
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 15 November 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1289671
 Oldřich Bartoš1*†
Oldřich Bartoš1*† Běla Klimešová2†
Běla Klimešová2† Karolina Volfová2
Karolina Volfová2 Martin Chmel1,3
Martin Chmel1,3 Jiří Dresler1
Jiří Dresler1 Petr Pajer1Hana Kabíčková1
Petr Pajer1Hana Kabíčková1 Peter Adamík4,5
Peter Adamík4,5 David Modrý6,7,8Alena Myslivcová Fučíková9
David Modrý6,7,8Alena Myslivcová Fučíková9 Jan Votýpka2,6
Jan Votýpka2,6Bartonelloses are neglected emerging infectious diseases caused by facultatively intracellular bacteria transmitted between vertebrate hosts by various arthropod vectors. The highest diversity of Bartonella species has been identified in rodents. Within this study we focused on the edible dormouse (Glis glis), a rodent with unique life-history traits that often enters households and whose possible role in the epidemiology of Bartonella infections had been previously unknown. We identified and cultivated two distinct Bartonella sub(species) significantly diverging from previously described species, which were characterized using growth characteristics, biochemical tests, and various molecular techniques including also proteomics. Two novel (sub)species were described: Bartonella grahamii subsp. shimonis subsp. nov. and Bartonella gliris sp. nov. We sequenced two individual strains per each described (sub)species. During exploratory genomic analyses comparing two genotypes ultimately belonging to the same species, both factually and most importantly even spatiotemporally, we noticed unexpectedly significant structural variation between them. We found that most of the detected structural variants could be explained either by prophage excision or integration. Based on a detailed study of one such event, we argue that prophage deletion represents the most probable explanation of the observed phenomena. Moreover, in one strain of Bartonella grahamii subsp. shimonis subsp. nov. we identified a deletion related to Bartonella Adhesin A, a major pathogenicity factor that modulates bacteria-host interactions. Altogether, our results suggest that even a limited number of passages induced sufficient selective pressure to promote significant changes at the level of the genome.
Bacteria of the genus Bartonella are facultative intracellular pathogens that parasitize erythrocytes and endothelial cells (Han et al., 2022). They have been described in a wide spectrum of mammalian hosts including rodents, carnivores, ungulates and humans (e.g., Cheslock and Embers, 2019). Members of the genus Bartonella are causative agents of rather neglected emerging human infectious zoonotic diseases altogether called bartonelloses.
While infections in natural hosts are mostly asymptomatic even though chronic bacteremia is developed (Jacomo et al., 2002; Harms and Dehio, 2012), the described symptoms of bartonelloses in humans range from mild symptoms such as fever to severe ones such as encephalitis or arthritis (Okaro et al., 2017; Krügel et al., 2022). Although most of the human cases are connected primarily with only three species (B. bacilliformis, B. quintana, and B. henselae; Okaro et al., 2017), it has been hypothesized that virtually any Bartonella species can have zoonotic potential (Breitschwerdt and Kordick, 2000; Breitschwerdt et al., 2010; Okaro et al., 2017). Due to the rather difficult diagnosis, only recent studies have shown how common not only seroprevalence, but also active bacteremia can be in humans (Pitassi et al., 2015; Portillo et al., 2020). Infection of vertebrate hosts is mediated primarily through a bite or infected feces of blood-sucking arthropods such as fleas or lice (e.g., Iannino et al., 2018).
In recent years, there has been a rapid increase in the number of recognized Bartonella species (Breitschwerdt, 2017). Currently, there are roughly more than 100 recognized (sub)species, although some of them yet lack a formal description (Parte et al., 2020). Most of them resulted from adaptive radiation triggered by acquisition of a key molecular system that is directly related to their unique ability to colonize diverse mammalian hosts (Jacomo et al., 2002; Engel et al., 2011). Thus, individual Bartonella species adapted/specialized to distinct hosts which has ultimately resulted in their compelling species diversity, especially in rodents (Gutiérrez et al., 2015). The prevalence of bartonellae in various small rodents typically varies from 30% to 90% and we are not aware of any study that has reported a naive rodent population (Knap et al., 2007; Tołkacz et al., 2018; Mardosaitė-Busaitienė et al., 2019).
Within this study we focused on edible dormice (Glis glis), small rodents with nocturnal activity and obligatory seasonal hibernation (Kryštufek, 2010); distributed in Europe and Asia Minor (Amori et al., 2021). Edible dormice are known for their long lifespan (up to 10 years) as well as their intermittent breeding strategy, which make their life-history traits unique among rodents (Pilastro et al., 2003). Whereas in the past edible dormice were considered a delicacy and hunted or bred, now they enter households voluntarily and come into frequent contact with people, which can pose a risk of transmission of various etiological agents (Büchner et al., 2018).
The main aim of this study is to describe and characterize novel isolates of Bartonella (species and subspecies) cultivated from the blood of edible dormice captured in the Czech Republic. Furthermore, we describe our experience with Bartonella cultivation and most importantly, it’s almost immediate effects on the bacterial genomes, when only a few passages appear to be sufficient to initiate (selective) changes.
Edible dormice were sampled as a part of a long-term ecological project in mixed forest stands close to Dlouhá Loučka, Olomouc district, Czech Republic (49°49′ N, 17° 12′ E). Altogether, 31 adult animals were captured in August 2019 in nest boxes that they use as resting and breeding sites (for details on the study site see Gazárková and Adamík, 2016). The animals were narcotized with an intramuscular application of ketamine/medetomidine, and the blood was sampled into microtubes with EDTA from the inferior vena cava cranialis using a sterilized hypodermic needle. Thereafter, the dormice were aroused from narcosis using Revertor and put back into their original nest boxes. The dormice were handled on the basis of permission issued by the Regional Authority of the Olomouc Region (KUOK 61548/2017). Field study protocols were approved by the Ethical Committees of Palacky University and the Ministry of Education (1/2011, 5525/2008-30).
The recommendations described in Gutiérrez et al. (2017) were followed for Bartonella cultivation (see also Riess et al., 2008). The cultivation medium consisting of Schneider’s Insect Medium (Sigma Aldrich) supplemented with fetal calf serum (10%), saccharose (5%), and Amphotericin B (0.1%) was filtered through a 0.22 μm filter (MILEX) into a new tube before use. The medium thus prepared was added to the blood samples. Such dilution in liquid media has been shown to improve the cultivation of bartonellae by limiting the overgrowth and influence of co-infecting bacteria that are common in wild animals (Gutiérrez et al., 2017). The samples were inoculated on chocolate agar plates and cultivated at 37°C in an atmosphere enriched with 5% CO2 for up to 6 weeks. Bartonella presence was initially determined by the morphology of the colonies and a total of 32 candidate colonies per blood sample were further examined by PCR. To obtain clones of particular Bartonella strains, a single colony growing on the original plate was inoculated onto a new agar plate. The same process was repeated two more times and these cultures (passaged at total three times consecutively; grown four times on agar plates) were considered to be a clone of a single Bartonella strain. These retrieved clonal lineages were used for further analysis and characterization.
DNA extracted from selected colonies and all of the original blood samples were subjected to PCR screening of the citrate synthase gene (gltA, 750 bp). DNeasy Blood & Tissue Kit (QIAGEN) was used for DNA extraction according to the product manual. For detailed information about the PCR including cycling conditions and sequences of primer pairs see Majerová et al. (2021). All Bartonella positive PCR products were purified using the ExoSAP-IT™ (Thermo Scientific) and sequenced by the BIOCEV sequencing laboratory of the Faculty of Science, Charles University, Prague.
All sequence manipulations and analyses took place in the Geneious Prime environment v2022.1.1.1 The amplicon sequencing data were visually controlled and adjusted, including end trimming and correction for mis-called nucleotides (e.g., Crossley et al., 2020). Preliminary taxonomic classification of such sequences was retrieved through BLASTn against the NCBI GenBank database. Next, we selected representative reference sequences for phylogenetic analysis, particularly with respect to the phylogenetic context. The phylogenetic inference was performed using PhyML (Guindon et al., 2010) and model selection was based on the jModelTest (Posada, 2008).
One strain of each novel (sub)species was used for a morphological examination by electron microscopy. The samples were prepared by negative staining, formvar-coated carbon-reinforced grids were used, and the samples were stained with a 2% solution of phosphotungstic acid. In addition, fixation and embedding were also used as another method of sample preparation for electron microscopy. The samples were fixated in the solution of 2.5% glutaraldehyde (Polysciences) and 5 mM CaCl2 in 0.1 M cacodylate buffer (pH 7.2), dehydrated and subsequently embedded in Epon-Durcupan. The embedded samples were cut into ultrathin sections, placed on a copper grid, and contrasted with lead citrate and uranyl acetate. The samples prepared by embedding and negative staining were visualized using a transmission electron microscope (JEM 200CX Jeol). The length and width of the bacteria were measured, mean estimates of length and width were calculated.
Biochemical characterization was performed on one-week old cultures of the type strains of the novel (sub)species and the reference strain of B. henselae CNCTC 5656 (serving as a control), provided by the National Institute of Public Health (Czech Republic). For each strain, several colonies from a clonal lineage were resuspended in sterile distilled water and transferred into the standard commercial biochemical panel (API 20 E; bioMérieux, Marcy l’Etoile, Lyon, France) that was used according to the manufacturer’s instructions (incubation at 37°C for 24 h). The results were interpreted using the API reading scales according to the manufacturer’s directions. Catalase activity was tested by immersion of a bacteria in 3% H2O2. Catalase-positivity was assessed by the appearance of oxygen bubbles.
Based on results from capillary sequencing of several genes, two possibly novel (sub)species were selected for Whole Genome Sequencing (WGS) and subsequent analyses, each represented by two strains/genotypes (i.e., biological replicates).
Libraries for Illumina sequencing were prepared using the Nextera XT DNA Library Preparation Kit. DNA concentrations were measured by a Qubit™ fluorometer (Invitrogene) device in combination with a Qubit™ dsDNA HS Assay Kit (Invitrogene). Library length profiles were measured using the Qsep 1 Capillary Electrophoresis System (BiOptic). Prepared libraries were sequenced on the Illumina iSeq 100 Sequencing System (2 × 150 paired-end).
