 Agnieszka Bogut
Agnieszka Bogut Piotr Koper2
Piotr Koper2- 1Chair and Department of Medical Microbiology, Medical University of Lublin, Lublin, Poland
- 2Department of Genetics and Microbiology, Institute of Biological Sciences, Maria Curie-Skłodowska University, Lublin, Poland
- 3Chair and Department of Forensic Medicine, Medical University of Lublin, Lublin, Poland
Phenotype switching from a wild type (WT) to a slow-growing subpopulation, referred to as small colony variants (SCVs), supports an infectious lifestyle of Staphylococcus epidermidis, the leading cause of medical device-related infections. Specific mechanisms underlying formation of SCVs and involved in the shaping of their pathogenic potential are of particular interest for stable strains as they have been only rarely cultured from clinical specimens. As the SCV phenotype stability implies the existence of genetic changes, the whole genome sequence of a stable, hemin-dependent S. epidermidis SCV strain (named 49SCV) involved in a late prosthetic joint infection was analyzed. The strain was isolated in a monoculture without a corresponding WT clone, therefore, its genome was compared against five reference S. epidermidis strains (ATCC12228, ATCC14990, NBRC113846, O47, and RP62A), both at the level of the genome structure and coding sequences. According to the Multilocus Sequence Typing analysis, the 49SCV strain represented the sequence type 2 (ST2) regarded as the most prominent infection-causing lineage with a worldwide dissemination. Genomic features unique to 49SCV included the absence of the Staphylococcal Cassette Chromosome (SCC), ~12 kb deletion with the loss of genes involved in the arginine deiminase pathway, and frameshift-generating mutations within the poly(A) and poly(T) homopolymeric tracts. Indels were identified in loci associated with adherence, metabolism, stress response, virulence, and cell wall synthesis. Of note, deletion in the poly(A) of the hemA gene has been considered a possible trigger factor for the phenotype transition and hemin auxotrophy in the strain. To our knowledge, the study represents the first genomic characterization of a clinical, stable and hemin-dependent S. epidermidis SCV strain. We propose that previously unreported indels in the homopolymeric tracts can constitute a background of the SCV phenotype due to a resulting truncation of the corresponding proteins and their possible biological dysfunction. Streamline of genetic content evidenced by the loss of the SCC and a large genomic deletion can represent a possible strategy associated both with the SCV phenotype and its adaptation to chronicity.
Introduction
Staphylococcus epidermidis (SE), a common member of the human skin microbiota, has emerged as an important causative agent of infections related to implanted medical devices (Post et al., 2017; Both et al., 2021; Fernández-Rodríguez et al., 2021; Liu et al., 2021). Infectious lifestyle of this microorganism is supported by phenotype switching from a wild type (WT) to a slow-growing subpopulation characterized by distinctive pathogenic traits and referred to as small colony variants (SCVs) (Proctor et al., 2006; Liu et al., 2021). The study of Edwards (Edwards, 2012) demonstrated that this transition occurs via a constitutive mechanism depending upon bacterial replication. It leads to the generation of a dynamic, antibiotic-resistant subpopulation able to revert to its parental phenotype. Additionally, a variety of environmental pressures including low temperature, antibiotic and disinfectant exposure, pH and oxidative stress as well as a prolonged growth under nutrient limiting conditions and intracellular environment have been reported to promote staphylococcal SCV formation (Vesga et al., 1996; Schaaff et al., 2003; Bayston et al., 2007; Onyango et al., 2013; Vestergaard et al., 2016; Perez and Patel, 2017, 2018; Lee et al., 2020; Islam et al., 2022).
According to the definition proposed by Tuchscherr et al. (2020), SCVs represent bacterial subpopulation formed within host cells, characterized by a variable phenotypic stability, slow growth rate, decreased expression of virulence factors and multifaceted alterations in metabolic pathways and/or global regulatory genes. Although there is no common metabolic change typical for SCVs, many clinical and laboratory-derived variants can be tied by a common thread which are alterations in an electron transport. These defects lead to a reduced amount of ATP available for many cellular processes including the cell wall biosynthesis, uptake of amino acids and carbohydrates; the resultant effect is a lower growth rate and production of smaller colonies (Proctor et al., 2006; Kahl et al., 2016; Proctor, 2019). Additionally, interruption of electron transport results in the accumulation of the NADH dinucleotide, which downregulates the citric acid cycle enzymes (Proctor, 2019). The decreased electron transport activity has also been related to antibiotic resistance, since an electrochemical gradient is required for the import of positively charged molecules including aminoglycosides into bacterial cells (Proctor et al., 2006). Mutations in the genes encoding for enzymes involved in the electron transport, such as those controlling menaquinone and cytochrome biosynthesis, result in the formation of respiratory-deficient SCVs which are auxotrophic for menadione and heme, respectively (Proctor, 2019; Bogut and Magryś, 2021).
There is increasing evidence that staphylococcal SCVs, including those produced by SE, can evolve from the WT clone during an infectious process (Baddour et al., 1988; Adler et al., 2003; Tuchscherr et al., 2011, 2016; Lin et al., 2016; Liu et al., 2021). Selection of SCVs is considered a bet-hedging strategy (Edwards, 2012; Liu et al., 2021) due to their higher adaptiveness compared to WTs and survival advantage associated with an ability to invade and maintain within host cells (Magryś et al., 2015; Perez and Patel, 2018).
Epigenetic, genetic and transcriptional factors are assumed to be implicated in the switch of the WT to its persistent SCV form (Bui and Kidd, 2015). Many SCVs isolated from clinical specimens can revert to a rapidly growing parental state. Non-stable (“dynamic”) SCVs are assumed to originate from regulatory mechanisms involving global regulators and non-defined mutations in response to changing environmental conditions. Intracellular milieu promotes later adaptations and formation of permanent (stable) SCVs which can carry specific mutations in their metabolic pathways (Tuchscherr et al., 2020; Liu et al., 2021).
Mechanisms underlying formation of SCVs by SE and involved in the shaping of their pathogenic potential are of particular interest for stable variants as they have been only rarely isolated from clinical samples (Liu et al., 2021). Stability, in turn, implies the existence of genetic changes. To the best of our knowledge, the genetic background of stable, clinically derived SCVs has been unveiled for only two staphylococcal strains, one represented by Staphylococcus aureus (SA) (Loss et al., 2019) and one strain of SE (Liu et al., 2021). Additionally, Bui and Kidd (2015) published a genomic comparison and transcriptomic analysis of a clinical SA isolate and the corresponding stable SCV which was induced experimentally by a prolonged exposure of the parental form to an inflammatory environment and steady-state growth conditions with low nutrients (Bui et al., 2015a,b).
In an attempt to investigate genetic mechanisms standing behind the SCV phenotype in the clinical context, we determined the whole genome sequence of a stable clinical SE SCV (named 49SCV) strain and compared it against five reference SE genomes. The strain was isolated from the sonicate fluid. The sample derived from a patient who had undergone a prosthetic hip joint revision surgery due to a clinical diagnosis of an aseptic implant loosening. Microbiological investigation revealed that the patient fulfilled criteria of a late prosthetic joint infection (PJI) (Parvizi et al., 2011). General characteristics of 49SCV were described previously. Briefly, the strain demonstrated hemin auxotrophy and gentamicin resistance (MIC >256 μg/mL) and was the icaADBC operon-positive, proficient biofilm producer under in vitro conditions (Bogut et al., 2014). A failure to cultivate the WT counterpart of 49SCV as well as its stability implied a furthest degree of chronicization of the infectious process leading to conversion of the original invading strain to the SCV variant and the existence of specific genetic changes involved in the phenotype transition.
This study represents the first genomic characterization of a stable and hemin-dependent SE SCV strain of clinical origin. We propose novel genetic determinants that can be involved in the phenotype switching and adaptation of SE to chronic, biofilm-associated and clinically indolent PJIs.
Materials and methods
Whole-genome sequencing
Genomic DNA of the 49SCV strain was extracted using the Genomic Micro AX Staphylococcus Gravity kit (A&A Biotechnology, Poland) according to the manufacturer’s protocol (including lysostaphin at a concentration of 4 U/μl and proteinase K at a concentration of 20 mg/mL). The purity and concentration of the DNA sample were verified using the BioTek Synergy LX spectrophotometer (Agilent Technologies, US).
The DNA sequencing was performed using the MiSeq benchtop sequencer (Illumina, San Diego, CA, USA). Sequencing of pair-end 150 nt libraries resulted in 1,708,902 reads. The reads were trimmed for quality and adaptor sequences were removed using Trimmomatic (Bolger et al., 2014). Genome assembly was carried out using the Unicycler tool (Wick et al., 2017). The genome sequence is available in the GenBank database with the accession number GCF_024505205.1.
The PGAP (Tatusova et al., 2016) tool was used to annotate the genome and the process was a part of submitting the genome to the GenBank database. Similarly, genome completeness was verified as a part of the submissions using the CheckM (Parks et al., 2015) tool.
Phylogenetic analysis
The AutoMLST (Alanjary et al., 2019) web server was used to determine the phylogenetic position of 49SCV. In addition, individual contigs were scanned against the PubMLST typing schemes using the mlst (Seemann, 2023a) tool. The iTol was used for the tree visualization (Letunic and Bork, 2019).
Comparative genomics
A full comparative analysis of the 49SCV genome was conducted in relation to five well-annotated SE reference genomes: ATCC 12228 (GCA_000007645.1), ATCC 14990 (GCA_006094375.1), NBRC 113846 (GCA_020181395.1), O47 (GCA_013317125.1), and RP62A (GCA_000011925.1). Briefly, the 49SCV genome contigs were reordered against the O47 reference and then compared to each other using the progressive Mauve (Darling et al., 2010) algorithm. In parallel, using the same genomes, the pangenome at the level of proteins of the analyzed strains was determined using the anvi’o (Eren et al., 2021) software package.
To determine the presence of transposons in the 49SCV genome, blastn (Zhang et al., 2000) and sequences of 11 transposons found in SE genomes (Tn551, Tn552, Tn554, Tn558, Tn559, Tn4001, Tn4003, Tn5404, Tn5406, Tn5801, and Tn6072) were used as queries. The strategy was also used to search for the insertion sequence (IS) elements, except that the ISFinder database was used (Siguier et al., 2006).
For the SNP analysis filtered, clean reads were mapped against each of the referenced genomes using Snippy (Seemann, 2023b).
Other bioinformatics
CRISPR sequences were searched for using the CRISPRfinder tool (Grissa et al., 2007). Prophages were found using the Phigaro (Starikova et al., 2020) and checked for completeness using CheckV (Nayfach et al., 2021). Unless otherwise stated, individual tools have been used with default parameters.
Results and discussion
General genomic features of 49SCV
The genome of the 49SCV strain was sequenced de novo. The trimmed and filtered reads were assembled, which resulted in a draft genome composed of 39 contigs and an average coverage of 165X. The genome size was estimated to be 2,386,088 bp, while the N50 (the sequence length of the shortest contig at 50% of the total assembly length) and L50 (the smallest number of contigs whose total length is half of the estimated genome size) parameter values were 135,189 bp and 5, respectively. A total number of 2,297 genes were predicted in the genome (versus 2,432 and 2,359 genes for ATCC 12228 and ATCC 14990, respectively), of which 2,161 were the protein-coding genes (versus 2,278 and 2,233 genes for ATCC 12228 and ATCC 14990, respectively). The percentage of GC pairs was 32.1%, which was very similar to the analogous parameters in other publicly available SE genomes. The CheckM analysis estimated the 49SCV strain genome as highly complete (98.53%) and free of contamination, which indicates a high quality of the obtained data.
Phylogenetic analysis
The AutoMLST tool revealed that the closest related genome for 49SCV was Staphylococcus spp. strain HMSC070A07 (Figure 1). A search of the analyzed genome against the PubMLST database allowed to assign 49SCV to the sequence type 2 (ST2), considered as the most prominent infection-causing SE lineage with a worldwide dissemination (Miragaia et al., 2007; Du et al., 2013; Post et al., 2017; Lee et al., 2018). It has been regarded as a hospital adapted SE line, circulating within health-care facilities, being transferred from fomites or health care personnel to patients and able to subsequently cause biomaterial-associated infections (Du et al., 2013; Lee et al., 2018; Both et al., 2021). Interestingly, the catheter-sourced and stable SE SCV representing the ST2 has already been reported (Liu et al., 2021).

Figure 1. Maximum-likelihood phylogenomic tree based on 70 concatenated core genes indicating the phylogenetic positions of SE 49SCV strain with other related species. Sulfurihydrogenibium subterraneum DSM 15120 was used as an outgroup. The GenBank genome accession numbers are shown in parentheses.
The ST2 typically carries pathogenesis-relevant genotypic (icaADBC, IS256) and phenotypic (biofilm formation) traits (Du et al., 2013; Lee et al., 2018; Both et al., 2021). Moreover, it tends to harbor a plethora of antimicrobial and antiseptic resistance genes (Lee et al., 2016, 2018; Both et al., 2021). Indeed, our previous study revealed that 49SCV was the icaADBC-positive, proficient biofilm producer (Bogut et al., 2014). The ica operon is involved in the synthesis of the polysaccharide intercellular adhesin (PIA) which contributes to the biofilm accumulation in many clinically significant SE (Fey and Olson, 2010; Rohde et al., 2010). The presence of a complete icaADBC cluster and a divergently transcribed regulatory icaR gene in the strain was further evidenced in this work (contig 22; NPW32_RS11125, NPW32_RS11120, NPW32_RS11115, NPW32_RS11110, and NPW32_RS11130, respectively). All ica genes showed a very high degree of similarity within the set of compared genomes. Nevertheless, the 49SCV strain did not meet the multiresistant profile suggested above. It demonstrated resistance to gentamicin in relation to its haemin-dependence (Bogut et al., 2014), a defective electron transport chain and the resultant decreased membrane potential necessary for the drug uptake.
Clustered regularly interspaced short palindromic repeats (CRISPR) sequences
The presence of CRISPR sequence clusters in SE leads to a reduction in the intensity of conjugation and transformation by plasmid DNA (Marraffini and Sontheimer, 2008). The CRISPR-related genes (cas1, cas2, cas6) were not found in the genome of 49SCV, similarly to the genomes of the reference ATCC 12228 and O47 strains (Raue et al., 2020). However, the CRISPRfinder tool enabled the selection of four candidate sequences, a phenomenon typical of SE genomes (Rossi et al., 2017).
Comparative genomics
The genomes of 49SCV and the two reference strains O47 and ATCC 12228 were aligned. Visualization of the alignment performed with the Mauve tool is demonstrated in Figure 2. As the 49SCV genome was sequenced in the draft form and available as 39 contigs, it was not possible to draw firm conclusions about the full structure of its genome. However, there were clear blocks of colinearity with the reference genomes, particularly O47, within the contigs. Interestingly, the O47 strain belongs to the same ST (ST2) (Raue et al., 2020) as 49SCV and was originally isolated in an analogous clinical context, that is from a patient with an orthopedic device-associated infection. The most striking differences between the 49SCV and O47 strains included the lack of Staphylococcal Cassette Chromosome (SCC) (see further in the text), the absence of genomic islands such as vSe2 or vSe7, and the lack of a sequence similar to the φO47A prophage in the former strain. It should be noted that a sequence similar to the φO47B was present, but at a different position in the genome and in the opposite orientation (Figure 2).
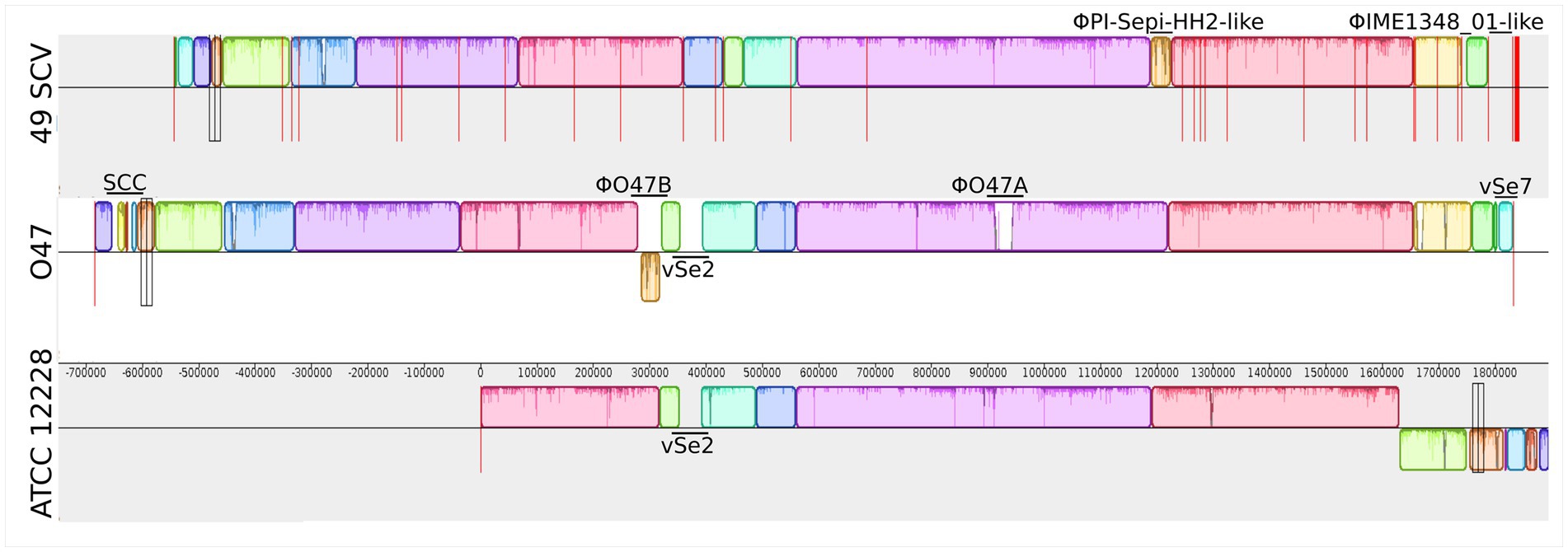
Figure 2. Mauve synteny alignment analysis of 49SCV and the reference O47 and ATCC 12228 genome sequences. Boxes of different colours represent the locally collinear blocks (LCBs). The LCB above the central horizontal line indicates that the sequence direction of the aligned genome is the same as the reference sequence, and the LCB below the reference line indicates that the sequence direction of the aligned genome is opposite to the reference sequence. White areas represent genome-specific sequence elements that may contain genome-specific sequence elements and genomic locations not sufficiently aligned between the selected genomes. Vertical red lines mark the boundaries of the contigs. The Staphylococcal Cassette Chromosome (SCC), genomic islands vSe2 and vSe7, as well as putative prophages (φO47A, φO47B, ΦPI-Sepi-HH2-like, ΦIME1348_01-like) were marked in the individual genomes.
