 Peng Wu1,2,3†
Peng Wu1,2,3† Qianqian Yuan3,4†Tingting Cheng1,2,3†
Qianqian Yuan3,4†Tingting Cheng1,2,3† Yifan Han2,3†
Yifan Han2,3† Wei Zhao2,3†
Wei Zhao2,3† Xiaoping Liao3,4Lu Wang1,2,3
Xiaoping Liao3,4Lu Wang1,2,3 Jingyi Cai3,4Qianqian He2,3
Jingyi Cai3,4Qianqian He2,3 Ying Guo2,3
Ying Guo2,3 Xiaoxia Zhang2,3Fuping Lu1
Xiaoxia Zhang2,3Fuping Lu1 Jingjing Wang2,3*
Jingjing Wang2,3* Hongwu Ma3,4*
Hongwu Ma3,4* Zhiyong Huang2,3*
Zhiyong Huang2,3*- 1College of Bioengineering, Tianjin University of Science and Technology, Tianjin, China
- 2Tianjin Key Laboratory for Industrial Biological Systems and Bioprocessing Engineering, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin, China
- 3National Technology Innovation Center of Synthetic Biology, Tianjin, China
- 4Biodesign Center, Key Laboratory of Systems Microbial Biotechnology, Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences, Tianjin, China
Sulfur-oxidizing bacteria play a crucial role in various processes, including mine bioleaching, biodesulfurization, and treatment of sulfur-containing wastewater. Nevertheless, the pathway involved in sulfur oxidation is highly intricate, making it complete comprehension a formidable and protracted undertaking. The mechanisms of sulfur oxidation within the Acidithiobacillus genus, along with the process of energy production, remain areas that necessitate further research and elucidation. In this study, a novel strain of sulfur-oxidizing bacterium, Acidithiobacillus Ameehan, was isolated. Several physiological characteristics of the strain Ameehan were verified and its complete genome sequence was presented in the study. Besides, the first genome-scale metabolic network model (AMEE_WP1377) was reconstructed for Acidithiobacillus Ameehan to gain a comprehensive understanding of the metabolic capacity of the strain.The characteristics of Acidithiobacillus Ameehan included morphological size and an optimal growth temperature range of 37-45°C, as well as an optimal growth pH range of pH 2.0-8.0. The microbe was found to be capable of growth when sulfur and K2O6S4 were supplied as the energy source and electron donor for CO2 fixation. Conversely, it could not utilize Na2S2O3, FeS2, and FeSO4·7H2O as the energy source or electron donor for CO2 fixation, nor could it grow using glucose or yeast extract as a carbon source. Genome annotation revealed that the strain Ameehan possessed a series of sulfur oxidizing genes that enabled it to oxidize elemental sulfur or various reduced inorganic sulfur compounds (RISCs). In addition, the bacterium also possessed carbon fixing genes involved in the incomplete Calvin-Benson-Bassham (CBB) cycle. However, the bacterium lacked the ability to oxidize iron and fix nitrogen. By implementing a constraint-based flux analysis to predict cellular growth in the presence of 71 carbon sources, 88.7% agreement with experimental Biolog data was observed. Five sulfur oxidation pathways were discovered through model simulations. The optimal sulfur oxidation pathway had the highest ATP production rate of 14.81 mmol/gDW/h, NADH/NADPH production rate of 5.76 mmol/gDW/h, consumed 1.575 mmol/gDW/h of CO2, and 1.5 mmol/gDW/h of sulfur. Our findings provide a comprehensive outlook on the most effective cellular metabolic pathways implicated in sulfur oxidation within Acidithiobacillus Ameehan. It suggests that the OMP (outer-membrane proteins) and SQR enzymes (sulfide: quinone oxidoreductase) have a significant impact on the energy production efficiency of sulfur oxidation, which could have potential biotechnological applications.
Highlights
– Discovered a new sulfur-oxidizing bacterium Acidithiobacillus Ameehan.
–First experimentally verified genome-scale reconstruction metabolic network of Acidithiobacillus Ameehan.
– Clarify the metabolic mechanism of sulfur oxidation pathway coupled with carbon metabolism.
1. Introduction
Sulfur-oxidizing microorganisms (SOM) can oxidize elemental sulfur or various reduced inorganic sulfur compounds (RISCs) to sulfuric acid or higher valence sulfides. SOM not only play a vital role in the sulfur biogeochemical cycle, but are also widely used in the metallurgical industry, environmental engineering and agriculture (Chen et al., 2022). Several taxa of sulfur-oxidizing bacteria have been isolated and identified, including Green sulfur bacteria (GSB), Purple sulfur bacteria (PSB), Purple non-sulfur bacteria (PNSB), and Colorless sulfur bacteria (CSB; Yang and Shao, 2018). It is worth noting that Acidithiobacillus, one of the CSB, play a major role in bioleaching, biodesulfurization, sulfur-containing wastewater treatment, and so on (Wen et al., 2016; Singh et al., 2019). The genus Acidithiobacillus comprises eight recognized species, including Acidithiobacillus ferrooxidans, Acidithiobacillus ferrivorans, Acidithiobacillus ferridurans, Acidithiobacillus ferriphilus, Acidithiobacillus ferrianus, Acidithiobacillus sulfuriphilus, Acidithiobacillus thiooxidans (including Acidithiobacillus albertensis), and Acidithiobacillus caldus (also called Fervidacidithiobacillus caldus; Moya-Beltran et al., 2021). However, there are many species yet to be discovered (Poole, 2012; Moya-Beltran et al., 2021). All these Acidithiobacillus are aerobic, chemolithoautotrophic, gram-negative, non-sporulating, rod-shaped microorganisms (Kumar P. et al., 2019). They oxidize various RISCs to obtain electrons for their autotrophic growth. Several sulfur oxidation pathways have been speculated in Acidithiobacillus (Mangold et al., 2011; Talla et al., 2014; Wu et al., 2017; Moya-Beltran et al., 2021). In Acidithiobacillus caldus, extracellular elemental sulfur (S8) is transported into the periplasm by outer membrane proteins (OMP), where sulfur is oxidized to SO32−. The SO32− can form S2O32− via a non-enzymatic reaction between SO32− and a sulfur atom. The SO32− and S2O32− can enter the sulfur oxidizing enzyme system (sox) pathway to form SO42− and elemental sulfur. The S2O32− can also be catalyzed by thiosulfate quinone oxidoreductase (TQO) to form S4O62− that is further hydrolyzed by tetrathionate hydrolase (TetH). The H2S generated in the transport of S8 is oxidized to elemental sulfur by sulfide quinone oxidoreductase (SQR) located in the inner membrane. The elemental sulfur is oxidized to SO32− by sulfur dioxygenase (SDO) or SOR-TST-HDR (SOR, sulfur oxygenase reductase; TST, rhodanese; HDR, HDR-like complex) located in the cytoplasm. The SO32− is further oxidized to SO42− via the APS pathway and SAT (ATP sulfurylase). Acidithiobacillus ferrooxidans does not have Sox system; its sulfur oxidation pathway is completely different from that of Acidithiobacillus caldus. Acidithiobacillus ferrooxidans equipped with Thiosulfate dehydrogenase (TSD), which could serve as an alternative Sox system. The quinol pool (QH2), located in the inner membrane, accepts electrons from TQO, HDR, and SQR. These electrons are then transferred to the terminal oxidases bd or bo3 for ATP production, or to NADH dehydrogenase (complex I) for NADH production (Wang et al., 2018). However, the sulfur oxidation pathway, particularly within the Acidithiobacillus genus, is a complex process that presents a significant challenge to fully comprehend. Despite some progress in characterizing certain genes and proteins, a comprehensive understanding of this pathway and its related metabolic processes remains elusive. Further research is needed to elucidate these mechanisms and the associated energy production (Hold et al., 2009; Koch and Dahl, 2018; Wang et al., 2018).
Genome-scale metabolic models (GEMs) are mathematical representations of metabolism for organisms and provide extensive gene–reaction–metabolite connectivity (Passi et al., 2022). GEMs combined with constraint-based algorithms, such as flux balance analysis (FBA; Orth et al., 2010), can be used to formulate mechanistic predictions of metabolic physiology (Orth et al., 2010; Larsen et al., 2012; Ang et al., 2018; Fang et al., 2020; Antoniewicz, 2021). There are over 6000 GEMs reconstructed for archaea, bacteria, and eukaryotes, which not only elucidate new biological knowledge and understanding (Gu et al., 2019; Chung et al., 2021; Oftadeh et al., 2021), but also help to design and engineer cellular metabolism (Kumar M. et al., 2019). Few GEMs have been used to analyze sulfur metabolism in Acidithiobacillus. The iMC507 model of Acidithiobacillus ferrooxidans ATCC 23270 is the only genome-scale metabolic network model (GEM) of Acidithiobacillus (Campodonico et al., 2016). TheiMC507 successfully elucidated the stoichiometry of proton translocation, electron transfer, and carbon flux distributions during chemolithoautotrophic growth of Acidithiobacillus ferrooxidans ATCC 23270 using Fe2+, S4O62−, and S2O32−. However, the iMC507 model only contain a fraction of the sulfur oxidation pathways of the Acidithiobacillus genus. Specifically, it includes the interconversion between S4O62− and S2O32−, which generates an electron flow for cell growth and CO2 fixation. Moreover, the scope of this model is quite limited, comprising merely 615 reactions, 573 metabolites, and 461 genes. The pathway of element sulfur oxidation has not been thoroughly examined in iMC507 model. This is far from sufficient as a reference knowledge base for the extensive Acidithiobacillus genus. Furthermore, the primary focus of this model is on iron oxidation rather than sulfur oxidation. Therefore, there is an urgent need to develop a novel, more comprehensive model. Further investigation using GEM that involve additional sulfur oxidation reactions beyond what is represented in iMC507. This will improve our understanding of sulfur metabolism in Acidithiobacillus.
In this study, we isolated sulfur-oxidizing bacteria from a previous consortium 5Biol (Han et al., 2013). The isolated bacteria were subjected to various physical and chemical characterization tests, and then their entire genome was sequenced and analyzed. Furthermore, we employed the GEM to further investigate the sulfur oxidation pathway. This will provide a basis for future strain studies as well as aid in the investigation of the exact pathway of sulfur metabolism.
2. Materials and methods
2.1. Isolation of sulfur-oxidizing bacterium
Sulfur-oxidizing bacterium (SOB) was isolated from the pyrolusite leaching microbial community 5Biol (Han et al., 2013) using the 9 K-S solid medium at 37°C. The 9 K-S solid medium was composed of separately sterilized solution A, solution B, and 10.0 g/L sulfur powder (sterilized at 105°C for 24 h). To prepare solution A, 15.0 g agar was added to 250 mL 9 K medium (Silverman and Lundgren, 1959), and the pH was adjusted to 7.0 with H2SO4 (10 M) and NaOH (5 M). To prepare solution B, the pH of 750 mL 9 K medium lowered to 2.0.
2.2. Morphological characterization
Scanning electron microscopy (Hitachi SU8010, Japan) was used to perform morphological observation of the SOB. The SOB was grown aerobically in 9 K-S liquid medium with an initial pH of 2.0 at 37°C until the mid-exponential phase. The 9 K-S liquid medium was prepared by sterilizing 9 K medium and adding 10.0 g/L sulfur powder separately. To perform scanning electron microscopy (Hitachi SU8010, Japan), the samples were subjected to cell fixation, gradient dehydration, critical point drying (Leica EM CPD030, Germany), and gold coating (Hitachi E-1045, Japan) before being observed (Yang et al., 2023).
2.3. Conditions for optimal growth
The SOB with a 10% inoculation was cultured in 9 K-S liquid medium at various temperatures (15, 20, 25, 30, 37, 45, and 50°C) and with an initial pH of 2.0, maintaining a speed of 180 r/min. This experiment aimed to discover the optimal growth temperature of the strain. During the experiment, the OD600 was measured every 2 days to assess cell growth.