Libraries for Oxford Nanopore Technologies (ONT) sequencing were prepared from a high molecular weight DNA using a Ligation Sequencing Kit (SQK-LSK109) and Native Barcoding Expansion 1–12 and 13–24 kits (EXP-NBD104 and EXP-NBD114). Libraries were sequenced on the ONT GridION platform using the R9.4 chemistry (Flow-Cell). Sequencing data were base-called, i.e., transmission from physical changes in the electric current signal measured by the ONT sequencing device to biologically relevant bases, using Guppy v5.1.13 (e.g., Wick et al., 2019).
Basic characteristics of the Illumina libraries were checked using the FastQC tool (Andrews, 2010). Considering all four samples, combined coverage both from Illumina as well as from the ONT sequencing reached values >>100× (see Supplementary Table 1), which shall be sufficient for assembly of a rather small bacterial genome. Genomes were assembled using the MaSuRCA v4.0.9 hybrid assembler, which can utilize both the long error-prone reads as well as short accurate Illumina reads data in parallel (Zimin et al., 2017). Gene annotation was originally performed using Prokka v1.14.6 (Seemann, 2014) and finally by the NCBI Proteomic Genome Annotation Pipeline v2022-10-03 (Tatusova et al., 2016). The resulting genome sequences were deposited into NCBI GenBank repositories.
We deliberately selected representative sequences across bartonellae to set our samples into the phylogenetic context (see Supplementary Table 2). For selected species/strains we downloaded protein sequences from NCBI databases. We utilized OrthoFinder v2.5.4 to identify single copy orthologous genes present in all samples (Emms and Kelly, 2019). Next, Multiple Sequence Alignments were conducted in the software MAFFT v7.505 (Katoh and Standley, 2013). In order to estimate the species tree, we concatenated individual genes using the AMAS tool (Borowiec, 2016). Nevertheless, beyond the species tree itself, all individual Maximum Likelihood gene trees were reconstructed by IQ-TREE v2.2.0 (Nguyen et al., 2015) using the extended model selection with a free rate of heterogeneity in combination with 1,000 ultrafast bootstrap replicates (Nguyen et al., 2015; Kalyaanamoorthy et al., 2017; Hoang et al., 2018). Finally, both gene Concordance Factors (gCF) and site Concordance Factors (sCF) were evaluated (Minh et al., 2020). The resulting tree was visualized using iTOL v5 (Letunic and Bork, 2021).
The average nucleotide identity, i.e., overall sequence similarity, of the novel strains to their nearest relatives was estimated from the genomic sequences using the OrthoANI tool (Lee et al., 2016).
Further, we utilized the R package micropan designed for microbial Pan-genome analyses and visualizations (Snipen and Liland, 2015; R Core Team, 2021). However, instead of relying on Prodigal annotations (Hyatt et al., 2010) as suggested by the micropan pipeline, we utilized original NCBI and Prokka annotations, which seemed to provide less problematic/ambiguous results. All genes of all species were clustered to gene families into a so-called pan-matrix. This allowed us to visualize the relation between pan-genome and core-genome. Next, we estimated whether the pan-genome is open or closed based on a Heaps law model (Tettelin et al., 2008). Further, we computed the principal component analysis (PCA) using the pan-matrix, which clustered individual species based on similarity of their gene contents (presence/absence).
Given that we have for each (sub)species sequenced two independent strains, it allowed us to assess their intra-specific variability. The overall similarity of the strains to each other was estimated by the OrthoANI tool and further assessed by the phylogenomic approach. We mapped ONT long reads from one strain to the other using the rather restrictive mapper Winnowmap2 (Jain et al., 2022). Structural Variations (long deletions) were estimated using Sniffles v2.0.7 (Sedlazeck et al., 2018). Similarly, we inferred such events from Illumina data using the pipeline consisting of BWA-MEM v0.7.17 (Li, 2013), gammaBOriS (Sperlea et al., 2020) and CNV-BAC (Wu et al., 2020). The detected events were controlled visually using the Integrative Genomics Viewer (Robinson et al., 2011). Only substantial events of size > 1 kb were considered.
Furthermore, we screened the genomes for prophages using the Prophage Hunter web-tool (Song et al., 2019). The genome collinearity was assessed using the Gepard tool (Krumsiek et al., 2007). The presence of coiled-coil segments within selected proteins was assessed using the Quick2D online-tool (Delorenzi and Speed, 2002; Gabler et al., 2020). We utilized GenomeTools to visualize annotation features (Gremme et al., 2013).
We provide a brief summary of the methods used here; detailed information about the methods may be found in the Supplementary material. Samples of both novel (sub)species, cultivated on agar plates as described above, were analyzed by proteomic techniques. Several colonies per each sample were harvested and lysed for the purposes of protein extraction and digestion. Isolated peptides were subject to the shotgun Label Free Quantification (LFQ) proteomic analysis. All samples were processed using the UltiMate 3,000 RS Liquid Chromatography Nano System Flow Meter with the Orbitrap Mass Spectrometer (Thermo Scientific) and the acquired data were evaluated using MaxQuant software (Cox and Mann, 2008).
Bartonella cultivation was successful for 23 out of the 31 edible dormice individuals captured in August 2019 (for one animal not enough blood was taken and only DNA isolation was performed, three animals were Bartonella-negative and in four samples other bacterial contamination prevented Bartonella positivity assessment by the cultivation method). Two different Bartonella (sub)species were identified: in 14 dormice specimens, infection by a single Bartonella (sub)species was observed whereas in nine of them, colonies of the two Bartonella (sub)species occurred alongside one another.
However, PCR analysis of the original host blood samples revealed an even higher positivity (30/31; 96.8%) and proportion of mixed infections by the two Bartonella (sub)species (20/30; 66.7%).
Several Bartonella clonal lineages were acquired during the cultivation. We selected two clonal lineages per each presumably novel (sub)species which were further extensively analyzed. Specifically, the strains denoted as GG3s1 and GG23s2 represented B. grahamii subsp. shimonis subsp. nov., whereas the strains denoted as GG20g1 and GG6g2 represented B. gliris sp. nov.
Considering all of the isolated strains, optimal colony growth was observed at 37°C with 5% of CO2, i.e., the conditions commonly used for Bartonella species cultivation (Brenner et al., 1993; Gutiérrez et al., 2017). Any other experimental conditions led to slower or mostly suppressed growth (see Supplementary Figure 1). We did not observe any considerable differences in growth rates between the studied isolates.
The morphology of the colonies did not systematically differ between the individual strains or even individual (sub)species. Instead, we noticed some variability common to all of them. Overall, the colonies were round with cleanly cut edges of white to yellow color, ranging from 0.6 to 3 mm in diameter. The colonies differed in their consistency from hard colonies attached to the surface to creamy ones without an attachment. The morphology of the colonies is comparable to that of other Bartonella species (Molia et al., 2016; Gutiérrez et al., 2020).
Transmission electron microscopy revealed rod-shaped bacilli without any flagella or other noticeable structures (see Supplementary Figure 2). The size of Bartonella grahamii subsp. shimonis subsp. nov. ranged in length from 0.83 to 1.43 μm (mean = 1.1 μm, SD = 0.06 μm) and the width from 0.26 to 0.41 μm (mean = 0.34 μm, SD = 0.03 μm). The size of Bartonella gliris sp. nov. ranged in length from 0.76 to 1.26 μm (mean = 1 μm, SD = 0.14 μm) and the width from 0.3 to 0.51 μm (mean = 0.38 μm, SD = 0.06 μm). In general, the strains in the study fell within the expected size range and did not possess any apparent morphological traits that would differentiate them from other related Bartonella species (Breitschwerdt and Kordick, 2000; Minnick and Anderson, 2015).
All three tested Bartonella species (each represented by a single strain/genotype) were negative for the catalase activity test and all of the tests included in the commercial API 20E panel, except for production of the enzyme gelatinase, which was positive for Bartonella gliris sp. nov. and B. henselae.
The results for the reference strain B. grahamii subsp. shimonis subsp. nov. partially contradict former reports of various type strains of B. grahamii for which, for example, Voger-Proskauer test activity has been reported (Birtles et al., 1995). However, they are in agreement with other reports of Bartonella spp. (do Amaral et al., 2022). In general, standard biochemical tests can not characterize or even distinguish members of the genus Bartonella (Diddi et al., 2013; Celebi et al., 2021), and thus no further tests were performed.
Already the primary phylogenetic analyses revealed that the strains under study are possibly novel, i.e., they formed separate clusters in the inferred phylogenies (see Supplementary Figure 3). In particular, one (sub)species clustered with the B. grahamii species group whereas the other appeared as a sister lineage to B. washoensis. These results were comparable with the phylogenomic approach that utilized sequence data from 501 genes (see Figure 1). The branches separating the analyzed (sub)species from their nearest relatives are well supported by the Concordance Factors metrics, both gCF and sCF, which also applies to the most branches of the entire phylogeny.
