 Duhyung Lee
Duhyung Lee Dong Hyun Kim
Dong Hyun Kim Hyejun Seo1,2,4
Hyejun Seo1,2,4 Bum-Joon Kim
Bum-Joon Kim- 1Department of Microbiology and Immunology, College of Medicine, Seoul National University, Seoul, Republic of Korea
- 2Cancer Research Institute, College of Medicine, Seoul National University, Seoul, Republic of Korea
- 3Department of Biomedical Sciences, College of Medicine, Seoul National University, Seoul, Republic of Korea
- 4Seoul National University Medical Research Center, Seoul, Republic of Korea
- 5Brain Korea 21 FOUR Biomedical Science Project, Seoul National University College of Medicine, Seoul, Republic of Korea
- 6Liver Research Institute, College of Medicine, Seoul National University, Seoul, Republic of Korea
Despite the great diversity of malonate semialdehyde decarboxylases (MSADs), one of five subgroups of the tautomerase superfamily (TSF) found throughout the biosphere, their distribution among strains within the genus Mycobacterium remains unknown. In this study, we sought to investigate the phylogenetic distribution of MSAD genes of mycobacterial species via genome analysis of 192 different reference Mycobacterium species or subspecies retrieved from NCBI databases. We found that in a total of 87 of 192 strains (45.3%), MSAD-1 and MSAD-2 were distributed in an exclusive manner among Mycobacterium species except for 12 strains, including Mycobacterium chelonae members, with both in their genome. Of note, Mycobacterium strains better adapted to the host and of high virulence potential, such as the Mycobacterium tuberculosis complex, Mycobacterium leprae, Mycobacterium marinum, Mycobacterium ulcerans, and Mycobacterium avium subsp. paratuberculosis, had no orthologs of MSAD in their genome, suggesting MSAD loss during species differentiation in pathogenic slow-growing Mycobacterium. To investigate the MSAD distribution among strains of M. avium subspecies, the genome sequences of a total of 255 reference strains from the four subspecies of M. avium (43 of subspecies avium, 162 of subspecies hominissuis, 49 of subspecies paratuberculosis, and 1 of subspecies silvaticum) were further analyzed. We found that only 121 of 255 strains (47.4%) had MSADs in their genome, with none of the 49 M. avium subsp. paratuberculosis strains having MSAD genes. Even in 13 of 121 M. avium strains with the MSAD-1 gene in their genome, deletion mutations in the 98th codon causing premature termination of MSAD were found, further highlighting the occurrence of MSAD pseudogenization during species or subspecies differentiation of M. avium. In conclusion, our data indicated that there are two distinct types of MSADs, MSAD-1 and MSAD-2, among strains in the Mycobacterium genus, but more than half of the strains, including pathogenic mycobacteria, M. tuberculosis and M. leprae, have no orthologs in their genome, suggesting MSAD loss during host adaptation of pathogenic mycobacteria. In the future, the role of two distinct MSADs, MSAD-1 and MSAD-2, in mycobacterial pathogenesis or evolution should be investigated.
1. Introduction
The genus Mycobacterium is composed of more than 200 established and validated species and subspecies belonging to the phylum Actinobacteria, defined by a rod-shaped morphology, acid fastness, unusual cell walls containing mycolic acids, and relatively high genomic DNA G+C contents (∼ 61 to 71%) (Vincent Lévy-Frébault and Portaels, 1992; Jankute et al., 2015; Gupta et al., 2018). Generally, Mycobacterium can be separated into two groups according to their pathogenic potential: human pathogens, including Mycobacterium tuberculosis and Mycobacterium leprae, which cause tuberculosis and leprosy, respectively, and nontuberculous Mycobacterium (NTM), which are environmental mycobacteria that do not cause tuberculosis, as the name suggests, and are often nonpathogenic to humans and animals (Gagneux, 2018; Johansen et al., 2020; van Hooij and Geluk, 2021). The genus can be further separated into two groups, slow-growing Mycobacterium (SGM) (i.e., requiring more than 7 days to form visible colonies on solid agar) and rapid-growing Mycobacterium (RGM) requiring <7 days to form colonies (Lotti and Hautmann, 1993). Although most NTMs are found in the environment, such as soil or natural and drinking water sources, a few species, including the Mycobacterium avium complex (MAC) and Mycobacterium abscessus, often cause serious lung diseases through infections in humans (Nishiuchi et al., 2017; Johansen et al., 2020; Ratnatunga et al., 2020). The increases in immunosuppressive drug use, broad-spectrum antibiotic therapy, and patients with underlying lung diseases, including cystic fibrosis and bronchiectasis, have contributed to the recent rise in the global incidence of NTM infections in developed countries (Petrini, 2006; Chalmers et al., 2018; To et al., 2020).
The tautomerase superfamily (TSF) consists of more than 11,000 members throughout the biosphere, which can be classified into five major categories: 4-oxalocrotonate tautomerase (4-OT), 5-(carboxymethyl)-2-hydroxymuconate isomerase (CHMI), macrophage migration inhibitory factor (MIF), cis- and trans-3-chloroacrylic acid dehalogenase (cis-CaaD and CaaD, respectively), and malonate semialdehyde decarboxylase (MSAD) (Poelarends et al., 2008a; Davidson et al., 2018). Despite some exceptions, they share a β-α-β structure with an unusual catalytic amino-terminal proline as a general base. MSAD not only catalyzes the decarboxylation of malonate semialdehyde to produce acetaldehyde but also participates in degradative pathways of 1,3-dichloropropene, a soil fumigant (Poelarends and Whitman, 2004).
To date, structural studies regarding MSAD have been mainly focused on nine organisms, originating from various species such as Pseudomonas pavonaceae 170 (PpMSAD) (Poelarends et al., 2003, 2004, 2005; Almrud et al., 2005), Coryneform bacterium strain fg41 (FG41 MSAD) (Poelarends et al., 2008b; Guo et al., 2013), Lactobacillus casei strain BL23 (IolK) (Poelarends et al., 2008b), Bacillus subtilis strain 168 (YusQ, YodA, and YrdN) (Poelarends et al., 2005), Burkholderia phymatum strain STM815 (Bp4401) (Huddleston et al., 2014), Calothrix sp. PCC 6303(437) (Lancaster et al., 2022), and Rivularia sp. PCC 7116(JJ3) (Lancaster et al., 2022). However, research on the MSADs of Mycobacterium strains is limited.
Comparative genomic studies have revealed that overt human-pathogenic Mycobacterium species, including M. tuberculosis, M. leprae, and Mycobacterium ulcerans, have undergone genome reduction and gene loss since their evolution from the ancestor (Gutierrez et al., 2005; Marri et al., 2006; Gómez-Valero et al., 2007; Qi et al., 2009). Since MSAD plays a key role in bacterial metabolism, it may have had distinct effects on the evolutionary scenario of Mycobacterium species in terms of their groups, pathogenic or environmental strains and slow-growing or rapid-growing status. Therefore, investigation of the phylogenetic distribution of MSAD genes among Mycobacterium strains would provide novel insight into their evolution and pathogenesis.
In the present study, we sought to investigate the phylogenetic distribution of MSAD genes of mycobacterial species via genome analysis of 192 different reference Mycobacterium species or subspecies retrieved from NCBI databases. In addition, to further test our hypothesis of MSAD loss in Mycobacterium strains more adapted to host-associated life, we further checked the distribution of MSADs among 255 MAC strains of the four subspecies of M. avium retrieved from NCBI databases.
2. Materials and methods
2.1. Mycobacterium type strain database
A total of 192 type strains of Mycobacterium with whole genome sequences present in the NCBI taxonomy browser, ATCC (American Type Culture Collection), DSMZ (German Collection of Microorganisms and Cell Cultures), and JCM (Japan Collection of Microorganisms) were selected (Figure 1). Of these, 129 type strains were referenced from a previous study (Gupta et al., 2018), and the other 67 type strains were selected from the ATCC (21 strains), DSMZ (34 strains), and JCM (4 strains). In addition, we selected four newly identified Mycobacterium species, Mycobacterium dioxanotrophicus, Mycolicibacterium nivoides, Mycobacterium terramassiliense, and Mycobacterium senriense (He et al., 2017; Bouam et al., 2018; Dahl et al., 2021; Abe et al., 2022), with their whole genome sequences registered with the NCBI and added them to the database. The whole-genome sequencing project names and accession numbers are listed in Supplementary Table 1.
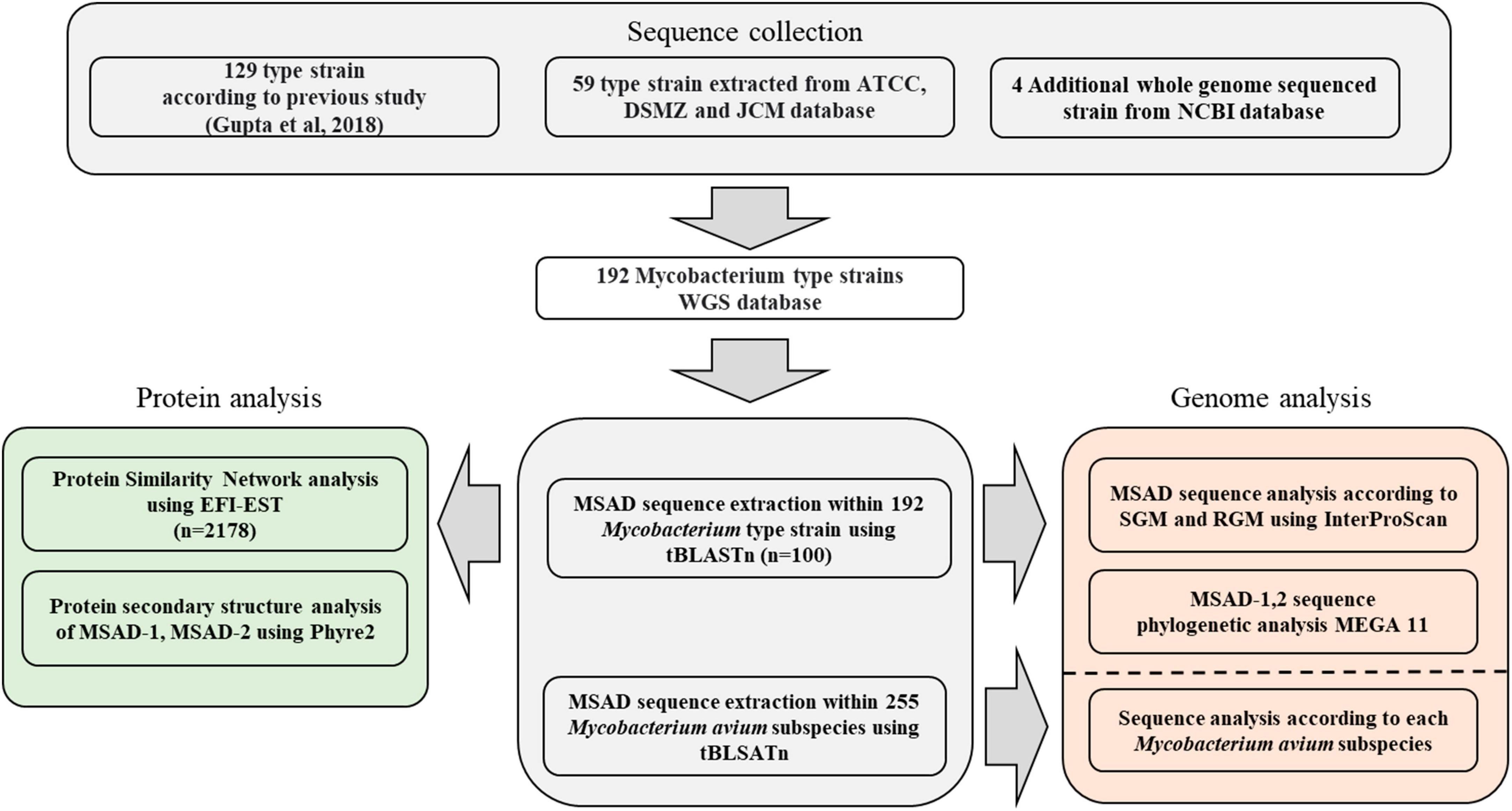
Figure 1. Workflow representing the construction of the Mycobacterium genome sequence database and software used for each analysis in this study.
