 Paola Vacca1
Paola Vacca1 Cecilia Fazio1
Cecilia Fazio1 Arianna Neri1
Arianna Neri1 Luigina Ambrosio1
Luigina Ambrosio1 Anna Carannante1Florigio Lista2
Anna Carannante1Florigio Lista2 Silvia Fillo2Andrea Ciammaruconi2Antonella Fortunato2
Silvia Fillo2Andrea Ciammaruconi2Antonella Fortunato2 Paola Stefanelli1*
Paola Stefanelli1*- 1Department of Infectious Diseases, Istituto Superiore di Sanità, Rome, Italy
- 2Scientific Department, Army Medical Centre of Rome, Rome, Italy
Background: In Italy the introduction of meningococcal C conjugate vaccine in 2005 has led to a significant reduction of invasive meningococcal disease (IMD) caused by Neisseria meningitidis of serogroup C (MenC). However, this serogroup is still responsible of sporadic cases, clusters and local outbreaks. The study aims to investigate the genotype and antimicrobial susceptibility profile of MenC isolates collected in Italy from 2000 to 2020.
Methods: Bacterial isolates and biological samples (blood or cerebrospinal fluid) from invasive meningococcal cases are collected and characterized at the National Reference Laboratory for IMD of Istituto Superiore di Sanità. Antimicrobial susceptibility was determined by MIC Test Strip Method and interpreted according to the EUCAST breakpoints guideline. Genotypic characteristics, including multi locus sequence typing (MLST), finetype, and antimicrobial resistance target genes were performed and analyzed using the PubMLST database. Genomic comparison of core genome MLST (cgMLST) of MenC genomes was also carried out.
Results: From 2000 to 2020, a total of 665 MenC isolates were investigated for antimicrobial susceptibility and 301 for genotyping. Over two decades, almost all MenC isolates resulted susceptible to antimicrobials with few isolates resulting resistant to ciprofloxacin (N = 2), penicillin G (N = 13), and rifampicin (N = 9), respectively. Molecular typing of MenC obtained from isolates or clinical specimens identified mostly the genotype C:P1.5-1,10-8:F3-6:ST-11(cc11). However, phylogenetic analysis, performed on genomes from MenC isolates, identified two sub lineages, 11.1 and 11.2, among cc11, of which the sub lineage 11.2 was the predominant.
Conclusion: Wider application of the genomic analysis and monitoring of antimicrobial susceptibility represent key aspects of IMD surveillance and to monitor the continued evolution of these hyperinvasive strains.
Introduction
Introduction of meningococcal conjugate vaccine against Neisseria meningitidis of serogroup C (MenC) contributed to control the disease after period where many countries had experienced of an increased incidence of invasive meningococcal disease (IMD) due to meningococcal of serogroup C (Pelton, 2016; Acevedo et al., 2019; Read, 2019).
In Italy, vaccination against MenC was firstly introduced into the National Immunization Plan (NIP) 2005–2007,1 with a single dose at 13–15 months of age, as recommended vaccination (Stefanelli et al., 2009; Pezzotti et al., 2018).
However, the emergence and the increased spread of hypervirulent MenC strains, particularly those belonging to the clonal complex cc11 (or lineage 11), represent a public health threat, even in those countries where effective treatment and prevention strategies are in place (Lucidarme et al., 2015; Tzeng et al., 2017). Invasive infections caused by meningococci of lineage cc11 (mostly of serogroups C and W, less frequently associated with serogroups B or Y) can be severe, with higher case-fatality rate and high proportion of sequelae (Lucidarme et al., 2015; Caugant and Brynildsrud, 2020). Moreover, events such as capsular switching and adaptation to new niches might contribute to the spread of this genotype (Tzeng et al., 2017; Acevedo et al., 2019; Caugant and Brynildsrud, 2020). Based on genomic analysis, lineage 11 can be split into two sub lineages: 11.1 and 11.2 (Lucidarme et al., 2015). The sub lineage 11.1, includes MenW, MenB and MenC. On the contrary, sub lineage 11.2, harboring the characteristic fumC polymorphism, is associated, for the data available so far, exclusively with MenC and MenB (Lucidarme et al., 2015).
