 Zhenhu Zhang
Zhenhu Zhang Guodong Zhang1†
Guodong Zhang1† Dong Wang
Dong Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 December 2023
Sec. Microorganisms in Vertebrate Digestive Systems
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1309596
This article is part of the Research Topic Roles of Gut Microbiota in Cancers of the Gastrointestinal Tract View all 10 articles
Background: Increasing evidence suggests that esophageal cancer (ESCA) may be correlated with gut flora. However, their causal connection remains unclear. This study aimed to evaluate potential causal linkages and gene–gut microbiome associations between the gut microbiota and ESCA using Mendelian randomization (MR).
Methods: We analyzed the data using genome-wide association studies. The exposure factor and outcome variable were the gut microbiota and ESCA, respectively. The MR-Egger method, weighted median, inverse-variance weighted method, heterogeneity test, sensitivity analysis, and multiplicity analysis were used for the MR analysis. And it was validated using an external dataset. Further meta-analysis was performed to validate the robustness of this relationship. Finally, we annotated single nucleotide polymorphisms in the gut microbiota that were causally associated with ESCA to explore possible host gene-gut microbiota correlations in patients with ESCA.
Results: We identified four species with potential associations with ESCA. Three of these species had a negative causal relationship with ESCA (odds ratio (OR): 0.961; 95% confidence interval (CI): 0.923–0.971; p = 0.047 for Romboutsia; OR: 0.972; 95% CI: 0.921–0.961; p = 0.018 for Lachnospira; OR: 0.948; 95% CI: 0.912–0.970; p = 0.032 for Eubacterium). A positive causal relationship was observed between one bacterial group and ESCA (OR: 1.105; 95% CI: 1.010–1.072; p = 0.018 for Veillonella). External datasets show the same trend. This is further supported by meta-analysis. None of the data showed pleiotropy, and leave-one-out analysis indicated the reliability of these findings. The gut microbiomes of patients with ESCA may correlate with the 19 identified genes.
Conclusion: Our data indicate a potential causal link between these four gut bacteria and ESCA and identify a correlation between host genes and gut microbiota in ESCA, offering novel therapeutic options.
Esophageal cancer (ESCA) is one of the most common cancers globally, with the sixth highest mortality rate according to global cancer data (Sung et al., 2021). Surgery is an effective treatment for ESCA, but in advanced ESCA, the 5-year survival rate of patients remains less than 25% even after surgery (Oppedijk et al., 2014). Chemotherapy is commonly used as a treatment for ESCA but has unavoidable side effects such as toxicity and drug resistance (He et al., 2021). Additionally, epidemiological data indicates that the incidence of ESCA is increasing annually, gravely endangering human health (Uhlenhopp et al., 2020). Therefore, identifying factors potentially associated with ESCA can provide an essential basis for the early prevention of ESCA.
Increasing evidence has shown that the gut microbiota and ESCA are closely related (Cheung et al., 2022; Muszynski et al., 2022; Ohkusa et al., 2022; Baba et al., 2023; Sugimoto et al., 2023). Further, significant variations have been reported in the composition and abundance of fecal microorganisms between patients with ESCA and healthy controls. Notably, these differences are closely correlated with the severity of the disease, suggesting that the gut microbiota may play a significant role in the development of ESCA (Li et al., 2022; Lin et al., 2022). Moreover, gut microbiota can alter genome-wide methylation levels in ESCA, which may be one of the mechanisms influencing the malignant behavior of ESCA cells (Baba et al., 2023). Exposure to antibiotics leads to changes in the gut microbiota, which in turn increase the risk of developing ESCA; the risk of developing the disease increases with the duration of antibiotic exposure (Thanawala et al., 2023). Therefore, investigating the potential link between the gut microbiota and disease to prevent and treat ESCA is crucial.
Although current research reveals that the gut microbiota and ESCA are related, the results are susceptible to confounding factors. Mendelian randomization (MR) is a genetic technique frequently used to investigate causal links between exposures and outcomes and prevents confounding variables in common observational studies because genetic variants are randomly assigned at conception (Smith and Ebrahim, 2003).
We investigated the potential causative relationship between gut microbiota and ESCA using MR to provide a proper theoretical foundation for understanding the interaction between ESCA and gut microbiota. We further identified genes related to the single nucleotide polymorphisms (SNPs) in the gut microbiota obtained by MR analysis. Our results may help identify novel therapeutic options for ESCA treatment.
In this study, heterogeneity, sensitivity, and multiplicity analyses were conducted in addition to MR analyses using genome-wide association studies (GWAS) information to evaluate the causal relationship between gut microbiota and ESCA. MR studies must satisfy three core assumptions of association, independence, and exclusivity: (i) the selected SNPs should be significantly associated with the exposure (intestinal microbiota); (ii) the SNPs must be independent of potential confounders between the exposure and the outcome; and (iii) there is no direct relationship between the SNPs and the outcome (ESCA), and the causal linkage can only be made through the intestinal flora. The workflow is illustrated in Figure 1.
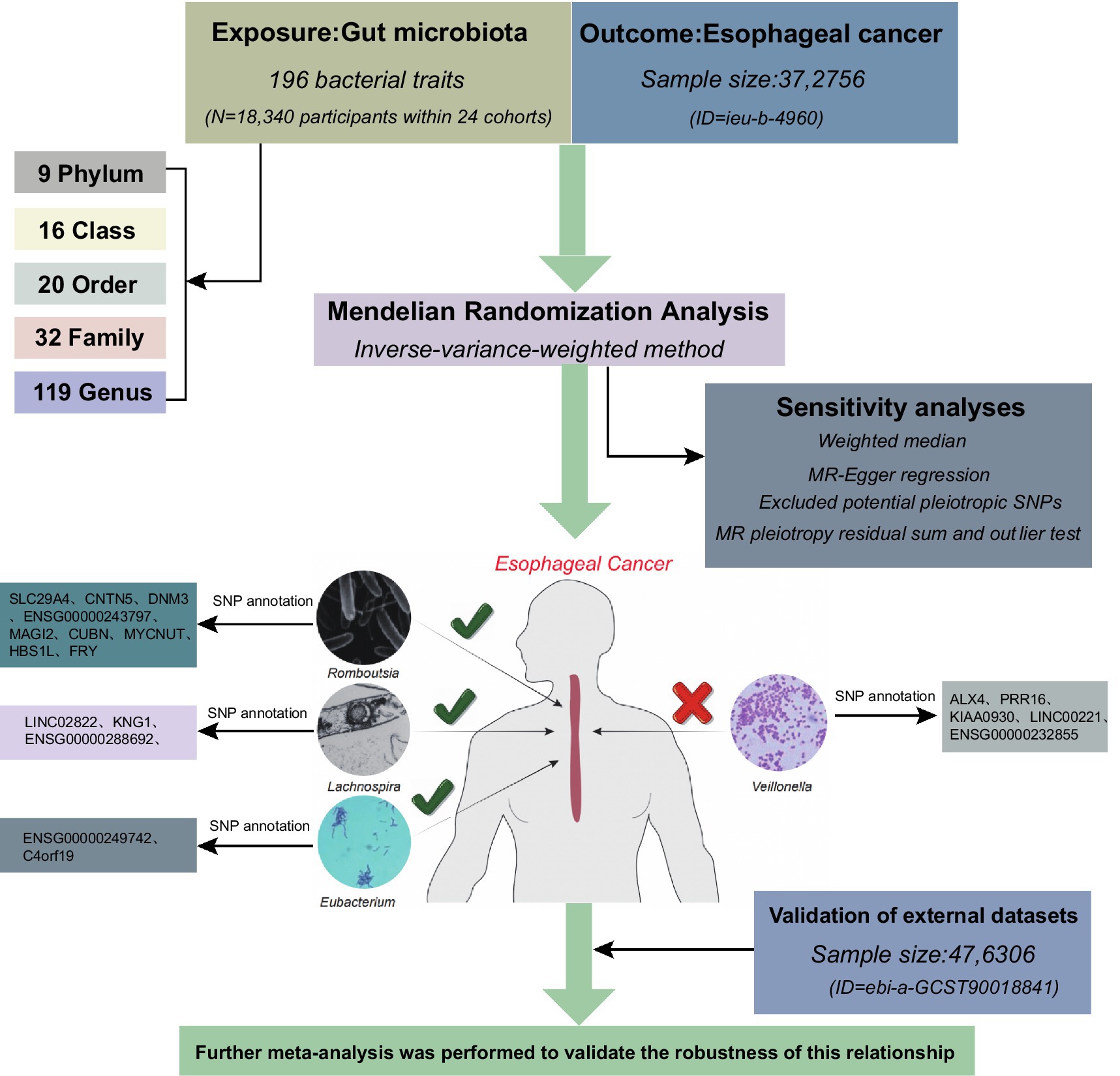
Figure 1. The workflow of the present study.