Mobile genetic elements and prophages
Using the ISfinder database, we found several sites where mobile elements were present in the genome of 49SCV. It should be noted that the draft genome makes a precise identification of the IS integration sites difficult. This is illustrated by the fact that three contigs in the studied genome [32–34] represent IS sequences (IS1182, IS110, and IS256). In addition, a truncated IS1272 was found in contig 1, between the genes NPW32_RS00680 (encoding a translational GTPase type A) and NPW32_RS00690 (encoding YlaI family protein). In the O47 genome, the IS1272 element was located at the same position. The second site was contig 2, where IS3 was located between the NPW32_RS02965 (uhpT gene) and NPW32_RS02975, coding for the SDR family oxidoreductase. The location was analogous to that in O47 (Raue et al., 2020). However, in both cases the transposase component of IS was a non-functional gene (in case of IS1272, it contained a number of nonsense mutations, while in IS3 there was a deletion leading to a frameshift).
Unlike the O47 genome, 49SCV contained a single complex transposon, Tn552 (Rowland and Dyke, 1990). Specifically, the transposon integration site was located within the gene NPW32_RS03445, which encodes a transcriptional regulator of the ArgR family, resulting in its truncation and the appearance of a stop codon after 83 codons. Most notably, the integration resulted in the deletion of a fragment of approximately 12 kbp in size, covering genes FHQ17_RS01225 to FHQ17_RS01275 for the O47 reference. A detailed description of the deleted genes can be found in subsequent sections.
Two prophage sequences were found in the 49SCV genome. One of them was assembled into a separate contig (ΦIME1348_01-like, locus tag: NPW32_RS09955 to NPW32_RS10275, contig 16). The second prophage sequence ΦPI-Sepi-HH2-like was located in contig 1 and was flanked by two genes encoding tRNA (NPW32_RS00090 - tRNA-Asn, NPW32_RS00095 - tRNA-Ser) on one side, and by a truncated gene encoding a plasmid recombination protein (NPW32_RS00320) on the other. Both sequences had a similar size of approximately 38 kbp, allowing them to be classified as Staphylococcus class II Siphoviridae phages (Deghorain and Van Melderen, 2012). The CheckV tool analysis characterized them as complete, which was consistent with the results described for the phage sequences present in the O47 genome. Blastn search against non-redundant nucleotide database showed that the first prophage sequence (ΦIME1348_01-like) was most similar to bacteriophage IME1348_01 (NC_055036), while the second one (ΦPI-Sepi-HH2-like) was most similar to PI-Sepi-HH2 (MT880871) (Figure 2).
As in other SE genomes, genomic islands were identified in 49SCV. However, only νSeγ, vSe3, and vSe5 islands were present in comparison with the reference genomes. Despite the overall similarity of 49SCV to the O47 genome, vSe2, vSe6, and vSe7 sequences were not identified here and their absence adds to the pool of numerous 49SCV deletions discussed in later sections.
Streamline of genetic content as a possible strategy associated with the SCV phenotype and its adaptation to chronicity
49SCV is devoid of the Staphylococcal Cassette Chromosome (SCC)
Comparative genomic analysis revealed the lack of the SCC element in 49SCV which was found in all the analyzed reference genomes (Figure 3). These genomic islands are disseminated among staphylococci and carry antimicrobial resistance and virulence-associated genes (Shore and Coleman, 2013). The most representative SCCs are the SCCmec elements harboring the mecA or mecC genes. These mobile genetic elements constitute a defining feature of methicillin-resistant staphylococci (MRS) which demonstrate a broad-spectrum beta-lactam resistance (Shore and Coleman, 2013; Lakhundi and Zhang, 2018). The SCCs carrying genes other than mec have also been identified and reported to harbor diverse genes, including those involved in fusidic acid resistance, capsule synthesis or mercury resistance. The non-mec SCCs share mutual characteristics with SCCmec by carrying cassette chromosome recombinase (ccr) genes in a ccr gene complex, integration at a specific integration site sequence (ISS) in the chromosome, and the occurrence of flanking direct repeat containing ISS (Lakhundi and Zhang, 2018). For example, the genome of O47, for which a high level of collinearity with 49SCV was revealed, contains a 54-kb non-mec SCC (Raue et al., 2020).
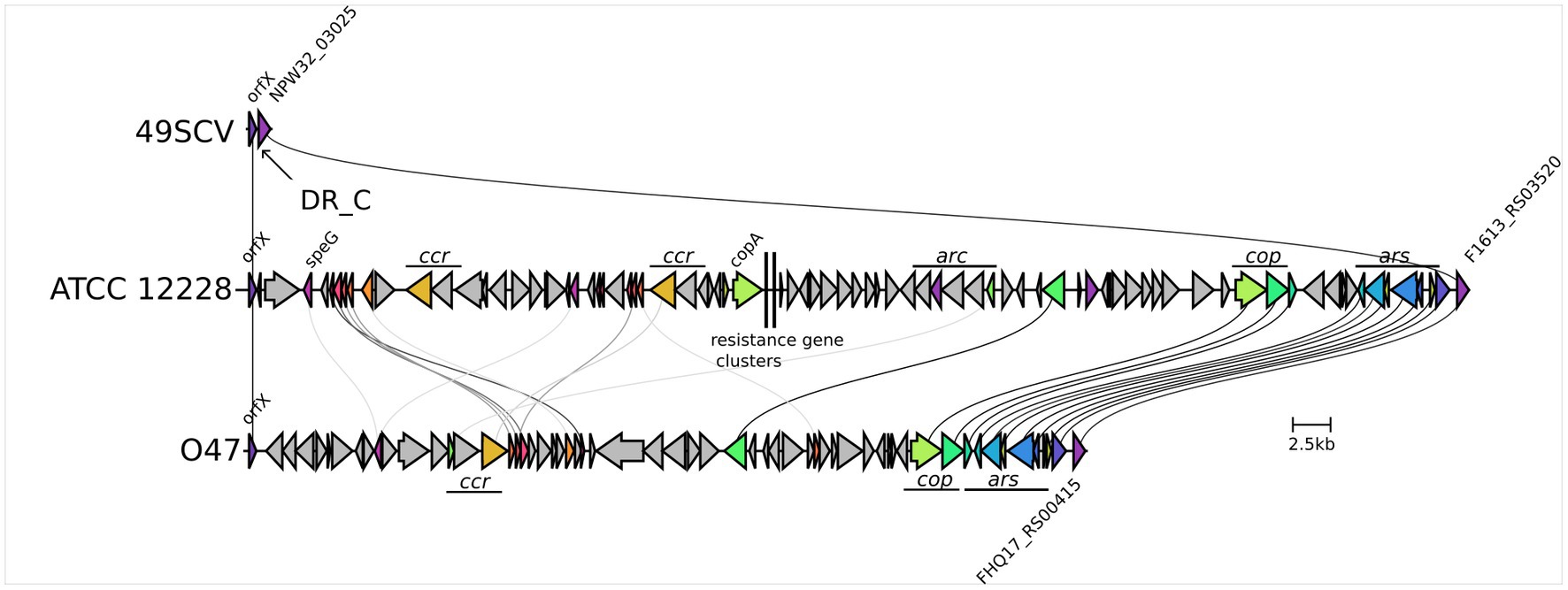
Figure 3. Genetic organisation of the SCC region in 49SCV and in the ATCC 12228 and O47 reference strains. The 49SCV strain is devoid of the SCC element, so only the excision site is marked, flanked on one side by the orfX (with the DR_C sequence marked) and by the NPW32_RS03025 gene on the other side. Sequence similarity of the individual genes greater than 30% was marked with lines connecting the individual CDSs. The same information was expressed by the common colours of the genes in question. In the case of ATCC 12228 and O47 strains significant functional blocks of genes specific to their SCC were also marked. The visualisation was created using the clinker tool.
Although the SCC has been considered to play a role in staphylococcal adaptation through the acquisition of additional antimicrobial resistance and virulence genes, its loss can be advantageous, as it may compensate for the fitness cost (Noto et al., 2008). Lin et al. (2016) reported isolation of daptomycin-resistant SA SCVs of the vancomycin-intermediate (VISA) form belonging to the hospital-acquired MRSA lineage during a long-term daptomycin treatment of septic arthritis. In addition to a stepwise development of ultrastructural changes and mutations in SCVs, the loss of SCCmec was observed. Conversion to vancomycin resistance has been reported to cause a decrease in the growth rate of MRSA in another study and shown to be partially compensated by the mecA deletion. Documented deletions within SCCmec during SE infection have also been published (Sendi et al., 2005; Weisser et al., 2010).
49SCV demonstrates a ~ 12 kb genomic deletion including the loss of genes involved in the arginine deiminase pathway (ADI)
Genomic comparisons revealed that 49SCV strain was devoid of a ~ 12 kb genomic region present in the analyzed reference strains. The deletion has been substituted with the Tn552 composite transposon. One of the most striking findings highlighting the potential biological significance of this deletion is the consequent loss of genes included in the ADI pathway, which is highly conserved in bacteria and represents a widespread anaerobic route for arginine degradation. The ADI pathway comprises three essential metabolic steps: conversion of L-arginine into L-citrulline and ammonia (1) catalyzed by arginine deiminase (ArcA); transfer of carbamoyl moiety of L-citrulline to phosphate (2) by catabolic ornithine transcarbamylase (ArcB/ArgF) which yields carbamoyl phosphate and L-ornithine; and phosphorylation of ADP (3) by carbamate kinase (ArcC) which occurs with the use of carbamoyl phosphate and results in the production of ATP, carbon dioxide, and ammonia. A key player in the ADI pathway is the L-arginine/L-ornithine antiporter (arcD gene) that catalyzes a stoichiometric exchange of one molecule of L-arginine and one molecule of L-ornithine thereby mediating a concomitant uptake of the substrate L-arginine and excretion of the end product L-ornithine. Since metabolic energy is not required for the transport reaction, ATP produced by the ADI pathway can be used for other energy-demanding purposes (Noens and Lolkema, 2017). This metabolic pathway functions as an alternative source of energy for bacterial growth, which is particularly important if neither glucose nor nitrate are available (Makhlin et al., 2007). Moreover, it has been considered a defense mechanism against acidification due to the production of ammonia (Lindgren et al., 2014; Noens and Lolkema, 2017).
Transcription of the arc operon is often arginine dependent via the ArgR proteins belonging to the Crp/Fnr-family of transcriptional regulators (Lindgren et al., 2014). Of note, a gene encoding for the ArgR regulator (NPW32_RS03445) has been found truncated in the 49SCV strain and followed by a Tn552-derived sequence (described above). The argR homolog was followed by three genes encoding components of the ADI pathway (arcA, argF, and arcD) and a gene encoding for the yet another Crp/Fnr-family transcriptional regulator in all reference SE genomes. Makhlin et al. (2007) demonstrated that transcription of the arc operon under anaerobic conditions depends strictly on the functional ArgR (Makhlin et al., 2007). Seggewiss et al. (2006) reported that a hypothetical gene of the Crp/Fnr family (SA2424) being a part of the ADI pathway showed significantly increased transcription in the laboratory-constructed hemB mutant of a clinically derived SA SCV strain. Their finding was correlated to up-regulation of the ADI pathway in the mutant strain. The 49SCV apparently lacked the function of both transcriptional regulators of the Crp/Fnr-family.
Genes encoding for enzymes involved in the metabolism of arginine are often duplicated in staphylococci, with a second copy of the arc operon contained within the majority of an arginine catabolic mobile element (ACME) allotypes (McManus et al., 2019). ACME is commonly collocated adjacent to SCCmec or SCC-associated genes in composite islands. Its carriage has been considered advantageous for bacterial transmission, persistence and survival (McManus et al., 2019). This mobile element was absent in 49SCV which was not surprising due to the lack of the SCC element to which ACME is commonly adjacent. Thus, while one or two genes encoding for the arginine deiminase were identified in the reference strains (core, +/− ACME derived), 49SCV was devoid of any homologous arcA genes. Only a residual genetic composition of the ADI pathway was detected in the strain, including the argF and arcC genes encoding for the ornithine transcarbamylase and carbamate kinase (NPW32_RS02705 and NPW32_RS02710), respectively, and detected elsewhere in the genome.
Our findings indicate the loss of activity of this energy-generating metabolic pathway and stand in contrast to several previous studies. Electron-deficient staphylococcal SCVs (including hemin- and menadione-dependent variants), have been reported to demonstrate an increased expression of the ADI pathway (Kohler et al., 2003, 2008; Seggewiss et al., 2006) as an alternative strategy for ATP production able to compensate for its loss due to defective utilization of a variety of carbon sources, including tricarboxylic cycle intermediates, and electron transport interruption (Kohler et al., 2003, 2008; Kriegeskorte et al., 2014). On the other hand, genes involved in the catabolism of arginine and ornithine were found among the most strongly down-regulated genes in a stable SA SCV strain (exhibiting no classical auxotrophy) isolated from a patient with a PJI relapse (Loss et al., 2019). The 49SCV strain was probably deficient in ATP production not only due to the lack of the functional ADI pathway but also due to a dysfunctional electron transport as it was hemin-dependent. The inhibition of respiratory pathways resulting from mutations in menadione and hemin biosynthesis genes exert physiological effects similar to those experienced in a low-oxygen environment. Since transcriptomic analyses were not performed in our study, we can speculate that other metabolic processes must have provided ATP levels necessary for the strain survival and growth. Accordingly, SCV mutations can be expected to cause upregulation of genes involved in anaerobic metabolism as an adaptive and compensatory response (Cao et al., 2017). Staphylococcal SCVs have been reported to generate ATP mainly from glucose or fructose by substrate phosphorylation which was evidenced by up-regulation of enzymes involved in glycolytic and fermentative pathways (Kohler et al., 2003, 2008; Seggewiss et al., 2006). Additionally, SCVs have been shown to use other optimizing reactions that result in production of ATP including the increased metabolism of glycerol, pyruvate, butanoate/acetoin, and nitrate (Proctor, 2019).
It is tempting to speculate that deletion of specific genetic elements including the SCC cassette and arc genes in 49SCV may represent a strategy adopted to streamline the genetic content and improve bacterial ability to infect and survive in the host. According to Both et al. (2021), hyper-variable regions of ACME and SCCmec may be a burden in infection and only confer an advantage in some conditions on the human skin as deletions in these regions seemed to occur preferentially in SE strains associated with infection.
Frameshift-generating events within homopolymeric tracts – phase variation in the background of the SCV phenotype
In order to obtain a complete view of the differences between the 49SCV strain and the reference genomes, especially within the mutated genes, the variant calling against five reference strains using the snippy tool (Seemann, 2023b), which identifies SNPs, MNPs, insertions, deletions and compound mutations was carried out.
Approximately 200 genes with frameshift mutations were identified in the 49SCV genome. Of note, a large proportion of the frameshift-generating events occurred within specific short sequence repeats (SSRs), namely poly(A) and poly(T) homopolymeric tracts (HTs). The SSRs are highly prone to insertion/deletion (indel) errors due to Slipped-Strand Mispairing (SSM) that occurs during DNA replication or Mismatch Repair (MMR). Variations in the length of SSRs account for one of the three major genetic mechanisms of bacterial phase variation (PV), in addition to the DNA inversion and recombination. The PV, defined as an adaptive mechanism based on the reversible gene expression switch, enables microorganisms to meet the challenge of fluctuating pressures existing in their environment (Gor et al., 2021). Gor et al. found that more than 700 genes in the genome of a highly virulent community-acquired methicillin-resistant SA strain (MW2) contained at least one poly(A) or poly(T) SSR, with a substantial number containing 3–4 SSRs (Gor et al., 2021). In an extensive study of 99 prokaryotic genomes, Orsi et al. (2010) revealed that poly(A) and poly(T) HTs were overrepresented in prokaryotic genes, similarly to our findings. In the analyzed 49SCV genome, 4,365 poly(A) and poly(T) HTs equal to or longer than 6 bp were identified in 1710 coding sequences (CDSs) [2.55 HTs per CD on average], whereas 1,483 of them were localized in intergenic regions (Figure 4).