To investigate the optimal growth pH of SOB, the strain was grown at a constant temperature of 37°C and a rate of 180 r/min, using varied initial pH levels in the 9 K-S liquid medium (1.0, 2.0, 3.0, 4.0, 5.0, 6.0, 7.0, 8.0, and 9.0). As in the previous experiment, the OD600 was measured every 2 days to evaluate cell growth.
2.4. Sulfur oxidizing ability
To determine the sulfur-oxidizing ability of SOB, the strain was cultured in 9 K-S liquid medium with the optimal pH and temperature for 16 days. The samples were collected every 4 days. The SO42− was detected by a sulfate reagent powder pillow (HACH 2106769), and the pH was measured by a pH meter (pH meter FE20, China).
The SOB was cultured in 9 K medium containing a range of energy sources such as S (5.0 g/L), Na2S2O3 (10.0 g/L), K2S4O6 (1.5 g/L), FeS2 (10.0 g/L), FeSO4·7H2O (5.0 g/L), glucose (1.0 g/L), and yeast extract (0.2 g/L). The utilization of energy sources was assessed by monitoring the growth of the strain.
2.5. Growth experiments on various carbon sources
The ability of the strain to grow on different carbon sources was tested using the Biolog GEN III MicroPlate. According to the manufacturer’s instructions, a pure culture of the strain was incubated at 37°C, and then suspended in a special inoculating fluid at the predetermined cell density (90–98% transmittance). Then, 100 μL of the cell suspension was inoculated into each well of the GEN III MicroPlate™. The microplate was incubated at 37°C for 240 h, during which time kinetic information was recorded and quantified using Biolog’s GEN III OmniLog II ComboPlus kinetic software (Biolog, United States) followed by data analysis (Wragg et al., 2014). In the case of substrate utilization, photographic measurements of color intensity resulting from dye reduction were represented in OmniLog units (OU). The substrates with a Biolog value higher than 114 were considered positive.
2.6. Whole-genome sequencing
10 mL of the SOB in the mid-exponential phase was centrifuged at 10,000 rpm for 15 min. The resulting pellet was then washed twice with a sterile PBS solution containing 8.0 g/L NaCl, 0.2 g/L KCl, 1.44 g/L Na2HPO4, and 0.24 g/L KH2PO4. Genomic DNA was extracted from the pellet using the MOBIO PowerSoil® DNA Isolation kit. The quality and integrity of the extracted DNA were analyzed using NanoDrop2000 and 1% (w/v) agarose gel electrophoresis. The PacBio System based on single-molecule real-time (SMRT) sequencing technology (Wuhan Institute of biotechnology) was used for whole-genome sequencing. The sequencing data were assembled using SPAdes (v3.8.1; Bankevich et al., 2012). The whole genome sequence was deposited at NCBI GenBank under the accession number (CP118747–CP118752). All protein-coding genes were functionally annotated BLASTP (V2.2.3; Alex and Antunes, 2015) against the public protein sequence databases Kyoto Encyclopedia of Genes and Genomes (KEGG; Kanehisa et al., 2017) with an E-value ≤ 1e−5.
2.7. Phylogenomic analysis
To determine the ecological status of the strain, a phylogenetic tree was constructed using MEGA (version 11.0; Tamura et al., 2021) based on the 16S rRNA sequence. The evolutionary history was inferred using Maximum Likelihood (Tamura et al., 2004), based on the best-fitting model of nucleotide substitution using a General Time Reversible (GTR) model (Nguyen et al., 2015), and bootstrapped using 1,000 replicates. The inference was performed in MEGA, running for 1,000,000 generations and saving trees every 10,000 generations.
The average nucleotide identity (ANI; Arahal, 2014) was calculated using the FastANI tool (Jain et al., 2018). The results were used to generate the phylogenetic tree and heat map using the Seaborn and Matplotlib packages in Python (Hunter, 2007; Waskom, 2020). Euclidean was selected as the distance metric to use on the data. Selected “average” as the linking method to use to calculate clusters. Selected “1” for “standard_scaleint” to standardize the columns by subtracting the minimum and dividing each by its maximum.
2.8. Reconstruction of the genome-scale metabolic network
Four main steps were involved in constructing the GEM (Thiele and Palsson, 2010). To begin with, the genome of strain AMEEHan was annotated using the classicRAST and RASTtk pipelines available at https://rast.nmpdr.org (Overbeek et al., 2005; Brettin et al., 2015). The annotation results from two pipelines were automatically used for reconstructing models using ModelSEED available at https://modelseed.org (Overbeek et al., 2014). By integrating the model created by RASTtk annotations with the model created by classicRAST annotations, the draft model was reconstructed.
However, the draft model produced metabolites and ATP without providing any energetic substrate. The Parsimonious flux balance analysis (pFBA; Lewis et al., 2010) algorithm was utilized to identify pathways that resulted in a net production of ATP without supplying any carbon substrate, and the reversibility of reactions in draft model was corrected referring the MetaCyc database (Caspi et al., 2020). Afterward, the ATP production rate was examined when substrates were input.
The biomass composition of the gram-negative representative strain E. coli iML1515 (Monk et al., 2017) was adopted to represent the strain AMEEHan’s biomass. For simulating biomass synthesis, the same cell growth-associated and non-growth-associated ATP maintenance values (GAM and NGAM, respectively) as those used in iML1515 were assumed. After modifying the biomass composition, the biomass compositions that cannot be synthesized were identified. The optimal target for FBA calculation was set as the demand reaction of each biomass composition. A result of zero indicates that the biomass composition cannot be synthesized due to gaps in its synthesis pathway. The gaps for each composition were filled using the weight-added pFBA gap filling program (Luo et al., 2022).
Gap-filling was conducted for substrates that were applicable in the Biology experiment but were not available in the model. Two categories were identified: (1) Substrates that lack exchange reactions from extracellular to intracellular, which were filled manually. (2) Substrates that lack a utilization pathway, which were filled using the gap-filling program.
2.9. Validating model and simulating sulfur oxidation pathway
The uptake rate of the sulfur was set to 1.5 mmol/gDW/h, while the rates for other energy sources input were set to 0. The consistency and annotation of the model were evaluated using the metabolic model-testing suite (MEMOTE; Lieven et al., 2020). Flux balance analysis (FBA; Orth et al., 2010) was used to optimize a pre-defined objective function under specified metabolic constraints (Antoniewicz, 2021). If not otherwise specified, the objective function considered was the maximization of the biomass production rate. We selected parsimonious flux balance analysis (pFBA; Lewis et al., 2010) to analyze the biosynthesis pathway of the product. We conducted these methods using COBRApy (0.18.1; Ebrahim et al., 2013). The optimization solvers GLPK and Gurobi were used for linear and quadratic programming (Meindl and Templ, 2013).
An optimal sulfur oxidation pathway was predicted by maximizing the ATP production rate using FBA optimization. The model was iteratively computed by closing one of the reactions in the optimal sulfur oxidation pathway, which led to other possible differences in energy metabolic pathways.
3. Results
3.1. Physiological characterization of the SOB
A sulfur-oxidizing bacterium, designated as strain AMEEHan, was successfully isolated from the pyrolusite leaching microbial community 5Biol (Han et al., 2013) using the 9 K-S solid medium at 37°C. The isolation was small, round, yellow, dome-shaped, and had a smooth surface with a distinct edge (Figure 1A). Scanning electron microscopy results demonstrated that the dimensions of the strain were approximately 1 μm × 2 μm and that it had a short rod-shaped (Figure 1B).

Figure 1. Physiological characterization of the strain AMEEHan. (A) The morphology of the strain. (B) Scanning electron microscopy (SEM) image of the strain. Effects of different temperatures (C) and pH (D) on the growth of the strain. Changes of SO42− concentration (E) and pH (F) during the strain growing in the 9 K-S liquid medium. CK, non-inoculation; AM, inoculated with the strain AMEEHan.
The strain exhibited growth within a temperature range of 15–45°C, with an increased growth rate as the temperature was sequentially raised from 15 to 45°C. The optimal growth temperatures for growth were found to be 37 and 45°C (Figure 1C). In terms of pH, the strain was able to grow within a range of pH 1.0–8.0. Growth was observed to be similar from pH 2.0 to 8.0, but was inhibited at pH 1.0 and pH 9.0 (Figure 1D).
When cultured under optimal conditions, the concentration of SO42− increased from 17 to 30 g/L while the pH decreased from 2.0 to 0.9 (Figures 1E,F). This indicated that approximately 50% sulfur was oxidized by the strain within 16 days.
The strain was found to be capable of growing on sulfur and K2O6S4, but not on Na2S2O3, FeS2, FeSO4·7H2O, glucose, and yeast extract (Table 1). Furthermore, it exhibited significantly enhanced growth on sulfur compared to K2O6S4, as indicated by higher OD600 values for sulfur compared to K2O6S4 (Supplementary Table S1).
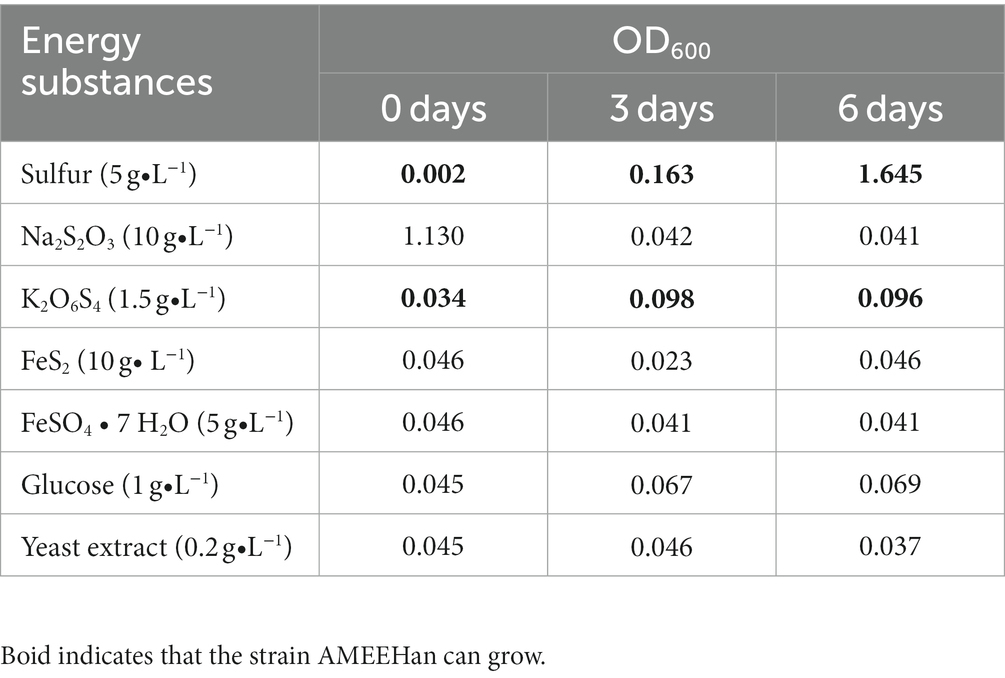
Table 1. The energy utilization of strain AMEEHan.
3.2. Genome analysis of strain AMEEHan
The genome length of the strain consisted of a single circular chromosome of 2,630,391 bp with a G + C content of 58.6% (Table 2; Figure 2A). The genome encoded five plasmids, six ribosomal operons, 49 tRNA genes, and one tmRNA gene. A total of 2,582 protein-coding genes (CDSs) were predicted, of which 1,941 (75.2%) were assigned a putative function.
