 Kexin Liu1,2†
Kexin Liu1,2† Yajie Wang
Yajie Wang Jianguo Chen
Jianguo Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 04 December 2023
Sec. Systems Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1297451
This article is part of the Research TopicMicrobiome and Machine Learning, Volume IIView all 16 articles
Although MALDI-TOF mass spectrometry (MS) is widely known as a rapid and cost-effective reference method for identifying microorganisms, its commercial databases face limitations in accurately distinguishing specific subspecies of Bifidobacterium. This study aimed to explore the potential of MALDI-TOF MS protein profiles, coupled with prediction methods, to differentiate between Bifidobacterium longum subsp. infantis (B. infantis) and Bifidobacterium longum subsp. longum (B. longum). The investigation involved the analysis of mass spectra of 59 B. longum strains and 41 B. infantis strains, leading to the identification of five distinct biomarker peaks, specifically at m/z 2,929, 4,408, 5,381, 5,394, and 8,817, using Recurrent Feature Elimination (RFE). To facilate classification between B. longum and B. infantis based on the mass spectra, machine learning models were developed, employing algorithms such as logistic regression (LR), random forest (RF), and support vector machine (SVM). The evaluation of the mass spectrometry data showed that the RF model exhibited the highest performace, boasting an impressive AUC of 0.984. This model outperformed other algorithms in terms of accuracy and sensitivity. Furthermore, when employing a voting mechanism on multi-mass spectrometry data for strain identificaton, the RF model achieved the highest accuracy of 96.67%. The outcomes of this research hold the significant potential for commercial applications, enabling the rapid and precise discrimination of B. longum and B. infantis using MALDI-TOF MS in conjunction with machine learning. Additionally, the approach proposed in this study carries substantial implications across various industries, such as probiotics and pharmaceuticals, where the precise differentiation of specific subspecies is essential for product development and quality control.
Bifidobacterium longum subsp. infantis (B. infantis) and Bifidobacterium longum subsp. longum (B. longum), the most abundant Bifidobacterium species in the intestinal flora of infants, are essential for their immune development. Human breast milk contains a large amount of human milk oligosaccharides (HMOs), which cannot be digested by infant due to a lack of necessary glucosidases. However, the positive effects of HMOs on newborns’ health are attributed to the “beneficial” microorganisms that specialize in metabolizing HMOs. In contrast to B. longum, B. infantis typically harbors all the genes required for utilizing HMOs (Duar et al., 2020) and can digest various types of HMOs, including 3′-SL, 6′-SL, 2′-FL, 3′-FL, LNnT, and LacNAc (Zhang et al., 2022). The absence of Bifidobacterium and HMO utilization genes in the gut microbiota is associated with inflammation and immune imbalances in early life (Henrick et al., 2021). B. infantis is commonly found in breastfed infants in countries with a low prevalence of immune-mediated diseases, such as Bangladesh (Vatanen et al., 2022) and Malawi, but is rare in Europe and North America (Casaburi et al., 2021). However, supplementation with B. infantis EVC001, by remodelling the gut microbiome of breastfed infants, reduced intestinal inflammation (Henrick et al., 2019), decreased intestinal Th2 and Th17 cytokines and up-regulated IFNβ, favouring immune development in early life (Henrick et al., 2021). Therefore, accurate identification of B. longum and B. infantis is essential for efficient screening, functional studies and application development of B. infantis.
The current methods used to identify Bifidobacteria include PCR, SNP, cgMLST, and MALDI-TOF MS. MALDI-TOF MS is particularly advantageous due to its high throughput, fast speed, and low cost, making it widely used for identifying clinical pathogenic microorganisms and general microorganisms (Gato et al., 2021; Heilbronner and Foster, 2021; Wang H. Y. et al., 2022). However, the successful identification of bacteria using MALDI-TOF MS heavily relies on databases that contain spectra of known organisms and most of the biomarker peaks are in the range m/z 2,000–10,000 (Carvalho et al., 2022; Topić Popović et al., 2023). Most commercial databases only identify bacteria at the species level and lack the ability to accurately differentiate closely related subspecies, such as B. longum and B. infantis. Although six biomarker peaks have been reported to differentiate between B. longum and B. infantis, these peaks have not been commercially applied due to their high mass peaks (>15,000 m/z) (Sato et al., 2011), low reproducibility, and lack of availability in commercial databases. Recently, machine learning techniques have been used to accurately identify strains that cannot be distinguished using commercial databases by analyzing protein mass spectra obtained through MALDI-TOF MS (Weis et al., 2022; Kim et al., 2023).
Machine learning (ML) technology encompasses various algorithms such as random forest (RF), support vector machines (SVM), logistic regression (LR) and decision trees (DT) (Weis et al., 2020). ML enables rapid and precise identification of species-specific biomarkers from MALDI-TOF MS spectra, which has been widely implemented to analyze microbial signatures and construct classification models. Recently, the combination of MALDI-TOF MS and ML has gained popularity in classifying clinically pathogenic and drug-resistant bacteria, including Escherichia coli (van Oosten and Klein, 2020), Staphylococcus aureus (Rodríguez-Temporal et al., 2022), Klebsiella pneumoniae (Yu et al., 2023), Brucella melitensis (Dematheis et al., 2022), and Campylobacter spp. (Feucherolles et al., 2021). However, there is a lack of identification schemes for Bifidobacterium subspecies within a specific taxon in these studies. Hence, there is an urgent need to develop a combined machine learning and MALDI-TOF MS method for rapid and accurate identification of Bifidobacterium subspecies.
In the present study, we first screened for robust variations in subspecies-specific features between B. longum and B. infantis based on MALDI-TOF MS analysis and a combination of machine learning methods such as LR, SVM, and RF (Figure 1). The objective of this research was to develop a fast classification tool using Machine-learning-combined MALDI-TOF MS to accurately distinguish between B. longum and B. infantis.
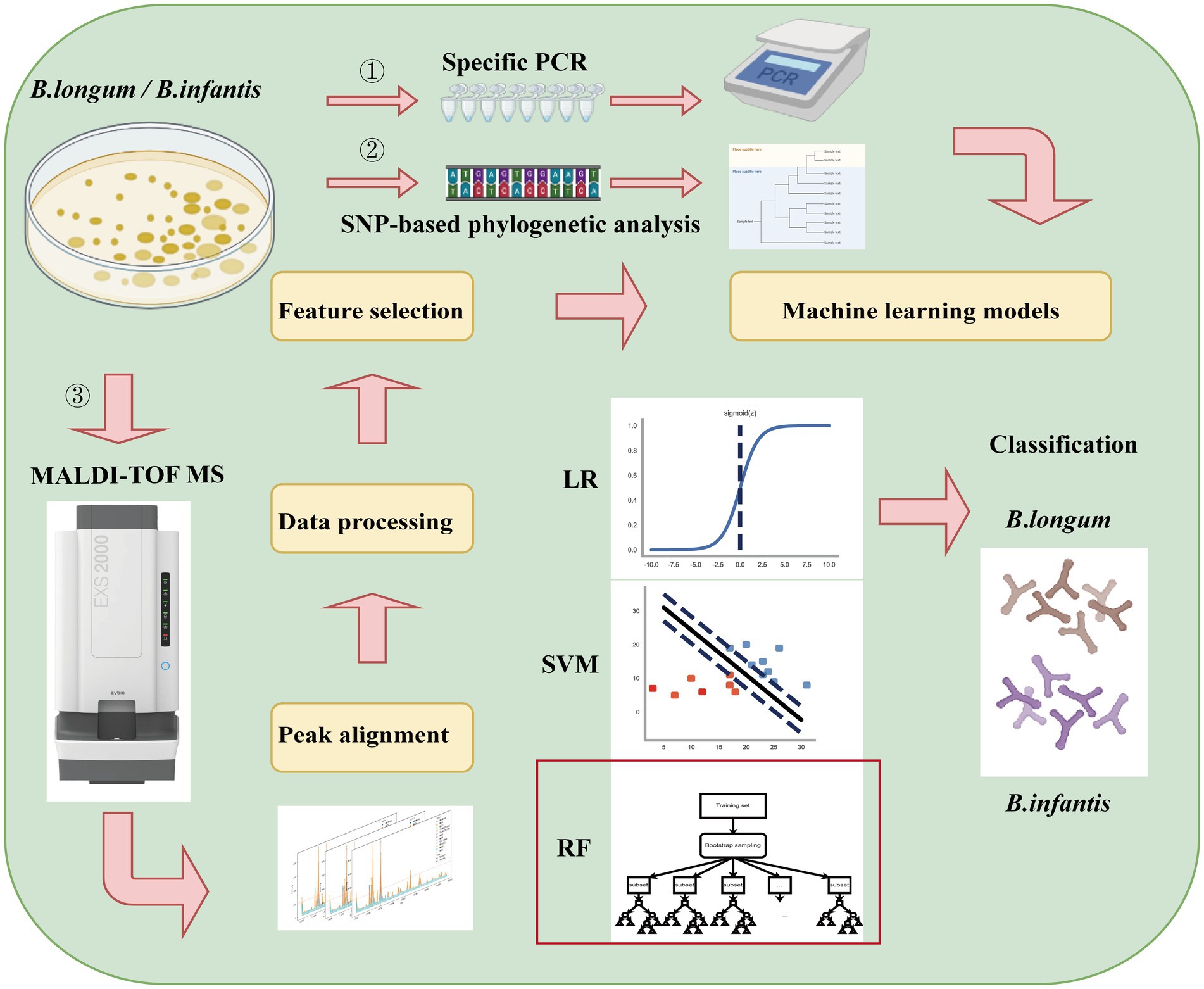
Figure 1. Experimental flow chart of Bifidobacterium longum subspecies discrimination based on MALDI-TOF MS and machine learning in this study.
