
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 21 December 2023
Sec. Microbial Symbioses
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1276438
This article is part of the Research Topic Insights in Microbial Symbioses: 2022/2023 View all 8 articles
 Nahui Olin Medina-Chávez1,2*
Nahui Olin Medina-Chávez1,2* Abigail Torres-Cerda3
Abigail Torres-Cerda3 Jeremy M. Chacón4
Jeremy M. Chacón4 William R. Harcombe1,2
William R. Harcombe1,2 Susana De la Torre-Zavala3
Susana De la Torre-Zavala3 Michael Travisano1,2,5
Michael Travisano1,2,5Microbial syntrophy, a cooperative metabolic interaction among prokaryotes, serves a critical role in shaping communities, due to the auxotrophic nature of many microorganisms. Syntrophy played a key role in the evolution of life, including the hypothesized origin of eukaryotes. In a recent exploration of the microbial mats within the exceptional and uniquely extreme Cuatro Cienegas Basin (CCB), a halophilic isolate, designated as AD140, emerged as a standout due to its distinct growth pattern. Subsequent genome sequencing revealed AD140 to be a co-culture of a halophilic archaeon from the Halorubrum genus and a marine halophilic bacterium, Marinococcus luteus, both occupying the same ecological niche. This intriguing coexistence hints at an early-stage symbiotic relationship that thrives on adaptability. By delving into their metabolic interdependence through genomic analysis, this study aims to uncover shared characteristics that enhance their symbiotic association, offering insights into the evolution of halophilic microorganisms and their remarkable adaptations to high-salinity environments.
Syntrophy, or cross-feeding, is widely distributed and occurs when a microbial strain consumes substrates produced by other microbial strains, promoting the growth of the cross-fed strain (Escalante et al., 2015). It is central in structuring microbial communities because most microorganisms are auxotrophic (Zengler and Zaramela, 2018), and cross-fed substrates provide essential resources that would be otherwise unavailable. Without syntrophic interactions, most microbial communities would collapse to relatively few prototrophic species. Syntrophic interactions were central in the evolutionary history of life, as eukaryotes are hypothesized to have evolved from a mutually beneficial syntrophy between an archaeon and a bacterium. Archaea and Bacteria are highly divergent, having long phylogenetically distinct evolutionary histories, and archaea are thought to be especially dependent upon syntrophic interactions (López-García and Moreira, 2019; López-García and Moreira, 2020). Even so, species from both domains share many features, size, morphology, movement, and defense mechanisms and were long thought to be one taxonomic group (Medina-Chávez and Travisano, 2022). Disentangling syntrophies between archaea and bacteria will shed light on current microbial communities and potentially on the evolutionary origins of eukaryotes.
Recent research has unveiled the remarkable diversity within microbial mats from the Cuatro Cienegas Basin (CCB) (Espinosa-Asuar et al., 2022), where similar to other extreme environments, the presence of archaea and extremophilic bacteria is prominent. CCB is an oasis in the Chihuahuan Desert in Coahuila, Mexico, characterized by its oligotrophic states, which involve phosphorus depletion, high salinity, high pH, and high radiation levels (Souza et al., 2008; Medina-Chávez et al., 2020). Within CCB, a plethora of microbial diversity has been described in previous reports (Bonilla-Rosso et al., 2012; Peimbert et al., 2012; Arocha-Garza et al., 2017). Due to its geographic isolation, species have evolved without interruption, resulting in a long and stable period of metabolic compatibility and adaptation for community assembly (Souza et al., 2018). Notably, one halophilic isolate designated as AD140 exhibits a distinctive growth pattern on agar plates. First, yellow-colored colonies grow over a two-week span, followed by red colonies 4 weeks after the initial striking. This is unique since the red colonies do not grow on top of the agar media but rather directly on top the yellow biomass. This is in contrast to other isolates in co-culture where they do not grow in physical contact with each other. Through genome sequencing, strain AD140 was identified as a co-culture between a halophilic archaeon from the Halorubrum genus (red colonies) and a marine halophilic bacterium, Marinococcus luteus (yellow colonies). Despite numerous attempts over the years to separate both strains in axenic cultures, with the use of antibiotics, serial-dilution to extinction, spent media, different media composition, varying ph or temperature, neither microorganism thrive in isolation. These two lineages occupy the same abiotic ecological niche and have developed comparable mechanisms of adaptation and stress response. In addition to thriving in salt-saturated conditions, they demonstrate resilience during periods of desiccation and fluctuations in other environmental factors (Medina-Chávez et al., 2023). The co-occurrence of these two strains as a sufficient symbiont interaction in laboratory settings led us to propose it as an early-stage symbiosis, where their symbiotic continuum depends on their adaptability in an eco-evolutionary framework.
The objective of this study was to explore the genomic features of a halophilic archaea and marine bacteria consortium through comparative genomic analysis, with the aim of understanding the metabolic co-dependence that shapes the early stages of symbiosis. Symbiosis through metabolic cross-feeding is enhanced by genome features such as genomic plasticity, horizontal gene transfer events, symbiosis-specific genes, and regulatory mechanisms, leading to changes in the organization of functional elements in the genome (Hentschel et al., 2000; Ochman and Moran, 2001; Moran, 2007; Juhas et al., 2009). Through comprehensive genomic analysis and metabolic network prediction, we describe the characteristics of these two microorganisms and their close interaction based on a set of shared traits. These findings have the potential to illuminate the convergent evolution of halophilic microorganisms and provide insights into the genetic basis of their adaptations to high-salinity environments.
Environmental sampling of water and sediment was collected in Cuatro Cienegas, Coahuila (26° 49′ 41.7″ N, 102° 01′ 28.7″ O), with ~10 cm of depth in sterile Falcon tubes.
For the first stage of culture (primary isolation) AM2 culture media was designed and used. AM2 contains ([Per liter]): 21.2 g bacteriological agar, 2.4 g dextrose, 5 g peptone (Oxoid), 5 g yeast extract, 3 g Na3C6H5O7, 0.2 g CaCl2 2H2O, 50 g MgCl2 6H2O, 20 g MgSO4 7H2O, 2 g KCl, 250 g NaCl. For the second stage of isolation, ATCC 213 media was used ([Per liter]): 25 g bacteriological agar, 3.1 g tryptone, 12.5 g yeast extract, 0.4 g CaCl2 2H2O, 50 g MgCl2 6H2O, 12.5 g MgSO4 7H2O, 6.2 g KCl, 250 g NaCl. Both media were adjusted at pH = 7.4. Liquid and agar media were used, and incubation was held at 28°C.
Samples of 1 mL from the cultures were extracted aseptically from the tubes to assess growth via optical density (O.D.) measurements at 600 nm using an Agilent Technologies Cary 100 UV–Visible Model G9821A spectrophotometer. This experiment was replicated three times for accuracy.
To assess the interdependency between archaea and bacteria, we conducted an antibiotic susceptibility test. We initiated the test by inoculating 100 μL of co-culture into 10 glass tubes with 6 mL of 213 media and 6 μL of ampicillin (final concentration 50 mg/mL, Roche), active against M. luteus, but not Halorubrum sp. followed by incubation at 28° C, 250 rpm, during 5 weeks. For positive controls we maintained another 10 tubes of 213 media without ampicillin and 100 μL of AD140 culture, as negative controls we used five media tubes with no antibiotic and without inoculum.
To test for biofilm production, we grew the co-culture at 28° C, using 6 mL of liquid 213 media and a 200 μL inoculum, which corresponds of approximately 5 × 106 cells, until full culture growth. We did not used the standard 48 or 96-well assay plate due to the large amount of salt in the media, the long period of static state and the small volume commonly used in well-assay plates, leading to evaporation of the media and precipitation of salts.
To identify the isolated strains at the species taxonomic level, genomic DNA extraction was performed. Phenol-Chloroform (1:1) protocol was used with the next modifications: biomass pellet was obtained from agar plates and resuspended in 400 μL phenol-chloroform solution and 400 μL of QTP lysis buffer which contained [Per liter] (40 mL Triton X-100, 100 mL SDS 20%, 20 mL NaCl 5 M, 10 mL Tris 2 M pH = 8,7 mL EDTA pH = 8, and milliQ water was added up to 10 mL), into 2 mL Eppendorf tubes with 0.2 μL of glass beads (0.1 mm Ø). The tubes were shaken in a rotational disruptor (MagNA Lyser Instrument, Roche Diagnostics) for 1 min at 5,600 rpm and incubated for 1 min in ice (4° C, 3× times). For DNA precipitation, the liquid phase was taken and placed into a new 2 mL Eppendorf tube adding 25 μL sodium acetate, 95% ethanol to the top, and incubated at −20° C overnight. The tubes were centrifugated recovering the pellet and washing it twice with 70% ethanol. DNA was resuspended in 30 μL of TE buffer + RNAse and stored at −20° C. Sequencing was done by CINVESTAV-LANGEBIO Irapuato, Mexico, using Illumina MiSeq technology 2 × 300 with a 150X depth.
Read quality control was performed with FastQC v0.11.9 (Andrews, 2010). The reads were cleaned and trimmed using Trimmomatic v0.33 (Bolger et al., 2014) using the parameters as it follows: trimmomatic-PE-phred33 input_forward.fq.gz input_reverse.fq.gz output_forward_paired.fq.gz output_forward_unpaired.fq.gz output_reverse_paired.fq.gz output_reverse_unpaired.fq.gz ILLUMINACLIP:TruSeq3-PE.fa:2:30:10 LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36. Once cleaned and trimmed, the sequences were assembled de novo with MetaSpades v.3.15.5 (Nurk et al., 2017) using 21, 31, 51, 61, and 71 k-mers. The quality analysis of the assembly was performed using MetaQUAST v5.2.0 (Mikheenko et al., 2015).
To retrieve individual bins, the assembly obtained with MetaSpades was analyzed with MaxBin2 v2.2.7 (Wu et al., 2014). To make sure it was not a contamination, we check genome quality using CheckM v.2 using default parameters. Genomic fasta sequences and abundance values from Halorubrum sp. and Marinococcus luteus were retrieved.
Genomes of Halorubrum sp. and Marinococcus luteus were annotated using Prokka v1.14.6 software (Seemann, 2014), establishing in the parameters the use of an archaea genome data bank from RefSeq, NCBI (see full annotation in Supplementary Tables 8, 9). Also, RASTtkv1.3.0 was used for additional annotation (Brettin et al., 2015).
To predict biosynthetic clusters, we used antiSMASH 7.0 with default parameters as it follows: antismash --cb-general --cb-knownclusters --cb-subclusters --asf --pfam2go --smcog-trees --tigrfam --output-dir antismash/prokka_06092021.merged.gbk.Genomic islands prediction was performed using GYPSy, and AlienHunter. GYPSy algorithm compares the genome of interest with a reference genome, considering only the regions that are absent in the reference genome; as for Alien hunter, it employs the Interpolated Variable Order Motifs (IVOMs) method to forecast potential HGT regions based solely on sequence composition, without needing a reference genome.
The protein sequences retrieved from Prokka were analyzed with BlastKOALA (Kanehisa et al., 2016) to obtain the KEGG Orthology assignation (KO) of each gene. The KO output was introduced into KEGG Mapper Reconstruct (Kanehisa et al., 2022) to annotate the enzymes and metabolic pathways from both genomes.