The epidemic potential of these hypervirulent strains, which are associated with high rates of mortality and morbidity, together with their ability to modify the antigenic profiles, imposes an ongoing surveillance and genomic analysis (Dickinson et al., 2012; Lucidarme et al., 2015; Caugant and Brynildsrud, 2020).
Nevertheless, serogroup C remains the second capsular serogroup after MenB, as well as the main responsible of local outbreaks occurred in Italy over the years (Fazio et al., 2009; Neri et al., 2015). To date, in Italy, MenC is usually responsible of sporadic cases, small clusters, and regional outbreaks, mostly due to cc11 (Stefanelli et al., 2009, 2012, 2016a, 2016b, 2019).
This study provides a retrospective analysis of MenC causing IMD collected in Italy within an extended surveillance period of 20 years (from 2000 to 2020). Antimicrobial susceptibility phenotypes and their target genes were also investigated. The whole genome sequences were used for genotyping and to identify the genetic lineages. The comparative genomic analysis was performed to identify genetic relatedness of invasive MenC over time.
Materials and methods
Collection of isolates and biological samples
All MenC (isolates or biological samples) were collected from 2000 to 2020 within the National Surveillance System of IMD, coordinated by the Istituto Superiore di Sanità (ISS) in collaboration with the Italian Ministry of Health in the framework of DPCM 3/3/2017. The case definition of IMD is based on the EU Commission Decision 2018/945 of 22 June 2018.2
Meningococcal isolate serogroups were confirmed by slide agglutination with commercial antisera (Remel Europe, Ltd., UK) or by multiplex PCR (Zhu et al., 2012) on heat-inactivated bacterial suspensions in case of negative or doubtful results by agglutination test. For biological specimens [blood or cerebrospinal fluid (CSF)], meningococcal DNA was extracted using the QiAmp mini kit (Qiagen, Hilden, Germany), following the manufacturer’s procedure. Species identification and genogrouping were performed by RT-PCR using the MenSerogroup kit (Diagenode, Belgium).
Antimicrobial susceptibility tests
Antimicrobial gradient strip diffusion method was used to define the Minimum Inhibitory Concentrations (MIC) of meningococcal isolates.
Etest (Biomerieux, Sweden) and MIC test strips (Liofilchem, Diagnostici, Italy) were used on Mueller Hinton agar (Oxoid, Ltd., England) plates supplemented with 5% of sheep blood. Meningococcal isolates were tested for a panel of antimicrobials: cefotaxime, ceftriaxone, ciprofloxacin, penicillin G and rifampicin.
MIC values were interpreted according to the clinical breakpoints criteria edited by European Committee Antimicrobial Susceptibility Testing (EUCAST) (v. 13.0).3
Primers for the PCR amplification and sequencing used for the main antimicrobial resistance target genes (gyrA for ciprofloxacin, penA for penicillin G, and rpoB for rifampicin) on clinical specimens were performed, as previously described (Taha et al., 2007, 2010; Hong et al., 2013).
Molecular typing and whole genome sequencing (WGS)
Chromosomal DNA of MenC was extracted using the QiAmp mini kit (Qiagen, Hilden, Germany) from an overnight culture, following the manufacturer’s procedure. WGS was performed only on meningococcal isolates, as previously described (Stefanelli et al., 2016a). PCR and Sanger sequencing were performed on culture-negative meningococcal specimens to determine the genotypes, as previously described (Birtles et al., 2005).
Molecular characterization on MenC was available for those isolates/biological samples collected starting from 2012. Genomes or gene sequences of meningococci were uploaded on PubMLST.org4 to obtain the finetype—molecular typing of the variable regions of two outer membrane proteins: VR1 and VR2 of the porin A (PorA) and the variable region of the ferric enterobactin transport (FetA)–and the multi locus sequence typing (MLST).