Specific primers-based PCR could differentiate between B. longum and B infantis. Thus, this method was employed to confirm the taxonomic classification of all the strains in study. The specificity and sensitivity of the PCR assay using specific primers for distinguishing the two subspecies were confirmed by successfully differentiation of 11 representative strains. Out of the 89 isolates analyzed, 54 were identified as B. longum and 35 were identified as B. infantis. For additional confirmation, SNP information obtained from 100 genome sequences were utilized to construct a phylogenetic tree. The tree effectively separated the sequences into two distinct branches. The phylogenetic tree revealed that 59 B. longum strains, comprising five typical strains and 54 isolates, clustered together with a blue background, while 41 B. infantis strains formed a distinct group with a red background (Figure 2). These findings underscore the efficacy of using phylogenetic tree features for precise classification and identification of B. longum and B. infantis, which align with the outcomes obtained from specific PCR genotyping (Supplementary Table S1).
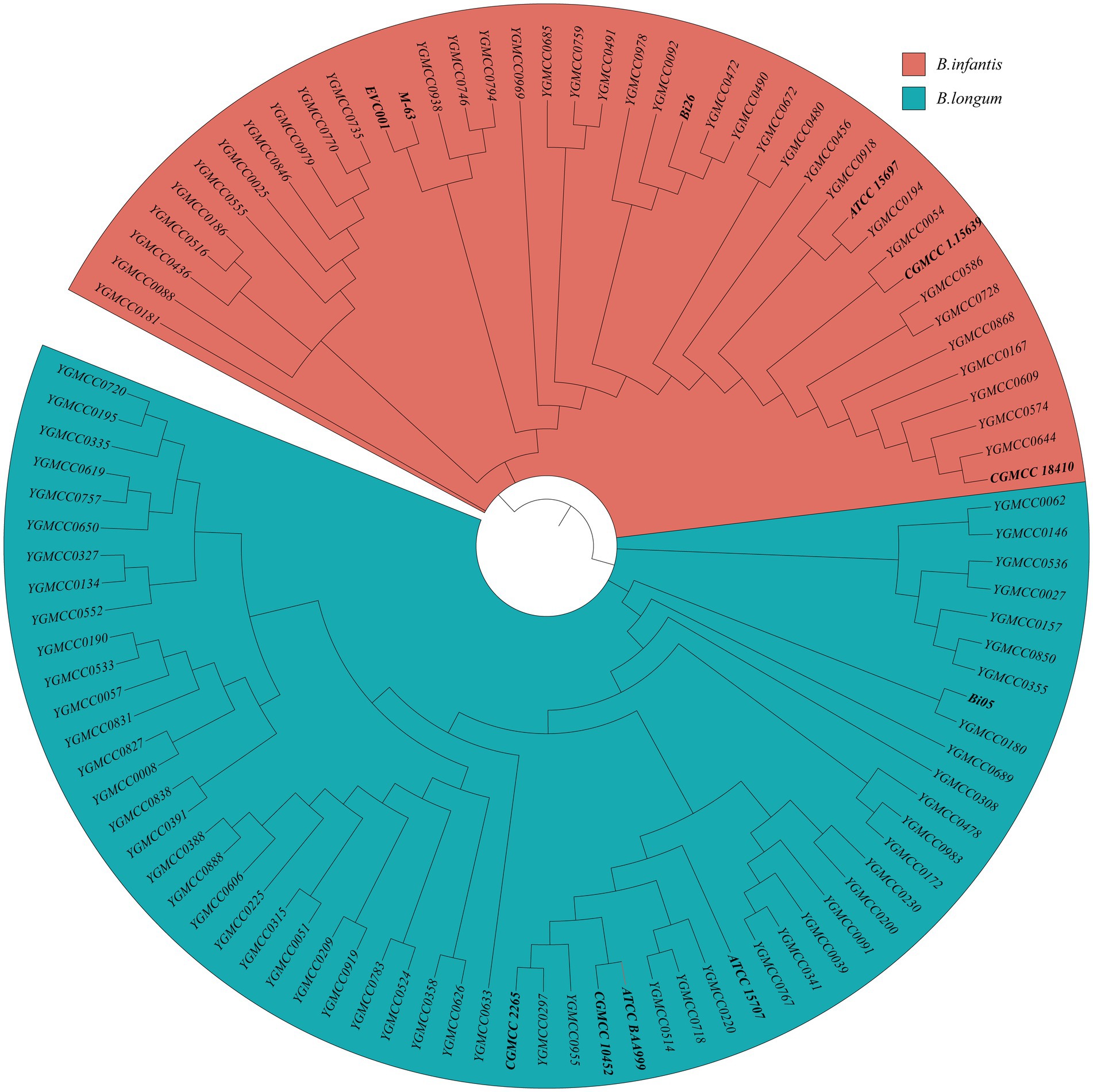
Figure 2. Identification of 41 B. infantis strains and 59 B. longum strains based on the phylogenetic analysis. The red and blue background represent B. infantis or B. longum strains, respectively.
Mass spectrometry results indicated the presence of numerous identical mass spectral peaks for both B. longum and B. infantis, making accurate differentiation challenging when relying solely on commercial databases (Figure 3A; Supplementary Table S1). However, further analysis unveiled six species-specific peaks that exhibited a high degree of conservation and could serve as potential biomarkers for identification. As shown in Figures 3B–D, peaks at m/z 4448.52 (94.9%, 56/59), 5394.35 (100.0%), and 8789.47 (100.0%) were exclusively found in the spectrogram of B. longum. Conversely, peaks at m/z 4408.42 (95.1%, 39/41), 5381.06, and 8817.28 (100.0%) were observed solely in the spectrogram of B. infantis. These findings reveal the potential of MALDI-TOF MS to differentiate between B. longum and B. infantis based on specific peaks with the protein fingerprint profile.