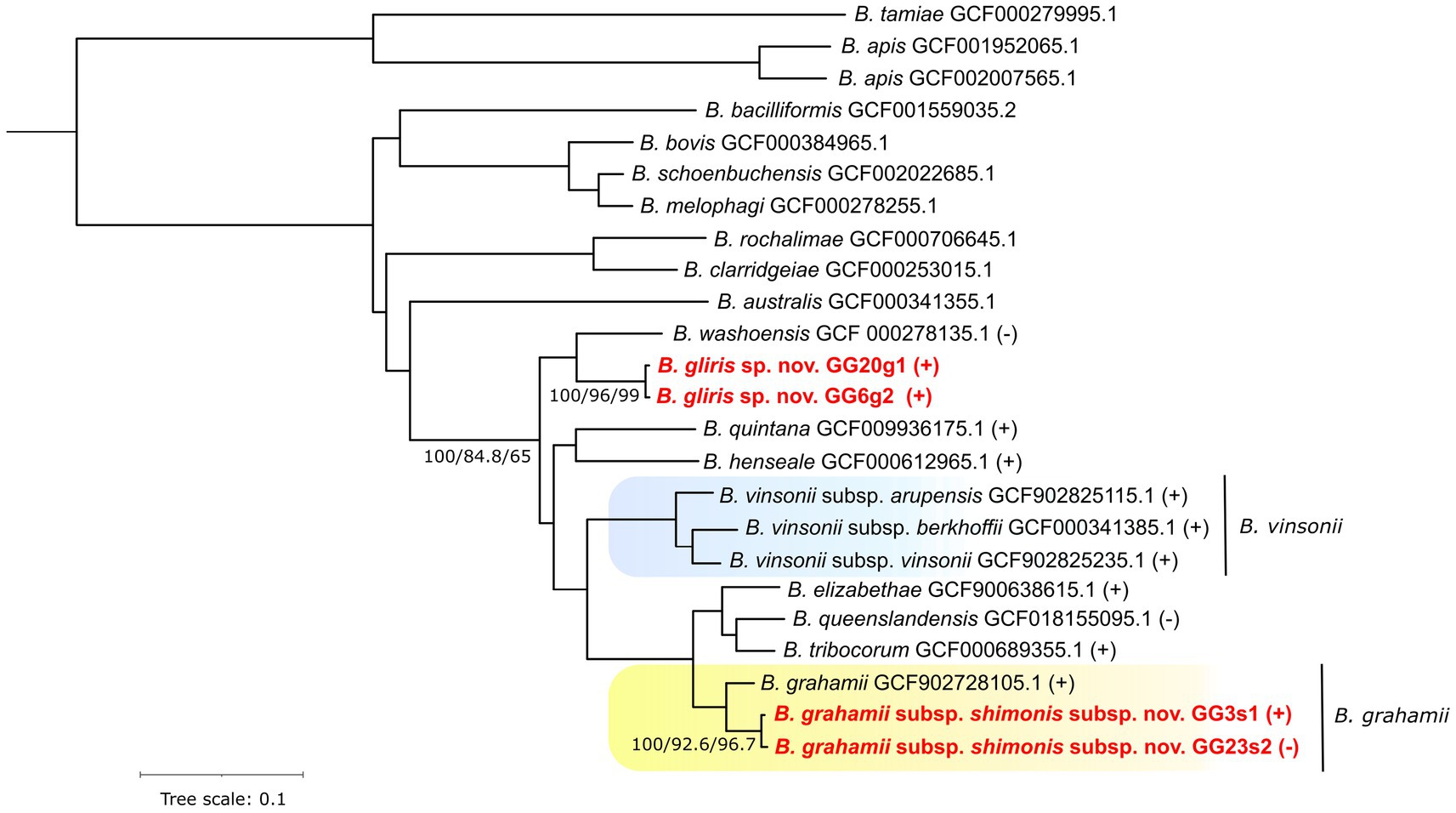
Figure 1. Phylogenomic species tree of Bartonella species/strains estimated from 501 single copy orthologous protein sequences. Branch supports expressed as Bootstrap values, gene Concordance Factors and site Concordance Factors are provided for selected branches. Symbols (+) and (−) indicate “Wolbachia endosymbiont wVitA of Nasonia vitripennis phage WOVitA1” prophage presence or absence, respectively, within a given species or strain. Newly described strains are highlighted by red font color. Background color indicates species-subspecies clusters.
The average nucleotide identity (ANI) scores estimated from the genomic sequences for the isolates GG3s1 and GG23s2 related to B. grahamii were 93.6% and 93.7%, respectively. The ANI estimates for the isolates GG3s1 and GG23s2 that appeared as sister lineage to B. washoensis only reached values of 88.6% and 88.9%, respectively (all estimates are based minimally on comparison of contigs > 1.45Mbp). The ANI values estimated between each of these strains and the rest of the entire phylogeny can be found in Supplementary Table 3. Basic statistics of the assembled genomes may be found in Supplementary Table 4.
Analysis of the pangenome showed that the core genome is relatively small, consisting of about 674 genes, while the pangenome is relatively large, probably exceeding more than 5,036 genes (see Figure 2). Based both on the shape of the pan-genome graph and the calculation according to Tettelin et al. (2008) (alpha estimate = 0.582), the pan-genome appears to be open. The PCA analysis that was based upon presence/absence of individual genes, contrary to phylogenetic estimates that are virtually based on sequence (dis)similarity, to some extent reflected patterns of the phylogenomic inference/history. In particular, the phylogenetically most distant (outgroup) species, i.e., B. apis and B. tamiae, are clearly separated from the rest of the samples (see Figure 3). In fact, the difference of those species was so significant that it hampered any comparisons within the ingroup species/samples, and therefore we excluded them from the analysis.
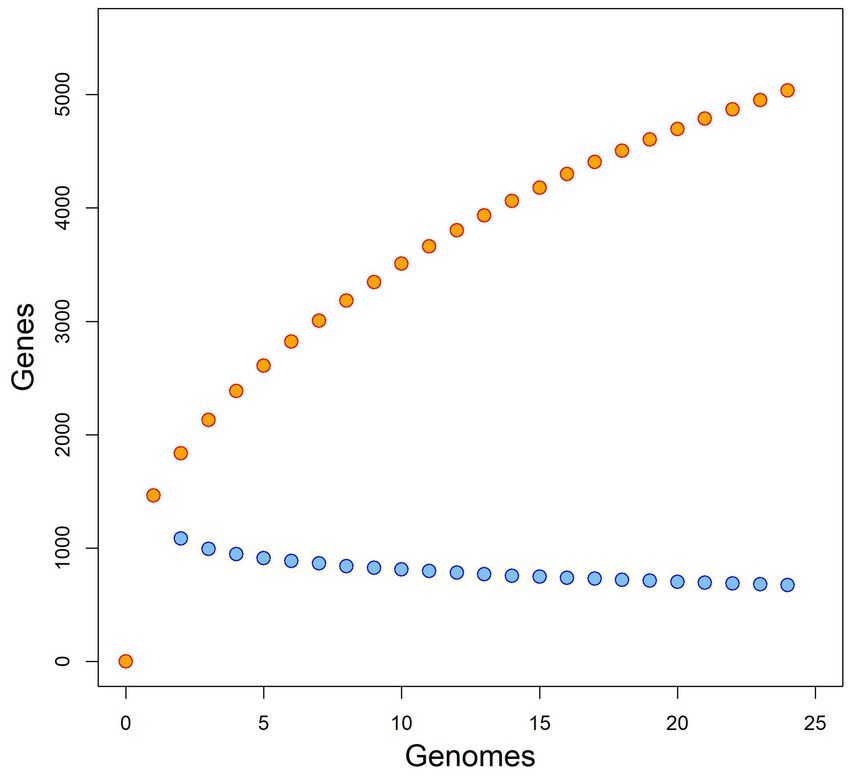
Figure 2. Pangenome and core-genome size for Bartonella spp., rarefaction “curves” plotted in orange and blue color, respectively.
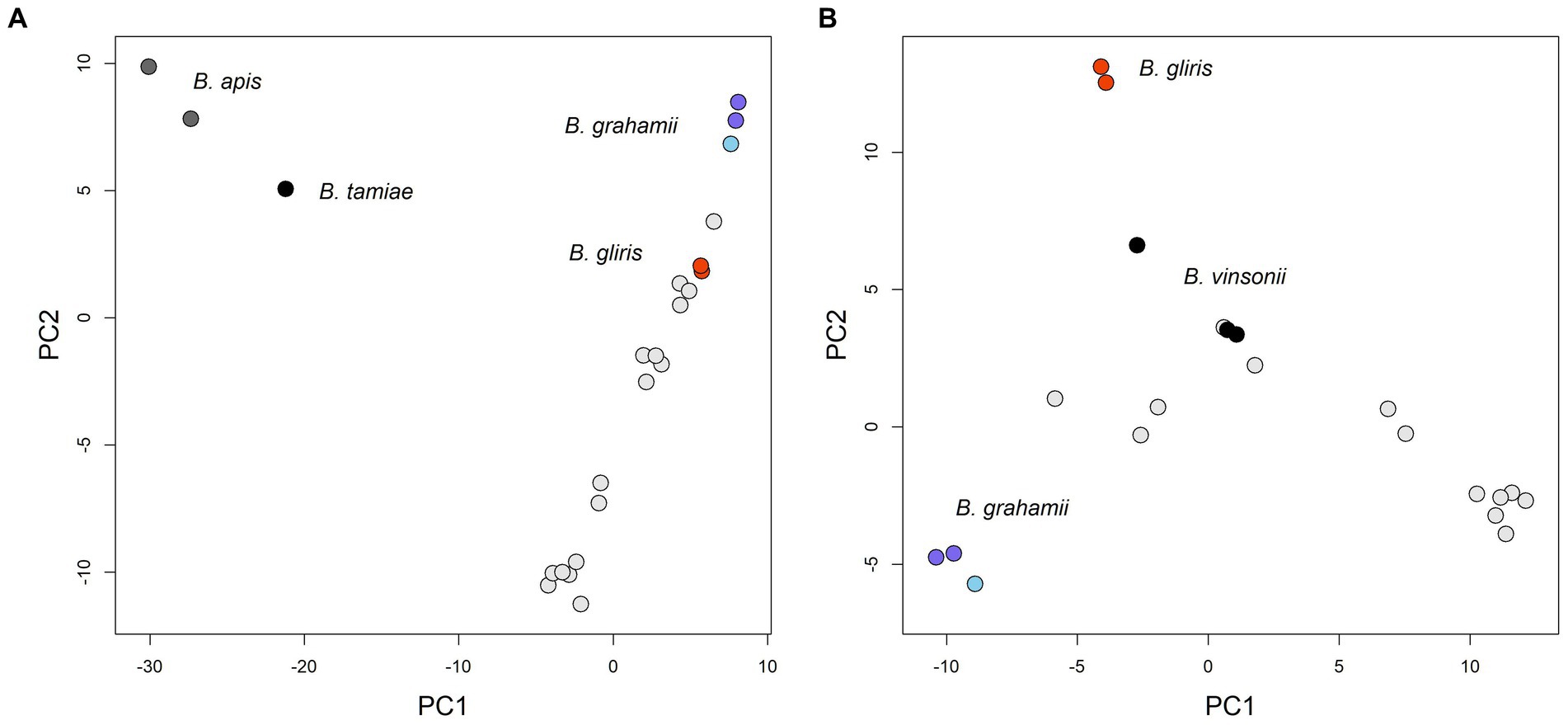
Figure 3. PCA figures based on similarity of gene contents between individual strains (gene presence/absence). Only several species could be highlighted both by distinct color and caption in the figures. Within the Bartonella grahamii group, B. grahamii subsp. shimonis subsp. nov. samples are depicted by darker blue. (A) The phylogenetically most distant species, i.e., B. apis and B. tamiae, appeared as clear outliers compared to the rest of samples in this analysis and factually dominated the Principal Component 1 (PC1); PC1 explained 28.0% of the total variability and PC2 13.0%. (B) We excluded B. apis and B. tamiae, which resulted in finer separation of the rest of the samples; PC1 explained 23.0% of the total variability and PC2 10.4%.