Individual genome-scale metabolic models were generated using the CarveMe pipeline (Machado et al., 2018), using the.fasta files of the protein sequences obtained from Prokka annotation and media composition used in the culture.
Metabolic requirements of each genome-scale metabolic model were determined by applying flux-balance analysis using the python tool COBRApy (Ebrahim et al., 2013). To determine basic needs, each model was initially provided with all nutrients the model is capable of taking up. Then each nutrient was removed one-at-a-time, and nutrients whose removal prevented growth were deemed essential.
To predict potential minimal media, we used the “minimal_medium” function in COBRApy, set to minimize the number of nutrients required (as opposed to minimizing total uptake flux). We did the 1e6 times to try to find all possible minimal media (i.e., we used the argument minimize_components = 1,000,000).
To test whether each model had the potential to synthesize the nutrients the other model required environmentally, we used flux balance analysis with production of the metabolite in question as the objective function. Specifically, a model was provided with one of the media identified in the minimal media analysis. Then, a sink reaction was added to the model which could draw one specific metabolite out of the intracellular space. This was done one-at-a-time for each non-ion needed by the other species. Then, flux-balance analysis was used with the sink reaction as the objective function. If the solution >1e-6, we state that the model could produce this nutrient.
Complete genome data is available at NCBI BioProject PRJNA1003106.
When cultivated in liquid media, it exhibits rapid growth at 28°C with agitation at 250 rpm, in 18–20 days. The AD140 consortium shows optimal growth in 213 and M2 media, reaching a maximum at day 29 and 31, respectively, after which it entered stationary phase (Figure 1). On agar plates, circular colonies with a yellow color begin to emerge after 14 days, subsequently identified as the bacterium, Marinococcus luteus. Over the course of 25–28 days, circular colonies with a red color appear atop the initial yellow colonies, which are identified as the archaeon, Halorubrum sp. (Figure 2).
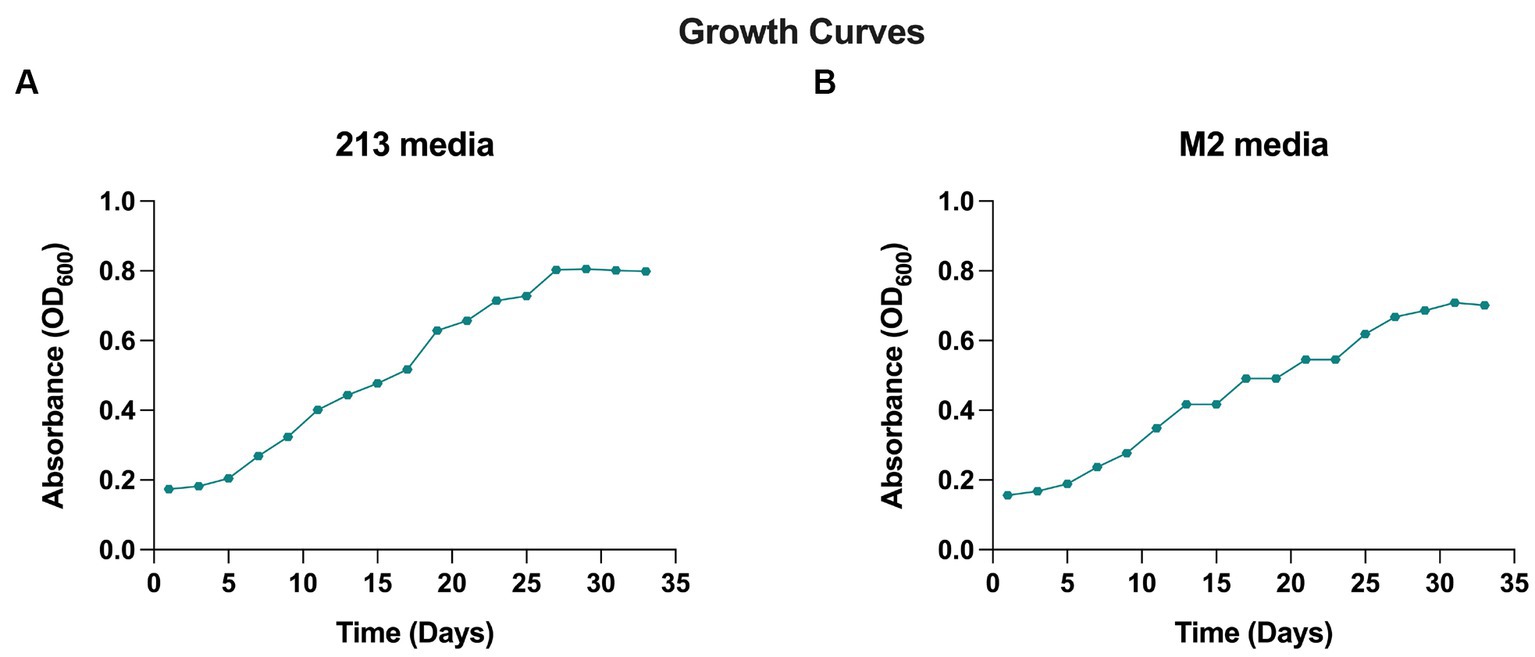
Figure 1. Growth curve of AD140 co-culture, in (A) 213 media and (B) M2 media (25% Salinity, temperature 28°C, pH = 7.4).
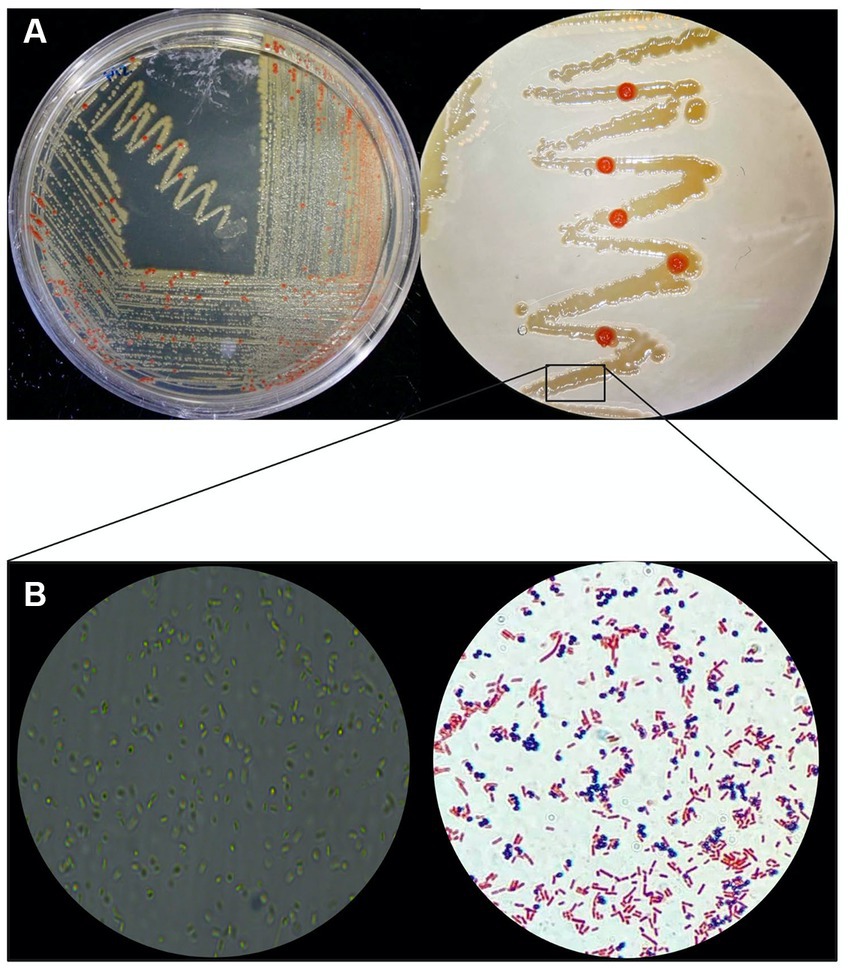
Figure 2. Phenotypic macro and microscopic characterization of AD140 co-culture. (A) Halorubrum sp. (red colonies) and Marinococcus luteus (yellow colonies) distinctive growing pattern on 213 media plates. (B) Microscopy and gram stain of the consortium (Eclipse Ci Microscope, 100× magnification), taken from a smear of the yellow biomass showing gram-negative rods and gram-positive cocci belonging to Halorubrum sp. and Marinococcus luteus, respectively. Created with BioRender.com.
To validate the mutualistic relationship, an antibiotic test was conducted making sure to target one of the microorganisms. The distinctive variances in the cell walls of archaea and bacteria allow for selective susceptibility to antibiotics. Ampicillin, a beta-lactam antibiotic, is widely used against gram-positive bacteria and has no effect on archaea. In the tubes designated for antibiotic exposure, either no visible growth or minimal growth was observed, declining afterwards. The absence of visible growth or minimal growth in these tubes indicates that the antibiotics tested successfully inhibited the proliferation of Marinococcus luteus. This outcome suggests that the microorganism is sensitive to the antibiotics used in the assay, as they effectively suppressed its growth or completely prevented its replication, but more importantly, Halorubrum sp. did not grow when the bacteria was eliminated from the culture. This shows that the propagation of Halorubrum is dependent from Marinococcus (Figure 3).

Figure 3. (A) Tubes containing ampicillin (1 μg/mL) show no growth to minimum up until 15 days and continue to decline afterwards. Halorubrum sp., does not grow while Marinococcus luteus is eliminated from the culture. While the tubes with no antibiotic developed as expected. (B) Biofilm formation was observed at the bottom of the culture tube during the first 15 day of culture under static conditions, meanwhile aggregation and attachment of the biofilm was observed from day 22 to day 32 under agitation regime.
Whole genome sequencing was performed to identify the halophilic isolate. The number of raw reads obtained after pair-end sequencing was 4,489,067 reads. The first assembly draft of the AD140 isolate obtained from Spades, resulted in a 7,796,468 bp genome with 2,729 contigs. The GC% average was found to be 58.81%, and the functional annotation revealed 7,860 coding genes, 147 tRNAs and 9 rRNAs. Compared to other haloarchaea genome sizes, we observed that the larger genome size of the AD140 genome was not typical for this taxon. The significant increase of ~2,310,000 bp, the abundance of genes, and the unique phenotypic growth led us to hypothesize that the AD140 culture could hold more than one specie. To confirm the presence of several microorganisms in the same sample, we performed assembly using MetaSpades and subsequently utilized MaxBin for automatic binning. The process yielded two distinct genomes, confirming the identity of Halorubrum sp. an archaeon, and Marinococcus luteus a bacterium. The presence of both members was supported by genome quality check, showing the Archaea genome had a completeness of 99.07% and contamination ratio of 4.48, while the Bacteria, showed a completeness 98.28% and contamination ratio of 0. The genomic features of both genomes are presented in Table 1.