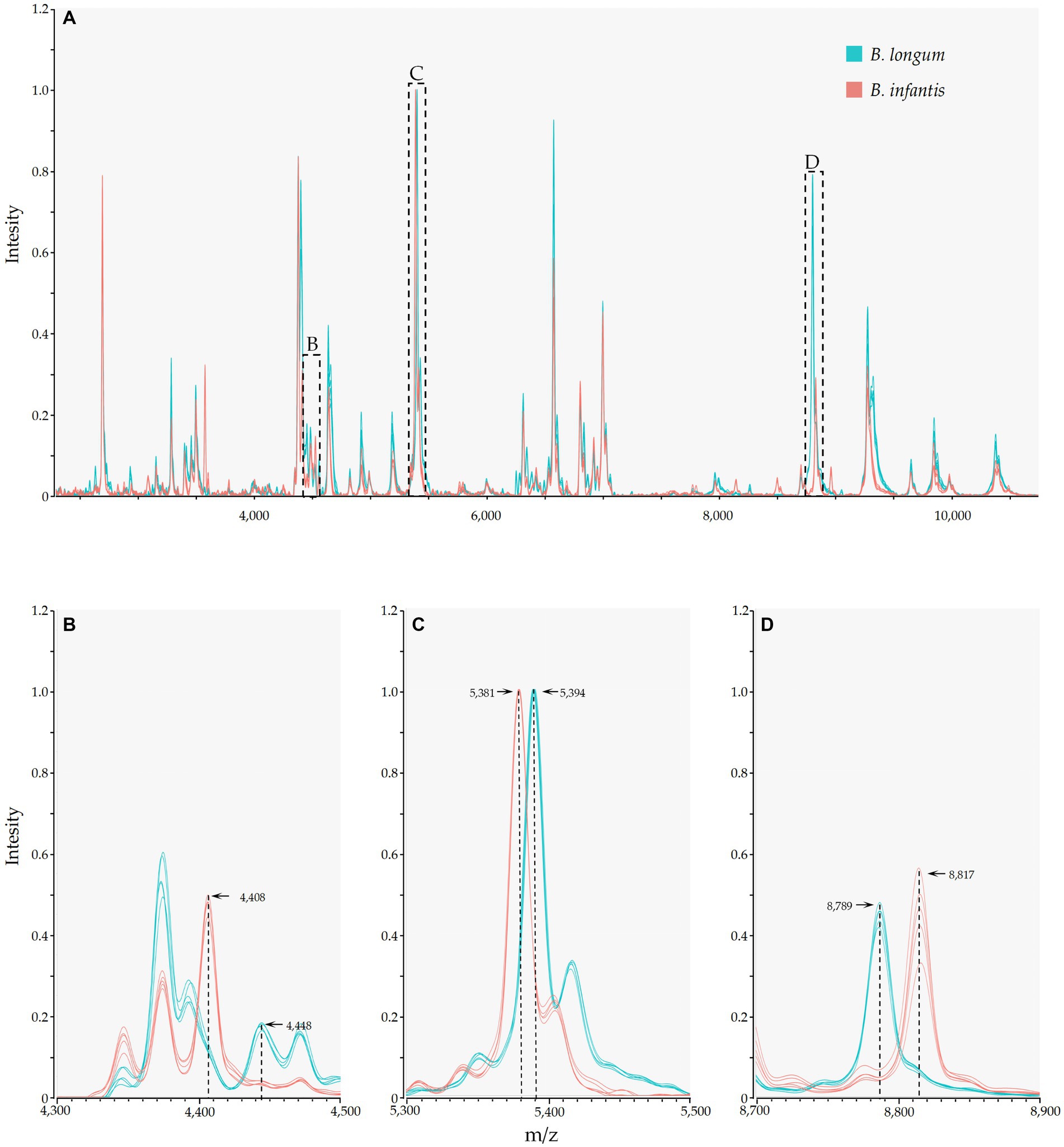
Figure 3. MALDI-TOF MS and species-specific peaks of B. longum (Orange) and B. infantis (Purple). The y-axis represents the intensity of the peaks, while the x-axis represents the m/z values; (A) depicts stowage diagram of B. longum and B. infantis; (B−D) display enlarged views of subspecies-specific peaks as depicted in A.
To investigate the applicability of MALDI-TOF MS for discriminating B. longum and B. infantis, we performed redundant removal, smoothing, and alignment of 400 spectra from 100 strains using OpenMS software. We identified some potential characteristic peaks and constructed a mass spectrometry data matrix for further analysis. To further investigate the distinguishing features, we performed a more specific heatmap clustering analysis of the mass spectrometry data matrix (Figure 4A). Then we performed the principal component analysis (PCA) of the mass spectrum data matrix obtained from the above method. The PCA plot clearly showed the distinct clustering patterns of the two subspecies (Figure 4B), indicating their potential for differentiation. Finally, 18 potential discriminatory peaks were identified, with 11 peaks specific to the B. infantis, including the 3,088 m/z, 3,573 m/z, 4,408 m/z, 5,338 m/z, 5,381 m/z, 6,820 m/z, 6,910 m/z, 8,131 m/z, 8,817 m/z, 9,963 m/z, 10,360 m/z. B. longum with seven specific peaks, respectively, are located at the 2,929 m/z, 3,152 m/z, 4,448 m/z, 4,479 m/z, 5,394 m/z, 7,051 m/z, 8,789 m/z. These discriminatory peaks are expected to serve as potential features for constructing the classifiers. Furthermore, to assess the importance of features, we analyzed between 18 feature peaks and drew bar graphs (Figure 4C) and found higher SHAP values for feature peaks at 4408 m/z, 5,381 m/z, 5,394 m/z and 8,817 m/z. This suggests that these peaks seem particularly well suited for building classifiers.
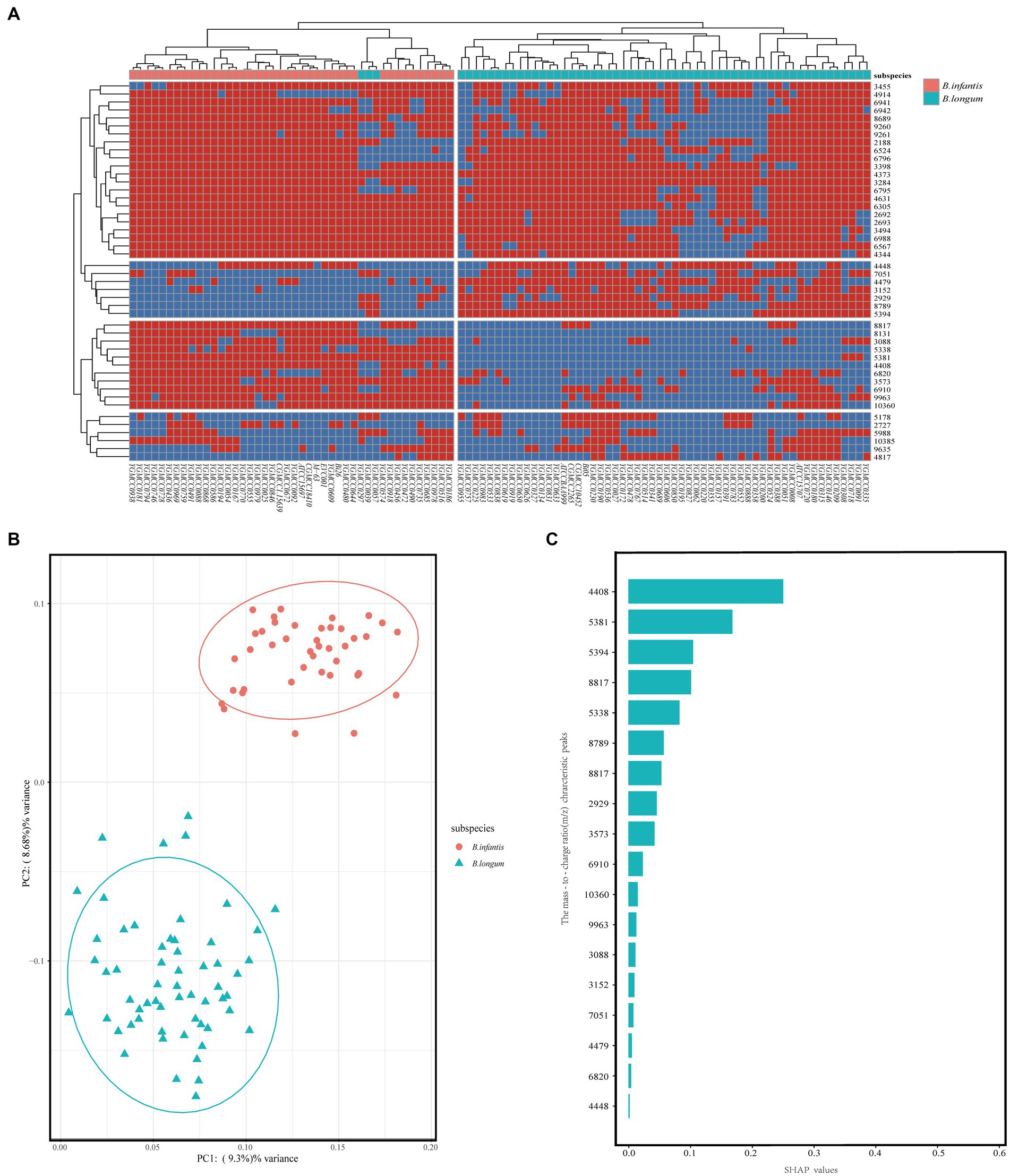
Figure 4. Unsupervised analysis and feature importance evaluation. (A) After mass spectral alignment, heat maps were plotted and clustered based on the absence/presence of common characteristic peaks in the top 50% of effective p values within subspecies. (B) PCA of Bifidobacterium longum subsp. Each dot on the PCA plot represents the average spectrum of each strain, blue for B. longum, and red for B. infantis. (C) Assessment of feature importance in a RF model for distinguishing between B. longum and B. infantis.