2.2. MSAD sequence data collection
Using two MSAD sequences (MSAD-1: NCBI accession number OLT81431.1 and MSAD-2: OLT83119.1) of Mycobacterium chelonae subsp. chelonae ATCC 35752T as query sequences, the presence of MSAD proteins was confirmed in 192 Mycobacterium type strains through the tBLASTn algorithm (Supplementary Table 1; Altschul et al., 1990, 1997). Among the BLAST search results, sequences were extracted based on a cutoff of 90% for query coverage and 10–20 for the E-value. MSAD sequences ranging in length from 119 to 137 amino acids were collected. A total of 100 MSAD sequences from 192 Mycobacterium type strains were extracted. The extracted protein sequences of MSAD orthologs are stored in FASTA format (Supplementary Data 1) and their DNA sequences are listed in Supplementary Table 2.
2.3. Protein similarity network of the total MSAD family
A total of 2,078 MSAD sequences, which are widely distributed throughout the biosphere, are registered in the SFLD (Superfamily of Ligand-Binding Protein Database) (Akiva et al., 2014; Davidson et al., 2018). They were trimmed and aligned before being submitted to the Enzyme Function Initiative enzyme similarity tool (EFI-EST) webserver for analysis (Zallot et al., 2019) (Job ID 96068). The EFI-EST was used to create the sequence similarity network (Oberg et al., 2023). We aligned total MSAD protein sequences (2,078 sequences) and mycobacterial MSADs (100 sequences) together and submitted them to the EFI-EST algorithm. Merged MSAD sequences were stored in FASTA format (Supplementary Data 2). In this study, the E-value for SSN Edge calculation was set to 5, and the convergence ratio was set to 0.743. This value decreases from 1.0 for sequences that are very similar (identical) to 0.0 for sequences that are very different (unrelated). Constructed networks were then transferred to the Color SSN utility for Representative Node (RepNode) Networks (Job ID 96072). The 100% identity RepNode network was stored as Supplementary Data 3, and 50% identical sequences were grouped to reduce the total number of nodes. The colored SSN was visualized in Cytoscape (Shannon et al., 2003) (version 3.9.1).
2.4. Protein structure prediction of MSAD
Four MSAD sequences of M. avium (MSAD-1 of slow growing mycobacteria), M. abscessus (MSAD-1 of rapid growing mycobacteria), Mycobacterium terrae (MSAD-2 of slow growing mycobacteria), and Mycobacterium fortuitum (MSAD-2 of rapid growing mycobacteria) were submitted to the phyre2 webserver1 for protein function prediction (Kelley et al., 2015). The structures of four stains of MSAD (two strains of MSAD-1 and two strains of MSAD-2) were determined based on their protein sequences (Supplementary Data 1). The constructed MSAD 3D models were then compared to PDB-registered proteins. The protein derived from the C. bacterium strain fg41 registered in the PDB (PDB ID: 3MJZ) was used as a template to align MSAD sequence of M. abscessus and M. terrae. And the PpMSAD derived from the P. pavonaceae registered in the PDB (PDB ID: 2AAL) was used as a template to align MSAD sequence of M. avium and M. fortuitum. The protein structures created with phyre were analyzed with the PyMOL Molecular Graphics System Schrödinger, Inc. (version 2.5.5). The identified Mycobacterium type strains were used to predict the structure and function of MSAD sequences using InterProScan (Jones et al., 2014).
2.5. MSAD sequence alignment and phylogenetic analysis
A total of 57 MSAD-1 sequences and 43 MSAD-2 sequences from Mycobacterium reference strains were aligned using the MUSCLE method for each DNA sequence and amino acid sequence through the MEGA 11 program (Tamura et al., 2021). The DNA and protein sequence-based phylogenetic trees of MSAD-1 and MSAD-2 were constructed through the maximum-likelihood method. Branch support value was calculated through 100 bootstrap replications. Phylogenetic trees based on 644 bp hsp65 sequences are often used to classify and identify Mycobacterium species (Kim et al., 2005). The hsp65 sequence of M. tuberculosis H37Rv (GenBank accession number M15467) was used to extract the hsp65 sequence from whole genome sequences of the type strains through BLAST (Supplementary Data 4). In this study, two hsp65 sequence-based trees (one for MSAD-1 sequences from 56 mycobacterial strains and the other for MSAD-2 sequences from 43 strains) were constructed through MUSCLE alignment and the maximum-likelihood method. Branch support value was calculated through 100 bootstrap replications. YrdN (MSAD of B. subtilis strain 168) was used as an outgroup in the MSAD tree, while Tsukamurella paurometabola KCTC 9821T (GenBank accession number UHIQ01000001.1) was used as an outgroup in the hsp65 tree.
2.6. Mycobacterium avium subspecies strain MSAD sequence analysis
To analyze MSAD protein retention within M. avium subspecies, a total of 255 whole genome sequences were obtained from the NCBI database, including 43 M. avium subsp. avium sequences, 162 M. avium subsp. hominissuis sequences, 49 M. avium subsp. paratuberculosis sequences, and 1 M. avium subsp. silvaticum sequence. In total, 121 MSAD sequences were extracted from the whole genomes of M. avium subspecies using tBLASTn (Altschul et al., 1997). The MSAD sequences were aligned and analyzed using the MUSCLE method through MEGA 11 (Tamura et al., 2021). Information regarding the accession numbers, metadata, and all publicly available assemblies for the whole genome sequences was also extracted from the NCBI database (Supplementary Data 5). Extracted MSAD sequences were stored in FASTA format (Supplementary Data 6).
2.7. Preparation of Mycobacterium abscessus MSAD-1 protein
Recombinant MSAD-1 protein of M. abscessus (NCBI accession number OLT57519.1) were purified from Escherichia coli as previously described with minor modification (Jeong et al., 2022). Briefly, the DNA sequence of MSAD-1 was amplified from M. abscessus ATCC 19977T using PCR with following primer sets (forward primer, 5′-TTT GGA TCC ATG CCA TTG GTG CGC ATC GAC CTC-3′; reverse primer, 5′-AAA AAG CTT GTG CGC CTG CGG CGG GCA C-3′), and cloned into pET-28a. The expression and purification of MSAD-1 were commercially commissioned by Bionics (Seoul, Republic of Korea). In detail, the protein expression was induced in E. coli Rosetta2 (DE3) strains (Novagen, WI, USA) transformed with pET28a-MSAD-1 by adding 1 mM isopropyl β-D-thiogalactopyranoside (IPTG) at 26°C for 6 h. Cultured bacterial cells were harvested and sonicated for 30 cycles at 70% amplitude. After centrifuge, the supernatant was purified with HisTrap™ HP His tag protein purification columns (Cytiva, MA, USA) for Ni-NTA affinity chromatography via ÄKTA go system (Cytiva, MA, USA). Purified proteins were subjected to endotoxin removal using Pierce™ high-capacity endotoxin removal spin columns (Thermo Scientific, MA, USA) and quantified by Pierce™ chromogenic endotoxin quant kit (Thermo Scientific, MA, USA).
2.8. Induction of pro-inflammatory cytokines, TNF-α and IL-6 on J774A.1 cells by MSAD-1
The murine macrophage cell line, J774A.1, was maintained at 37°C with 5% CO2 in RPMI 1640 supplemented with 10% (v/v) fetal bovine serum (FBS) and 1% penicillin-streptomycin (PS). To induce protein uptake efficiently, J774A.1 cells were starved in reduced serum medium, opti-MEM (Gibco, MT, USA), at 37°C for 1 h. Subsequently, they were incubated with various concentrations of MSAD-1 or 100 ng/ml LPS (Sigma, MO, USA) in RPMI 1640 supplemented with 2% (v/v) FBS and 1% PS at 37°C for either 24 or 48 h. The culture medium was used to measure the levels of the cytokines using an enzyme-linked immunosorbent assay (ELISA) kit (Invitrogen, MA, USA) according to the manufacture’s instructions.
3. Results
3.1. Construction of a novel MSAD protein similarity network including 100 mycobacterial MSAD sequences
To examine the distribution of a total of 100 mycobacterial MSAD orthologs extracted from genome sequences of 192 strains in this study among all the biospheres, we constructed a novel protein similarity network including a total of 2,178 MSAD protein sequences from established MSAD protein sequences (2,078 sequences) (Akiva et al., 2014) and mycobacterial MSADs (100 sequences) extracted in this study and submitted to the EFI-EST algorithm (Zallot et al., 2019; Figure 1). All 2,178 MSADs are grouped into 12 clusters of 472 nodes, which are connected by 7,677 edges, and further clustered into two major groups (colored red and blue) (Figure 2). While one group (colored red) consists of 328 larger nodes of 1,633 sequences, including PpMSAD (Poelarends et al., 2003) and FG41 MSAD (Poelarends et al., 2008b), which have been widely studied for the elucidation of MSAD function, the other group consists of 81 smaller nodes of 453 MSAD sequences. We decided to designate these two groups as MSAD-1 and MSAD-2, respectively. Generally, all 100 mycobacterial MSAD sequences extracted in this study also belonged to these two major groups, MSAD-1 and MSAD-2 (Figure 2). However, there are some discrepancies in their distribution in the protein similarity network. Strains of MSAD-1 are more widely scattered among 13 nodes compared with those of MSAD-2, found at 4 nodes, suggesting more sequence divergence between MSAD-1 strains than between MSAD-2 strains (Figure 2).