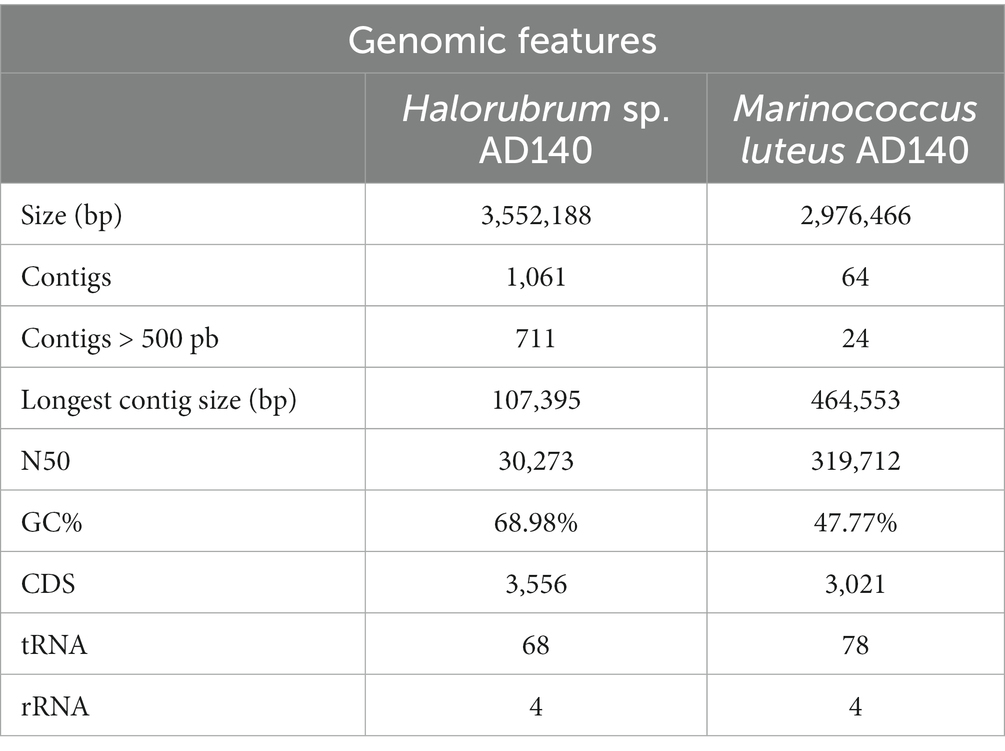
Table 1. Genomic features of Halorubrum sp. AD140 and Marinococcus luteus AD140.
Preliminary functional annotation was conducted using the RAST Server: Rapid Annotation using Subsystems Technology (Brettin et al., 2015). The annotation process involved the identification of protein-encoding, rRNA and tRNA genes through homology, and assigning these proteins to specific metabolic subsystems (Figure 4). The construction of these models was performed for both genomes. Complete functional annotation using prokka v1.14.6, allow to explore gene regulatory systems and transposable elements associated with Horizontal Gene Transfer (Table 2).
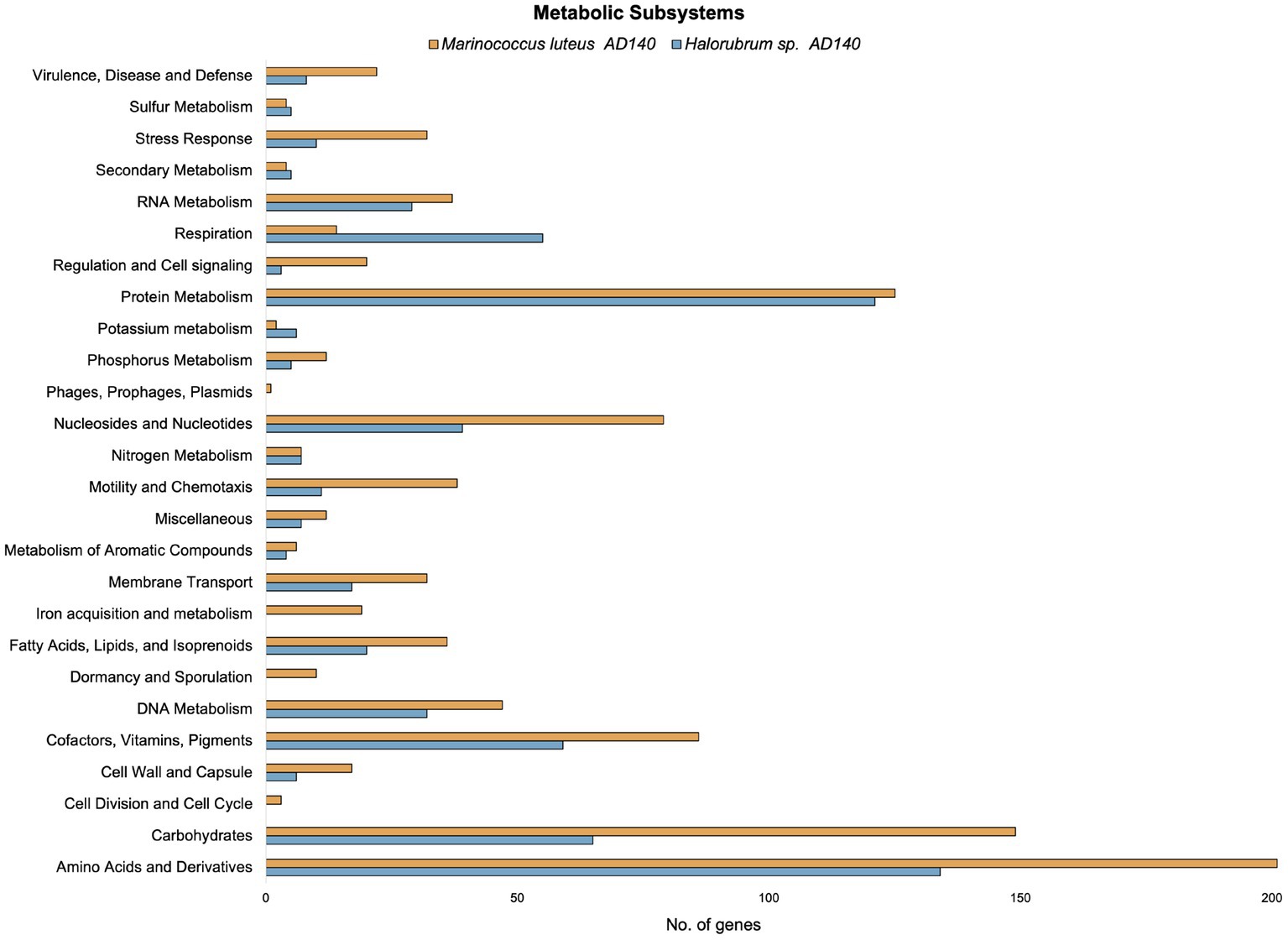
Figure 4. Comparison of the number of genes in different metabolic subsystems based on RAST annotation of Marinococcus luteus (yellow) and Halorubrum sp. (blue).
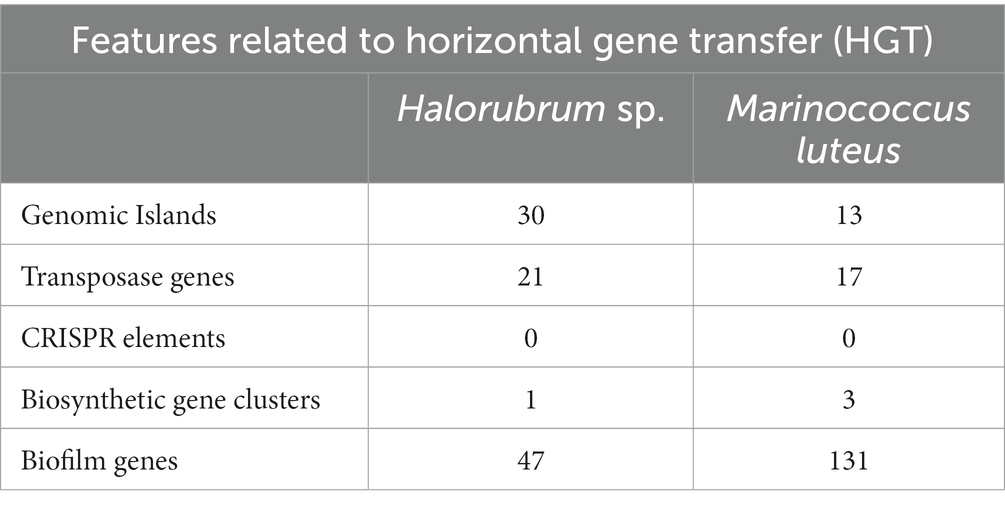
Table 2. Features related to Horizontal Gene Transfer (HGT) in Halorubrum sp. AD140 and Marinococcus luteus AD140.
A comprehensive analysis revealed the presence of 30 genomic islands within the genome of Halorubrum sp., while M. luteus exhibited 13 genomic islands. These islands were predominantly located in regions characterized by low %GC content, indicating their likely acquisition from other organisms. Notably, these islands harbored genes associated with the mobilization of genomic material, such as integrases, recombinases, transposases, and exonucleases. In the case of Halorubrum sp., mobile elements were specifically identified within islands 6, 8, 9, 18, 20, 22, and 29 (Table 3), whereas for M. luteus, were identified in islands 1, 4, 5, 6, 7, 9, 10, and 13 (Table 4). Complete genomic islands descriptions can be found in Supplementary Tables 3, 4, respectively.
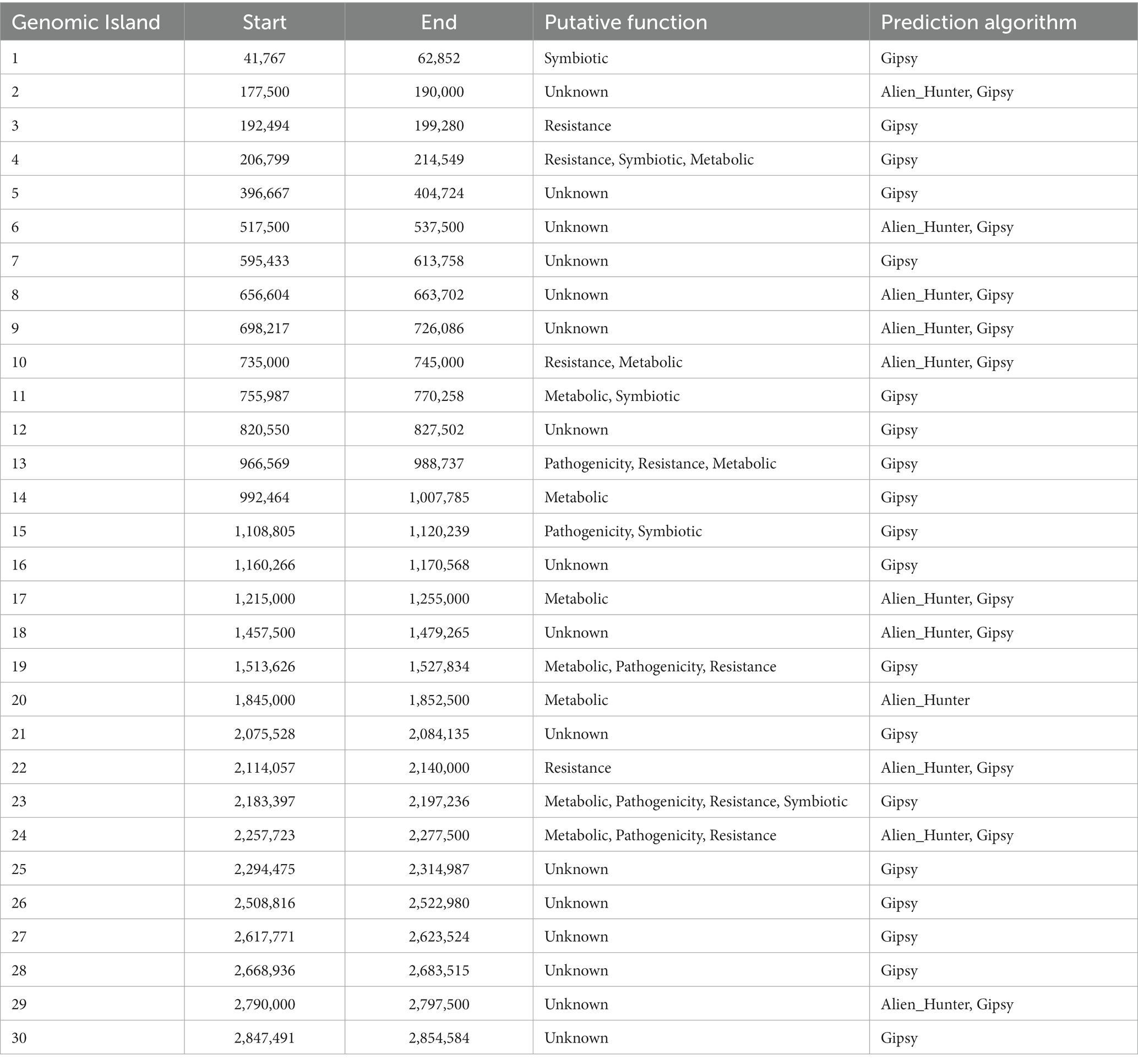
Table 3. Description of Genomic Islands of Halorubrum sp. AD140.
