a significantly lower relative abundance of Ruminococcaceae members in non-obese patients with NASH than in non-obese controls (Wang B. et al., 2016; Duarte et al., 2018). Lee et al. (2020) reported that the severity of liver fibrosis in non-obese patients with NAFLD was negatively related to the abundance of Ruminococcaceae. In addition, Faecalibacterium prausnitzii (Ruminococcaceae member) was associated with decreased adipose tissue inflammation in high-fat diet-fed mice (Munukka et al., 2017), and Ruminococcus faecalis mitigated fibrosis in diet-induced NAFLD models (Lee et al., 2020). The discrepancies in conclusions may be attributed to the different inclusion criteria for the participants, including the presence of comorbid obesity.
The role of NB1n in NAFLD has not been previously reported, and to the best of our knowledge, this study is the first to identify NB1n as a harmful bacterium in NAFLD. While NB1n has been associated with infections (Lyu et al., 2023; Yan et al., 2023) and urolithiasis (Zhang L. et al., 2023), its role in the development of NAFLD remains unclear. On the other hand, the specific role of Oxalobacteraceae in NAFLD is also completely unclear. Oxalic acid is ingested through the diet or endogenously produced by the liver and metabolized and degraded in the gut by microorganisms, including Oxalobacteraceae members (Ermer et al., 2023). Notably, there is a significant correlation between steatosis severity and oxalate excretion in overweight children and adolescents and higher liver production and urinary oxalate excretion in ob/ob mice (Gianmoena et al., 2021). Furthermore, steatotic hepatocytes are responsible for glyoxalate detoxification (Gianmoena et al., 2021). A recent study revealed that patients with obesity have a significantly higher liver expression of Oxalobacteraceae-specific genes than lean individuals; the genus Massilia (Oxalobacteraceae member) was only detected in patients with obesity (Suppli et al., 2021). However, whether Oxalobacteraceae members naturally colonize the liver or translocate from the intestine remains unclear, and their role in oxalic acid synthesis in the liver requires further investigation.
We also demonstrated that the eight target gut bacteria mainly regulate the expression of three key genes in the liver during NAFLD: CSF2RB, HSD17B14, and FUT2. Compared with the control group, intrahepatic CSF2RB mRNA expression increased in the NAFLD group, whereas that of HSD17B14 and FUT2 decreased. CSF2RB, also called granulocyte-macrophage colony-stimulating factor/interleukin-3/interleukin-5 receptor common β-subunit, was highly activated in the liver of mice with NASH, aggravating liver tissue inflammation by promoting the production and differentiation of macrophages and granulocytes (Wessendarp et al., 2022). Moreover, the intrahepatic expression of HSD17B14 (also known as DHRS10, a member of the 17 be-ta-hydroxysteroid dehydrogenase superfamily) was negatively correlated with NASH severity and involved in the regulation of retinol and fatty acid metabolism (Govaere et al., 2020). A large GWAS-based analysis revealed that FUT2 (rs2519093) mutations are closely associated with low-density lipoprotein levels (Hoffmann et al., 2018). Notably, this gene is highly expressed in the digestive tract (intestine and gallbladder) of both humans and mice and is primarily responsible for encoding alpha-1, 2-focusing transferase (Franks, 2011) and synthesizing H antigen (a receptor for adhesion molecules) (Xing and Guo, 2000). Approximately 20% of White individuals lack the functional FUT allele (i.e., the non-secretor), resulting in structural changes in the gut microbiota and disrupting mucosal immunity, which in turn increases the risk of diseases, including inflammatory bowel disease and primary sclerosing cholangitis (McGovern et al., 2010; Folseraas et al., 2012; Jostins et al., 2012). Zhou et al. (2021) have reported that Western diets decrease FUT2 expression in the intestinal epithelium of mice and that the loss of Fut2 activity mitigates diet-induced bile acid accumulation, insulin sensitivity, and hepatic steatosis. The specific roles of CSF2RB, HSD17B14, and FUT2 in the enterohepatic dialog of NAFLD are unknown, warranting further studies to determine how the eight gut bacteria identified in our study regulate the involvement of these three genes in NAFLD progression.
However, this study has some limitations, necessitating exploration in future research. First, our genome-wide data is limited to European populations, and validating our findings in more diverse populations is critical. Second, several factors, including lifestyle, metabolic factors, age, and sex, that influence the gut microbiota need to be considered (Redondo-Useros et al., 2020; Yuan et al., 2022). Third, the sample size of transcriptome sequencing data needs to be further expanded. Finally, the roles of the eight gut microbes and the related key genes in NAFLD development require further evaluation in animal models.