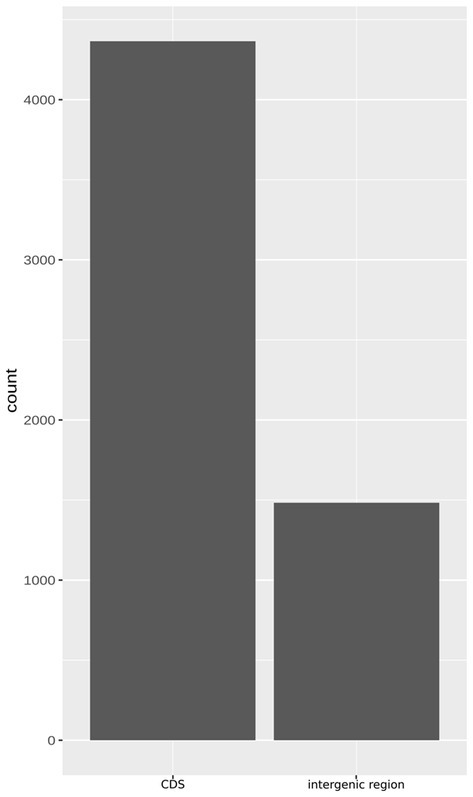
Figure 4. The bar plot demonstrating the abundance of poly(A) and poly(T) homopolymer tracts (HTs), understood as six or more nucleotides of the same type, within gene and intergenic sequences in the genome of 49SCV.
While the HTs predominated in the CDSs, indels within the HTs were found predominantly in the intergenic regions in the 49SCV genome, irrespective of the reference genome used for comparison. At the same time, however, 38–51 mutations in CDSs were also identified (Figure 5). Most of these frameshift-generating mutations in the CDSs were insertions, followed by deletions and compound (two different independent changes within one HT) mutations (Figure 5). The predominance of insertions over other types of mutations was less pronounced for intergenic regions (Figure 5). Indels in the SSRs can result in frameshifts that have an ON↔OFF effect on gene expression or protein function, depending on the SSR’s location (Gor et al., 2021). If alterations in the length of the SSRs occur in the promoter regions, inhibition of the RNA polymerase binding or a gradation effect on the gene expression can occur (Gor et al., 2021). A shift in the translational reading frame usually results in the synthesis of a truncated and a non-functional protein. In particular, indels at the 5′ end of a coding sequence usually lead to expression of short, non-functional peptides (Orsi et al., 2010). Orsi et al. (2010) revealed the preferential location of HTs at the 5′ ends of genes, indicating selective pressure for the presence of these HTs consistent with their role as regulatory elements. In line with these findings, approximately 70% of indels detected in the poly(A)/poly(T) HTs in 49SCV carried mutations at the 5′-end. Moreover, results of our study revealed that most indels occurred in the poly(A) HTs (Table 1), similarly to the findings of Gor et al. (2021).
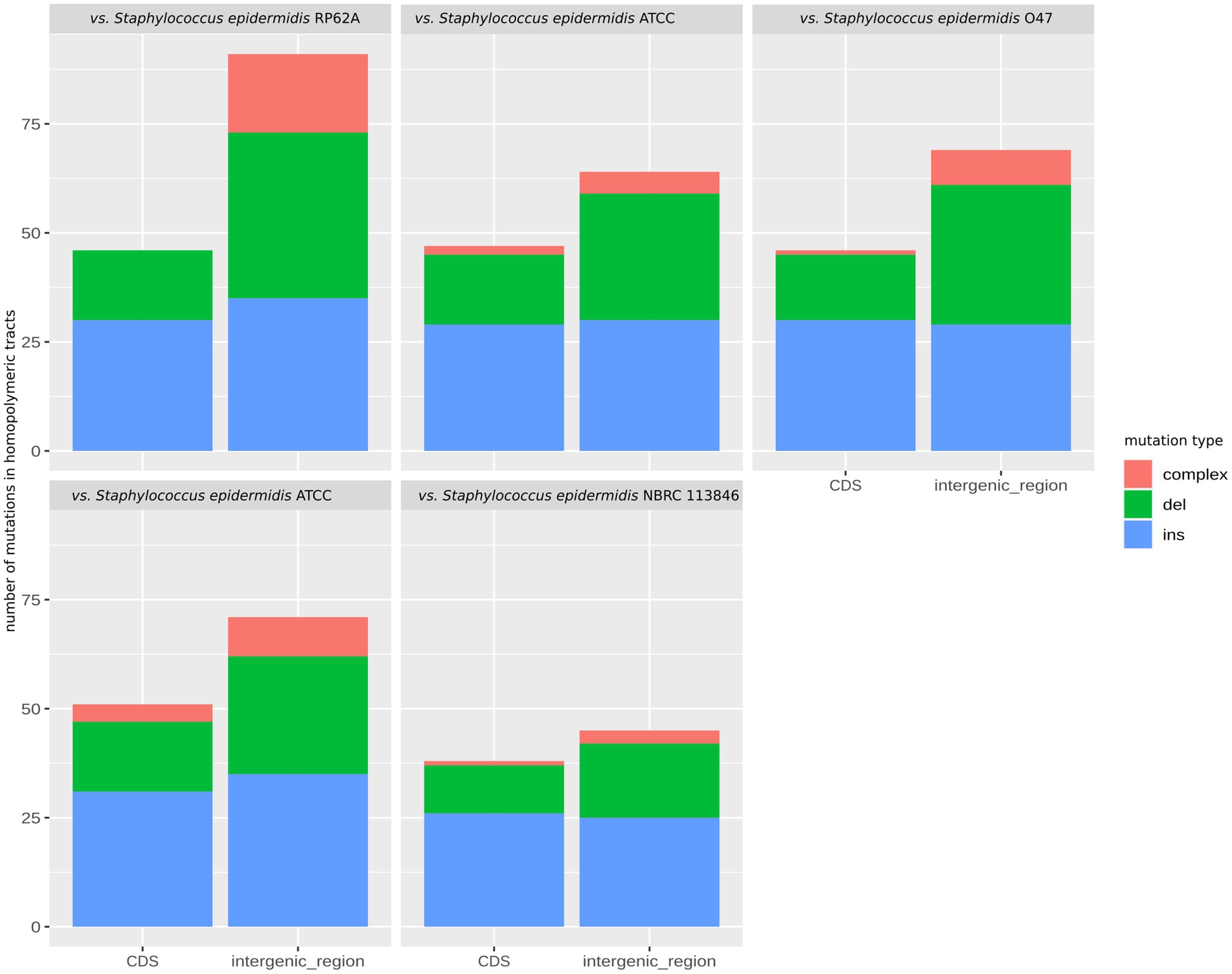
Figure 5. The faceted stacked bar chart showing the number of mutations in the homopolymeric tracts categorized as deletions, insertions, and complex mutations in the genome of 49SCV. The bars represent mutations within coding sequences (CDS) and intergenic regions, while the facets depict mutations observed when comparing with different reference genomes.
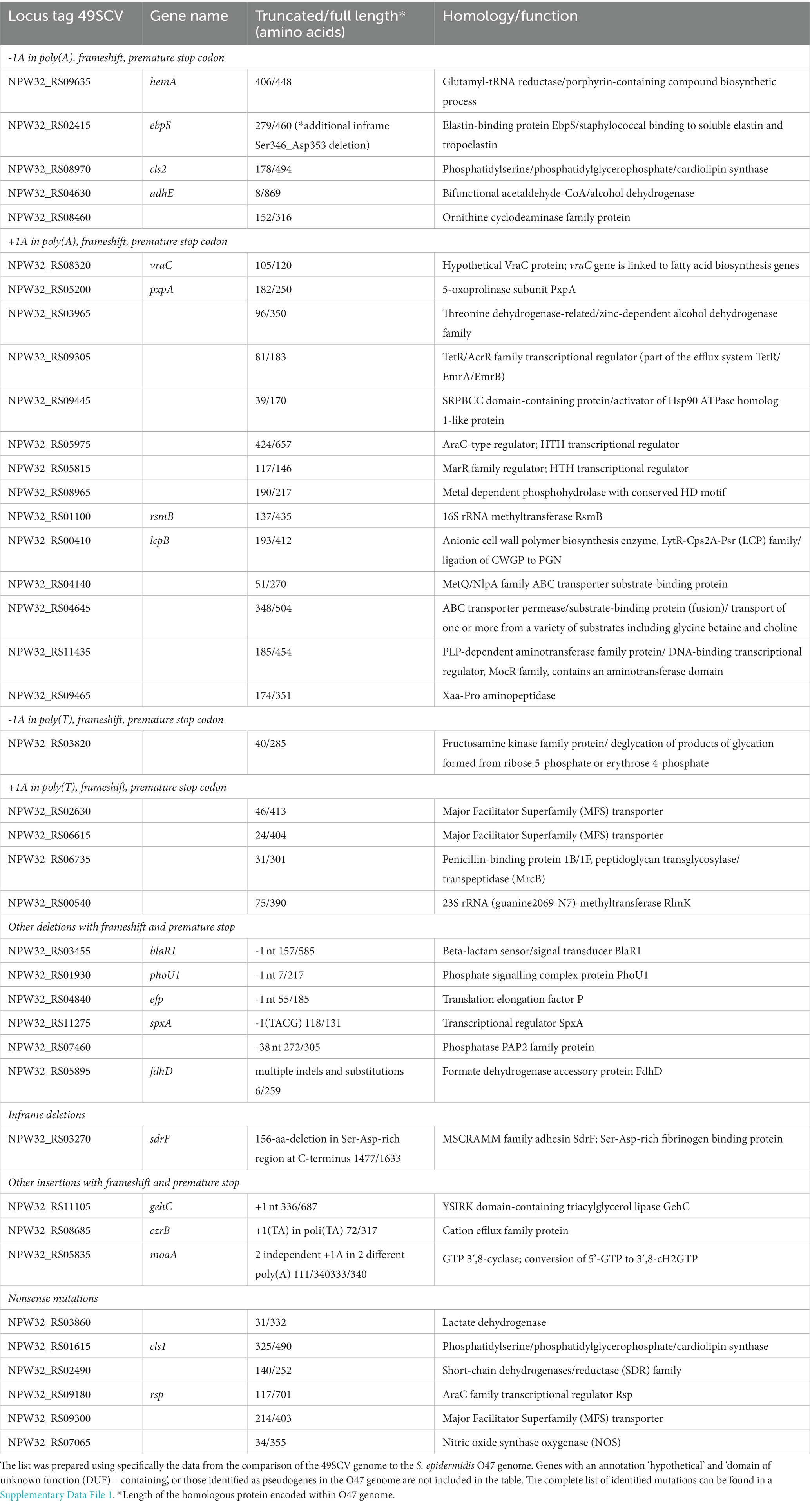
Table 1. Genes in the 49SCV genome encoding proteins that are truncated due to insertions/deletions within poly(A)/poly(T) tracts and di/tetranucleotide repeats, deletions beyond HTs, internal in-frame deletions or nonsense mutations.
Detection of frameshift-generating mutations within the HTs can suggest an association between PV events and the SCV phenotype. To the best of our knowledge, PV occurring by insertion or deletion of tandem repeats has not been described to occur for staphylococcal SCVs so far. The only investigation linking formation of the SCV phenotype to genetic events reminiscent of the PV phenomena has been published by Kleinert et al. (2017). As evidenced experimentally in their study, transposition and subsequent excision of IS256 can mediate formation and fast reversion of SA SCV-like phenotypes, respectively. Introduction of IS256 into a laboratory strain followed by cultivation in the presence of antibiotics resulted in the isolation of SCVs that possessed IS256 insertions in guaA and hemY genes and displayed increased resistance to vancomycin and aminoglycosides, respectively.
Genes subject to the PV typically encode cell-surface associated proteins including adhesins and pili but also virulence factors and secreted proteins (Orsi et al., 2010; Gor et al., 2021). It should be noted that indels in the HTs of 49SCV genome occurred in the loci whose biological functions have been reported to be associated with bacterial adherence, metabolism, stress response, virulence, and cell wall synthesis (Table 1). Although we cannot provide the evidence of a direct impact of these genetic events on the expression of proteins, we propose that indels in the HTs within open reading frames (ORFs) can constitute an important background of the SCV phenotype, since a resulting truncation of the corresponding proteins makes their biological dysfunction highly probable.
Deletion in the poly(A) of the hemA gene as a possible trigger factor for phenotype switching
One-nucleotide (1-nt) deletion in the poly(A) HT was detected at the 3′-end of the hemA gene (NPW32_RS09635) leading to the generation of a premature stop codon and shortening of the encoded protein by 42 amino acids (Table 1). This gene encodes a glutamyl-tRNA reductase involved in the first step of porphyrin biosynthesis. HemA dimer, which is a probable functional form of the enzyme, forms a complex with glutamate−1-semialdehyde 2,1-aminomutase, the second enzyme in the pathway, suggesting metabolic channeling of a reactive intermediate glutamate-1-semialdehyde. Mutants of hemA are dependent on δ-aminolevulinic acid (ALA) for growth and are unable to produce ALA from either glutamate or glutamyl-tRNA (EcoCyc, 2013; Kahl et al., 2016). We hypothesize that truncation of the hemA ORF and a consequent synthesis of a dysfunctional HemA protein was a trigger factor for the phenotype switching in 49SCV and a primary reason for the development of hemin auxotrophy in the strain. Functional or genetic defects in the biosynthesis of heme, which is used in the cytochrome biosynthesis, lead to defects in electron transport. As a result, the amount of ATP that is available for the cell wall biosynthesis is drastically reduced. Consequently, electron transport-defective SCVs reveal a lower growth rate and small colonies on agar media (Kahl et al., 2016). Moreover, hemA truncation in 49SCV and the subsequent dysfunction of the electron transport system is the most conceivable explanation for a high gentamicin MIC (>256 mg/L) reported previously (Bogut et al., 2014). Uptake of this drug is dependent on the membrane potential, which is significantly reduced when the flow of electrons in the electron transport chain is impaired (Vestergaard et al., 2016). Mutations in the hemA gene have been reported in SCVs but only in laboratory-derived mutants of SA. Schaaf et al. characterized genes of the hem operon in a stable gentamicin-induced, hemin auxotrophic SA SCV and revealed a deletion in hemH and a frameshift at the 3′ end of hemA. This frameshift mutation led to truncation of the protein by 54 amino acids (Schaaff et al., 2003). Lannergård et al. (2011) analyzed FusE mutants selected from a drug-susceptible laboratory wild-type SA strain displaying fusidic acid resistance and the SCV phenotype. In addition to mutations in the rplF gene (encoding for a ribosomal protein L6), four hemin-auxotrophic strains had missense mutations in hemA, hemB or hemH genes. Truncation of the hemA gene in 49SCV is unique due to a deletion in a poly(A) tract. It is worth noting that in comparison to the ortholog from the O47 strain, the hemA gene also carried three missense mutations: Ile84Met, Glu89Gln, and Ala357Val (Table 2), which might have further contributed to its dysfunction.
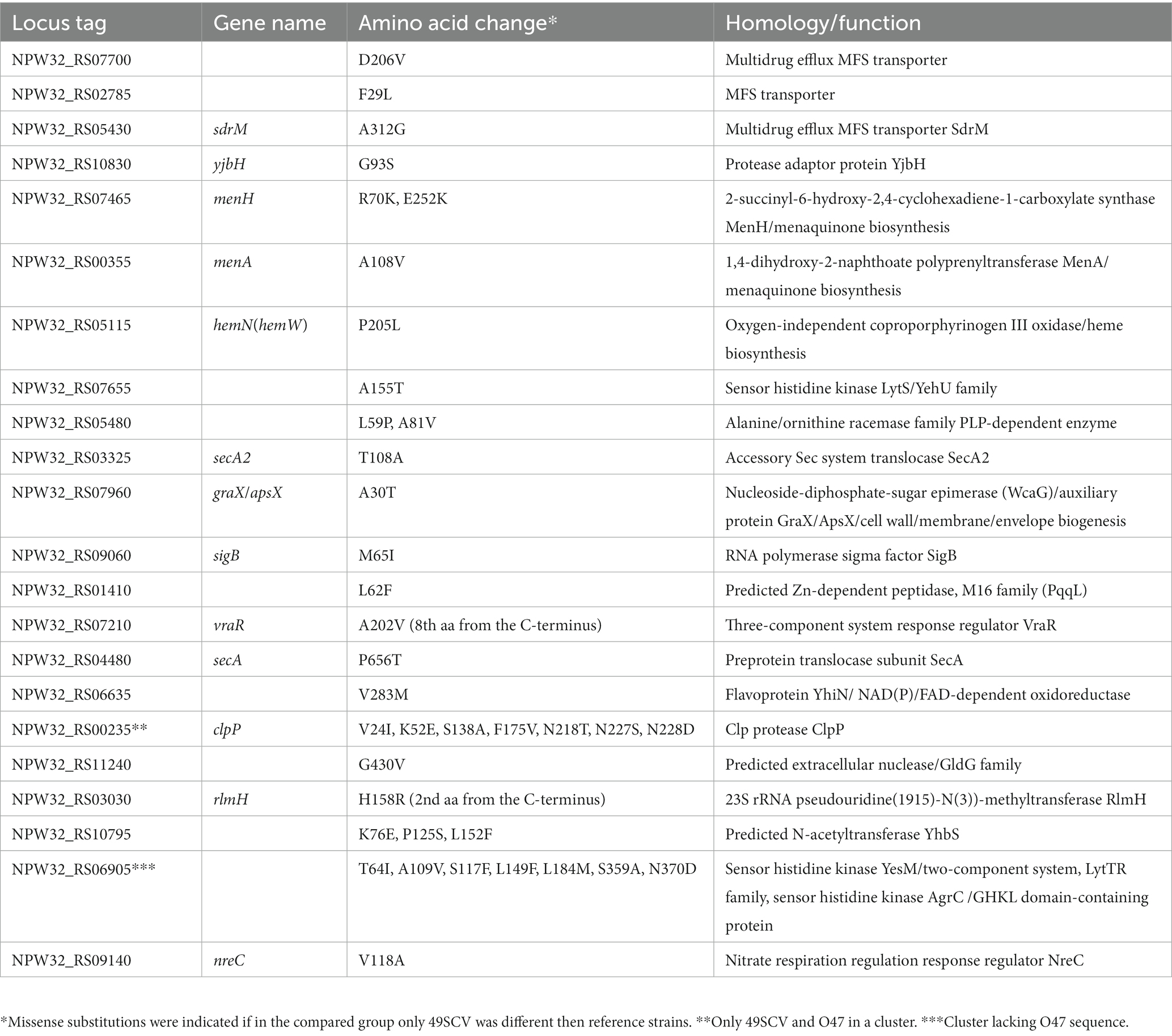
Table 2. Genes, belonging to pangenomic gene clusters, for which the predicted amino acid sequence was specific only to the strain 49SCV when compared to the five reference strains.