To gain insights into the identities of these characteristic peaks, we conducted a comparison between the experimental m/z values and genomic data. This analysis suggested that the ion peaks at m/z 5,381 and 5,394 corresponded to the 50S ribosomal protein L34. Additionally, peaks at m/z 8,817 and 7,051 were associated with 50S ribosomal proteins L27 and L30, respectively. The peak at 4408 m/z indicated the presence of the 30S ribosomal protein S5. Moreover, we identified matches with proteins from the DUF (domain of unknown function) family, including m/z 4,479, 8,789, and 9,963. Proteins belonging to the DUF family are characterized by a conserved EYA motif and a length ranging from 66 to 95 amino acids. However, their functional roles remain elusive due to the lack of annotation.
The 18 feature peaks obtained above were conducted recursive feature elimination using a logistic regression algorithm with cross-validation to determine the optimal feature set. Figure 5A illustrated that the highest cross-validation score of 0.945 was achieved when using five features. These five optimal features include m/z 2,929, 4,408, 5,381, 5,394, and 8,817. Among them, m/z 2,929 and 5,394 were characteristic peaks of B. longum, while the remaining peaks were specific to B. infantis. The significance of the five selected features was presented using a boxplot (Figure 5B), and the results indicated that the p-values of the five features, as determined by Fisher’s exact test, were all less than 0.001. In addition, individual ROC curves were plotted for the five selected features (Figure 5C). The AUC values ranged from 0.777 for m/z 2,929 to 0.917 for m/z 5,381. It indicates that the features obtained after recursive elimination can contribute to achieving the best classification performance.

Figure 5. Recursive feature elimination. Line plot of 18 characteristic peaks and cross-validation fractions after REFCV (A), and boxplot of mass-to-charge ratio and relative intensity of 5 optimal characteristic peaks between the two subspecies of Bifidobacterium longum (**** represents the p value of the difference < 0.0001). (B). ROC curve and AUC value of the five optimal characteristic peaks (C).
We developed three commonly used machine learning models: LR, SVM, and RF, for microbial discrimination. The dataset utilized for model construction consisted of 100 strains, with their subspecies verified through PCR and phylogenetic analysis. This dataset was randomly divided into a training set for building the models and a test set for evaluation their performance. Based on the results obtained from the test set, we calculated performance metrics such as sensitivity, specificity, accuracy, Youden coefficient, and AUC value (see Table 1).
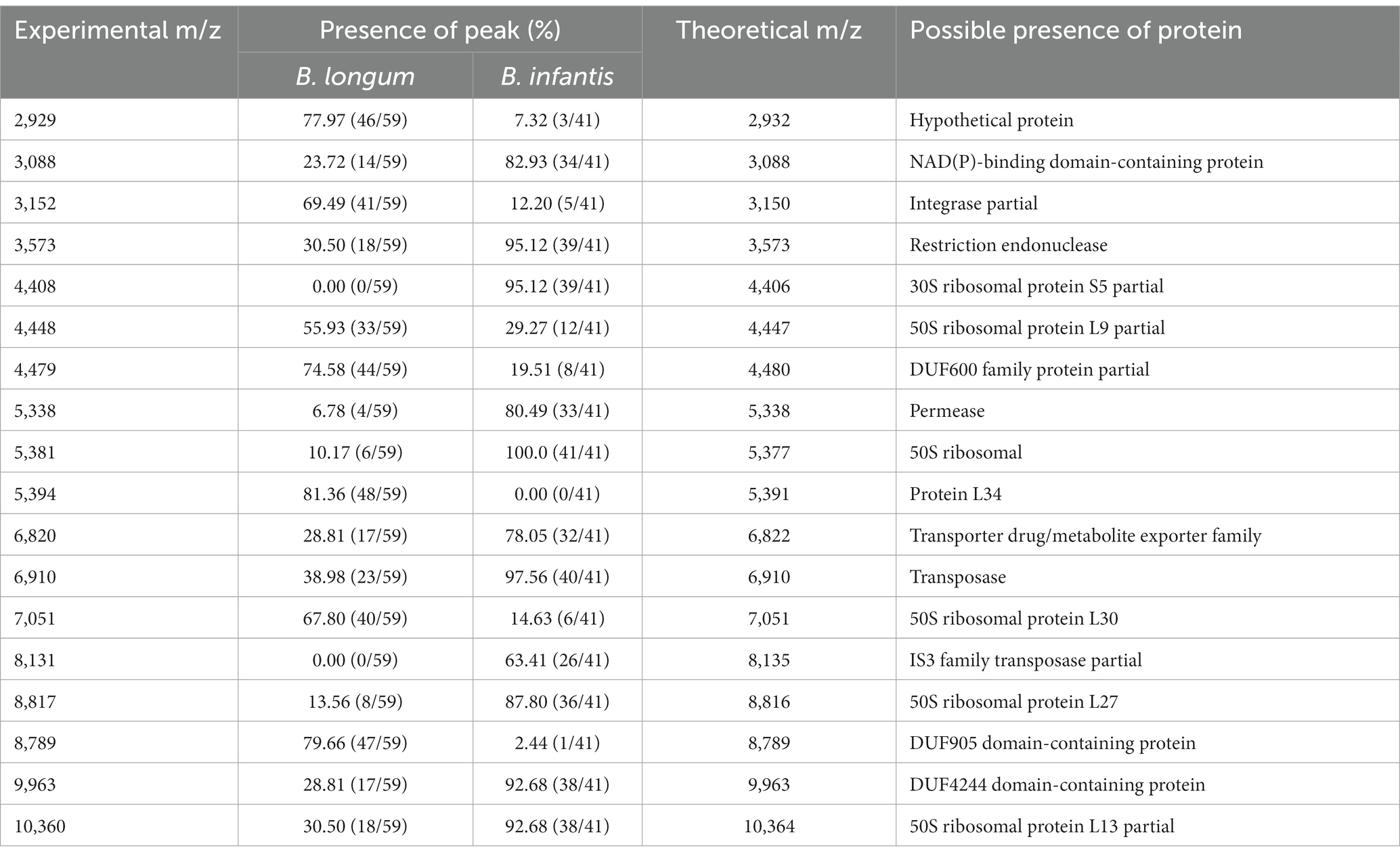
Table 1. Frequencies and assignments of species-specific peaks for B. longum and B. infantis.
The classification performance parameters of the three models are shown in Table 2. Among them, RF achieved the highest accuracy, AUC, and Youden coefficient, all equal to 1.0, indicating its superior ability to discriminate between the two subspecies. The sensitivity of all three models was 1.0, which means that they could correctly identify all the positive cases. The RF model demonstrated the highest specificity with a value of 1.0, whereas the LR and SVM models exhibited a specificity of 0.931. The RF model also has the highest AUC value of 1.0, demonstrating excellent classification performance. The SVM model’s AUC was slightly better than that of the LR model, with values of 0.995, and 0.993, respectively. The Youden coefficient, reflecting the overall efficiency of the RF model, was 1.0, while for the SVM and LR models, it was 0.931.
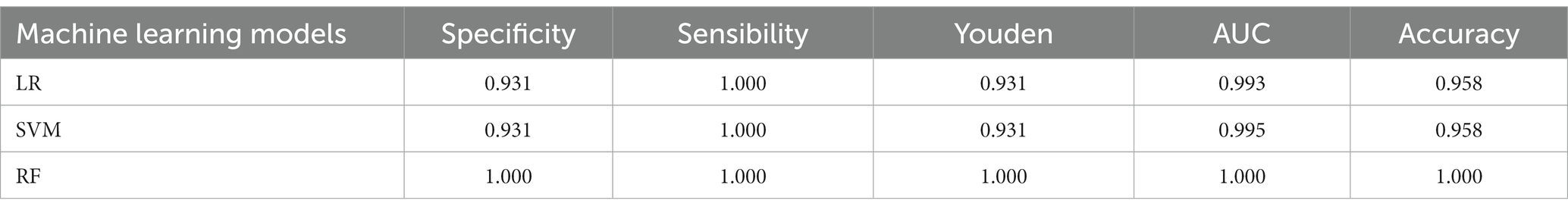
Table 2. Model result metrics for three machine learning models in validation dataset.
An external dataset comprising 240 spectra obtained from 60 Bifidobacterium longum strains was collected. These isolates were obtained under identical experimental conditions. To validate the model’s effectiveness, the three trained models were utilized to predict the subspecies of these 60 strains.
Among the three models, both LR and SVM model exhibited a specificity of 0.983, while it was 0.967 for the RF model. However, the LR model demonstrated a higher sensitivity (0.942) compared to the SVM model (0.883) and the RF model (0.900). Regarding accuracy, the RF model outperformed the SVM model and the LR model, achieving an accuracy rate of 0.954. To provide a more intuitive comparison of the models performance, we plotted the ROC curve (Figure 6A) and calculate the AUC values. All three models exhibited very similar AUC values, accurately measured at 0.984. The RF model had the highest Youden index (0.908), surpassing the SVM model (0.867) and the LR model (0.883). Figure 6B illustrated the distribution of prediction scores indicating the likelihood of being B. infantis strains for the two subspecies, as determined by the three models.