5 Conclusion
Our results suggest a causal relationship between eight gut microbes and NAFLD, highlighting three key SNP-harboring genes (CSF2RB, FUT2, and HSD17B14) for specific microbiome regulation. This study suggests that increasing the abundance of Lactobacillaceae, Christensenaceae R7 group, and Intestinibacter may alleviate NAFLD and that decreasing that of enterogenous Actinomycetales, NB1n, Actinomycetaceae, Oxalobacteraceae, and Ruminococcaceae UCG005 may be potential therapeutic targets for NAFLD. In addition, inhibiting the expression of CSF2RB or upregulating that of FUT2 and HSD17B14 may be another beneficial approach to alleviate NAFLD.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by the Research Ethics Committee of the University of Tartu and Danish Ethics Committee. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. The animal study was approved by Institutional Animal Care and Use Committee of Wenzhou Medical University. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
TP: Conceptualization, Writing – review & editing. LS: Data curation, Writing – original draft. YZ: Writing – original draft. FY: Data curation, Writing – original draft. YC: Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the National Natural Science Foundation of China (Grant number: 82070593) and the Natural Science Foundation of Zhejiang Province (Grant number: LD21H030002).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1320279/full#supplementary-material
Footnotes
References
Albillos, A., de Gottardi, A., and Rescigno, M. (2020). The gut-liver axis in liver disease: pathophysiological basis for therapy. J. Hepatol. 72, 558–577. doi: 10.1016/j.jhep.2019.10.003
Anstee, Q. M., Darlay, R., Cockell, S., Meroni, M., Govaere, O., Tiniakos, D., et al. (2020). Genome-wide association study of non-alcoholic fatty liver and steatohepatitis in a histologically characterised cohort. J. Hepatol. 73, 505–515. doi: 10.1016/j.jhep.2020.04.003
Bonder, M. J., Kurilshikov, A., Tigchelaar, E. F., Mujagic, Z., Imhann, F., Vila, A. V., et al. (2016). The effect of host genetics on the gut microbiome. Nat. Genet. 48, 1407–1412. doi: 10.1038/ng.3663
Boursier, J., Mueller, O., Barret, M., Machado, M., Fizanne, L., Araujo-Perez, F., et al. (2016). The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 63, 764–775. doi: 10.1002/hep.28356
Bowden, J., Davey Smith, G. D., and Burgess, S. (2015). Mendelian randomization with invalid instruments: effect estimation and bias detection through egger regression. Int. J. Epidemiol. 44, 512–525. doi: 10.1093/ije/dyv080
Burgess, S., Small, D. S., and Thompson, S. G. (2017). A review of instrumental variable estimators for Mendelian randomization. Stat. Methods Med. Res. 26, 2333–2355. doi: 10.1002/jrsm.1346
Canfora, E. E., Meex, R. C. R., Venema, K., and Blaak, E. E. (2019). Gut microbial metabolites in obesity, NAFLD and T2DM. Nat. Rev. Endocrinol. 15, 261–273. doi: 10.1038/s41574-019-0156-z
Cheng, Q., Yang, Y., Shi, X., Yeung, K. F., Yang, C., Peng, H., et al. (2020). MR-LDP: a two-sample Mendelian randomization for GWAS summary statistics accounting for linkage disequilibrium andhorizontal pleiotropy. NAR Genom. Bioinform. 2:lqaa028. doi: 10.1093/nargab/lqaa028
Craven, L., Rahman, A., Nair Parvathy, S. N., Beaton, M., Silverman, J., Qumosani, K., et al. (2020). Allogenic fecal microbiota transplantation in patients with nonalcoholic fatty liver disease improves abnormal small intestinal permeability: a randomized control trial. Am. J. Gastroenterol. 115, 1055–1065. doi: 10.14309/ajg.0000000000000661
Curtis, S. W., Cobb, D. O., Kilaru, V., Terrell, M. L., Marder, M. E., Barr, D. B., et al. (2019). Environmental exposure to polybrominated biphenyl (PBB) associates with an increased rate of biological aging. Aging 11, 5498–5517. doi: 10.18632/aging.102134