Other mutations within the HTs and beyond them – their possible association with the SCV phenotype
Genes associated with adhesion and growth rate
Staphylococci possess a wide range of structurally-related surface proteins with adhesive properties and many of them belong to a structurally related family of microbial surface components recognizing adhesive matrix molecules (MSCRAMMs), that facilitate colonization step in the pathogenesis of prosthetic device infections (Arrecubieta et al., 2009). Two genes encoding for the MSCRAMMs (ebpS, sdrF) have been found truncated in 49SCV which can suggest an association with the strain phenotype.
A conservative inframe deletion of an internal gene fragment encoding 156 alternating amino acid residues in a specific, C-terminal Ser-Asp-rich domain was identified in the sdrF gene (NPW32_RS03270). SdrF produced by SE binds collagen and has been reported to play a significant role in the initiation of ventricular assist device driveline infections (Arrecubieta et al., 2009). Another work reported the involvement of SdrF in the adherence to human keratin and suggested its role in facilitating skin colonization by SE (Trivedi et al., 2017). Hence, its role in SE pathogenesis remains to be elucidated.
Genetic changes in the ebpS gene (NPW32_RS02415) encoding an elastin-binding protein (EbpS) included a conservative inframe deletion of several codons and one-nucleotide deletion in the poly(A) resulting in shortening of the protein by 181 amino acids (Table 1). We assume that accumulation of mutations in this gene might have had a potential biological significance. The EbpS protein has been reported to mediate staphylococcal binding to soluble elastin and tropoelastin (Campoccia et al., 2009) but one of the previous studies demonstrated that the adherence to immobilized elastin in SA is rather mediated by fibronectin-binding proteins, and inactivation of the ebpS has a minimal effect on bacterial binding to elastin peptides (Roche et al., 2004). Interestingly, Campoccia et al. (2009) identified a group of new variant forms of the ebpS gene in SA clinical strains isolated from implant-related orthopedic infections which were shortened for 180 bp. The corresponding protein amino acid sequence lacked an entire peptide segment of 60 amino acids implicating the disappearance of its entire hydrophobic domain (Campoccia et al., 2009). Nakakido et al. (2014) suggested that despite its relatively weak contribution to adhesion, EbpS may play other important roles including the regulation of the biofilm formation that are zinc concentration dependent. Importantly, the authors observed a considerably decreased growth rate of the EbpS-deficient mutant strain. Similarly, the EbpS expression was correlated with the ability of cells to grow to a higher density in liquid culture in another study, implying its role in regulating cell growth (Downer et al., 2002). It can be hypothesized that truncation of the EbpS protein might have an association with a slowly growing phenotype of 49SCV. One of the most recent works suggested that downregulation of genes encoding surface proteins can be beneficial for immune evasion and survival of SCVs due to reduced adhesion to phagocytic cells (Liu et al., 2023). It can support our assumptions regarding the possible involvement of the ebpS and sdrF mutations in the SCV phenotype.
The elongation factor P (EF-P) is a universally conserved translation factor that alleviates ribosome pausing at polyproline motifs by facilitating peptide bond formation. In the absence of EF-P, a polyproline peptide bond formation can limit translation rate, leading to pleiotropic phenotypes including slowed growth, increased antibiotic sensitivity, and the loss of virulence (Balibar et al., 2013; Tollerson et al., 2018). The efp gene has been classified among genes important for survival and growth of SA (Chaudhuri et al., 2009). The efp ortholog in 49SCV (NPW32_RS04840) was found to carry 1-nt deletion resulting in a frameshift and protein shortening by 130 aa which can suggest a loss-of-function effect. Due to the critical role of EF-P in cell growth and virulence its truncation could have been involved in the 49SCV phenotype.
The pxpA homolog in NPW32_RS05200 locus encoded a protein truncated by 68aa when compared to its homolog in the genome of the O47 strain. The pxpA-encoded 5-oxoprolinase catalyzes the cleavage of 5-Oxo-L-proline (OP) to form L-glutamate which is coupled to the hydrolysis of ATP to ADP and inorganic phosphate (UniProtKB|UniProt, n.d.). The OP is considered one of the major metabolite damage products. Its accumulation is deleterious and it can lead to a bacterial growth inhibition (Park et al., 2001). In the study of Niehaus et al. inactivation of pxpA, pxpB, or pxpC genes in Bacillus subtilis slowed growth, caused OP accumulation in cells and medium, and prevented its use as a nitrogen source (Niehaus et al., 2017). These results are suggestive of a possible contribution of the pxpA gene truncation to the slowed growth of 49SCV.
Genes associated with the cell membrane and cell wall composition
Genetic events that might have led to dysfunction of the corresponding proteins were detected in two cardiolipin (CL) synthase genes, namely cls1 and cls2. The NPW32_RS08970 locus was found to carry a cls2 homolog encoding a truncated protein due to a deletion in the poly(A) HT which generated a frameshift and a premature stop codon (178 aa/494 aa). The other gene (NPW32_RS01615) – cls1 homolog - encoded a truncated protein variant due to a nonsense mutation (325aa/490aa) (Table 1). The CL synthase mediates condensation of two phosphatidylglycerol (PG) molecules to yield cardiolipin and glycerol. The phospholipid composition of bacterial membranes undergoes changes triggered by the growth phase or environmental stressors. In actively growing SA, the predominant phospholipid is PG, whereas CL becomes predominant in a stationary growth phase, with a corresponding decline in the PG content. Similar changes have been observed under conditions of osmotic stress, energy deprivation, or after phagocytosis by polymorphonuclear leukocytes (PMN) (Koprivnjak et al., 2011; Ohniwa et al., 2013). Both cls1 and cls2 participate in the CL accumulation in SA, where the cls2 gene serves a housekeeping function and the cls1 is active under stress conditions including high salinity or low pH (Tsai et al., 2011; Ohniwa et al., 2013). Deletion of both cls genes was found to abolish the CL synthesis (Koprivnjak et al., 2011; Tsai et al., 2011). Membrane composition can be modulated and other components such as the PG can compensate for the stalled function of the cls genes, which might have been the case for the 49SCV strain. The PG level was observed to be increased in mutants that did not accumulate CL (Tsai et al., 2011).
The vraC gene homolog in NPW32_RS08320 locus encodes a protein truncated by 15C-terminal amino acid residues (Table 1). The gene is a part of the cluster of five genes transcribed in the same direction: the gene encoding a HAD family hydrolase (NPW32_RS08335), vraA encoding a long chain fatty acid CoA ligase or acyl-CoA synthetase (EC 6.2.1.3; NPW32_RS08330), vraB encoding an acetyl-CoA C-acetyltransferase (EC 2.3.1.9; NPW32_RS08325), vraC (with no particular annotation), and a gene encoding a hypothetical protein (NPW32_RS08315). This gene cluster is highly syntenic in staphylococci, and proteins encoded in NPW32_RS08320 - NPW32_RS08315 loci are reminiscent of the recently described VraC and VraP proteins (Wang and Sun, 2021). One study showed that vraC was co-transcribed with vraP located downstream of vraC and that encoded proteins may form a stable complex to play a key role in the cell wall metabolism. VraCP promoted the expression of the cell wall synthesis (glyS, sgtB, ddl and alr2) and hydrolysis (sceD, lytM and isaA) genes which influenced the cell wall thickness, antibiotic resistance and autolysis rate. Deletion of vraC, vraP and vraCP led to phenotypic alterations, including increased susceptibility to the cell wall-associated antibiotics, reduced cell wall thickness and decreased autolysis (Wang and Sun, 2021). It can, therefore, be assumed that VraC truncation in 49SCV could have played a role in the cell wall remodeling reported previously for SCVs (Onyango et al., 2013).
A hypothetical threonine dehydrogenase/zinc-dependent alcohol dehydrogenase family encoding gene in the NPW32_RS03965 locus was found disrupted by insertion in the poly(A) leading to truncation of a resultant protein (96aa/350aa) (Table 1). The enzyme L-threonine dehydrogenase was shown to be the first enzyme of the pathway converting threonine to glycine (Newman et al., 1976). The dehydrogenase pathway degrades threonine in two steps to acetyl-CoA and glycine. Acetyl-CoA is required for carbon source utilization, whereas glycine and its subsequent degradation are required for nitrogen source utilization (Reitzer, 2014). Glycine also represents the principal ingredient of bridges linking peptidoglycan units in staphylococci (Liu et al., 2021). Liu et al. (2021) reported that genetic mutations in a stable catheter-derived SE SCV were detected in genes involved in an array of cellular metabolic processes including glycine and threonine metabolism. While contents of most detected amino acids increased significantly in the SCV strain, glycine demonstrated a significantly lower level. The reduced glycine concentration was interlinked with the frameshift in SERP1287 gene, whose product is the alanine-glyoxylate aminotransferase (AGXT1) that catalyzes synthesis of glycine from alanine or glyoxylate (Liu et al., 2021). Their results make a potential association between the frameshift-generated truncation of the threonine dehydrogenase and dysfunction of one of the pathways of glycin biosynthesis in 49SCV conceivable. The resultant shortage in glycine might, in turn, have a negative influence on the growth of the strain by decreasing the peptidoglycan-linking rate.
It was also revealed that 49SCV harbored three lcp genes (NPW32_RS01825, NPW32_RS06035 and NPW32_RS00410 loci encoding for LcpA, LcpC, and LcpB proteins, respectively) whose products are essential for the optimal cell separation. The lcp-encoded LytR-CpsA-Psr (LCP) transferases are involved in the final ligation step of the cell wall glycopolymers to the peptidoglycan backbone (Stefanović et al., 2021). The lcpB gene had an insertion within the poly(A) resulting in frameshifting and the protein truncation (193aa/412aa). Dysfunctional LcpB could have been linked to the SCV phenotype and impaired growth rate. Following characterization of single-, double- and triple-deletion lcp mutants in SA, distinct phenotypes for each of the three proteins were suggested, including LcpA as the one involved in cell separation and septum formation, LcpB protecting cells from autolysis, and LcpC enhancing the properties of lcpA and lcpB mutants when deleted (Stefanović et al., 2021). Deletion of all three LCP genes resulted in complete teichoic acid loss in staphylococcal cell walls whereas deletion of any of individual LCP genes disturbed their attachment (Chan et al., 2013). Previous studies showed that the loss of one or more of the lcp genes can be interlinked with an aberrant septum and biofilm formation, increased susceptibility to β-lactam antibiotics, autolysis, induction of the cell wall stress responses, and reduced levels of envelope phosphate (Hübscher et al., 2009; Over et al., 2011; Dengler et al., 2012; Chan et al., 2013). A severely defective growth phenotype was observed in the triple lcp mutant. On the other hand, bacterial growth could be rescued to varying degrees by any one of the three proteins, suggesting some functional redundancy and flexibility in maintaining cell division (Over et al., 2011).
Undecaprenyl phosphate (UP) is an essential compound in the biosynthesis of bacterial extracellular polysaccharides including peptidoglycan and teichoic acids. It is produced by the dephosphorylation of undecaprenyl diphosphate (UPP) via de novo synthetic and recycling pathways. Gram-positive bacteria contain remarkable amounts of undecaprenol, which is phosphorylated to UP. Dephosphorylation of UPP is catalyzed by the BacA homolog and the type-2 phosphatidic acid phosphatase (PAP2) homolog. The presence of one of these UPP phosphatases is essential for bacterial growth (Kawakami and Fujisaki, 2018). The 49SCV strain had three genes annotated as the pap2 homologs (NPW32_RS02135, NPW32_RS07460, NPW32_RS10675) among which NPW32_RS07460 showed a deletion resulting in protein truncation by 23 amino acids. Dysfunctional protein might have led to impairment of the cell wall synthesis in the strain.
Spherical cocci synthesize peptidoglycan at the septum only. This process involves two essential penicillin-binding proteins (PBPs), namely PBP1 (transpeptidase) and a bifunctional PBP2 (transpeptidase and transglycosylase) (Pinho et al., 2013). The NPW32_RS06735 locus in 49SCV encoded a protein with a similarity to penicillin-binding proteins 1B/1F and peptidoglycan transglycosylase/transpeptidase family (MrcB). It can be speculated that a substantial shortening (31 aa/301 aa) of this protein due to a 1 nt insertion in the poly(T) and a premature stop codon might have had an effect on the peptidoglycan synthesis and cell division in this strain. One of the previous studies showed that the loss of PBP1 or its C-terminal domains, which can bind peptidoglycan and potentially coordinate the cell division process, result in cessation of division at the point of septal plate formation. The loss of PBP1 transpeptidase activity was linked to a phenotype of thickened and aberrant septa (Wacnik et al., 2022).
Genes associated with virulence
A 1-nt insertion resulting in the frameshift in the gehC gene (NPW32_RS11105) was detected in 49SCV. The gene encodes the YSIRK domain-containing triacylglycerol lipase. The insertion led to a premature codon and the protein truncation (336 aa/687 aa). It was previously reported (Longshaw et al., 2000) that, in addition to GehC, SE can produce a second lipase, designated GehD. Both genes were found present in isolates from both clinical and non-clinical backgrounds. Moreover, GehD could partially compensate for the GehC enzyme in a laboratory-derived mutant. The GehC shortening resulting in a loss of lipase activity in the O47 strain was reported in the study of Raue et al. (2020). Since 49SCV encoded a truncated, and most probably dysfunctional GehC protein, it is conceivable that its virulence related to the lipase activity could have been reduced, even if partially present due to an intact gehD gene (NPW32_RS02885).
Xaa-Pro aminopeptidases, enzymes widely distributed in bacteria, catalyze the release of dipeptides from the amino termini of polypeptides containing a proline or an alanine at the penultimate position (Ge et al., 2009). A presumed Xaa-Pro aminopeptidase has been recently reported as a novel virulence determinant in Campylobacter jejuni. Its deficiency in the mutant strain corresponded to a significantly lower virulence in a murine model of campylobacteriosis (Heimesaat et al., 2020). Xaa-Pro aminopeptidases have also been reported in E. coli (Zheng et al., 2005), streptococci (Goldstein et al., 2001; De et al., 2016) or Porphyromonas gingivalis (Yagishita et al., 2001) and linked to their virulence. The NPW32_RS09465 gene in 49SCV was annotated as the Xaa-Pro aminopeptidase and appeared to be disturbed by insertion in the poly(A) HT leading to the protein truncation (174 aa/ 351 aa) and dysfunction. No other gene encoding a homologous protein was found in the 49SCV genome, suggesting that dysfunction of the NPW32_RS09465 may be linked with a decreased virulence.
Genes associated with metabolism
Deletion of adenine in the poly(A) HT leading to a premature stop codon (8aa/869aa) was identified in the adhE gene (synonymous gene name adhC; NPW32_RS04630) encoding a hypothetical bifunctional acetaldehyde-CoA dehydrogenase and an iron-dependent alcohol dehydrogenase. The AdhE catalyzes the sequential reduction of acetyl-CoA to acetaldehyde and then to ethanol under fermentative conditions (EcoCyc, 2022). Additionally, a nonsense mutation detected in the NPW32_RS03860 locus, leading to truncation (31aa/332aa) of the corresponding D-lactate dehydrogenase enzyme, allowed us to hypothesize that a possible dysfunction of the two enzymes could have been related to redirection of the 49SCV metabolic activity.
Under growth-limiting conditions that exist during an infectious process, coupled with an inability of electron transport-deficient SCVs to produce energy via oxidative phosphorylation, ATP production can be sustained by the substrate level phosphorylation. It was previously reported in the (Kohler et al., 2003) that proteins involved in the glycolytic and fermentation pathways were increased in all phases of growth of the hemB mutant SCV of SA. Alcohol dehydrogenase (Adh) and L-lactate dehydrogenase (L-Ldh) were reported among proteins induced by the hemB mutation with lactate being the main fermentation product. Since mutations were detected in the aforementioned loci encoding for the Adh and Ldh enzymes in the genome of 49SCV, it can be assumed that either their possible dysfunction could have been compensated by the corresponding enzymes encoded by alternative genes or that metabolic pathways other than those based on the ethanol or lactate production could have been implicated in the metabolism of the strain. Interestingly, Kriegeskorte et al. (2014) reported that, in contrast to the hemB mutant and gentamicin-induced SCVs of SA, clinically derived SCVs showed no prominent up-regulation of glycolytic proteins.
Another deletion was detected in the poly(A) HT of the NPW32_RS08460 locus encoding for a hypothetical ornithine cyclodeaminase/μ-crystallin (OCD/CRYM) family protein. The OCD/CRYM superfamily consists of enzymes involved in the metabolism of amino acids (Uma Mahesh et al., 2022). Since the enzymes are functionally diverse and the gene annotation in 49SCV was not sufficient to identify the exact enzyme and its biological function, it is impossible to predict the possible effect of this hypothetical protein truncation (152aa/316aa) on the metabolism of the strain.
Similarly, a nonsense mutation detected in the NPW32_RS02490 locus of 49SCV leading to truncation (140aa/252aa) of the corresponding protein makes speculations about its probable dysfunction conceivable but impossible to link to any specific metabolic process. The protein is a member of a large NAD(P)(H)-dependent short-chain dehydrogenases/reductase (SDR) family. The family includes enzymes that catalyze reactions on a wide range of substrates and play critical roles in lipid, amino acid, carbohydrate, cofactor, and xenobiotic metabolism as well as in redox sensor mechanisms (Lord et al., 2014).
