 Electra Sofou1†
Electra Sofou1† Glykeria Gkoliou1,2†
Glykeria Gkoliou1,2† Nikolaos Pechlivanis1,3
Nikolaos Pechlivanis1,3 Konstantinos Pasentsis1Kimon Chatzistamatiou4
Konstantinos Pasentsis1Kimon Chatzistamatiou4 Fotis Psomopoulos1
Fotis Psomopoulos1 Theodoros Agorastos5‡
Theodoros Agorastos5‡ Kostas Stamatopoulos1,6*‡
Kostas Stamatopoulos1,6*‡- 1Institute of Applied Biosciences, Centre for Research and Technology Hellas, Thessaloniki, Greece
- 2Department of Molecular Biology and Genetics, Democritus University of Thrace, Alexandroupoli, Greece
- 3Department of Genetics, Development and Molecular Biology, Faculty of Biology, Aristotle University of Thessaloniki, Thessaloniki, Greece
- 41st Department of Obstetrics and Gynecology, Aristotle University of Thessaloniki, Papageorgiou General Hospital, Thessaloniki, Greece
- 5St. Luke’s Hospital, Thessaloniki, Greece
- 6Department of Molecular Medicine and Surgery, Karolinska Institute, Stockholm, Sweden
Increasing evidence supports a role for the vaginal microbiome (VM) in the severity of HPV infection and its potential link to cervical intraepithelial neoplasia. However, a lot remains unclear regarding the precise role of certain bacteria in the context of HPV positivity and persistence of infection. Here, using next generation sequencing (NGS), we comprehensively profiled the VM in a series of 877 women who tested positive for at least one high risk HPV (hrHPV) type with the COBAS® 4,800 assay, after self-collection of a cervico-vaginal sample. Starting from gDNA, we PCR amplified the V3–V4 region of the bacterial 16S rRNA gene and applied a paired-end NGS protocol (Illumina). We report significant differences in the abundance of certain bacteria compared among different HPV-types, more particularly concerning species assigned to Lacticaseibacillus, Megasphaera and Sneathia genera. Especially for Lacticaseibacillus, we observed significant depletion in the case of HPV16, HPV18 versus hrHPVother. Overall, our results suggest that the presence or absence of specific cervicovaginal microbial genera may be linked to the observed severity in hrHPV infection, particularly in the case of HPV16, 18 types.
Introduction
Infection with the human papillomavirus (HPV) is one of the most common sexually transmitted infections (STIs) worldwide (Schiffman et al., 2016). Although the vast majority of infections resolve without disease progression, a smaller -yet significant- number of infected individuals develop HPV-associated cancer. In fact, HPV accounts for about 600,000 new cancer cases annually and is considered as the main cause of invasive cervical cancer (ICC); moreover, it is implicated in many other cancers, including oropharyngeal cancers (OPCs) and anal cancer (Chaturvedi et al., 2011; Forman et al., 2012; Roden and Stern, 2018). Several genotypes of HPV have been associated with high-risk infections, i.e., 16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, and 59 (Reid et al., 1982; Muñoz et al., 2003). HPV16 and 18 are the most common carcinogenic (high-risk) HPV types involved in cervical cancer pathogenesis (Reid et al., 1982; Muñoz et al., 2004), responsible for low-grade squamous intraepithelial lesions (LSIL), high-grade squamous intraepithelial lesions (HSIL), adeno- and squamous cell carcinoma (SCC) (Lazare et al., 2019). Collectively, HPV16 and HPV18 infections account for ~70% of cervical cancer cases worldwide (Pimple and Mishra, 2022).
An increasing number of studies support the significant role of the vaginal and the cervical microbiome in HPV infection persistence and the formation of precancerous lesions in the cervix (Cervical Intraeptithelial Neoplasia, CIN) (Mirabello et al., 2016; Cheng et al., 2020; Lin et al., 2022). The healthy, cervicovaginal microbial flora is normally dominated by Lactobacillus spp., a genus known for its key role in the maintenance of the cervicovaginal eubiosis by producing bacteriocins, lactic acid and hydrogen peroxide, exerting a protective effect on the cervix/vagina from pathogen infections (Ravel et al., 2011; Lebeau et al., 2022). However, HPV infections can disturb the balance of vaginal and cervical microenvironment, leading to abnormal changes in microbiota, including Lactobacillus spp. depletion and predominance of Lactobacillus iners and anaerobic bacteria (such as Gardnerella, Prevotella, Sneathia, Megasphaera) (Lee et al., 2013; Bik et al., 2019; Borgogna et al., 2020; Caselli et al., 2020; Usyk et al., 2020; Wei et al., 2021; Lebeau et al., 2022). These bacteria may destroy the cervical epithelial barrier and promote HPV infection by producing enzymes and metabolites which act on several cellular pathways that enable persistence of viral infection and disease progression (Li et al., 2012).
Studies have demonstrated that abnormal changes in the composition of the vaginal microbiome, which lead to reduced mucus production and consequent clearance of pathogenic bacteria, can be associated with the late clearance of HPV infection and progression to CIN (Watts et al., 2005; Brotman et al., 2014; Audirac-Chalifour et al., 2016; Lin et al., 2022). Furthermore, the development of squamous intraepithelial lesions (SIL) has been associated with an increased vaginal microbial diversity (Curty et al., 2020; Lin et al., 2022). Altogether, all these findings allude to a significant role of the cervicovaginal microbiome in HPV-associated CIN. However, to-date there are no direct links of specific genera/species to the infection by specific HPV types or to the development of CIN.
Here, we investigated the vaginal and cervical microbiota composition and its potential role in HPV infection, employing amplicon-based Next Generation Sequencing (NGS) of cervicovaginal specimens from a large series of HPV-infected women. Our aim was to characterize in detail the resident cervical and vaginal microbial flora under conditions of HPV infection and their potential link to CIN.
Materials and methods
Study cohort and design of the study
This is a retrospective study analyzing samples from a cohort of 877 non-pregnant women who were diagnosed with high-risk (hr) HPV infection in the context of GRECOSELF, a nationwide observational cross-sectional study aiming to suggest a way to implement HPV-DNA testing in conjunction with self-sampling for cervical cancer screening in Greece (Agorastos et al., 2019).
In brief, upon written informed consent, cervico-vaginal samples were self-collected with a vaginal dry swab (Roche Molecular Systems) and then stored and transported as previously described (Agorastos et al., 2019). HPV infection was confirmed by the detection of viral DNA using the Cobas 4,800 HPV test (Roche, United States). This assay is specifically designed to identify 14 hrHPVs, namely HPV16, 18, 31, 33, 35, 39, 45, 51, 52, 56, 58, 59, 66, 68 and also performs partial genotyping of the HPV16, HPV18 types among the other hrHPV types.
Women who tested positive for at least one hrHPV type were referred for colposcopy. Overall, we acquired colposcopic data from 636/877 (72.5%) women, whereas the remainder 241/877 (27.5%) for various reasons did not comply with their colposcopy referral upon their HPV test positive result. In cases with abnormal or suspicious colposcopic findings, multiple focal biopsies, and/or endocervical curettage (ECC), were taken from the abnormal area of the cervix. ECC was performed in case of a transformation zone type III.