Gut microbiota data from the most recent GWAS meta-analysis, comprising 24 cohorts and 18,340 participants, were used in this investigation (Kurilshikov et al., 2021). ESCA data for the experimental and validation groups were obtained from the UK Biospecimen Repository,1 the experimental group included 372,756 samples and the validation group included 476,306 samples (Table 1).
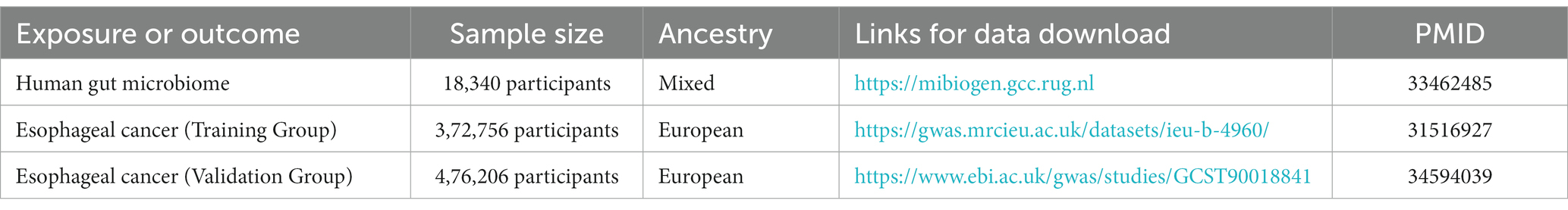
Table 1. Details of the genome-wide association studies and datasets used in our analyses.
A total of 196 bacterial traits, including 9 phyla, 16 classes, 20 orders, 32 families, and 119 genera, were retained after initially removing 15 bacterial qualities without specific names. SNPs significantly related to the gut microbiota were chosen at the genome-wide level (p < 1.0 × 10−5, R2 < 0.001, and clumping distance = 10,000 kb) to fulfill the first MR hypothesis that SNPs must be strongly associated with the gut microbiota. Second, to ensure that the genetic variants were not associated with potential confounders (smoking, heavy alcohol consumption, hot beverages, pickles), a query was performed in the Phenoscanner database.2 This step was performed to ensure that the SNPs were not associated with known confounders and, ultimately, to obtain SNPs significantly associated with the gut microbiota, serving as instrumental variables. Next, we calculated the proportion of variance (R2). We calculated the F-statistic using the following formula: R = 2 × MAF × (1-MAF) × β2, F = R2 (n-k-1)/K (1-R2), where “MAF” is the minor allele frequency, “N” denotes the exposed GWAS sample size, and “K” is the number of SNPs. F > 10 confirmed the absence of a mild instrumental variable bias. The process was completed by annotating the SNPs using an internet database.3
The weighted median approach, MR-Egger method, and random effects inverse variance weighting (IVW) method were used in the MR analysis. Statistical significance was set at p < 0.05. The IVW approach was primarily employed to analyze these studies (Burgess and Thompson, 2017), and methods such as MR-Egger and weighted median were used to complement the IVW method (Verbanck et al., 2018). Additionally, we applied Cochran’s Q method to evaluate heterogeneity among SNPs. We performed several heterogeneity tests, including the MR-Egger intercept test and a sensitivity analysis, to ensure the robustness of our results. The leave-one-out test was used for the sensitivity analysis to determine outliers among the final SNPs. All data were analyzed using the R packages “Two-Sample-MR” and “MR-PRESSO” in R software (version 4.3.0).
The F-statistics for the 196 bacterial characteristics, ranging from 21.63 to 144.84 and with mean values exceeding 10, suggested a robust connection with exposure. We screened for SNPs strongly associated with the gut microbiota (p < 1.0 × 10−5), and the linkage disequilibrium parameter was set (R2 < 0.001, kb = 10,000). Thereafter, the data of the final ESCA were extracted from GWAS, and 196 intestinal flora were merged with the final ESCA sequentially. SNPs with palindromic sequences were excluded from the analysis. Finally, we obtained four gut microbiota samples with potential associations with ESCA using the IVW method (Figure 2).
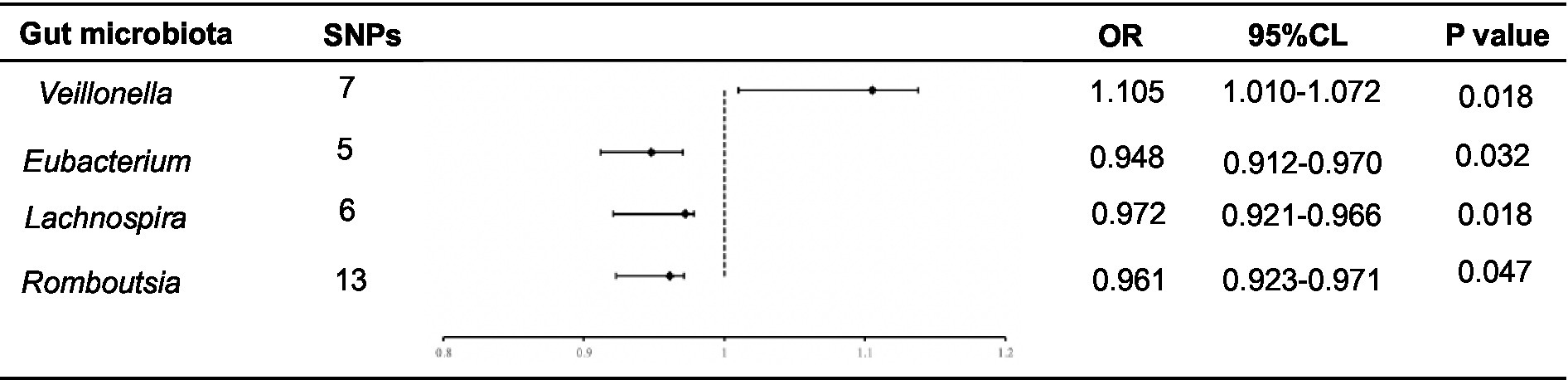
Figure 2. Forest plot of the associations between genetically determined 4 bacterial traits with the ESCA.
Using the IVW method, we found that Romboutsia was negatively associated with ESCA (odds ratio (OR): 0.961; p = 0.047). After screening for F-statistics and excluding chain imbalances, 13 SNPs related to ESCA were included (Supplementary Table S1). In the weighted median approach, the results for the association between Romboutsia and ESCA remained stable (p = 0.02). To evaluate the stability of these findings, we performed the MR-Egger test on the loci of the included SNPs. No possible horizontal pleiotropy was found (p = 0.96), indicating that instrumental variables did not significantly alter the outcomes through mechanisms other than exposure. Cochran’s Q test results showed no significant heterogeneity among the SNPs (p = 0.45).
Similarly, Lachnospira was negatively associated with ESCA (OR = 0.972; p = 0.018). After screening, six SNPs associated with ESCA were included (Supplementary Table S2). In the weighted median approach, the genus Lachnospira was weakly associated with ESCA (p = 0.08). The SNPs were then subjected to an MR-Egger test, which revealed no apparent level of multiple effects (p = 0.96). According to Cochran’s Q analysis (p = 0.45), no discernible heterogeneity was observed among the selected SNPs. Additionally, using the IVW method, we discovered that the genus Eubacterium was adversely linked with ESCA (OR: 0.948; p = 0.032). Overall, five SNPs associated with ESCA were included (Supplementary Table S3). Again, no pleiotropy (p = 0.60) or heterogeneity (p = 0.79) was observed. In contrast, using the IVW method, we found that Veillonella was positively associated with ESCA (OR: 1.105; p = 0.018). The seven SNPs associated with ESCA identified after screening (Supplementary Table S4) were free of pleiotropy (p = 0.87) and heterogeneity (p = 0.41).
