
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 08 December 2023
Sec. Extreme Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1275665
 Romain Gastineau1*
Romain Gastineau1* Sara Harðardóttir2,3,4
Sara Harðardóttir2,3,4 Caroline Guilmette3
Caroline Guilmette3 Claude Lemieux5,6
Claude Lemieux5,6 Monique Turmel5,6
Monique Turmel5,6 Christian Otis5,7
Christian Otis5,7 Brian Boyle5,7
Brian Boyle5,7 Roger C. Levesque5
Roger C. Levesque5 Jeff Gauthier5
Jeff Gauthier5 Marianne Potvin5,6
Marianne Potvin5,6 Connie Lovejoy3,5*
Connie Lovejoy3,5*Introduction: There is little information on evolutionarily ancient eukaryotes, which are often referred to as basal eukaryotes, in Arctic waters. Despite earlier studies being conducted in the Russian White Sea, only few have been reported.
Methods: Following a shotgun sequence survey of diatom cultures from Sugluk Inlet off the Hudson Strait in Northern Québec, we obtained the complete mitochondrial genome and the operon of nuclear ribosomal RNA genes from a strain that matches that of Ancyromonas sigmoides (Kent, 1881).
Results: The sequence of the mitogenome retrieved was 41,889 bp in length and encoded 38 protein-coding genes, 5 non-conserved open-reading frames, and 2 rRNA and 24 tRNA genes. The mitogenome has retained sdh2 and sdh3, two genes of the succinate dehydrogenase complex, which are sometimes found among basal eukaryotes but seemingly missing among the Malawimonadidae, a lineage sister to Ancyromonadida in some phylogenies. The phylogeny inferred from the 18S rRNA gene associated A. sigmoides from Sugluk Inlet with several other strains originating from the Arctic. The study also unveiled the presence of a metagenomic sequence ascribed to bacteria in GenBank, but it was clearly a mitochondrial genome with a gene content highly similar to that of A. sigmoides, including the non-conserved open-reading frames.
Discussion: After re-annotation, a phylogeny was inferred from mitochondrial protein sequences, and it strongly associated A. sigmoides with the misidentified organism, with the two being possibly conspecific or sibling species as they are more similar to one another than to species of the genus Malawimonas. Overall our phylogeny showed that the ice associated ancryomonads were clearly distinct from more southerly strains.
Ancyromonas sigmoides (Kent, 1881) is a zooflagellate protist whose identification and classification have been controversial for more than a century. According to WORMS, the genus Ancyromonas encompasses 13 different species from marine and freshwater environments, with seven of them being taxonomically accepted and six with an uncertain status. Ancyromonas sigmoides, along with the introduction of the genus Ancyromonas, was initially described by Kent (1880–1882) in his “Manual of the Infusoria.” The samples that were observed by Kent consisted in ‘decaying fucus’ described as Fucus silicosa, probably referring to Fucus siliquosus (Linnaeus 1753), today considered a synonym of the taxonomically accepted name Halidrys siliquosa (Linnaeus) Lyngbye 1819. These macroalgal samples were picked in the vicinities of Saint Helier, the capital city of the Channel Island Jersey (49.11°N). Since then, several reports have noted the presence of this species worldwide, but solely based on light microscopy, as listed in Cavalier-Smith et al. (2008).
In 1990, Mylnikov (1990) described a colorless flagellate isolated from the Island of Sredni in the White Sea, Republic of Karelia, Russia. He successfully cultivated this strain and shared it with other researchers, including Thomas Cavalier-Smith, and culture collections. Cavalier-Smith et al. carried out the first molecular barcoding of the strain coupled with ultrastructural analyses using transmission electron microscopy (Cavalier-Smith et al., 2008). Based on these results and their suspicions of previous misidentifications, they introduced a taxonomic order called Planomonadida, belonging to the phylum Apusozoa, and redescribed Mylnikov’s strain as Planomonas mylnikovi Cavalier-Smith, a species name that was meant to replace A. sigmoides.
This reclassification sparked some debate in the community. Heiss et al. (2010) questioned the validity of the genus Planomonas, proposed to reverse the transfer of Planomonas spp. to their original genera, and declared P. mylikovi as a junior synonym of A. sigmoides. This new classification was partly accepted by Glücksman et al. (2013) in an article co-authored by Thomas Cavalier-Smith, in which authors agreed to use the species name A. sigmoides instead of P. mylnikovi to avoid further confusion, but retained the validity of the genus Planomonas. Currently, the accepted name of this taxon is A. sigmoides, with most of the subsequent work on this species having been conducted on Mylnikov’s strain, generally labeled as B70 or CCAP 1958/3 (e.g., Heiss et al., 2010; Brown et al., 2018).
Finding the phylogenetic position of A. sigmoides and to a larger extent of the Ancyromonadida has been challenging. A recent phylogenomic study based on transcriptomic analyses (Brown et al., 2018) has revealed some new insights. The Maximum-Likelihood phylogeny derived from 351 proteins associated the three cultivated species of Ancyromonadida: A. sigmoides, Cavalier-Smith 1997 emend Atkins 2000, Nutomonas longa Cavalier-Smith & Glücksman 2013, and Fabomonas tropica Glücksman and Cavalier-Smith 2013. The phylogeny tended to associate this cluster with Malawimonadidae O’Kelly and Nerad 1999, represented by Malawimonas jakobiformis O’Kelly and Nerad 1999, the informally named Malawimonas californiana, and a specimen labeled as Malawimonad sp. 249, later formerly described as Gefionella okellyi (Heiss et al., 2018). However, the Bayesian inference of phylogeny was not decisive, and Brown et al. (2018) remained cautious with their conclusions, emphasizing the importance of including ancyromonads in further phylogenetic analysis. Missing from these analyses is the genomic information on the mitochondrial genomes of this and other Ancromonadida strains. As mitochondria are a defining feature of eukaryotic cells, knowledge of their phylogeny and genetic architecture is evolutionarily informative and provides basic information on ecology in submarginal habitats (Roger et al., 2017).
In 2018, a strain of diatom identified from light microscopy as Haslea crucigeroides (Hustedt) Simonsen, 1974 was isolated from the Sugluk Inlet near the hamlet of Salluit, Nunavik, Québec, Canada. Sagluk Inlet is located along the Hudson Strait, which connects Hudson Bay to Baffin Bay. Genomic sequencing conducted on DNA extracts of the culture revealed the presence of other protists in the culture. A large sequence matching the 18S rRNA gene of A. sigmoides was found along with another large contig suspected to represent the mitochondrial genome. Since no reference was available for mitochondrial genes of A. Sigmoides, the assignment of this mitogenome to this species was confirmed using previously published transcriptomic data (Brown et al., 2018). Following the annotation of this mitogenome and a general search for similar sequence in GenBank, a sequence derived from the metagenomic analysis that followed the Tara Ocean expedition carried out in open oceans and in the Mediterranean Sea (Tully et al., 2018) was a close match. The sequence had been automatically classified as an “unknown bacteria” in the genus Rhodovulum and annotated as such. However, the similarity with the mitogenome of A. sigmoides suggested otherwise, and we speculated that it could belong to another species of Ancyromonadida. This sequence was re-annotated and found to have a very similar gene content and phylogenetic proximity with A. sigmoides. To the best of our knowledge, the finding of the sequence represents the first confirmed record using molecular techniques of a putative Ancryomonadida in the Mediterranean Sea.