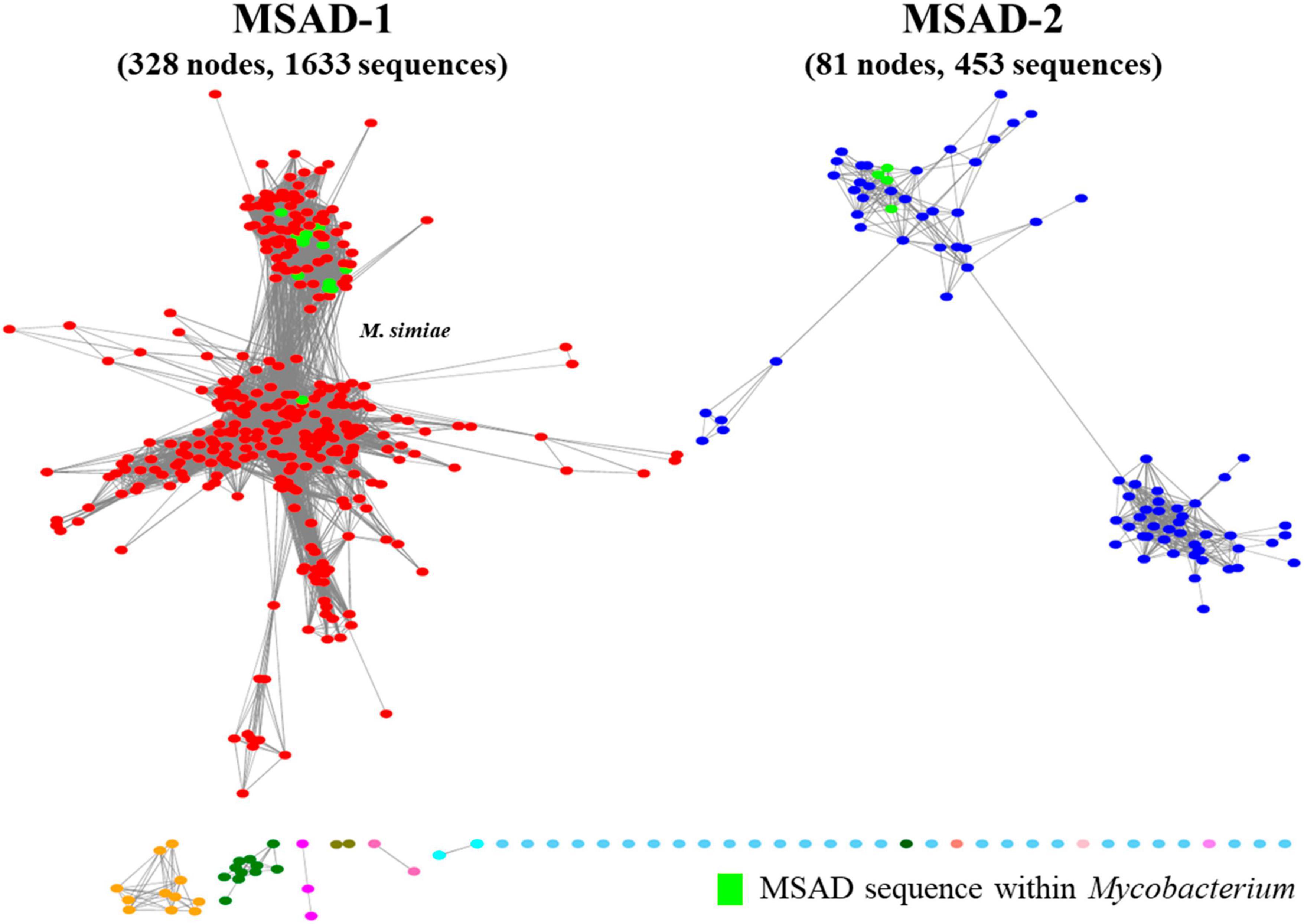
Figure 2. A new malonate semialdehyde decarboxylase (MSAD) protein similarity network including mycobacterial MSADs constructed in this study. Visualized protein similarity network of MSADs within the biosphere. Total MSAD family protein sequences (2,078 sequences) and mycobacterial MSADs (100 sequences) were used to construct the network and the network was visualized through the EFI-EST webserver. MSADs consist of two independent branches, MSAD-1 (red) and MSAD-2 (blue). The MSAD-1 branch consists of 328 nodes with 1,633 sequences. The MSAD-2 branch consists of 81 nodes with 453 sequences. All mycobacterial MSADs were scattered among the MSAD-1 and MSAD-2 groups.
3.2. The distribution of MSAD genes among Mycobacterium strains
Of 192 Mycobacterium reference strains, only fewer than half (87, 45.3%) had MSAD genes in their genomes (105 strains without any MSAD orthologs in their genomes) (Figure 3). The DNA sequences and accession numbers of all 100 MSADs from 87 Mycobacterium strains are presented in Supplementary Table 1. Of all 100 MSAD sequences, 57 and 43 belonged to MSAD-1 and MSAD-2, respectively. Most Mycobacterium strains (75 strains) have a single MSAD in their genomes, MSAD-1 or MSAD-2, in an exclusive manner (74 strains have single copy of MSAD gene and Mycobacterium simiae with two copies of MSAD-1). The remaining 12 species, including the three subspecies M. chelonae subsp. chelonae, M. chelonae subsp. bovis (Kim et al., 2017), and M. chelonae subsp. gwanakae (Kim et al., 2018), have both types of MSADs, MSAD-1 and MSAD-2, in their genomes (Supplementary Table 1). Of note, overt slow-grower human pathogens, including M. tuberculosis, M. leprae, Mycobacterium marinum, M. ulcerans, and M. avium subsp. paratuberculosis, do not have any MSAD genes in their genomes (Supplementary Table 1 and Figure 3). In addition, of 49 slow-growing strains with an MSAD gene, all of those from the Tuberculosis-Simiae clade (emended genus Mycobacterium) have the MSAD-1 type but not the MSAD-2 type in their genomes. However, all members of the Terrae clade (Mycolicibacter gen. nov.) and Triviale clade (Mycolicibacillus gen. nov.) have MSAD-2 but not MSAD-1. Our finding of no MSAD genes in the genomes of more than half the Mycobacterium species, particularly in overt human pathogens, suggests gene loss during the evolutionary adaptation of Mycobacterium towards host-associated lifestyles, particularly in slow-growing human pathogens. In addition, the finding that two slow grower groups, the Tuberculosis-Simiae clade and the M. terrae complex group, including the Terrae and Triviale clades, have distinct MSAD types, MSAD-1 and MSAD-2, respectively (Supplementary Table 1 and Figure 3), suggests the distinct roles of the two MSAD types in slow-grower evolution.
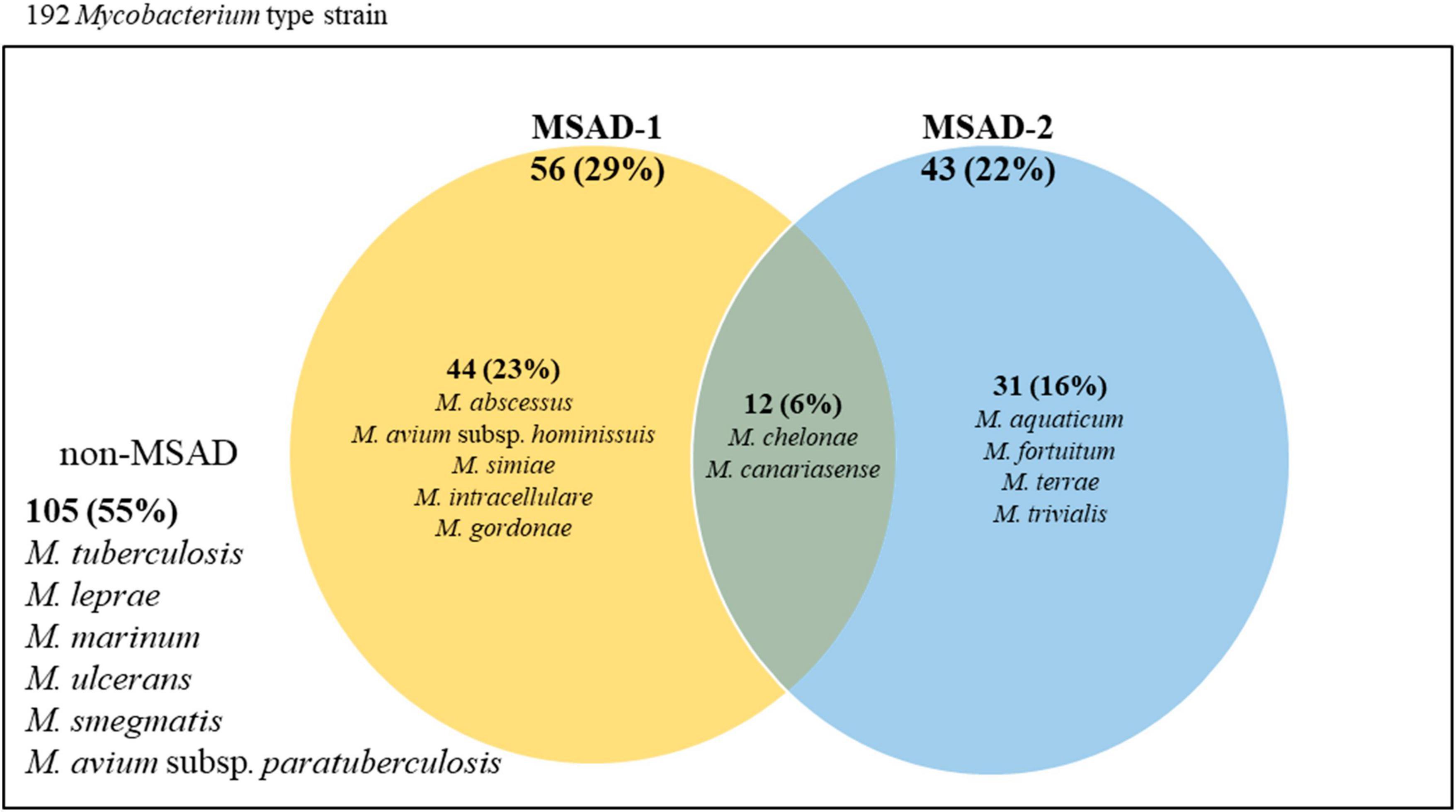
Figure 3. Venn diagram representing the distributions of MSAD-1 and MSAD-2 among strains within the genus Mycobacterium. Of all 192 Mycobacterium reference strains, only 87 (45.3%) strains have MSAD genes in their genome. Of these, 74 strains have either MSAD-1 or MSAD-2 genes in their genome, and 12 strains, including three from subspecies of M. chelonae, have both MSAD-1 and MSAD-2 genes. The remaining 105 strains, including strains of M. tuberculosis and M. leprae, do not have MSAD-like genes.
3.3. Distinct primary structures between MSAD-1s of slow-grower, MSAD-1s of rapid-grower and MSAD-2s
Our protein similarity network analysis indicated that all 100 mycobacterial MSADs consisted of a total of 17 nodes, with 13 nodes for MSAD-1s and 4 nodes for MSAD-2s (Figure 2). The predicted structures of both MSAD types of M. chelonae were identified as consistent with the MSAD model (PDB FG41) with 100% confidence, despite low protein percent identity between strains of MSAD-1 and MSAD-2 (75 and 25%, respectively). Both MSAD-1s and MSAD-2s share the characteristic structures of MSADs such as the β-α-β structure and proline-1 sequences of the TSF signature (Figure 4A; Davidson et al., 2018).