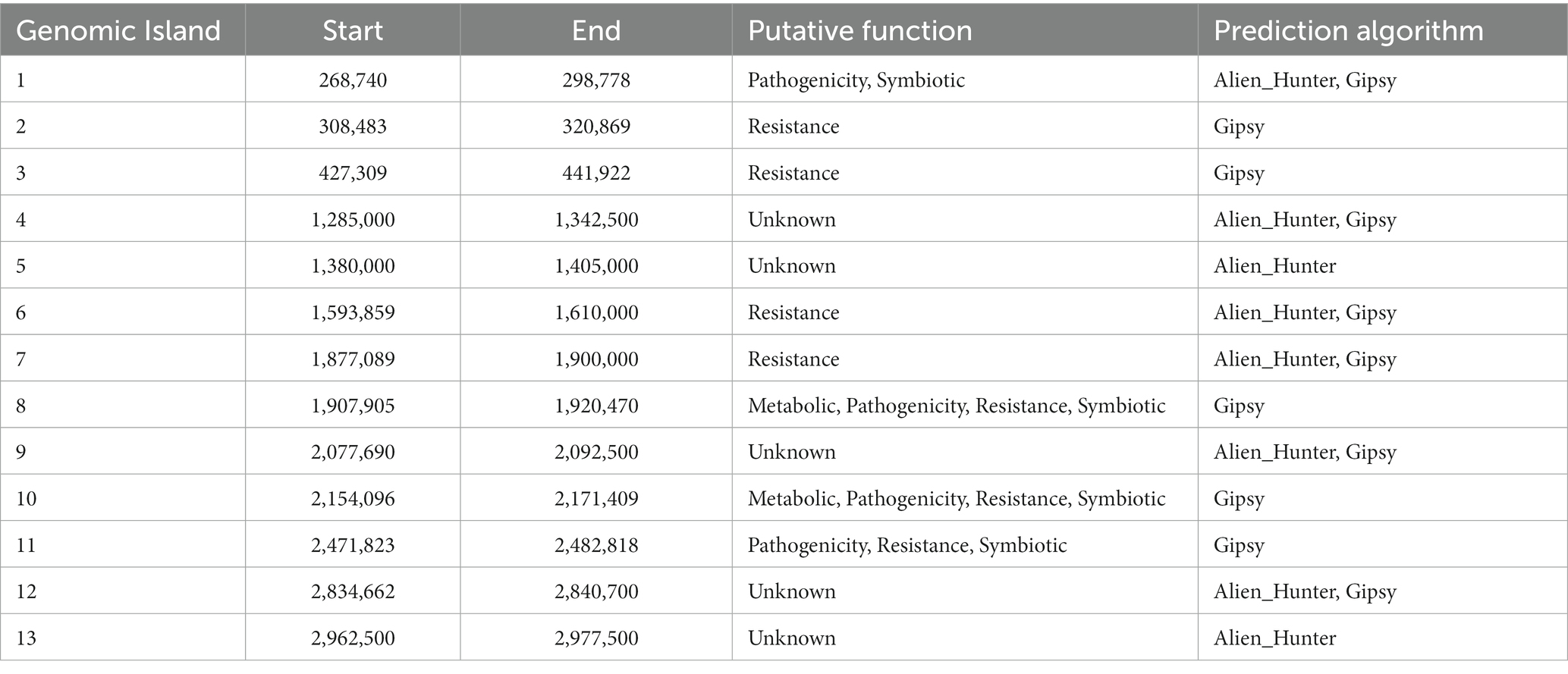
Table 4. Description of Genomic Islands of Marinococcus luteus sp. AD140.
Phage infection was identified as the probable mechanism of acquisition for specific islands, namely islands 6, 8, and 22 in Halorubrum, and island 4 in M. luteus, as these islands contained genes encoding phage proteins. Moreover, both microorganisms have genomic islands containing resistance genes. The archaeon, Halorubrum sp., possess islands associated with cadmium resistance (island 3) and arsenic resistance (island 24). Furthermore, island 13 harbors genes encoding metallo-beta-lactamase, while island 22 contains genes for multidrug resistance proteins. Similarly, to Halorubrum sp., Marinococcus luteus, has an island (island 7) carrying an arsenic resistance gene. Additionally, it exhibits genes related to fluoroquinolone resistance in island 2.
In Halorubrum sp., several islands containing genes involved in metabolite synthesis were identified. For instance, island 4 is associated with succinoglycan synthesis, island 14 with thiazole synthesis, and island 20 with cisthiatonine synthesis. Additionally, islands with metabolic features were discovered, including island 10 (associated with amilase production), island 13 (related to thiaminase activity) and island 17 (involved in sulfur metabolism) On the other hand, M. luteus, genomic islands with genes related to metal metabolism were also identified. Specifically, island 8 contains genes related to iron metabolism, as well as genes encoding nitrous oxidoreductase, an enzyme involved in the production of nitrogen (N2) from nitrous oxide (N2O).
Biosynthetic gene clusters were identified with antiSMASH. Only one biosynthetic cluster was found in Halorubrum sp., while three clusters were found in Marinococcus luteus (Figure 5). Both species have a terpene biosynthesis clusters, noteworthy, the bacterium terpene cluster did not have transport related genes, but several additional biosynthetic genes, with a very distinctive arrangement. The terpene biosynthesis cluster in Halorubrum sp. contains a phytoenesynthase gene (HAD140_00435), three additional biosynthetic genes: rutB (isochorismatase), gph_2 (HAD hydrolase), and malE_1 (sugar-binding lipoprotein), and one transport-related gene: btuD_2 (phosphonate ABC transporter). While the terpene biosynthesis cluster in Marinococcus luteus also contains a phytoene synthase gene (crtB), the other genes in the cluster are different from the ones found in the terpene cluster in Halorubrum sp. The cluster in M. luteus does not have any transport genes and has six additional biosynthetic genes: pepT, MLAD140_01902, crtN_2, crtNc, yvaM, and pld1.
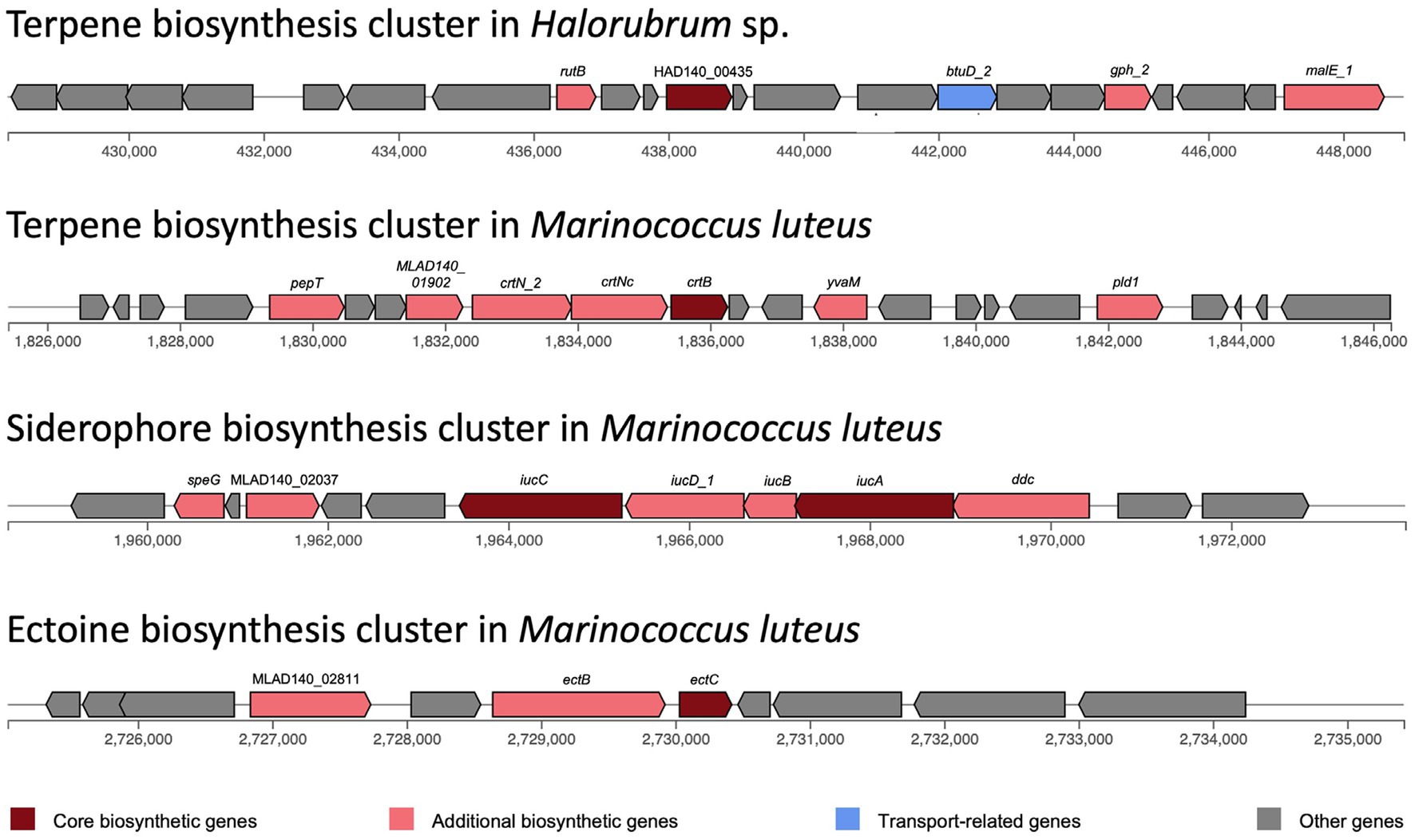
Figure 5. Biosynthesis clusters predicted in Halorubrum sp. and Marinococcus luteus AD140, using antiSMASH v6.0.0. Each block represents a gene.