A diatom identified as H. crucigeroides was sampled and isolated from the Sugluk Inlet (62°16′27.66” N, 75°32′3.10” W) in March 2018. The diatom was isolated by micropipette and cultivated in F/2 medium at the Takuvik Laboratory of Université Laval in a Caron culture cabinet model 7901-33-2 (Marietta, OH, United States). The culture cabinet had been modified for cultivating ice algae using low light with continuous irradiance of 9 μmol photons m−2 s−1 maintained at 0°C. Subsequently, heterotrophic protists were found to be associated with the culture, and they were identified following shotgun sequencing of DNA from the culture. The most prominent co-cultured organism was identified as A. sigmoides.
DNA was extracted with the CTAB protocol following Doyle and Doyle (1990). The library was prepared at the Plateforme d’Analyse génomique, Institut de Biologie Intégrative et des Systèmes, Université Laval (Québec, Canada)1 by shearing 500 ng of DNA with a Covaris M220 (Covaris, Woburn MA, United States) and using the dedicated NEBNext Ultra II DNA Library Prep Kit Illumina from New England Biolab (Ipswich, MA, United States). The library was sent to Génome Québec (Montreal QC)2 for sequencing on an Illumina (San Diego, CA, United States) NovaSeq 6,000 platform. After sequencing, reads were trimmed using fastp (Chen et al., 2018; Chen, 2023) with the default parameters. After quality trimming, a total of nearly 221 M clean reads remained. Reads were assembled using SPAdes 3.15.4 with the meta mode on and a k-mer of 85, which was found to be optimal in the multispecies culture and provided a good coverage of the A. sigmoides mitogenome. The contigs corresponding to the nuclear rRNA cluster and mitochondrial genome of A. sigmoides were identified by datamining using standalone blastn and blastx analyses (Boratyn et al., 2012) with customized databases containing nucleotide and protein sequences from diatoms, prepared using the make blastdb function of the NCBI package. All bioinformatic analyses were conducted on the superdome flex server at IBIS, Université Laval.
The RNA-Seq data from Brown et al. (2018) on A. sigmoides B70/CCAP 1958/3 (SRR5997436) were downloaded from the NCBI Sequence Read Archive (SRA). After conversion to fastq format by SRA Toolkit,3 reads were assembled using RNASPAdes 3.15.4 (Bushmanova et al., 2019) with a k-mer of 35. The resulting contig file was datamined by a command line blastn analysis using the putative cox1 gene sequence of A. sigmoides from Sugluk Inlet.
The rRNA cluster was analyzed using Rfam (Kalvari et al., 2021). Annotation of the protein coding genes (PCG) of mitochondrial genomes was done with genetic code 11 using the findORF tools developed at Université Laval (Gagnon, 2004) and used to annotate organellar genomes in several recent publications (for, e.g., Hamedi et al., 2019; Gastineau et al., 2021; and Solak et al., 2021). The database on which findORF operates contains sequences from the gene-rich mitochondrial genome of the early emerging (basal) protists Jakoba libera (Ruinen) D.J. Patterson 1990 ATCC 50422 (GenBank: KC353355), Reclinomonas americana Flavin & Nerad, 1993 (GenBank: KC353356), and Histiona aroides Pascher 1943 (GenBank: KC353353), from Leigh and Lang (2004), Seif et al. (2006), and Burger et al. (2013). The positions of tRNA genes were found using ARWEN (Laslett and Canbäck, 2008). Finally, the maps of the mitochondrial genomes were drawn using the OrganellarGrenomeDRAW (OGDRAW v. 1.3.1) portal (Greiner et al., 2019).
For the 18S inferred phylogeny, the dataset from Glücksman et al. (2013) was appended with the 18S gene of A. sigmoides from Sugluk Inlet. Sequences were aligned using MAFFT 7 (Katoh and Standley, 2013; auto option) and trimmed using trimAl (Capella-Gutiérrez et al., 2009; automated1 option); the resulting alignment was 1,613 bp long. The best model of evolution (TrN + I + G4 model) was obtained on this alignment using Model-Test-NG (Darriba et al., 2020) with the default parameters. This model works well for datasets originating from different types of genomes and is used to find the best model on separate alignments before concatenation. The Maximum-Likelihood phylogenetic analysis was conducted using IQ-TREE v.2.2.0 (Minh et al., 2020) with 10,000 ultra-fast bootstrap replicates. Following the protocol in Glücksman et al. (2013), the tree was rooted with taxa representing Mantamonadida and Apusomonadida.
For the mitochondrial protein inferred phylogeny, amino-acid sequences of the proteins encoded by atp6, atp8, atp9, cob, cox2, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, and nad9 were extracted from the mitogenomes of A. sigmoides, M. jakobiformis (AF295546), M. californiana (KP165387), C. okellyi (represented by two contigs, KP165390 + KP165391), and J. libera (KC353355) as well as from the sequence registered as PALT01000012 on GenBank after it had been curated based on our findORF analysis, as explained in the Results section. The protein sequences used for the phylogeny were selected according to three criteria: they are present in all species (excluding, for example, genes of the complex II), not fused into a single open-reading frame (thus excluding cox1 and cox3), and easily alignable (excluding, for example, atp4). The protein sequences were aligned separately using MAFFT 7, trimmed using trimAl (identical options), and then concatenated using Phyutility 2.7.1 (Smith and Dunn, 2008). The size of the resulting alignment was 3,482 amino acids. The best model of evolution, the MTZOA+G4 + F model, was obtained with Model-Test-NG using the default parameters on the concatenated alignment. The phylogeny was inferred using IQ-TREE v.2.2.0 with 1,000 ultra-fast bootstrap replicates.
We then searched the nine files containing the metagenomic co-assemblies obtained in the frame of the TARA project from the Arctic Ocean and sub-Arctic North Atlantic Ocean samples (project PRJEB41575) downloaded from ENA.4 Each file was submitted to a blastn query using the mitogenome of A. sigmoides with a E-value filter of 1e−100. Contigs of interest were extracted from the assembly files and submitted to megablast queries online to test for possible extended northern distribution of the species.
After assembly and trimming, a 5,062 bp contig was obtained and its sequence was deposited on GenBank (OR380967). The sizes of the different components of the operon as identified by Rfam are listed in Table 1. It is unclear whether the 28S gene was complete, but we failed at extending the fragment further.