Duarte, S. M. B., Stefano, J. T., Miele, L., Ponziani, F. R., Souza-Basqueira, M., Okada, L. S. R. R., et al. (2018). Gut microbiome composition in lean patients with NASH is associated with liver damage independent of caloric intake: a prospective pilot study. Nutr. Metab. Cardiovasc. Dis. 28, 369–384. doi: 10.1016/j.numecd.2017.10.014
Ermer, T., Nazzal, L., Tio, M. C., Waikar, S., Aronson, P. S., and Knauf, F. (2023). Oxalate homeostasis. Nat. Rev. Nephrol. 19, 123–138. doi: 10.1038/s41581-022-00643-3
Folseraas, T., Melum, E., Rausch, P., Juran, B. D., Ellinghaus, E., Shiryaev, A., et al. (2012). Extended analysis of a genome-wide association study in primary sclerosing cholangitis detects multiple novel risk loci. J. Hepatol. 57, 366–375. doi: 10.1016/j.jhep.2012.03.031
Franks, I. (2011). Gut microbiota: FUT2 genotype influences the gut microbiota in patients with Crohn’s disease and healthy individuals. Nat. Rev. Gastroenterol. Hepatol. 9:2. doi: 10.1038/nrgastro.2011.237
Ghodsian, N., Abner, E., Emdin, C. A., Gobeil, É., Taba, N., Haas, M. E., et al. (2021). Electronic health record-based genome-wide meta-analysis provides insights on the genetic architecture of non-alcoholic fatty liver disease. Cell Rep. Med. 2:100437. doi: 10.1016/j.xcrm.2021.100437
Gianmoena, K., Gasparoni, N., Jashari, A., Gabrys, P., Grgas, K., Ghallab, A., et al. (2021). Epigenomic and transcriptional profiling identifies impaired glyoxylate detoxification in NAFLD as a risk factor for hyperoxaluria. Cell Rep. 36:109526. doi: 10.1016/j.celrep.2021.109526
González-Lozano, E., García-García, J., Gálvez, J., Hidalgo-García, L., Rodríguez-Nogales, A., Rodríguez-Cabezas, M. E., et al. (2022). Novel horizons in postbiotics: Lactobacillaceae extracellular vesicles and their applications in health and disease. Nutrients 14:5296. doi: 10.3390/nu14245296
Govaere, O., Cockell, S., Tiniakos, D., Queen, R., Younes, R., Vacca, M., et al. (2020). Transcriptomic profiling across the nonalcoholic fatty liver disease spectrum reveals gene signatures for steatohepatitis and fibrosis. Sci. Transl. Med. 12:eaba4448. doi: 10.1126/scitranslmed.aba4448
Hariton, E., and Locascio, J. J. (2018). Randomised controlled trials—the gold standard for effectiveness research: study design: randomised controlled trials. Br. J. Obstet. Gynaecol. 125:1716. doi: 10.1111/1471-0528.15199
Hemani, G., Zheng, J., Elsworth, B., Wade, K. H., Haberland, V., Baird, D., et al. (2018). The MR-base platform supports systematic causal inference across the human phenome. elife 7:e34408. doi: 10.7554/eLife.34408
Hoffmann, T. J., Theusch, E., Haldar, T., Ranatunga, D. K., Jorgenson, E., Medina, M. W., et al. (2018). A large electronic-health-record-based genome-wide study of serum lipids. Nat. Genet. 50, 401–413. doi: 10.1038/s41588-018-0064-5
Jostins, L., Ripke, S., Weersma, R. K., Duerr, R. H., McGovern, D. P., Hui, K. Y., et al. (2012). Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 491, 119–124. doi: 10.1038/nature11582
Juárez-Fernández, M., Porras, D., Petrov, P., Román-Sagüillo, S., García-Mediavilla, M. V., Soluyanova, P., et al. (2021). The synbiotic combination of Akkermansia muciniphila and quercetin ameliorates early obesity and NAFLD through gut microbiota reshaping and bile acid metabolism modulation. Antioxidants 10:2001. doi: 10.3390/antiox10122001
Kurilshikov, A., Medina-Gomez, C., Bacigalupe, R., Radjabzadeh, D., Wang, J., Demirkan, A., et al. (2021). Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165. doi: 10.1038/s41588-020-00763-1
Lee, G., You, H. J., Bajaj, J. S., Joo, S. K., Yu, J., Park, S., et al. (2020). Distinct signatures of gut microbiome and metabolites associated with significant fibrosis in non-obese NAFLD. Nat. Commun. 11:4982. doi: 10.1038/s41467-020-18754-5
Li, Y., Liang, X., Lyu, Y., Wang, K., Han, L., Wang, Y., et al. (2023). Association between the gut microbiota and nonalcoholic fatty liver disease: a two-sample Mendelian randomization study. Dig. Liver Dis. 55, 1464–1471. doi: 10.1016/j.dld.2023.07.014