Histopathological examination was performed by expert histopathologists. Based on the grade of CIN, cases were classified in groups, namely (from low grade to high grade CIN) CIN1 < CIN2 < CIN3 or worse (CIN3+). Women with no evidence of CIN, were classified as ‘No detectable disease (NDD)’ in terms of cervical histopathologic findings.
Samples analyzed for this study included 877 self-collected vaginal samples, each corresponding to a different woman, who tested positive for either HPV16, or HPV18 types or another high-risk type, namely “HPV other” (Figure 1). Moreover, for 60/877 women we analyzed paired samples, one self-collected (vaginal) and one collected by a physician prior to colposcopy (cervico-vaginal).
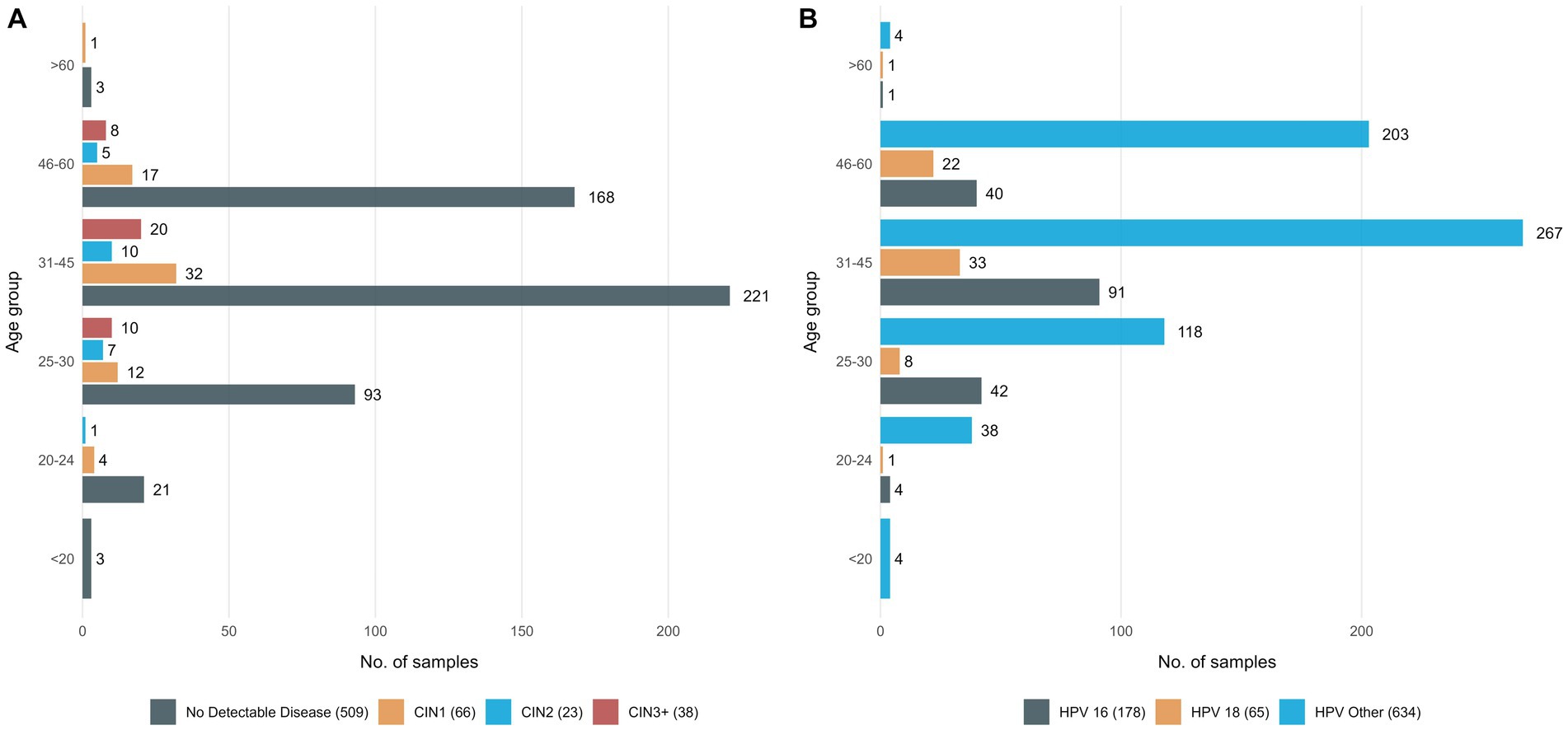
Figure 1. Detailed information on the study group (A) 636 women classified in 4 groups based on their histopathologic findings and age group, (B) all 877 women who tested positive with the Cobas assay classified in 4 groups, based on the HPV type responsible for the infection and age group.
Ethical approval and clinical data management
All procedures were in accordance with the standards set by the 1964 Declaration of Helsinki, and were also approved by the Ethics Committees of the Aristotle University of Thessaloniki and the Centre for Research and Technology Hellas. Next Generation Sequencing, as well as clinical data were stored and handled in accordance with the ISO/IEC 27001:2013 guidelines.
16S V3–V4 amplification and high-throughput sequencing methodology
Starting with 12.5 ng of total DNA as template, we PCR amplified the V3–V4 region of the bacterial 16S rRNA gene using primers with overhang adapters (forward primer with overhang adapter).
5’-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG-3′; reverse primer with overhang adapter.
5’-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC-3′.
Both the amplicon PCR, as well as the index PCR reactions were carried out using the KAPA HiFi HotStart DNA polymerase 2X ReadyΜix (Roche, United States) and SYTO9 (Thermo Fisher Scientific, United States) to monitor the amplification in real-time. Regarding thermal cycling conditions, 16S amplification was initiated by denaturation at 98°C for 20s, followed by 30 cycles of denaturation at 98°C for 20s to annealing of the primers at 55°C for 30s, and 72°C for 30s with a final extension at 72°C for 5 min.
NGS libraries were prepared by direct incorporation of unique index primer pairs (Nextera XT, Illumina) to 1 μL of the adapter tailed PCR amplicons. Thermal cycling conditions included denaturation at 98°C for 2 min, followed by 10 cycles of ramping from 98°C for 15 s to 64°C for 60 s. Paired-end NGS was performed with the MiSeq reagent v3 kit (2 × 300 bp) in a MiSeq Benchtop Sequencer (Illumina, United States) according to the manufacturer’s instructions.
Bioinformatics, statistical analysis, and data visualization
NGS raw data were analyzed using the QIIME 2020.8.0 software (Bolyen et al., 2019). In detail, adapter trimming of the demultiplexed paired-end sequences was performed using the Cutadapt software (included in the QIIME 2 suite). Trimmed sequences were error-filtered and truncated using the DADA2 software. Based on the quality score summary, R1 reads were truncated at 290 bp while R2 reads at 230 bp (Figures 2A,B). The resulting paired-end sequences were merged and searched for chimeras. The minimum abundance of potential ancestral sequences of a given merged sequence being tested as chimeric was increased to 8. An overview of DADA2 processing steps is given in Figure 2C. For comparison with existing Operational Taxonomic Unit (OTU) tables, open-reference OTU picking was applied against SILVA 123, using a threshold of 99% identity. Finally, taxonomy classification was performed with the RDP classifier. During this process, an assignment confidence cutoff of 80% was applied.