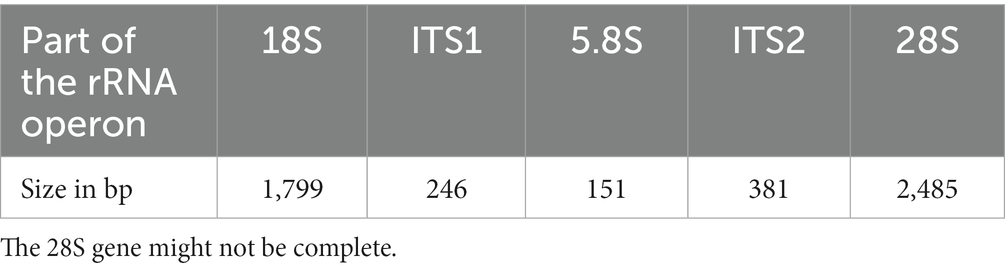
Table 1. Sizes of the different genes and internal transcribed spacers in the rRNA operon of Ancyromona sigmoides from the Sugluk Inlet.
The BLASTn analysis of the 18S gene showed 98.66% similarity (2 polymorphisms) with the sequence EU349231 obtained by Cavalier-Smith et al. (2008) from the B70/CCAP 1958/3 reference strain. The whole cluster was 98.66% similar to the sequence MW872731, which corresponds to the partial 3,363 bp rRNA operon cluster from the same strain as the EU349231 sequence. Most of the differences were within the internal transcribed spacers 1 (92.21% similarity) and 2 (92.61% similarity) and consisted of both SNPs and indels.
A detailed tree of available Apusomonadida 18S rRNA sequences (available as a Newick file; see data availability statement) distinguished a highly supported branch (cluster) containing Ancyromonas spp. (Figure 1). It clearly associates A. sigmoides from Sugluk Inlet with other strains, including the type material from Cavalier-Smith et al. (2008). As previously observed by Glücksman et al. (2013), specimens from the Northern Hemisphere, including the Baltic Sea, the vicinities of Svalbard, Sugluk Inlet, and the White Sea can be distinguished from other strains that were described as A. sigmoides from the Angola Basin in Scheckenbach et al. (2005) and Wylezich et al. (2010). The latter strains appear closer to Ancyromonas kenti Glücksman and Cavalier-Smith as defined by Glücksman et al. (2013). It should be noted that the names (nomenclature) in the tree used for the species correspond to the original GenBank references and were not modified, as mentioned by Glücksman et al. (2013).

Figure 1. Ancyromonas spp. phylogenetic position, derived from the 18S rRNA gene-inferred Maximum-Likelihood tree. The values at the nodes indicate the support. The scale represents the number of substitutions per site. The complete phylogeny was performed with 34 taxa (doi: 10.5281/zenodo.8208290). The complete tree was rooted with the cluster containing Apusomonadida and Mantomonadida.
As the small multiple copies of mitogenomes can be retained in RNAseq data, we performed a BLASTn analysis using the putative cox1 gene sequence of A. sigmoides as a query against the assembled transcriptome from Brown et al. (2018) returned a 5,466 bp sequence with a coverage of 345.26X. This contig derived from the assembled transcriptome perfectly aligned with our assembled mitogenome. For the corresponding cox1 gene, a total of 12 SNPs out of 1986 bp were found, leading to a single amino-acid change in the predicted protein sequence.
The contig identified as the mitogenome of A. sigmoides is 41,889 bp long (GenBank: OR393254). Because of the suspected presence of repetitions at its ends, the complete mitogenome may be larger; however, to facilitate reading, we have represented the mitogenome as circular (Figure 2). Annotation of the protein coding genes (PCG) was done using code 11, with all PCG starting with ATG except for rpl1 (starts with GTG). The genome is gene-rich and encodes 38 PCG and five non-conserved open-reading frames (ORF). Two rRNA genes were detected (rns and rnl) but we failed to identify a putative 5S rRNA gene. A total of 24 tRNA genes were also found. The detailed gene composition is provided in Table 2 and Figure 2.
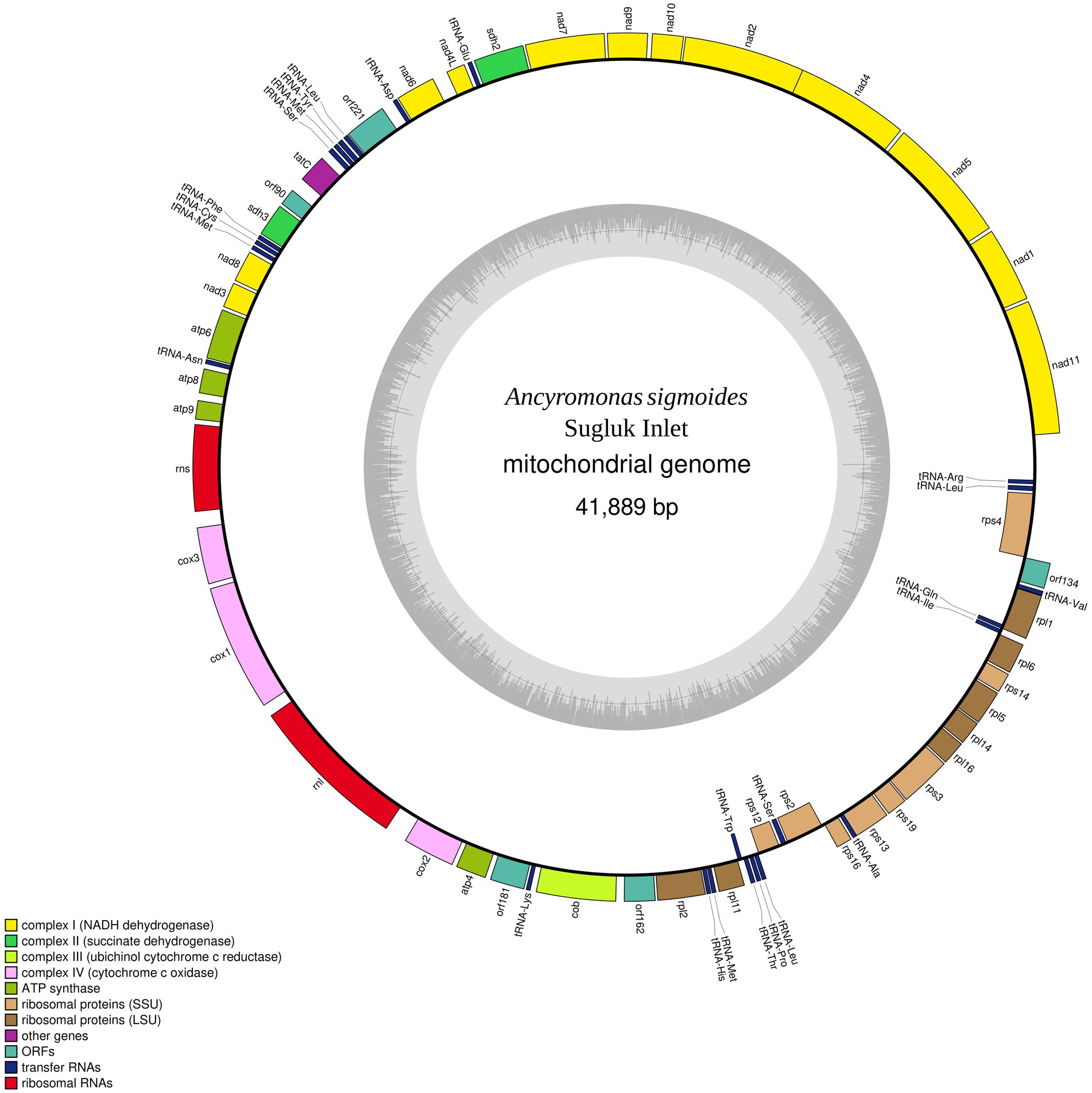
Figure 2. Map of the mitochondrial genome of Ancyromonas sigmoides from the Sugluk Inlet.