Lyu, B., Ma, J., Bai, Y., and Feng, Z. (2023). Casual effects of gut microbiota on risk of infections: a two-sample Mendelian randomization study. Front. Microbiol. 14:1284723. doi: 10.3389/fmicb.2023.1284723
McGovern, D. P. B., Jones, M. R., Taylor, K. D., Marciante, K., Yan, X., Dubinsky, M., et al. (2010). Fucosyltransferase 2 (FUT2) non-secretor status is associated with Crohn’s disease. Hum. Mol. Genet. 19, 3468–3476. doi: 10.1093/hmg/ddq248
Munukka, E., Rintala, A., Toivonen, R., Nylund, M., Yang, B., Takanen, A., et al. (2017). Faecalibacterium prausnitzii treatment improves hepatic health and reduces adipose tissue inflammation in high-fat fed mice. ISME J. 11, 1667–1679. doi: 10.1038/ismej.2017.24
Pan, Z., Mao, B., Zhang, Q., Tang, X., Yang, B., Zhao, J., et al. (2022). Postbiotics prepared using Lactobacillus paracasei CCFM1224 prevent nonalcoholic fatty liver disease by modulating the gut microbiota and liver metabolism. Int. J. Mol. Sci. 23:13522. doi: 10.3390/ijms232113522
Parker, D. C., Bartlett, B. N., Cohen, H. J., Fillenbaum, G., Huebner, J. L., Kraus, V. B., et al. (2020). Association of blood chemistry quantifications of biological aging with disability and mortality in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 75, 1671–1679. doi: 10.1093/gerona/glz219
Redondo-Useros, N., Nova, E., González-Zancada, N., Díaz, L. E., Gómez-Martínez, S., and Marcos, A. (2020). Microbiota and lifestyle: a special focus on diet. Nutrients 12:1776. doi: 10.3390/nu12061776
Riazi, K., Azhari, H., Charette, J. H., Underwood, F. E., King, J. A., Afshar, E. E., et al. (2022). The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. Lancet Gastroenterol. Hepatol. 7, 851–861. doi: 10.1016/S2468-1253(22)00165-0
Shang, R., Liu, Y., Xin, Z., Guo, W., Guo, Z., Hao, B., et al. (2013). Synthesis and antibacterial evaluation of novel pleuromutilin derivatives. Eur. J. Med. Chem. 63, 231–238. doi: 10.1016/j.ejmech.2013.01.048
Suppli, M. P., Bagger, J. I., Lelouvier, B., Broha, A., Demant, M., Kønig, M. J., et al. (2021). Hepatic microbiome in healthy lean and obese humans. JHEP Rep. 3:100299. doi: 10.1016/j.jhepr.2021.100299
Tavella, T., Rampelli, S., Guidarelli, G., Bazzocchi, A., Gasperini, C., Pujos-Guillot, E., et al. (2021). Elevated gut microbiome abundance of Christensenellaceae, Porphyromonadaceae and Rikenellaceae is associated with reduced visceral adipose tissue and healthier metabolic profile in Italian elderly. Gut Microb. 13, 1–19. doi: 10.1080/19490976.2021.1880221
Tilg, H., Adolph, T. E., Dudek, M., and Knolle, P. (2021). Non-alcoholic fatty liver disease: the interplay between metabolism, microbes and immunity. Nat. Metab. 3, 1596–1607. doi: 10.1038/s42255-021-00501-9
Tsai, M. C., Liu, Y. Y., Lin, C. C., Wang, C. C., Wu, Y. J., Yong, C. C., et al. (2020). Gut microbiota dysbiosis in patients with biopsy-proven nonalcoholic fatty liver disease: a cross-sectional study in Taiwan. Nutrients 12:820. doi: 10.3390/nu12030820
Verbanck, M., Chen, C. Y., Neale, B., and Do, R. (2018). Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698. doi: 10.1038/s41588-018-0099-7
Wang, B., Jiang, X., Cao, M., Ge, J., Bao, Q., and Tang, L. (2016). Altered fecal microbiota correlates with liver biochemistry in nonobese patients with non-alcoholic fatty liver disease. Sci. Rep. 6:32002. doi: 10.1038/srep32002
Wang, J., Thingholm, L. B., Skiecevičienė, J., Rausch, P., Kummen, M., Hov, J. R., et al. (2016). Genome-wide association analysis identifies variation in vitamin D receptor and other host factors influencing the gut microbiota. Nat. Genet. 48, 1396–1406. doi: 10.1038/ng.3695
Wessendarp, M., Watanabe-Chailland, M., Liu, S., Stankiewicz, T., Ma, Y., Kasam, R. K., et al. (2022). Role of GM-CSF in regulating metabolism and mitochondrial functions critical to macrophage proliferation. Mitochondrion 62, 85–101. doi: 10.1016/j.mito.2021.10.009
Xing, L., and Guo, L. H. (2000). FUT2 gene involved in expression of H blood group antigen on surface of human tumor cell lines BEL-7404, SGC-7901, and SPC-A-1. Acta Pharmacol. Sin. 21, 997–1001.