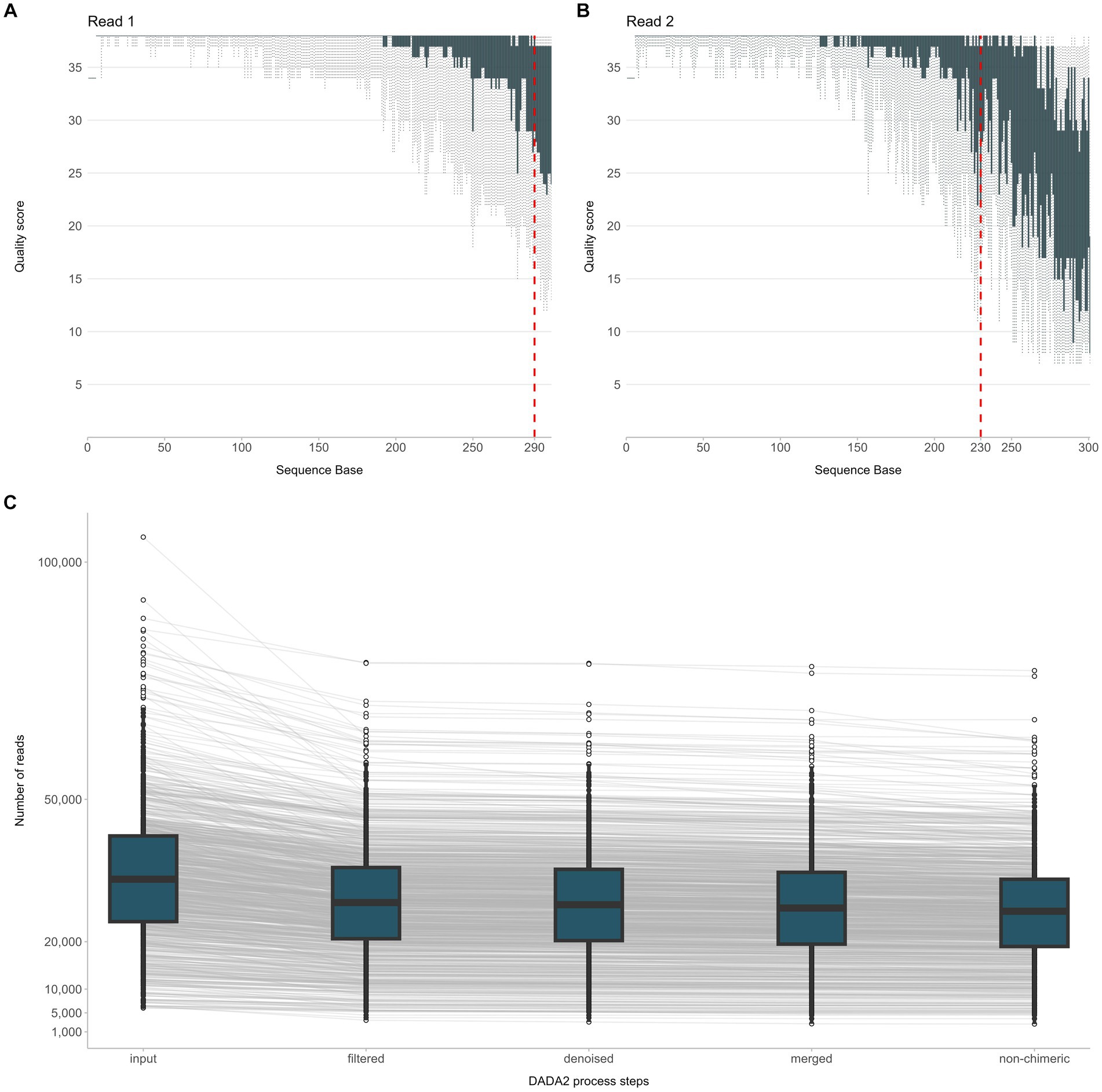
Figure 2. Overview of the NGS raw data processing steps from quality filtering of sequencing reads R1, R2 (A,B) to quality assessment of the joined reads and exclusion of chimeric sequences (C).
Meta-analysis of the QIIME 2 results was performed using R v4.2.0. BIOM files were processed using the {rbiom} package, while alpha diversity was calculated using the {vegan} package. Relative abundance analysis was performed with DESeq2 (Love et al., 2014). All plots were created using the {ggplot2} library (Wickham, 2016). The OTU table was annotated by keeping the genus labels from the RDP taxonomy results. Any OTUs (genus) or samples that have zero counts across the table were removed. In addition, samples with less than 10,000 reads were also removed from the rest of the analysis. All comparisons were performed at genus level taking into account the relative abundance of each genus in each sample.
Assessment of statistical significance was performed with the packages incorporated in the aforementioned bioinformatics tools.
Results
Comparison of cervicovaginal microbial composition between self-collected and physician-collected samples reveals significant overlap
In total, 38,328,108 raw reads (median 33,133 reads/sample) were obtained for the 937 sequenced PCR amplicons. After quality filtering, 30,353,253 non-chimeric reads (median 26,516 reads/sample) were further processed for taxonomic assignment and meta-data analysis (Figure 2C).
First, we assessed how self-collected samples compared to the respective physician-collected ones in regard to microbial diversity and composition. To that end, we compared paired self- versus physician-collected samples from 60 women and compared alpha diversity and taxonomic assignment. Despite differences in the sampling methods, alpha microbial diversity did not differ significantly between the two sample types (median Shannon Index 0.9 and 0.7 for self-versus physician-collected samples, respectively, p = 0.4) (Figure 3A). In addition, in both sample types the same microbial populations dominated the cervicovaginal flora, with Lactobacillus being the predominant genus, followed by anaerobic, Community State Type IV (CST-IV) bacteria associated with bacterial vaginosis, such as Gardnerella, Prevotella, Sneathia, and Megasphaera. Altogether, the dominant genera in both samples accounted for an average 57.7% of the cervicovaginal microbiome, with each bacterial genus displaying similar relative frequencies in both the self- and the physician-collected samples (Figure 3B).