We conducted a “leave-one-out” sensitivity analysis to confirm the impact of each SNP on the overall causation. The findings revealed that none of the SNPs exhibited significant differences when one SNP was excluded (Supplementary Figure S1). Finally, the results were plotted using a pattern map (Figure 3). Using an online database, we identified 19 genes that may be associated with the gut microbiota of patients with ESCA (Table 2).
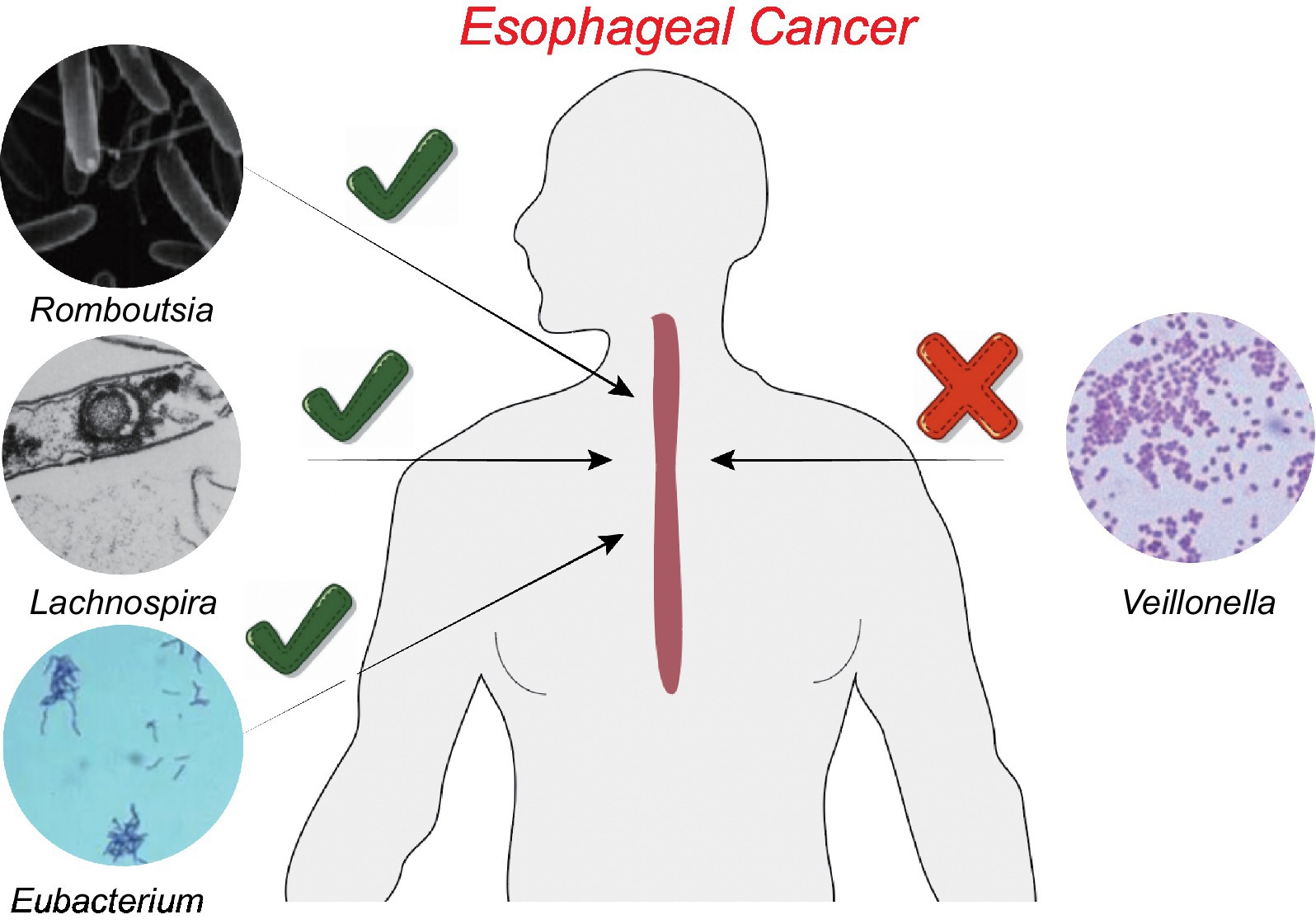
Figure 3. Patterns of the relationship between four gut microbiota and ESCA.
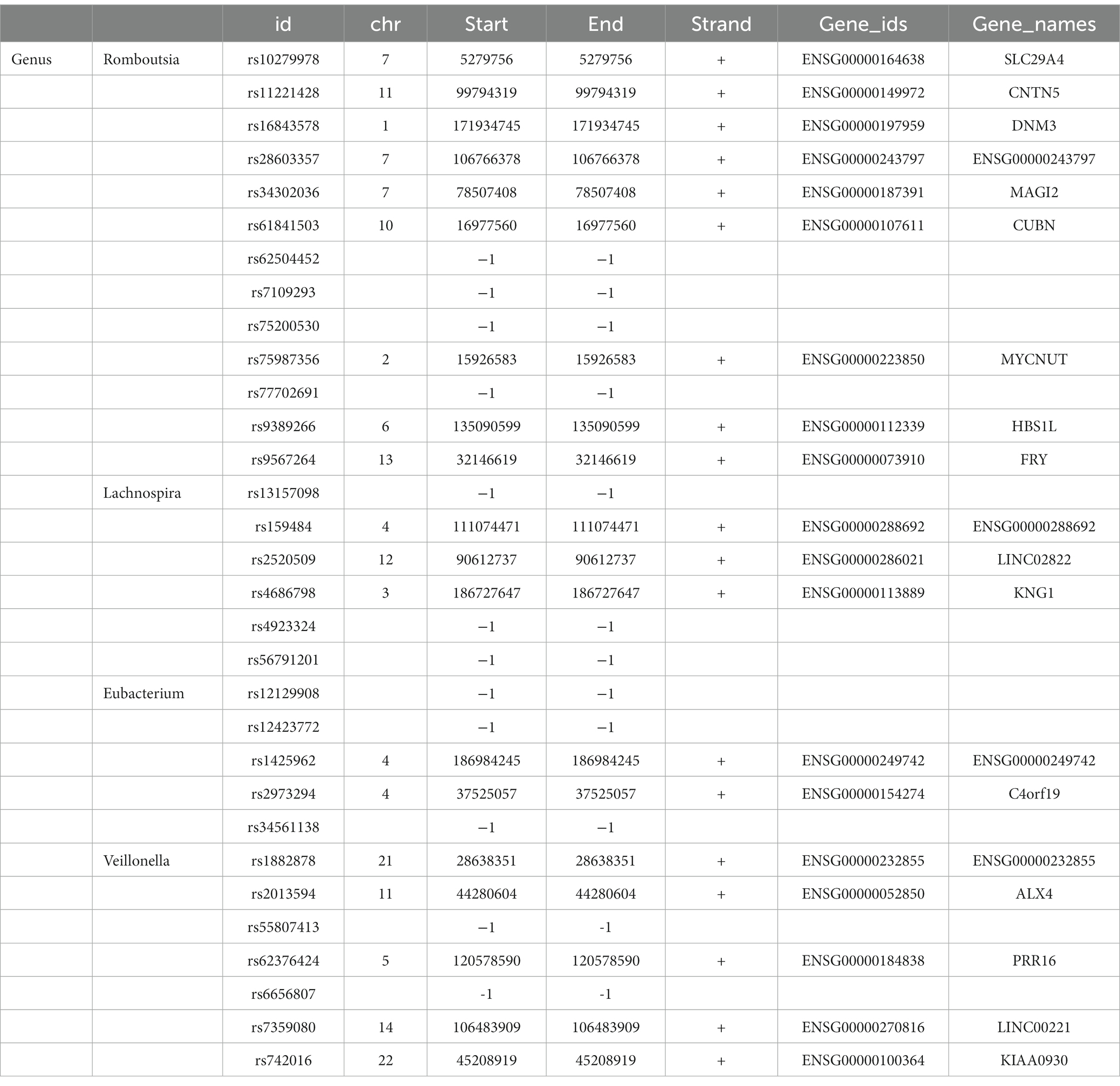
Table 2. SNP annotation of intestinal flora IVs.