Yan, M., Ouyang, Y. L., Xiao, L. Y., Ao, M., Gosau, M., Friedrich, R. E., et al. (2023). Correlations between gut microbiota and lichen planus: a two-sample Mendelian randomization study. Front. Immunol. 14:1235982. doi: 10.3389/fimmu.2023.1235982
Younossi, Z. M., Koenig, A. B., Abdelatif, D., Fazel, Y., Henry, L., and Wymer, M. (2016). Global epidemiology of nonalcoholic fatty liver disease-meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 64, 73–84. doi: 10.1002/hep.28431
Yu, J. S., Youn, G. S., Choi, J., Kim, C. H., Kim, B. Y., Yang, S. J., et al. (2021). Lactobacillus lactis and Pediococcus pentosaceus-driven reprogramming of gut microbiome and metabolome ameliorates the progression of non-alcoholic fatty liver disease. Clin. Transl. Med. 11:e634. doi: 10.1002/ctm2.634
Yuan, S., Chen, J., Li, X., Fan, R., Arsenault, B., Gill, D., et al. (2022). Lifestyle and metabolic factors for nonalcoholic fatty liver disease: Mendelian randomization study. Eur. J. Epidemiol. 37, 723–733. doi: 10.1007/s10654-022-00868-3
Zhang, L., Wang, Z., Zhang, X., Zhao, L., Chu, J., Li, H., et al. (2022). Alterations of the gut microbiota in patients with diabetic nephropathy. Microbiol. Spectr. 10:e0032422. doi: 10.1128/spectrum.00324-22
Zhang, R., Zhao, W., Zhao, R., Zhao, Y., Zhang, Y., and Liang, X. (2023). Causal relationship in gut microbiota and upper urinary urolithiasis using Mendelian randomization. Front. Microbiol. 14:1170793. doi: 10.3389/fmicb.2023.1170793
Zhang, L., Zi, L., Kuang, T., Wang, K., Qiu, Z., Wu, Z., et al. (2023). Investigating causal associations among gut microbiota, metabolites, and liver diseases: a Mendelian randomization study. Front. Endocrinol. 14:1159148. doi: 10.3389/fendo.2023.1159148
Zhao, Z. H., Wang, Z. X., Zhou, D., Han, Y., Ma, F., Hu, Z., et al. (2021). Sodium butyrate supplementation inhibits hepatic steatosis by stimulating liver kinase B1 and insulin-induced gene. Cell. Mol. Gastroenterol. Hepatol. 12, 857–871. doi: 10.1016/j.jcmgh.2021.05.006
Zhao, L., Zhang, Q., Ma, W., Tian, F., Shen, H., and Zhou, M. (2017). A combination of quercetin and resveratrol reduces obesity in high-fat diet-fed rats by modulation of gut microbiota. Food Funct. 8, 4644–4656. doi: 10.1039/c7fo01383c
Keywords: gut microbiota, nonalcoholic fatty liver disease, Mendelian randomization, colonystimulating factor 2 receptor β, fucosyltransferase 2, 17-beta-hydroxysteroid dehydrogenase 14
Citation: Pan T, Su L, Zhang Y, Yi F and Chen Y (2024) Impact of gut microbiota on nonalcoholic fatty liver disease: insights from a leave-one-out cross-validation study. Front. Microbiol. 14:1320279. doi: 10.3389/fmicb.2023.1320279
Edited by:
Giovanni Tarantino, University of Naples Federico II, ItalyReviewed by:
Jingfeng Chen, First Affiliated Hospital of Zhengzhou University, ChinaJianan Zhao, Shanghai Guanghua Rheumatology Hospital, China
Copyright © 2024 Pan, Su, Zhang, Yi and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yongping Chen, Y3lwQHdtdS5lZHUuY24=
†These authors have contributed equally to this work and share first authorship