The overall similarity between both strain pairs expressed as Average Nucleotide Identity was 99.18% for B. grahamii subsp. shimonis subsp. nov. and 99.28% for B. gliris sp. nov. isolates. Furthermore, genome collinearity is almost fully preserved in both cases (see Supplementary Figures 4, 5), with the sole exception of deletion events (ranging in size typically from ~20 to ~55 kb).
Except for one such deletion event, which will be further described in detail, virtually all of the substantial deletions are linked with prophages. Moreover, such prophages are typically predicted with a high degree of certainty and denoted as “Active.” This suggests that the prophages most likely maintained their ability to excise from the genome (under certain conditions). As an example, we can describe probable loss of “Wolbachia endosymbiont wVitA of Nasonia vitripennis phage WOVitA1” in the B. grahamii subsp. shimonis subsp. nov. GG23s2 strain. Interestingly, this prophage contains a gene for Lysin sensu (Low et al., 2011), whose role in a bacteria/prophage is speculative. On one hand, it might serve the prophage leaving the cell by lysis of the cell wall (Fischetti, 2008), on the other hand, the presence of prophage Lysin affects the phenotype of a bacteria (Aucouturier et al., 2018). The presence of this prophage seems to be typical within the considered part of Bartonella phylogeny (see Figure 1). The only species in which the prophage was not detected were Bartonella washoensis and Bartonella quintana. The latter one has further a quite small (reduced) genome size (~1.6 Mbp) compared to a typical Bartonella species (~2 Mbp).
The prophage locus is bounded on both sides by an identical short sequence motif (“TCCCTCTCTCTCCGCCAT”). Whereas in the strain where the prophage was presumably excised, only a single copy of this motif was found. This suggests that the phage excision was probably achieved through site-specific tyrosine recombinase/integrase that is actually encoded by the prophage (e.g., Van Houdt et al., 2012).
The presumable deletion event that cannot be evidenced as prophage excision, also occurred in the B. grahamii subsp. shimonis subsp. nov. GG23s2 strain, moreover, in close proximity to the described prophage excision site. Interestingly, this ~22 kb long sequence contained Bartonella Adhesin A (BadA) sensu (Thibau et al., 2022), i.e., ~4,000 amino acids long protein coding gene with defined internal repetitive motifs. Moreover, the locus contained several accessory BadA subunits (or remnants). BadA is known to represent a key virulence factor (Thibau et al., 2022).
The lost copy of BadA clearly represents an ortholog of BadA sensu (Thibau et al., 2022), but beyond that, two other copies of BadA-like sequences were identified (see Figure 4). BadA-like protein 1 is sequentially related to the BadA gene, but differs in length and structure. It is substantially shorter and does not contain the typical repetitive motifs (coiled coils). BadA-like protein 1 could be due to its structural properties as well as topological position considered to be orthologous to a sequence described as BadA pseudogene by Thibau et al. (2022). BadA-like protein 0 differs considerably sequentially from the two other sequences.

Figure 4. Presumable wild type Bartonella Adhesin A (BadA) locus identified in Bartonella grahamii subsp. shimonis GG3s1 strain. The locus contains three BadA(-like) genes that are well diversified at the sequence level and structural properties. The rightmost copy represents the BadA sensu (Thibau et al., 2022), i.e., the presumed functional copy of the Bartonella Adhesin A with described structure and properties.
Further, we revealed the mechanistic basis of BadA locus deletion. BadA and BadA-like protein 1 are in general quite divergent (63% identity), however they share perfect homology at a part of the C-terminal domain (~460 aa). We noticed that BadA-like protein 1 in the genome with the deleted BadA locus is “chimeric,” i.e., its C-terminal domain comes from the BadA. This suggests that the BadA locus was excised through recombination between the conserved C-terminal domains (see Figure 5).
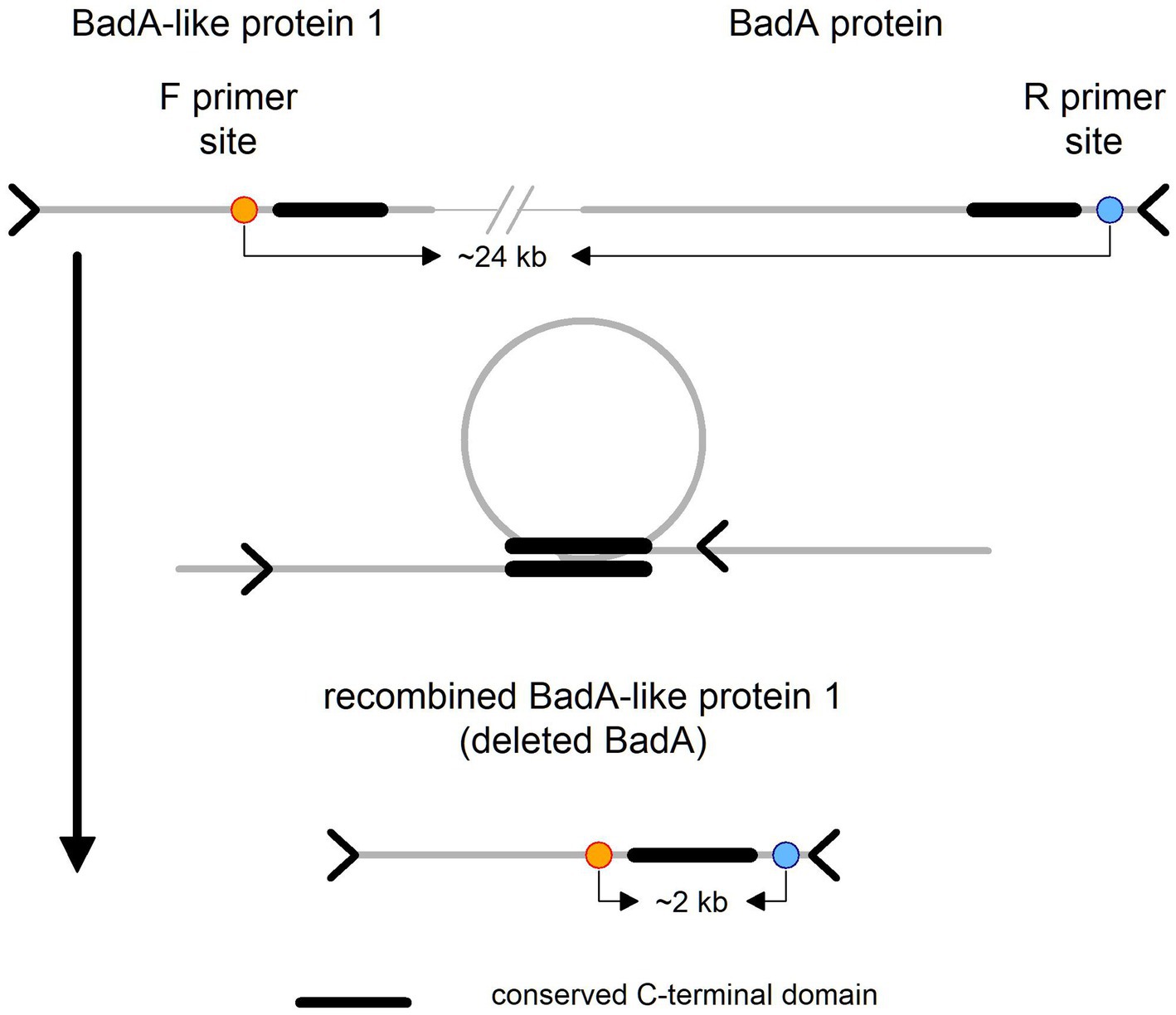
Figure 5. Schematic representation of BadA-like 1 and BadA proteins. On the top of the figure is illustrated the wild type locus variant with primer sites, forward on the left and reverse on the right side. In the middle is depicted the probable cause of the deletion event, i.e., recombination between the conserved C-terminal domains, which has resulted in the recombined (“chimeric”) variant of BadA-like protein 1 depicted at the bottom of the figure.
The architecture of the BadA locus itself is complicated and most importantly repetitive. This fact has complicated the development of a simple PCR test that could theoretically prove the presence/absence of the BadA deleted phenotype within the host. We designed primers that should yield a product of size ~2 kb in the deletion phenotype and no product in the wild type variant (primers are ~24 kb apart). Further, we developed B. grahamii subsp. shimonis subsp. nov. specific primers that should yield a product of similar size as a positive control. We optimized and confirmed the function of the primers using high-quality bacterial DNA (also utilized for sequencing). Surprisingly, we observed that even the presumably wild type strain of B. grahamii subsp. shimonis subsp. nov. (GG20g1) contains the deletion phenotype, however its frequency was several fold lower compared to the wild type variant. This observation is based on the fact that while the deletion phenotype was undetectable in the sequencing data, the PCR analysis weakly detected it (see Supplementary Figure 6).
Finally, we used such primers on the DNA extracted directly from the blood of two dormouse specimens from which the strains were originally isolated (and cultivated). The PCR reaction required different conditions (more cycles) compared to pure bacterial DNA (detailed information including primer sequences is provided in the Supplementary material). Nevertheless, in both cases we clearly detected the positive control PCR product, but we did not reliably detect the deletion phenotype marker (see Supplementary Figure 6). However, in one of the host samples, the results of this assay were inconclusive because we weakly detected the product at about the size of the expected marker. But there were also other products that were not detected in any other reaction.