The combination of capsular group, finetype and MLST defines the genotypic profile, as follows: capsular group: PorA (P1). VR1, VR2: FetA (F)VR: sequence type (ST) (clonal complex).4 Alleles of the main antimicrobial resistance target genes–as gyrA, encoding the subunit A of DNA gyrase, penA, encoding the penicillin binding protein 2 (PBP2), and rpoB, encoding for the RNA polymerase β chain–were also analyzed.
Genomic comparison was performed on meningococcal genomes collected between 2012 and 2020, using the Genome Comparator tool available on PubMLST.5 The resulting distance matrix was visualized as a Neighbor-Net network in Split Tree4 (version 4.13.1). Incomplete loci were automatically removed from the distance matrix calculation for the neighbor-net graphs. All genome sequences were submitted to PubMLST Neisseria database4 (see Supplementary Table 1 for the isolate details). Additionally, a subset of European and non-European genomes belonging to MenC:cc11 (accessible in the Neisseria PubMLST4) were used for comparison versus Italian MenC:cc11 genomes.
European and non-European genomes were selected according to serogroup, year of isolation, ST, cc and lineages (see Supplementary Table 2 for the isolate details).
Results
Antimicrobial susceptibility profile
From 2000 to 2020, 665 MenC isolates were analyzed for antimicrobial susceptibility (Table 1). All meningococci were susceptible to ceftriaxone (MIC ≤ 0.125 mg/L) and to cefotaxime (MIC ≤ 0.125 mg/L). The latter was introduce in the panel of antimicrobials starting from 2014.
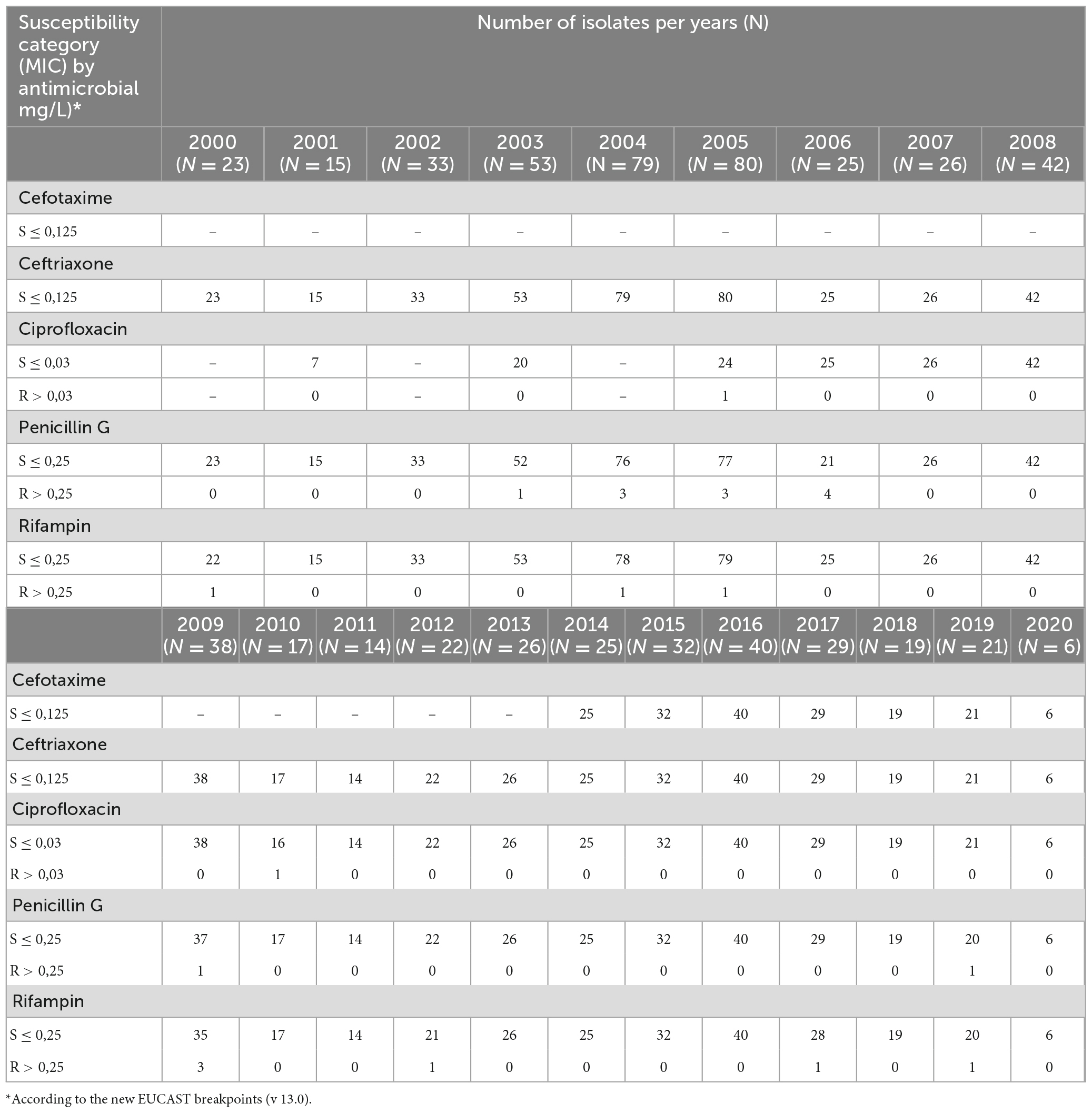
Table 1. Antimicrobial susceptibility of MenC isolates collected in Italy, 2000–2020.
According to the EUCAST breakpoints,3 98% (648/665) of isolates were susceptible to penicillin G (MICS < 0.25 mg/L); however, more than half showed MIC values close to the resistance breakpoint (MIC values between 0.094 and 0.25 mg/L).