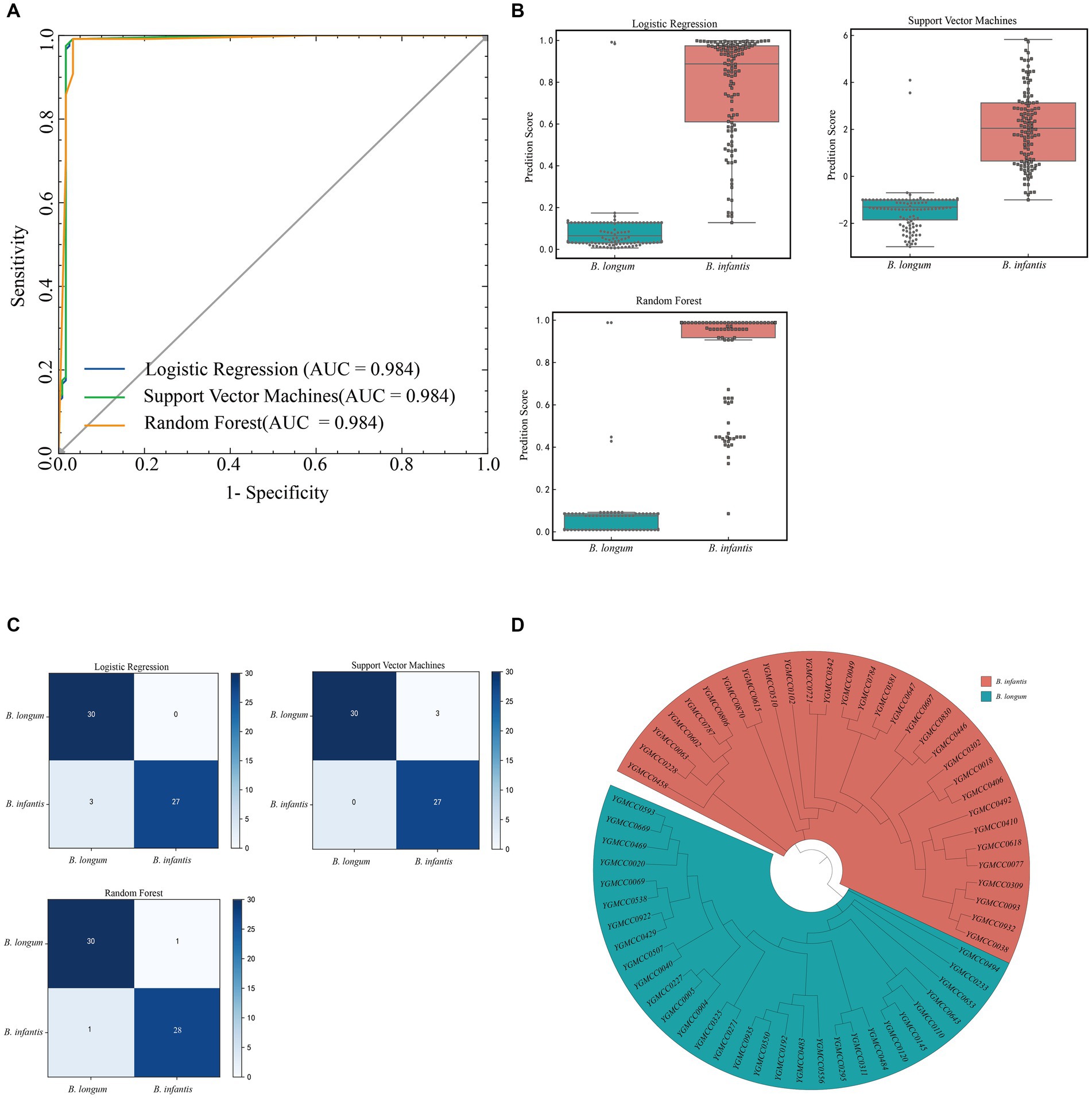
Figure 6. Model evaluation in test dataset. (A) ROC curves and AUC of three machine learning models in the test dataset. (B) Boxplot of the verification score of the three machine learning models on the spectral data of the external test set. (C) Confusion matrix of external strain identification results for three models. (D) Cluster analysis of isolated B. infantis or B. longum strains used for external validation set based on phylogenetic analysis. The red and blue background represnt B. infantis or B. longum strains, respectively.
Based on the four data points results, we established the prediction conditions for the strain subspecies model. A confusion matrix for external strain identification was calculated based on the voting results (Figure 6C). Specific PCR test results and phylogenetic analysis results (Figure 6D) showed consistency. The results from specific PCR tests and phylogenetic analysis (Figure 6D) were consistent with these findings (Supplementary Table S1). Among them, in the LR model, the identification of B. longum was in line with PCR and phylogenetic results. However, for the B. infantis, specifically YGMCC0271, YGMCC0192, and YGMCC0550, there was inconsistency, with an accuracy of 95%. In the SVM model, the identification of YGMCC0618, YGMCC0063, and YGMCC0038 did not align with PCR and phylogenetic results, resulting in an accuracy rate of 95%. Lastly, in the RF model, the identification of YGMCC0063 and YGMCC0120 differed from PCR and SNP results, achieving an accuracy rate of 96.67%. Based on the external strain identification results, the RF model emerged as the optimal choice.
Genome-based taxonomy is a more standard method of classifying microorganisms than traditional methods (Parks et al., 2018). However, it is time-consuming, expensive, and labor-intensive, and fails to meet the demand for rapid and high-throughput identification of microorganisms. In recent years, MALDI-TOF MS has gained increasing importance in clinical microbial taxonomy as a fast, high-throughput, and robust method for microbial identification. It relies on the detection of microbial housekeeping and ribosomal proteins (Kim et al., 2022a; Haider et al., 2023). Nonetheless, while MALDI-TOF MS can identify bacteria at the species level, it struggles to accurately distinguish closely related species or subspecies. Machine learning algorithms have the capability to identify specific information in mass spectrometry data and analyze relationships among different features, enabling more precise analysis (Weis et al., 2020). By combining machine learning with MALDI-TOF MS, it becomes possible to accurately identify closely related microorganisms at the subspecies level (De Bruyne et al., 2011; Rodríguez-Temporal et al., 2023). Recent studies have demonstrated the application of machine learning techniques in overcoming the limitations of mass spectrometry, such as detecting antibiotic-resistant microorganisms (Yoon and Jeong, 2021), analyzing antimicrobial resistance (Feucherolles et al., 2021), and distinguishing closely related species. By utilizing features obtained from MALDI-TOF MS, SVM algorithms have successfully differentiated clinically resistant strains of carbapenem, methicillin, and β-lactam antibiotics, as well as predicted resistance phenotypes with high accuracy (Ho et al., 2017; Wang J. et al., 2022). Furthermore, the combination of MALDI-TOF MS and machine learning is commonly used to distinguish closely related foodborne microorganisms. For example, an SVM-RBF model achieved a prediction accuracy of approximately 100% in accurately identifying W. cibaria and W. confusa (Kim et al., 2023).
In our research, we have found that distinguishing closely related species using MALDI-TOF MS can be challenging due to the similarities in their protein fingerprints. MALDI-TOF MS generates a report of the ten closest matches for an unknown species based on mass spectra and the consistency of reference strains in the database. However, when different species within the same genus or different subspecies within the same species have high scores among the top ten matches, accurately identifying the microorganism becomes difficult. Previous studies have attempted to distinguish between Bifidobacterium longum subspecies (Kim et al., 2022b) and Bifidobacterium animalis subspecies (Jahan et al., 2021) using MALDI-TOF MS. However, these studies had limitations in terms of sample size, unsystematic markers, and lack of validation data, and have not been commercially applied. In this study, our focus was specifically on identifying B. longum and B. infantis using MALDI-TOF MS. We discovered that commercial databases were unable to accurately differentiate between these two subspecies, which aligns with previous findings (Yahiaoui et al., 2020; Jahan et al., 2021; Kim et al., 2022b).