We brought the above four intestinal flora into the external dataset for further analysis, and by plotting scatter plots for the training group (Figure 4A) and the validation group (Figure 4B), we found that the four intestinal flora in the validation group had the same tendency to have the same effect on ESCA when compared with the experimental group, which further validated our results. Meanwhile, we further counted the heterozygosity and pleiotropy of the training and validation group analyses (Figure 5), which showed that none of them had pleiotropy, indicating the credibility of our results.
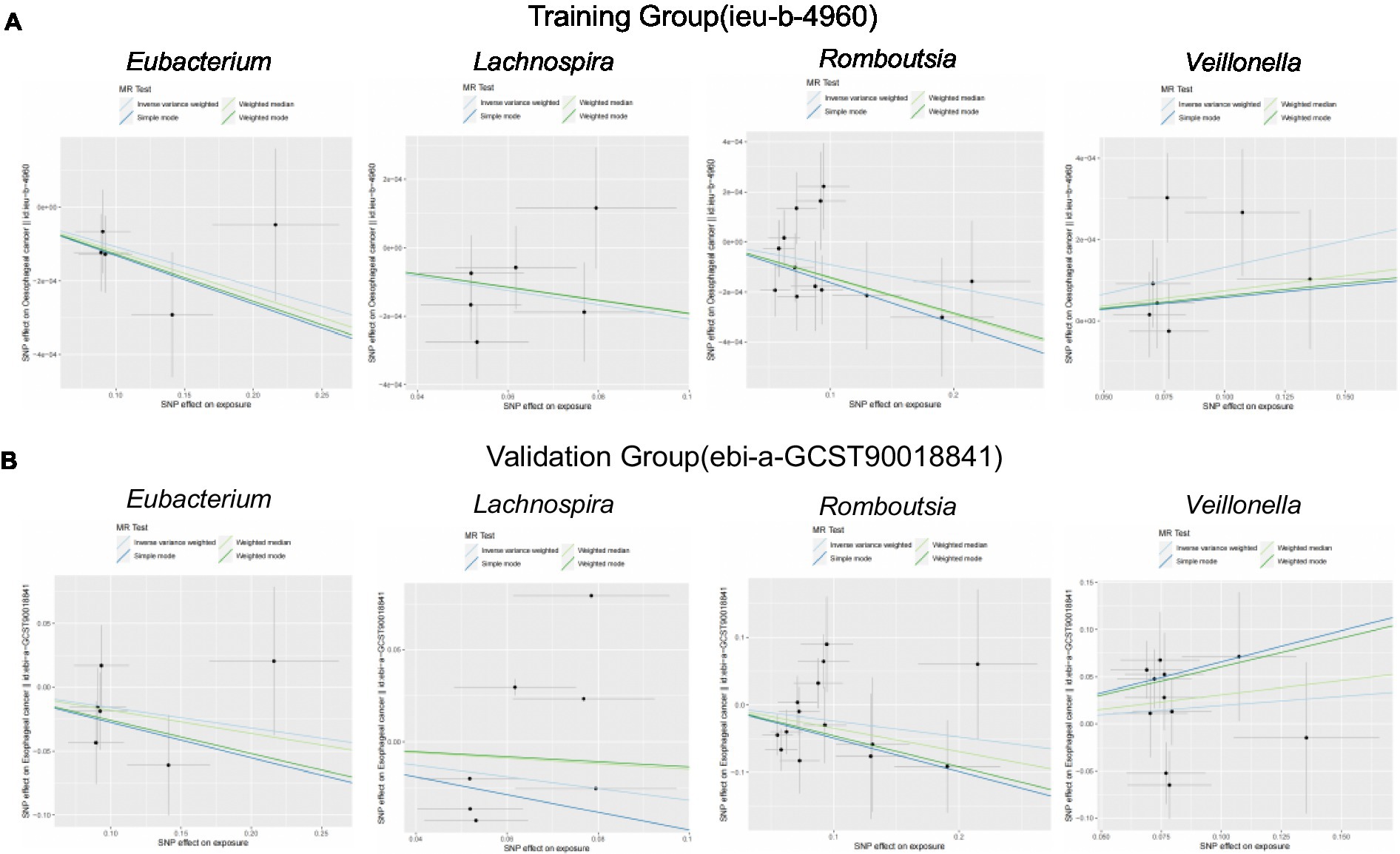
Figure 4. Scatterplot of correlation between training and validation groups. (A) Scatterplot of the causal relationship between the four intestinal flora and ESCA in the training group. (B) Scatterplot of the causal relationship between the four intestinal flora and ESCA in the validation group.
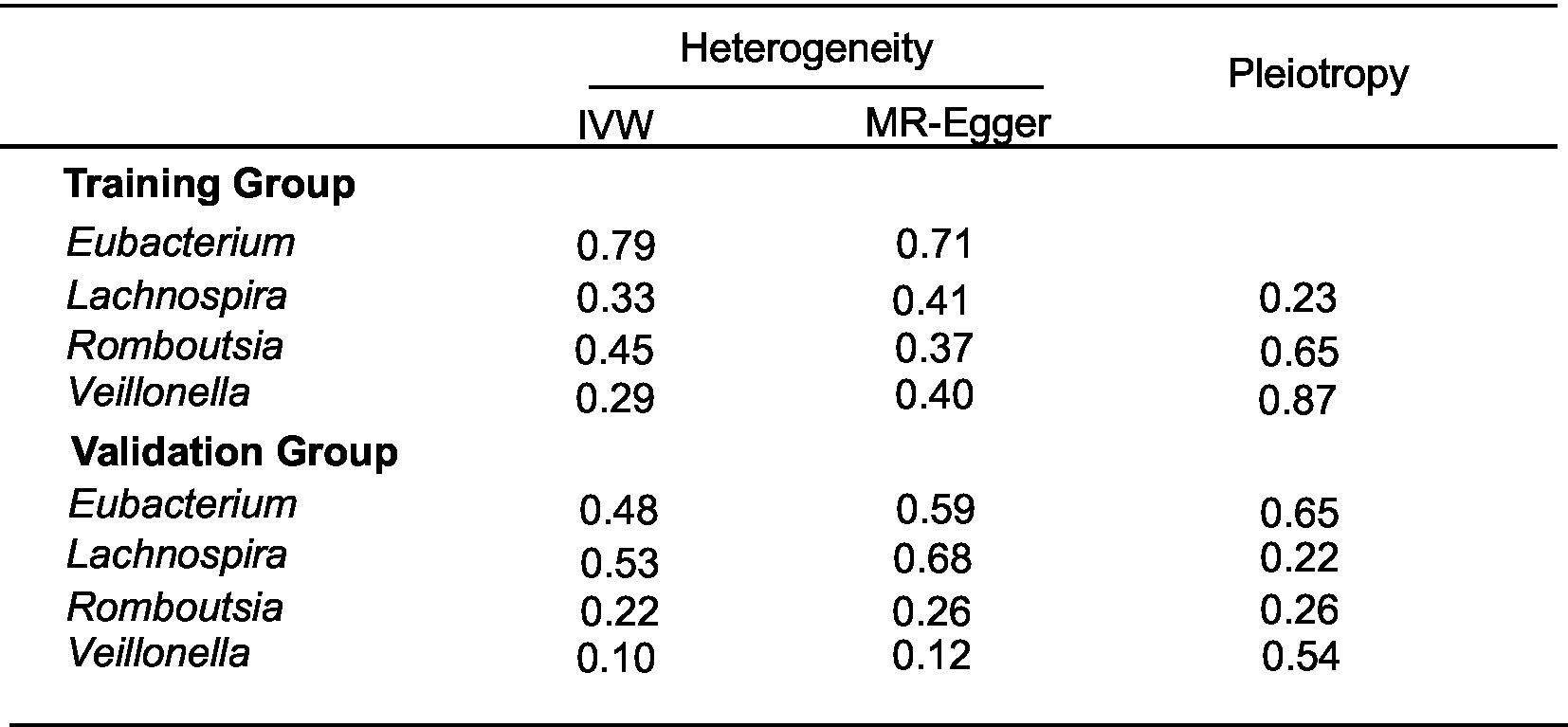
Figure 5. Heterogeneity and pleiotropy results of training and validation group analyses.