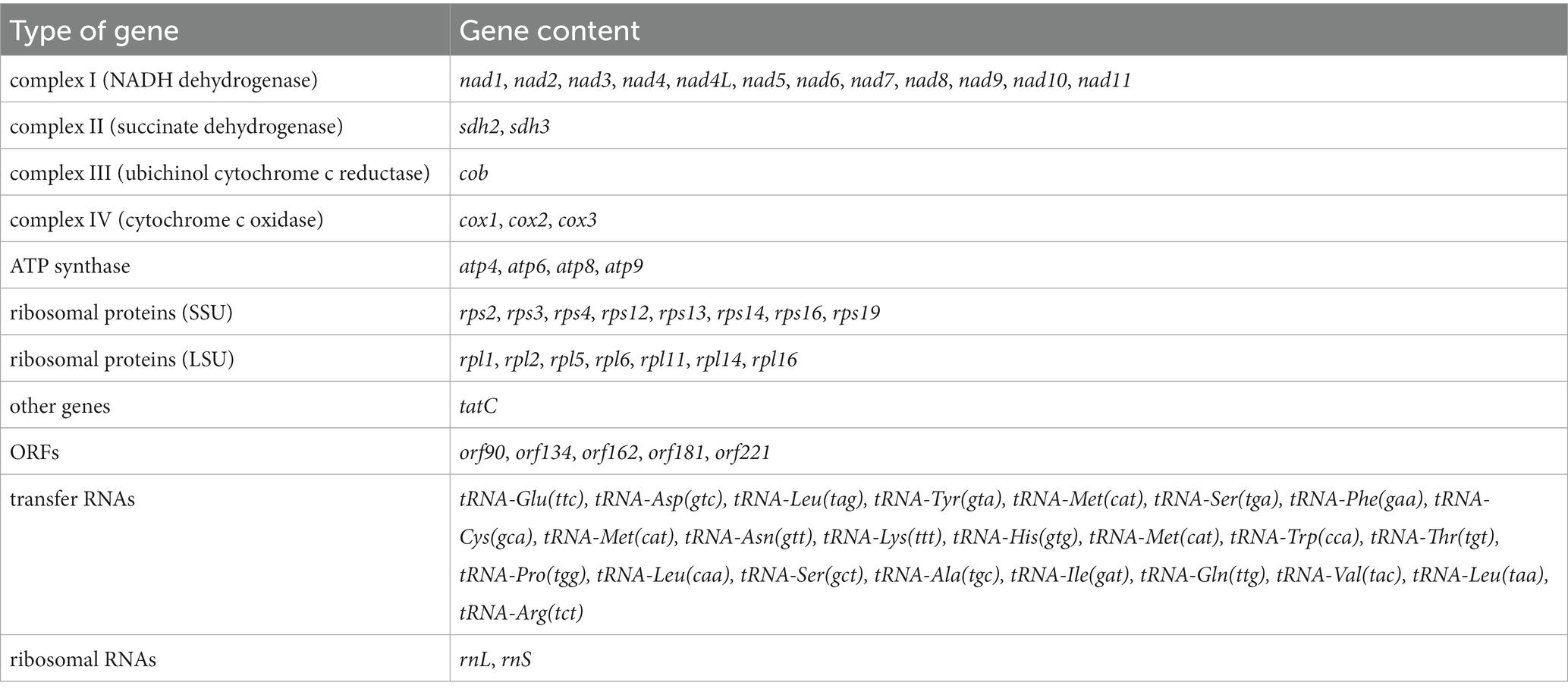
Table 2. Gene composition of the mitochondrial genome of Ancyromona sigmoides from Sugluk Inlet.
Among the 24 tRNA gene products, 19 had a normal cloverleaf structure and 5 had a long variable loop (2 tRNA-Leu, 2 tRNA-Ser, and 1 tRNA-Tyr). It is worth noting that three tRNA-Leu (different anticodons), three tRNA-Met (identical anticodons), and two tRNA-Ser (different anticodons) were detected.
No inverted repeat-like structure was detected in A. sigmoides, unlike the mitogenome of M. jakobiformis.5 The A. sigmoides mitogenome was richer than any of its homologs in the Malawimonadidae, in terms of NADH dehydrogenase subunit genes, as it also contained nad7, nad8, nad10, and nad11. The number of ribosomal protein genes appears to be variable, with the lowest number being recorded for G. okellyi. A. sigmoides appears to be the only species to encode rps16 and rpl1. A tatC gene, coding for a putative SecY-independent transporter protein, was found only in A. sigmoides and M. jakobiformis. Both species of Malawimonas spp. contain ccmF and ccmC genes. All four species (A. sigmoides, M. jakobiformis, M. californiana and G. okellyi) display the three cytochrome c oxidase genes, cox1, cox2, and cox3; however, G. okellyi is the only one in which cox1 and cox3 are fused into a single ORF. Finally, A. sigmoides is the only species in the phylogeny whose mitogenome has retained two genes related to the succinate dehydrogenase complex, namely, sdh2 and sdh3.
Our blastp queries of the Sdh2 and Sdh3 proteins from A. sigmoides returned as the best-matched proteins ascribed to Rhodovulum sp., with e-values/identities of 7e-147/78.12% and 9e-23/35.26%, respectively. A deeper enquiry on the genome sequence from which these proteins were derived showed that it corresponds to a 39,016 bp contig of a metagenomic assembly (PALT01000012) based on the Mediterranean Sea samples collected in the frame of the Tara Ocean (Tully et al., 2018). The TARA data were automatically annotated using the NCBI Prokaryotic Genome Annotation Pipeline. The contig’s size, gene content, and gene organization strongly suggested that it might instead belong to the mitochondrial genome of a basal eukaryote, likely an Ancyromonadida. We annotated this sequence using the same tools and parameters as for A. sigmoides. The gene content appeared nearly identical when compared to the A. sigmoides mitogenome (Figure 3). All genes of the complexes I to IV were shared by the two mitogenomes. The differences were observed for the ribosomal proteins, as rps10 was not found in A. sigmoides by the annotation tool but possibly corresponding to orf162. In contrast, rps4 and rpl1 were not found in PALT01000012 but could possibly correspond to orf200 in the case of rpl1. In addition, we noted that some unidentified ORFs clustered with conserved protein-coding genes within both genomes. This is the case for orf221/orf177, orf181/orf194, and orf90/orf83 in A. sigmoides/PALT01000012. Although poorly informative, this might still be an indication that these ORFs correspond to functional genes that remain to be identified (Table 3).