The siderophore biosynthesis cluster in M. luteus contains two core biosynthesis genes: iucA and iucC. This cluster also has five additional biosynthetic genes: speG, MLAD140_02037, iucD_1, iucB and ddc. Moreover, an ectoine biosynthesis cluster was identified in M. luteus. This cluster has one core biosynthetic gene (ectC) and two additional biosynthetic genes: ectB and MLAD140_02811.
A total of 9 CRISPR elements were found in the genome of Halorubrum sp. using CRISPRCasFinder. Nevertheless, these elements were predicted with level 1 evidence, the lowest prediction level of CRISPRCasFinder. Complete features of predicted CRISPR elements are found in Supplementary Table 5. Meanwhile, Marinococcus luteus did not show any CRISPR element.
Biofilms found in hypersaline environments are found to hold archaea and bacteria thriving together. These biofilms facilitate genetic exchange, nutrient availability, and provide protection for microorganisms residing within them. The genome of Halorubrum sp. AD140 exhibits numerous biofilm gene homologs to Halorubrum lacusprofundi and Haloferax volcanii genomes. Among the queried genes, only HVO_2056 has no homolog in Halorubrum sp. AD140, which contributes to the biogenesis of the low-salt tetrasaccharide sugar withing the EPS operon (Kaminski et al., 2013). Genes involved in EPS synthesis include sugar and amino acid metabolizing enzymes such as glmU, glmS, dapH, arnB, iolG and wecC. Additionally, two genes were identified with the N-terminal domain of the type IV pili: HAD140_00545 and HAD140_02714. The presence of che genes, responsible for chemotaxis and regulation of archaella and flagella motility, was also observed. Among the genes related to archaella and flagella, flaI, flaC, flaH, flaJ, flaG, flaF and the archaellins flgA1 and flgA2 were identified. Furthermore, four S-layer genes involved in surface adhesion were found, as depicted in Figure 6. These genes collectively enable Halorubrum sp. AD140 to undergo biofilm formation, including surface adhesion, microcolony formation, and biofilm maturation. Finally, 4 S-layer genes were found, which participate in surface adhesion, indicated in blue in Figure 6. All these genes interact so that Halorubrum sp. AD140 can perform the process of biofilm formation, from surface adhesion to microcolony formation and biofilm maturation.
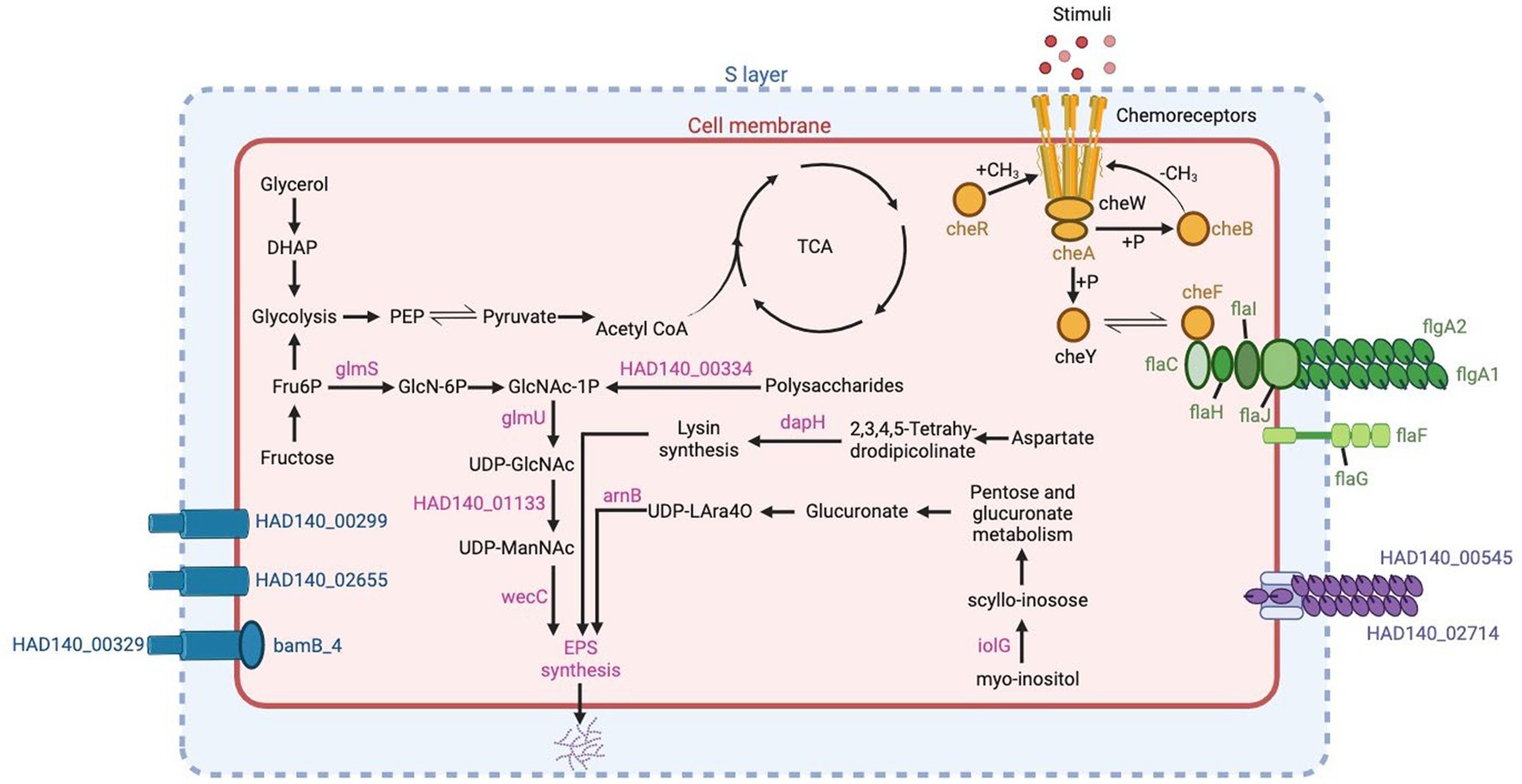
Figure 6. Genes related to biofilm production in Halorubrum sp. AD140 metabolic pathways. Blue: S-layer genes, pink: EPS synthesis genes, yellow: chemotaxis genes, green: archaella and flagella genes, purple: pili genes. Created with BioRender.com.
The genes homologs identified in Marinococcus luteus were based on previous annotations from different Halomonas and Marinobacter species. As expected, Marinococcus luteus does not have genes related to S-layer, and archaellin which are inner characteristics of Archaea domain. However, compared to Halorubrum sp. AD140, it contains ~64% more EPS biosynthesis genes, subdivided in nucleotide sugar synthesis, kps system, and ATP-binding cassette (transporters) genes, 74% more in chemotaxis and quorum sensing and ~ 80% more genes related to flagella. Marinococcus also possess similar genes related to flagella, although the operon component arrangement is different from the archaeon. Unlike Halorubrum, Marinococcus has five genes responsible for biofilm regulation, csrA, cpdA, uvrY and wspA. Detailed description of the prokka annotation followed by BLAST search performed to identify biofilm genes in Halorubrum sp. and Marinococcus lutes are found on Supplementary Tables 6, 7, respectively. Evaluation of biofilm formation of the AD140 co-culture was performed (Figure 3B), observing a continuous growth over time of an extracellular matrix at the bottom of the tube in static culture. Moreover, we observed biofilm formation and natural aggregation and attachment to the tube wall while the culture grows with agitation, visible at day 20.
We generated genome-scale metabolic models for each species, using the prokka-annotated genomes retrieved from maxbin. Since we do not know any defined media upon which either of these strains can grow independently, we did not gap-fill beyond the basic requirement that growth must be possible when all environmental metabolites the model can uptake are present, including the composition of the 213 and M2 media. We first used these models to predict essential nutrients for both species. As expected, each requires many of the same basic ions as Cl, K, Ca2, Mg2, Mn2, Cobalt2, Zn2, Cu2. Beyond these, there are differences in nutrient requirements. The models predict that Halorubrum and Marinococcus are each auxotrophic for different amino acids (glutamine and asparagine, respectively), that Marinococcus requires nicotinamide from the environment, and Halorubrum requires multiple environmental nutrients, including thiamin. Nonetheless, in the two functional annotations (prokka and RAST) of each microorganism, it was found in Halorubrum elements from a pathway to synthesize nicotinamide-D-ribonucleotide which is used by the bacteria as a precursor for N-ribosyl-nicotinamide synthesis. Conversely, Marinococcus, synthetizes thiamin monophosphate by coupling two components thiazole and pyrimidine in separate pathways, and becoming active as thiamin diphosphate with a thiamine phosphate kinase (Figure 7).
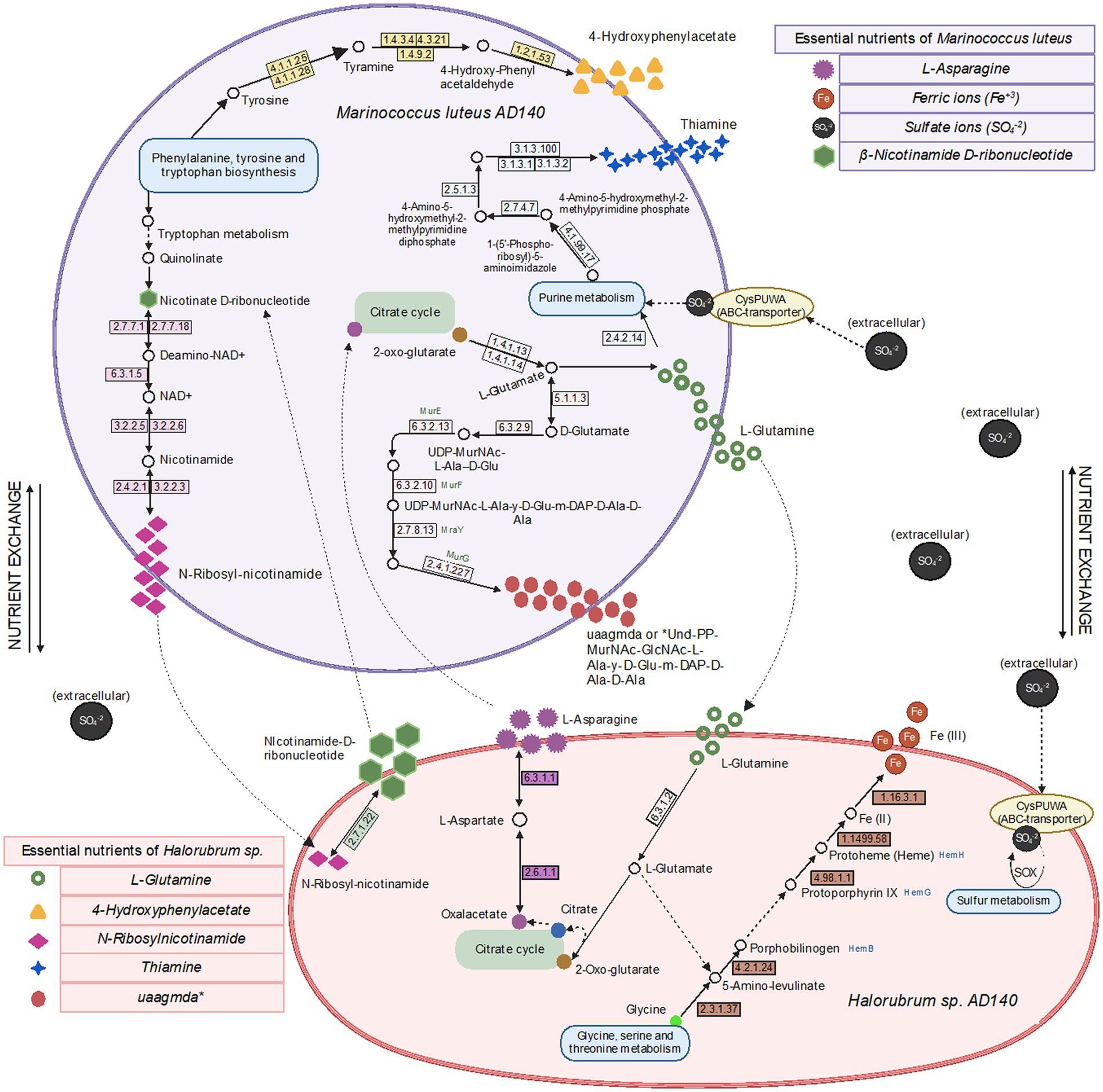
Figure 7. Metabolic pathways, metabolite exchange and essential nutrients between Halorubrum sp. and Marinococcus luteus model predicted using drop-out flux balance analysis. Extracellular Sulfate ions comes from one of the components of 213 and M2 media, MgSO4 7H2O, available in the culture environment. Created with BioRender.com.