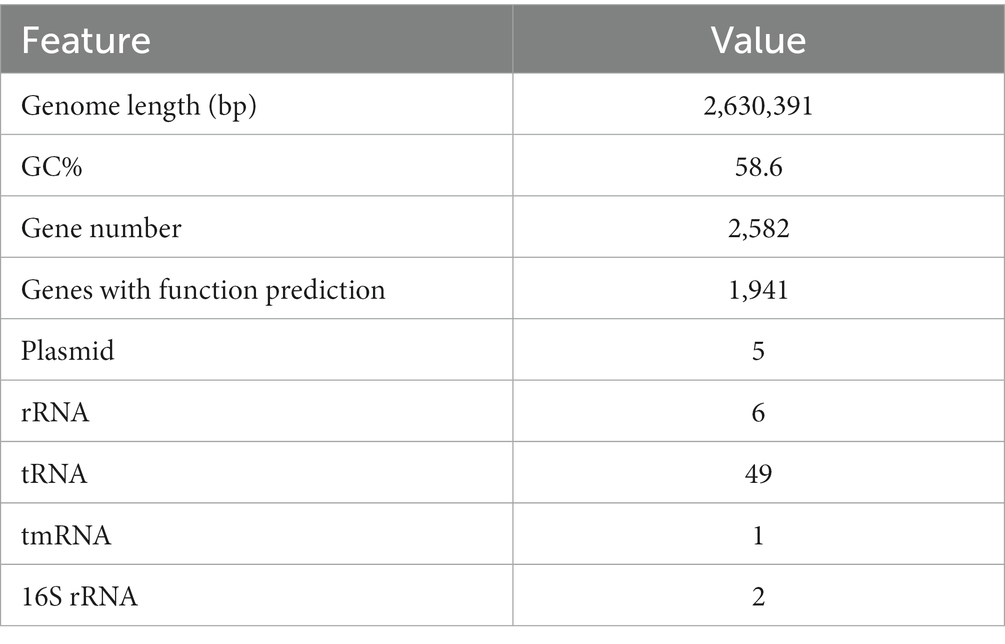
Table 2. Basic characteristics of the genome of strain AMEEHan.
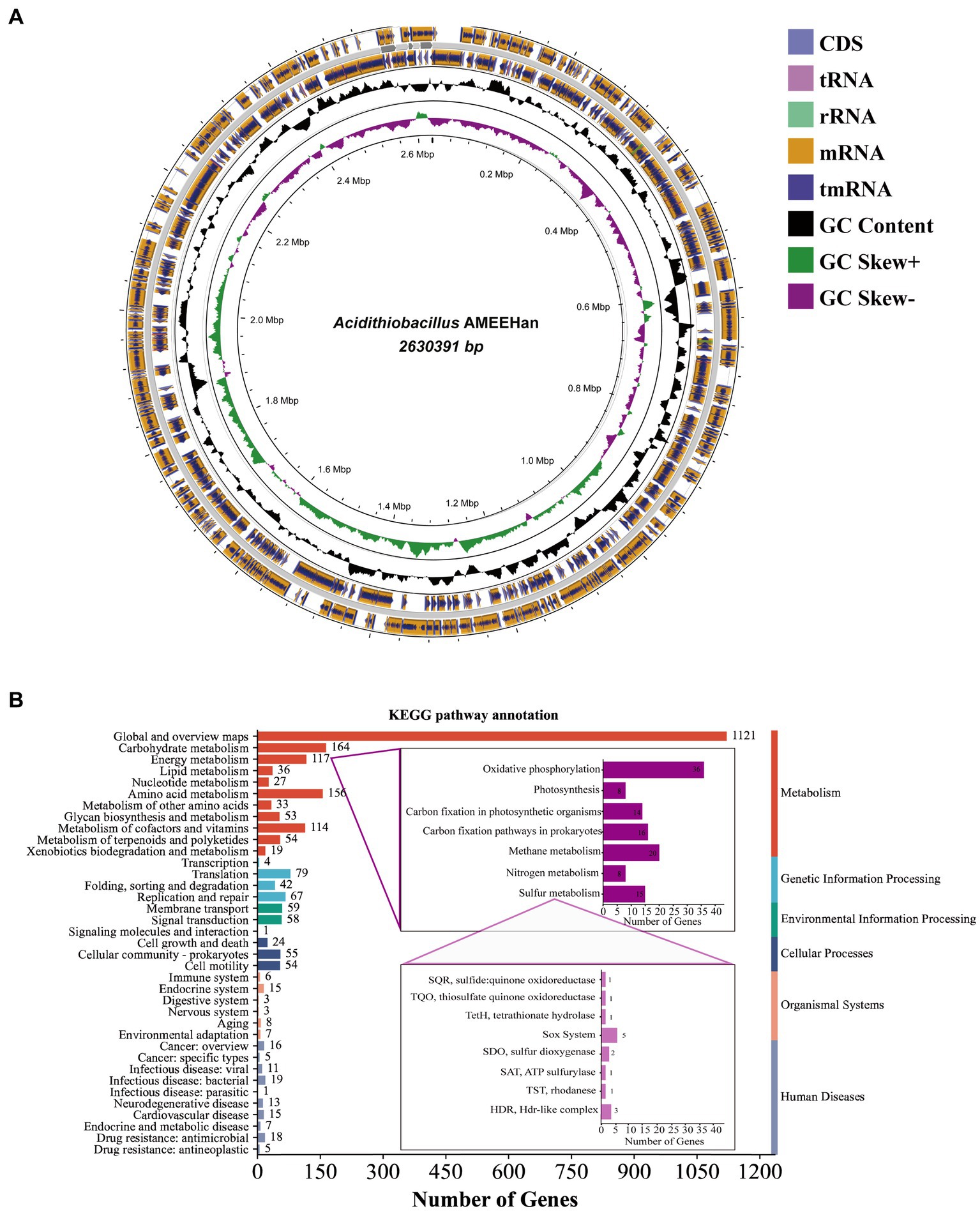
Figure 2. The genome of strain AMEEHan. (A) Circular genome map of the strain. The outermost circle represents the coding region on the positive and negative strands and the structural RNA genes, the innermost circle represents the variation in GC content in different regions of the genome, and the innermost circle represents the GC skew, where green indicates skew+ and purple indicates skew−. (B) A bar diagram of the number of genes involved in different functions from the KEGG pathway annotation.
The majority of genes involved in global, carbohydrate, amino acid, energy, cofactors, and vitamins metabolism. In terms of energy metabolism, it primarily contained oxidative phosphorylation, methane metabolism, carbon fixation pathways in prokaryotes, and sulfur metabolism (Figure 2B). Fifteen genes were identified in sulfur metabolism, including SQR (sulfide: quinone oxidoreductase), thiosulfate quinone oxidoreductase (TQO), tetrathionate hydrolase (TetH), sox system, sulfur dioxygenase (SDO), SAT (ATP sulfurylase), TST (rhodanese), and HDR (HDR-like complex). These gene compositions suggested that the strain had the capacity to convert sulfur into sulfate. Additionally, it also exhibited the capacity for carbon fixation, as demonstrated by the presence of eight genes, which include fructose-bisphosphate aldolase (FBA), transketolase (tKt), ribose 5-phosphate isomerase A (rpiA), phosphoribulokinase (prk), Rubisco (ribulose-bisphosphate carboxylase small chain), phosphoglycerate kinase (pgk), glyceraldehyde 3-phosphate dehydrogenase (gap), and fructose-1,6-bisphosphatase (FBP; Supplementary Table S2).
Sixteen Acidithiobacillus, sharing high genetic similarity with strain AMEEHan, were utilized to create the 16S rRNA phylogenetic tree. The results illustrated that it had the highest degree of relatedness to Acidithiobacillus caldus SM1, with a similarity of 95.93% (Figure 3A). Furthermore, the same set of 16 Acidithiobacillus species that were used for the 16S rRNA analysis were evaluated for the ANI analysis. The ANI analysis indicated that the strain had the highest similarity to Acidithiobacillus caldus MTH 04 and Acidithiobacillus caldus SM-1, with ANI values of 70.38 and 70.29%, respectively (Figure 3B). The similarity of the strain to other strains was below 70% (Supplementary Table S3).

Figure 3. Classification of the strain AMEEHan. (A) Phylogenetic tree based on 16S rRNA gene by the Maximum Likelihood method. (B) Average Nucleotide Identity (ANI; %) based on the whole genomes.
3.3. Reconstruction GEM of Acidithiobacillus AMEEHan
The draft model of Acidithiobacillus AMEEHan was constructed by combining two models: one created with RASTtk annotation (589 genes, 1,191 metabolites, and 1,083 reactions), which covered 22% (589/2,708) of the annotated ORFs (Supplementary material; RASTtk, https://github.com/wupeng1998/Acidithiobacillus-Ameehan/tree/main/RASTtk) and the other with classicRAST annotation (702 genes, 1,214 metabolites, and 1,163 reactions), which covered 27% (702/2,582) of the annotated ORFs (Supplementary material; classicRAST, https://github.com/wupeng1998/Acidithiobacillus-Ameehan/tree/main/classicRAST). The information of charge and formula for the metabolites from ModelSEED were added to the draft model. The draft model comprised 723 genes, 1,293 metabolites, and 1,220 reactions, accounting for 28% (723/2,582) of the annotated ORFs (Figure 4C). Further analysis was conducted to assess the energy generation of the draft model. However, the simulated results showed the production of ATP without the supply of any carbon substrate (Figure 4C). To avoid a net production of ATP and correct the occurrence of energy-generating cycles (Supplementary Figure S1), 213 reactions were modified (Supplementary Table S4).

Figure 4. The reconstruction process of genome-scale metabolic network workflow for Acidithiobacillus AMEEHan. (A) Whole genome sequencing of the strain, perform whole genome sequencing of samples to obtain gene sequence data. (B) Analysis of bacterial whole genome, analyze genome sequences to obtain gene function annotation information. (C) Draft model generation and brief information, predict initial draft metabolic network model containing some gaps based on genome annotation. (D) Model_01 generation from Draft Model, refine and correct wrong reactions in Draft model using knowledge bases like Metacyc to produce Model_01. (E) Model_02 generation from Model_01, further integrate literature knowledge and add some known biochemical reaction pathways to generate Model_02. (F) Model_03 generation from Model_02, perform gap-filling on the Model_02 using the pFBA algorithm to produce Model_03. (G) Final model Model_04, based on Biolog experimental results, expand Model_03 to generate the final high-quality model Model_04.
Following the above modifications, Model_01 was developed (Figure 4D). However, the ATP generation rate calculated by Model_01 was 0.0 mmol/gDW/h, when the sulfur uptake rate was 1.5 mmol/gDW/h. It indicated that there were gaps in the energy metabolism pathways. Through the integration of the physiological characteristics of the strain, genetic data, and an examination of the ATP synthesis pathway, it was ascertained that Model_01 exhibited a malfunctioning sulfur oxidation pathway. Therefore, the Model_01 was extended to generate Model_02 by incorporating 21 additional reactions and seven new metabolites (Supplementary Table S5) based on the genome of the strain and other reports (Wang et al., 2018). The optimum ATP generation rate calculated by Model_02 was 14.8 mmol/gDW/h with the sulfur uptake rate of 1.5 mmol/gDW/h (Figure 4E).
The growth rate simulated by Model_02 was zero with the same sulfur uptake rate. However, the experimentally measured growth rate of Acidithiobacillus AMEEHan was 0.0379 h−1 when cultured in minimal medium (Supplementary Table S4). Upon analyzing the biomass composition, it was revealed that the inability to synthesize certain biomass precursors was the reason why Model_02 failed to simulate growth accurately. In order to tackle this problem, the synthetic pathway for nine precursors was completed by adding a total of 68 reactions (Supplementary Table S6), which resulted in the development of Model_03 (Figure 4F). Model_03 predicted a growth rate of 0.0384 h−1, which was close to the actual in vivo value of 0.0379 h−1 (Supplementary Figure S2).