Genes encoding transcriptional regulators
Rapid, adaptive responses triggered by regulatory proteins are crucial for bacterial survival under challenging conditions existing in the host. They respond to specific environmental and cellular signals leading to modulation of transcription, translation, or some other events in gene expression. As a result, physiological responses are modified appropriately (Ramos et al., 2005). The most abundant groups of transcriptional regulators encoded in bacterial genomes include the LysR, TetR/AcrR, AraC/XylS, and Lrp families (Kotecka et al., 2021).
The NPW32_RS09305 locus of 49SCV harbored a gene encoding a hypothetical protein with TetR/AcrR homology. An insertion detected in the poly(A) of this gene was associated with a substantial truncation of the ORF. Interestingly, the gene in the adjacent locus NPW32_RS09300, encoding a hypothetical MFS (Major Facilitator Superfamily) transporter, was also disrupted due to a nonsense mutation leading to the protein truncation (Table 1). The TetR/AcrR family proteins generally function as transcriptional repressors and regulate a wide range of cellular activities, including osmotic stress, homeostasis, biosynthesis of antibiotics, multidrug resistance, efflux pumps, enzymes implicated in different catabolic pathways, virulence and pathogenicity of bacteria (Ramos et al., 2005; Deng et al., 2013). Prediction on the role of TetR/AcrR in 49SCV will not be possible without further genetic experiments and metabolic modelling.
Insertion in the poly(A) leading to truncation of the encoded protein was also detected in the locus NPW32_RS05975 annotated as the AraC-type transcriptional regulator. The AraC family regulators mostly act as activators of gene expression in bacteria. They control diverse cellular functions including carbon metabolism, type III secretion systems, stress response, quorum sensing and virulence (Kotecka et al., 2021). For example, AraC-type transcriptional regulator Rbf was found to promote the icaADBC operon expression by a negative regulation of expression of sarR encoding for the ica operon repressor. The Rbf regulator was also able to indirectly repress the ica operon activator, SarX. A complicated interplay between Rbf and the two Sar family proteins in the regulation of the biofilm phenotype has been suggested. However, in the hierarchy of biofilm regulators, IcaR was found dominant over the Rbf-SarR-SarX axis (Rowe et al., 2016). The NPW32_RS05975 locus in 49SCV was found adjacent to genes encoding a hypothetical transcriptional regulator (NPW32_RS05970), a hypothetical PH domain-containing protein (NPW32_RS05965), and SarR transcriptional regulator (subfamily within SarA-family; NPW32_RS05960). The gene encoding a global transcriptional regulator SarA itself was located elsewhere in the genome (NPW32_RS07800). The NPW32_RS05975 encoded protein was only 22%/44% identical/similar to Rbf encoded by SA0622 in Staphylococcus aureus subsp. aureus N315. However, there was another gene encoding an AraC-type transcriptional regulator in 49SCV. Similarity/identity of NPW32_RS08005-encoded protein to Rbf was 49%/68%, suggesting its possible role as a functional homolog. Altogether, the Rbf regulatory function did not seem to be affected in 49SCV.
Li et al. (2015) reported that the AraC-type transcriptional regulator, Rsp, promotes the production of key toxins while repressing major biofilm-associated genes and biofilm formation in SA. Their study indicated that upregulation of the accessory gene regulator (Agr) and downregulation of the ica operon were central to the regulatory impact of Rsp on virulence. The Rsp protein was found to directly bind to the agrP2 and icaADBC promoters, resulting in increased levels of the Agr-controlled toxins, phenol-soluble modulins (PSMs) and alpha-toxin, as well as a reduced production of PIA. The Rsp can, therefore, be regarded as an essential regulator for the development of acute SA infections. Its role in the regulation of gene expression in SE remains unsolved but a truncation of the protein encoded by the rsp ortholog in 49SCV (NPW32_RS09180) due to a nonsense mutation was suggestive of the Rsp protein dysfunction, which might have been correlated to the PIA-mediated proficient biofilm production.
Additionally, a deletion of one out of the two tetranucleotides TACG in the spxA gene (NPW32_RS11275) was observed in 49SCV. The encoded protein has been annotated as a negative transcriptional regulator of the biofilm formation in SE. It affects the PIA production by regulating transcription of the icaADBC in an icaR-independent manner. ClpP protease has also been involved in this process as evidenced by its ability to degrade Spx and enhance biofilm formation (Wang et al., 2010). Since the ica-positive 49SCV strain was reported as a proficient biofilm producer under in vitro conditions (Bogut et al., 2014), inactivity of this regulatory pathway is conceivable, taking into account genetic changes detected both in the spxA (truncation of the protein by 13 amino acids) and clpP (missense mutations) genes (Table 1).
The 1-nt insertion was also detected in the poly(A) HT in the NPW32_RS11435 locus, generating frameshift and a premature stop codon (185aa/454aa) in the gene encoding the PLP (pyridoxal 5′-phosphate - a biologically active form of vitamin B6) - dependent aminotransferase family protein/ DNA-binding transcriptional regulator belonging to the MocR family. The MocR-like transcription factors (MocR-TFs) represent a group of understudied chimeric proteins formed by the fusion between DNA-binding proteins and PLP-dependent enzymes (Tramonti et al., 2018). The MocR-TFs subfamily was predicted to control transcription of bacterial genes involved in diverse processes including vitamin B6, gamma aminobutyric acid (GABA), taurine, and ectoine metabolism. MocR-TF regulators were also suggested to control genes encoding enzymes involved in reduction/oxidation processes, various transporters and PLP-dependent enzymes (Suvorova and Rodionov, 2016). Genomic analyses demonstrated that MocR-TFs are widespread among eubacteria, implying their essential role in the metabolism but knowledge on these regulators is still scarce as only few of them have been experimentally characterized (Tramonti et al., 2018). Prediction on the role of MocR-TF in 49SCV will not be possible without further genetic experiments and metabolic modelling.
Genes encoding for transport system proteins
The disrupted [1 nt insertion in the poly(A)] gene encoding a truncated (51 aa/270 aa) MetQ/NlpA family ABC transporter substrate-binding protein (NPW32_RS04140) was located downstream of the NPW32_RS04145 (encoding an ABC transporter permease), and NPW32_RS04150 (encoding a methionine ABC transporter ATP-binding protein) loci. All three genes are transcribed in the same direction and most probably make up an operon. Interestingly, a regulatory S-adenosylmethionine-binding riboswitch class I sequence was predicted upstream of the first NPW32_RS4150 gene. The hypothetical protein encoded by the disrupted NPW32_RS04140 gene demonstrates characteristics of lipoproteins. In silico analyses with tools devoted to lipoprotein prediction (LipoP for Gram-negative and PRED-LIPO for Gram-positive bacteria) revealed the presence of a lipoprotein signal sequence with a cleavage site LAA(19)↓C(20)G. Methionine plays a key role in the protein biosynthesis initiation and many cellular processes with a stabilizing role in protein structure (Beavers et al., 2021). At least two transport systems can serve for the entry of methionine into E. coli (Kadner and Watson, 1974), and there was another possible methionine transport system in 49SCV encoded by the genes: NPW32_RS10720 (gmpC) annotated as a dipeptide ABC transporter glycylmethionine-binding lipoprotein, NPW32_RS10715, encoding an ABC transporter permease, and NPW32_RS10710 annotated as a methionine ABC transporter ATP-binding protein. Given that many bacteria including staphylococci have evolved mechanisms to synthesize methionine de novo due to its importance for viability (Schoenfelder et al., 2013) and that the second methionine transport system that can compensate for dysfunction of the other was identified, the negative impact of the mutation in NPW32_RS04140 in 49SCV is questionable.
Another transport system whose element was found truncated in 49SCV was the ABC transporter permease/substrate-binding protein (fused permease and substrate binding protein) involved in the transport of one or more from a variety of substrates including glycine betaine and choline (NPW32_RS04645). The ABC transporters are multisubunit complexes composed of integral membrane proteins that function as a permease, peripheral membrane ATP binding proteins able to hydrolyze ATP, and extracellular substrate binding proteins (SBPs) acting as receptors for the substrate to be transported. Although structurally conserved, these transporters play a role in the uptake of a diverse range of molecules (Williams et al., 2004). Choline and glycine betaine were reported to act as osmoprotectants enhancing staphylococcal growth at high osmolarity (Graham and Wilkinson, 1992). Stimeling et al. (1994) provided evidence for two glycine betaine transport systems in SA which cannot exclude their mutually compensatory roles. Similarly, another gene with annotation as encoding a betaine/proline/choline family ABC transporter ATP-binding protein (NPW32_RS02740; contig 2) was identified in the 49SCV genome. Hence, effective osmoprotection occuring in SCVs including 49SCV in spite of a truncated osmoprotectant transport system cannot be ruled out.
We also identified three genes encoding for hypothetical Major Facilitator Superfamily (MFS) proteins to be disrupted either by insertion in poly(T) (NPW32_RS02630, NPW32_RS06615) or through a nonsense mutation (NPW32_RS09300). However, lack of specific annotation or similarity unables to comment on the possible contribution of these mutations to 49SCV phenotype.
Genes associated with stress response
Fructosamine kinase family (FN3K) comprises proteins involved in the removal of fructosamines produced during a spontaneous reaction of glucose with amines. As a result, FN3K protects proteins against damage caused by high glucose concentrations. Its translation in SA was reported to remain under control of RsaI - a small non-coding RNA. Following glucose consumption, translation of permease involved in the uptake of glucose and the FN3K enzyme is repressed by RsaI (Bronesky et al., 2019). A possible dysfunction of FN3K enzyme in 49SCV (NPW32_RS03820) caused by a one-nucleotide deletion within the gene poly(T) HT and truncation of the resultant protein may suggest the existence of genetically based metabolic disturbances in terms of glucose processing that might have had influence on its slow growth. On the other hand, it may seem conceivable that truncation of the protein that plays its detoxification role at high glucose concentrations may represent one of the possible ways to streamline genetic content to adapt the microorganism to nutrient deficiency in the depths of the implant-associated biofilm structure.
The 49SCV genome also harbored a 2 nt insertion in the poli(TA) of the czrB (NPW32_RS08685) gene leading to truncation of the corresponding protein (72 aa/317 aa). The czrB gene is a component of the czrAB operon suggested to play a role in the transport of zinc across the cell membrane (Kuroda et al., 1999). The operon functions to maintain appropriate intracellular concentrations of this element, which is a prosthetic factor of many intracellular enzymes. The czrB, coding for a 36 kDa membrane spanning protein, was found to be homologous to the czcD gene, cobalt, zinc and the cadmium-resistant factor of Bacillus subtilis and Alcaligenes eutrophus. An association between truncation of CzrB in 49SCV and its phenotype is disputable, but possible, taking into account the fact that at higher concentrations, which may result from dysfunction of the regulatory CzrAB operon, zinc inhibits bacterial growth rate (Kuroda et al., 1999).
A nonsense mutation was detected in the NPW32_RS07065 locus encoding a truncated (34 aa/355 aa) nitric oxide synthase oxygenase (NOS) enzyme. Wang et al. suggested that the NOS gene can negatively regulate biofilm formation in SE. Disruption of the NOS gene resulted in an enhanced biofilm formation coupled with its weakened dispersal, as well as a slight retardation of bacterial growth and a decreased autolysis rate (Wang et al., 2022). Nitric oxide was also reported to increase bacterial resistance to antibiotics, which can be achieved both by the chemical modification of toxic compounds and the alleviation of the oxidative stress imposed by antibiotics (Gusarov et al., 2009). Kinkel et al. showed that NOS produced by SA, in concert with a NO·-metabolising flavohaemoprotein, regulates electron transfer by targeting haem-containing cytochrome oxidases under microaerobic conditions to maintain membrane bioenergetics (Kinkel et al., 2016). Following these literature reports, we can assume that disruption of the nos gene in 49SCV could have been related both to its strong biofilm production capability, but also to sensitivity to antibiotics, with the exception of gentamicin resistance. The 49SCV strain has been assumed to develop its gentamicin resistance due to genetically-based defects in electron transport. A supporting role of NOS dysfunction in this regard, however, cannot be excluded.
Single nucleotide polymorphisms (SNPs) identified in 49SCV
Point mutations have been evidenced to have a limited impact on cellular metabolism of SE SCV compared to the frameshift mutations (Liu et al., 2021). Using the reference O47 genome, the set of nonsynonymous single-nucleotide variations were identified in 49SCV. The set covered 713 genes, involved in a wide range of metabolic processes. Based on the amino acid sequence, COG categories were assigned to these genes. Mutated genes were distributed into 20 of the 26 functional categories. The overrepresented COG categories included: amino acid transport and metabolism (E), carbohydrate transport and metabolism (G), cell wall/membrane/envelope biogenesis (M), inorganic ion transport and metabolism (P), general function prediction (R), and unknown function (S) (Figure 6).
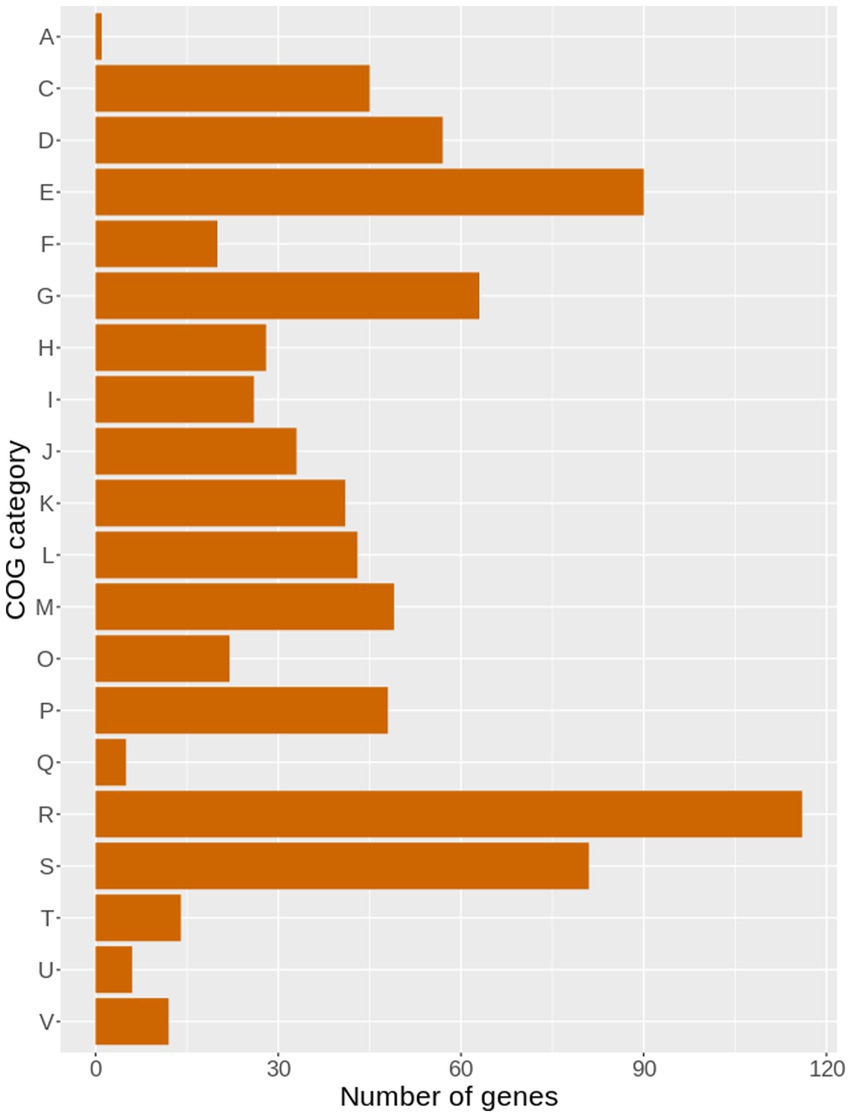
Figure 6. The bar plot illustrating the distribution of genes with single nucleotide polymorphisms (SNPs) in the genome of 49SCV, compared to the reference O47 genome, and clustered within different COG categories.
As the WT counterpart of the 49SCV strain was not cultured from the patient, we assume that a great number of SNPs identified against the O47 strain could be ascribed to the natural level of variability between the strains. In order to obtain a more complete picture and to find SNPs hypothesized to be directly responsible for the SCV phenotype formation, we analyzed the pangenome constructed from the genome of 49SCV and the five reference SE genomes. This analysis was conducted in the search of gene clusters for which the predicted amino acid sequences were specific only to 49SCV and remained constant in the other reference strains. We identified 178 variable gene clusters (Supplementary File 2), among which 22 had a unique sequence at the amino acid level in 49SCV in comparison to the reference strains (Table 2).
Substitutions identified as unique to the 49SCV genome, can be associated either with the loss or gain of gene function. Although metabolic analyses will be necessary to ascertain whether these genetic mutations reprogrammed the metabolism of the stable 49SCV strain, several hypotheses regarding the biological significance of identified SNPs in 49SCV can be corroborated by the results of phenotypic analysis of 49SCV published previously (Bogut et al., 2014).