Next, we predicted potential minimal media for each species, specifically aiming at minimizing the number of compounds, rather than minimizing the summed uptake of the compounds. Over 1e6 iterations, the algorithm identified only two alternative minimal media for Halorubrum, and four for Marinococcus. Halorubrum alternative minimal media were composed of differed minimal ways to uptake sugar (ribose or gluconate), phosphate (Sn-Glycero-3-phospho-1-inositol or glycerol-3-phosphate), and sulfate (sulfate or choline sulfate). Marinococcus’ alternative media were also composed in part of different ways to uptake an energy source, phosphate, and in one case, oxygen (Figure 8).
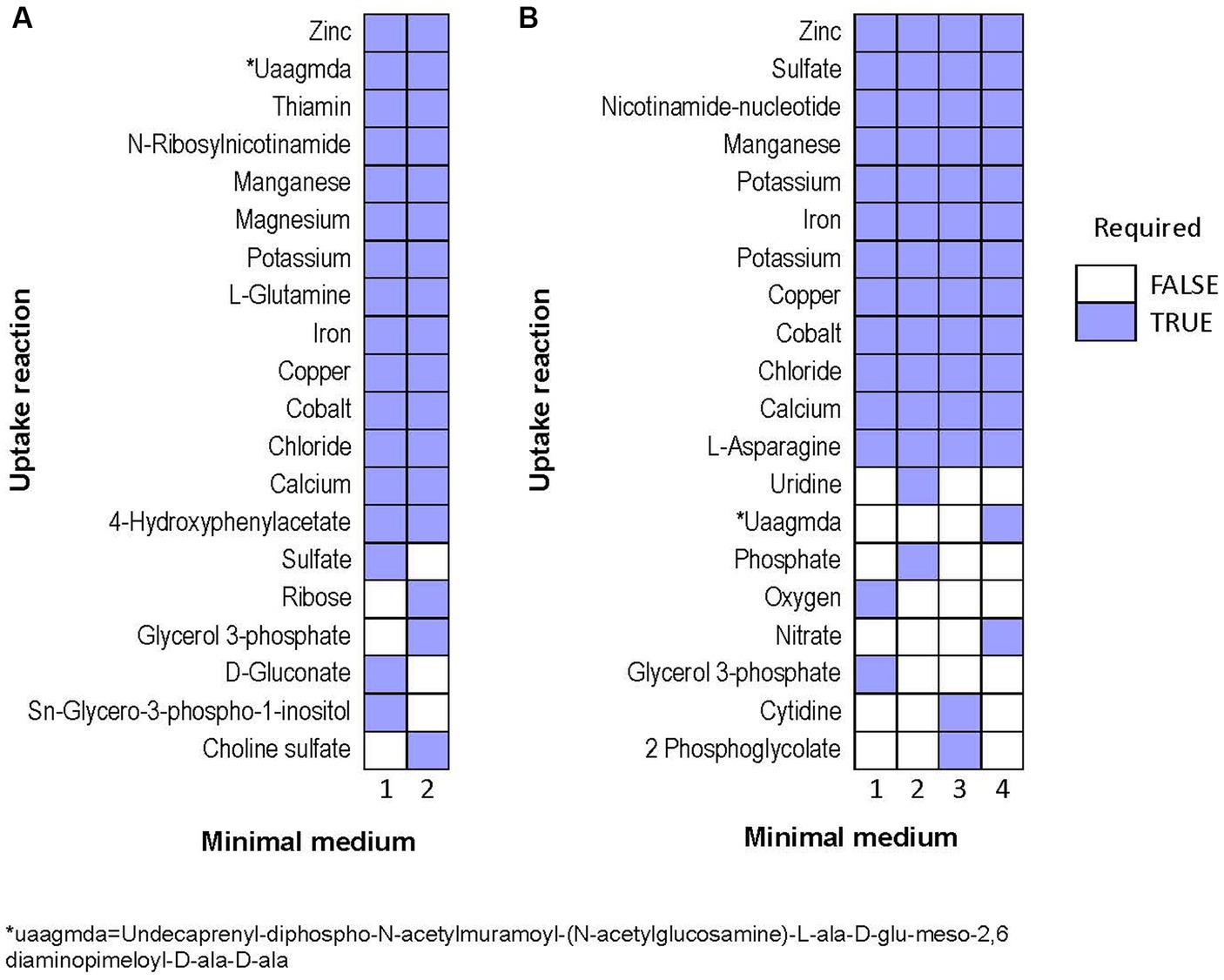
Figure 8. Alternative minimal media identified for (A) Halorubrum sp. and (B) Marinococcus luteus. Each column is a different set of nutrients which allow growth in flux balance analysis.
Syntrophic interactions are central to microbial communities and were foundational in the history of complex life. Despite this centrality in microbial ecology and evolution, the initial evolution and genomic basis of cross-feeding are challenging to determine. The vast majority of microorganisms cannot be cultured, limiting experimental investigation on most syntrophic interactions. And even for mixed species microbial cultures involving syntrophic interactions, determining shared substrates is daunting as microbes alter their environments in numerous diverse ways. Using a genomic approach through Next Generation Sequencing (NGS) technologies, we assessed the community context for growth of a microbial species pair. Obtained as a single isolate, neither species can be cultivated alone. When grown together in nutrient rich media, the culture reaches a high density in liquid and shows abundant growth on plates. A metabolic network analysis suggests that most auxotrophies in one species could be complemented by crossfeeding from the other species, but not all. This mix of auxotrophies and syntrophies suggests that the source microbial community contains a web of diffuse beneficial syntrophic interactions. The complexity of source microbial community is also reflected in other aspects of their genomes, including genes and genomic islands associated with secondary metabolite production, cell attachment, and horizontal gene transfer. These results suggest that the structure of microbial communities involves numerous competitive and mutualistic interactions, providing opportunities for beneficial symbioses to evolve.
Comparing the genomes of symbiotic organisms by identifying conserved genes or specific adaptations unique to symbiotic partners, along with metabolic network predictions, can aid in addressing the remaining questions about symbiotic interactions like: “How do organisms in symbiotic relationships recognize and select suitable partners?” “What are the molecular mechanisms and signaling pathways involved in the recognition and establishment of symbiotic associations?” What are the evolutionary origins of symbiotic relationships? How did these complex interactions among organisms arise, and how have they evolved? Implementing these opens a new perspective into long-standing evolutionary questions about spatio-temporal microbial symbiosis and how cross-feeding affects diversity and speciation.
Cultures of halophilic archaea and bacteria, like Halorubrum and Marinococcus, can be challenging to cultivate in laboratory settings. In this work, we proposed this syntrophic relation between Halorubrum sp. and Marinococcus luteus as an early-stage symbiosis due to their consistent co-occurrence in laboratory conditions, evolving to thrive as consortia, contributing to the community’s resilience and adaptability to harsh salinity conditions. One of the central questions in this work was whether these species engage in an obligate mutualistic relationship. To address their close relationship, we performed metabolic network modeling. It was found that each species had unique nutrient requirements. Importantly, most of these nutrient requirements could be produced by other species, though that was not true of all Halorubrum requirements. In the model, Marinococcus could not produce 4-hydroxyphenylacetate or thiamine. However, functional annotations coupled with media composition use to gap-fill the network, shows that Marinococcus, has the complete pathway to synthetize thiamine and 4-hydroxyphenylacetate. Furthermore, while both species had the capacity to produce the amino acid that the other species was auxotrophic for, this production itself depended upon the environmental availability of an amino acid that the producer itself was auxotrophic for. For instance, Marinococcus could only produce glutamine when provided with asparagine, and conversely, Halorubrum relied on asparagine to produce glutamine. This cooperative metabolic interaction can lead to increased growth rates, improved resource utilization, expanding ecological niches for the symbiotic microorganisms. Moreover, the prediction of the essential nutrients alone, suggests either the archaea or the bacteria requirements are partially fulfilled by the other, this representation might be attributed to poor annotation in archaeal references available. Archaea are distinguished by the existence of distinctive, modified versions of conventional pathways like the Entner-Doudoroff (ED), Embden-Meyerhof-Parnas (EMP), and novel pentose-degrading and CO2-fixing pathways catalyzed by new enzymes (Sato and Atomi, 2011; Bräsen et al., 2014).
Interestingly, the number of alternative media predicted to allow optimal growth is higher in M. luteus than the archaeon; given the genome plasticity, and the higher number of genes within the metabolic subsystems (Figure 4), M. luteus is probably more efficient utilizing nutrients present in the media.
Next, we asked whether each model could produce the compounds which the other is not capable of producing, by optimizing a sink reaction for that compound (see methods). In the model, we found that Marinococcus could produce L-Glutamate (gln__L_c), Undecaprenyl-diphospho-N-acetylmuramoyl-N-acetylglucosamine-L-ala-D-glu-meso-2,6-diaminopimeloyl-D-ala-D-ala (uaagmda_c) and N-Ribosylnicotinamide (rnam_c), but not 4 hydroxyphenylacetate (4hphac_c), or Thiamin (thm_c), however, the genome annotation show the presence of the elements needed for the pathway. Moreover, it could only produce as much N-Ribosylnicotinamide (rnam_c) as it was supplied with (nmn_e), potentially suggesting that this precursor is limiting to both species. On the other hand, Halorubrum can produce both L-Asparagine (asn__L_c) and N1-methylnicotinamide (nmn_e) during growth in a minimal medium. However, it could only produce as much N1-methylnicotinamide (nmn_e) as it was supplied with N-Ribosylnicotinamide (rnam_e).