The aim of this study was to evaluate the ability of MALDI-TOF MS combined with machine-learning methods to rapidly and accurately discriminate between the closely related B. longum and B. infantis. We employed advanced machine learning algorithms and a larger sample size to enhance statistical significance. We ensured systematic biomarker collection and data analysis to improve the reliability and repeatability of our findings. We examined 400 mass spectra from 100 Bifidobacterium longum strains and used a logistic regression model with recursive feature elimination to identify the five most significant mass peaks. Among these peaks, the masses at 2929 and 5,394 m/z were specific to B. longum, while the masses at 4408, 5381, and 8,817 m/z were specific to B. infantis. These mass peaks can potentially serve as biomarkers for distinguishing between these two species. Using these biomarkers, we developed machine learning models employing LR, RF, and SVM algorithms. All three models exhibited excellent performance in identifying the spectrogram, with the RF model demonstrating high accuracy in discriminating between B. longum and B. infantis. Furthermore, after evaluating mass spectrum identification results through voting, the RF model achieved the highest accuracy in practical strain identification applications (see Table 3).
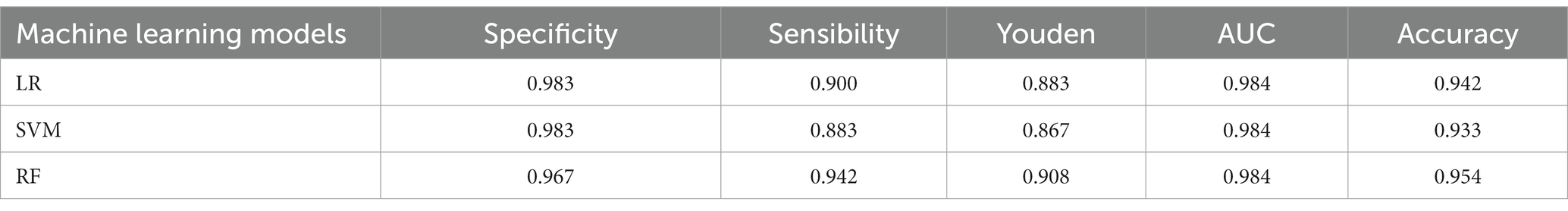
Table 3. Model result parameters for three machine learning models on an test dataset.
Twelve reference strains and eighty-nine strains of Bifidobacterium longum subspecies, isolated at Beijing Yujing Pharmaceutical Co., Ltd., were selected to explore potential biomarkers (Table 4). The bifidobacterial strains were incubated for 48 h at 37°C under anaerobic conditions. E. coli ATCC 25922 incubated for 24 h at 37°C in Luria-Bertani (Solarbio, Beijing, China) agar was applied to external calibration of MALDI-TOF MS.
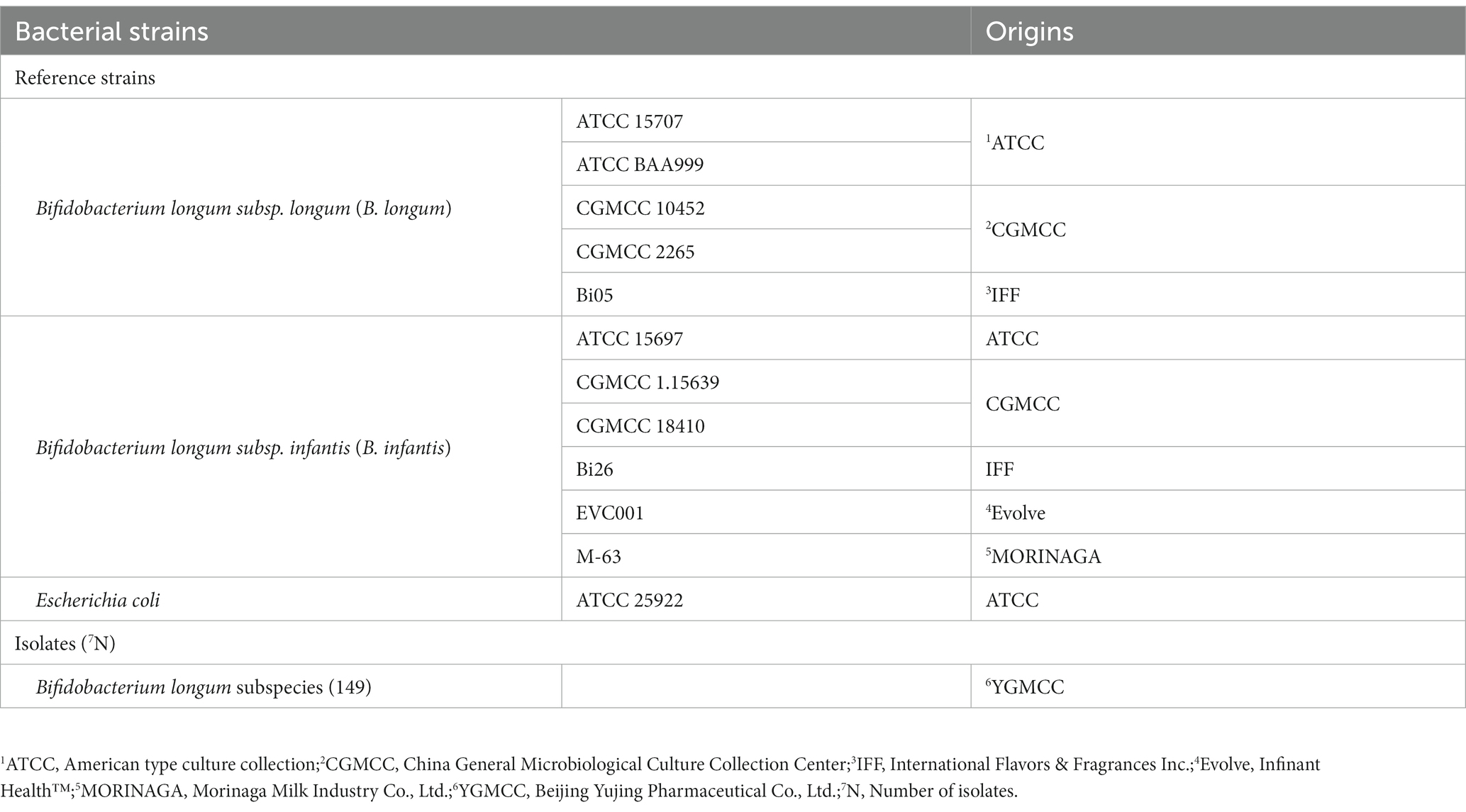
Table 4. Strain information used in this study.
Proteins from B. longum and B. infantis were extracted using the ethanol-formic acid-extraction method (Cuénod et al., 2023). Concisely, fresh bacterial culture was suspended in 300 μL of ddH2O to which 900 μL of ethanol was added. The bacterial suspension was centrifuged at high speed (10,000× g) for 2 min, the supernatant was removed to completely discard the residual ethanol and recentrifuged. The resulting pellet was resuspended in 20 μL of 70% formic acid to which an equal volume of acetonitrile was added. After centrifugation at 10,000× g for 2 min, 1 μL of each supernatant was transferred to the 96-position MALDI-TOF target plate, allowed to air dry, and then overlaid with 1 μL of the matrix solution (10 mg/mL of α-cyano-4-hydroxy-cinnamic acid (HCCA) in 50% (v/v) acetonitrile with 2.5% (v/v) trifluoroacetic acid).
The mass spectra were acquired using an EXS2000 MALDI-TOF MS (Zybio Inc., Chongqing, China) equipped with a 200 Hz smart-beam solid-state laser and operated in positive linear mode (Xiong et al., 2023). Mass spectra were automatically recorded within a mass range of 2–20 kDa with a total of 200 laser shots. E. coli ATCC 25922 was used for mass calibration and instrument parameter optimization, with an average deviation of molecular weight less than 300 ppm after correction. MS data were analyzed using MDT Master (version 1.1). log scores ≥2.0 were accepted for the identification at the species level, and log scores <2.0 and ≥ 1.7 were used for identification at the genus level or the presumptive species level. Log scores below 1.7 were considered unreliable. For establishing stable machine learning models, four high-quality mass spectra (log scores ≥2.3, stable benchmarks, abundant protein peaks, and uniform distribution) were selected in each strain.
For the identification of the isolates, genomic DNA was extracted using Easy Pure Bacteria Genomic DNA Kit (Trans, Beijing, China) in accordance with the manufacturer’s instructions. Then, 1 μL of supernatant was used for the following PCR reaction, the reaction mixture contained 10 μL of SapphireAmp® Fast PCR Master Mix (TaKaRa, Beijing, China), 0.5 μL of each primer (10 μM), 1 μL of DNA template, and 8 μL of ddH2O. Specific primers were listed in Table 5. PCR reactions were conducted as follows: one cycle of initial denaturation at 98°C for 3 min, followed by 35 cycles of 98°C for 10 s, 55°C for 10 s, and 72°C for 5 s, and a final extension at 72°C for 2 min. The PCR products were observed by an Agarose gel imaging system (Tanon, Shanghai, China).