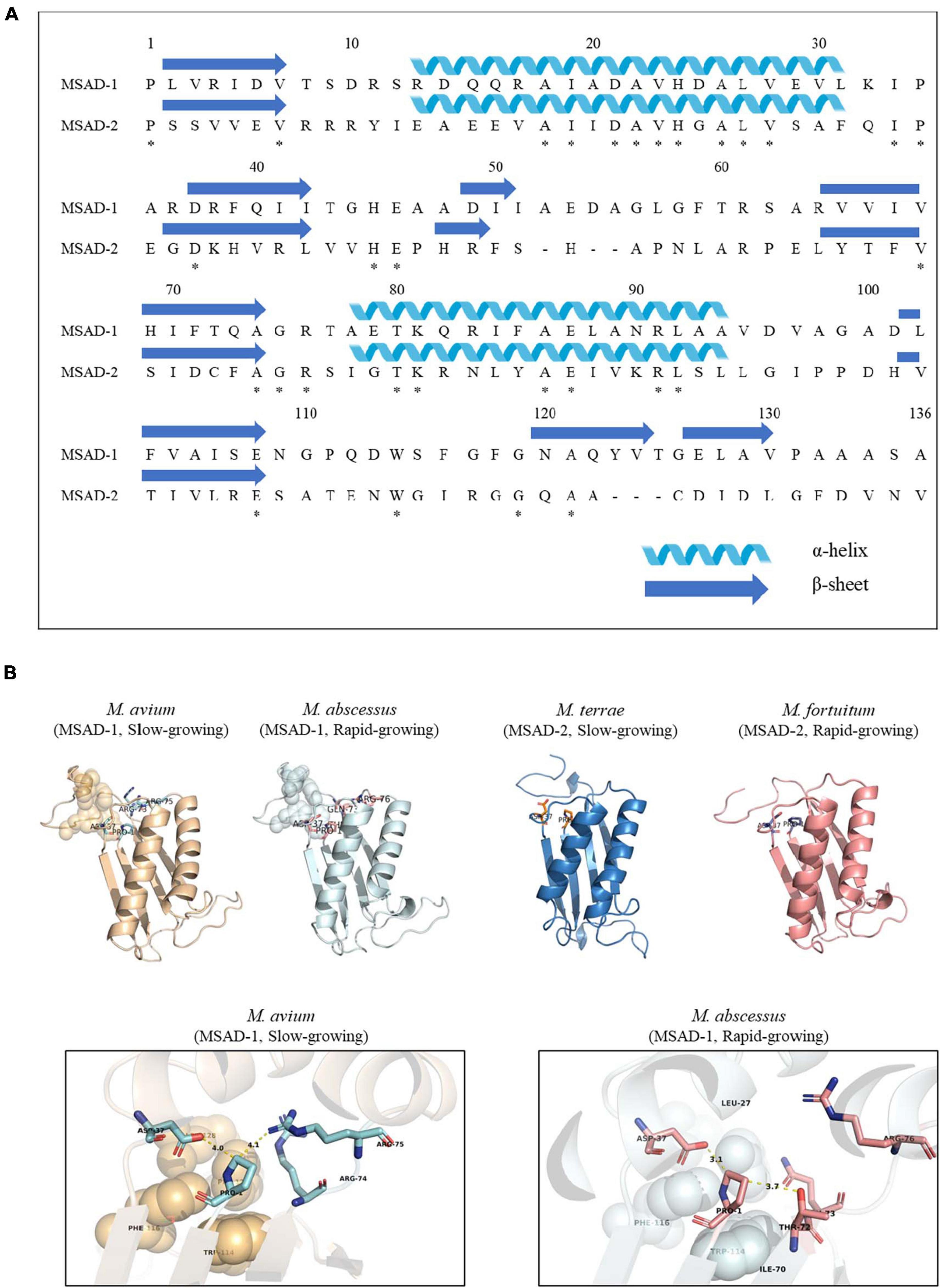
Figure 4. The primary and tertiary structures of mycobacterial MSAD-1 and MSAD-2 proteins. This number corresponds to the codon number of the P. pavonaceae MSAD protein. (A) Primary structure of MSAD-1 and MSAD-2 of M. chelonae ATCC 35752T (MSAD-1: NCBI accession number OLT81431.1 and MSAD-2: OLT83119.1). The arrow indicates the β-sheet structure, while the spiral indicates the α-helix structure. (B) Ribbon diagram representing predicted tertiary structures of MSAD-1 of M. avium (slow-grower, left), MSAD-1 of M. abscessus (rapid-grower, center left), MSAD-2 of M. terrae (slow-grower, center right), and MSAD-2 of M. fortuitum (rapid-grower, right). MSAD-1 types showed a hydrophobic wall (shown as sphere) behind the active site (shown as stick) of Pro-1 and Asp-37. The structure of MSAD-2 is the same as MSAD-1 in that the positions of Pro-1 and Asp-37 located. *Identical amino acids between two MSADs.
Tertiary structure prediction was performed for several Mycobacterium species according to each type. One of the template MSADs originating from P. pavonaceae (PpMSAD) have critical amino acids that facilitate binding to substrates (Poelarends et al., 2003, 2004, 2005; Almrud et al., 2005). Briefly, the Pro-1 and β-α-β structure shape is required for MSAD enzyme activity. Asp-37 and a pair of arginines, Arg-73 and Arg-75, are thought to act as linkers in the enzyme–substrate complex. Trp-114, Phe-116, Phe-123, and Leu-128 also participate in the stabilization of the substrate by forming a hydrophobic wall in the MSAD homotrimer (Almrud et al., 2005). Meanwhile, one of the other template MSAD originated from C. bacterium strain fg41 (FG41 MSAD) has a distinct primary structure compared with that of PpMSAD (Poelarends et al., 2008b; Guo et al., 2013). Despite also having Pro-1, a β-α-β structure shape, and Asp-37, similar to PpMSAD, FG41 MSAD has different signature protein sequences, indicating that the mechanisms of the two MSADs are different. Side chains of Thr-72, Gln-73, Arg-76, and Tyr-123 replace the pair of arginine residues in FG41 MSAD (Figure 4B).
Our data showed that all 57 mycobacterial MSAD-1 proteins have Pro-1, Asp-37, Trp-114, Phe-116, and Leu-128 (Figure 5). However, there are differences between rapid-growers and slow-growers in the substrate interaction region. While the MSAD-1 of rapid-growers has Thr-72 and Gln-73 except in Mycobacterium aubagnense and Mycobacterium phocaicum, the MSAD-1s of slow-growers have the Arg-73 and Arg-75 pair without exception, similar to PpMSAD (Figures 5A, B).
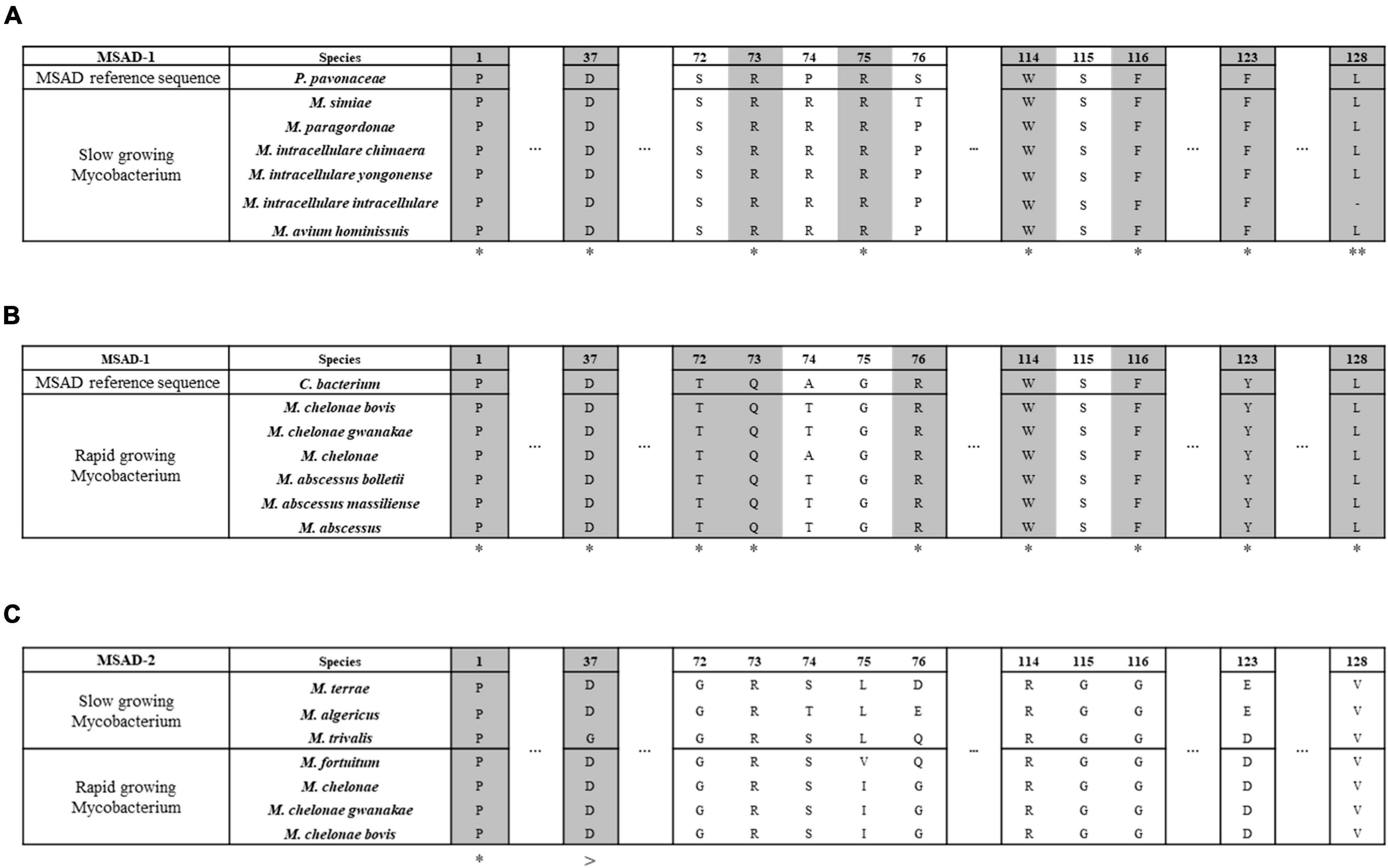
Figure 5. Distinct primary structures between mycobacterial MSAD-1 of slow-growers and rapid-growers and MSAD-2 of slow-growers and rapid-growers. This number corresponds to the codon number of the P. pavonaceae MSAD protein (PpMSAD). (A) The primary structure of MSAD-1 of slow-growers. MSAD-1 of slow-growers has structural characteristics similar to those of P. pavonaceae MSAD (PpMSAD), such as a pair of arginine (Arg-73 and Arg-75) residues and a hydrophobic wall (Trp-114, Phe-116, Phe-123, and Leu-128). (B) The primary structure of MSAD-1 of rapid-growers. MSAD-1 of rapid-growers has structural characteristics similar to those of Coryneform bacterium MSAD (FG41 MSAD), such as side chains (Thr-72, Gln-73, and Tyr-123) and a hydrophobic wall (Trp-114, Phe-116, and Leu-128). (C) The primary structure of MSAD-2. MSAD-2 does not have any specific sequence related to the enzymatic reaction of MSAD, regardless of the growth characteristics of Mycobacterium. *Indicates that all type strains have the same nucleotide. **Means that most of them have the same nucleotide. >Is the same nucleotide except for the trivalis clade.
We also found that mycobacterial MSAD-2 has a distinct primary structure compared with that of MSAD-1. All MSAD-2 groups also start with Pro-1, and most of them have Asp-37 (Figure 5C). However, the MSAD-2 of Mycobacterium koreense, Mycobacterium parakoreense, and Mycobacterium trivialis belonging to the Triviale clade encodes Asn-37 instead of Asp-37. Moreover, unlike MSAD-1, MSAD-2 has no other signature sequences, including the pair of arginines (Arg-73 and Arg-75) or sequences related to the hydrophobic wall (Figure 5C). Our data showed that sequences of MSAD-2 are more conserved between strains (61.6–100%) than those of the MSAD-1 group (29.7–100%) (Supplementary Figure 1A). Together, our data indicated that there are three distinct primary protein structures of MSADs between Mycobacterium strains (MSAD-1 of slow-growers, MSAD-1 of rapid-growers, and MSAD-2), suggesting their distinct roles in the evolution and pathogenesis of Mycobacterium.