The Biolog phenotype microarray experiments were conducted, revealing that 16 of 71 carbon sources can be used by Acidithiobacillus AMEEHan (Supplementary Table S7). However, 15 of the 16 carbon sources could not be utilized by Model_03 due to the lack of transport reactions or utilization pathways. For instance, D-Mannose had an exchange reaction in model_03, but its metabolic pathway was missing one step, “D-mannose-6-phosphate = > D-fructose-6-phosphate.” Model_04 was generated after supplementing 70 reactions (Figure 4G; Supplementary Table S8). Model_04 demonstrated an 87.5% agreement with the experimental data, yielding 11 true positive (substrates that can be used both in vivo experiments and simulations) and 52 true negative (substrates that cannot be used both in vivo experiments and simulations) results (Figure 5; Supplementary Table S7).

Figure 5. The Biolog carbon source validation results in models and experiments. Purple circles indicate a positive value, while blue circles indicate a boundary value. Blank circles indicate negative values, and semi-blue circles indicate boundary values. Wired circles suggest that the comparison results with Corynebacterium pilosum in the Biolog GEN III MicroPlate™ library are false positive or false negative. However, as there are no Acidithiobacillus strains present in the Biolog GEN III MicroPlate™ library, false negative values are considered negative and false positive values are considered positive.
The final model Model_04, named AME_WP1377 (Model_04), was comprised of 744 genes, 1,374 metabolites, and 1,377 reactions that are distributed among the extracellular, periplasmic, and cytoplasmic compartments1. The model of AME_WP1377 was evaluated by MEMOTE (Lieven et al., 2020; Supplementary material—MemoteReportApp.html). The annotation process evaluated the assignment of appropriate SBO terms to model instances using the Systems Biology Ontology (SBO) terminology. The SBO annotation achieved a high score of 91%. The annotations for reactions and metabolites achieved scores of 82 and 86%, respectively. The model consistency check consisted of tests to evaluate stoichiometric consistency, mass and charge balance, metabolite connectivity, and unbounded fluxes in the default medium. Moreover, the consistency score of AME_WP1377 was high at 98%. These results indicate a clear advantage of AME_WP1377 with a total score of 90% (Figure 6).
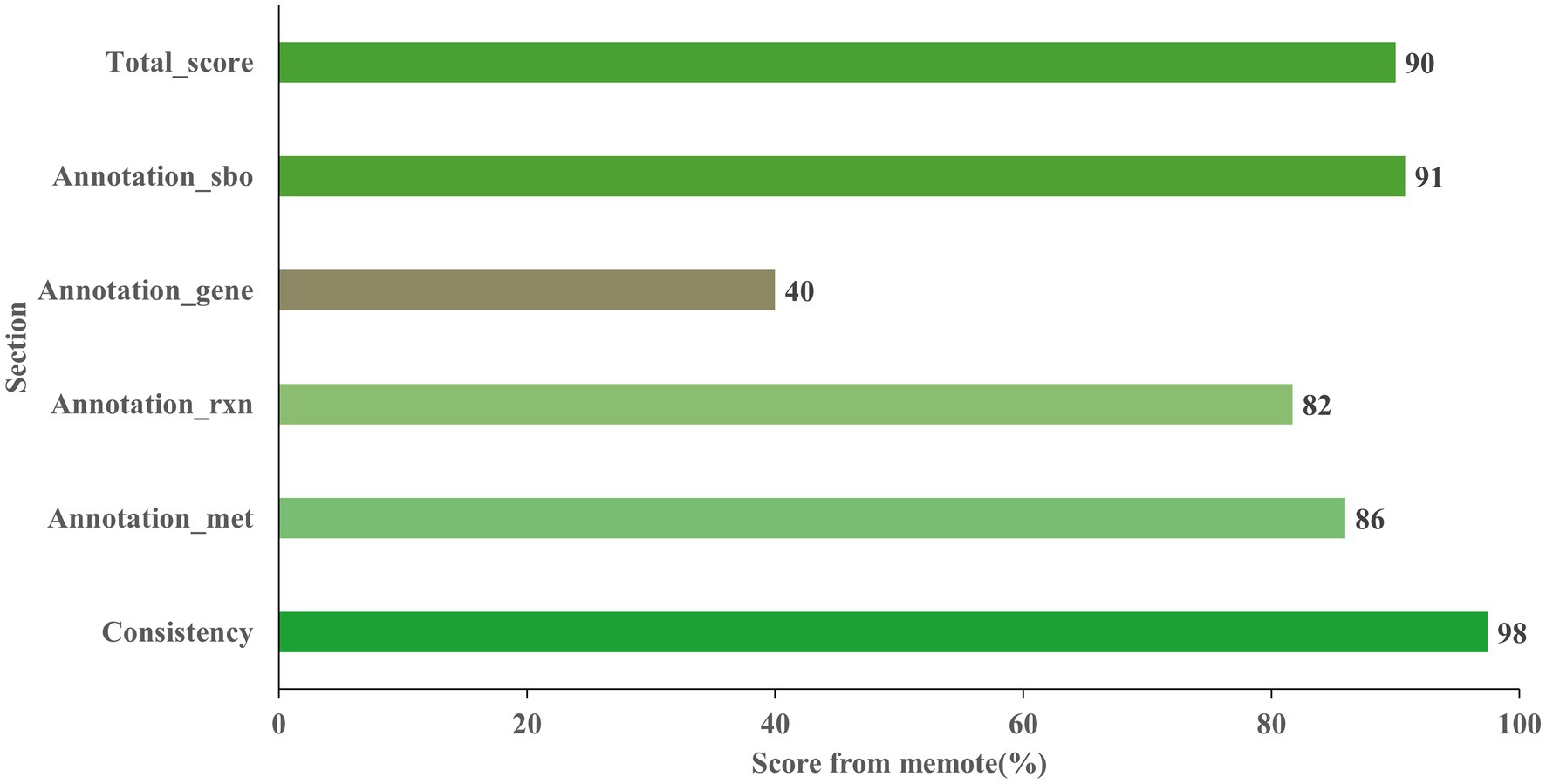
Figure 6. Evaluation the Acidithiobacillus AMEEHan model AME_WP1377 by MEMOTE.
3.4. Simulating optimal sulfur oxidation pathway of Acidithiobacillus AMEEHan
Through model simulation, the AME_WP1377 model demonstrated the existence of five potential pathways for sulfur oxidation in Acidithiobacillus AMEEHan when using sulfur as substrates (Table 3). By consuming 1.5 mmol/gDW/h sulfur, it in the path I can produce 14.81 mmol/gDW/h ATP, 5.76 mmol/gDW/h NADH/NADPH, and fixation CO2 at a rate of 1.575 mmol/gDW/h with the growth rate of 0.0384 mmol/gDW/h. In path II, it can produce 6.56 mmol/gDW/h ATP, 2.76 mmol/gDW/h NADH/NADPH and fix CO2 at a rate of 0.694 mmol/gDW/h with the growth rate of 0.0169 mmol/gDW/h. In path III, it can produce 5.63 mmol/gDW/h ATP, 2.13 mmol/gDW/h NADH/NADPH, and fix CO2 at a rate of 0.598 mmol/gDW/h with the growth rate of 0.0146 mmol/gDW/h. In path IV, it can produce 4.69 mmol/gDW/h ATP and 1.50 mmol/gDW/h NADH/NADPH and fix CO2 at a rate of 0.499 mmol/gDW/h with the growth rate of 0.0121 mmol/gDW/h. In path V, it can produce 2.25 mmol/gDW/h ATP and 0.95 mmol/gDW/h NADH/NADPH and fix CO2 at a rate of 0.238 mmol/gDW/h with the growth rate of 0.0058 mmol/gDW/h.
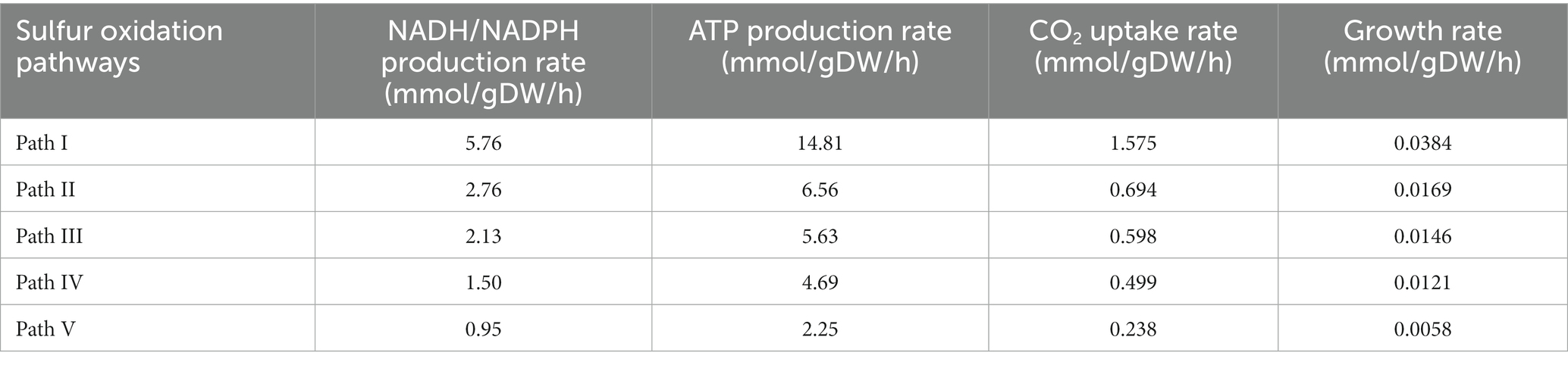
Table 3. The calculated five sulfur oxidation pathways of Acidithiobacillus AMEEHan.
By model simulation, from the potential sulfur oxidation pathways in Acidithiobacillus AMEEHan (Figure 7A), an optimal sulfur oxidation pathway (Table 3, Path I; Figure 7B) had been predicted. In Path I, the extracellular elemental sulfur (S) was activated to sulfide and transported by special outer-membrane proteins (OMP) with sulfide into the periplasm. In the periplasm, the sulfide was oxidized to produce H2S, which was further oxidized by the SQR enzyme (sulfide: quinone oxidoreductase), located in the inner membrane, to generate sulfur and QH2. The sulfur reacted with SO32− in a conventional chemical reaction to S2O32−. Subsequently, the TST enzyme (rhodanese enzyme) and HDR enzymes (Hdr-like complex enzymes) catalyze the S2O32− to produce SO32− and QH2. Finally, SO32− is processed through the APS pathway to generate ATP and SO42−, and the SO42− was secreted out of the cell. Additionally, the electron produced by sulfur oxidation were stored in QH2, which reduced O2 through the downhill pathway of the ubiquinone cytochrome oxidoreductase complex (cytochrome bd, bo3, and aa3 complex), and reduces NAD/NADP through the uphill pathway of NADHI, resulting in the production of reducing power NADH. The produced NADH participated in the incomplete Calvin–Benson–Bassham (CBB) cycle to fix environmental CO2 (Figure 7B).

Figure 7. Sulfur oxidation pathway of Acidithiobacillus AMEEHan in silico predictions. The green line showed the common sulfur oxidation reactions found in all pathways while the yellow lines indicated intracellular and extracellular transport of substances. The purple dots represented the corresponding metabolites. Arrows indicate the direction of enzymatic activity and the arrow thicknesses are proportional to the flux through each reaction (a thicker arrow has a larger flux). (A) The potential sulfur oxidation pathways in Acidithiobacillus AMEEHan. (B) The red line denoted the optimal pathway predicted for sulfur oxidation. (C) The blue line denoted the second pathway predicted for sulfur oxidation. (D) The brown line denoted the third pathway predicted for sulfur oxidation. (E) The purple line denoted the fourth pathway predicted for sulfur oxidation. (F) The pink line denoted the fifth pathway predicted for sulfur oxidation. S, Sulfur; SSH, Sulfane sulfur atom of glutathione persulfide; OMP, Outer-membrane proteins; TQO, Thiosulfate quinone oxidoreductase; TetH, Tetrathionate hydrolase; SoxABXYZ, Sulfur oxidizing enzyme system; SQR, Sulfide: quinone oxidoreductase; SDO, Sulfur dioxygenase; TST, Rhodanese; HDR, HDR-like complex; SAT, ATP sulfurylase; aa3, bd, bo3, terminal oxidases; QH2, Quinol pool; NADHI, NADH dehydrogenase complex I; and ATPs, ATP hydrolytic enzyme.