Table 5. Specific primer information used in this study.
Total 149 unknown Bifidobacterium longum strains were cultured anaerobically at 37°C for 24 h, then the cultured liquid (50 mL) was centrifuged at 12,000 × g and 4°C for 10 min to collect the cell biomass. Genomic DNA of 149 unknown Bifidobacterium longum strains were extracted using a Wizard® Genomic DNA Purification Kit (Promega, United States). Purified genomic DNA was quantified using a TBS-380 fluorometer (Turner BioSystems Inc., Sunnyvale, CA, United States). High-quality DNA (OD260/280 = 1.8–2.0, ≥10 μg) was used for further research. Genomic DNA was sequenced using Illumina sequencing (Illumina, Inc.). The data generated from Illumina platforms were used for bioinformatics analysis.
The phylogenetic analysis included the comparison of genomic sequences from 5 standard strains of B. infantis, 6 standard strains of B. longum, and an additional 149 unknown B. longum strains from our laboratory. These sequences were compared with the genomic sequence of ASM19655v1, which served as the reference genome. The analysis was performed using the Parsnp software, focusing on the core genome (Treangen et al., 2014; Wang et al., 2023). The iTOL (Interactive Tree of Life) tool was utilized to visualize and explore the phylogenetic tree (Letunic and Bork, 2019; Pereira et al., 2023), facilitating the identification and classification of B. longum subspecies based on their phylogenetic positions.
To investigate the significance of using unique peaks from mass spectrum data as biomarkers, we conducted genomic data mining using publicly available databases. The genome sequences of B. longum and B. infantis were obtained from the National Center for Biotechnology Information (NCBI) database. To annotate the selected protein biomarkers, the web-based ProtParam tool1 was utilized to calculate their theoretical molecular weights based on the translated amino acid sequences. Subsequently, a custom script was employed to filter and align the selected proteins, identifying the most relevant proteins enriched in the vicinity of the characteristic peaks.
The MS data obtained using openMS (v2.8) software exhibited high quality, allowing for alignment of peaks obtained from different batches. The processed peak map data matrix was subjected to PCA to access the potential of the features. In addition, a heatmap was drawn for cluster analysis using the R language (v4.2.2). After obtaining the cluster branches of the potential feature peaks, the importance parameters of the features and evaluate the importance of the features.
The dataset consisting of 400 spectra from 59 B. longum and 41 B. infantis was randomly divided into 70% training and 30% test datasets. The data of subspecies type was binarized, with 0 representing the long subspecies and 1 representing the infant subspecies. All peaks (features) were scaled using Min-Max scalar to ensure variables at different scales contributed equally to the model fitting process.
Firstly, feature selection was carried by a meta-converter approach based on a logistic regression classifier with scikit-learn (v1.3.0). Recursive feature elimination with 5x cross-validation (RFECV) was applied to discard irrelevant features and improve the model’s generalization ability.
Secondly, SHAP (SHapley Additive exPlanations) was used to interpret predictions. SHAP is a unified framework that assigns importance values to each feature for a specific prediction and identifies which feature is most important, facilitating the understanding of a machine learning model’s decision-making process (Lundberg and Lee, 2017).
Thirdly, three machine learning algorithms including random forest (RF), logistic regression (LR), and support vector machine (SVM) were used to construct the distinguishing models using the scikit-learn package. The performances of the models were evaluated by generating the confusion matrix on the test dataset. The ROC curve was plotted using the Matplotlib package, and the area under the subject operating characteristic curve (AUROC) was calculated as a measure of classifier performance. The Youden index was utilized to determine the optimal cutoff threshold and calculate the sensitivity, specificity, and accuracy metrics for the model.
To assess the practical applicability of the model in strain identification, we performed an external validation using a new dataset. Each strain in this dataset was accompanied by four mass spectra collected under identical experimental conditions. Subsequently, we compared the identification outcomes with those obtained through specific PCR detection and phylogenetic analysis.
In our research, we successfully demonstrated the effectiveness of combining MALDI-TOF-MS with machine learning to accurately discriminate between B. longum and B. infantis. We identified everything from protein fingerprints to potential biomarkers, and developed three spectral map identification models using the ML algorithm, and finally evaluated the various performance metrics and voted to find the optimal algorithm. The algorithm is highly reliable and accurate in distinguishing the two subspecies. This approach has the potential to be applied in various industries, such as the food or pharmaceutical industry, for rapid and cost-effective identification of B. longum and B. infantis. Furthermore, the identification strategy presented in this study can also be extended to other closely related species.
The names of the repository/repositories and accession number(s) can be found below: NCBI; PRJNA1020989.
KL: Investigation, Methodology, Validation, Writing – original draft. YW: Investigation, Methodology, Validation, Writing – original draft. MZ: Data curation, Investigation, Methodology, Validation, Writing – original draft. GX: Software, Validation, Writing – original draft. AW: Data curation, Writing – review & editing. WW: Supervision, Validation, Writing – review & editing. LX: Data curation, Methodology, Supervision, Writing – review & editing. JC: Project administration, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
KL, GX, AW, and LX were employed by Hotgen Biotechnology Inc. MZ and JC were employed by Beijing YuGen Pharmaceutical Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1297451/full#supplementary-material
Carvalho, M., Sands, K., Thomson, K., Portal, E., Mathias, J., Milton, R., et al. (2022). Antibiotic resistance genes in the gut microbiota of mothers and linked neonates with or without sepsis from low- and middle-income countries. Nat. Microbiol. 7, 1337–1347. doi: 10.1038/s41564-022-01184-y
Casaburi, G., Duar, R. M., Brown, H., Mitchell, R. D., Kazi, S., Chew, S., et al. (2021). Metagenomic insights of the infant microbiome community structure and function across multiple sites in the United States. Sci. Rep. 11:1472. doi: 10.1038/s41598-020-80583-9
Cuénod, A., Aerni, M., Bagutti, C., Bayraktar, B., Boz, E. S., Carneiro, C. B., et al. (2023). Quality of MALDI-TOF mass spectra in routine diagnostics: results from an international external quality assessment including 36 laboratories from 12 countries using 47 challenging bacterial strains. Clin. Microbiol. Infect. 29, 190–199. doi: 10.1016/j.cmi.2022.05.017
De Bruyne, K., Slabbinck, B., Waegeman, W., Vauterin, P., De Baets, B., and Vandamme, P. (2011). Bacterial species identification from MALDI-TOF mass spectra through data analysis and machine learning. Syst. Appl. Microbiol. 34, 20–29. doi: 10.1016/j.syapm.2010.11.003
Dematheis, F., Walter, M. C., Lang, D., Antwerpen, M., Scholz, H. C., Pfalzgraf, M.-T., et al. (2022). Machine learning algorithms for classification of MALDI-TOF MS spectra from phylogenetically closely related species Brucella melitensis, Brucella abortus and Brucella suis. Microorganisms 10:8. doi: 10.3390/microorganisms10081658
Duar, R. M., Henrick, B. M., Casaburi, G., and Frese, S. A. (2020). Integrating the ecosystem services framework to define Dysbiosis of the breastfed infant Gut: the role of B. infantis and Human Milk Oligosaccharides. Front. Nutr. 7:33. doi: 10.3389/fnut.2020.00033
Feucherolles, M., Nennig, M., Becker, S. L., Martiny, D., Losch, S., Penny, C., et al. (2021). Combination of MALDI-TOF mass spectrometry and machine learning for rapid antimicrobial resistance screening: the case of Campylobacter spp. Front. Microbiol. 12:804484. doi: 10.3389/fmicb.2021.804484
Gato, E., Constanso, I. P., Candela, A., Galán, F., Rodiño-Janeiro, B. K., Arroyo, M. J., et al. (2021). An improved matrix-assisted laser desorption ionization-time of flight mass spectrometry data analysis pipeline for the identification of Carbapenemase-producing Klebsiella pneumoniae. J. Clin. Microbiol. 59:e0080021. doi: 10.1128/JCM.00800-21
Haider, A., Ringer, M., Kotroczó, Z., Mohácsi-Farkas, C., and Kocsis, T. (2023). The current level of MALDI-TOF MS applications in the detection of microorganisms: a short review of benefits and limitations. Microbiol. Res. 14, 80–90. doi: 10.3390/microbiolres14010008
Heilbronner, S., and Foster, T. J. (2021). Staphylococcus lugdunensis: a skin commensal with invasive pathogenic potential. Clin. Microbiol. Rev. 34:2. doi: 10.1128/CMR.00205-20
Henrick, B. M., Chew, S., Casaburi, G., Brown, H. K., Frese, S. A., Zhou, Y., et al. (2019). Infantis EVC001 modulates enteric inflammation in exclusively breastfed infants. Pediatr. Res. 86, 749–757. doi: 10.1038/s41390-019-0533-2
Henrick, B. M., Rodriguez, L., Lakshmikanth, T., Pou, C., Henckel, E., Arzoomand, A., et al. (2021). Bifidobacteria-mediated immune system imprinting early in life. Cells 184, 3884–3898.e11. doi: 10.1016/j.cell.2021.05.030
Ho, P.-L., Yau, C.-Y., Ho, L.-Y., Chen, J. H. K., Lai, E. L. Y., Lo, S. W. U., et al. (2017). Rapid detection of cfiA metallo-β-lactamase-producing Bacteroides fragilis by the combination of MALDI-TOF MS and CarbaNP. J. Clin. Pathol. 70, 868–873. doi: 10.1136/jclinpath-2017-204335
Jahan, N. A., Godden, S. M., Royster, E., Schoenfuss, T. C., Gebhart, C., Timmerman, J., et al. (2021). Evaluation of the matrix-assisted laser desorption ionization time of flight mass spectrometry (MALDI-TOF MS) system in the detection of mastitis pathogens from bovine milk samples. J. Microbiol. Methods 182:106168. doi: 10.1016/j.mimet.2021.106168
Kim, E., Yang, S.-M., Cho, E.-J., and Kim, H.-Y. (2022b). Evaluation of matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for the discrimination of Lacticaseibacillus species. Food Microbiol. 107:104094. doi: 10.1016/j.fm.2022.104094
Kim, E., Yang, S.-M., Jung, D.-H., and Kim, H.-Y. (2023). Differentiation between Weissella cibaria and Weissella confusa using machine-learning-combined MALDI-TOF MS. Int. J. Mol. Sci. 24:11009. doi: 10.3390/ijms241311009
Kim, E., Yang, S.-M., Kim, H.-J., and Kim, H.-Y. (2022a). Differentiating between Enterococcusfaecium and Enterococcuslactis by matrix-assisted laser desorption ionization time-of-flight mass spectrometry. Foods 11:7. doi: 10.3390/foods11071046
Letunic, I., and Bork, P. (2019). Interactive tree of life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Lundberg, S., and Lee, S. I., A unified approach to interpreting model predictions. 31st Conference on Neural Information Processing Systems (2017), 1–10.