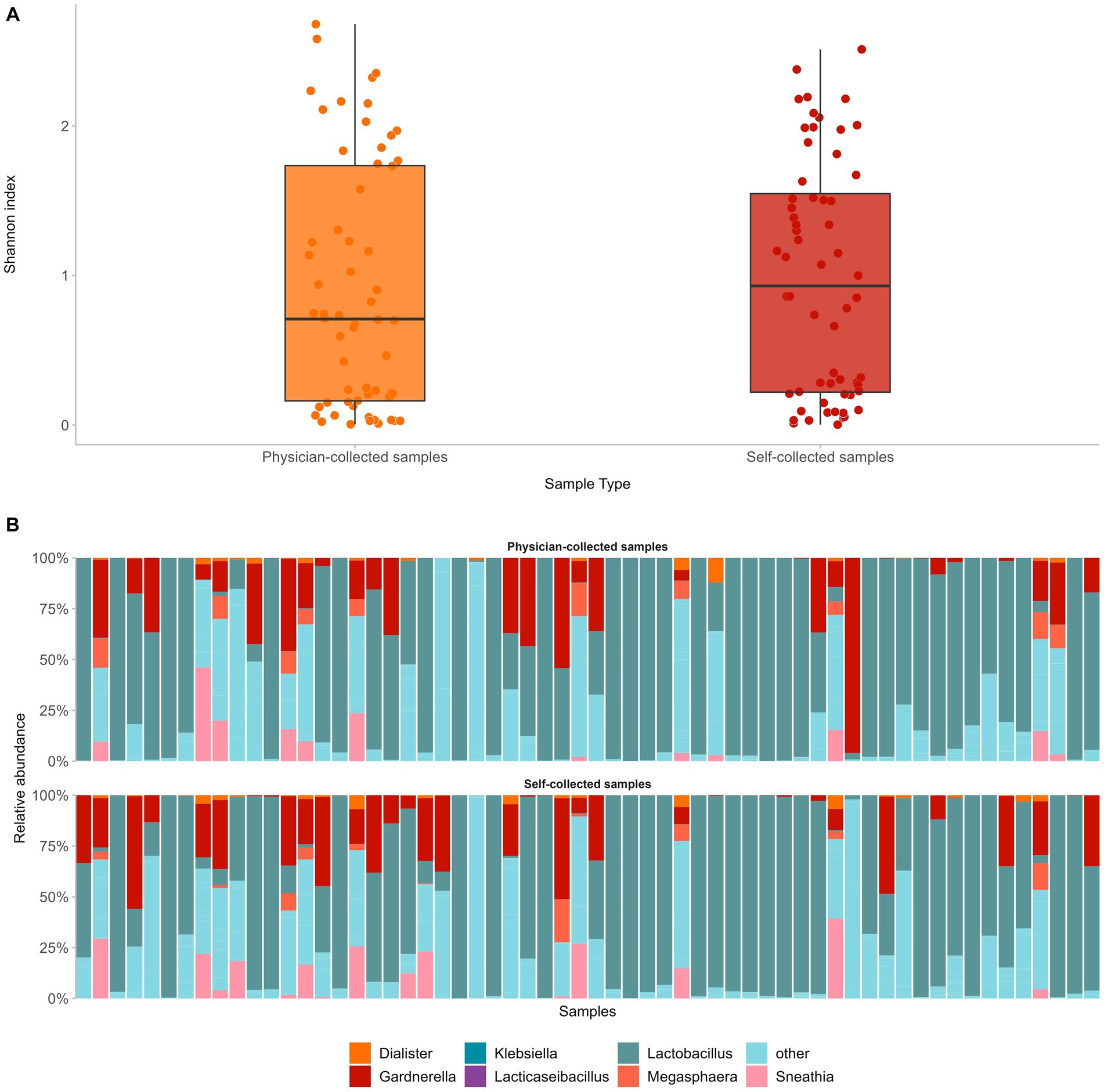
Figure 3. Comparison of paired self- versus physician-collected samples showed no significant differences in terms of vaginal microbial diversity (A), as well as composition of the predominant bacterial genera in both sample types (B).
Operational taxonomic unit assignment and comparison between different CIN groups and HPV types
Taxonomic assignment was performed with the RDP classifier, and vaginal microbial composition was compared in women infected with different hrHPV types, namely HPV16, HPV18, HPV other, as well as women who exhibited precancerous cervical lesions after abnormal colposcopic findings. For the latter comparison, groups were as defined above, i.e., CIN1, CIN2, CIN3, or, in the absence of any lesions, NDD (no detectable disease).
Overall, 10,788 bacterial operational taxonomic units (OTUs) were identified, of which 4,860 matched with 305 different and well-characterized genera. In all groups, we observed a Lactobacillus-dominated vaginal flora with a median relative abundance of 60%. Besides Lactobacillus, the majority of the vaginal microbiome in HPV-infected women consisted of CST-IV bacteria common in all groups irrespective of the HPV type responsible for the infection, such as Gardnerella (median frequency 11.5%), Prevotella (median frequency 3.8%), Sneathia and Megasphaera (median frequencies 1.8 and 0.6%, respectively), amongst others.
Vaginal microbial diversity and richness is independent of the type of HPV or the presence of cervical lesions, however associates with age
Alpha diversity was calculated at the OTU level for the different groups with the Shannon Index. No significant differences were found in terms of microbial diversity for women infected with different HPV types. In fact, although CIN2 and CIN3 groups tended to be slightly more diverse than the CIN1 or NDD groups, such comparisons did not reach statistical significance. Indeed, the CIN2 and CIN3 groups displayed a higher Shannon index (Figure 4C), however comparison with the CIN1 or NDD groups did not reach statistical significance (p = 0.6), meaning that regardless the histopathology status, all women had a similar microbial diversity.