Therefore, we sequenced the products in order to definitively prove or disprove the presence of the deletion marker in the original host blood samples (ONT GridION platform). While in the positive control we got mostly reads with length corresponding to the given marker (~1,970 bp), in the questionable sample we got a mixture of reads without obvious peak and also without any clear identification (estimated by BLASTn; data not shown). We can only speculate that these are probably repetitive sequences from the host genome, but we certainly did not detect the expected marker.
Using the Label Free Quantification (LFQ) proteomic approach, a total of 523 quantifiable proteins were detected. The observed quantities of the proteins are expressed as log2 transformation of LFQ intensities (Cox et al., 2014). Values ranging from 19 to 34 reflect the dynamic range of the mass spectrometry-based workflow. LFQ intensities below this value were considered as non-analyzable by the implemented qualitative test (Cox et al., 2011). We focused especially on analysis of Bartonella Adhesin A(-like) proteins. But since these proteins are similar to some extent, we considered only peptides that could be unambiguously mapped exclusively to a single gene. While the proteomic evidence for expression of the BadA protein is rather weak, i.e., based on a single peptide (“VEGDSLVKQDK”), the expression of BadA-like protein 1 was evident (see Supplementary Figure 7). We did not detect any peptide originating from BadA-like protein 0.
The cells are rod-shaped bacilli without any additional structure with a length of 0.8 to 1.4 μm and a width of 0.3 to 0.4 μm. Within 10 days on chocolate agar plates under optimal conditions (37°C with 5% CO2), the white-yellow colonies appear round and vary in size (0.6 to 3 mm). The type strain GG3s1 was negative for b-galactosidase (ONPG hydrolysis), decarboxylation of arginine, lysine, and ornithine, utilization of citrate, hydrogen sulfide production, urease, tryptophan deaminase, indole production from tryptophan, production of the enzyme gelatinase, Voges–Proskauer reactions, catalase activity, and fermentation of glucose, mannose, inositol, sorbitol, rhamnose, sucrose, melibiose, amygdalin, and arabinose. The type strain carried a single circular chromosome with a size of 2.26 Mb and GC content of 37.92%. The closest species with a published genome is B. grahamii (93.6% ANI). The type strain GG3s1, isolated from the blood of edible dormice (Glis glis) sampled in the Olomouc district of the Czech Republic, was deposited in the Department of Parasitology, Charles University, Prague, and in the Military Health Institute, Prague (sample ID #000522).
The main phenotypic characteristics are identical to those of the genus Bartonella. Within 10 days on chocolate agar plates under optimal conditions (37°C with 5% CO2), the white-yellow colonies appear round and vary in size (0.6 to 3 mm). The cells are rod-shaped bacilli without any additional structure with a length of 0.8 to 1.3 μm and a width of 0.3 to 0.5 μm. The type strain GG6g2 is positive for the enzyme gelatinase and negative for b-galactosidase (ONPG hydrolysis), decarboxylation of arginine, lysine, and ornithine, utilization of citrate, hydrogen sulfide production, urease, tryptophan deaminase, indole production from tryptophan, Voges–Proskauer reactions, catalase activity, and fermentation of glucose, mannose, inositol, sorbitol, rhamnose, sucrose, melibiose, amygdalin, and arabinose. The type strain carried a single circular chromosome a size of 1.95 Mb and GC content of 39.26%. The closest species with a published genome is B. washoensis (88.9% ANI). The type strain GG6g2, isolated from the blood of edible dormice (Glis glis) sampled in the Olomouc district of the Czech Republic, was deposited in the Department of Parasitology, Charles University, Prague, and in the Military Health Institute, Prague (sample ID #000521).
Species recognition is somewhat tricky in bacteria. In recent history, even the very existence of bacterial species was questioned, mainly due to the exaggerated importance of a horizontal gene transfer (HGT) that could melt the presumed species boundaries (Riley and Lizotte-Waniewski, 2009). Species concept and its definition in microbiology remains to be problematic till now (Bobay, 2020). Instead, so-called Core Genome Hypothesis (CGH) was proposed to solve the apparent paradox of HGT and rapid gene flow in general on one hand and the objective existence of distinct bacterial species on the other hand (Riley and Lizotte-Waniewski, 2009). Basically, the CGH defines a stable part of a genome (core), which is responsible for a maintaining of the species identity, whereas the rest of the genome allows for rapid adaptation to an ever-changing environment. Thus far, only CGH offers a reasonable perspective on the fact that analysis of over 2,000 genomes of E. coli resulted in a pan-genome size reaching 90,000 genes (Land et al., 2015; Iranzadeh and Mulder, 2019).
Nowadays, the standard for species delimitation is represented by a rather pragmatic threshold of average nucleotide identity (ANI) estimated from genomic sequences (Bobay, 2020). As a rule of thumb, if a strain shows ANI to any nearest described species lower than 95%, it might be considered a novel species (Chan et al., 2012; Bobay, 2020). So, the values estimated in the analyzed strains (88.9% and 93.7%), demonstrated that they represent well established and independent evolutionary entities. Nevertheless, even the threshold of 95% does not apply to current phylogenies categorically, which might be explained by historic or other reasons (Welch et al., 1999). Particularly, many species/taxa descriptions come from the times when genome sequencing was not as common as it is today and to obtain exact estimates of species divergence was complicated (e.g., Brenner et al., 1982; Richter and Rosselló-Móra, 2009) and/or the authors may have had other reasons for deciding not to adopt the species rank (e.g., Kordick et al., 1996; Welch et al., 1999). If we estimate the ANI values of B. vinsonii subsp. berkhoffii and B. vinsonii subsp. arupensis to B. vinsonii (and even between each other) today, they fall significantly below the 95% threshold, 94 and 93%, respectively. This observation was recently acknowledged by do Amaral et al. (2022), who indeed suggested taxonomic reclassification of the genus Bartonella. In summary, we also considered other criteria beyond the ANI estimates.
Specifically, we considered the results of the pangenome analysis, which suggested that while the core genome of Bartonella is rather small, the pangenome appears to be open. This characteristic does not seem to be of major importance considering the above-mentioned study of E. coli pan-genome (Land et al., 2015), however, not all bacteria share this feature (Hemsley et al., 2019). Both HGT as well as gene loss and duplication were described to play a role in Bartonella genome dynamics (Québatte and Dehio, 2019; Banerjee et al., 2020). The PCA results showed two important things. First, that Bartonella gliris sp. nov., seems to represent a unique genome composition that is not directly related to any other species. And second, that Bartonella grahamii subsp. shimonis subsp. nov. closely clustered together with B. grahamii. Moreover, even after inclusion of this new subspecies, the B. grahamii (sub)species would still form a compact and distinct cluster, especially when compared with the B. vinsonii (sub)species.
Furthermore, based on long-term monitoring of the prevalence of Bartonella infections in small mammals carried out in the Czech Republic (Obiegala et al., 2019; Majerová et al., 2021), we noticed an important biological difference between the described (sub)species. Bartonella grahamii subsp. shimonis subsp. nov. displays rather low host specificity and is be found in a broad range of mammalian hosts similarly to B. grahamii (Berglund et al., 2010). Within the study of Majerová et al. (2021) the newly described Bartonella grahamii subsp. shimonis subsp. nov. was already detected in hedgehogs (Erinaceus europaeus and E. roumanicus) and squirrels (Sciurus vulgaris), based upon PCR screening of the gltA and rpoB genes, and the detected genotype was provisionally designated as Bartonella sp. SCIER (see Supplementary Figure 3). On the other hand, Bartonella gliris sp. nov. has been detected solely in edible dormice to date; nevertheless, further investigations would be needed to confirm this observation. But if Bartonella gliris sp. nov. has truly adapted to a single host, then it would represent a potentially interesting example of specific host adaptation (Withenshaw et al., 2016; Québatte and Dehio, 2019).
In the past, the role of prophages and the effects of their residence within bacterial genomes were in the past considered simply as parasitism (Bondy-Denomy and Davidson, 2014). Nowadays, it is clear that this point of view was an oversimplification and in reality prophages might be beneficial for their bacterial host (Wang et al., 2010; Bobay et al., 2014). Despite the fact that we were not able to experimentally prove a direct causal progression of individual deletion/excision events (due to technical reasons/limitations), we think that the presented evidence strongly supports our interpretation, which furthermore represents the most parsimonious explanation of the observed phenomena.
Further, genome streamlining was recently described as a nearly universal response to naturally occurring environmental stress conditions (Simonsen, 2022). On the other hand, the study of Simonsen (2022) did not consider the role of phages/prophages, which seem to play a dominant role in our case. Actually, it has been shown that gene acquisition through prophage integration is a major evolutionary route for facultative bacterial pathogens (Busby et al., 2013). Greenrod et al. (2022) emphasized not only the role of prophages as carriers of auxiliary genes with a potential role in bacteria metabolism and gene regulation. But most importantly, they demonstrated that host evolution is virtually mirrored by phage evolution in certain cases, a phenomenon which could have resulted (only) from a shared coevolutionary history. In other words, phage possession might represent a typical feature of a certain bacterial clade, as we evidenced for the “Wolbachia endosymbiont wVitA of Nasonia vitripennis phage WOVitA1.” Therefore, we interpret its absence in one of the analyzed B. grahamii subsp. shimonis subsp. nov. strains as an excision event.
Similarly, the absence of Bartonella Adhesin A (BadA) locus in Bartonella grahamii subsp. shimonis subsp. nov. strain GG23s2 is considered a deletion event. BadA is known to represent a key virulence factor which mediates bacteria-host interactions and might even contribute to host immune evasion (Thibau et al., 2022). This adhesin was also identified as a key factor for adherence to host cells and biofilm formation (Müller et al., 2011; Okaro et al., 2019). Adhesins are generally a (hyper)variable class of proteins. Their variability stems from never-ending interactions between the pathogen and an adaptive host immune system (May and Brown, 2011). Their sequence as well as topological variability has already been documented in Bartonella henselae strains (Thibau et al., 2022). However, the presumable deletion of the ~22 kb long fragment is far beyond the scope of what Thibau et al. (2022) describe as variation and adaptation. Such an irreversible loss of the genetic repertoire can hardly be considered adaptive. Instead, it is in line with previous studies which have reported that “extensive passaging” of Bartonella henselae strains led to deletion of ~10 kb of the BadA locus (Riess et al., 2004). Long-term survival and proliferation of the bacteria within a host without the BadA machinery is improbable and therefore, its irreversible loss provides the strongest evidence for cultivation-induced genomic changes. This interpretation is also clearly supported by the results of PCR analysis of host blood, where we did not detect the deletion phenotype, conversely to both agar plates cultivated Bartonella grahamii subsp. shimonis subsp. nov. strains. This result on the other hand cannot exclude the possibility of natural occurrence of the deletion phenotype, but it proves that the wild type variant is dominant within a host.