The second alternative sulfur oxidation pathway (Table 3, Path II) is the same as in the first pathway from S to S2O32−, followed by oxidation of S2O32− by the Sox system (SoxABXYZ) in the periplasmic space to monomeric sulfur in the cytoplasmic for sulfur oxidation in the first pathway. As for electron transfer to generate proton pumps, NADH regeneration and CO2 fixation are consistent with the first sulfur oxidation pathway (Figure 7C).
A third potential pathway (Table 3, Path III) for sulfur oxidation is almost similar to the second pathway, except that extracellular elemental sulfur enters the cell and is simultaneously oxidized by SQR and SDO into the periplasmic space for sulfur oxidation (Figure 7D).
The fourth alternative sulfur oxidation pathway (Table 3, Path IV) is activation of extracellular elemental sulfur through OMP and transport to the periplasm, where it is converted to SO32− by SDO enzyme. The resulting SO32− is transported from the periplasm to the cytoplasm by a transport system. As for electron transport, H2S in the cytoplasm is oxidized by the SQR, located in the inner membrane, to produce sulfur and QH2. The SSH is oxidized by SDO enzyme in the cytoplasm to produce SO32−. Finally, the SO32− is processed through the APS pathway to generate ATP and SO42−, and the SO42− is secreted out of the cell through the transport system (Figure 7E).
The fifth alternative sulfur oxidation pathway (Table 3, Path V) involves activation of extracellular elemental sulfur through OMP and transport to the periplasm, where it is oxidized by the SDO enzyme to SO32−. The sulfur reacts with SO32− in a conventional chemical reaction to S2O32−. This compound is further oxidized by TQO to S4O62− and QH2. The S4O62− is converted by TetH to S2O32−, sulfur and SO42−. Finally, the SO42− is secreted out of the cell through the transport system (Figure 7F).
4. Discussion
4.1. The Acidithiobacillus AMEEHan is a new species
The physicochemical properties of strain AMEEHan were significantly different from other species of the genus Acidithiobacillus (Table 4). The growth pH of the strain ranged from 2.0 to 8.0, while other species of the genus Acidithiobacillus ranged from 1.7 to 4.0 (Kelly and Wood, 2000; Kupka et al., 2007; Valdes et al., 2011; Wang et al., 2018; Sriaporn et al., 2021). This broader optimal pH range suggested that strain AMEEHan possessed a heightened ability to adapt to varying pH conditions, thereby enhancing its competitive advantage across different growth pH levels, which would provide great application potential for its future application. More importantly, all species of the genus Acidithiobacillus but not Acidithiobacillus AMEEHan were able to utilize S2O32− as energy source (Bryant et al., 1983; Johnson, 1998; Janiczek et al., 2007; Valdes et al., 2008; Beard et al., 2011; Chen et al., 2012; Hedrich and Johnson, 2013a,b; Yin et al., 2014; Falagán and Johnson, 2016; Miyauchi et al., 2018; Zhan et al., 2019; Zhang et al., 2020; Moya-Beltran et al., 2021; Zhao et al., 2021). Five plasmids were detected in the strain, while there were less than four plasmids in other Acidithiobacillus (Acidithiobacillus ferrooxidans YQ-N3 contained five plasmids; Valdes et al., 2009; You et al., 2011; Moya-Beltran et al., 2021). These indicated that the strain might be a new species of the genus Acidithiobacillus.
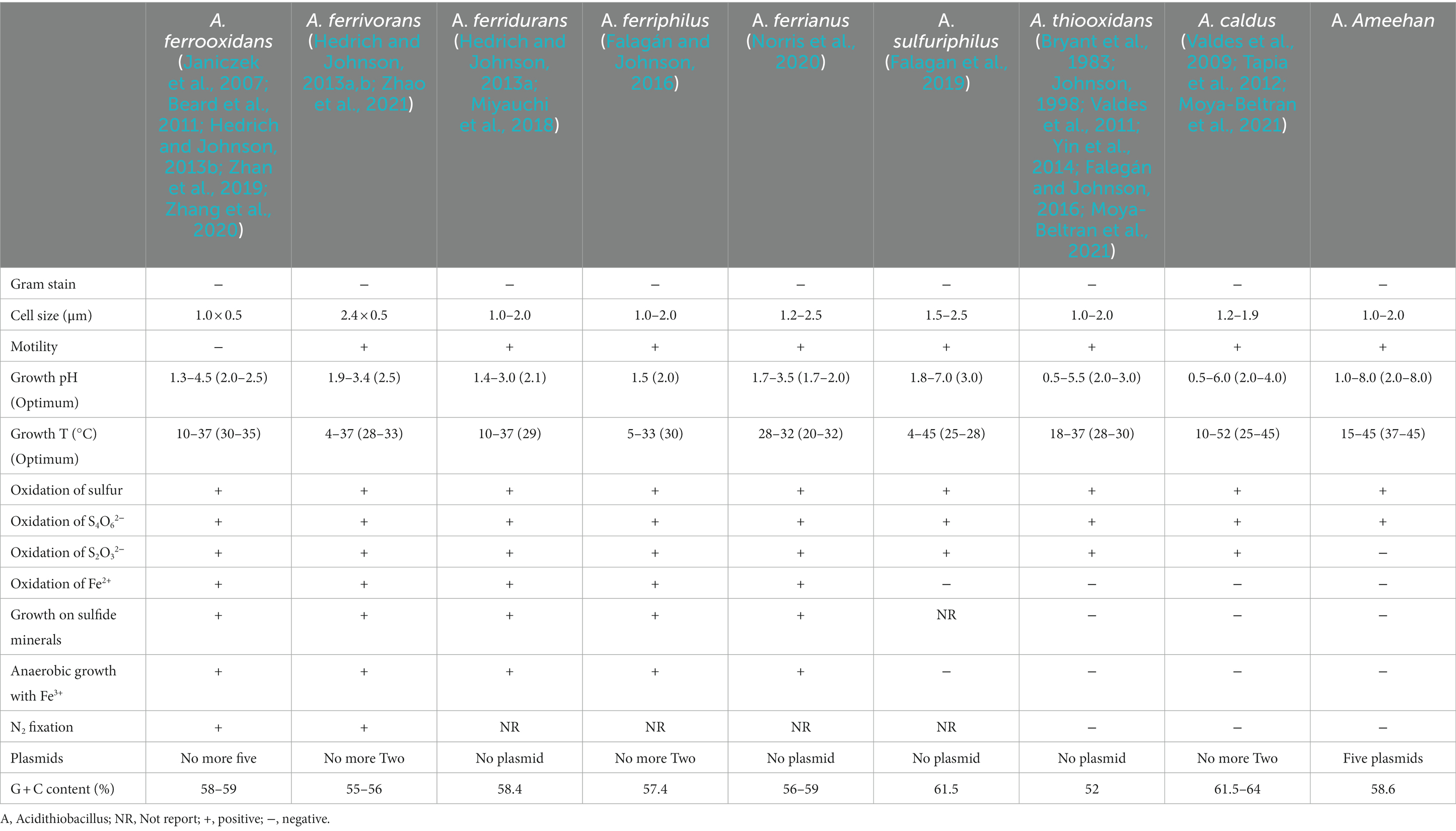
Table 4. Growth phenotypic features of species in the genus of Acidithiobacillus.
The 16S rRNA gene phylogenetic tree showed that Acidithiobacillus AMEEHan was closest to Acidithiobacillus caldus SM-1 with 95.68% (Figure 3A). However, the 16S rRNA genes with more than 97% or even 99% similarity represent the same species (Gevers et al., 2005; Edgar, 2018).
Although the 16S rRNA gene has been a useful marker for species classification, this conserved gene approach has limitations in large-scale taxonomy (Huse et al., 2008; Klindworth et al., 2013; Jeong et al., 2021). To address these limitations, we use the ANI value to compare genomes in pairs, which has higher resolution and stronger correlation (Richter and Rossello-Mora, 2009; Vandervalk et al., 2015). The ANI comparisons showed that the maximum similarity between the strain and the genus Acidithiobacillus were no more than 70%. However, the ANI values of the same species should be higher than 95% (Arahal, 2014; Yoon et al., 2017). The strain AMEEHan is a novel species in the genus Acidithiobacillus, as demonstrated by these results, which highlights its significant divergence from other strains in the genus. We have therefore named it Acidithiobacillus Ameehan.
4.2. Acidithiobacillus Ameehan had unique functional genes and pathways
Similar to Acidithiobacillus thioooxidans and Acidithiobacillus caldus, Acidithiobacillus Ameehan lacks the ability to oxidize iron and fix nitrogen (Table 4). A putative nitrogenase gene cluster (nifDHK) of Acidithiobacillus Ameehan was found similar to that of most other Acidithiobacillus except Acidithiobacillus caldus and Acidithiobacillus thiooxidans (Figure 8). These genes may encode the nitrogenase complex and proteins involved in the synthesis of the nitrogenase cofactor.
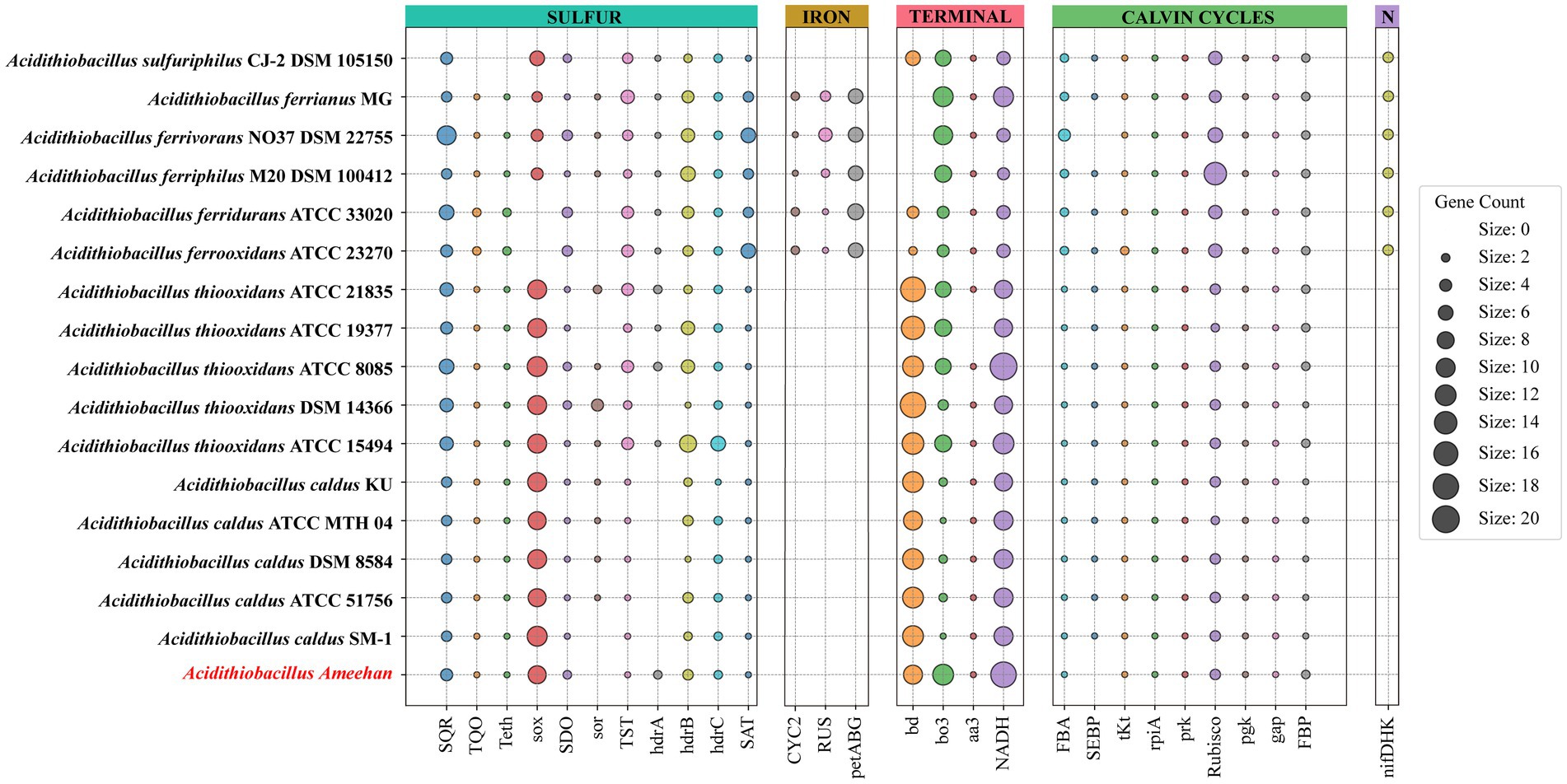
Figure 8. Metabolic characteristics of Acidithiobacillus Ameehan and other species of the Acidithiobacillus. The genes annotated for each strain of the genus Acidithiobacillus in sulfur metabolism, carbohydrate metabolism, metal iron oxidation and nitrogen fixation.