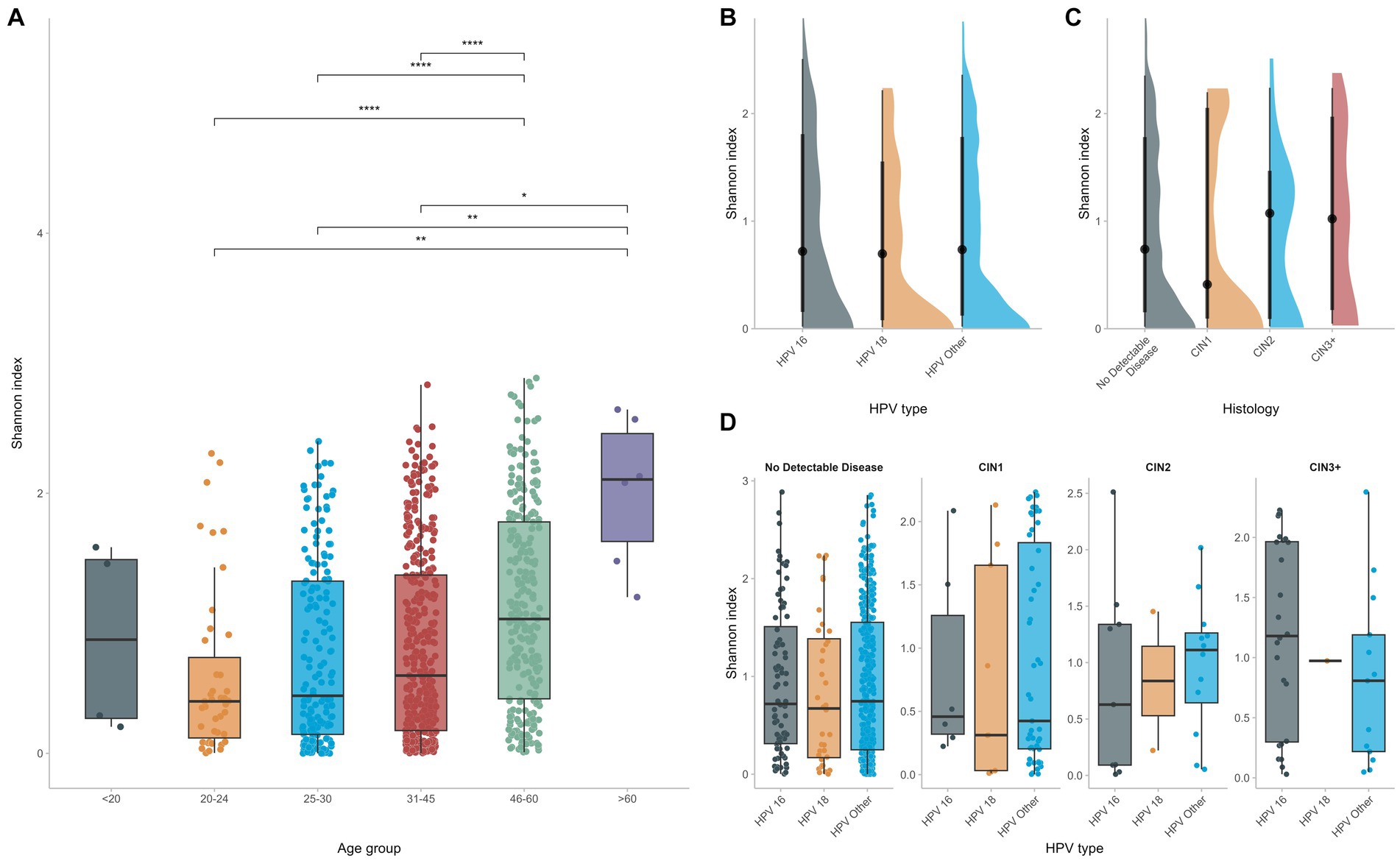
Figure 4. Alpha diversity was calculated and compared among different groups (A). HPV infection does not affect significantly vaginal microbial diversity (B–D) whereas increased age seems to be the predominant factor for increased vaginal diversity.
On the contrary, when considering the age of the study participants, the vaginal flora of women who were of 45 years of age or older was significantly more diverse than that of younger women (p < 0.0001), supporting that, even in the context of HPV, age at infection is the dominant determinant of vaginal diversity and richness (Figure 4A).
HPV infection displays distinct vaginal microbial signatures compared among HPV types
Having identified the main bacterial genera that populate the vaginal microenvironment, next we explored if their relative abundance differed between the different groups. Differential analysis revealed pronounced differences in the abundance of certain bacterial genera between HPV types. In detail, all three HPV-positive groups exhibited increased frequencies of the CST-IV genera Megasphaera and Sneathia. Especially for Lacticaseibacillus, we observed a significant depletion in the case of types HPV16, HPV18 compared to cases infected with other high-risk HPV type(s) (p < 0.0001) (Figure 5). Similar comparisons were also performed among groups based on the grade of CIN in order to explore if the abundance of the aforementioned or other bacterial genera differed significantly in the presence of precancerous lesions: however, no significant differences were identified.
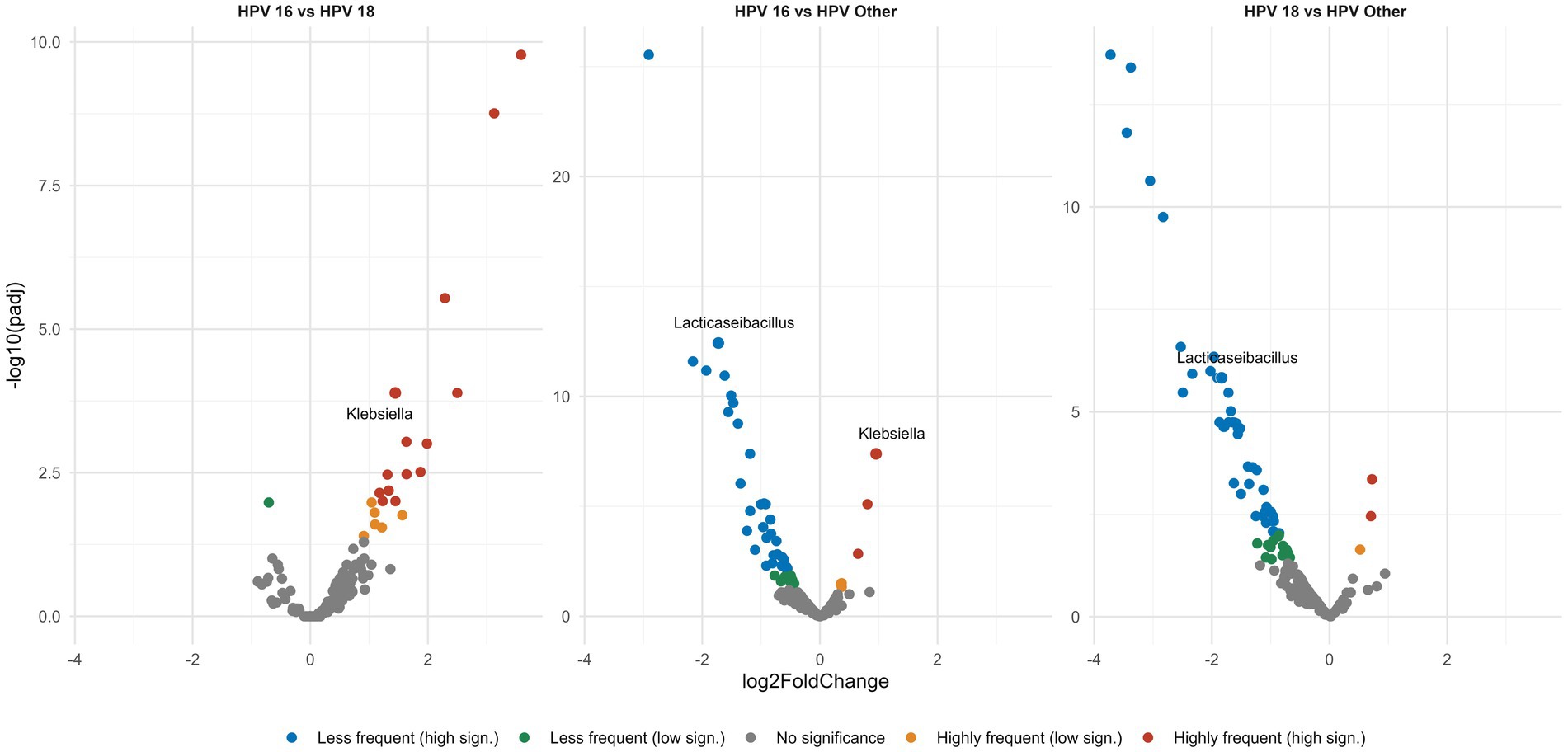
Figure 5. Volcano plots show pairwise comparisons of the relative abundance of the different genera. Each dot represents a specific genus and is plotted on a Cartesian coordinate system, where the x axis represents the fold change in the relative abundance between the compared groups and the y axis represents the statistical significance describing the low frequency (green, blue dots) or high frequency (orange, red dots) of each genus in a given group. All HPV-positive groups display significant Lacticaseibacillus depletion. Among the three HPV-positive groups the HPV16, 18 types show the greatest Lacticaseibacillus depletion of all.