3.4. Phylogenetic analysis based on MSAD-1 and MSAD-2 sequences
To assess the phylogenetic relationships of MSAD-1s and MSAD-2s among Mycobacterium strains, we performed phylogenetic analysis based on mycobacterial MSAD-1 and MSAD-2 sequences. First, the mycobacterial MSAD-1 phylogenetic tree was constructed from DNA sequences of 57 mycobacterial MSAD-1s from 56 Mycobacterium strains (two independent MSAD-1s in M. simiae) with DNA lengths ranging from 348 to 411 bp. The G+C content of MSAD-1 ranges from 56.1 to 68.4%. We found that most Mycobacterium strains can be separated at the species level, showing sequence similarity levels ranging from 29.7 to 100% (Supplementary Figure 1A). In general, the MSAD-1 DNA sequence-based phylogenetic tree revealed natural relationships between Mycobacterium, as shown in the hsp65-based tree (Supplementary Figure 2), clearly including separation between slow-growers (Tuberculosis-Simiae clade) and rapid-growers and separation between the Fortuitum-Vaccae clade and Abscessus-Chelonae clade. Of note, our MSAD-1-based phylogenetic analysis showed that the MSAD of five strains (M. simiae, M. dioxanotrophicus, Mycobacterium farcinogenes, Mycobacterium senegalense, and Mycobacterium agri) did not belong to the Mycobacterium clade, suggesting that their MSAD-1s was laterally transferred from another bacterial group (Figure 6A). In parallel, our further BLASTn analysis also supports LGT transfer of their MSAD-1 genes (Supplementary Table 3). Phylogenetic analysis based on MSAD-1 protein sequences also showed a topology similar to that based on MSAD-1 DNA sequences (Supplementary Figure 3).
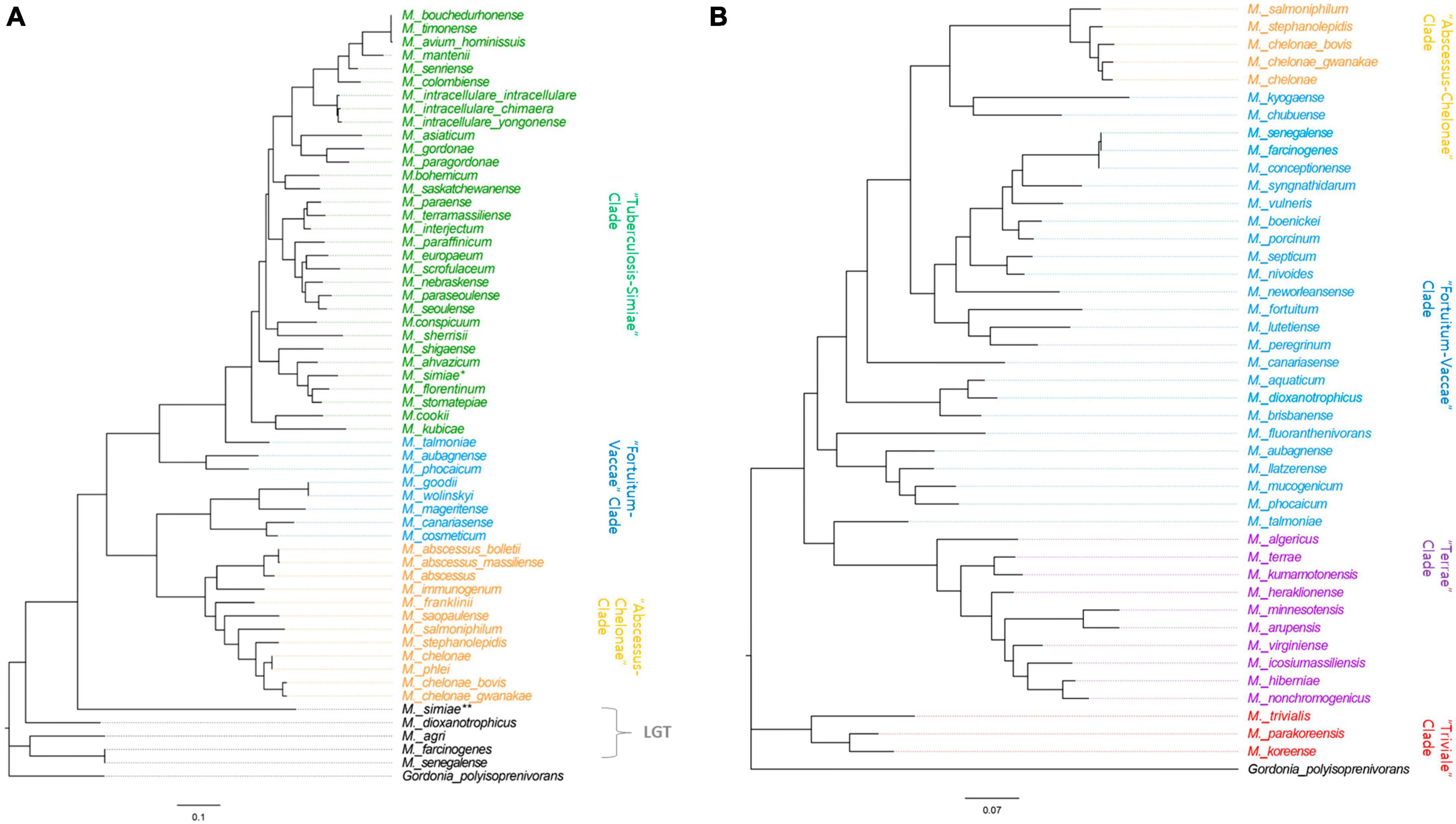
Figure 6. Phylogenetic trees based on the MSAD-1 (348–411 bp) and MSAD-2 (357–435 bp) sequences. (A) The mycobacterial MSAD-1 phylogenetic tree was constructed from DNA sequences of 57 mycobacterial MSAD-1s from 56 mycobacterial strains (two independent MSAD-1s in M. simiae) with DNA lengths ranging from 348 to 411 bp. (B) The MSAD-2 phylogenetic analysis was constructed using 43 DNA sequences of mycobacterial MSAD-2s, with DNA sequence lengths ranging from 357 to 435 bp.
MSAD-2 phylogenetic analysis was performed using 43 DNA sequences of mycobacterial MSAD-2, with DNA sequence lengths ranging from 357 to 435 bp. The G+C content of MSAD-2 is slightly higher than that of MSAD-1, ranging from 58 to 69.5%, and most strains can be separated at the species level, showing sequence similarity levels ranging from 61.6 to 100%, indicating that MSAD-2 is more conserved than MSAD-1 (Supplementary Figure 1B). Our MSAD-2 phylogenetic analysis also reveals natural relationships between mycobacterial strains, including separation between four clades, the Fortuitum-Vaccae and Abscessus-Chelonae clades of rapid-growers and Terrae and Triviale clades of slow-growers (Figure 6B), which is also shown in the MSAD-2 protein-based phylogeny (Supplementary Figure 3B). Together, our findings show that MSAD-1 and MSAD-2 sequences basically reflect the phylogenetic relationships between strains within the genus Mycobacterium except for some strains subject to LGT of their MSAD-1 gene, suggesting their pivotal role in the pathogenesis and evolution of Mycobacterium speciation.
3.5. MSAD-1 distribution between strains of Mycobacterium avium subspecies
Mycobacterium avium (Ma) is one of the most virulent NTM species, causing a broad spectrum of diseases in humans and ruminant animals as a member of the MAC, and it consists of four subspecies, namely, M. avium (Maa), M. hominissuis (Mah), M. paratuberculosis (Map), and M. silvaticum (Mas) (Thorel et al., 1990). Due to their distinct pathogenic potentials, their subspecies separation has recently gained great attention (Turenne et al., 2007). In this study, to investigate MSAD distributions between strains of Ma subspecies, a total of 255 genome sequences of four Ma subspecies were analyzed using tBLASTn. Of note, none of the 49 Map strains and 1 Mas strain had the MSAD gene in their genomes (Table 1). Only some Maa strains and Mah strains have MSAD-1 orthologs in their genomes. Of the 43 Maa strains, only 17 strains (39.53%) have MSAD-1 genes, showing 99.0–100% sequence similarity values between strains. In particular, a type strain of Maa, ATCC 25291T, does not have MSAD in its genome. In the case of Mah, 104 of 162 strains (64.19%) have MSADs in their genomes, showing 98.7–100% sequence similarity values between strains (Table 1). Of note, among the 121 Ma strains with MSADs in their genomes, 13 (11 from Mah and 2 from Maa) have identical types of mutations in their MSAD genes, including a total of three mutations, two types of silent mutations, C81T and G87T, and a one-letter deletion at site 98T, which causes premature termination of the MSAD protein (Figure 7). Interestingly, 9 of 13 Ma strains with truncated MSADs were isolated from domestic pigs in Japan (Table 2; To et al., 2020; Komatsu et al., 2021), highlighting their potential roles in the pathogenesis or epidemiology of swine mycobacteriosis.
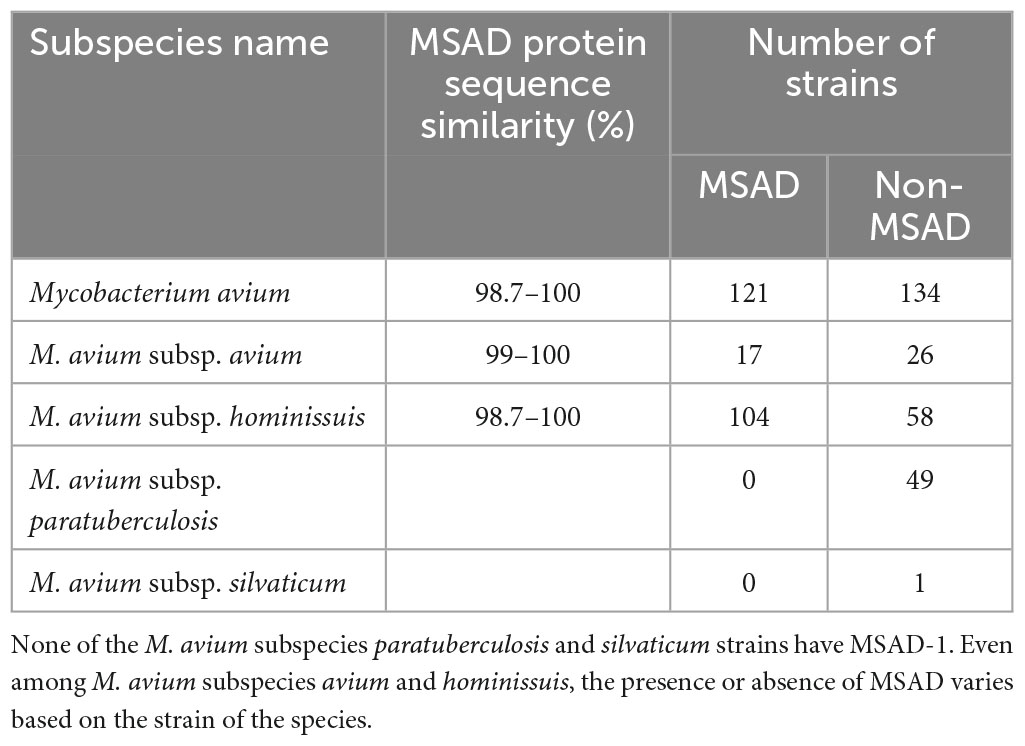
Table 1. Distribution of MSAD-1 among strains of Mycobacterium avium subspecies.