We further meta-analyzed and plotted and mapped the correlation data of the training and validation groups with the four intestinal flora in a forest plot (Figures 6A–D), which showed that the above relationship was robust. For ease of analysis, we summarized the results into a three-line table (Figure 6E), which showed less statistical heterogeneity and statistically significant results, which further justified our conclusions above.
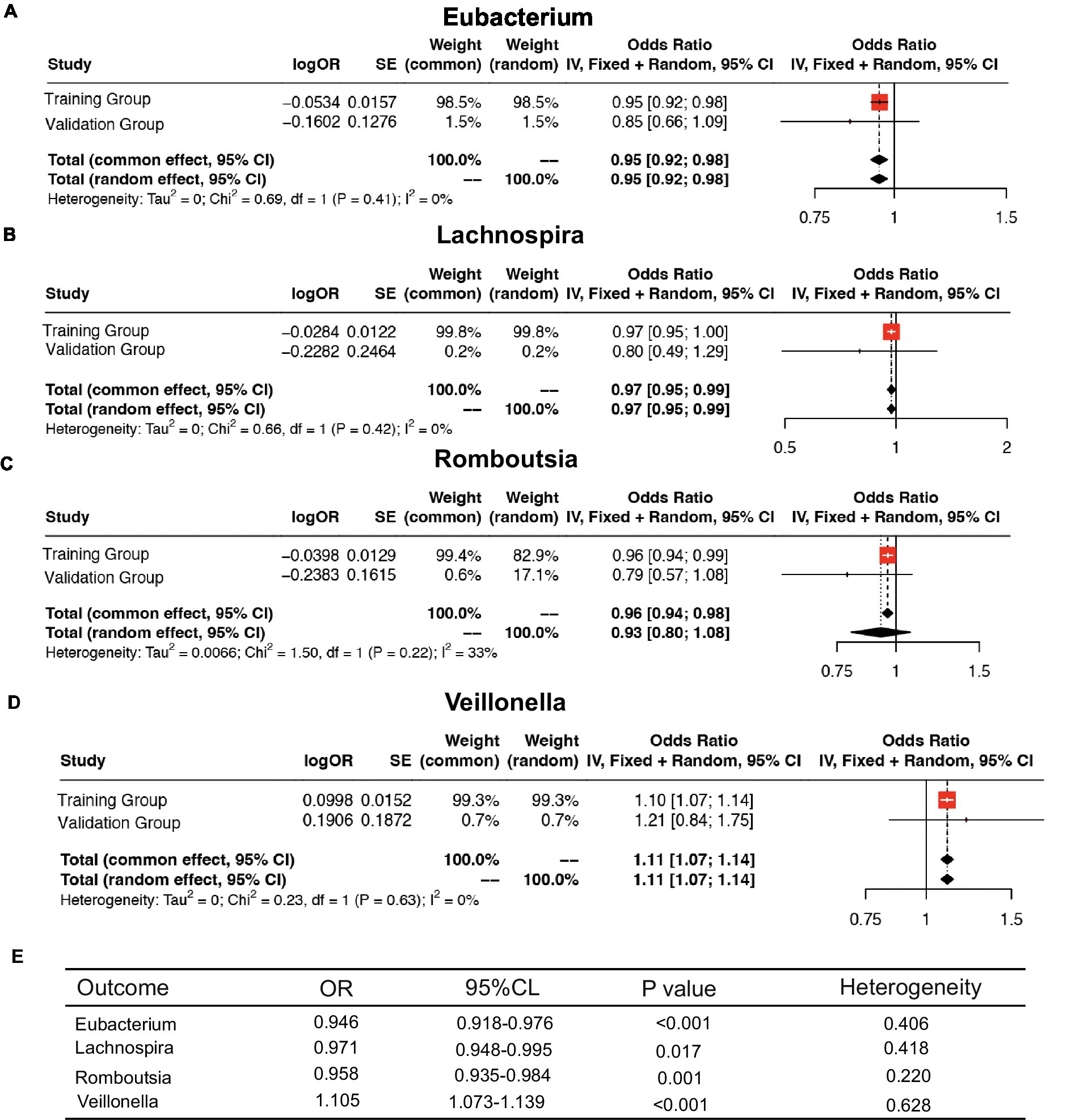
Figure 6. Meta-analysis of the causal association between host gene–gut microbiota and ESCA. (A–D) Meta-analysis of the causal relationship between four intestinal flora and ESCA. (E) Meta-analysis results.
The esophagus in the gastrointestinal tract is colonized by various microorganisms and is not sterile (Laserna-Mendieta et al., 2021). Healthy individuals have a relatively stable pH (approximately 7) that provides a stable environment for microbial survival (Hasan et al., 2021). The gut microbiota can directly or indirectly affect human health and disease and is considered a new “organ” (Baquero and Nombela, 2012). Gut microbes significantly impact cell formation, differentiation, metabolism, and growth. Dysbiotic gut microbiota may contribute to the body’s carcinogenic process (Zhou et al., 2021; Gou et al., 2023; Guevara-Ramirez et al., 2023). Notably, the gut microbiota may be involved in the development of ESCA (Lv et al., 2019; Zhou et al., 2021). For example, human papillomavirus (HPV) and alterations in intestinal bacteria may cause ESCA (Meng et al., 2018). Moreover, in regions with a high incidence of ESCA, a high prevalence of HPV has often been reported (Yano et al., 2021). Deng et al. studied 23 patients with ESCA and 23 healthy individuals and observed that patients with ESCA have higher levels of Firmicutes and Actinobacteria and lower levels of Bacteroidetes (Deng et al., 2021). This suggests that the gut microbiota and development of ESCA are closely related. In addition, changes in the gut microbiota can increase the levels of pro-inflammatory cytokines and immune cells, thereby inducing tumorigenesis (Proano-Vasco et al., 2021). Notably, gut microbiota can induce the overexpression of nitric oxide synthase, potentially leading to ESCA (Gillespie et al., 2021). The gut microbiota can also interact with the host by secreting bioactive substances, such as vitamins (Malesza et al., 2021), that can be beneficial or harmful for the organism (Riwes and Reddy, 2018). These explanations help to clarify how the gut microbiota and ESCA are related.
In this study, MR analysis was used to assess the causal relationship between gut microbiota and ESCA for providing a theoretical foundation for the interactions between ESCA and gut microbiota, given the lack of conclusive evidence to support the potential relationship between gut microbiota and ESCA. The results of this study showed that four intestinal microbiota were potentially associated with ESCA. Specifically, the genera Romboutsia, Lachnospira, and Eubacterium were negatively associated with ESCA, whereas Veillonella had a positive causal relationship with ESCA.