Discussion
The increasingly evident role of the cervicovaginal microbiome in HPV infection persistence and cervical cancer pathogenesis alongside the diverse applications of high-throughput sequencing methodologies in metagenomics analyses, pose an exciting challenge for the use of NGS towards identifying microbial markers that will advance our understanding of HPV-associated cervical cancer ontogeny. Identifying specific bacterial species as microbial biomarkers of high-grade and lowgrade dysplasia could conceivably assist in triaging individuals with pronounced chances of lesion regression or progression (Kudela et al., 2021). In that regard, the assessment of microbial composition and associated risk factors constitutes the critical background for preventive, predictive, and personalized medicine (3P medicine) improving state-of-the-art medical care in patients with cervical cancer (Kudela et al., 2021).
In this context, we analyzed the vaginal and cervical microbial composition using high-throughput, amplicon-based metagenomics and investigated its relationship with HPV infection, using self- and physician-collected samples obtained from a large cohort of HPV positive women.
Comparison of paired self- versus physician-collected samples showed no significant differences in terms of either the vaginal microbial diversity or the composition of the predominant bacterial genera. The same microbial populations dominated the cervicovaginal flora, displaying similar relative frequencies in both self-collected and physician-collected specimens, with Lactobacillus being the predominant genus, followed by anaerobic bacteria, associated with bacterial vaginosis, such as Gardnerella, Prevotella, Sneathia, Megasphaera (Caselli et al., 2020; Wei et al., 2021; Lin et al., 2022). The aforementioned similarities between the two different sample types are noteworthy, especially in light of increasing evidence on the value of self-sampling as an equally informative approach for HPV testing as sampling by a healthcare professional. Our results reinforce this notion, showing for the first time that self-sampling can be implemented not only for HPV testing but also for other molecular-based applications, including microbiome analysis in the context of HPV infection.
Comparison of the vaginal microbiota between groups of women infected by different HPV types showed no significant differences in terms of microbial diversity, with Lactobacillus being the dominant genus in the vaginal flora of all groups. Similar to previous studies (Chen et al., 2020; Usyk et al., 2020; Wei et al., 2021), the majority of the vaginal microbiome in HPV-infected women consisted of CST-IV bacteria common in all groups irrespective of the HPV type responsible for the infection, revealing species assigned to Gardnerella, Prevotella, Sneathia and Megasphaera genera, amongst others. Previous studies have offered evidence that the presence of Gardnerella species in the vaginal microbiome is associated with progression to CIN2+ (Usyk et al., 2020; Xu et al., 2022). This might be attributed to the release of vaginal cytolysin and production of sialidase in the vaginal microenvironment, inducing tissue destruction, immune response evasion and bacterial invasion (Usyk et al., 2020; Xu et al., 2022). Similarly, other bacteria such as Prevotella, Bacteroides and Mobiluncus may inhibit the protective effect of the vaginal mucosal barrier by producing sialidase (Dong et al., 2022). The presence of these bacteria in our cohort further supports that HPV infections appear to go hand-in-hand with an overall dysbiotic vaginal microenvironment. However, it remains unclear whether it is the HPV infection itself that disrupts the balance of the vaginal microenvironment, or a pre-existing vaginal dysbiosis sets the ground for a persistent viral infection.
Although several reports mention an increased microbial diversity and richness of the vaginal flora in the context of HPV infections (Usyk et al., 2020; López-Filloy et al., 2022), we did not observe any significant differences in microbial diversity between groups of women infected by different hrHPV types, including women with precancerous cervical lesions. However, significant differences in the vaginal microbial diversity were observed depending on the age of HPV infected women. In fact, the vaginal flora of women who were of 45 years of age or older was significantly more diverse than that of younger women. This finding indicates that age is a major determinant of increased vaginal diversity, perhaps more significant than HPV infection status.
Finally, despite the overall similarities in the composition of the vaginal microbiome between the different examined groups, we found differences in the abundance of certain bacterial genera in the vaginal microenvironment among the HPV-infected women. In accordance with previous studies (Ravel et al., 2011; Zhou et al., 2019; Caselli et al., 2020), Lacticaseibacillus was significantly depleted in all HPV-positive women, while increased frequencies were also seen for the CST-IV genera Megasphaera and Sneathia. HPV infections can disturb the balance of vaginal and cervical microenvironment, leading to abnormal changes in microbiota, including Lactobacillus spp. depletion and predominance of Lactobacillus iners and anaerobic bacteria (such as Gardnerella, Prevotella, Sneathia, Megasphaera) (Lee et al., 2013; Bik et al., 2019; Borgogna et al., 2020; Caselli et al., 2020; Usyk et al., 2020; Wei et al., 2021; Lebeau et al., 2022), allowing the persistence of viral infection and disease progression (Li et al., 2012). Studies have demonstrated that abnormal changes in the composition of the vaginal microbiome, which lead to reduced mucus production and consequent clearance of pathogenic bacteria, can be associated with the late clearance of HPV infection (Li et al., 2012).
A major finding of the present study concerned the significant differences among different HPV types regarding the abundance of Lacticaseibacillus. In particular, Lacticaseibacillus was significantly depleted in the case of women infected by types HPV16, HPV18 compared to cases infected by other high-risk HPV type(s). This was particularly intriguing, especially in view of published evidence that a Lacticaseibacillus species, namely Lacticaseibacillus casei LH23 strains, inhibits the expression of the E6 and E7 viral proteins, which play a crucial role in viral replication during HPV infection (Tomaić, 2016). In this context, this novel finding could potentially be relevant in explaining the high risk for cervical cancer posed by the HPV16, 18 types. On the contrary, we found no significant differences in the abundance of certain bacteria (including the aforementioned genera) when comparing groups based on the presence or not of precancerous lesions. This indicates that although microbial profiling could provide insightful information regarding the clinical manifestations of certain hrHPV types, it appears as less helpful for triaging patients by risk of CIN/cancer development.