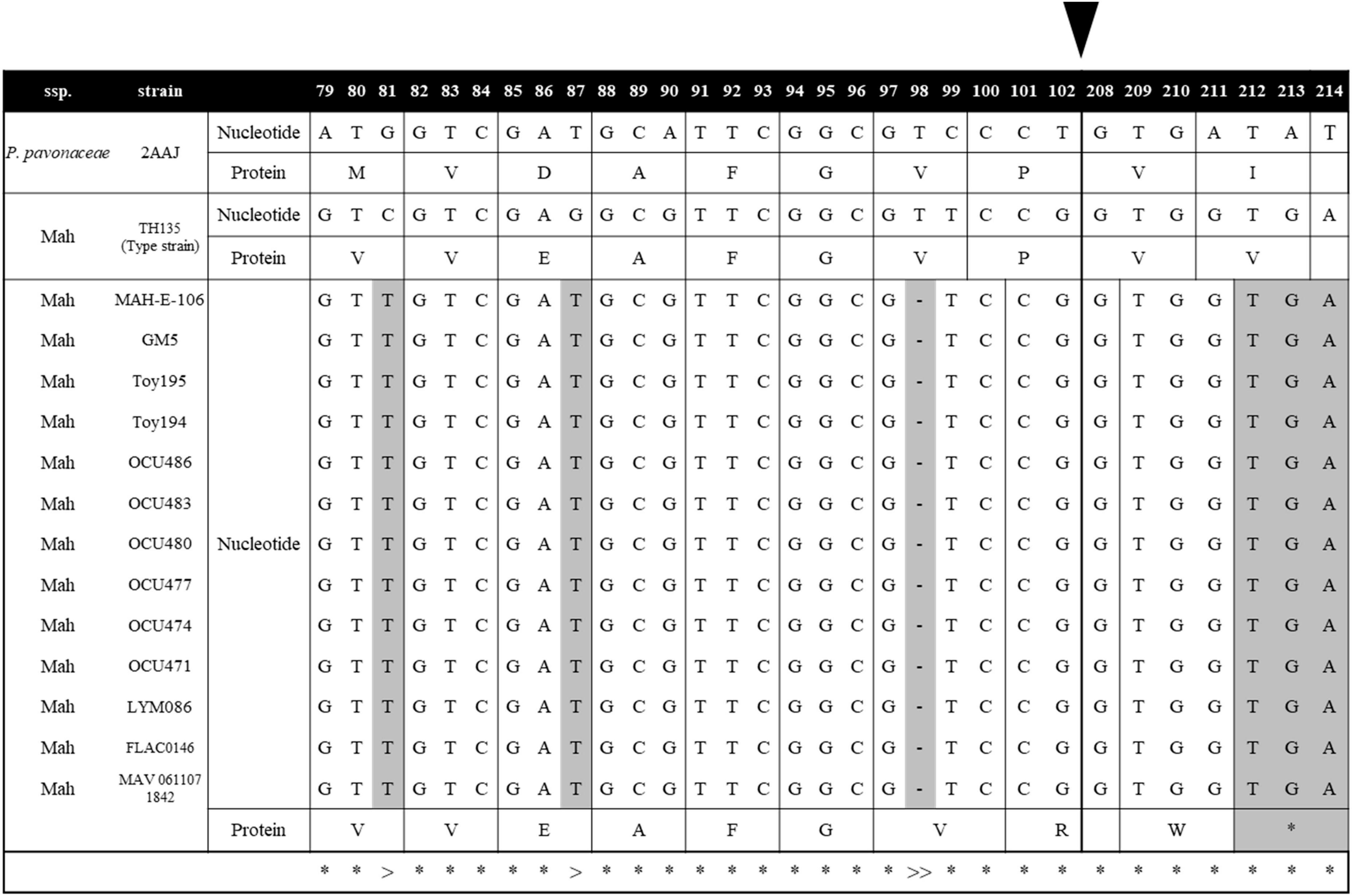
Figure 7. MSAD-1 sequence alignments of 13 Mycobacterium avium strains with truncated MSADs. The 13 M. avium strains with truncated MSADs have mutations at C81T and G87T and a one-letter gene deletion at 98T. The MSAD protein of M. avium consists of 130 amino acids, but the 13 strains above were truncated to 71 amino acids due to frameshift by deletion. The symbol “*” indicates that all mycobacterium nucleotides are the same. The symbol “>” represents the site where synonymous mutation occurred and the symbol “>>” where deletion occurs and a frameshift occurs. The black arrow indicates a skip from the 102nd nucleotide to the 208th nucleotide. This number corresponds to the codon number of the P. pavonaceae MSAD protein.
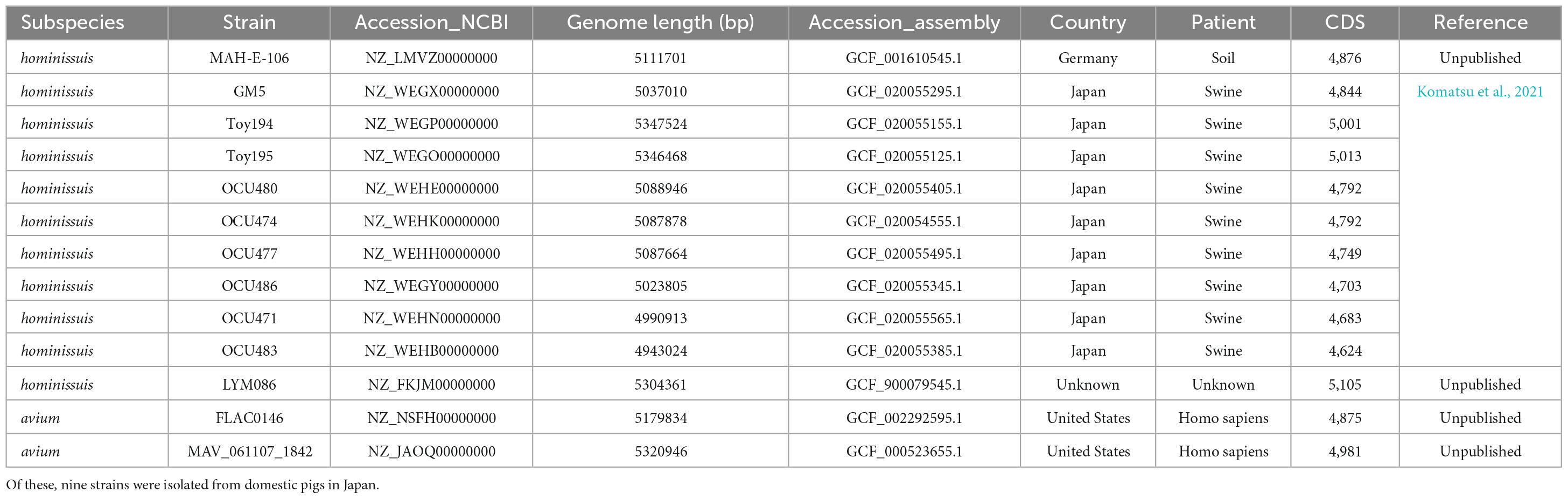
Table 2. Summary information of 13 M. avium strains with truncated MSAD-1 genes.
Together with our finding that there are no MSAD orthologs in the genome of Map strains, the finding that 13 of the 121 Ma strains with an MSAD-1 gene in their genome have mutated MSAD genes with premature translation termination further highlights the role of MSAD pseudogenization in species or subspecies differentiation of Ma.
3.6. Induction of pro-inflammatory cytokines in murine macrophage, J774A.1 cell by MSAD-1 protein of Mycobacterium abscessus
Our findings showing that loss of MSAD gene in the genome of pathogenic mycobacteria during their evolutionary adaptation prompt us to hypothesize that MSAD loss could contribute to their pathogenesis via evading immune responses of innate cells, macrophage, or dendritic cells. To address this hypothesis, we evaluated effect of MSAD-1 of M. abscessus on production of pro-inflammatory cytokines in murine macrophage, J774A.1 cells (Figure 8). We found that MSAD-1 treatment exerted enhanced productions of two inflammatory cytokines, TNF-α and IL-6 in J774A.1 cells in a dose dependent manner, suggesting that it could elicit immune response of innate cells in infection of mycobacteria.

Figure 8. Induction of pro-inflammatory cytokines, TNF-α, and IL-6, in murine macrophage, J774A.1 cells, by MSAD-1 protein of Mycobacterium abscessus. (A) The levels of TNF-α induced by MSAD-1 were measured by ELISA in the culture medium after 24 or 48 h. (B) The levels of IL-6 induced by MSAD-1 were measured by ELISA in the culture medium after 24 or 48 h. Data represent means ± SEM (standard error of mean) from quadruplicate samples and are representative of at least two independent experiments. Statistical analysis was performed using one-way ANOVA with Tukey’s multiple comparisons test, and statistical significance are denoted by asterisks (*P < 0.05, ***P < 0.001). An asterisk above the bar indicates statistical significance compared to the PBS control group, and an asterisk with a line indicates statistical significance compared to the indicated group.
4. Discussion
Comparative genomic analysis has revealed reductive genome evolution over the course of mycobacterial speciation, particularly in pathogenic slow-growing strains with strict host-associated lifestyles, including M. tuberculosis (Gagneux and Small, 2007; ten Bokum et al., 2008; Gagneux, 2018), M. leprae (Gómez-Valero et al., 2007; van Hooij and Geluk, 2021), and M. ulcerans (Stinear et al., 2007; Röltgen et al., 2012). Despite its extensive distribution throughout the biosphere, MSAD is not a housekeeping gene; thus, its distribution between mycobacterial strains with broad-spectrum lifestyles from environmental saprophytes to strict host-restricted pathogens could provide a deep understanding of their evolution and pathogenesis.
There are several noteworthy findings in this study. First, we found that there are no MSAD orthologs in the genome of more than half of the analyzed reference mycobacterial strains (105 of 192 strains, 54.7%) (Figure 3). In particular, highly virulent strains such as M. tuberculosis, M. leprae, M. ulcerans, M. marinum, Map, and Mycobacterium kansasii, which are regarded as overt human pathogens, do not have MSADs in their genomes (Supplementary Table 1), consistent with previous findings of their reductive genome evolution (Brosch et al., 2001; Veyrier et al., 2011; Wee et al., 2017). Moreover, we did not find any MSAD orthologs in any strains of Map, which is more virulent than other Ma subspecies, including Maa and Mah (Turenne et al., 2007) (Figure 7). In addition, we confirmed pseudogenization events in the MSAD genes of 13 strains of Maa and Mah by one-letter gene deletion causing frameshift events (Figure 7), further supporting our hypothesis of MSAD gene loss in more pathogenic mycobacteria.