The observed phenomenon could be to some extent compared to the cultivation induced genomic changes of Coxiella burnetii Nine Mile strain, i.e., another facultative intracellular pathogenic bacteria. For which it has been repeatedly demonstrated that cultivation ultimately leads to loss (deletion) of a defined section of the genome (Hoover et al., 2002). Moreover, this systematic deletion of several genes significantly negatively affects the pathogenicity of concerned strains compared to the wild-type (Kersh et al., 2011).
Using the proteomic approach, we demonstrated that BadA-like protein 1 was expressed in both B. grahamii subsp. shimonis sp. nov. strains and therefore cannot be considered as a pseudogene as suggested by Thibau et al. (2022). Instead, it is possible that under natural conditions some BadA variants might be preferred under specific conditions connected with the life cycle of the bacteria, i.e., vector-host dynamics.
Due to the frequent close contact of dormice with humans, the newly detected and described Bartonella (sub)species may pose a risk of zoonotic infection. Therefore, more attention should be paid to these species and their vectors.
The presumed deletion events, especially the deletion of Bartonella Adhesin A (BadA) locus, described in this study suggest that even relatively short-term laboratory cultivation with a low number of passages seems to represent sufficient selective force. In other words, it seems to induce enough stress to promote substantial changes at the level of the genome. Although we broadly focused on the BadA deletion within this study, it is worth to note that the importance of (presumed) prophage excision events should not be overlooked. However, in contrast to well-described genes, the significance and implications of these phenomena are very difficult to interpret.
We emphasize that a novel model that would allow us to study Bartonella spp. in their natural form and conditions needs to be established to confirm the findings presented in this study. Also, the role of individual Bartonella Adhesin A-like genes should be reviewed as we have clearly demonstrated that they are expressed during cultivation. Nevertheless, further research would be needed to reveal their possible role and function under natural conditions.
The raw sequencing data generated for this study can be found in the NCBI SRA archives. Similarly, the genome sequence data as well as the annotations may be accessed in the NCBI GenBank archives. All the data may be found under the NCBI BioProject PRJNA909980. The proteomic datasets generated and analyzed during the current study have been deposited in the ProteomeXchange Consortium via the PRIDE (Perez-Riverol et al., 2019) partner repository with the dataset identifier PXD046060 (https://www.ebi.ac.uk/pride/archive/projects/PXD046060).
The animal study was approved by Ethical Committees of Palacky University and the Ministry of Education. The study was conducted in accordance with the local legislation and institutional requirements.
OB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Supervision, Visualization, Writing – original draft, Writing – review & editing. BK: Investigation, Methodology, Writing – original draft. KV: Investigation, Methodology, Visualization, Writing – review & editing, Writing – original draft. MC: Data curation, Investigation, Methodology, Writing – original draft. JD: Funding acquisition, Investigation, Methodology, Writing – original draft. PP: Funding acquisition, Investigation, Writing – original draft. HK: Investigation, Methodology, Writing – original draft. PA: Investigation, Resources, Writing – original draft. DM: Investigation, Resources, Writing – original draft. AF: Investigation, Writing – original draft. JV: Conceptualization, Formal analysis, Investigation, Supervision, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by funding from projects of the Ministry of Interior, Czech Republic (VJ01010107: Extending and characterizing the collection of high-risk and risk biological causative agents of dangerous infectious diseases and implementing new procedures for working with them).
The authors would like to express their great appreciation to Pavel Dvouletý from the Military Health Institute in Prague for his long-term management and support of our team. The authors would also like to acknowledge Kateřina Kybicová (The National Institute of Public Health, Czech Republic) for her help with biochemical characterization of Bartonella isolates.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1289671/full#supplementary-material
Amori, G., Hutterer, R., Kryštufek, B., Yigit, N., Mitsainas, G., Muñoz, L., et al. (2021). Glis glis (amended version of 2016 assessment). The IUCN red list of threatened species 2021: e.T39316A197292692. Available at: https://dx.doi.org/10.2305/IUCN.UK.2021-1.RLTS.T39316A197292692.en. (Accessed 06 December 2022).
Andrews, S. (2010). FastQC: A quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/
Aucouturier, A., Chain, F., Langella, P., and Bidnenko, E. (2018). Characterization of a prophage-free derivative strain of Lactococcus lactis ssp. Lactis IL1403 reveals the importance of prophages for phenotypic plasticity of the host. Frontiers in. Microbiology 9:2032. doi: 10.3389/fmicb.2018.02032
Banerjee, R., Shine, O., Rajachandran, V., Krishnadas, G., Minnick, M. F., Paul, S., et al. (2020). Gene duplication and deletion, not horizontal transfer, drove intra-species mosaicism of Bartonella henselae. Genomics 112, 467–471. doi: 10.1016/j.ygeno.2019.03.009
Berglund, E. C., Ellegaard, K., Granberg, F., Xie, Z., Maruyama, S., Kosoy, M. Y., et al. (2010). Rapid diversification by recombination in Bartonella grahamii from wild rodents in Asia contrasts with low levels of genomic divergence in northern Europe and America. Mol. Ecol. 19, 2241–2255. doi: 10.1111/j.1365-294X.2010.04646.x
Birtles, R. J., Harrison, T. G., Saunders, N. A., and Molyneux, D. H. (1995). Proposals to unify the genera Grahamella and Bartonella, with descriptions of Bartonella talpae comb, nov., Bartonella peromysci comb. nov., and three new species, Bartonella grahamii sp. nov., Bartonella taylorii sp. nov., and Bartonella doshiae sp. nov. Int. J. Syst. Evol. Microbiol. 45, 1–8. doi: 10.1099/00207713-45-1-1
Bobay, L.-M. (2020). “The prokaryotic species concept and challenges” in The Pangenome. eds. H. Tettelin and D. Medini (Cham, Switzerland: Springer International Publishing), 21–49.
Bobay, L.-M., Touchon, M., and Rocha, E. P. C. (2014). Pervasive domestication of defective prophages by bacteria. Proc. Natl. Acad. Sci. 111, 12127–12132. doi: 10.1073/pnas.1405336111
Bondy-Denomy, J., and Davidson, A. R. (2014). When a virus is not a parasite: the beneficial effects of prophages on bacterial fitness. J. Microbiol. 52, 235–242. doi: 10.1007/s12275-014-4083-3
Borowiec, M. L. (2016). AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4:e1660. doi: 10.7717/peerj.1660
Breitschwerdt, E. B. (2017). “Bartonellosis, one health and all creatures great and small” in Advances in veterinary dermatology. eds. S. M. F. Torres and P. Roudebush (Hoboken, New Jersey, U.S.: John Wiley & Sons, Ltd), 111–121.
Breitschwerdt, E. B., and Kordick, D. L. (2000). Bartonella infection in animals: Carriership, reservoir potential, pathogenicity, and zoonotic potential for human infection. Clin. Microbiol. Rev. 13, 428–438. doi: 10.1128/CMR.13.3.428
Breitschwerdt, E. B., Maggi, R. G., Chomel, B. B., and Lappin, M. R. (2010). Bartonellosis: an emerging infectious disease of zoonotic importance to animals and human beings. J. Vet. Emerg. Crit. Care 20, 8–30. doi: 10.1111/j.1476-4431.2009.00496.x
Brenner, D. J., McWhorter, A. C., Knutson, J. K. L., and Steigerwalt, A. G. (1982). Escherichia vulneris: a new species of Enterobacteriaceae associated with human wounds. J. Clin. Microbiol. 15, 1133–1140. doi: 10.1128/jcm.15.6.1133-1140.1982
Brenner, D. J., O’Connor, S. P., Winkler, H. H., and Steigerwalt, A. G. (1993). Proposals to unify the genera Bartonella and Rochalimaea, with descriptions of Bartonella quintana comb. Nov., Bartonella vinsonii comb. Nov., Bartonella henselae comb. Nov., and Bartonella elizabethae comb. Nov., and to remove the family Bartonellaceae from the order Rickettsiales. Int. J. Syst. Bacteriol. 43, 777–786. doi: 10.1099/00207713-43-4-777
Büchner, S., Trout, R., and Adamík, P. (2018). Conflicts with Glis glis and Eliomys quercinus in households: a practical guideline for sufferers (Rodentia: Gliridae). Lynx 49, 19–26. doi: 10.2478/lynx-2018-0003
Busby, B., Kristensen, D. M., and Koonin, E. V. (2013). Contribution of phage-derived genomic islands to the virulence of facultative bacterial pathogens: genomics update. Environ. Microbiol. 15, 307–312. doi: 10.1111/j.1462-2920.2012.02886.x
Celebi, B., Anani, H., Zgheib, R., Carhan, A., Raoult, D., and Fournier, P.-E. (2021). Genomic characterization of the novel Bartonella refiksaydamii sp. isolated from the blood of a Crocidura suaveolens (Pallas, 1811). Vector Borne Zoonotic Dis. 21, 432–440. doi: 10.1089/vbz.2020.2626
Chan, J. Z.-M., Halachev, M. R., Loman, N. J., Constantinidou, C., and Pallen, M. J. (2012). Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol. 12:302. doi: 10.1186/1471-2180-12-302
Cheslock, M. A., and Embers, M. E. (2019). Human bartonellosis: an underappreciated public health problem?. Trop. Med. Infect. Dis. 4, 69. doi: 10.3390/tropicalmed4020069
Cox, J., Hein, M. Y., Luber, C. A., Paron, I., Nagaraj, N., and Mann, M. (2014). Accurate proteome-wide label-free quantification by delayed normalization and maximal peptide ratio extraction, Termed MaxLFQ. Mol. Cell. Proteomics 13, 2513–2526. doi: 10.1074/mcp.M113.031591
Cox, J., and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. doi: 10.1038/nbt.1511
Cox, J., Neuhauser, N., Michalski, A., Scheltema, R. A., Olsen, J. V., and Mann, M. (2011). Andromeda: a peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 10, 1794–1805. doi: 10.1021/pr101065j
Crossley, B. M., Bai, J., Glaser, A., Maes, R., Porter, E., Killian, M. L., et al. (2020). Guidelines for sanger sequencing and molecular assay monitoring. J. Vet. Diagn. Invest. 32, 767–775. doi: 10.1177/1040638720905833
Delorenzi, M., and Speed, T. (2002). An HMM model for coiled-coil domains and a comparison with PSSM-based predictions. Bioinformatics 18, 617–625. doi: 10.1093/bioinformatics/18.4.617
Diddi, K., Chaudhry, R., Sharma, N., and Dhawan, B. (2013). Strategy for identification & characterization of Bartonella henselae with conventional & molecular methods. Indian J. Med. Res. 137, 380–387. Available at: https://journals.lww.com/ijmr/Fulltext/2013/37020/Strategy_for_identification___characterization_of.18.aspx.