A limitation of our study concerns the lack of information regarding antibiotic usage by the study participants. This was due to the epidemiological nature of the GRECOSELF study which provided the samples. Although all participants stated that they were in good health and were not under any medication, we cannot a priori exclude the possibility of any alterations in the microbial composition due to potential antibiotic activity. Another limitation concerns the lack of a healthy control group consisting of women without an HPV infection, thus precluding the possibility to perform comparisons versus hr-HPV infected women. This was due to the retrospective nature of the study, whereby the analyzed samples were collected in the context of the GRECOSELF project (Agorastos et al., 2019), that investigated the feasibility of implementing secondary prevention of cervical cancer in Greece through HPV-DNA testing on self-collected cervicovaginal samples. In GRECOSELF, all samples that tested negative for HPV during GRECOSELF, were excluded from any downstream procedure and no material was stored for further use, eventually leading to the absence of a control group in the present study. Also worth mentioning is the fact that we were unable to assess the potential relationship between microbial composition and the cytological results in the women of our cohort. That was due the fact that in the context of the GRECOSELF study, upon confirmation of HPV infection, women were referred to their ob/gyn for a PAP smear. However, most participants did not follow up with their physician, thus leading to significant lack of information regarding the cytological results of our cohort, and hampering our ability to perform a robust statistical analysis regarding the correlation of the microbial composition and the cytological results.
In conclusion, we performed in-depth, high-throughput cervicovaginal microbiome profiling in one of the largest series of HPV infected women reported so far offering evidence for a distinct cervicovaginal microbial composition in HPV-infected women in relation to hrHPV types. Although our findings do not allow definitive conclusions to be drawn regarding their clinical relevance, still they provide the grounds for further exploration of links between differential cervicovaginal microenvironments and different types of HPV infection, their persistence and progression to cervical precancer and cancer.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/ena, PRJEB59411.
Ethics statement
The studies involving humans were approved by Ethics Committees of the Aristotle University of Thessaloniki and the Centre for Research and Technology Hellas. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
ES: Formal analysis, Methodology, Writing – original draft. GG: Formal analysis, Methodology, Writing – original draft. NP: Formal analysis, Visualization, Writing – original draft. KP: Methodology, Writing – review & editing. KC: Investigation, Supervision, Writing – review & editing. FP: Formal analysis, Writing – review & editing. TA: Investigation, Supervision, Writing – review & editing. KS: Investigation, Supervision, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research was supported in part by the project “CerHPVix: an integrated biomolecular approach towards innovative triage of high-risk HPV positive women for the prevention of cervical cancer”, funded by Roche Hellas S. A. (2019–2021); by the European Regional Development Fund of the European Union and Greek national funds through the Operational Program Competitiveness, Entrepreneurship and Innovation, under the call RESEARCH-CREATE-INNOVATE (GenOptics, project code: T2E1 K-00407).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Agorastos, T., Chatzistamatiou, K., Tsertanidou, A., Mouchtaropoulou, E., Pasentsis, K., Kitsou, A., et al. (2019). Implementation of HPV-based cervical cancer screening combined with self-sampling using a midwifery network across rural Greece: the GRECOSELF study. Cancer Prev. Res. 12, 701–710. doi: 10.1158/1940-6207.CAPR-19-0192
Audirac-Chalifour, A., Torres-Poveda, K., Bahena-Román, M., Téllez-Sosa, J., Martínez-Barnetche, J., Cortina-Ceballos, B., et al. (2016). Cervical microbiome and cytokine profile at various stages of cervical cancer: a pilot study. PLoS One 11:e0153274. doi: 10.1371/journal.pone.0153274
Bik, E. M., Bird, S. W., Bustamante, J. P., Leon, L. E., Nieto, P. A., Addae, K., et al. (2019). A novel sequencing-based vaginal health assay combining self-sampling, HPV detection and genotyping, STI detection, and vaginal microbiome analysis. PLoS One 14, 1–25. doi: 10.1371/journal.pone.0215945
Bolyen, E., Rideout, J. R., Dillon, M. R., Bokulich, N. A., Abnet, C. C., al-Ghalith, G. A., et al. (2019). Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 37, 852–857. doi: 10.1038/s41587-019-0209-9
Borgogna, J. C., Shardell, M. D., Santori, E. K., Nelson, T. M., Rath, J. M., Glover, E. D., et al. (2020). The vaginal metabolome and microbiota of cervical HPV-positive and HPV-negative women: a cross-sectional analysis. BJOG 127, 182–192. doi: 10.1111/1471-0528.15981
Brotman, R. M., Shardell, M. D., Gajer, P., Tracy, J. K., Zenilman, J. M., Ravel, J., et al. (2014). Interplay between the temporal dynamics of the vaginal microbiota and human papillomavirus detection. J. Infect. Dis. 210, 1723–1733. doi: 10.1093/infdis/jiu330
Caselli, E., D’Accolti, M., Santi, E., Soffritti, I., Conzadori, S., Mazzacane, S., et al. (2020). Vaginal microbiota and cytokine microenvironment in HPV clearance/persistence in women surgically treated for cervical intraepithelial neoplasia: an observational prospective study. Front. Cell. Infect. Microbiol. 10, 1–15. doi: 10.3389/fcimb.2020.540900
Chaturvedi, A. K., Engels, E. A., Pfeiffer, R. M., Hernandez, B. Y., Xiao, W., Kim, E., et al. (2011). Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J. Clin. Oncol. 29, 4294–4301. doi: 10.1200/JCO.2011.36.4596
Chen, Y., Qiu, X., Wang, W., Li, D., Wu, A., Hong, Z., et al. (2020). Human papillomavirus infection and cervical intraepithelial neoplasia progression are associated with increased vaginal microbiome diversity in a Chinese cohort. BMC Infect. Dis. 20, 1–12. doi: 10.1186/s12879-020-05324-9
Cheng, L., Norenhag, J., Hu, Y., Brusselaers, N., Fransson, E., Ährlund-Richter, A., et al. (2020). Vaginal microbiota and human papillomavirus infection among young Swedish women. NPJ Biofilms Microbiomes. 6, 1–10. doi: 10.1038/s41522-020-00146-8
Curty, G., de Carvalho, P. S., and Soares, M. A. (2020). The role of the cervicovaginal microbiome on the genesis and as a biomarker of premalignant cervical intraepithelial neoplasia and invasive cervical cancer. Int. J. Mol. Sci. 21:10222. doi: 10.3390/ijms21010222