The genomes of Acidithiobacillus species contained genes encoding enzymes and electron transfer proteins predicted to be involved in the oxidation of RISCs (Figure 8; Supplementary Table S2). Notably, the functional genes of Acidithiobacillus Ameehan were significantly different from other Acidithiobacillus species. Acidithiobacillus Ameehan, similar to Acidithiobacillus caldus SM-1 and Acidithiobacillus thiooxidans ATCC 19377, possessed 10 out of 11 genes related to sulfur metabolism, with the exception of the sor gene. The sor gene was a key enzyme in archaea that was often used for energy production from inorganic sulfur oxidation, but was found to be nonessential in Acidithiobacillus (Chen et al., 2012; Ferreira et al., 2022). The sor gene deletion was also simulated in the AME_WP1377 model. It had no effect on the growth of the strain when the sor gene (4 H2O + O2 + 4 Elemental-Sulfur = > 4 H+ + 2 Sulfite +2 H2S) was added to the model (Supplementary Table S9). Therefore, the loss of this unnecessary gene in Acidithiobacillus Ameehan is likely a result of long-term natural evolution and adaptation to its specialized niche.
The genus Acidithiobacillus fixed CO2 via the CBB reductive pentose phosphate cycle, using energy and reducing power derived from the oxidation of iron or sulfur. However, it is worth noting that both Acidithiobacillus Ameehan and Acidithiobacillus ferrivorans NO37 DSM 22755 were found to lack the enzyme SEBP (sedoheptulose-1,7-bisphosphatase) in the CBB. The enzyme should catalyze the reaction of sedoheptulose-1,7-bisphosphatase to sedoheptulose-7-phosphate during the regeneration of RuBP in the third steps of the CBB cycle (Supplementary Figure S3). This absence suggests that these organisms may have alternative mechanisms to compensate for the lack of SEBP function. Interestingly, it was observed that the enzyme 2.2.1.2, which converts erythrose-4-phosphate to sedoheptulose-7-phosphate, known as dihydroxyacetone transferase, could potentially fulfill the role of SEBP, and provide a viable substitute in the metabolic pathway (Figure 8). Model simulations can provide a solid explanation for this discrepancy. In model simulations, we discovered that when Acidithiobacillus Ameehan used the incomplete CBB cycle, they were able to fix more CO2, and the growth rate was better than the classical CBB cycle (Table 5). This discovery may provide some theoretical insight into the evolution of the strain.
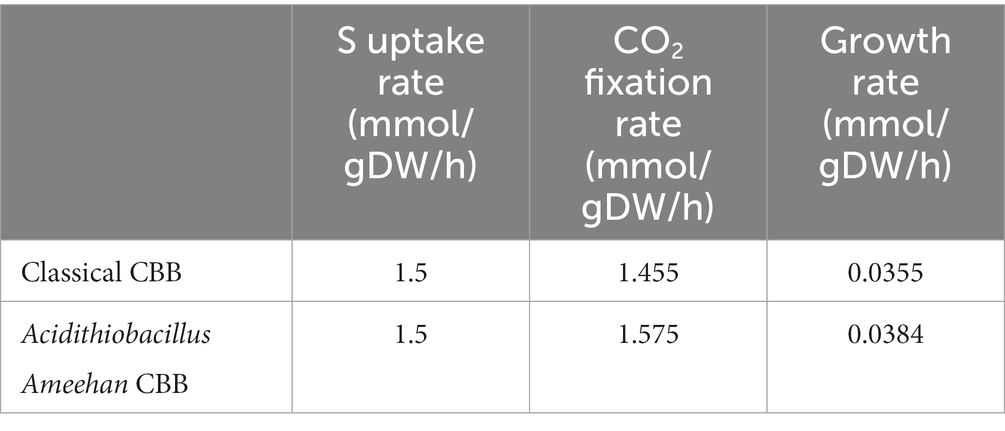
Table 5. Differences between classical CBB and incomplete CBB cycles.
And for K2S4O6 utilization, when simulated with K2S4O6 as the energy substrate at the same CO2 uptake rate (1.575 mmol/gDW/h), the model showed that the K2S4O6 uptake rate was 0.76 mmol/gDW/h. This was equivalent to elemental sulfur at a rate of 3.04 mmol/gDW/h. It indicated that Acidithiobacillus Ameehan had a relatively low efficiency in the utilization of K2S4O6, which was consistent with the experimental results (Table 1). Comparing the energy consumed by the AMEE_WP1377 model and the Acidithiobacillus ferrooxidans ATCC 23270 model iMC507 (Campodonico et al., 2016) under the same fixed CO2 rate, we found that the oxidation efficiency of Acidithiobacillus Ameehan for RISCs was higher than that of Acidithiobacillus ferrooxidans ATCC 23270. The oxidation efficiency of Acidithiobacillus Ameehan on S4O62− (S4O62− uptake rate was 0.717 mmol/gDW/h) was about six times higher than that of Acidithiobacillus ferrooxidans ATCC 23270 (S4O62− uptake rate was 4.45 mmol/gDW/h; Eccleston and Kelly, 1978; Campodonico et al., 2016). These results further demonstrated the high application value of Acidithiobacillus Ameehan in bioleaching.
4.3. Simulation of the optimal sulfur oxidation pathway of Acidithiobacillus Ameehan
The sulfur oxidation pathway is extremely complex, and unraveling it will be a long and difficult endeavor. Although some genes and proteins involved in sulfur oxidation have been characterized, there is still a lack of accurate and comprehensive analysis of the sulfur oxidation pathway and its related metabolic pathways (Wang et al., 2018; Yang et al., 2019). However, by using GEMs, we can obtain an optimal sulfur oxidation pathway (Figure 7B), which provides a clearer understanding of the sulfur oxidation pathway in the study of Acidithiobacillus Ameehan. By examining the changes in the oxidation states of elemental sulfur during the energy production process in these five calculated sulfur metabolic pathways, we discovered that the first pathway produced the highest amount of energy. This could be due to its ability to completely oxidize all elemental sulfur to the final sulfate state. However, the other proposed pathways did not completely oxidize all elemental sulfur to the final sulfate state. In addition, the OMP protein played a critical role in all sulfur oxidation pathways because all sulfur oxidation pathways required it to transport extracellular elemental sulfur into the cell. Meanwhile, enzymes in other pathways could discover possible alternative pathways or alternative enzymes, increasing the resilience of the pathways. In addition, we found that SQR enzymes were involved in the top three pathways with the highest energy production efficiency. This suggests that SQR enzymes have a significant impact on the energy production efficiency of sulfur oxidation. It uses inorganic electron donor (sulfur) to generate reducing power to drive light-dependent CO2 fixation via the incomplete CBB cycle. This discovery could serve as a valuable breakthrough and reference point for future strain modifications aimed at improving sulfur oxidation efficiency.
While the above results are based on computational modeling, further experimental validation will be critical in the future. The genome-scale metabolic modeling approach allowed for the identification of key enzymes and the optimal sulfur oxidation pathway, providing valuable hypotheses to explore experimentally. Future investigations should prioritize validation of the predicted sulfur oxidation pathway and quantification of flux through said pathway. The functions of crucial enzymes, including SQR, need to be verified through experiments, such as enzyme assays and gene knockouts. Moreover, the comparative energetics of various putative sulfur oxidation pathways could be assessed by measuring growth and ATP production in mutants that lack different enzymes. Following these experimental approaches, apart from confirming computational results, can also provide additional insights into sulfur oxidation mechanisms in Acidithiobacillus Ameehan. Combining genome-scale modeling with targeted experiments will enhance our understanding of this intricate and ecologically significant metabolic process.
5. Conclusion
In this research investigation, a sulfur-oxidizing bacterium was isolated from the 5biol colony. The complete genome sequence of this bacterium was determined and subsequently used to confirm various physiological traits of the isolation. These characteristics included a small, round, yellow, dome-shaped, and had a smooth surface with obvious edges. The strain had a size of 1 μm × 2 μm, and a short rod-shaped. Moreover, it demonstrated an optimal growth temperature range of 37–45°C and an optimal growth pH range of pH 2.0–8.0. The microbe was able to grow on sulfur and K2O6S4, but could not grow on Na2S2O3, FeS2, FeSO4·7H2O, glucose, or yeast extract. Phylogenetic tree construction based on the 16S rRNA and whole-genome average nucleotide identity (ANI) values of the strain allowed us to identify the isolated strain as a novel strain, named Acidithiobacillus Ameehan. Furthermore, the KEGG pathway annotated several genes associated with distinct functions. This strain was capable of oxidizing elemental sulfur or different RISCs, with the exception of Na2S2O3. Additionally, it could fix CO2 using the incomplete CBB cycle. However, it did not have the capacity to oxidize iron and fix nitrogen. We also constructed the first high-quality metabolic model for Acidithiobacillus Ameehan. The model is comprised of 744 genes, 1,374 metabolites, and 1,377 reactions that are distributed among the extracellular, periplasmic, and cytoplasmic compartments, which allowed us to investigate the unique sulfur oxidation pathways and the interconnected carbon fixation pathways of the strain. Through rigorous validation against experimental data and existing literature, we observed a high degree of agreement between the model and experimental results (88.7% agreement with experimental Biolog data). The model showed five potential sulfur oxidation pathways for Acidithiobacillus Ameehan using sulfur as a substrate. The optimal sulfur oxidation pathway showed the highest ATP production rate of 14.81 mmol/gDW/h and NADH/NADPH production rate of 5.76 mmol/gDW/h, with CO2 consumption of 1.575 mmol/gDW/h and sulfur consumption of 1.5 mmol/gDW/h. By analyzing the commonalities among these sulfur oxidation pathways, we found two crucial genes, OMP and SQR, which played critical roles in cellular sulfur oxidation and significantly enhanced sulfur oxidation efficiency. These findings provided valuable insights into the growth and metabolism of various substances by Acidithiobacillus Ameehan. Moreover, the constructed model serves as a convenient platform for future engineering efforts aimed at modifying the strain.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary material. The data presented have been deposited in GitHub: https://github.com/wupeng1998/Acidithiobacillus-Ameehan.