Of note, SNPs were detected in the menA, menH, and hemN(hemW) genes whose products are involved in menaquinone and haem biosynthesis, respectively. A frameshift mutation in the HT of the hemA gene was also detected, which, as discussed earlier, could have been sufficient to develop an electron-transport-deficient SCV phenotype. Although it is conceivable that the above mentioned SNPs might have had their contribution to the phenotype transition, our previous study (Bogut et al., 2014) revealed that 49SCV did not demonstrate menadione auxotrophy. The potential role of SNP detected in the hemN, encoding for an oxygen-independent coproporphyrinogen-III oxidase, is also under speculation. Oxidative decarboxylation of coproporphyrinogen III to protoporphyrinogen IX mediated by HemN in E. coli can be catalyzed by the hemF gene product as well, which is active in the presence of oxygen (EcoCyc, 2020). The E. coli hemN mutant was found defective for the cytochrome b and cytochrome c synthesis only under anaerobic growth conditions (Tyson et al., 1997) which makes a compensatory role of HemF oxidase conceivable.
SNP detected in the gene encoding for the alternative sigma factor B (SigB; σB) represents a genetic event whose contribution to 49SCV phenotype and its related characteristics could indicate an intact or even increased biological activity. SigB is an important transcription factor in staphylococci that is associated with responses to environmental stresses via redirection of transcriptional priorities (Tuchscherr et al., 2015). SigB activity has been described to influence the production of virulence factors, biofilm formation and intracellular persistence in SCVs (Mitchell et al., 2013). SigB may positively influence the appearance of SA SCVs and the production of biofilm upon aminoglycoside exposure (Mitchell et al., 2010). Tuchscherr et al. (2015) demonstrated that SigB enables SA to switch from the highly aggressive phenotype involved in acute infections to the SCV phenotype associated with a long-term intracellular persistence. In their study, ΔsigB-mutants failed to generate SCVs and were completely cleared by the host cells within a few days. SigB was also reported to silence the agr system which is involved in the enhanced inflammatory activity (Bischoff et al., 2001). The homology of the nucleotide sequence and the organization of the sigB operon in SE suggest a general function similar to that observed for SA (Knobloch et al., 2001). Several reports described a crucial role for σB in SE pathogenesis with the prominent example of biofilm formation and the σB-dependent expression of PIA (Knobloch et al., 2001, 2004). Positive regulation of the biofilm formation in SE by σB was reported to rely on transcriptional activation of icaADBC and assumed to be mediated by a negative transcriptional control of IcaR (Conlon et al., 2002; Handke et al., 2007). Hence, in view of a prominent PIA-dependent biofilm production in 49SCV (Bogut et al., 2014), it can be speculated that the amino acid substitution in SigB either did not affect or even enhanced its functionality, e.g., through modulation of interactions with specific promoter or regulatory proteins.
As many as seven SNPs were identified in the 49SCV clpP gene which may indicate the loss or impairment of the biological activity of its product. The gene encodes a protease involved in the regulation of the toxin-antitoxin (TA) system. The toxin and antitoxin genes are coexpressed to form a TA complex. MazEF is an important example of the TA module in SA (Fu et al., 2007). MazE represents an antitoxin protein that binds to the toxin MazF, thereby preventing its activity. MazF has RNase activity, which targets selected mRNAs (including sigB, hla, spa mRNAs) in SA. The ATP-dependent protease pair, ClpPC, serves as the functional unit for the degradation of the MazE antitoxin, thereby ensuring MazF toxin release and activation. Overproduction of MazF toxin was reported to arrest bacterial growth and assumed to be involved in persistence (Magnuson, 2007; Donegan and Cheung, 2009; Donegan et al., 2010; Proctor et al., 2014). On the other hand, low levels of ATP and ClpP as well as the higher levels σB in electron transport SCVs suggest that the MazEF system may not contribute to persistence in electron transport SCVs (Proctor et al., 2014). The latter assumption is in line with our hypothesis on the genetically induced dysfunction of the ClpP protease and an intact SigB (discussed above) in the 49SCV electron-deficient strain. It is conceivable that the dysfunctional ClpP is not capable of the MazE antitoxin cleavage; hence, the MazF toxin probably cannot be released and exert its action on the sigB mRNA.
SNPs were detected in the genes encoding for the protein export systems SecA and SecA2. Since Sec proteins are involved in delivering proteins from the cytoplasm to the cell envelope or extracellular environment, their role is considered critical in terms of bacterial viability, physiology and pathogenesis. Proteins whose transport is mediated by the Sec pathway are synthesized as preproteins equipped with N-terminal signal peptides that are recognized by the Sec machinery. These peptides are cleaved during export across the inner membrane to produce mature proteins. SecA is a key component of the general protein secretion machinery. It participates in recruitment and delivery of suitable substrates to the SecYEG channel and functions as an ATPase motor to provide energy for the export. The main catalytic moiety of SecA is the ‘DEAD’ motor formed by the nucleotide binding domain and the intramolecular regulator of ATPase activity 2 domain. The ATP binding site of SecA is located at the interface of the two domains (Chaudhary et al., 2015). SecA2s is another transport-associated ATPase. It is a part of the accessory Sec (aSec) system that includes SecY2 (a paralogue of SecY) and accessory Sec proteins (Asps). Of note, the aSec system mediates the export of serine-rich repeat glycoproteins that function as adhesins (Chatzi et al., 2014; Jin et al., 2018; Braunstein et al., 2019; Ambroziak et al., 2021). In the study of Siboo et al. (2005), disruption of the secA2 in SA resulted in the near complete loss of SraP adhesin expression on the cell surface. The loss of this platelet-binding adhesin expression has been linked to a reduced SA virulence in a model of endovascular infection (Siboo et al., 2005). Amino acid substitution in the SecA protein of 49SCV was found distant from the DEAD motor encoding region, near its C-terminal domain. However, the C-terminal domain along with the preprotein binding domain (PBD) contribute to the SecA substrate specificity (Chaudhary et al., 2015). Substitution in the SecA2 protein was detected within the ATP-binding site. The ATP-binding properties of the protein domains were reported to be essential for SecA-dependent translocation ATPase and protein translocation in E. coli (Mitchell and Oliver, 1993). Amino acid substitutions in the Walker A motif of SecA2 affected ATP binding in vitro in the study of Mycobacterium tuberculosis (Hou et al., 2008). We assume that SNPs in the genes representing the Sec system components might have led to the modulation of efficiency of the transport of Sec-dependent proteins making an association with a prolonged growth of 49SCV and chronicity of the PJI conceivable.
The two-component regulator system vraSR (vancomycin-resistance associated sensor/regulator) has been postulated to play several roles important for bacterial viability, virulence, and antibiotic resistance. It constitutes a positive regulator of the cell wall biosynthesis and is involved in the expression of beta-lactam and glycopeptide resistance in SA. Additionally, VraSR can regulate transcription of genes encoding stress-response proteins including proline/glycine-betaine (osmoprotectant) transporters (proP and opuD) (Kuroda et al., 2003; Cui et al., 2009). It the study of Dai et al. increase in the MICs of vancomycin was correlated with an increased vraR expression and a decreased expression of virulence genes (hla, hlb, and coa) and virulence-regulated genes (RNAIII, agrA, and saeR) suggesting that VraR might also regulate SA virulence (Dai et al., 2017). Interestingly, the vraRS genes were found among the down-regulated genes following a transcriptomic analysis of a stable SA SCV strain isolated from a patient with a PJI relapse. Downregulation of energy pathways and vraRS was considered in line with the emergence of the SCV phenotype and the switch from acute to chronic infection for isolates of clinical origin (Loss et al., 2019). The SNP-mediated modulation of the vraR gene function can also be assumed for the 49SCV strain which was additionally devoid of the functional ADI pathway and characterized by electron-deficiency due to hemin-auxotrophy.
Conclusion
This study demonstrates novel genetic mechanisms that can be involved in staphylococcal phenotype switching. We propose that previously unreported indels in the HTs can constitute a background of this phenotype due to a resulting truncation of the corresponding proteins and their possible biological dysfunction. The strongest evidence for our hypothesis is a deletion in the poly(A) tract of the hemA gene, considered a possible trigger factor for phenotype switching and hemin auxotrophy in 49SCV. Streamline of the genetic content evidenced by the absence of the SCC element and a large ~12 kb deletion can represent a strategy associated with the development of the SCV phenotype and its adaptation to chronicity. Future studies based on transcriptomic and proteomic analyses are expected to shed more light on the actual association between genetic changes and the expression of mutated genes. They will be important to improve our understanding of molecular mechanisms driving phenotype switching and pathogenesis of PJIs in which staphylococcal SCVs are involved.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
AB: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Visualization, Writing – original draft, Writing – review & editing. PK: Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing – original draft, Writing – review & editing. MM: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – original draft, Writing – review & editing. PC: Formal analysis, Investigation, Methodology, Software, Validation, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The research conducted in this article was funded under the project Miniatura 4 (number 2020/04/X/NZ6/00484) obtained by AB from the National Science Centre, Poland.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1289844/full#supplementary-material
References
Adler, H., Widmer, A., and Frei, R. (2003). Emergence of a teicoplanin-resistant small colony variant of Staphylococcus epidermidis during vancomycin therapy. Eur. J. Clin. Microbiol. Infect. Dis. 22, 746–748. doi: 10.1007/s10096-003-1029-9
Alanjary, M., Steinke, K., and Ziemert, N. (2019). AutoMLST: an automated web server for generating multi-locus species trees highlighting natural product potential. Nucleic Acids Res. 47, W276–W282. doi: 10.1093/nar/gkz282
Ambroziak, P., Rzepka, I., and Skorko-Glonek, J. (2021). SecA - a multidomain and multitask bacterial export protein. Acta Biochim. Pol. 68, 427–436. doi: 10.18388/abp.2020_5761
Arrecubieta, C., Toba, F. A., von Bayern, M., Akashi, H., Deng, M. C., Naka, Y., et al. (2009). SdrF, a Staphylococcus epidermidis surface protein, contributes to the initiation of ventricular assist device driveline-related infections. PLoS Pathog. 5:e1000411. doi: 10.1371/journal.ppat.1000411
Baddour, L. M., Simpson, W. A., Weems, J. J. Jr., Hill, M. M., and Christensen, G. D. (1988). Phenotypic selection of small-colony variant forms of Staphylococcus epidermidis in the rat model of endocarditis. J. Infect. Dis. 157, 757–763. doi: 10.1093/infdis/157.4.757
Balibar, C. J., Iwanowicz, D., and Dean, C. R. (2013). Elongation factor P is dispensable in Escherichia coli and Pseudomonas aeruginosa. Curr. Microbiol. 67, 293–299. doi: 10.1007/s00284-013-0363-0
Bayston, R., Ashraf, W., and Smith, T. (2007). Triclosan resistance in methicillin-resistant Staphylococcus aureus expressed as small colony variants: a novel mode of evasion of susceptibility to antiseptics. J. Antimicrob. Chemother. 59, 848–853. doi: 10.1093/jac/dkm031
Beavers, W. N., DuMont, A. L., Monteith, A. J., Maloney, K. N., Tallman, K. A., Weiss, A., et al. (2021). Staphylococcus aureus peptide methionine sulfoxide reductases protect from human whole-blood killing. Infect. Immun. 89:e0014621. doi: 10.1128/IAI.00146-21
Bischoff, M., Entenza, J. M., and Giachino, P. (2001). Influence of a functional sigB operon on the global regulators sar and agr in Staphylococcus aureus. J. Bacteriol. 183, 5171–5179. doi: 10.1128/JB.183.17.5171-5179.2001
Bogut, A., and Magryś, A. (2021). The road to success of coagulase-negative staphylococci: clinical significance of small colony variants and their pathogenic role in persistent infections. Eur. J. Clin. Microbiol. Infect. Dis. 40, 2249–2270. doi: 10.1007/s10096-021-04315-1
Bogut, A., Niedźwiadek, J., Kozioł-Montewka, M., Strzelec-Nowak, D., Blacha, J., Mazurkiewicz, T., et al. (2014). Characterization of Staphylococcus epidermidis and Staphyloccocus warneri small-colony variants associated with prosthetic-joint infections. J. Med. Microbiol. 63, 176–185. doi: 10.1099/jmm.0.066068-0
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Both, A., Huang, J., Qi, M., Lausmann, C., Weißelberg, S., Büttner, H., et al. (2021). Distinct clonal lineages and within-host diversification shape invasive Staphylococcus epidermidis populations. PLoS Pathog. 17:e1009304. doi: 10.1371/journal.ppat.1009304
Braunstein, M., Bensing, B. A., and Sullam, P. M. (2019). The two distinct types of SecA2-dependent export systems. Microbiol. Spectr. 7.
Bronesky, D., Desgranges, E., Corvaglia, A., François, P., Caballero, C. J., Prado, L., et al. (2019). A multifaceted small RNA modulates gene expression upon glucose limitation in Staphylococcus aureus. EMBO J. 38:e99363. doi: 10.15252/embj.201899363
Bui, L. M. G., Hoffmann, P., Turnidge, J. D., Zilm, P. S., and Kidd, S. P. (2015a). Prolonged growth of a clinical Staphylococcus aureus strain selects for a stable small-colony-variant cell type. Infect. Immun. 83, 470–481. doi: 10.1128/IAI.02702-14
Bui, L. M. G., and Kidd, S. P. (2015). A full genomic characterization of the development of a stable Small Colony Variant cell-type by a clinical Staphylococcus aureus strain. Infect. Genet. Evol. 36, 345–355. doi: 10.1016/j.meegid.2015.10.011
Bui, L. M. G., Turnidge, J. D., and Kidd, S. P. (2015b). The induction of Staphylococcus aureus biofilm formation or Small Colony Variants is a strain-specific response to host-generated chemical stresses. Microbes Infect. 17, 77–82. doi: 10.1016/j.micinf.2014.09.009
Campoccia, D., Montanaro, L., Ravaioli, S., Cangini, I., Speziale, P., and Arciola, C. R. (2009). Description of a new group of variants of the Staphylococcus aureus elastin-binding protein that lacks an entire DNA segment of 180 bp. Int. J. Artif. Organs 32, 621–629. doi: 10.1177/039139880903200911
Cao, S., Huseby, D. L., Brandis, G., and Hughes, D. (2017). Alternative evolutionary pathways for drug-resistant small colony variant mutants in Staphylococcus aureus. mBio 8:e00358-17. doi: 10.1128/mBio.00358-17
Chan, Y. G. Y., Frankel, M. B., Dengler, V., Schneewind, O., and Missiakas, D. (2013). Staphylococcus aureus mutants lacking the LytR-CpsA-Psr family of enzymes release cell wall teichoic acids into the extracellular medium. J. Bacteriol. 195, 4650–4659. doi: 10.1128/JB.00544-13
Chatzi, K. E., Sardis, M. F., Economou, A., and Karamanou, S. (2014). SecA-mediated targeting and translocation of secretory proteins. Biochim. Biophys. Acta 1843, 1466–1474. doi: 10.1016/j.bbamcr.2014.02.014
Chaudhary, A. S., Chen, W., Jin, J., Tai, P. C., and Wang, B. (2015). SecA: a potential antimicrobial target. Future Med. Chem. 7, 989–1007. doi: 10.4155/fmc.15.42
Chaudhuri, R. R., Allen, A. G., Owen, P. J., Shalom, G., Stone, K., Harrison, M., et al. (2009). Comprehensive identification of essential Staphylococcus aureus genes using Transposon-Mediated Differential Hybridisation (TMDH). BMC Genomics 10:291. doi: 10.1186/1471-2164-10-291
Conlon, K. M., Humphreys, H., and O’Gara, J. P. (2002). icaR encodes a transcriptional repressor involved in environmental regulation of ica operon expression and biofilm formation in Staphylococcus epidermidis. J. Bacteriol. 184, 4400–4408. doi: 10.1128/jb.184.16.4400-4408.2002
Cui, L., Neoh, H.-M., Shoji, M., and Hiramatsu, K. (2009). Contribution of vraSR and graSR point mutations to vancomycin resistance in vancomycin-intermediate Staphylococcus aureus. Antimicrob. Agents Chemother. 53, 1231–1234. doi: 10.1128/AAC.01173-08
Dai, Y., Chang, W., Zhao, C., Peng, J., Xu, L., Lu, H., et al. (2017). VraR binding to the promoter region of agr inhibits its function in vancomycin-intermediate Staphylococcus aureus (VISA) and heterogeneous VISA. Antimicrob. Agents Chemother. 61:61. doi: 10.1128/AAC.02740-16
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
De, A., Lupidi, G., Petrelli, D., and Vitali, L. A. (2016). Molecular cloning and biochemical characterization of Xaa-Pro dipeptidyl-peptidase from Streptococcus mutans and its inhibition by anti-human DPP IV drugs. FEMS Microbiol. Lett. 363:fnw066. doi: 10.1093/femsle/fnw066
Deghorain, M., and Van Melderen, L. (2012). The Staphylococci phages family: an overview. Viruses 4, 3316–3335. doi: 10.3390/v4123316
Deng, W., Li, C., and Xie, J. (2013). The underling mechanism of bacterial TetR/AcrR family transcriptional repressors. Cell. Signal. 25, 1608–1613. doi: 10.1016/j.cellsig.2013.04.003
Dengler, V., Meier, P. S., Heusser, R., Kupferschmied, P., Fazekas, J., Friebe, S., et al. (2012). Deletion of hypothetical wall teichoic acid ligases in Staphylococcus aureus activates the cell wall stress response. FEMS Microbiol. Lett. 333, 109–120. doi: 10.1111/j.1574-6968.2012.02603.x
Donegan, N. P., and Cheung, A. L. (2009). Regulation of the mazEF toxin-antitoxin module in Staphylococcus aureus and its impact on sigB expression. J. Bacteriol. 191, 2795–2805. doi: 10.1128/JB.01713-08
Donegan, N. P., Thompson, E. T., Fu, Z., and Cheung, A. L. (2010). Proteolytic regulation of toxin-antitoxin systems by ClpPC in Staphylococcus aureus. J. Bacteriol. 192, 1416–1422. doi: 10.1128/jb.00233-09
Downer, R., Roche, F., Park, P. W., Mecham, R. P., and Foster, T. J. (2002). The elastin-binding protein of Staphylococcus aureus (EbpS) is expressed at the cell surface as an integral membrane protein and not as a cell wall-associated protein. J. Biol. Chem. 277, 243–250. doi: 10.1074/jbc.M107621200
Du, X., Zhu, Y., Song, Y., Li, T., Luo, T., Sun, G., et al. (2013). Molecular analysis of Staphylococcus epidermidis strains isolated from community and hospital environments in China. PLoS One 8:e62742. doi: 10.1371/journal.pone.0062742
EcoCyc (2022). Escherichia coli K-12 substr. MG1655 adhE. Available at: https://biocyc.org/gene?orgid=ECOLI&id=EG10031 (Accessed July 11, 2023).