Dong, B., Huang, Y., Cai, H., Chen, Y., Li, Y., Zou, H., et al. (2022). Prevotella as the hub of the cervicovaginal microbiota affects the occurrence of persistent human papillomavirus infection and cervical lesions in women of childbearing age via host NF-κB/C-myc. J. Med. Virol. 94, 5519–5534. doi: 10.1002/jmv.28001
Forman, D., de Martel, C., Lacey, C. J., Soerjomataram, I., Lortet-Tieulent, J., Bruni, L., et al. (2012). Global burden of human papillomavirus and related diseases. Vaccine 30, F12–F23. doi: 10.1016/j.vaccine.2012.07.055
Kudela, E., Liskova, A., Samec, M., Koklesova, L., Holubekova, V., Rokos, T., et al. (2021). The interplay between the vaginal microbiome and innate immunity in the focus of predictive, preventive, and personalized medical approach to combat HPV-induced cervical cancer. EPMA J. 12, 199–220. doi: 10.1007/s13167-021-00244-3
Lazare, C., Xiao, S., Meng, Y., Wang, C., Li, W., Wang, Y., et al. (2019). Evaluation of cervical intraepithelial neoplasia occurrence following the recorded onset of persistent high-risk human papillomavirus infection: a retrospective study on infection duration. Front. Oncol. 9:976. doi: 10.3389/fonc.2019.00976
Lebeau, A., Bruyere, D., Roncarati, P., Peixoto, P., Hervouet, E., Cobraiville, G., et al. (2022). HPV infection alters vaginal microbiome through down-regulating host mucosal innate peptides used by lactobacilli as amino acid sources. Nat. Commun. 13:1076. doi: 10.1038/s41467-022-28724-8
Lee, J. E., Lee, S., Lee, H., Song, Y. M., Lee, K., Han, M. J., et al. (2013). Association of the vaginal microbiota with human papillomavirus infection in a Korean twin cohort. PLoS One 8:e63514. doi: 10.1371/journal.pone.0063514
Li, J., McCormick, J., Bocking, A., and Reid, G. (2012). Importance of vaginal microbes in reproductive health. Reprod. Sci. 19, 235–242. doi: 10.1177/1933719111418379
Lin, W., Zhang, Q., Chen, Y., Dong, B., Xue, H., Lei, H., et al. (2022). Changes of the vaginal microbiota in HPV infection and cervical intraepithelial neoplasia: a cross-sectional analysis. Sci. Rep. 12, 1–14. doi: 10.1038/s41598-022-06731-5
Lin, S., Zhang, B., Lin, Y., Lin, Y., and Zuo, X. (2022). Dysbiosis of cervical and vaginal microbiota associated with cervical intraepithelial neoplasia. Front. Cell. Infect. Microbiol. 12, 1–9. doi: 10.3389/fcimb.2022.767693
López-Filloy, M., Cortez, F. J., Gheit, T., Cruz y Cruz, O., Cruz-Talonia, F., Chávez-Torres, M., et al. (2022). Altered vaginal microbiota composition correlates with human papillomavirus and mucosal immune responses in women with symptomatic cervical ectopy. Front. Cell. Infect. Microbiol. 12:884272. doi: 10.3389/fcimb.2022.884272
Love, M. I., Huber, W., and Anders, S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15:550. doi: 10.1186/s13059-014-0550-8
Mirabello, L., Yeager, M., Cullen, M., Boland, J. F., Chen, Z., Wentzensen, N., et al. (2016). HPV16 sublineage associations with histology-specific Cancer risk using HPV whole-genome sequences in 3200 women. J. Natl. Cancer Inst. 108, djw100–djw109. doi: 10.1093/jnci/djw100
Muñoz, N., Bosch, F. X., Castellsagué, X., Díaz, M., de Sanjose, S., Hammouda, D., et al. (2004). Against which human papillomavirus types shall we vaccinate and screen? The international perspective. Int. J. Cancer 111, 278–285. doi: 10.1002/ijc.20244
Muñoz, N., Bosch, F. X., de Sanjosé, S., Herrero, R., Castellsagué, X., Shah, K. V., et al. (2003). Epidemiologic classification of human papillomavirus types associated with cervical cancer. N. Engl. J. Med. 348, 518–527. doi: 10.1056/NEJMoa021641
Pimple, S., and Mishra, G. (2022). Cancer cervix: epidemiology and disease burden. Cyto J. 19:21. doi: 10.25259/CMAS_03_02_2021
Ravel, J., Gajer, P., Abdo, Z., Schneider, G. M., Koenig, S. S. K., McCulle, S. L., et al. (2011). Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. U. S. A. 108, 4680–4687. doi: 10.1073/pnas.1002611107
Reid, R., Stanhope, C. R., Herschman, B. R., Booth, E., Phibbs, G. D., and Smith, J. P. (1982). Genital warts and cervical cancer. I. Evidence of an association between subclinical papillomavirus infection and cervical malignancy. Cancer 50, 377–387. doi: 10.1002/1097-0142(19820715)50:2<377::AID-CNCR2820500236>3.0.CO;2-A
Roden, R. B. S., and Stern, P. L. (2018). Opportunities and challenges for human papillomavirus vaccination in cancer. Nat. Rev. Cancer 18, 240–254. doi: 10.1038/nrc.2018.13
Schiffman, M., Doorbar, J., Wentzensen, N., de Sanjosé, S., Fakhry, C., Monk, B. J., et al. (2016). Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers. 2:16086. doi: 10.1038/nrdp.2016.86
Tomaić, V. (2016). Functional roles of E6 and E7 oncoproteins in HPV-induced malignancies at diverse anatomical sites. Cancers 8:95. doi: 10.3390/cancers8100095
Usyk, M., Zolnik, C. P., Castle, P. E., Porras, C., Herrero, R., Gradissimo, A., et al. (2020). Cervicovaginal microbiome and natural history of HPV in a longitudinal study. PLoS Pathog. 16, 1–20. doi: 10.1371/journal.ppat.1008376
Watts, D. H., Fazarri, M., Minkoff, H., Hillier, S. L., Sha, B., Glesby, M., et al. (2005). Effects of bacterial vaginosis and other genital infections on the natural history of human papillomavirus infection in HIV-1-infected and high-risk HIV-1-uninfected women. J. Infect. Dis. 191, 1129–1139. doi: 10.1086/427777
Wei, Z. T., Chen, H. L., Wang, C. F., Yang, G. L., Han, S. M., and Zhang, S. L. (2021). Depiction of vaginal microbiota in women with high-risk human papillomavirus infection. Front. Public Health 8:587298. doi: 10.3389/fpubh.2020.587298
Xu, X., Zhang, Y., Yu, L., Shi, X., Min, M., Xiong, L., et al. (2022). A cross-sectional analysis about bacterial vaginosis, high-risk human papillomavirus infection, and cervical intraepithelial neoplasia in Chinese women. Sci. Rep. 12:6609. doi: 10.1038/s41598-022-10532-1
Keywords: HPV, vaginal microbiome, cervical microbiome, next generation sequencing (NGS), Lacticaseibacillus
Citation: Sofou E, Gkoliou G, Pechlivanis N, Pasentsis K, Chatzistamatiou K, Psomopoulos F, Agorastos T and Stamatopoulos K (2023) High risk HPV-positive women cervicovaginal microbial profiles in a Greek cohort: a retrospective analysis of the GRECOSELF study. Front. Microbiol. 14:1292230. doi: 10.3389/fmicb.2023.1292230
Edited by:
Efthymia Giannitsioti, University General Hospital Attikon, GreeceReviewed by:
Erik Kudela, Comenius University, SlovakiaCharalampos D. Moschopoulos, University General Hospital Attikon, Greece
Nikolaos Spernovasilis, University of Crete, Greece
Copyright © 2023 Sofou, Gkoliou, Pechlivanis, Pasentsis, Chatzistamatiou, Psomopoulos, Agorastos and Stamatopoulos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kostas Stamatopoulos, a29zdGFzLnN0YW1hdG9wb3Vsb3NAY2VydGguZ3I=
†These authors have contributed equally to this work and share first authorship
‡These authors share senior authorship