The shift from a free-living lifestyle to symbiotic interaction is occasionally facilitated by horizontal gene transfer (HGT) events, which frequently carry traits that enable the rapid exploitation of the environment (Madsen et al., 2012; Drew et al., 2021). Archaea and Bacteria are usually found in mixed population biofilms, offering nutrient supply, protection, and boosting genetic variation through HGT events (Fröls, 2013). HGT processes can be involved in sympatric speciation and in the high rate of genetic recombination on halophilic communities (Ram Mohan et al., 2014). We found genomic evidence of biofilm production in both strains, since there are some gene homologs that participate in chemotaxis, EPS production, pili, and flagella.
The initial recognition between the two strains can involve surface interactions through adhesins or pili-like appendages that facilitate attachment to each other or a common surface. These genes were identified in Halorubrum, with the N-terminal domain of the type IV pili from six adhesion pili PilA1-6. Other studies have demonstrated that the expression of PilA1, PilA2, or PilA6 allows the microcolony formation, while Pil3 or Pil4 enhance surface adhesion (Esquivel et al., 2016). Additionally, archaellum genes were identified, along with FlaF and FlaG gene-complex, which is required for archaellum assembly and motility, plus four accessory motor components, FlaC, FlaH, FlaI, and FlaJ (Shahapure et al., 2014; Tsai et al., 2020). As for Marinococcus, two motility-related gene clusters fliEFGHIJKLMNOPQR and flgBCDEFGHIJKL were identified, along with their own regulatory systems. Moreover, similar Che gene sets were present, CheW and CheA led to the activation of CheY gene, which acts upon MotAB a flagellar motor complex, in charge of the rotation of bacterial flagella through conformational alterations (Liu and Ochman, 2007; Athmika et al., 2021).
Interestingly, several biofilm genes found in both strains have more than one copy, this duplication of the genome content offers an abundance of genetic material for evolution and is suggested to be crucial in adapting to novel environments and promoting diversification (Xu et al., 2020). Meanwhile, some genes belonging to operon-complexes were not found, probably due to adaptive evolution trough gene loss, as a rapid response to environmental fluctuations.
Genomic islands are also part of those exchanged elements, and another example of their interaction capacity is observed in the genome content of Halorubrum sp. and M. luteus, with accessory functions of symbiosis, secondary metabolites production, and antibiotic resistance (see Supplementary Tables 3, 4). Particularly, Halorubrum comprises seven genomic islands related to symbiosis, while Marinococcus possesses four. According to the metabolic prediction analysis, thiamine constitutes one of the essential nutrients for the Halorubrum strain. Noteworthy, it was identified the presence of a set of islands with thiamine and sugar catabolism capacity, allowing the archaea to acquire and metabolize energy from compounds found in the environment (Lacour et al., 2006).
One of the singularities, is the presence of arsenic and cadmium resistant genes in both, Halorubrum sp. (island 19) and Marinococcus sp. (island 3). Previous studies have observed the abundance of arsenic and cadmium in the CCB (Valdespino-Castillo et al., 2018; García-Ulloa et al., 2022).
Arsenic resistance genomic islands have been characterized particularly in microbes that inhabit oligotrophic environments. Phosphate and arsenate are chemical analogs, substituting each other in chemical reactions (Strawn, 2018). The arsenic metabolism genes can help the microorganisms to use low concentrations of phosphorus under high arsenic concentrations (Li et al., 2013). The scarcity of phosphorus can reinforce arsenic uptake, while high concentrations impede arsenic uptake (Strawn, 2018). Oligotrophic conditions are a main characteristic of the Cuatro Cienegas Basin environment (Souza et al., 2008), strongly marked by a phosphorus limitation. Therefore, the evidence of genomic islands related to arsenic metabolism, might indicate an alternative way of Halorubrum sp. to survive in the CCB environment by up taking the arsenic available. Interestingly, both cadmium resistance and arsenic resistance often co-occur (Parsons et al., 2020).
The presence of the highly genetically divergent species Halorubrum sp., and M. luteus in CCB suggests that both microorganisms have acquired similar traits, using unique metabolic pathways and osmoregulation mechanisms, allowing them to thrive in high salt concentrations and fluctuating environments through convergent evolution events by evolving specialized transporters, ion channels, or regulatory systems. The Cuatro Cienegas Basin (CCB) has often shown the surprising presence of microorganisms limited to certain distinct environments, and the extant marine environment, along with its community, has been preserved (Souza et al., 2006, 2018). However, given the ancestral marine state of CCB, we might be able to explain the presence of some unexpected taxa, such as M. luteus, commonly found in salt lakes (Wang et al., 2009; Fox-Powell and Cockell, 2018). The functional annotation of both strains revealed the presence of an osmotic regulation system composed of elements coding for compatible solutes such as trehalose, glycerol, glutamate synthesis. These solutes help maintain cell turgor to counteract osmotic stress. Independently, Halorubrum sp. has a complete Trk gene system in charge of potassium uptake, meanwhile, M. luteus has ectoine and betaine biosynthesis, as well as choline uptake regulation pathways, regulated by gbuA, gbuB, gbuC and nsmX genes.
While identifying adaptative events can be challenging, the independent evolution of similar traits in multiple lineages is one of the strongest indications that adaptation has occurred. Despite the challenges and limitations of identifying adaptive events, this criterion can help us to understand better the mechanisms driving biological evolution and the relationship between microorganisms and their environment. Part of this suggested evidence is the presence of terpene biosynthetic clusters in both, Halorubrum and Marinococcus strains. This cluster is distinctive for each one; although both clusters have a phytoene synthase gene, the gene content and gene arrangement is different. Phytoene synthase gene is conserved in all terpene and carotenoid-producing archaea and in most carotenoid-producing bacteria. However, carotenoid and terpene biosynthesis pathways are more diverse in bacteria. The study of secondary metabolites in archaea is still rare, and most archaea only have one or two BGCs, usually a terpene or a bacteriocin BGC (Wang et al., 2019). Most of the genes in bacteria associated with terpene biosynthesis belong to the crt gene family (Sandmann, 2021). This correlates with the observed genes in the terpene BGC in M. luteus, as most genes in this cluster belong to the crt family. The terpene biosynthesis is directly involved in the carotenoid biosynthesis pathways, and carotenoid production is common in halophilic archaea and bacteria, as one of its functions stabilizes the cell membrane (Sandmann, 2021).
Perhaps surprisingly, we also observed little, or no, evidence of CRISPR-Cas elements. No CRISPR elements were observed in the Marinococcus genome. And in the Halorubrum genome, we have only a low evidence level, which can be sequencing artifacts (see Supplementary Table 5). Although the incidence of CRISPR-Cas elements in archaea is high, it has been reported some Halorubrum archaea do not have them. This might be due to the deterioration of CRISPR arrangements in the genome, which can occur during DNA repair processes or because the need for the system is not constant (Fullmer et al., 2014). Likewise, the low concentrations of phosphorus present in the CCB can limit the plasticity of the genome, in addition to the fact that the transformation is reduced under these conditions (Souza et al., 2008).
The comparative genomic analysis revealed intriguing similarities and differences between the halophilic Halorubrum sp. and M. luteus. Both organisms exhibit convergence in terms of the genetic components involved in their adaptation to high-salt environments. The shared presence of gene clusters related to osmo-adaptation and ion transport suggests convergent evolution in response to salinity. However, the presence of distinct genomic elements highlights the influence of their respective evolutionary histories. Along with the metabolic prediction, the data suggest that the two species partially complement each other’s metabolism, as well as greater complexity and environmental dependency than a simple, direct cross-feeding arrangement. Noteworthy, other organisms thriving in the same niche, such as bacteria, archaea, fungi, and protozoa, may also improve the survival, fitness, and metabolism of these strains. Moreover, models derived from metabolic network analysis improve understanding of the ecological complexity and dynamics within microbial communities, offering new insights into metabolism and novel culture-media alternatives. Future work to delve into the dynamic behavior of the co-culture is needed. To increase experimental tractability, time-lapse microscopy can enable us to visualize the physical interactions between archaea and bacteria, providing insights into spatial arrangements and cellular associations. Moreover, conducting stable isotope labeling techniques will help trace the flow of specific nutrients and the transfer of resources.
Complete genome data is available at NCBI, BioProject PRJNA1003106.
NM-C: Conceptualization, Formal analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. AT-C: Data curation, Formal analysis, Investigation, Methodology, Software, Visualization, Writing – review & editing. JC: Methodology, Validation, Writing – review & editing, Data curation, Formal analysis, Investigation, Software, Visualization. WH: Supervision, Validation, Visualization, Writing – review & editing. ST-Z: Methodology, Validation, Project administration, Supervision, Writing – review & editing. MT: Supervision, Validation, Visualization, Writing – review & editing, Conceptualization, Funding acquisition, Project administration, Resources, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Funds for this work were provided by a Conacyt Postdoctoral Fellowship 516728 (NM-C), NSF EF-1724011 (MT), and NASA IDEAS16002 (MT).
The authors want to thank Caleb M. Smithberg for the biofilm and antibiotic test pictures and to Travisano Lab members for their critical review of the manuscript, their valuable support, and their critical observations throughout the project.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1276438/full#supplementary-material
Andrews, S. (2010). "FastQC: A quality control tool for high throughput sequence data ". Babraham Bioinformatics, Babraham Institute, Cambridge, United Kingdom.