EcoCyc (2013). Escherichia coli K-12 substr. MG1655 hemA. Available at: https://biocyc.org/gene?orgid=ECOLI&id=EG10427 (Accessed July 9, 2023).
EcoCyc (2020). Escherichia coli K-12 substr. MG1655 hemF. Available at: https://biocyc.org/gene?orgid=ECOLI&id=EG12189 (Accessed July 10, 2023).
Edwards, A. M. (2012). Phenotype switching is a natural consequence of Staphylococcus aureus replication. J. Bacteriol. 194, 5404–5412. doi: 10.1128/jb.00948-12
Eren, A. M., Kiefl, E., Shaiber, A., Veseli, I., Miller, S. E., Schechter, M. S., et al. (2021). Community-led, integrated, reproducible multi-omics with anvi’o. Nat. Microbiol. 6, 3–6. doi: 10.1038/s41564-020-00834-3
Fernández-Rodríguez, D., Colín-Castro, C. A., Hernández-Durán, M., López-Jácome, L. E., and Franco-Cendejas, R. (2021). Staphylococcus epidermidis small colony variants, clinically significant quiescent threats for patients with prosthetic joint infection. Microbes Infect. 23:104854. doi: 10.1016/j.micinf.2021.104854
Fey, P. D., and Olson, M. E. (2010). Current concepts in biofilm formation of Staphylococcus epidermidis. Future Microbiol. 5, 917–933. doi: 10.2217/fmb.10.56
Fu, Z., Donegan, N. P., Memmi, G., and Cheung, A. L. (2007). Characterization of MazFSa, an endoribonuclease from Staphylococcus aureus. J. Bacteriol. 189, 8871–8879. doi: 10.1128/jb.01272-07
Ge, J., Feng, Y., Ji, H., Zhang, H., Zheng, F., Wang, C., et al. (2009). Inactivation of dipeptidyl peptidase IV attenuates the virulence of Streptococcus suis serotype 2 that causes streptococcal toxic shock syndrome. Curr. Microbiol. 59, 248–255. doi: 10.1007/s00284-009-9425-8
Goldstein, J. M., Banbula, A., Kordula, T., Mayo, J. A., and Travis, J. (2001). Novel extracellular x-prolyl dipeptidyl-peptidase (DPP) from Streptococcus gordonii FSS2: an emerging subfamily of viridans Streptococcal x-prolyl DPPs. Infect. Immun. 69, 5494–5501. doi: 10.1128/IAI.69.9.5494-5501.2001
Gor, V., Ohniwa, R. L., and Morikawa, K. (2021). No change, no life? What we know about phase variation in Staphylococcus aureus. Microorganisms 9:244. doi: 10.3390/microorganisms9020244
Graham, J. E., and Wilkinson, B. J. (1992). Staphylococcus aureus osmoregulation: roles for choline, glycine betaine, proline, and taurine. J. Bacteriol. 174, 2711–2716. doi: 10.1128/jb.174.8.2711-2716.1992
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Gusarov, I., Shatalin, K., Starodubtseva, M., and Nudler, E. (2009). Endogenous nitric oxide protects bacteria against a wide spectrum of antibiotics. Science 325, 1380–1384. doi: 10.1126/science.1175439
Handke, L. D., Slater, S. R., Conlon, K. M., O'Donnell, S. T., Olson, M. E., Bryant, K. A., et al. (2007). σB and SarA independently regulate polysaccharide intercellular adhesin production in Staphylococcus epidermidis. Can. J. Microbiol. 53, 82–91. doi: 10.1139/w06-108
Heimesaat, M. M., Schmidt, A.-M., Mousavi, S., Escher, U., Tegtmeyer, N., Wessler, S., et al. (2020). Peptidase PepP is a novel virulence factor of Campylobacter jejuni contributing to murine campylobacteriosis. Gut Microbes 12:1770017. doi: 10.1080/19490976.2020.1770017
Hou, J. M., D'Lima, N. G., Rigel, N. W., Gibbons, H. S., McCann, J. R., Braunstein, M., et al. (2008). ATPase activity of Mycobacterium tuberculosis SecA1 and SecA2 proteins and its importance for SecA2 function in macrophages. J. Bacteriol. 190, 4880–4887. doi: 10.1128/jb.00412-08
Hübscher, J., McCallum, N., Sifri, C. D., Majcherczyk, P. A., Entenza, J. M., Heusser, R., et al. (2009). MsrR contributes to cell surface characteristics and virulence in Staphylococcus aureus. FEMS Microbiol. Lett. 295, 251–260. doi: 10.1111/j.1574-6968.2009.01603.x
Islam, S., Callender, A. C., Ho, Q. N., and Wakeman, C. A. (2022). Iron restriction induces the small-colony variant phenotype in Staphylococcus aureus. Front. Microbiol. 13:978859. doi: 10.3389/fmicb.2022.978859
Jin, J., Hsieh, Y.-H., Chaudhary, A. S., Cui, J., Houghton, J. E., Sui, S., et al. (2018). SecA inhibitors as potential antimicrobial agents: differential actions on SecA-only and SecA-SecYEG protein-conducting channels. FEMS Microbiol. Lett. 365:fny145. doi: 10.1093/femsle/fny145
Kadner, R. J., and Watson, W. J. (1974). Methionine transport in Escherichia coli: physiological and genetic evidence for two uptake systems. J. Bacteriol. 119, 401–409. doi: 10.1128/jb.119.2.401-409.1974
Kahl, B. C., Becker, K., and Löffler, B. (2016). Clinical significance and pathogenesis of staphylococcal small colony variants in persistent infections. Clin. Microbiol. Rev. 29, 401–427. doi: 10.1128/CMR.00069-15
Kawakami, N., and Fujisaki, S. (2018). Undecaprenyl phosphate metabolism in gram-negative and gram-positive bacteria. Biosci. Biotechnol. Biochem. 82, 940–946. doi: 10.1080/09168451.2017.1401915
Kinkel, T. L., Ramos-Montañez, S., Pando, J. M., Tadeo, D. V., Strom, E. N., Libby, S. J., et al. (2016). An essential role for bacterial nitric oxide synthase in Staphylococcus aureus electron transfer and colonization. Nat. Microbiol. 2, 16224–16227. doi: 10.1038/nmicrobiol.2016.224
Kleinert, F., Kallies, R., Hort, M., Zweynert, A., Szekat, C., Nagel, M., et al. (2017). Influence of IS256 on genome variability and formation of small-colony variants in Staphylococcus aureus. Antimicrob Agents Chemother 61:e00144-17. doi: 10.1128/AAC.00144-17
Knobloch, J. K., Bartscht, K., Sabottke, A., Rohde, H., Feucht, H. H., and Mack, D. (2001). Biofilm formation by Staphylococcus epidermidis depends on functional RsbU, an activator of the sigB operon: differential activation mechanisms due to ethanol and salt stress. J. Bacteriol. 183, 2624–2633. doi: 10.1128/JB.183.8.2624-2633.2001
Knobloch, J. K.-M., Jäger, S., Horstkotte, M. A., Rohde, H., and Mack, D. (2004). RsbU-dependent regulation of Staphylococcus epidermidis biofilm formation is mediated via the alternative sigma factor sigmaB by repression of the negative regulator gene icaR. Infect. Immun. 72, 3838–3848. doi: 10.1128/IAI.72.7.3838-3848.2004
Kohler, C., von Eiff, C., Liebeke, M., McNamara, P. J., Lalk, M., Proctor, R. A., et al. (2008). A defect in Menadione biosynthesis induces global changes in gene expression in Staphylococcus aureus. J. Bacteriol. 190, 6351–6364. doi: 10.1128/jb.00505-08
Kohler, C., von Eiff, C., Peters, G., Proctor, R. A., Hecker, M., and Engelmann, S. (2003). Physiological characterization of a Heme-deficient mutant of Staphylococcus aureus by a proteomic approach. J. Bacteriol. 185, 6928–6937. doi: 10.1128/jb.185.23.6928-6937.2003
Koprivnjak, T., Zhang, D., Ernst, C. M., Peschel, A., Nauseef, W. M., and Weiss, J. P. (2011). Characterization of Staphylococcus aureus cardiolipin synthases 1 and 2 and their contribution to accumulation of cardiolipin in stationary phase and within phagocytes. J. Bacteriol. 193, 4134–4142. doi: 10.1128/JB.00288-11
Kotecka, K., Kawalek, A., Kobylecki, K., and Bartosik, A. A. (2021). The AraC-type transcriptional regulator GliR (PA3027) activates genes of glycerolipid metabolism in Pseudomonas aeruginosa. Int. J. Mol. Sci. 22:5066. doi: 10.3390/ijms22105066
Kriegeskorte, A., Grubmüller, S., Huber, C., Kahl, B. C., von Eiff, C., Proctor, R. A., et al. (2014). Staphylococcus aureus small colony variants show common metabolic features in central metabolism irrespective of the underlying auxotrophism. Front. Cell. Infect. Microbiol. 4:141. doi: 10.3389/fcimb.2014.00141
Kuroda, M., Hayashi, H., and Ohta, T. (1999). Chromosome-determined zinc-responsible operon czr in Staphylococcus aureus strain 912. Microbiol. Immunol. 43, 115–125. doi: 10.1111/j.1348-0421.1999.tb02382.x
Kuroda, M., Kuroda, H., Oshima, T., Takeuchi, F., Mori, H., and Hiramatsu, K. (2003). Two-component system VraSR positively modulates the regulation of cell-wall biosynthesis pathway in Staphylococcus aureus. Mol. Microbiol. 49, 807–821. doi: 10.1046/j.1365-2958.2003.03599.x
Lakhundi, S., and Zhang, K. (2018). Methicillin-resistant Staphylococcus aureus: molecular characterization, evolution, and epidemiology. Clin. Microbiol. Rev. 31, e00020–e00018. doi: 10.1128/CMR.00020-18
Lannergård, J., Cao, S., Norström, T., Delgado, A., Gustafson, J. E., and Hughes, D. (2011). Genetic complexity of fusidic acid-resistant small colony variants (SCV) in Staphylococcus aureus. PLoS One 6:e28366. doi: 10.1371/journal.pone.0028366
Lee, J. Y. H., Monk, I. R., Gonçalves da Silva, A., Seemann, T., Chua, K. Y. L., Kearns, A., et al. (2018). Global spread of three multidrug-resistant lineages of Staphylococcus epidermidis. Nat. Microbiol. 3, 1175–1185. doi: 10.1038/s41564-018-0230-7
Lee, J. Y. H., Monk, I. R., Pidot, S. J., Singh, S., Chua, K. Y. L., Seemann, T., et al. (2016). Functional analysis of the first complete genome sequence of a multidrug resistant sequence type 2 Staphylococcus epidermidis. Microb. Genomics 2:e000077. doi: 10.1099/mgen.0.000077
Lee, J., Zilm, P. S., and Kidd, S. P. (2020). Novel research models for Staphylococcus aureus small Colony variants (SCV) development: co-pathogenesis and growth rate. Front. Microbiol. 11:321. doi: 10.3389/fmicb.2020.00321
Letunic, I., and Bork, P. (2019). Interactive Tree Of Life (iTOL) v4: recent updates and new developments. Nucleic Acids Res. 47, W256–W259. doi: 10.1093/nar/gkz239
Li, T., He, L., Song, Y., Villaruz, A. E., Joo, H.-S., Liu, Q., et al. (2015). AraC-type regulator Rsp adapts Staphylococcus aureus gene expression to acute infection. Infect. Immun. 84, 723–734. doi: 10.1128/IAI.01088-15
Lin, Y.-T., Tsai, J.-C., Yamamoto, T., Chen, H.-J., Hung, W.-C., Hsueh, P.-R., et al. (2016). Emergence of a small colony variant of vancomycin-intermediate Staphylococcus aureus in a patient with septic arthritis during long-term treatment with daptomycin. J. Antimicrob. Chemother. 71, 1807–1814. doi: 10.1093/jac/dkw060
Lindgren, J. K., Thomas, V. C., Olson, M. E., Chaudhari, S. S., Nuxoll, A. S., Schaeffer, C. R., et al. (2014). Arginine deiminase in Staphylococcus epidermidis functions to augment biofilm maturation through pH homeostasis. J. Bacteriol. 196, 2277–2289. doi: 10.1128/jb.00051-14
Liu, S., Chen, H., Chen, J., Wang, T., Tu, S., Zhang, X., et al. (2023). Transcriptome and proteome of methicillin-resistant Staphylococcus aureus small-colony variants reveal changed metabolism and increased immune evasion. Microbiol. Spectr. 11:e0189822. doi: 10.1128/spectrum.01898-22
Liu, J., Shen, Z., Tang, J., Huang, Q., Jian, Y., Liu, Y., et al. (2021). Extracellular DNA released by glycine-auxotrophic Staphylococcus epidermidis small colony variant facilitates catheter-related infections. Commun. Biol. 4, 904–914. doi: 10.1038/s42003-021-02423-4
Longshaw, C. M., Farrell, A. M., Wright, J. D., and Holland, K. T. (2000). Identification of a second lipase gene, gehD, in Staphylococcus epidermidis: comparison of sequence with those of other staphylococcal lipases. Microbiology 146, 1419–1427. doi: 10.1099/00221287-146-6-1419
Lord, D. M., Baran, A. U., Wood, T. K., Peti, W., and Page, R. (2014). BdcA, a protein important for Escherichia coli biofilm dispersal, is a short-chain dehydrogenase/reductase that binds specifically to NADPH. PLoS One 9:e105751. doi: 10.1371/journal.pone.0105751
Loss, G., Simões, P. M., Valour, F., Cortês, M. F., Gonzaga, L., Bergot, M., et al. (2019). Staphylococcus aureus Small Colony Variants (SCVs): news from a chronic prosthetic joint infection. Front. Cell. Infect. Microbiol. 9:363. doi: 10.3389/fcimb.2019.00363
Magnuson, R. D. (2007). Hypothetical functions of toxin-antitoxin systems. J. Bacteriol. 189, 6089–6092. doi: 10.1128/jb.00958-07
Magryś, A., Paluch-Oleś, J., Bogut, A., Kiełbus, M., Plewik, D., and Kozioł-Montewka, M. (2015). The role of programmed death ligand 1 pathway in persistent biomaterial-associated infections. J. Microbiol. 53, 544–552. doi: 10.1007/s12275-015-5022-7
Makhlin, J., Kofman, T., Borovok, I., Kohler, C., Engelmann, S., Cohen, G., et al. (2007). Staphylococcus aureus ArcR controls expression of the arginine deiminase operon. J. Bacteriol. 189, 5976–5986. doi: 10.1128/JB.00592-07
Marraffini, L. A., and Sontheimer, E. J. (2008). CRISPR interference limits horizontal gene transfer in staphylococci by targeting DNA. Science 322, 1843–1845. doi: 10.1126/science.1165771
McManus, B. A., O’Connor, A. M., Egan, S. A., Flanagan, P. R., and Coleman, D. C. (2019). First description of arginine catabolic mobile element (ACME) type VI harboring the kdp operon only in Staphylococcus epidermidis using short and long read whole genome sequencing: further evidence of ACME diversity. Infect. Genet. Evol. 71, 51–53. doi: 10.1016/j.meegid.2019.03.008
Miragaia, M., Thomas, J. C., Couto, I., Enright, M. C., and de Lencastre, H. (2007). Inferring a population structure for Staphylococcus epidermidis from multilocus sequence typing data. J. Bacteriol. 189, 2540–2552. doi: 10.1128/JB.01484-06
Mitchell, G., Brouillette, E., Séguin, D. L., Asselin, A.-E., Jacob, C. L., and Malouin, F. (2010). A role for sigma factor B in the emergence of Staphylococcus aureus small-colony variants and elevated biofilm production resulting from an exposure to aminoglycosides. Microb. Pathog. 48, 18–27. doi: 10.1016/j.micpath.2009.10.003
Mitchell, G., Fugère, A., Pépin Gaudreau, K., Brouillette, E., Frost, E. H., Cantin, A. M., et al. (2013). SigB is a dominant regulator of virulence in Staphylococcus aureus small-colony variants. PLoS One 8:e65018. doi: 10.1371/journal.pone.0065018
Mitchell, C., and Oliver, D. (1993). Two distinct ATP-binding domains are needed to promote protein export by Escherichia coli SecA ATPase. Mol. Microbiol. 10, 483–497. doi: 10.1111/j.1365-2958.1993.tb00921.x
Nakakido, M., Aikawa, C., Nakagawa, I., and Tsumoto, K. (2014). The staphylococcal elastin-binding protein regulates zinc-dependent growth/biofilm formation. J. Biochem. 156, 155–162. doi: 10.1093/jb/mvu027
Nayfach, S., Camargo, A. P., Schulz, F., Eloe-Fadrosh, E., Roux, S., and Kyrpides, N. C. (2021). CheckV assesses the quality and completeness of metagenome-assembled viral genomes. Nat. Biotechnol. 39, 578–585. doi: 10.1038/s41587-020-00774-7
Newman, E. B., Kapoor, V., and Potter, R. (1976). Role of L-threonine dehydrogenase in the catabolism of threonine and synthesis of glycine by Escherichia coli. J. Bacteriol. 126, 1245–1249. doi: 10.1128/jb.126.3.1245-1249.1976
Niehaus, T. D., Elbadawi-Sidhu, M., de Crécy-Lagard, V., Fiehn, O., and Hanson, A. D. (2017). Discovery of a widespread prokaryotic 5-oxoprolinase that was hiding in plain sight. J. Biol. Chem. 292, 16360–16367. doi: 10.1074/jbc.M117.805028