Second, our SSN and phylogenetic analysis revealed two distinct MSAD types, MSAD-1 and MSAD-2, in Mycobacterium (Figures 2–5). We found that in 87 of 192 strains (45.3%), MSAD-1s (found in 57 sequences from 56 slow-growing strains and rapid-growing strains, two distinct MSAD-1s in M. simiae) and MSAD-2s (found in 43 strains in the M. terrae complex and rapid-growing strains) were distributed in an exclusive manner among Mycobacterium species. However, 12 rapid-growing strains, including three M. chelonae members, had both types of MSADs, MSAD-1 and MSAD-2, in their genomes (Supplementary Table 2). These findings suggest an evolutionary event from a common ancestor with both types of MSADs, possibly belonging to the rapid-growing group, into the strain with either a single MSAD-1 or a single MSAD-2 during Mycobacterium speciation. Both types of MSADs show distinct distributions among Mycobacterium strains. In particular, among slow-growing strains with an MSAD gene, the Tuberculosis-Simiae clade, including most pathogenic slow-growing strains, has only MSAD-1 (Figure 6), suggesting a pivotal role of MSAD-1 in the pathogenesis and evolution of pathogenic slow-grower. In contrast, the Triviale and Terrae clades have only the MSAD-2 type in their genomes (Figure 6), highlighting the role of MSAD-2 in their pathogenesis and evolution. In rapid-growers, the MSAD distribution is more complex than that in slow-grower. Rapid-growing strains with the MSAD gene could be divided into three groups according to their MSAD distributions: a group with only the MSAD-1 type, including three subspecies of M. abscessus, namely, M. abscessus, M. massiliense and M. bolleti, a group with only the MSAD-2 type, including most of the Fortuitum-Vaccae clade, and a group with both types of MSADs, MSAD-1 and MSAD-2, including three members of the M. chelonae subspecies (Figure 6 and Supplementary Table 1), suggesting distinct roles of the two MSAD types in pathogenesis and evolution in terms of respective rapid-growing groups. Given the close phylogenetic relationships between M. chelonae and M. abscessus strains (Adékambi and Drancourt, 2004; Tortoli et al., 2017), it is tempting to speculate on MSAD-2 loss during speciation from the common ancestor of two species with both types of MSADs into M. abscessus, which may have contributed to its pathogenesis.
Third, our data showed that the MSAD-1s of the slow-growers and rapid-growers and MSAD-2s have distinct primary signature sequences that can play a crucial role in their function. The MSAD-1s of all slow-growers have Arg-73, Arg-75, and Asp-37, which play a critical role in substrate reactions, and Trp-114, Phe-116, Phe-123, and Leu-128, which are essential for the hydrophobic wall, as shown in P. pavonaceae strain 170’s MSAD (PpMSAD) (Figure 5A and Supplementary Data 1; Almrud et al., 2005), suggesting a similar role of the MSADs of slow-growers with that of the latter. On the other hand, the MSAD-1s of rapid-growers have a primary signature sequence of Thr-72, Gln-73, and Tyr-123 (Guo et al., 2013) instead of a pair of arginines reacting with the substrate, distinct from the pattern in slow-growers, as shown in the MSADs of C. bacterium strain FG41 (FG41 MSAD) (Figure 5B and Supplementary Data 1; Guo et al., 2013), suggesting distinct functional roles or distinct evolutionary selection pressures between the MSAD-1s of slow-growers and rapid-growers. Interestingly, all the strains with MSAD-2 have primary sequences distinct from MSAD-1, suggesting distinct functional roles of MSAD-2s from slow-growers and rapid-growers MSAD-1s (Figure 5C and Supplementary Data 1). Further research is needed to determine the exact role of MSAD-2s in enzyme function.
Our phylogenetic analysis showed that MSAD-2 is more resistant than MSAD-1 to LGT events (Figure 6). Comparing the results with phylogenetic trees based on MSAD-1, MSAD-2, and hsp65 sequences, we could not find any LGT events in 43 strains with MSAD-2, but 5 of 53 strains had MSAD-1s (9.4%) (the second MSAD-1 type of M. simiae, M. dioxanotrophicus, M. farcinogenes, M. senegalense, and M. agri), which may have been laterally transferred from another bacterial group (Figure 6A and Supplementary Table 3). Of these, 4 strains (M. dioxanotrophicus, M. farcinogenes, M. senegalense, and M. agri) belong to the rapid-growers of the Fortuitum-Vaccae clade. Interestingly, we found that while one (accession number BBX43605.1) of two MSAD-1 genes of M. simiae belongs to the clade of Tuberculosis-Simiae by MSAD-1-based phylogenetic analysis, the other MSAD-1 of M. simiae (accession number BBX40959.1) did not (Figure 6A). Our protein similarity network analysis indicated that it has sequence similarity with the MSAD from the Methylobacterium phylum (Supplementary Table 3), suggesting acquisition by M. simiae of the second MSAD-1 gene via LGT.
Our in vitro experiment using MSAD-1 protein indicated that MSAD-1 could evoke inflammatory response from innate cells in mycobacteria infection, suggesting that MSAD-1 loss in pathogenic mycobacteria could contribute into their chronic infection or pathogenesis via evading immune response of innate cell. However, the role of two distinct MSADs, MSAD-1 and MSAD-2, in mycobacterial pathogenesis or evolution must be proved in the future via further in vitro and in vivo studies using MSAD gene knock out or reinforced mutant.
5. Conclusion
In conclusion, our data revealed two distinct types of MSADs, MSAD-1 and MSAD-2, among strains in the Mycobacterium genus, but more than half of the strains, including strains of pathogenic mycobacteria such as M. tuberculosis, M. leprae, M. marinum, M. ulcerans and Map, have no MSAD orthologs in their genomes. Furthermore, in 13 Ma strains, MSAD-1 pseudogenization was found, suggesting MSAD-1 loss during host adaptation of pathogenic mycobacteria. Loss of MSAD during speciation could contribute to their pathogenicity via escape from host innate immune cells.
There are several limitations to this study. Study regarding the role of MSAD in mycobacteria evolution and pathogenesis is mainly focused on the bioinformatics prediction. Biochemical and structural evidence based on actual enzyme activities of MSAD-1 and MSAD-2 have not been introduced. So, these limitations should be addressed in the future.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
Author contributions
B-JK: Conceptualization, Data curation, Funding acquisition, Project administration, Validation, Writing – original draft, Writing – review and editing. DL: Conceptualization, Data curation, Investigation, Methodology, Software, Validation, Visualization, Writing – original draft, Writing – review and editing. DK: Data curation, Methodology, Software, Validation, Visualization, Writing – review and editing. HS: Conceptualization, Formal analysis, Writing – review and editing. SC: Data curation, Methodology, Validation, Visualization, Writing – review and editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The study was supported by grant from the Seoul National University Hospital (SNUH) Research Fund (Grant No. 03-2020-0180) and Korea Health Industry Development Institute (KHIDI), funded by Ministry of Health and Family Welfare, Republic of Korea (Grant No. HI22C0312). The funder was not involved in the study design, data analysis and writing of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1275616/full#supplementary-material
Footnotes
References
Abe, Y., Fukushima, K., Matsumoto, Y., Niitsu, T., Nabeshima, H., Nagahama, Y., et al. (2022). Mycobacterium senriense sp. nov., a slowly growing, non-scotochromogenic species, isolated from sputum of an elderly man. Int. J. Syst. Evol. Microbiol. 72:005378. doi: 10.1099/ijsem.0.005378
Adékambi, T., and Drancourt, M. (2004). Dissection of phylogenetic relationships among 19 rapidly growing Mycobacterium species by 16S rRNA, hsp65, sodA, recA and rpoB gene sequencing. Int. J. Syst. Evol. Microbiol. 54, 2095–2105. doi: 10.1099/ijs.0.63094-0
Akiva, E., Brown, S., Almonacid, D., Barber, A. II, Custer, A., Hicks, M., et al. (2014). The structure–function linkage database. Nucleic Acids Res. 42, D521–D530. doi: 10.1093/nar/gkt1130
Almrud, J., Poelarends, G., Johnson, W., Serrano, H., Hackert, M., and Whitman, C. (2005). Crystal structures of the wild-type, P1A mutant, and inactivated malonate semialdehyde decarboxylase: A structural basis for the decarboxylase and hydratase activities. Biochemistry 44, 14818–14827. doi: 10.1021/bi051383m
Altschul, S., Gish, W., Miller, W., Myers, E., and Lipman, D. (1990). Basic local alignment search tool. J, Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Altschul, S., Madden, T., Schäffer, A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bouam, A., Armstrong, N., Levasseur, A., and Drancourt, M. (2018). Mycobacterium terramassiliense, Mycobacterium rhizamassiliense and Mycobacterium numidiamassiliense sp. nov., three new Mycobacterium simiae complex species cultured from plant roots. Sci. Rep. 8:9309. doi: 10.1038/s41598-018-27629-1
Brosch, R., Pym, A., Gordon, S., and Cole, S. (2001). The evolution of mycobacterial pathogenicity: Clues from comparative genomics. Trends Microbiol. 9, 452–458. doi: 10.1016/S0966-842X(01)02131-X
Chalmers, J., Aksamit, T., Carvalho, A., Rendón, A., and Franco, I. (2018). Non-tuberculous mycobacterial pulmonary infections. Pulmonology 24, 120–131. doi: 10.1016/j.pulmoe.2017.12.005
Dahl, J., Gatlin, I. W., Tran, P., and Sheik, C. (2021). Mycolicibacterium nivoides sp. Nov. isolated from a peat bog. Int. j. Syst. Evol. Microbiol. 71:004438. doi: 10.1099/ijsem.0.004438
Davidson, R., Baas, B., Akiva, E., Holliday, G., Polacco, B., LeVieux, J., et al. (2018). A global view of structure–function relationships in the Tautomerase superfamily. J. Biol. Chem. 293, 2342–2357. doi: 10.1074/jbc.M117.815340
Gagneux, S. (2018). Ecology and evolution of Mycobacterium tuberculosis. Nat. Rev. Microbiol. 16, 202–213. doi: 10.1038/nrmicro.2018.8
Gagneux, S., and Small, P. (2007). Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect. Dis. 7, 328–337. doi: 10.1016/S1473-3099(07)70108-1
Gómez-Valero, L., Rocha, E., Latorre, A., and Silva, F. (2007). Reconstructing the ancestor of Mycobacterium leprae: The dynamics of gene loss and genome reduction. Genome Res. 17, 1178–1185. doi: 10.1101/gr.6360207