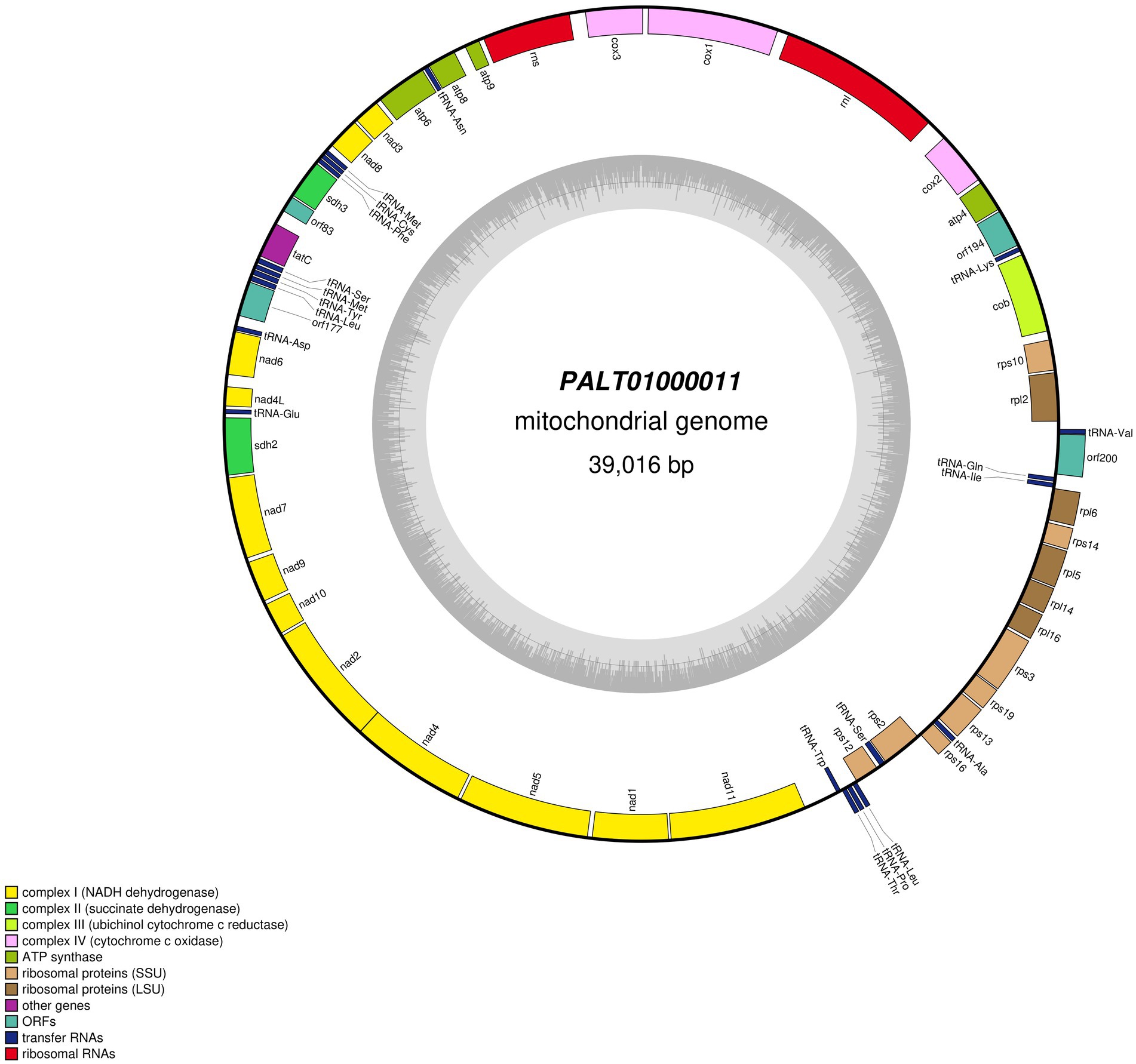
Figure 3. Map of the mitochondrial genome of the sequence labeled in GenBank as PALT01000011 after re-annotation.
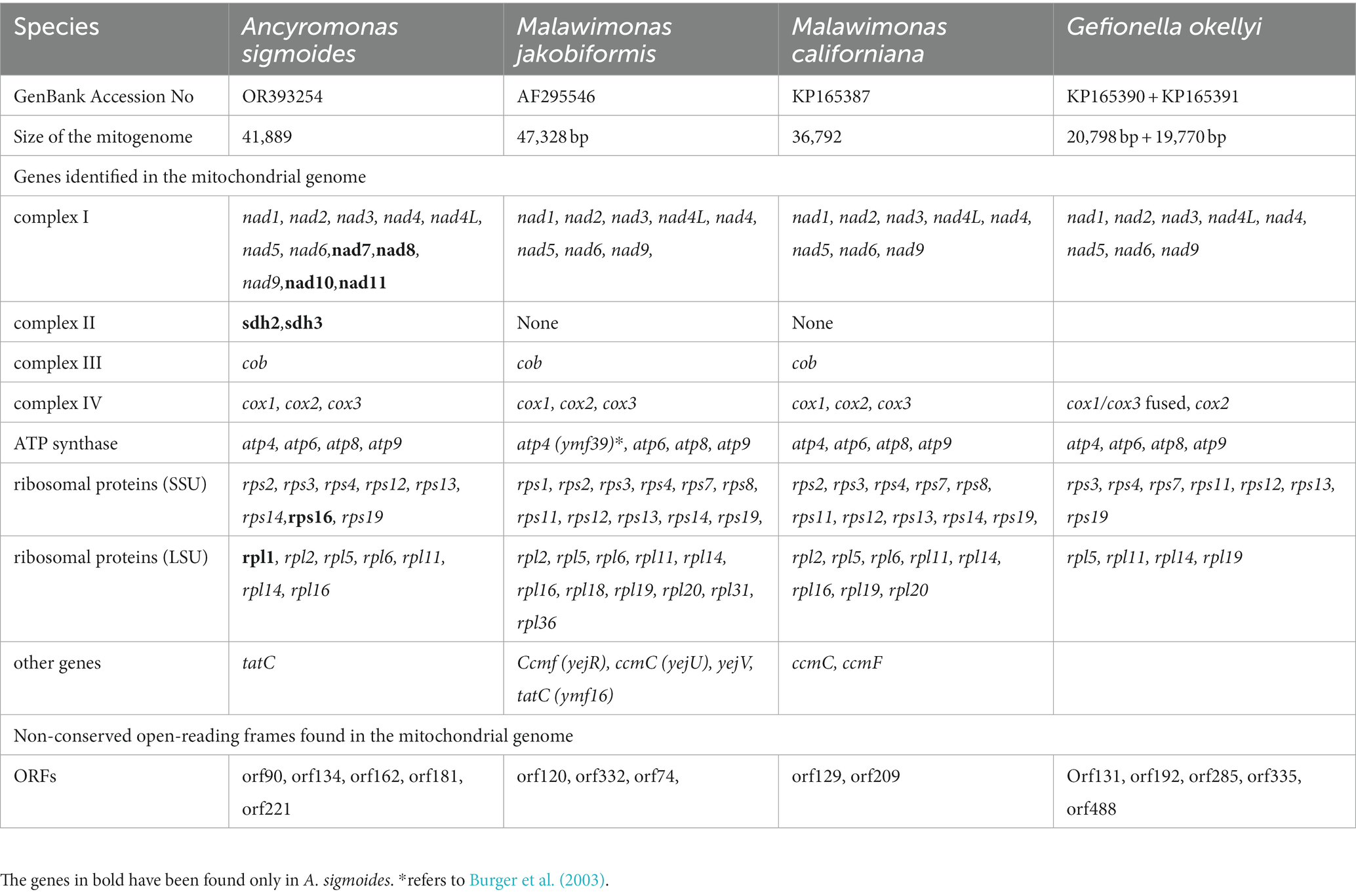
Table 3. Comparison between the protein-coding genes identified and open-reading frames found in the mitochondrial genomes of Ancyromonas sigmoides, Malawimonas jakobiformis, Malawimonas californiana, and Gefionella okellyi.