The genus Romboutsia is a group of gram-positive bacteria first proposed in 2014 by Ricaboni et al. (2016) from the right half of the human colon using colonoscopy. Most studies concluded that Romboutsia belongs to the natural gut microbial community and plays an essential role in host health. For example, Lactobacillus acidophilus ameliorates colitis by increasing the abundance of Romboutsia (Han et al., 2023). Several studies have found that Romboutsia is significantly less abundant in patients with inflammatory bowel disease (e.g., Crohn’s disease, ulcerative colitis) (Qiu et al., 2020; Wang et al., 2023), and is associated with hepatocellular liver cancer and postherpetic neuralgia (Feng et al., 2023; Jiao et al., 2023). Consequently, Romboutsia may influence immune regulation and intestinal health (Liu et al., 2023). At the same time Romboutsia is able to increase the antioxidant capacity of the body (Zhang et al., 2023), attenuates intestinal inflammatory damage and inhibits endoplasmic reticulum stress, which may be the mechanism by which it affects human health (Li et al., 2023). Moreover, Romboutsia is closely associated with esophageal epithelial atrophy (Pan et al., 2021), an essential stage in the development of ESCA. The potential relationship between Romboutsia and ESCA identified in this study is consistent with the above findings, which suggests that our analysis is logical.
Lachnospira is integral to the gut microbiota and colonizes the intestinal lumen from birth (Vacca et al., 2020). It is a group of potentially beneficial bacteria involved in the metabolism of various carbohydrates. Fermentation produces acetic and butyric acids, which provide energy to the host (Devillard et al., 2007; Wong et al., 2014). Lachnospira has been implicated in various diseases, including obesity (Natividad et al., 2018), liver disease (De Minicis et al., 2014), and chronic kidney disease (Yasuno et al., 2023), and can also lead to depression via the gut–brain axis (Bonaz et al., 2018). The Lachnospira spp. have anti-inflammatory and antioxidant effects and can protect the intestinal mucosal barrier by inhibiting inflammatory responses and scavenging free radicals, which may be a mechanism for preventing esophageal diseases (Mukherjee et al., 2020). A cohort study showed that Lachnospira, which has anti-inflammatory properties, was significantly reduced in ESCA patients compared to the normal group (Cheung et al., 2022). This is consistent with our findings in the current study.
Eubacterium has been found to be associated with age-related macular degeneration (AMD) (Mao et al., 2023), female infertility (Xi et al., 2023), multiple sclerosis (MS) (Vacaras et al., 2023) and other diseases. Although a previous Mendelian analysis done by Yang et al. showed that Eubacterium reduced the risk of Barrett’s esophagus (Yang et al., 2022), the literature exploring the relationship between Eubacterium spp. and ESCA remains scarce to date. This is the innovative finding of our present study, and it also suggests an interesting research direction for us. Now, our experiments on Eubacterium for the prevention of ESCA are in progress, and the results will be published in a follow-up study.
The genus Veillonella includes gram-negative, anaerobic, non-motile, and non-spore-forming coccus bacteria (Djais et al., 2019). Veillonella is strongly associated with the development of several diseases, and has been found to promote the proliferation of lung adenocarcinoma (Zeng et al., 2023), whilst Veillonella activates macrophages to promote inflammatory responses via the LPS-TLR4 pathway (Zhan et al., 2022), for example, Veillonella correlated with the severity of radiation esophagitis (Lin et al., 2022), and inflammation is one of the factors leading to esophageal cancer, suggesting that Veillonella may indirectly contribute to esophageal carcinogenesis through inflammation. In our study, only the genus Veillonella positively correlated with ESCA. An extensive body of literature describes the pathogenic role of Veillonella in the esophagus. For example, a study using whole genome sequencing (wGS) and RNA sequencing (rNAseq) of tumors from 61 patients with ESCA found a high abundance of Veillonella in ESCA (Nomburg et al., 2022). It has also been shown that Veillonella levels gradually increase during the development of esophageal reflux (GR)-Barrett’s esophagus (BE)-esophageal adenocarcinoma (EA) (Di Pilato et al., 2016; Hao et al., 2022). Gram-negative bacteria are mainly associated with esophageal abnormalities, e.g., Veillonella is mostly associated with BE (Lv et al., 2019; Park and Lee, 2020), which aligns with our findings. These findings have important implications for research in ESCA in the future and may inspire new prevention and treatment strategies for this disease.
Many previous studies have demonstrated a potential relationship between the gut microbiota and ESCA (Baba et al., 2023; Co et al., 2023; Sugimoto et al., 2023). However, most of these studies were observational and susceptible to confounding factors. In contrast, the present study used a genetic epidemiological approach to minimize the influence of confounding factors and provide a compelling insight into the relationship between gut microbiota and ESCA. And the use of external datasets to validate trends and Meta-analysis of the results ensures maximum stability of the results.
Gut flora plays a dual role in cancer development, and the use of gut flora in conjunction with traditional antitumor treatment strategies, as well as the use of probiotics, FMT, and dietary control, can improve the efficacy of anticancer treatments, while reducing the incidence of side effects and improving prognosis (Sun et al., 2023). However, this study had certain limitations. First, most of the participants were European. Additional research is required to determine whether these conclusions apply to other ethnic groups. Second, the flora in this study was limited to the genus level and was not further subdivided.
We analyzed the potential relationship between 196 common intestinal microbiota and ESCA. We found that the genera Romboutsia, Lachnospira, Eubacterium, and Veillonella may be causally associated with ESCA, which may provide new ideas for ESCA research and treatment.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
Ethical approval was not required for this study, because data were obtained from publicly available databases and no identifiable data was published.
ZZ: Writing – original draft. GZ: Conceptualization, Writing – review & editing. ZH: Methodology, Writing – review & editing. YS: Investigation, Writing – review & editing. DW: Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Science Foundation of Shandong Province of China (no. ZR2019MH092).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1309596/full#supplementary-material
SUPPLEMENTARY FIGURE 1
Leave-one-out analysis forest plot.
SUPPLEMENTARY TABLE 1
13 SNPs associated with ESCA in Romboutsia.
SUPPLEMENTARY TABLE 2
6 SNPs associated with ESCA in Lachnospira.
SUPPLEMENTARY TABLE 3
5 SNPs associated with ESCA in Eubacterium.
SUPPLEMENTARY TABLE 4
7 SNPs associated with ESCA in Veillonella.
BE, Barrett’s esophagus; CI, Confidence interval; ESCA, Esophageal cancer; GWAS, Genome-wide association studies; HPV, Human papillomavirus; IVW, Inverse-variance weighted; MAF, Minor allele frequency; ME, Mendelian randomization; OR, Odds ratio; SNPs, Single nucleotide polymorphisms.