Guo, Y., Serrano, H., Poelarends, G., Johnson, W. Jr., Hackert, M., and Whitman, C. (2013). Kinetic, mutational, and structural analysis of malonate semialdehyde decarboxylase from Coryneform bacterium strain FG41: mechanistic implications for the decarboxylase and hydratase activities. Biochemistry 52, 4830–4841. doi: 10.1021/bi400567a
Gupta, R., Lo, B., and Son, J. (2018). Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front. Microbiol. 9:67. doi: 10.3389/fmicb.2018.00067
Gutierrez, M., Brisse, S., Brosch, R., Fabre, M., Omaïs, B., Marmiesse, M., et al. (2005). Ancient origin and gene mosaicism of the progenitor of Mycobacterium tuberculosis. PLoS Pathog 1:e5. doi: 10.1371/journal.ppat.0010005
He, Y., Mathieu, J., Yang, Y., Yu, P., da Silva, M., and Alvarez, P. (2017). 1, 4-Dioxane biodegradation by Mycobacterium dioxanotrophicus PH-06 is associated with a group-6 soluble di-iron monooxygenase. Environ. Sci. Technol. Lett. 4, 494–499. doi: 10.1021/acs.estlett.7b00456
Huddleston, J., Burks, E., and Whitman, C. (2014). Identification and characterization of new family members in the tautomerase superfamily: analysis and implications. Arch. Biochem. Biophys. 564, 189–196. doi: 10.1016/j.abb.2014.08.019
Jankute, M., Cox, J., Harrison, J., and Besra, G. (2015). Assembly of the mycobacterial cell wall. Annu. Rev. Microbiol. 69, 405–423. doi: 10.1146/annurev-micro-091014-104121
Jeong, H., Lee, S., Seo, H., Kim, D., Lee, D., and Kim, B. (2022). Potential of Mycobacterium tuberculosis chorismate mutase (Rv1885c) as a novel TLR4-mediated adjuvant for dendritic cell-based cancer immunotherapy. Oncoimmunology 11:2023340. doi: 10.1080/2162402X.2021.2023340
Johansen, M., Herrmann, J., and Kremer, L. (2020). Non-tuberculous mycobacteria and the rise of Mycobacterium abscessus. Nat. Rev. Microbiol. 18, 392–407. doi: 10.1038/s41579-020-0331-1
Jones, P., Binns, D., Chang, H., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kelley, L., Mezulis, S., Yates, C., Wass, M., and Sternberg, M. (2015). The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015.053
Kim, B., Kim, B., Jeong, J., Lim, J., Park, S., Lee, S., et al. (2018). A description of Mycobacterium chelonae subsp. gwanakae subsp. Nov., a rapidly growing mycobacterium with a smooth colony phenotype due to glycopeptidolipids. Int. J. Syst. Evol. Microbiol. 68, 3772–3780. doi: 10.1099/ijsem.0.003056
Kim, B., Kim, G., Kim, B., Jeon, C., Jeong, J., Lee, S., et al. (2017). Description of Mycobacterium chelonae subsp. bovis subsp. nov., isolated from cattle (Bos taurus coreanae), emended description of Mycobacterium chelonae and creation of Mycobacterium chelonae subsp. chelonae subsp. nov. Int. J. Syst. Evol. Microbiol. 67, 3882–3887. doi: 10.1099/ijsem.0.002217
Kim, H., Kim, S., Shim, T., Kim, M., Bai, G., and Park, Y. (2005). Differentiation of Mycobacterium species by analysis of the heat-shock protein 65 gene (hsp65). Int. J. Syst. Evol. Microbiol. 55, 1649–1656. doi: 10.1099/ijs.0.63553-0
Komatsu, T., Ohya, K., Ota, A., Nishiuchi, Y., Yano, H., Matsuo, K., et al. (2021). Genomic features of Mycobacterium avium subsp. hominissuis isolated from pigs in Japan. Gigabyte 2021:gigabyte33. doi: 10.46471/gigabyte.33
Lancaster, E., Yang, W., Johnson, W. Jr., Baas, B., Zhang, Y., and Whitman, C. (2022). Kinetic, Inhibition, and Structural Characterization of a Malonate Semialdehyde Decarboxylase-like Protein from Calothrix sp. PCC 6303: A Gateway to the non-Pro1 Tautomerase Superfamily Members. Biochemistry 61, 1136–1148. doi: 10.1021/acs.biochem.2c00101
Lotti, T., and Hautmann, G. (1993). Atypical mycobacterial infections: a difficult and emerging group of infectious dermatoses. Int. J. Dermatol. 32, 499–501. doi: 10.1111/j.1365-4362.1993.tb02832.x
Marri, P., Bannantine, J., and Golding, G. (2006). Comparative genomics of metabolic pathways in Mycobacterium species: gene duplication, gene decay and lateral gene transfer. FEMS Microbiol. Rev. 30, 906–925. doi: 10.1111/j.1574-6976.2006.00041.x
Nishiuchi, Y., Iwamoto, T., and Maruyama, F. (2017). Infection sources of a common non-tuberculous mycobacterial pathogen. Mycobacterium avium complex. Front. Med. 4:27. doi: 10.3389/fmed.2017.00027
Oberg, N., Zallot, R., and Gerlt, J. (2023). EFI-EST, EFI-GNT, and EFI-CGFP: Enzyme Function Initiative (EFI) Web Resource for Genomic Enzymology Tools. J. Mol. Biol. 2023:168018. doi: 10.1016/j.jmb.2023.168018
Petrini, B. (2006). Mycobacterium abscessus: an emerging rapid-growing potential pathogen. Apmis 114, 319–328. doi: 10.1111/j.1600-0463.2006.apm_390.x
Poelarends, G., Johnson, W., Murzin, A., and Whitman, C. (2003). Mechanistic characterization of a bacterial malonate semialdehyde decarboxylase: identification of a new activity in the tautomerase superfamily. J. Biol. Chem. 278, 48674–48683. doi: 10.1074/jbc.M306706200
Poelarends, G., Serrano, H., Johnson, W., Hoffman, D., and Whitman, C. (2004). The hydratase activity of malonate semialdehyde decarboxylase: mechanistic and evolutionary implications. J. Am. Chem. Soc. 126, 15658–15659. doi: 10.1021/ja044304n
Poelarends, G., Serrano, H., Johnson, W., and Whitman, C. (2005). Inactivation of malonate semialdehyde decarboxylase by 3-halopropiolates: Evidence for hydratase activity. Biochemistry 44, 9375–9381. doi: 10.1021/bi050296r
Poelarends, G., Serrano, H., Person, M., Johnson, W. Jr., and Whitman, C. (2008a). Characterization of Cg10062 from Corynebacterium glutamicum: implications for the evolution of cis-3-chloroacrylic acid dehalogenase activity in the tautomerase superfamily. Biochemistry 47, 8139–8147. doi: 10.1021/bi8007388
Poelarends, G., Veetil, V., and Whitman, C. (2008b). The chemical versatility of the β–α–β fold: Catalytic promiscuity and divergent evolution in the tautomerase superfamily. Cell. Mol. Life Sci. 65, 3606–3618. doi: 10.1007/s00018-008-8285-x
Poelarends, G., and Whitman, C. (2004). Evolution of enzymatic activity in the tautomerase superfamily: mechanistic and structural studies of the 1, 3-dichloropropene catabolic enzymes. Bioorgan. Chem. 32, 376–392. doi: 10.1016/j.bioorg.2004.05.006
Qi, W., Käser, M., Röltgen, K., Yeboah-Manu, D., and Pluschke, G. (2009). Genomic diversity and evolution of Mycobacterium ulcerans revealed by next-generation sequencing. PLoS Pathog 5, e1000580. doi: 10.1371/journal.ppat.1000580
Ratnatunga, C., Lutzky, V., Kupz, A., Doolan, D., Reid, D., Field, M., et al. (2020). The rise of non-tuberculosis mycobacterial lung disease. Front. Immunol. 11:303. doi: 10.3389/fimmu.2020.00303
Röltgen, K., Stinear, T., and Pluschke, G. (2012). The genome, evolution and diversity of Mycobacterium ulcerans. Infect. Genet. Evol. 12, 522–529. doi: 10.1016/j.meegid.2012.01.018
Shannon, P., Markiel, A., Ozier, O., Baliga, N., Wang, J., Ramage, D., et al. (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. doi: 10.1101/gr.1239303
Stinear, T., Seemann, T., Pidot, S., Frigui, W., Reysset, G., Garnier, T., et al. (2007). Reductive evolution and niche adaptation inferred from the genome of Mycobacterium ulcerans, the causative agent of Buruli ulcer. Genome Res. 17, 192–200. doi: 10.1101/gr.5942807
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
ten Bokum, A., Movahedzadeh, F., Frita, R., Bancroft, G., and Stoker, N. (2008). The case for hypervirulence through gene deletion in Mycobacterium tuberculosis. Trends in microbiology. 16, 436–441. doi: 10.1016/j.tim.2008.06.003
Thorel, M., Krichevsky, M., and Vincent Lévy-Frébault, V. (1990). Numerical taxonomy of mycobactin-dependent mycobacteria, emended description of Mycobacterium avium, and description of Mycobacterium avium subsp. avium subsp. nov., Mycobacterium avium subsp. paratuberculosis subsp. nov., and Mycobacterium avium subsp. silvaticum subsp. nov. Int. J. Syst. Evol. Microbiol. 40, 254–260. doi: 10.1099/00207713-40-3-254
To, K., Cao, R., Yegiazaryan, A., Owens, J., and Venketaraman, V. (2020). General overview of nontuberculous mycobacteria opportunistic pathogens: Mycobacterium avium and Mycobacterium abscessus. J. Clin. Med. 9, 2541. doi: 10.3390/jcm9082541
Tortoli, E., Fedrizzi, T., Meehan, C., Trovato, A., Grottola, A., Giacobazzi, E., et al. (2017). The new phylogeny of the genus Mycobacterium: the old and the news. Infect. Genet. Evol. 56, 19–25. doi: 10.1016/j.meegid.2017.10.013
Turenne, C., Wallace, R. Jr., and Behr, M. (2007). Mycobacterium avium in the postgenomic era. Clin. Microbiol. Rev. 20, 205–229. doi: 10.1128/CMR.00036-06
van Hooij, A., and Geluk, A. (2021). In search of biomarkers for leprosy by unraveling the host immune response to Mycobacterium leprae. Immunol. Rev. 301, 175–192. doi: 10.1111/imr.12966
Veyrier, F., Dufort, A., and Behr, M. (2011). The rise and fall of the Mycobacterium tuberculosis genome. Trends Microbiol. 19, 156–161. doi: 10.1016/j.tim.2010.12.008
Vincent Lévy-Frébault, V., and Portaels, F. (1992). Proposed minimal standards for the genus Mycobacterium and for description of new slowly growing Mycobacterium species. Int. J. Syst. Evol. Microbiol. 42, 315–323. doi: 10.1099/00207713-42-2-315
Wee, W., Dutta, A., and Choo, S. (2017). Comparative genome analyses of mycobacteria give better insights into their evolution. PLoS One. 12, e0172831. doi: 10.1371/journal.pone.0172831
Keywords: malonate semialdehyde decarboxylase (MSAD), Mycobacterium, phylogenetic distribution, MSAD-1, MSAD-2, Mycobacterium avium subsp. paratuberculosis
Citation: Lee D, Kim DH, Seo H, Choi S and Kim B-J (2023) Phylogenetic distribution of malonate semialdehyde decarboxylase (MSAD) genes among strains within the genus Mycobacterium: evidence of MSAD gene loss in the evolution of pathogenic mycobacteria. Front. Microbiol. 14:1275616. doi: 10.3389/fmicb.2023.1275616
Received: 10 August 2023; Accepted: 29 September 2023;
Published: 13 October 2023.
Edited by:
Digvijay Verma, Babasaheb Bhimrao Ambedkar University, IndiaReviewed by:
Sadiya Parveen, Johns Hopkins University, United StatesLeny Jose, Indiana University–Purdue University Indianapolis, United States
Copyright © 2023 Lee, Kim, Seo, Choi and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bum-Joon Kim, a2J1bWpvb25Ac251LmFjLmty
†These authors have contributed equally to this work and share first authorship