do Amaral, R. B., Cardozo, M. V., Varani, A. d. M., Furquim, M. E. C., Dias, C. M., Assis, W. O., et al. (2022). First report of Bartonella spp. in marsupials from Brazil, with a description of Bartonella harrusi sp. Nov. and a new proposal for the taxonomic reclassification of species of the genus Bartonella. Microorganisms 10:1609. doi: 10.3390/microorganisms10081609
Emms, D. M., and Kelly, S. (2019). OrthoFinder: phylogenetic orthology inference for comparative genomics. Genome Biol. 20, 238–214. doi: 10.1186/s13059-019-1832-y
Engel, P., Salzburger, W., Liesch, M., Chang, C.-C., Maruyama, S., Lanz, C., et al. (2011). Parallel evolution of a type IV secretion system in radiating lineages of the host-restricted bacterial pathogen Bartonella. PLoS Genet. 7:e1001296. doi: 10.1371/journal.pgen.1001296
Fischetti, V. A. (2008). Bacteriophage lysins as effective antibacterials. Curr. Opin. Microbiol. 11, 393–400. doi: 10.1016/j.mib.2008.09.012
Gabler, F., Nam, S., Till, S., Mirdita, M., Steinegger, M., Söding, J., et al. (2020). Protein sequence analysis using the MPI bioinformatics toolkit. Curr. Protoc. Bioinformatics 72:108. doi: 10.1002/cpbi.108
Gazárková, A. H., and Adamík, P. (2016). Timing of breeding and second litters in edible dormouse (Glis glis). Folia Zool. 65, 165–168. doi: 10.25225/fozo.v65.i2.a12.2016
Greenrod, S. T. E., Stoycheva, M., Elphinstone, J., and Friman, V.-P. (2022). Global diversity and distribution of prophages are lineage-specific within the Ralstonia solanacearum species complex. BMC Genomics 23:689. doi: 10.1186/s12864-022-08909-7
Gremme, G., Steinbiss, S., and Kurtz, S. (2013). GenomeTools: a comprehensive software library for efficient processing of structured genome annotations. IEEE/ACM Trans. Comput. Biol. Bioinform. 10, 645–656. doi: 10.1109/TCBB.2013.68
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Gutiérrez, R., Krasnov, B., Morick, D., Gottlieb, Y., Khokhlova, I. S., and Harrus, S. (2015). Bartonella infection in rodents and their flea Ectoparasites: an overview. Vector Borne Zoonotic Dis. 15, 27–39. doi: 10.1089/vbz.2014.1606
Gutiérrez, R., Shalit, T., Markus, B., Yuan, C., Nachum-Biala, Y., Elad, D., et al. (2020). Bartonella kosoyi sp. Nov. and Bartonella krasnovii sp. Nov., two novel species closely related to the zoonotic Bartonella elizabethae, isolated from black rats and wild desert rodent-fleas. Int. J. Syst. Evol. Microbiol. 70, 1656–1665. doi: 10.1099/ijsem.0.003952
Gutiérrez, R., Vayssier-Taussat, M., Buffet, J.-P., and Harrus, S. (2017). Guidelines for the isolation, molecular detection, and characterization of Bartonella species. Vector Borne Zoonotic Dis. 17, 42–50. doi: 10.1089/vbz.2016.1956
Han, H. J., Li, Z. M., Li, X., Liu, J. X., Peng, Q. M., Wang, R., et al. (2022). Bats and their ectoparasites (Nycteribiidae and Spinturnicidae) carry diverse novel Bartonella genotypes, China. Transbound. Emerg. Dis. 69, e845–e858. doi: 10.1111/tbed.14357
Harms, A., and Dehio, C. (2012). Intruders below the radar: molecular pathogenesis of Bartonella spp. Clin. Microbiol. Rev. 25, 42–78. doi: 10.1128/CMR.05009-11
Hemsley, C. M., O’Neill, P. A., Essex-Lopresti, A., Norville, I. H., Atkins, T. P., and Titball, R. W. (2019). Extensive genome analysis of Coxiella burnetii reveals limited evolution within genomic groups. BMC Genomics 20:441. doi: 10.1186/s12864-019-5833-8
Hoang, D. T., Chernomor, O., Von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2018). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hoover, T. A., Culp, D. W., Vodkin, M. H., Williams, J. C., and Thompson, H. A. (2002). Chromosomal DNA deletions explain phenotypic characteristics of two antigenic variants, phase II and RSA 514 (crazy), of the Coxiella burnetii nine mile strain. Infect. Immun. 70, 6726–6733. doi: 10.1128/IAI.70.12.6726-2733.2002
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Iannino, F., Salucci, S., Di Provvido, A., Paolini, A., and Ruggieri, E. (2018). Bartonella infections in humans dogs and cats. Vet. Ital. 54, 63–72. doi: 10.12834/VetIt.398.1883.2
Iranzadeh, A., and Mulder, N. J. (2019). “Bacterial Pan-genomics” in Microbial genomics in sustainable agroecosystems. eds. V. Tripathi, P. Kumar, P. Tripathi, and A. Kishore (Singapore: Springer Singapore), 21–38.
Jacomo, V., Kelly, P. J., and Raoult, D. (2002). Natural history of Bartonella infections (an exception to Koch’s postulate). Clin. Vaccine Immunol. 9, 8–18. doi: 10.1128/CDLI.9.1.8-18.2002
Jain, C., Rhie, A., Hansen, N. F., Koren, S., and Phillippy, A. M. (2022). Long-read mapping to repetitive reference sequences using Winnowmap2. Nat. Methods 19, 705–710. doi: 10.1038/s41592-022-01457-8
Kalyaanamoorthy, S., Minh, B. Q., Wong, T. K., Von Haeseler, A., and Jermiin, L. S. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kersh, G. J., Oliver, L. D., Self, J. S., Fitzpatrick, K. A., and Massung, R. F. (2011). Virulence of pathogenic Coxiella burnetii strains after growth in the absence of host cells. Vector Borne Zoonotic Dis. 11, 1433–1438. doi: 10.1089/vbz.2011.0670
Knap, N., Duh, D., Birtles, R., Trilar, T., Petrovec, M., and Avšic-Županc, T. (2007). Molecular detection of Bartonella species infecting rodents in Slovenia. FEMS Immunol. Med. Microbiol. 50, 45–50. doi: 10.1111/j.1574-695X.2007.00226.x
Kordick, D. L., Swaminathan, B., Greene, C. E., Wilson, K. H., Whitney, A. M., O’Connor, S., et al. (1996). Bartonella vinsonii subsp. Berkhoffii subsp. Nov., isolated from dogs; Bartonella vinsonii subsp. Vinsonii; and emended description of Bartonella vinsonii. Int. J. Syst. Bacteriol. 46, 704–709. doi: 10.1099/00207713-46-3-704
Krügel, M., Król, N., Kempf, V. A., Pfeffer, M., and Obiegala, A. (2022). Emerging rodent-associated Bartonella: a threat for human health? Parasit. Vectors 15:113. doi: 10.1186/s13071-022-05162-5
Krumsiek, J., Arnold, R., and Rattei, T. (2007). Gepard: a rapid and sensitive tool for creating dotplots on genome scale. Bioinformatics 23, 1026–1028. doi: 10.1093/bioinformatics/btm039
Land, M., Hauser, L., Jun, S.-R., Nookaew, I., Leuze, M. R., Ahn, T.-H., et al. (2015). Insights from 20 years of bacterial genome sequencing. Funct. Integr. Genomics 15, 141–161. doi: 10.1007/s10142-015-0433-4
Lee, I., Ouk Kim, Y., Park, S.-C., and Chun, J. (2016). OrthoANI: an improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 66, 1100–1103. doi: 10.1099/ijsem.0.000760
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Li, H. (2013). Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv [Preprint].
Low, L. Y., Yang, C., Perego, M., Osterman, A., and Liddington, R. (2011). Role of net charge on catalytic domain and influence of Cell Wall binding domain on bactericidal activity, specificity, and host range of phage Lysins. J. Biol. Chem. 286, 34391–34403. doi: 10.1074/jbc.M111.244160
Majerová, K., Gutiérrez, R., Fonville, M., Hönig, V., Papežík, P., Hofmannová, L., et al. (2021). Hedgehogs and squirrels as hosts of zoonotic Bartonella species. Pathogens 10:686. doi: 10.3390/pathogens10060686
Mardosaitė-Busaitienė, D., Radzijevskaja, J., Balčiauskas, L., Bratchikov, M., Jurgelevičius, V., and Paulauskas, A. (2019). Prevalence and diversity of Bartonella species in small rodents from coastal and continental areas. Sci. Rep. 9:12349. doi: 10.1038/s41598-019-48715-y
May, M., and Brown, D. R. (2011). Diversity of expressed vlhA Adhesin sequences and intermediate Hemagglutination phenotypes in Mycoplasma synoviae. J. Bacteriol. 193, 2116–2121. doi: 10.1128/JB.00022-11
Minh, B. Q., Hahn, M. W., and Lanfear, R. (2020). New methods to calculate concordance factors for Phylogenomic datasets. Mol. Biol. Evol. 37, 2727–2733. doi: 10.1093/molbev/msaa106
Minnick, M. F., and Anderson, B. E. (2015). “Bartonella” in Molecular Medical Microbiology Eds. Y-W. Tang, M. Sussman, D. Liu, I. Poxton, and J. Schwartzman (Academic Press resides in Cambridge, Massachusetts: Elsevier), 1911–1939.