Noens, E. E. E., and Lolkema, J. S. (2017). Convergent evolution of the arginine deiminase pathway: the ArcD and ArcE arginine/ornithine exchangers. MicrobiologyOpen 6:e00412. doi: 10.1002/mbo3.412
Noto, M. J., Fox, P. M., and Archer, G. L. (2008). Spontaneous deletion of the methicillin resistance determinant, mecA, partially compensates for the fitness cost associated with high-level vancomycin resistance in Staphylococcus aureus. Antimicrob. Agents Chemother. 52, 1221–1229. doi: 10.1128/AAC.01164-07
Ohniwa, R. L., Kitabayashi, K., and Morikawa, K. (2013). Alternative cardiolipin synthase Cls1 compensates for stalled Cls2 function in Staphylococcus aureus under conditions of acute acid stress. FEMS Microbiol. Lett. 338, 141–146. doi: 10.1111/1574-6968.12037
Onyango, L. A., Dunstan, R. H., Roberts, T. K., Macdonald, M. M., and Gottfries, J. (2013). Phenotypic variants of staphylococci and their underlying population distributions following exposure to stress. PLoS One 8:e77614. doi: 10.1371/journal.pone.0077614
Orsi, R. H., Bowen, B. M., and Wiedmann, M. (2010). Homopolymeric tracts represent a general regulatory mechanism in prokaryotes. BMC Genomics 11:102. doi: 10.1186/1471-2164-11-102
Over, B., Heusser, R., McCallum, N., Schulthess, B., Kupferschmied, P., Gaiani, J. M., et al. (2011). LytR-CpsA-Psr proteins in Staphylococcus aureus display partial functional redundancy and the deletion of all three severely impairs septum placement and cell separation. FEMS Microbiol. Lett. 320, 142–151. doi: 10.1111/j.1574-6968.2011.02303.x
Park, C. B., Lee, S. B., and Ryu, D. D. Y. (2001). L-pyroglutamate spontaneously formed from l-glutamate inhibits growth of the hyperthermophilic archaeon Sulfolobus solfataricus. Appl. Environ. Microbiol. 67, 3650–3654. doi: 10.1128/AEM.67.8.3650-3654.2001
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Parvizi, J., Zmistowski, B., Berbari, E. F., Bauer, T. W., Springer, B. D., Della Valle, C. J., et al. (2011). New definition for periprosthetic joint infection: from the Workgroup of the Musculoskeletal Infection Society. Clin. Orthop. Relat. Res. 469, 2992–2994. doi: 10.1007/s11999-011-2102-9
Perez, K., and Patel, R. (2017). Staphylococcusepidermidis small-colony variants are induced by low pH and their frequency reduced by lysosomal alkalinization. J. Infect. Dis. 215, 488–490. doi: 10.1093/infdis/jiw503
Perez, K., and Patel, R. (2018). Survival of Staphylococcus epidermidis in fibroblasts and osteoblasts. Infect. Immun. 86:e00237-18. doi: 10.1128/IAI.00237-18
Pinho, M., Kjos, M., and Veening, J. W. (2013). How to get (a)round: mechanisms controlling growth and division of coccoid bacteria. Nat. Rev. Microbiol. 11, 601–614. doi: 10.1038/nrmicro3088
Post, V., Harris, L. G., Morgenstern, M., Mageiros, L., Hitchings, M. D., Méric, G., et al. (2017). Comparative genomics study of Staphylococcus epidermidis isolates from orthopedic-device-related infections correlated with patient outcome. J. Clin. Microbiol. 55, 3089–3103. doi: 10.1128/JCM.00881-17
Proctor, R. (2019). Respiration and small colony variants of Staphylococcus aureus. Microbiol. Spectr. 7. doi: 10.1128/microbiolspec.GPP3-0069-2019
Proctor, R. A., Kriegeskorte, A., Kahl, B. C., Becker, K., Löffler, B., and Peters, G. (2014). Staphylococcus aureus Small Colony Variants (SCVs): a road map for the metabolic pathways involved in persistent infections. Front. Cell. Infect. Microbiol. 4:99. doi: 10.3389/fcimb.2014.00099
Proctor, R. A., von Eiff, C., Kahl, B. C., Becker, K., McNamara, P., Herrmann, M., et al. (2006). Small colony variants: a pathogenic form of bacteria that facilitates persistent and recurrent infections. Nat. Rev. Microbiol. 4, 295–305. doi: 10.1038/nrmicro1384
Ramos, J. L., Martínez-Bueno, M., Molina-Henares, A. J., Terán, W., Watanabe, K., Zhang, X., et al. (2005). The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69, 326–356. doi: 10.1128/mmbr.69.2.326-356.2005
Raue, S., Fan, S.-H., Rosenstein, R., Zabel, S., Luqman, A., Nieselt, K., et al. (2020). The genome of Staphylococcus epidermidis O47. Front. Microbiol. 11:2061. doi: 10.3389/fmicb.2020.02061
Reitzer, L. (2014). “Amino acid synthesis☆” in Reference module in biomedical sciences (Amsterdam: Elsevier)
Roche, F. M., Downer, R., Keane, F., Speziale, P., Park, P. W., and Foster, T. J. (2004). The N-terminal A domain of fibronectin-binding proteins A and B promotes adhesion of Staphylococcus aureus to elastin. J. Biol. Chem. 279, 38433–38440. doi: 10.1074/jbc.M402122200
Rohde, H., Frankenberger, S., Zähringer, U., and Mack, D. (2010). Structure, function and contribution of polysaccharide intercellular adhesin (PIA) to Staphylococcus epidermidis biofilm formation and pathogenesis of biomaterial-associated infections. Eur. J. Cell Biol. 89, 103–111. doi: 10.1016/j.ejcb.2009.10.005
Rossi, C. C., Souza-Silva, T., Araújo-Alves, A. V., and Giambiagi-deMarval, M. (2017). CRISPR-Cas systems features and the gene-reservoir role of coagulase-negative staphylococci. Front. Microbiol. 8:1545. doi: 10.3389/fmicb.2017.01545
Rowe, S. E., Campbell, C., Lowry, C., O'Donnell, S. T., Olson, M. E., Lindgren, J. K., et al. (2016). AraC-type regulator Rbf controls the Staphylococcus epidermidis biofilm phenotype by negatively regulating the icaADBC repressor SarR. J. Bacteriol. 198, 2914–2924. doi: 10.1128/jb.00374-16
Rowland, S. J., and Dyke, K. G. (1990). Tn552, a novel transposable element from Staphylococcus aureus. Mol. Microbiol. 4, 961–975. doi: 10.1111/j.1365-2958.1990.tb00669.x
Schaaff, F., Bierbaum, G., Baumert, N., Bartmann, P., and Sahl, H.-G. (2003). Mutations are involved in emergence of aminoglycoside-induced small colony variants of Staphylococcus aureus. Int. J. Med. Microbiol. 293, 427–435. doi: 10.1078/1438-4221-00282
Schoenfelder, S. M. K., Marincola, G., Geiger, T., Goerke, C., Wolz, C., and Ziebuhr, W. (2013). Methionine biosynthesis in Staphylococcus aureus is tightly controlled by a hierarchical network involving an initiator tRNA-specific T-box riboswitch. PLoS Pathog. 9:e1003606. doi: 10.1371/journal.ppat.1003606
Seemann, T. (2023a). mlst. Available at: https://github.com/tseemann/mlst (Accessed February 15, 2023).
Seemann, T. (2023b). Snippy. Available at: https://github.com/tseemann/snippy (Accessed January 19, 2023).
Seggewiss, J., Becker, K., Kotte, O., Eisenacher, M., Yazdi, M. R. K., Fischer, A., et al. (2006). Reporter metabolite analysis of transcriptional profiles of a Staphylococcus aureus strain with normal phenotype and its isogenic hemB mutant displaying the small-colony-variant phenotype. J. Bacteriol. 188, 7765–7777. doi: 10.1128/JB.00774-06
Sendi, P., Graber, P., and Zimmerli, W. (2005). Loss of mecA gene in Staphylococcus epidermidis after prolonged therapy with vancomycin. J. Antimicrob. Chemother. 56, 794–795. doi: 10.1093/jac/dki316
Shore, A. C., and Coleman, D. C. (2013). Staphylococcal cassette chromosome mec: recent advances and new insights. Int. J. Med. Microbiol. 303, 350–359. doi: 10.1016/j.ijmm.2013.02.002
Siboo, I. R., Chambers, H. F., and Sullam, P. M. (2005). Role of SraP, a serine-rich surface protein of Staphylococcus aureus, in binding to human platelets. Infect. Immun. 73, 2273–2280. doi: 10.1128/iai.73.4.2273-2280.2005
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Starikova, E. V., Tikhonova, P. O., Prianichnikov, N. A., Rands, C. M., Zdobnov, E. M., Ilina, E. N., et al. (2020). Phigaro: high-throughput prophage sequence annotation. Bioinformatics 36, 3882–3884. doi: 10.1093/bioinformatics/btaa250
Stefanović, C., Hager, F. F., and Schäffer, C. (2021). LytR-CpsA-Psr glycopolymer transferases: essential bricks in gram-positive bacterial cell wall assembly. Int. J. Mol. Sci. 22:908. doi: 10.3390/ijms22020908
Stimeling, K. W., Graham, J. E., Kaenjak, A., and Wilkinson, B. J. (1994). Evidence for feedback (trans) regulation of, and two systems for, glycine betaine transport by Staphylococcus aureus. Microbiology 140, 3139–3144. doi: 10.1099/13500872-140-11-3139
Suvorova, I. A., and Rodionov, D. A. (2016). Comparative genomics of pyridoxal 5′-phosphate-dependent transcription factor regulons in Bacteria. Microbial Genomics 2:e000047. doi: 10.1099/mgen.0.000047
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tollerson, R., Witzky, A., and Ibba, M. (2018). Elongation factor P is required to maintain proteome homeostasis at high growth rate. Proc. Natl. Acad. Sci. 115, 11072–11077. doi: 10.1073/pnas.1812025115
Tramonti, A., Nardella, C., di Salvo, M. L., Pascarella, S., and Contestabile, R. (2018). The MocR-like transcription factors: pyridoxal 5′-phosphate-dependent regulators of bacterial metabolism. FEBS J. 285, 3925–3944. doi: 10.1111/febs.14599
Trivedi, S., Uhlemann, A.-C., Herman-Bausier, P., Sullivan, S. B., Sowash, M. G., Flores, E. Y., et al. (2017). The surface protein SdrF mediates Staphylococcus epidermidis adherence to keratin. J. Infect. Dis. 215, 1846–1854. doi: 10.1093/infdis/jix213
Tsai, M., Ohniwa, R. L., Kato, Y., Takeshita, S. L., Ohta, T., Saito, S., et al. (2011). Staphylococcus aureus requires cardiolipin for survival under conditions of high salinity. BMC Microbiol. 11:13. doi: 10.1186/1471-2180-11-13
Tuchscherr, L., Bischoff, M., Lattar, S. M., Noto Llana, M., Pförtner, H., Niemann, S., et al. (2015). Sigma factor SigB is crucial to mediate Staphylococcus aureus adaptation during chronic infections. PLoS Pathog. 11:e1004870. doi: 10.1371/journal.ppat.1004870
Tuchscherr, L., Kreis, C. A., Hoerr, V., Flint, L., Hachmeister, M., Geraci, J., et al. (2016). Staphylococcus aureus develops increased resistance to antibiotics by forming dynamic small colony variants during chronic osteomyelitis. J. Antimicrob. Chemother. 71, 438–448. doi: 10.1093/jac/dkv371
Tuchscherr, L., Löffler, B., and Proctor, R. A. (2020). Persistence of Staphylococcus aureus: multiple metabolic pathways impact the expression of virulence factors in small-Colony variants (SCVs). Front. Microbiol. 11:1028. doi: 10.3389/fmicb.2020.01028
Tuchscherr, L., Medina, E., Hussain, M., Völker, W., Heitmann, V., Niemann, S., et al. (2011). Staphylococcus aureus phenotype switching: an effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol. Med. 3, 129–141. doi: 10.1002/emmm.201000115
Tyson, K., Metheringham, R., Griffiths, L., and Cole, J. (1997). Characterisation of Escherichia coli K-12 mutants defective in formate-dependent nitrite reduction: essential roles for hemN and the menFDBCE operon. Arch. Microbiol. 168, 403–411. doi: 10.1007/s002030050515
Uma Mahesh, M. V. N., Faidh, M. A., and Chadha, A. (2022). The ornithine cyclodeaminase/μ-crystallin superfamily of proteins: a novel family of oxidoreductases for the biocatalytic synthesis of chiral amines. Curr. Res. Biotechnol. 4, 402–419. doi: 10.1016/j.crbiot.2022.09.003
UniProtKB|UniProt pxpA - 5-oxoprolinase subunit A – Escherichia coli (strain 55989/EAEC) (n.d.). Available at: https://www.uniprot.org/uniprotkb/B7LAB9/entry (Accessed July 10, 2023).
Vesga, O., Groeschel, M. C., Otten, M. F., Brar, D. W., Vann, J. M., and Proctor, R. A. (1996). Staphylococcus aureus small colony variants are induced by the endothelial cell intracellular milieu. J. Infect. Dis. 173, 739–742. doi: 10.1093/infdis/173.3.739
Vestergaard, M., Paulander, W., Leng, B., Nielsen, J. B., Westh, H. T., and Ingmer, H. (2016). Novel pathways for ameliorating the fitness cost of gentamicin resistant small colony variants. Front. Microbiol. 7:1866. doi: 10.3389/fmicb.2016.01866
Wacnik, K., Rao, V. A., Chen, X., Lafage, L., Pazos, M., Booth, S., et al. (2022). Penicillin-binding protein 1 (PBP1) of Staphylococcus aureus has multiple essential functions in cell division. MBio 13, e00669–e00622. doi: 10.1128/mbio.00669-22
Wang, C., Fan, J., Niu, C., Wang, C., Villaruz, A. E., Otto, M., et al. (2010). Role of spx in biofilm formation of Staphylococcus epidermidis. FEMS Immunol. Med. Microbiol. 59, 152–160. doi: 10.1111/j.1574-695X.2010.00673.x
Wang, J., Rao, L., Huang, Z., Ma, L., Yang, T., Yu, Z., et al. (2022). The nitric oxide synthase gene negatively regulates biofilm formation in Staphylococcus epidermidis. Front. Cell. Infect. Microbiol. 12:1015859. doi: 10.3389/fcimb.2022.1015859
Wang, W., and Sun, B. (2021). VraCP regulates cell wall metabolism and antibiotic resistance in vancomycin-intermediate Staphylococcus aureus strain Mu50. J. Antimicrob. Chemother. 76, 1712–1723. doi: 10.1093/jac/dkab113
Weisser, M., Schoenfelder, S. M. K., Orasch, C., Arber, C., Gratwohl, A., Frei, R., et al. (2010). Hypervariability of biofilm formation and oxacillin resistance in a Staphylococcus epidermidis strain causing persistent severe infection in an immunocompromised patient. J. Clin. Microbiol. 48, 2407–2412. doi: 10.1128/JCM.00492-10
Wick, R. R., Judd, L. M., Gorrie, C. L., and Holt, K. E. (2017). Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol. 13:e1005595. doi: 10.1371/journal.pcbi.1005595
Williams, W. A., Zhang, R., Zhou, M., Joachimiak, G., Gornicki, P., Missiakas, D., et al. (2004). The membrane-associated lipoprotein-9 GmpC from Staphylococcus aureus binds the dipeptide GlyMet via side chain interactions. Biochemistry 43, 16193–16202. doi: 10.1021/bi048877o
Yagishita, H., Kumagai, Y., Konishi, K., Takahashi, Y., Aoba, T., and Yoshikawa, M. (2001). Histopathological studies on virulence of dipeptidyl aminopeptidase IV (DPPIV) of Porphyromonas gingivalis in a mouse abscess model: use of a DPPIV-deficient mutant. Infect. Immun. 69, 7159–7161. doi: 10.1128/IAI.69.11.7159-7161.2001
Zhang, Z., Schwartz, S., Wagner, L., and Miller, W. (2000). A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 7, 203–214. doi: 10.1089/10665270050081478
Keywords: small-colony variant (SCV), Staphylococcus epidemidis , prosthetic joint infection (PJI), auxotrophy, arginine deiminase pathway (ADI), Staphylococcal Cassette Chromosome, homopolymeric tracts, phase variation (PV)
Citation: Bogut A, Koper P, Marczak M and Całka P (2023) The first genomic characterization of a stable, hemin-dependent small colony variant strain of Staphylococcus epidermidis isolated from a prosthetic-joint infection. Front. Microbiol. 14:1289844. doi: 10.3389/fmicb.2023.1289844
Edited by:
María Guembe, Gregorio Marañón Hospital, SpainReviewed by:
John Jairo Aguilera-Correa, Health Research Institute Foundation Jimenez Diaz (IIS-FJD), SpainPablo Sanz-Ruiz, Gregorio Marañon Hospital, Spain
Copyright © 2023 Bogut, Koper, Marczak and Całka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Agnieszka Bogut, YWduaWVzemthLmJvZ3V0QHVtbHViLnBs