Author contributions
PW: Data curation, Formal Analysis, Methodology, Writing – original draft. QY: Formal Analysis, Methodology, Writing – original draft, Funding acquisition, Writing – review & editing. TC: Formal Analysis, Writing – original draft, Data curation. YH: Formal Analysis, Investigation, Methodology, Writing – original draft. WZ: Data curation, Formal Analysis, Investigation, Writing – original draft. XL: Formal Analysis, Writing – original draft, Methodology. LW: Formal Analysis, Writing – original draft, Data curation, Investigation. JC: Formal Analysis, Investigation, Writing – original draft, Funding acquisition. QH: Investigation, Writing – original draft, Data curation, Methodology. YG: Data curation, Investigation, Writing – original draft, Formal Analysis. XZ: Data curation, Formal Analysis, Writing – original draft. FL: Project administration, Resources, Writing – review & editing. JW: Project administration, Writing – review & editing, Funding acquisition, Methodology. HM: Funding acquisition, Project administration, Writing – review & editing. ZH: Funding acquisition, Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Key Research and Development Program of China (2018YFA0902200), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDA28030301 and XDB0480000), the Tianjin Natural Science Foundation (20JCYBJC01220), the Tianjin Synthetic Biotechnology Innovation Capacity Improvement Project (TSBICIP-IJCP-001 and TSBICIP-PTJS-001), the Science and Technology Partnership Program of Ministry of Science and Technology (KY202001017), National Nature Science Foundation of China (32100035), China Postdoctoral Science Foundation (2021M693351), and Ministry of Science of China and Youth Innovation Promotion Association CAS (292023000018).
Acknowledgments
The author would like to thank Yan Zhu (Tianjin Institute of Industrial Biotechnology, Chinese Academy of Sciences), and reviewers and editors for their constructive suggestions.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1277847/full#supplementary-material
References
Alex, A., and Antunes, A. (2015). Whole genome sequencing of the symbiont Pseudovibrio sp from the intertidal marine sponge Polymastia penicillus revealed a gene repertoire for host-switching permissive lifestyle. Genome Biol. Evol. 7, 3022–3032. doi: 10.1093/gbe/evv199
Ang, K. S., Lakshmanan, M., Lee, N. R., and Lee, D. Y. (2018). Metabolic modeling of microbial community interactions for health, environmental and biotechnological applications. Curr. Genomics 19, 712–722. doi: 10.2174/1389202919666180911144055
Antoniewicz, M. R. (2021). A guide to metabolic flux analysis in metabolic engineering: methods, tools and applications. Metab. Eng. 63, 2–12. doi: 10.1016/j.ymben.2020.11.002
Arahal, D. R. (2014). Whole-genome analyses: average nucleotide identity. New Approach. Prokaryot. System. 41, 103–122. doi: 10.1016/bs.mim.2014.07.002
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Beard, S., Paradela, A., Albar, J. P., and Jerez, C. A. (2011). Growth of Acidithiobacillus ferrooxidans ATCC 23270 in thiosulfate under oxygen-limiting conditions generates extracellular sulfur globules by means of a secreted tetrathionate hydrolase. Front. Microbiol. 2:79. doi: 10.3389/fmicb.2011.00079
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Bryant, R. D., Mcgroarty, K. M., Costerton, J. W., and Laishley, E. J. (1983). Isolation and characterization of a new acidophilic Thiobacillus species (Thiobacillus-Albertis). Can. J. Microbiol. 29, 1159–1170. doi: 10.1139/m83-178
Campodonico, M. A., Vaisman, D., Castro, J. F., Razmilic, V., Mercado, F., Andrews, B. A., et al. (2016). Acidithiobacillus ferrooxidans's comprehensive model driven analysis of the electron transfer metabolism and synthetic strain design for biomining applications. Metab. Eng. Commun. 3, 84–96. doi: 10.1016/j.meteno.2016.03.003
Caspi, R., Billington, R., Keseler, I. M., Kothari, A., Krummenacker, M., Midford, P. E., et al. (2020). The MetaCyc database of metabolic pathways and enzymes—a 2019 update. Nucleic Acids Res. 48, D445–D453. doi: 10.1093/nar/gkz862
Chen, J. J., Liu, Y. L., Diep, P., and Mahadevan, R. (2022). Genetic engineering of extremely acidophilic Acidithiobacillus species for biomining: Progress and perspectives. J. Hazard. Mater. 438:129456. doi: 10.1016/j.jhazmat.2022.129456
Chen, L. X., Ren, Y. L., Lin, J. Q., Liu, X. M., Pang, X., and Lin, J. Q. (2012). Acidithiobacillus caldus sulfur oxidation model based on transcriptome analysis between the wild type and sulfur oxygenase reductase defective mutant. PLoS One 7:e39470. doi: 10.1371/journal.pone.0039470
Chung, C. A. H., Lin, D. W., Eames, A., and Chandrasekaran, S. (2021). Next-generation genome-scale metabolic modeling through integration of regulatory mechanisms. Meta 11:606. doi: 10.3390/metabo11090606
Ebrahim, A., Lerman, J. A., Palsson, B. O., and Hyduke, D. R. (2013). COBRApy: COnstraints-based reconstruction and analysis for Python. BMC Syst. Biol. 7:74. doi: 10.1186/1752-0509-7-74
Eccleston, M., and Kelly, D. P. (1978). Oxidation kinetics and chemostat growth kinetics of Thiobacillus ferrooxidans on tetrathionate and thiosulfate. J. Bacteriol. 134, 718–727. doi: 10.1128/jb.134.3.718-727.1978
Edgar, R. C. (2018). Updating the 97% identity threshold for 16S ribosomal RNA OTUs. Bioinformatics 34, 2371–2375. doi: 10.1093/bioinformatics/bty113
Falagán, C., and Johnson, D. B. (2016). Acidithiobacillus ferriphilus sp. nov., a facultatively anaerobic iron- and sulfur-metabolizing extreme acidophile. Int. J. Syst. Evol. Microbiol. 66, 206–211. doi: 10.1099/ijsem.0.000698
Falagan, C., Moya-Beltran, A., Castro, M., Quatrini, R., and Johnson, D. B. (2019). Acidithiobacillus sulfuriphilus sp. nov.: an extremely acidophilic sulfur-oxidizing chemolithotroph isolated from a neutral pH environment. Int. J. Syst. Evol. Microbiol. 69, 2907–2913. doi: 10.1099/ijsem.0.003576
Fang, X., Lloyd, C. J., and Palsson, B. O. (2020). Reconstructing organisms in silico: genome-scale models and their emerging applications. Nat. Rev. Microbiol. 18, 731–743. doi: 10.1038/s41579-020-00440-4
Ferreira, P., Fernandes, P. A., and Ramos, M. J. (2022). The archaeal non-heme iron-containing sulfur oxygenase reductase. Coord. Chem. Rev. 455:214358. doi: 10.1016/j.ccr.2021.214358
Gevers, D., Cohan, F. M., Lawrence, J. G., Spratt, B. G., Coenye, T., Feil, E. J., et al. (2005). Re-evaluating prokaryotic species. Nat. Rev. Microbiol. 3, 733–739. doi: 10.1038/nrmicro1236
Gu, C. D., Kim, G. B., Kim, W. J., Kim, H. U., and Lee, S. Y. (2019). Current status and applications of genome-scale metabolic models. Genome Biol. 20:121. doi: 10.1186/s13059-019-1730-3
Han, Y. F., Ma, X. M., Zhao, W., Chang, Y. K., Zhang, X. X., Wang, X. B., et al. (2013). Sulfur-oxidizing bacteria dominate the microbial diversity shift during the pyrite and low-grade pyrolusite bioleaching process. J. Biosci. Bioeng. 116, 465–471. doi: 10.1016/j.jbiosc.2013.04.012
Hedrich, S., and Johnson, D. B. (2013a). Acidithiobacillus ferridurans sp nov., an acidophilic iron-, sulfur- and hydrogen-metabolizing chemolithotrophic gammaproteobacterium. Int. J. Syst. Evol. Microbiol. 63, 4018–4025. doi: 10.1099/ijs.0.049759-0
Hedrich, S., and Johnson, D. B. (2013b). Aerobic and anaerobic oxidation of hydrogen by acidophilic bacteria. FEMS Microbiol. Lett. 349, 40–45. doi: 10.1111/1574-6968.12290
Hold, C., Andrews, B. A., and Asenjo, J. A. (2009). A stoichiometric model of Acidithiobacillus ferrooxidans ATCC 23270 for metabolic flux analysis. Biotechnol. Bioeng. 102, 1448–1459. doi: 10.1002/bit.22183
Hunter, J. D. (2007). Matplotlib: a 2D graphics environment. Comput. Sci. Eng. 9, 90–95. doi: 10.1109/MCSE.2007.55
Huse, S. M., Dethlefsen, L., Huber, J. A., Welch, D. M., Relman, D. A., and Sogin, M. L. (2008). Exploring microbial diversity and taxonomy using SSU rRNA hypervariable tag sequencing. PLoS Genet. 4:e1000255. doi: 10.1371/journal.pgen.1000255
Jain, C., Rodriguez-R, L. M., Phillippy, A. M., Konstantinidis, K. T., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Janiczek, O., Zemanova, J., and Mandl, M. (2007). Purification and some properties of thiosulfate dehydrogenase from Acidithiobacillus ferrooxidans. Prep. Biochem. Biotechnol. 37, 101–111. doi: 10.1080/10826060701199015
Jeong, J., Yun, K., Mun, S., Chung, W. H., Choi, S. Y., Nam, Y. D., et al. (2021). The effect of taxonomic classification by full-length 16S rRNA sequencing with a synthetic long-read technology. Sci. Rep. 11:1727. doi: 10.1038/s41598-020-80826-9
Johnson, D. B. (1998). Biodiversity and ecology of acidophilic microorganisms. FEMS Microbiol. Ecol. 27, 307–317. doi: 10.1016/S0168-6496(98)00079-8
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kelly, D. P., and Wood, A. P. (2000). Reclassification of some species of Thiobacillus to the newly designated genera Acidithiobacillus gen. nov., Halothiobacillus gen. nov. and Thermithiobacillus gen. nov. Int. J. Syst. Evol. Microbiol. 50, 511–516. doi: 10.1099/00207713-50-2-511
Klindworth, A., Pruesse, E., Schweer, T., Peplies, J., Quast, C., Horn, M., et al. (2013). Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 41:e1. doi: 10.1093/nar/gks808
Koch, T., and Dahl, C. (2018). A novel bacterial sulfur oxidation pathway provides a new link between the cycles of organic and inorganic sulfur compounds. ISME J. 12, 2479–2491. doi: 10.1038/s41396-018-0209-7
Kumar, M., Ji, B., Zengler, K., and Nielsen, J. (2019). Modelling approaches for studying the microbiome. Nat. Microbiol. 4, 1253–1267. doi: 10.1038/s41564-019-0491-9
Kumar, P., Jyoti, B., Kumar, A., and Paliwal, A. (2019). “Chapter 8—biotechnological and microbial standpoint cahoot in bioremediation” in Smart Bioremediation Technologies. ed. P. Bhatt (Smart Bioremediation Technologies Academic Press), 137–158.