The phylogenetic tree clearly associated A. sigmoides with PALT01000012, separating them from Malawimonadidae, which formed a highly supported cluster of their own (Figure 4). However, the tree did not display bootstrap values for the node separating Ancyromonadida and Malawimonadidae. To check whether this result could be linked to the software used, a separate phylogenetic analysis of the same dataset was carried out using RAxML 8.0 (Stamatakis, 2014) with the MTZOA+G4 model of evolution. The best tree computed for 100 bootstrap replicates led to identical results. It is thus likely that, at this taxonomic scale, our dataset was not sufficient to resolve phylogenetic relationships, as pointed out by Brown et al. (2018). Nevertheless, the proximity between A. sigmoides and PALT01000012 supports our assumption that the latter sequence belongs to the mitochondrial genome of Ancyromonadida.
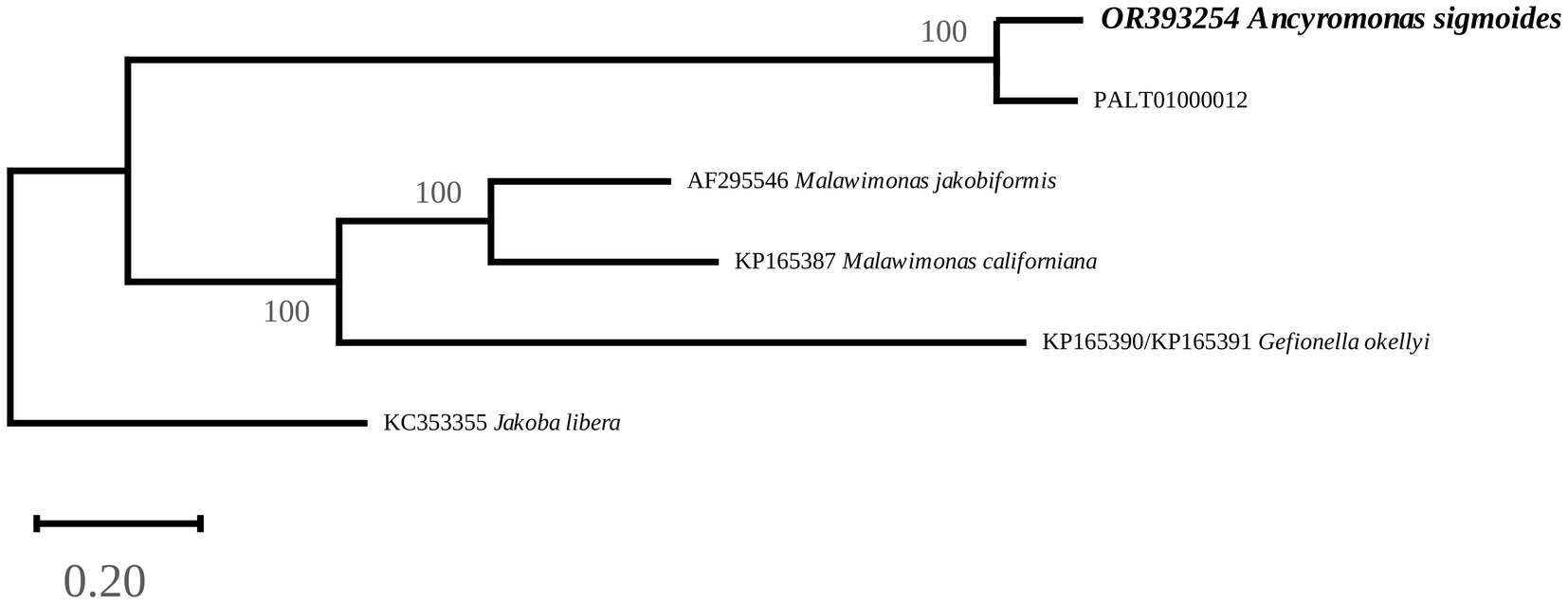
Figure 4. Maximum-Likelihood phylogenetic tree inferred from an alignment of 13 protein sequences. The values at the nodes indicate the support. The scale represents the number of substitutions per site. Jakoba libera was used as an outgroup.
We attempted to use the filtering parameters described above, to assess the presence of A. sigmoides among the metagenomes in PRJEB41575 from the Arctic Tara data. Most of the contigs returning positive mitochondria results belonged to various stramenopiles, including diatoms while others showed limited identity (approximately 85%) with an unidentified organism of the order Telonemida Cavalier-Smith 1993 (MN082145). A single fragment could possibly be ascribed to a protist related to Ancyromonadida. This fragment from the biosample SAMEA11025526, analysis ERZ4109989, was from co-assembly of multiple station mesopelagic and mixed layer samples. The contig labeled as k117_6354597 was 2,156 bp long and returned a best megablast query to A. sigmoides with e value 4e−110 and 85.43% identity. The contig contained three ORFs that correspond to nad9, nad10, and atp8. Blastp queries of these ORFs returned as best results A. sigmoides followed by P ALT01000012 as discussed above. It is worth noting that, although nad9 and nad10 are adjacent in A. sigmoides and PALT01000012, atp8 is not. Overall, the organism represented by contig k117_6354597 was distant from the two others.