Molia, S., Kasten, R. W., Stuckey, M. J., Boulouis, H. J., Allen, J., Borgo, G. M., et al. (2016). Isolation of Bartonella henselae, Bartonella koehlerae subsp. Koehlerae, Bartonella koehlerae subsp. Bothieri and a new subspecies of B. koehlerae from free-ranging lions (Panthera leo) from South Africa, cheetahs (Acinonyx jubatus) from Namibia and captive cheetahs from California. Epidemiol. Infect. 144, 3237–3243. doi: 10.1017/S0950268816001394
Müller, N. F., Kaiser, P. O., Linke, D., Schwarz, H., Riess, T., Schäfer, A., et al. (2011). Trimeric autotransporter adhesin-dependent adherence of Bartonella henselae, Bartonella quintana, and Yersinia enterocolitica to matrix components and endothelial cells under static and dynamic flow conditions. Infect. Immun. 79, 2544–2553. doi: 10.1128/IAI.01309
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Obiegala, A., Jeske, K., Augustin, M., Król, N., Fischer, S., Mertens-Scholz, K., et al. (2019). Highly prevalent bartonellae and other vector-borne pathogens in small mammal species from the Czech Republic and Germany. Parasit. Vectors 12:332. doi: 10.1186/s13071-019-3576-7
Okaro, U., Addisu, A., Casanas, B., and Anderson, B. (2017). Bartonella species, an emerging cause of blood-culture-negative endocarditis. Clin. Microbiol. Rev. 30, 709–746. doi: 10.1128/CMR.00013-17
Okaro, U., Green, R., Mohapatra, S., and Anderson, B. (2019). The trimeric autotransporter adhesin BadA is required for in vitro biofilm formation by Bartonella henselae. NPJ Biofilms Microbiomes 5:10. doi: 10.1038/s41522-019-0083-8
Parte, A. C., Sardà Carbasse, J., Meier-Kolthoff, J. P., Reimer, L. C., and Göker, M. (2020). List of Prokaryotic names with Standing in Nomenclature (LPSN) moves to the DSMZ. Int. J. Syst. Evol. Microbiol. 70, 5607–5612. doi: 10.1099/ijsem.0.004332
Perez-Riverol, Y., Csordas, A., Bai, J., Bernal-Llinares, M., Hewapathirana, S., Kundu, D. J., et al. (2019). The PRIDE database and related tools and resources in 2019: improving support for quantification data. Nucleic Acids Res. 47, D442–D450. doi: 10.1093/nar/gky1106
Pilastro, A., Tavecchia, G., and Marin, G. (2003). Long living and reproduction skipping in the fat dormouse. Ecology 84, 1784–1792. doi: 10.1890/0012-9658(2003)084[1784:LLARSI]2.0.CO;2
Pitassi, L. H. U., de Paiva Diniz, P. P. V., Scorpio, D. G., Drummond, M. R., Lania, B. G., Barjas-Castro, M. L., et al. (2015). Bartonella spp. bacteremia in blood donors from Campinas, Brazil. PLoS Neglected Trop Dis 9:e0003467. doi: 10.1371/journal.pntd.0003467
Posada, D. (2008). jModelTest: phylogenetic model averaging. Mol. Biol. Evol 25, 1253–1256. doi: 10.1093/molbev/msn083
Portillo, A., Maggi, R., Oteo, J. A., Bradley, J., García-Álvarez, L., San-Martín, M., et al. (2020). Bartonella spp. prevalence (serology, culture, and PCR) in sanitary workers in La Rioja Spain. Pathogens 9:189. doi: 10.3390/pathogens9030189
Québatte, M., and Dehio, C. (2019). Bartonella gene transfer agent: evolution, function, and proposed role in host adaptation. Cell. Microbiol. 21:e13068. doi: 10.1111/cmi.13068
R Core Team (2021). R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria.
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Riess, T., Andersson, S. G. E., Lupas, A., Schaller, M., Schäfer, A., Kyme, P., et al. (2004). Bartonella Adhesin a mediates a proangiogenic host cell response. J. Exp. Med. 200, 1267–1278. doi: 10.1084/jem.20040500
Riess, T., Dietrich, F., Schmidt, K. V., Kaiser, P. O., Schwarz, H., Schäfer, A., et al. (2008). Analysis of a novel insect cell culture medium-based growth medium for Bartonella species. Appl. Environ. Microbiol. 74, 5224–5227. doi: 10.1128/AEM.00621-08
Riley, M. A., and Lizotte-Waniewski, M. (2009). “Population genomics and the bacterial species concept” in Horizontal Gene Transfer. eds. M. B. Gogarten, J. P. Gogarten, and L. C. Olendzenski, vol. 532 (Totowa, New Jersey, U.S.: Humana Press), 367–377.
Robinson, J. T., Thorvaldsdóttir, H., Winckler, W., Guttman, M., Lander, E. S., Getz, G., et al. (2011). Integrative genomics viewer. Nat. Biotechnol. 29, 24–26. doi: 10.1038/nbt.1754
Sedlazeck, F. J., Rescheneder, P., Smolka, M., Fang, H., Nattestad, M., von Haeseler, A., et al. (2018). Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468. doi: 10.1038/s41592-018-0001-7
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Simonsen, A. K. (2022). Environmental stress leads to genome streamlining in a widely distributed species of soil bacteria. ISME J. 16, 423–434. doi: 10.1038/s41396-021-01082-x
Snipen, L., and Liland, K. H. (2015). Micropan: an R-package for microbial pan-genomics. BMC Bioinformatics 16:79. doi: 10.1186/s12859-015-0517-0
Song, W., Sun, H.-X., Zhang, C., Cheng, L., Peng, Y., Deng, Z., et al. (2019). Prophage hunter: an integrative hunting tool for active prophages. Nucleic Acids Res. 47, W74–W80. doi: 10.1093/nar/gkz380
Sperlea, T., Muth, L., Martin, R., Weigel, C., Waldminghaus, T., and Heider, D. (2020). gammaBOriS: identification and taxonomic classification of origins of replication in Gammaproteobacteria using motif-based machine learning. Sci. Rep. 10:6727. doi: 10.1038/s41598-020-63424-7
Tatusova, T., DiCuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, E. P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Tettelin, H., Riley, D., Cattuto, C., and Medini, D. (2008). Comparative genomics: the bacterial pan-genome. Curr. Opin. Microbiol. 11, 472–477. doi: 10.1016/j.mib.2008.09.006
Thibau, A., Hipp, K., Vaca, D. J., Chowdhury, S., Malmström, J., Saragliadis, A., et al. (2022). Long-read sequencing reveals genetic adaptation of Bartonella Adhesin a among different Bartonella henselae isolates. Front. Microbiol. 13:838267. doi: 10.3389/fmicb.2022.838267
Tołkacz, K., Alsarraf, M., Kowalec, M., Dwużnik, D., Grzybek, M., Behnke, J. M., et al. (2018). Bartonella infections in three species of Microtus: prevalence and genetic diversity, vertical transmission and the effect of concurrent Babesia microti infection on its success. Parasit. Vectors 11:491. doi: 10.1186/s13071-018-3047-6
Van Houdt, R., Leplae, R., Lima-Mendez, G., Mergeay, M., and Toussaint, A. (2012). Towards a more accurate annotation of tyrosine-based site-specific recombinases in bacterial genomes. Mob. DNA 3:6. doi: 10.1186/1759-8753-3-6
Wang, X., Kim, Y., Ma, Q., Hong, S. H., Pokusaeva, K., Sturino, J. M., et al. (2010). Cryptic prophages help bacteria cope with adverse environments. Nat. Commun. 1:147. doi: 10.1038/ncomms1146
Welch, D. F., Carroll, K. C., Hofmeister, E. K., Persing, D. H., Robison, D. A., Steigerwalt, A. G., et al. (1999). Isolation of a new subspecies, Bartonella vinsonii subsp. arupensis, from a cattle rancher: identity with isolates found in conjunction with Borrelia burgdorferi and Babesia microti among naturally infected mice. J. Clin. Microbiol. 37, 2598–2601. doi: 10.1128/JCM.37.8.2598-2601.1999
Wick, R. R., Judd, L. M., and Holt, K. E. (2019). Performance of neural network basecalling tools for Oxford Nanopore sequencing. Genome Biol. 20:129. doi: 10.1186/s13059-019-1727-y
Withenshaw, S. M., Devevey, G., Pedersen, A. B., and Fenton, A. (2016). Multihost Bartonella parasites display covert host specificity even when transmitted by generalist vectors. J. Anim. Ecol. 85, 1442–1452. doi: 10.1111/1365-2656.12568
Wu, L., Wang, H., Xia, Y., and Xi, R. (2020). CNV-BAC: copy number variation detection in bacterial circular genome. Bioinformatics 36, 3890–3891. doi: 10.1093/bioinformatics/btaa208
Keywords: Bartonella gliris, Bartonella grahamii subsp. shimonis, Bartonella Adhesin A, cultivation-related genomic changes, gene deletion
Citation: Bartoš O, Klimešová B, Volfová K, Chmel M, Dresler J, Pajer P, Kabíčková H, Adamík P, Modrý D, Fučíková AM and Votýpka J (2023) Two novel Bartonella (sub)species isolated from edible dormice (Glis glis): hints of cultivation stress-induced genomic changes. Front. Microbiol. 14:1289671. doi: 10.3389/fmicb.2023.1289671
Edited by:
Richard Allen White III, University of North Carolina at Charlotte, United StatesReviewed by:
Leonardo Gabriel Panunzi, CEA Saclay, FranceCopyright © 2023 Bartoš, Klimešová, Volfová, Chmel, Dresler, Pajer, Kabíčková, Adamík, Modrý, Fučíková and Votýpka. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oldřich Bartoš, MTI0NjAwQHNlem5hbS5jeg==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.