Kupka, D., Rzhepishevska, O. I., Dopson, M., Lindstrom, E. B., Karnachuk, O. V., and Tuovinen, H. (2007). Bacterial oxidation of ferrous iron at low temperatures. Biotechnol. Bioeng. 97, 1470–1478. doi: 10.1002/bit.21371
Larsen, P., Hamada, Y., and Gilbert, J. (2012). Modeling microbial communities: current, developing, and future technologies for predicting microbial community interaction. J. Biotechnol. 160, 17–24. doi: 10.1016/j.jbiotec.2012.03.009
Lewis, N. E., Hixson, K. K., Conrad, T. M., Lerman, J. A., Charusanti, P., Polpitiya, A. D., et al. (2010). Omic data from evolved E-coli are consistent with computed optimal growth from genome-scale models. Mol. Syst. Biol. 6:390. doi: 10.1038/msb.2010.47
Lieven, C., Beber, M. E., Olivier, B. G., Bergmann, F. T., Ataman, M., Babaei, P., et al. (2020). MEMOTE for standardized genome-scale metabolic model testing. Nat. Biotechnol. 38, 272–276. doi: 10.1038/s41587-020-0446-y
Luo, J., Yuan, Q., Mao, Y., Wei, F., Zhao, J., Yu, W., et al. (2022). Reconstruction of a genome-scale metabolic network for Shewanella oneidensis MR-1 and analysis of its metabolic potential for bioelectrochemical systems. Front. Bioeng. Biotechnol. 10:913077. doi: 10.3389/fbioe.2022.913077
Mangold, S., Valdes, J., Holmes, D. S., and Dopson, M. (2011). Sulfur metabolism in the extreme acidophile Acidithiobacillus caldus. Front. Microbiol. 2:17. doi: 10.3389/fmicb.2011.00017
Meindl, B., and Templ, M. (2013). Analysis of commercial and free and open source solvers for the cell suppression problem. Trans. Data Priv. 6, 147–159. doi: 10.5555/2612167.2612169
Miyauchi, T., Kouzuma, A., Abe, T., and Watanabe, K. (2018). Complete genome sequence of Acidithiobacillus ferridurans JCM 18981. Microbiol. Resour. Announc. 7, e01028–18. doi: 10.1128/MRA.01028-18
Monk, J. M., Lloyd, C. J., Brunk, E., Mih, N., Sastry, A., King, Z., et al. (2017). iML1515, a knowledgebase that computes Escherichia coli traits. Nat. Biotechnol. 35, 904–908. doi: 10.1038/nbt.3956
Moya-Beltran, A., Beard, S., Rojas-Villalobos, C., Issotta, F., Gallardo, Y., Ulloa, R., et al. (2021). Genomic evolution of the class Acidithiobacillia: deep-branching Proteobacteria living in extreme acidic conditions. ISME J. 15, 3221–3238. doi: 10.1038/s41396-021-00995-x
Nguyen, L. T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Norris, P. R., Falagan, C., Moya-Beltran, A., Castro, M., Quatrini, R., and Johnson, D. B. (2020). Acidithiobacillus ferrianus sp. nov.: an ancestral extremely acidophilic and facultatively anaerobic chemolithoautotroph. Extremophiles 24, 329–337. doi: 10.1007/s00792-020-01157-1
Oftadeh, O., Salvy, P., Masid, M., Curvat, M., Miskovic, L., and Hatzimanikatis, V. (2021). A genome-scale metabolic model of Saccharomyces cerevisiae that integrates expression constraints and reaction thermodynamics. Nat. Commun. 12:4790. doi: 10.1038/s41467-021-25158-6
Orth, J. D., Thiele, I., and Palsson, B. O. (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248. doi: 10.1038/nbt.1614
Overbeek, R., Begley, T., Butler, R. M., Choudhuri, J. V., Chuang, H. Y., Cohoon, M., et al. (2005). The subsystems approach to genome annotation and its use in the project to annotate 1000 genomes. Nucleic Acids Res. 33, 5691–5702. doi: 10.1093/nar/gki866
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Passi, A., Tibocha-Bonilla, J. D., Kumar, M., Tec-Campos, D., Zengler, K., and Zuniga, C. (2022). Genome-scale metabolic modeling enables in-depth understanding of big data. Meta 12:14. doi: 10.3390/metabo12010014
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U. S. A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Silverman, M. P., and Lundgren, D. G. (1959). Studies on the Chemoautotrophic Iron Bacterium Ferrobacillus-Ferrooxidans.2. Manometric Studies. J. Bacteriol. 78, 326–331. doi: 10.1128/jb.78.3.326-331.1959
Singh, P., Jain, K., Desai, C., Tiwari, O., and Madamwar, D. (2019). “Chapter 18—microbial community dynamics of extremophiles/extreme environment” in Microbial Diversity in the Genomic Era. eds. S. Das and H. R. Dash (Microbial Diversity in the Genomic Era Academic Press), 323–332.
Sriaporn, C., Campbell, K. A., Van Kranendonk, M. J., and Handley, K. M. (2021). Genomic adaptations enabling Acidithiobacillus distribution across wide-ranging hot spring temperatures and pHs. Microbiome 9:135. doi: 10.1186/s40168-021-01090-1
Talla, E., Hedrich, S., Mangenot, S., Ji, B. Y., Johnson, D. B., Barbe, V., et al. (2014). Insights into the pathways of iron- and sulfur-oxidation, and biofilm formation from the chemolithotrophic acidophile Acidithiobacillus ferrivorans CF27. Res. Microbiol. 165, 753–760. doi: 10.1016/j.resmic.2014.08.002
Tamura, K., Nei, M., and Kumar, S. (2004). Prospects for inferring very large phylogenies by using the neighbor-joining method. Proc. Natl. Acad. Sci. U. S. A. 101, 11030–11035. doi: 10.1073/pnas.0404206101
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tapia, P., Flores, F. M., Covarrubias, P. C., Acuna, L. G., Holmes, D. S., and Quatrini, R. (2012). Complete genome sequence of temperate bacteriophage AcaML1 from the extreme Acidophile Acidithiobacillus caldus ATCC 51756. J. Virol. 86, 12452–12453. doi: 10.1128/Jvi.02261-12
Thiele, I., and Palsson, B. O. (2010). A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121. doi: 10.1038/nprot.2009.203
Valdes, J., Ossandon, F., Quatrini, R., Dopson, M., and Holmes, D. S. (2011). Draft genome sequence of the extremely acidophilic biomining bacterium Acidithiobacillus thiooxidans ATCC 19377 provides insights into the evolution of the Acidithiobacillus genus. J. Bacteriol. 193, 7003–7004. doi: 10.1128/Jb.06281-11
Valdes, J., Pedroso, I., Quatrini, R., and Holmes, D. S. (2008). Comparative genome analysis of Acidithiobacillus ferrooxidans, A. Thiooxidans and A. caldus: insights into their metabolism and ecophysiology. Hydrometallurgy 94, 180–184. doi: 10.1016/j.hydromet.2008.05.039
Valdes, J., Quatrini, R., Hallberg, K., Dopson, M., Valenzuela, P. D. T., and Holmes, D. S. (2009). Draft genome sequence of the extremely acidophilic bacterium Acidithiobacillus caldus ATCC 51756 reveals metabolic versatility in the genus Acidithiobacillus. J. Bacteriol. 191, 5877–5878. doi: 10.1128/Jb.00843-09
Vandervalk, B. P., Yang, C., Xue, Z., Raghavan, K., Chu, J., Mohamadi, H., et al. (2015). Konnector v2.0: pseudo-long reads from paired-end sequencing data. BioMed Genom. 8:S1. doi: 10.1186/1755-8794-8-S3-S1
Wang, R., Lin, J. Q., Liu, X. M., Pang, X., Zhang, C. J., Yang, C. L., et al. (2018). Sulfur oxidation in the acidophilic autotrophic Acidithiobacillus spp. Front. Microbiol. 9:3290. doi: 10.3389/fmicb.2018.03290
Waskom, M. L. (2020). Seaborn: statistical data visualization. J. Open Source Softw. 6:3021. doi: 10.21105/joss.03021
Wen, J.-K., Chen, B.-W., Shang, H., and Zhang, G.-C. (2016). Research progress in biohydrometallurgy of rare metals and heavy nonferrous metals with an emphasis on China. Rare Metals 35, 433–442. doi: 10.1007/s12598-016-0739-y
Wragg, P., Randall, L., and Whatmore, A. M. (2014). Comparison of biolog GEN III MicroStation semi-automated bacterial identification system with matrix-assisted laser desorption ionization-time of flight mass spectrometry and 16S ribosomal RNA gene sequencing for the identification of bacteria of veterinary interest. J. Microbiol. Methods 105, 16–21. doi: 10.1016/j.mimet.2014.07.003
Wu, W., Pang, X., Lin, J. Q., Liu, X. M., Wang, R., Lin, J. Q., et al. (2017). Discovery of a new subgroup of sulfur dioxygenases and characterization of sulfur dioxygenases in the sulfur metabolic network of Acidithiobacillus caldus. PLoS One 12:e0183668. doi: 10.1371/journal.pone.0183668
Yang, Y., Huang, Y. R., Ren, A. Q., Wan, Y. L., and Liu, Y. (2023). Xylem development and phloem conductivity in relation to the stem mechanical strength of Paeonia lactiflora. J. Plant Physiol. 283:153963. doi: 10.1016/j.jplph.2023.153963
Yang, L. J., and Shao, Z. (2018). Advances in sulfur-oxidizing bacterial taxa and their sulfur oxidation pathways. Acta Microbiol Sin. 58, 191–201. doi: 10.13343/j.cnki.wsxb.20170138
Yang, L., Zhao, D., Yang, J., Wang, W. D., Chen, P., Zhang, S., et al. (2019). Acidithiobacillus thiooxidans and its potential application. Appl. Microbiol. Biotechnol. 103, 7819–7833. doi: 10.1007/s00253-019-10098-5
Yin, H. Q., Zhang, X., Li, X. Q., He, Z. L., Liang, Y. L., Guo, X., et al. (2014). Whole-genome sequencing reveals novel insights into sulfur oxidation in the extremophile Acidithiobacillus thiooxidans. BMC Microbiol. 14:179. doi: 10.1186/1471-2180-14-179
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., and Chun, J. (2017). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
You, X. Y., Guo, X., Zheng, H. J., Zhang, M. J., Liu, L. J., Zhu, Y. Q., et al. (2011). Unraveling the Acidithiobacillus caldus complete genome and its central metabolisms for carbon assimilation. J. Genet. Genomics 38, 243–252. doi: 10.1016/j.jgg.2011.04.006
Zhan, Y., Yang, M. R., Zhang, S., Zhao, D., Duan, J. G., Wang, W. D., et al. (2019). Iron and sulfur oxidation pathways of Acidithiobacillus ferrooxidans. World J. Microbiol. Biotechnol. 35:60. doi: 10.1007/s11274-019-2632-y
Zhang, Y., Zhang, S., Zhao, D., Ni, Y. Q., Wang, W. D., and Yan, L. (2020). Complete genome sequence of Acidithiobacillus ferrooxidans YNTRS-40, a strain of the ferrous Iron- and sulfur-oxidizing Acidophile. Microorganisms 8:2. doi: 10.3390/microorganisms8010002
Keywords: new sulfur-oxidizing bacteria, Acidithiobacillus Ameehan, genome-scale metabolic network model, pathways of cellular metabolism, constraint-based flux analysis
Citation: Wu P, Yuan Q, Cheng T, Han Y, Zhao W, Liao X, Wang L, Cai J, He Q, Guo Y, Zhang X, Lu F, Wang J, Ma H and Huang Z (2023) Genome sequencing and metabolic network reconstruction of a novel sulfur-oxidizing bacterium Acidithiobacillus Ameehan. Front. Microbiol. 14:1277847. doi: 10.3389/fmicb.2023.1277847
Edited by:
Yiman Qi, John Innes Center, United KingdomReviewed by:
Janaka N. Edirisinghe, Argonne National Laboratory (DOE), United StatesAnkita Chatterjee, National Chemical Laboratory (CSIR), India
Copyright © 2023 Wu, Yuan, Cheng, Han, Zhao, Liao, Wang, Cai, He, Guo, Zhang, Lu, Wang, Ma and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jingjing Wang, d2FuZ19qakB0aWIuY2FzLmNu; Hongwu Ma, bWFfaHdAdGliLmNhcy5jbg==; Zhiyong Huang, aHVhbmdfenlAdGliLmNhcy5jbg==
†These authors share first authorship