To the best of our knowledge, it was possible to obtain, characterize, and annotate the complete mitochondrial genome of a sub-arctic-Arctic A. sigmoides for Ancyromonadida. In the near future, we hope to isolate A. sigmoides from the diatom culture and cultivate it as a monoclonal strain for further investigation.
Unlike the mitogenomes of Malawimonadidae, the mitogenomes of A. sigmoides has retained two succinate dehydrogenase genes (sdh2 and sdh3). These genes have been found in the mitogenomes of other ancient lineages, such as the Jakobida, which include the previously mentioned J. libera, H. aroides, and R. americana. In these species, it is worth noting that three sdh genes are present, the third being sdh4. It is possible that one of the non-identified ORFs of A. sigmoides represents a poorly conserved version of this gene; however, in the absence of deeper investigation, it can also be speculated that this gene has been lost or more likely transferred to the nuclear genome. Sequencing additional mitochondrial genomes of Ancyromonadida should provide clues regarding the presence and number of succinate dehydrogenase genes among them.
This is not the first time that specimens ascribed to A. sigmoides have been found in cold waters of the North, as illustrated in our 18S-inferred phylogeny. Beyond the strain isolated by Mylnikov, a specimen labeled as A. sigmoides strain HFCC104 by Scheckenbach et al. (2005) and Wylezich et al. (2010) was isolated off the western coast of Svalbard. Based on our findings and the research conducted by Glücksman et al. (2013) it is evident that the Arctic and Northern Hemisphere strains are distinctly different from those found in the Angola Basin, which were labeled as A. sigmoides by Scheckenbach et al. (2005, 2006) but mentioned as Ancyromonas sp. by Glücksman et al. (2013). Similarly, the previous studies conducted by Scheckenbach et al. (2006) led to separate freshwater and seawater strains ascribed to A. sigmoides based on molecular phylogeny. It is possible that further studies will reveal the cryptic diversity among Ancyromonadida, with A. sigmoides having geographically restricted representatives in the Arctic and North Atlantic regions, as previously observed in algal isolates (Dorrell et al., 2022). The association with a sea-ice diatom culture is also intriguing and could suggest that this strain is potentially a mutualistic partner or first year ice specialist, and as such, of relevance for this threatened Arctic ecosystem. The use of a mitochondrial genome has advantages, as mitochondrial genes evolve faster than nuclear rRNA genes, thus enabling work at the population level of a species. With this aim, it would be informative to compare the mitogenomes of different strains of A. sigmoides from the Arctic and North Atlantic regions.
Finally, our study suggests that there might be unsuspected genomic information in GenBank that cannot be reached by taxonomical lineage queries on the database. When such results from general blastn searches are anomalous, a second deeper exploration of these sequences is worth carrying out, and the annotation could be verified with the tools dedicated to eukaryotes. In our case, we showed the conserved presence of the two succinate dehydrogenase genes and non-identified ORFs clustering with conserved PCG within the sequences of our Sugluk Inlet A. sigmoides and an environmental construct. Future studies could extend targeted datamining; searching for additional mitogenomes belonging to diverse protists in the TARA Arctic metagenomes and other large environmental databases.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, OR380967 and OR393254.
RG: Investigation, Writing – original draft. SH: Investigation, Writing – review & editing. CG: Investigation, Writing – review & editing. CLe: Investigation, Writing – review & editing. MT: Investigation, Writing – review & editing. CO: Investigation, Writing – review & editing. BB: Investigation, Writing – review & editing. RL: Investigation, Writing – review & editing. JG: Investigation, Writing – review & editing. MP: Investigation, Writing – review & editing. CLo: Funding acquisition, Project administration, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study received funding from the European Union’s Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement no: 846860 (grant to SH and CLo). CLe and MT were supported by the grant RGPIN-2017-04506 from the Natural Sciences and Engineering Research Council of Canada (NSERC). BB and CO were supported by the “Programme d’appui aux plateformes technologiques stratégiques” from the Ministère de l’Économie, de l’Innovation et de l’Énergie Québec. CLo received additional funds from NSERC Discovery and Northern Supplement, with support from the ArcticNet, a Network of Centers of Excellence of Canada, Canada First Research Excellence Fund to Sentinelle Nord and Fonds de Recherche du Québec Nature et Technologies (FRQNT) to Québec Océan. Comput Canada access is also acknowledged.
We thank Guillaume Massé for his contribution in the isolation and cultivation of the diatoms. Additionally, we extend our appreciation to the community of Salluit for their generous support, logistical assistance, and meaningful discussions.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. ^ https://www.ibis.ulaval.ca/en/services-2/genomic-analysis-platform/
2. ^ https://www.genomequebec.com/
3. ^ https://github.com/ncbi/sra-tools
4. ^ https://www.ebi.ac.uk/ena/browser/view/PRJEB41575
5. ^ https://megasun.bch.umontreal.ca/ogmp/projects/mjako/gen.html
Boratyn, G. M., Schäffer, A. A., Agarwala, R., Altschul, S. F., Lipman, D. J., and Madden, T. L. (2012). Domain enhanced lookup time accelerated BLAST. Biol. Direct 7, 12–14. doi: 10.1186/1745-6150-7-12
Brown, M. W., Heiss, A. A., Kamikawa, R., Inagaki, Y., Yabuki, A., Tice, A. K., et al. (2018). Phylogenomics places orphan protistan lineages in a novel eukaryotic super-group. Genome Biol. Evol. 10, 427–433. doi: 10.1093/gbe/evy014
Burger, G., Gray, M. W., Forget, L., and Lang, B. F. (2013). Strikingly bacteria-like and gene-rich mitochondrial genomes throughout jakobid protists. Genome Biol. Evol. 5, 418–438. doi: 10.1093/gbe/evt008
Burger, G., Lang, B. F., Braun, H. P., and Marx, S. (2003). The enigmatic mitochondrial ORF ymf39 codes for ATP synthase chain b. Nucleic Acids Res. 31, 2353–2360. doi: 10.1093/nar/gkg326