Arocha-Garza, H. F., Canales-Del Castillo, R., Eguiarte, L. E., Souza, V., and De la Torre-Zavala, S. (2017). High diversity and suggested endemicity of culturable Actinobacteria in an extremely oligotrophic desert oasis. PeerJ 5:e3247. doi: 10.7717/peerj.3247
Athmika, S. D., Ghate, A. B., Arun, S. S., Rao, S. T. A., Kumar, M. K., Kandiyil, M. K., et al. (2021). Genome analysis of a halophilic bacterium Halomonas malpeensis YU-PRIM-29(T) reveals its exopolysaccharide and pigment producing capabilities. Sci. Rep. 11:1749. doi: 10.1038/s41598-021-81395-1
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bonilla-Rosso, G., Peimbert, M., Alcaraz, L. D., Hernandez, I., Eguiarte, L. E., Olmedo-Alvarez, G., et al. (2012). Comparative metagenomics of two microbial mats at Cuatro Cienegas Basin II: community structure and composition in oligotrophic environments. Astrobiology 12, 659–673. doi: 10.1089/ast.2011.0724
Bräsen, C., Esser, D., Rauch, B., and Siebers, B. (2014). Carbohydrate metabolism in Archaea: current insights into unusual enzymes and pathways and their regulation. Microbiol. Mol. Biol. Rev. 78, 89–175. doi: 10.1128/mmbr.00041-13
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Drew, G. C., Stevens, E. J., and King, K. C. (2021). Microbial evolution and transitions along the parasite-mutualist continuum. Nat. Rev. Microbiol. 19, 623–638. doi: 10.1038/s41579-021-00550-7
Ebrahim, A., Lerman, J. A., Palsson, B. O., and Hyduke, D. R. (2013). COBRApy: COnstraints-based reconstruction and analysis for Python. BMC Syst. Biol. 7:74. doi: 10.1186/1752-0509-7-74
Escalante, A. E., Rebolleda-Gómez, M., Benítez, M., and Travisano, M. (2015). Ecological perspectives on synthetic biology: insights from microbial population biology. Front. Microbiol. 6:143. doi: 10.3389/fmicb.2015.00143
Espinosa-Asuar, L., Monroy-Guzman, C., Madrigal-Trejo, D., Navarro-Miranda, M., Sanchez-Perez, J., Buenrostro Munoz, J., et al. (2022). Diversity of an uncommon elastic hypersaline microbial mat along a small-scale transect. PeerJ 10:e13579. doi: 10.7717/peerj.13579
Esquivel, R. N., Schulze, S., Xu, R., Hippler, M., and Pohlschroder, M. (2016). Identification of Haloferax volcanii pilin N-Glycans with diverse roles in pilus biosynthesis, adhesion, and microcolony formation. J. Biol. Chem. 291, 10602–10614. doi: 10.1074/jbc.M115.693556
Fox-Powell, M. G., and Cockell, C. S. (2018). Building a geochemical view of microbial salt tolerance: halophilic adaptation of Marinococcus in a natural magnesium sulfate brine. Front. Microbiol. 9:739. doi: 10.3389/fmicb.2018.00739
Fröls, S. (2013). Archaeal biofilms: widespread and complex. Biochem. Soc. Trans. 41, 393–398. doi: 10.1042/bst20120304
Fullmer, M. S., Soucy, S. M., Swithers, K. S., Makkay, A. M., Wheeler, R., Ventosa, A., et al. (2014). Population and genomic analysis of the genus Halorubrum. Front. Microbiol. 5:140. doi: 10.3389/fmicb.2014.00140
García-Ulloa, M. 2nd, Souza, V., Esquivel-Hernández, D. A., Sánchez-Pérez, J., Espinosa-Asuar, L., Viladomat, M., et al. (2022). Recent differentiation of aquatic bacterial communities in a hydrological system in the cuatro ciénegas basin, after a natural perturbation. Frontiers in microbiology 13:825167. doi: 10.3389/fmicb.2022.825167
Hentschel, U., Steinert, M., and Hacker, J. (2000). Common molecular mechanisms of symbiosis and pathogenesis. Trends Microbiol. 8, 226–231. doi: 10.1016/S0966-842X(00)01758-3
Juhas, M., van der Meer, J. R., Gaillard, M., Harding, R. M., Hood, D. W., and Crook, D. W. (2009). Genomic islands: tools of bacterial horizontal gene transfer and evolution. FEMS Microbiol. Rev. 33, 376–393. doi: 10.1111/j.1574-6976.2008.00136.x
Kaminski, L., Guan, Z., Yurist-Doutsch, S., and Eichler, J. (2013). Two distinct N-glycosylation pathways process the Haloferax volcanii S-layer glycoprotein upon changes in environmental salinity. MBio 4, e00716–e00713. doi: 10.1128/mBio.00716-13
Kanehisa, M., Sato, Y., and Kawashima, M. (2022). KEGG mapping tools for uncovering hidden features in biological data. Protein Sci. 31, 47–53. doi: 10.1002/pro.4172
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Lacour, S., Gaillard, M., and Meer, J. R. V. D. (2006). “Genomic islands and evolution of catabolic pathways” in Prokaryotic diversity: Mechanisms and significance. eds. H. M. Lappin-Scott, N. A. Logan, and P. C. F. Oyston (Cambridge: Cambridge University Press), 255–274.
Li, H., Li, M., Huang, Y., Rensing, C., and Wang, G. (2013). In silico analysis of bacterial arsenic islands reveals remarkable synteny and functional relatedness between arsenate and phosphate. Front. Microbiol. 4:347. doi: 10.3389/fmicb.2013.00347
Liu, R., and Ochman, H. (2007). Origins of flagellar gene operons and secondary flagellar systems. J. Bacteriol. 189, 7098–7104. doi: 10.1128/jb.00643-07
López-García, P., and Moreira, D. (2019). Eukaryogenesis, a syntrophy affair. Nat. Microbiol. 4, 1068–1070. doi: 10.1038/s41564-019-0495-5
López-García, P., and Moreira, D. (2020). The Syntrophy hypothesis for the origin of eukaryotes revisited. Nat. Microbiol. 5, 655–667. doi: 10.1038/s41564-020-0710-4
Machado, D., Andrejev, S., Tramontano, M., and Patil, K. R. (2018). Fast automated reconstruction of genome-scale metabolic models for microbial species and communities. Nucleic Acids Res. 46, 7542–7553. doi: 10.1093/nar/gky537
Madsen, J. S., Burmølle, M., Hansen, L. H., and Sørensen, S. J. (2012). The interconnection between biofilm formation and horizontal gene transfer. FEMS Immunol. Med. Microbiol. 65, 183–195. doi: 10.1111/j.1574-695X.2012.00960.x
Medina-Chávez, N. O., De la Torre-Zavala, S., Arreola-Triana, A. E., and Souza, V. (2020). “Cuatro Ciénegas as an Archaean Astrobiology Park” in Astrobiology and Cuatro Ciénegas Basin as an analog of early earth. ed. V. Souza (Segura: Antígona, Foster, Jamie Springer International Publishing), 219–228.
Medina-Chávez, N. O., and Travisano, M. (2022). Archaeal communities: the microbial Phylogenomic frontier. Front. Genet. 12:693193. doi: 10.3389/fgene.2021.693193
Medina-Chávez, N. O., Viladomat-Jasso, M., Zarza, E., Islas-Robles, A., Valdivia-Anistro, J., Thalasso-Siret, F., et al. (2023). A transiently hypersaline microbial mat harbors a diverse and stable archaeal community in the Cuatro Cienegas basin, Mexico. Astrobiology 23, 796–811. doi: 10.1089/ast.2021.0047
Mikheenko, A., Saveliev, V., and Gurevich, A. (2015). MetaQUAST: evaluation of metagenome assemblies. Bioinformatics 32, 1088–1090. doi: 10.1093/bioinformatics/btv697
Moran, N. A. (2007). Symbiosis as an adaptive process and source of phenotypic complexity. Proc Natl Acad Sci USA 104, 8627–8633. doi: 10.1073/pnas.0611659104
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes: a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Ochman, H., and Moran, N. A. (2001). Genes lost and genes found: evolution of bacterial pathogenesis and symbiosis. Science 292, 1096–1099. doi: 10.1126/science.1058543
Parsons, C., Lee, S., and Kathariou, S. (2020). Dissemination and conservation of cadmium and arsenic resistance determinants in Listeria and other gram-positive bacteria. Mol. Microbiol. 113, 560–569. doi: 10.1111/mmi.14470
Peimbert, M., Alcaraz, L. D., Bonilla-Rosso, G., Olmedo-Alvarez, G., Garcia-Oliva, F., Segovia, L., et al. (2012). Comparative metagenomics of two microbial mats at Cuatro Cienegas Basin I: ancient lessons on how to cope with an environment under severe nutrient stress. Astrobiology 12, 648–658. doi: 10.1089/ast.2011.0694
Ram Mohan, N., Fullmer, M. S., Makkay, A. M., Wheeler, R., Ventosa, A., Naor, A., et al. (2014). Evidence from phylogenetic and genome fingerprinting analyses suggests rapidly changing variation in Halorubrum and Haloarcula populations. Front. Microbiol. 5:143. doi: 10.3389/fmicb.2014.00143
Sandmann, G. (2021). Diversity and origin of carotenoid biosynthesis: its history of coevolution towards plant photosynthesis. New Phytol. 232, 479–493. doi: 10.1111/nph.17655
Sato, T., and Atomi, H. (2011). Novel metabolic pathways in Archaea. Curr. Opin. Microbiol. 14, 307–314. doi: 10.1016/j.mib.2011.04.014
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Shahapure, R., Driessen, R. P. C., Haurat, M. F., Albers, S.-V., and Dame, R. T. (2014). The archaellum: a rotating type IV pilus. Mol. Microbiol. 91, 716–723. doi: 10.1111/mmi.12486
Souza, V., Eguiarte, L. E., Siefert, J., and Elser, J. J. (2008). Microbial endemism: does phosphorus limitation enhance speciation? Nat. Rev. Microbiol. 6, 559–564. doi: 10.1038/nrmicro1917
Souza, V., Espinosa-Asuar, L., Escalante, A. E., Eguiarte, L. E., Farmer, J., Forney, L., et al. (2006). An endangered oasis of aquatic microbial biodiversity in the Chihuahuan desert. Proc. Natl. Acad. Sci. U. S. A. 103, 6565–6570. doi: 10.1073/pnas.0601434103
Souza, V., Moreno-Letelier, A., Travisano, M., Alcaraz, L. D., Olmedo, G., and Eguiarte, L. E. (2018). The lost world of Cuatro Cienegas Basin, a relictual bacterial niche in a desert oasis. Elife 7:8278. doi: 10.7554/eLife.38278
Strawn, D. G. (2018). Review of interactions between phosphorus and arsenic in soils from four case studies. Geochem. Trans. 19:10. doi: 10.1186/s12932-018-0055-6
Tsai, C. L., Tripp, P., Sivabalasarma, S., Zhang, C., Rodriguez-Franco, M., Wipfler, R. L., et al. (2020). The structure of the periplasmic FlaG-FlaF complex and its essential role for archaellar swimming motility. Nat. Microbiol. 5, 216–225. doi: 10.1038/s41564-019-0622-3
Valdespino-Castillo, P. M., Hu, P., Merino-Ibarra, M., López-Gómez, L. M., Cerqueda-García, E., González-De Zayas, R., et al. (2018). Exploring Biogeochemistry and Microbial Diversity of Extant Microbialites in Mexico and Cuba. Frontiers in microbiology 9:510. doi: 10.3389/fmicb.2018.00510
Wang, Y., Cao, L.-L., Tang, S.-K., Lou, K., Mao, P.-H., Jin, X., et al. (2009). Marinococcus luteus sp. nov., a halotolerant bacterium isolated from a salt lake, and emended description of the genus Marinococcus. Int. J. Syst. Evol. Microbiol. 59, 2875–2879. doi: 10.1099/ijs.0.009670-0
Wang, S., Zheng, Z., Zou, H., Li, N., and Wu, M. (2019). Characterization of the secondary metabolite biosynthetic gene clusters in archaea. Comput. Biol. Chem. 78, 165–169. doi: 10.1016/j.compbiolchem.2018.11.019
Wu, Y.-W., Tang, Y.-H., Tringe, S. G., Simmons, B. A., and Singer, S. W. (2014). MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2:26. doi: 10.1186/2049-2618-2-26
Xu, S., Wang, J., Guo, Z., He, Z., and Shi, S. (2020). Genomic convergence in the adaptation to extreme environments. Plant Commun 1:100117. doi: 10.1016/j.xplc.2020.100117
Keywords: syntrophy, cross-feeding, metabolic exchange, archaea, bacteria, halophiles, extremophiles
Citation: Medina-Chávez NO, Torres-Cerda A, Chacón JM, Harcombe WR, De la Torre-Zavala S and Travisano M (2023) Disentangling a metabolic cross-feeding in a halophilic archaea-bacteria consortium. Front. Microbiol. 14:1276438. doi: 10.3389/fmicb.2023.1276438
Edited by:
Chih-Horng Kuo, Academia Sinica, TaiwanReviewed by:
Yu-Wei Wu, Taipei Medical University, TaiwanCopyright © 2023 Medina-Chávez, Torres-Cerda, Chacón, Harcombe, De la Torre-Zavala and Travisano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nahui Olin Medina-Chávez, bm1lZGluYWNAdW1uLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.