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P.-A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Pereira, C. R., Neia, R. C., Silva, S. B., Williamson, C. H. D., Gillece, J. D., O'Callaghan, D., et al. (2023). Comparison of Brucella abortus population structure based on genotyping methods with different levels of resolution. J. Microbiol. Methods 211:106772. doi: 10.1016/j.mimet.2023.106772
Rodríguez-Temporal, D., Díez, R., Díaz-Navarro, M., Escribano, P., Guinea, J., Muñoz, P., et al. (2022). Determination of the ability of matrix-assisted laser desorption ionization time-of-flight mass spectrometry to identify high-biofilm-producing strains. Front. Microbiol. 13:1104405. doi: 10.3389/fmicb.2022.1104405
Rodríguez-Temporal, D., Herrera, L., Alcaide, F., Domingo, D., Héry-Arnaud, G., van Ingen, J., et al. (2023). Identification of Mycobacterium abscessus subspecies by MALDI-TOF mass spectrometry and machine learning. J. Clin. Microbiol. 61:e0111022. doi: 10.1128/jcm.01110-22
Sato, H., Teramoto, K., Ishii, Y., Watanabe, K., and Benno, Y. (2011). Ribosomal protein profiling by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry for phylogenety-based subspecies resolution of Bifidobacterium longum. Syst. Appl. Microbiol. 34, 76–80. doi: 10.1016/j.syapm.2010.07.003
Topić Popović, N., Kazazić, S., Bojanić, K., Strunjak-Perović, I., and Čož-Rakovac, R. (2023). Sample preparation and culture condition effects on MALDI-TOF MS identification of bacteria: a review. Mass. Spectrom. Rev. 42, 1589–1603. doi: 10.1002/mas.21739
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
van Oosten, L. N., and Klein, C. D. (2020). Machine learning in mass spectrometry: a MALDI-TOF MS approach to phenotypic antibacterial screening. J. Med. Chem. 63, 8849–8856. doi: 10.1021/acs.jmedchem.0c00040
Vatanen, T., Ang, Q. Y., Siegwald, L., Sarker, S. A., Le Roy, C. I., Duboux, S., et al. (2022). A distinct clade of Bifidobacterium longum in the gut of Bangladeshi children thrives during weaning. Cells 185, 4280–4297.e12. doi: 10.1016/j.cell.2022.10.011
Wang, H. Y., Kuo, C. H., Chung, C. R., Lin, W. Y., Wang, Y. C., Lin, T. W., et al. (2022). Rapid and accurate discrimination of Mycobacterium abscessus subspecies based on matrix-assisted laser desorption ionization-time of flight Spectrum and machine learning algorithms. Biomedicine 11:45. doi: 10.3390/biomedicines11010045
Wang, J., Xia, C., Wu, Y., Tian, X., Zhang, K., and Wang, Z. (2022). Rapid detection of Carbapenem-resistant Klebsiella pneumoniae using machine learning and MALDI-TOF MS platform. Infect. Drug Resist. 15, 3703–3710. doi: 10.2147/IDR.S367209
Wang, Y. Y., Xie, L., Zhang, W. Z., Du, X. L., Li, W. G., Bia, L. L., et al. (2023). Application of a core genome sequence typing (cgMLST) pipeline for surveillance of Clostridioides difficile in China. Front. Cell. Infect. Microbiol. 13:1109153. doi: 10.3389/fcimb.2023.1109153
Weis, C., Cuénod, A., Rieck, B., Dubuis, O., Graf, S., Lang, C., et al. (2022). Direct antimicrobial resistance prediction from clinical MALDI-TOF mass spectra using machine learning. Nat. Med. 28, 164–174. doi: 10.1038/s41591-021-01619-9
Weis, C. V., Jutzeler, C. R., and Borgwardt, K. (2020). Machine learning for microbial identification and antimicrobial susceptibility testing on MALDI-TOF mass spectra: a systematic review. Clin. Microbiol. Infect 26, 1310–1317. doi: 10.1016/j.cmi.2020.03.014
Xiong, L., Long, X., Ni, L., Wang, L., Zhang, Y., Cui, L., et al. (2023). Comparison of autof Ms1000 and EXS3000 MALDI-TOF MS platforms for routine identification of microorganisms. Infect. Drug Resist. 16, 913–921. doi: 10.2147/IDR.S352307
Yahiaoui, R. Y., Goessens, W. H., Stobberingh, E. E., and Verbon, A. (2020). Differentiation between Streptococcus pneumoniae and other viridans group streptococci by matrix-assisted laser desorption/ionization time of flight mass spectrometry. Clin. Microbiol. Infect. 26, 1088.e1–1088.e5. doi: 10.1016/j.cmi.2019.11.024
Yoon, E.-J., and Jeong, S. H. (2021). MALDI-TOF mass spectrometry technology as a tool for the rapid diagnosis of antimicrobial resistance in Bacteria. Antibiotics (Basel) 10:982. doi: 10.3390/antibiotics10080982
Yu, J., Lin, Y.-T., Chen, W.-C., Tseng, K.-H., Lin, H.-H., Tien, N., et al. (2023). Direct prediction of carbapenem-resistant, carbapenemase-producing, and colistin-resistant Klebsiella pneumoniae isolates from routine MALDI-TOF mass spectra using machine learning and outcome evaluation. Int. J. Antimicrob. Agents 61:106799. doi: 10.1016/j.ijantimicag.2023.106799
Keywords: Bifidobacterium longum subspecies, MALDI-TOF MS, machine learning, identification, B. longum , B. infantis
Citation: Liu K, Wang Y, Zhao M, Xue G, Wang A, Wang W, Xu L and Chen J (2023) Rapid discrimination of Bifidobacterium longum subspecies based on MALDI-TOF MS and machine learning. Front. Microbiol. 14:1297451. doi: 10.3389/fmicb.2023.1297451
Edited by:
Aldert Zomer, Utrecht University, NetherlandsReviewed by:
Antonella Lupetti, University of Pisa, ItalyCopyright © 2023 Liu, Wang, Zhao, Xue, Wang, Wang, Xu and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianguo Chen, Y2lqMTIzQDEyNi5jb20=; Weijie Wang, d2Vpamlld2FuZ0BuY3N0LmVkdS5jbg==; Lida Xu, bGlkYS54dUBob3RnZW4uY29tLmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.