Baba, Y., Hara, Y., Toihata, T., Kosumi, K., Iwatsuki, M., Iwagami, S., et al. (2023). Relationship between gut microbiome fusobacterium nucleatum and LINE−1 methylation level in esophageal cancer. Esophagus 20, 704–712. doi: 10.1007/s10388-023-01009-9
Baquero, F., and Nombela, C. (2012). The microbiome as a human organ. Clin. Microbiol. Infect. 18, 2–4. doi: 10.1111/j.1469-0691.2012.03916.x
Bonaz, B., Bazin, T., and Pellissier, S. (2018). The Vagus nerve at the Interface of the microbiota-gut-brain Axis. Front. Neurosci. 12:49. doi: 10.3389/fnins.2018.00049
Burgess, S., and Thompson, S. G. (2017). Interpreting findings from Mendelian randomization using the MR-egger method. Eur. J. Epidemiol. 32, 377–389. doi: 10.1007/s10654-017-0255-x
Cheung, M. K., Yue, G. G. L., Lauw, S., Li, C. S. Y., Yung, M. Y., Ng, S. C., et al. (2022). Alterations in gut microbiota of esophageal squamous cell carcinoma patients. J. Gastroenterol. Hepatol. 37, 1919–1927. doi: 10.1111/jgh.15941
Co, E. L., Hameed, M., Sebastian, S. A., Garg, T., Sudan, S., Bheemisetty, N., et al. (2023). Narrative review of probiotic use on the recovery of postoperative patients with esophageal Cancer. Curr. Nutr. Rep. doi: 10.1007/s13668-023-00490-z
De Minicis, S., Rychlicki, C., Agostinelli, L., Saccomanno, S., Candelaresi, C., Trozzi, L., et al. (2014). Dysbiosis contributes to fibrogenesis in the course of chronic liver injury in mice. Hepatology 59, 1738–1749. doi: 10.1002/hep.26695
Deng, Y., Tang, D., Hou, P., Shen, W., Li, H., Wang, T., et al. (2021). Dysbiosis of gut microbiota in patients with esophageal cancer. Microb. Pathog. 150:104709. doi: 10.1016/j.micpath.2020.104709
Devillard, E., McIntosh, F. M., Duncan, S. H., and Wallace, R. J. (2007). Metabolism of linoleic acid by human gut bacteria: different routes for biosynthesis of conjugated linoleic acid. J. Bacteriol. 189, 2566–2570. doi: 10.1128/JB.01359-06
Di Pilato, V., Freschi, G., Ringressi, M. N., Pallecchi, L., Rossolini, G. M., and Bechi, P. (2016). The esophageal microbiota in health and disease. Ann. N. Y. Acad. Sci. 1381, 21–33. doi: 10.1111/nyas.13127
Djais, A. A., Theodorea, C. F., Mashima, I., Otomo, M., Saitoh, M., and Nakazawa, F. (2019). Identification and phylogenetic analysis of oral Veillonella species isolated from the saliva of Japanese children. F1000Res 8:616. doi: 10.12688/f1000research.18506.5
Feng, J., Wu, Y., Dai, P., Wang, D., Liu, L., and Chai, B. (2023). Gut microbial signatures of patients with primary hepatocellular carcinoma and their healthy first-degree relatives. J. Appl. Microbiol. 134:221. doi: 10.1093/jambio/lxad221
Gillespie, M. R., Rai, V., Agrawal, S., and Nandipati, K. C. (2021). The role of microbiota in the pathogenesis of esophageal adenocarcinoma. Biology (Basel) 10:697. doi: 10.3390/biology10080697
Gou, H., Su, H., Liu, D., Wong, C. C., Shang, H., Fang, Y., et al. (2023). Traditional medicine Pien Tze Huang suppresses colorectal tumorigenesis through restoring gut microbiota and metabolites. Gastroenterology 165, 1404–1419. doi: 10.1053/j.gastro.2023.08.052
Guevara-Ramirez, P., Cadena-Ullauri, S., Paz-Cruz, E., Tamayo-Trujillo, R., Ruiz-Pozo, V. A., and Zambrano, A. K. (2023). Role of the gut microbiota in hematologic cancer. Front. Microbiol. 14:1185787. doi: 10.3389/fmicb.2023.1185787
Han, M., Liao, W., Dong, Y., Fei, T., and Gai, Z. (2023). Sustained ameliorative effect of Lactobacillus acidophilus LA85 on dextran sulfate sodium-induced colitis in mice. J. Food Sci. 88, 3893–3904. doi: 10.1111/1750-3841.16723
Hao, Y., Karaoz, U., Yang, L., Yachimski, P. S., Tseng, W., Nossa, C. W., et al. (2022). Progressive dysbiosis of human orodigestive microbiota along the sequence of gastroesophageal reflux, Barrett's esophagus and esophageal adenocarcinoma. Int. J. Cancer 151, 1703–1716. doi: 10.1002/ijc.34191
Hasan, A., Hasan, L. K., Schnabl, B., Greytak, M., and Yadlapati, R. (2021). Microbiome of the Aerodigestive tract in health and esophageal disease. Dig. Dis. Sci. 66, 12–18. doi: 10.1007/s10620-020-06720-6
He, S., Xu, J., Liu, X., and Zhen, Y. (2021). Advances and challenges in the treatment of esophageal cancer. Acta Pharm. Sin. B 11, 3379–3392. doi: 10.1016/j.apsb.2021.03.008
Jiao, B., Cao, X., Zhang, C., Zhang, W., Yu, S., Zhang, M., et al. (2023). Alterations of the gut microbiota in patients with postherpetic neuralgia. AMB Express 13:108. doi: 10.1186/s13568-023-01614-y
Kurilshikov, A., Medina-Gomez, C., Bacigalupe, R., Radjabzadeh, D., Wang, J., Demirkan, A., et al. (2021). Large-scale association analyses identify host factors influencing human gut microbiome composition. Nat. Genet. 53, 156–165. doi: 10.1038/s41588-020-00763-1
Laserna-Mendieta, E. J., FitzGerald, J. A., Arias-Gonzalez, L., Ollala, J. M., Bernardo, D., Claesson, M. J., et al. (2021). Esophageal microbiome in active eosinophilic esophagitis and changes induced by different therapies. Sci. Rep. 11:7113. doi: 10.1038/s41598-021-86464-z
Li, N., Bai, C., Zhao, L., Ge, Y., and Li, X. (2022). Characterization of the fecal microbiota in gastrointestinal cancer patients and healthy people. Clin. Transl. Oncol. 24, 1134–1147. doi: 10.1007/s12094-021-02754-y
Li, K., Wu, J., Xu, S., Li, X., Zhang, Y., and Gao, X. J. (2023). Rosmarinic acid alleviates intestinal inflammatory damage and inhibits endoplasmic reticulum stress and smooth muscle contraction abnormalities in intestinal tissues by regulating gut microbiota. Microbiol. Spectr. 11:1914. doi: 10.1128/spectrum.01914-23
Lin, M. Q., Wu, Y. H., Yang, J., Lin, H. C., Liu, L. Y., Yu, Y. L., et al. (2022). Gut microbiota characteristics are associated with severity of acute radiation-induced esophagitis. Front. Microbiol. 13:883650. doi: 10.3389/fmicb.2022.883650
Liu, J., Cai, J., Fan, P., Dong, X., Zhang, N., Tai, J., et al. (2023). Salidroside alleviates dextran sulfate sodium-induced colitis in mice by modulating the gut microbiota. Food Funct. 14, 7506–7519. doi: 10.1039/D3FO01929B
Lv, J., Guo, L., Liu, J. J., Zhao, H. P., Zhang, J., and Wang, J. H. (2019). Alteration of the esophageal microbiota in Barrett's esophagus and esophageal adenocarcinoma. World J. Gastroenterol. 25, 2149–2161. doi: 10.3748/wjg.v25.i18.2149
Malesza, I. J., Malesza, M., Walkowiak, J., Mussin, N., Walkowiak, D., Aringazina, R., et al. (2021). High-Fat, western-style diet, systemic inflammation, and gut microbiota: A narrative review. Cells 10:3164. doi: 10.3390/cells10113164
Mao, D., Tao, B., Sheng, S., Jin, H., Chen, W., Gao, H., et al. (2023). Causal effects of gut microbiota on age-related macular degeneration: a Mendelian randomization study. Invest. Ophthalmol. Vis. Sci. 64:32. doi: 10.1167/iovs.64.12.32
Meng, C., Bai, C., Brown, T. D., Hood, L. E., and Tian, Q. (2018). Human gut microbiota and gastrointestinal Cancer. Genomics Proteomics Bioinformatics 16, 33–49. doi: 10.1016/j.gpb.2017.06.002
Mukherjee, A., Lordan, C., Ross, R. P., and Cotter, P. D. (2020). Gut microbes from the phylogenetically diverse genus Eubacterium and their various contributions to gut health. Gut Microbes 12:1802866. doi: 10.1080/19490976.2020.1802866
Muszynski, D., Kudra, A., Sobocki, B. K., Folwarski, M., Vitale, E., Filetti, V., et al. (2022). Esophageal cancer and bacterial part of gut microbiota – a multidisciplinary point of view. Front. Cell. Infect. Microbiol. 12:1057668. doi: 10.3389/fcimb.2022.1057668