Bushmanova, E., Antipov, D., Lapidus, A., and Prjibelski, A. D. (2019). rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. GigaScience 8:giz100. doi: 10.1093/gigascience/giz100
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics (Oxford, England) 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Cavalier-Smith, T., Chao, E. E., Stechmann, A., Oates, B., and Nikolaev, S. (2008). Planomonadida Ord. Nov. (Apusozoa): ultrastructural affinity with Micronuclearia podoventralis and deep divergences within Planomonas gen. Nov. Protist 159, 535–562. doi: 10.1016/j.protis.2008.06.002
Chen, S. (2023). Ultrafast one-pass FASTQ data preprocessing, quality control, and deduplication using fastp. iMeta 2:e107. doi: 10.1002/imt2.107
Chen, S., Zhou, Y., Chen, Y., and Gu, J. (2018). Fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics (Oxford, England) 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2020). ModelTest-NG: a new and scalable tool for the selection of DNA and protein evolutionary models. Mol. Biol. Evol. 37, 291–294. doi: 10.1093/molbev/msz189
Dorrell, R. G., Kuo, A., Füssy, Z., Richardson, E. H., Salamov, A., Zarevski, N., et al. (2022). Convergent evolution and horizontal gene transfer in Arctic Ocean microalgae. Life Science Alliance 6:e202201833. doi: 10.26508/lsa.202201833
Gagnon, J. (2004). Création d’outils pour l’automatisation d’analyses phylogénétiques de génomes d’organites [Development of metagenomics tools for the phylogenetic analyzes of organellar genomes] 2004. M.Sc. Dissertation. University of Laval. Available at: https://www.collectionscanada.gc.ca/obj/s4/f2/dsk3/QQLA/TC-QQLA-21635.pdf
Gastineau, R., Konucu, M., Tekdal, D., Lemieux, C., Turmel, M., Witkowski, A., et al. (2021). A gene-rich and compact chloroplast genome of the green alga Nephroselmis pyriformis (N. Carter) Ettl 1982 from the shores of Mersin (eastern Mediterranean Sea). Mitochondrial DNA Part B 6, 308–310. doi: 10.1080/23802359.2020.1866461
Glücksman, E., Snell, E. A., and Cavalier-Smith, T. (2013). Phylogeny and evolution of Planomonadida (Sulcozoa): eight new species and new genera Fabomonas and Nutomonas. Eur. J. Protistol. 49, 179–200. doi: 10.1016/j.ejop.2012.08.007
Greiner, S., Lehwark, P., and Bock, R. (2019). OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Hamedi, C., Gastineau, R., Lemieux, C., Turmel, M., Witkowski, A., Baba, H. M., et al. (2019). Complete chloroplast genome of the diatom Skeletonema pseudocostatum from the Western Mediterranean coast of Algeria. Mitochondrial DNA Part B 4, 1091–1092. doi: 10.1080/23802359.2019.1586481
Heiss, A. A., Kolisko, M., Ekelund, F., Brown, M. W., Roger, A. J., and Simpson, A. G. (2018). Combined morphological and phylogenomic re-examination of malawimonads, a critical taxon for inferring the evolutionary history of eukaryotes. R. Soc. Open Sci. 5:171707. doi: 10.1098/rsos.171707
Heiss, A. A., Walker, G., and Simpson, A. G. (2010). Clarifying the taxonomic identity of a phylogenetically important group of eukaryotes: Planomonas is a junior synonym of Ancyromonas. J. Eukaryot. Microbiol. 57, 285–293. doi: 10.1111/j.1550-7408.2010.00477.x
Kalvari, I., Nawrocki, E. P., Ontiveros-Palacios, N., Argasinska, J., Lamkiewicz, K., Marz, M., et al. (2021). Rfam 14: expanded coverage of etagenomics, viral and microRNA families. Nucleic Acids Res. 49, D192–D200. doi: 10.1093/nar/gkaa1047
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Laslett, D., and Canbäck, B. (2008). ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics (Oxford, England) 24, 172–175. doi: 10.1093/bioinformatics/btm573
Leigh, J., and Lang, B. F. (2004). Mitochondrial 3’ tRNA editing in the jakobid Seculamonas ecuadoriensis: A novel mechanism and implications for tRNA processing. RNA (New York, N.Y) 10, 615–621. doi: 10.1261/rna.5195504
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Mylnikov, A. P. (1990). Characteristic features of the ultrastructure of colourless flagellate Heteromita sp. Citologiâ 32, 567–571.
Roger, A. J., Muñoz-Gómez, S. A., and Kamikawa, R. (2017). The origin and diversification of mitochondria. Current 27, R1177–R1192. doi: 10.1016/j.cub.2017.09.015
Scheckenbach, F., Wylezich, C., Mylnikov, A. P., Weitere, M., and Arndt, H. (2006). Molecular comparisons of freshwater and marine isolates of the same morphospecies of heterotrophic flagellates. Appl. Environ. Microbiol. 72, 6638–6643. doi: 10.1128/AEM.02547-05
Scheckenbach, F., Wylezich, C., Weitere, M., Hausmann, K., and Arndt, H. (2005). Molecular identity of strains of heterotrophic flagellates isolated from surface waters and deep-sea sediments of the South Atlantic based on SSU rDNA. Aquat. Microb. Ecol. 38, 239–247. doi: 10.3354/ame038239
Seif, E., Cadieux, A., and Lang, B. F. (2006). Hybrid E. coli—Mitochondrial ribonuclease P RNAs are catalytically active. RNA 12, 1661–1670. doi: 10.1261/rna.52106
Smith, S. A., and Dunn, C. W. (2008). Phyutility: a phyloinformatics tool for trees, alignments and molecular data. Bioinformatics 24, 715–716. doi: 10.1093/bioinformatics/btm619
Solak, C. N., Gastineau, R., Lemieux, C., Turmel, M., Gorecka, E., Trobajo, R., et al. (2021). Nitzschia anatoliensis sp. Nov., a cryptic diatom species from the highly alkaline Van Lake (Turkey). PeerJ 9:e12220. doi: 10.7717/peerj.12220
Stamatakis, A. (2014). RaxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics (Oxford, England) 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tully, B. J., Graham, E. D., and Heidelberg, J. F. (2018). The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Scientific Data 5:170203. doi: 10.1038/sdata.2017.203
Keywords: Ancyromonas sigmoides , basal protist, Nunavik, Arctic, mitogenome, phylogeny
Citation: Gastineau R, Harðardóttir S, Guilmette C, Lemieux C, Turmel M, Otis C, Boyle B, Levesque RC, Gauthier J, Potvin M and Lovejoy C (2023) Mitochondrial genome sequence of the protist Ancyromonas sigmoides Kent, 1881 (Ancyromonadida) from the Sugluk Inlet, Hudson Strait, Nunavik, Québec. Front. Microbiol. 14:1275665. doi: 10.3389/fmicb.2023.1275665
Edited by:
Richard Allen White III, University of North Carolina at Charlotte, United StatesReviewed by:
Salvador Mirete, Spanish National Research Council (CSIC), SpainCopyright © 2023 Gastineau, Harðardóttir, Guilmette, Lemieux, Turmel, Otis, Boyle, Levesque, Gauthier, Potvin and Lovejoy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Romain Gastineau, cm9tYWluLmdhc3RpbmVhdUB1c3ouZWR1LnBs; Connie Lovejoy, Y29ubmllLmxvdmVqb3lAYmlvLnVsYXZhbC5jYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.