Natividad, J. M., Agus, A., Planchais, J., Lamas, B., Jarry, A. C., Martin, R., et al. (2018). Impaired aryl hydrocarbon receptor ligand production by the gut microbiota is a key factor in metabolic syndrome. Cell Metab. 28:e734, 737–749.e4. doi: 10.1016/j.cmet.2018.07.001
Nomburg, J., Bullman, S., Nasrollahzadeh, D., Collisson, E. A., Abedi-Ardekani, B., Akoko, L. O., et al. (2022). An international report on bacterial communities in esophageal squamous cell carcinoma. Int. J. Cancer 151, 1947–1959. doi: 10.1002/ijc.34212
Ohkusa, T., Nishikawa, Y., and Sato, N. (2022). Gastrointestinal disorders and intestinal bacteria: advances in research and applications in therapy. Front. Med. 9:935676. doi: 10.3389/fmed.2022.935676
Oppedijk, V., van der Gaast, A., van Lanschot, J. J., van Hagen, P., van Os, R., van Rij, C. M., et al. (2014). Patterns of recurrence after surgery alone versus preoperative chemoradiotherapy and surgery in the CROSS trials. J. Clin. Oncol. 32, 385–391. doi: 10.1200/JCO.2013.51.2186
Pan, F., Zhang, L. L., Luo, H. J., Chen, Y., Long, L., Wang, X., et al. (2021). Dietary riboflavin deficiency induces ariboflavinosis and esophageal epithelial atrophy in association with modification of gut microbiota in rats. Eur. J. Nutr. 60, 807–820. doi: 10.1007/s00394-020-02283-4
Park, C. H., and Lee, S. K. (2020). Exploring esophageal microbiomes in esophageal diseases: a systematic review. J. Neurogastroenterol. Motil. 26, 171–179. doi: 10.5056/jnm19240
Proano-Vasco, A., Baumeister, T., Metwaly, A., Reitmeier, S., Kleigrewe, K., Meng, C., et al. (2021). High-fructose diet alters intestinal microbial profile and correlates with early tumorigenesis in a mouse model of Barrett's esophagus. Microorganisms 9:2432. doi: 10.3390/microorganisms9122432
Qiu, X., Zhao, X., Cui, X., Mao, X., Tang, N., Jiao, C., et al. (2020). Characterization of fungal and bacterial dysbiosis in young adult Chinese patients with Crohn's disease. Ther. Adv. Gastroenterol. 13:175628482097120. doi: 10.1177/1756284820971202
Ricaboni, D., Mailhe, M., Khelaifia, S., Raoult, D., and Million, M. (2016). Romboutsia timonensis, a new species isolated from human gut. New Microbes New Infect. 12, 6–7. doi: 10.1016/j.nmni.2016.04.001
Riwes, M., and Reddy, P. (2018). Microbial metabolites and graft versus host disease. Am. J. Transplant. 18, 23–29. doi: 10.1111/ajt.14443
Smith, G. D., and Ebrahim, S. (2003). 'Mendelian randomization': can genetic epidemiology contribute to understanding environmental determinants of disease? Int. J. Epidemiol. 32, 1–22. doi: 10.1093/ije/dyg070
Sugimoto, T., Atobe, S., Kado, Y., Takahashi, A., Motoori, M., Sugimura, K., et al. (2023). Gut microbiota associated with the mitigation effect of synbiotics on adverse events of neoadjuvant chemotherapy in patients with esophageal cancer: a retrospective exploratory study. J. Med. Microbiol. 72:1723. doi: 10.1099/jmm.0.001723
Sun, J., Chen, F., and Wu, G. (2023). Potential effects of gut microbiota on host cancers: focus on immunity, DNA damage, cellular pathways, and anticancer therapy. ISME J. 17, 1535–1551. doi: 10.1038/s41396-023-01483-0
Sung, H., Ferlay, J., Siegel, R. L., Laversanne, M., Soerjomataram, I., Jemal, A., et al. (2021). Global Cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249. doi: 10.3322/caac.21660
Thanawala, S. U., Kaplan, D. E., Falk, G. W., Beveridge, C. A., Schaubel, D., Serper, M., et al. (2023). Antibiotic exposure is associated with a risk of esophageal adenocarcinoma. Clin. Gastroenterol. Hepatol. 21, 2817–2824.e4. doi: 10.1016/j.cgh.2023.03.012
Uhlenhopp, D. J., Then, E. O., Sunkara, T., and Gaduputi, V. (2020). Epidemiology of esophageal cancer: update in global trends, etiology and risk factors. Clin. J. Gastroenterol. 13, 1010–1021. doi: 10.1007/s12328-020-01237-x
Vacaras, V., Muresanu, D. F., Buzoianu, A. D., Nistor, C., Vesa, S. C., Paraschiv, A. C., et al. (2023). The role of multiple sclerosis therapies on the dynamic of human gut microbiota. J. Neuroimmunol. 378:578087. doi: 10.1016/j.jneuroim.2023.578087
Vacca, M., Celano, G., Calabrese, F. M., Portincasa, P., Gobbetti, M., and De Angelis, M. (2020). The controversial role of human gut Lachnospiraceae. Microorganisms 8:573. doi: 10.3390/microorganisms8040573
Verbanck, M., Chen, C. Y., Neale, B., and Do, R. (2018). Publisher correction: detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50:1196. doi: 10.1038/s41588-018-0164-2
Wang, K., Qin, L., Cao, J., Zhang, L., Liu, M., Qu, C., et al. (2023). Kappa-Selenocarrageenan oligosaccharides prepared by Deep-Sea enzyme alleviate inflammatory responses and modulate gut microbiota in ulcerative colitis mice. Int. J. Mol. Sci. 24:4672. doi: 10.3390/ijms24054672
Wong, J., Piceno, Y. M., DeSantis, T. Z., Pahl, M., Andersen, G. L., and Vaziri, N. D. (2014). Expansion of urease-and uricase-containing, indole-and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 39, 230–237. doi: 10.1159/000360010
Xi, Y., Zhang, C., Feng, Y., Zhao, S., Zhang, Y., Duan, G., et al. (2023). Genetically predicted the causal relationship between gut microbiota and infertility: bidirectional Mendelian randomization analysis in the framework of predictive, preventive, and personalized medicine. EPMA J. 14, 405–416. doi: 10.1007/s13167-023-00332-6
Yang, Z., Yu, R., Deng, W., and Wang, W. (2022). Roles of 21 genera of human gut microbiota in Barrett's esophagus risk: a Mendelian randomization study. Front. Genet. 13:894900. doi: 10.3389/fgene.2022.894900
Yano, Y., Etemadi, A., and Abnet, C. C. (2021). Microbiome and cancers of the esophagus: A Review. Microorganisms 9:764. doi: 10.3390/microorganisms9081764
Yasuno, T., Takahashi, K., Tada, K., Hiyamuta, H., Watanabe, M., Ito, K., et al. (2023). Dysbiosis of gut microbiota in patients with chronic kidney disease. Intern. Med. doi: 10.2169/internalmedicine.1602-23
Zeng, W., Wang, Y., Wang, Z., Yu, M., Liu, K., Zhao, C., et al. (2023). Veillonella parvula promotes the proliferation of lung adenocarcinoma through the nucleotide oligomerization domain 2/cellular communication network factor 4/nuclear factor kappa B pathway. Discov. Oncol. 14:129. doi: 10.1007/s12672-023-00748-6
Zhan, Z., Liu, W., Pan, L., Bao, Y., Yan, Z., and Hong, L. (2022). Overabundance of Veillonella parvula promotes intestinal inflammation by activating macrophages via LPS-TLR4 pathway. Cell Death Discov. 8:251. doi: 10.1038/s41420-022-01015-3
Zhang, P., Jiang, G., Wang, Y., Yan, E., He, L., Guo, J., et al. (2023). Maternal consumption of l-malic acid enriched diets improves antioxidant capacity and glucose metabolism in offspring by regulating the gut microbiota. Redox Biol. 67:102889. doi: 10.1016/j.redox.2023.102889
Keywords: esophageal cancer, gut microbiota, Mendelian randomization, single nucleotide polymorphism, meta-analysis
Citation: Zhang Z, Zhang G, Huang Z, Shi Y and Wang D (2023) Application of Mendelian randomization to assess host gene–gut microbiota correlations in patients with esophageal cancer. Front. Microbiol. 14:1309596. doi: 10.3389/fmicb.2023.1309596
Edited by:
William K. K. Wu, The Chinese University of Hong Kong, ChinaReviewed by:
Wang Shuai, Zhongshan Hospital, Fudan University, ChinaCopyright © 2023 Zhang, Zhang, Huang, Shi and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Wang, c2RwaHdkQDE2My5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.