Thirteen MenC were penicillin G resistant (MIC > 0.25 mg/L) with MIC values ranging from 0.38 to 0.50 mg/L. Analysis of penA gene revealed 16 alleles, of which 10 were characterized by polymorphisms in the C-terminal region of PBP2 (F504L, A510V, I515V, H541N, and I566V) and associated with reduced susceptibility to penicillin G. The remaining 6 alleles were considered wild-type because no alteration was present in PBP2 and were associated with susceptibility to penicillin G. Among the penA alleles found in this study, 4 isolates harbored penA327 having only 4 out of 5 alterations (F504L, A510V, I515V, H541N) in the corresponding amino acid sequence. These isolates showed MIC values ranging between 0.094 and 0.25 mg/L.
Overall, the penA248 (N = 100) was the most frequent allele associated with cc11 sub lineages.
Ciprofloxacin was introduced in the antimicrobial susceptibility test starting from 2005. Except for 2 isolates, resistant to ciprofloxacin (MIC values of 0.064 and 0.094 mg/L, respectively), the remaining were susceptible.
Molecular investigation of the target gene gyrA identified gyrA4 (N = 182), gyrA18 (N = 31), gyrA2 (N = 4), gyrA5 and gyrA12 (one isolates each one), as the main alleles.
Although the majority of isolates were susceptible to rifampicin, 9 MenC isolates resulted resistants (MIC > 0.25 mg/L), showing MIC values of 0.38 mg/L (N = 3), 0.5 mg/L (N = 1), 32 mg/L (N = 3), and 256 mg/L (N = 2), respectively. The presence of the amino acid substitution H552Y, in the rpoB gene, confirmed the antimicrobial resistant phenotype to rifampicin.
Genotyping
Genotyping was performed on 301 MenC of which 232 isolates and 69 biological samples, collected from 2012 to 2020 (Table 2). Overall, MenC were grouped in 13 ccs. Cc11 (248/301; 82%) was the predominant over the years, of them 94% (234/248) belonged to ST-11. Cc334 was the second most frequent clonal complex (34/301; 11%), mostly of ST-1031 (27/34). The remaining ccs were cc8, cc18, cc22, cc32, cc103, cc175, cc198, cc231, cc269, cc865, and cc10217. Four MenC belonged to ccs not assigned at the time of writing (unknown, UNK).
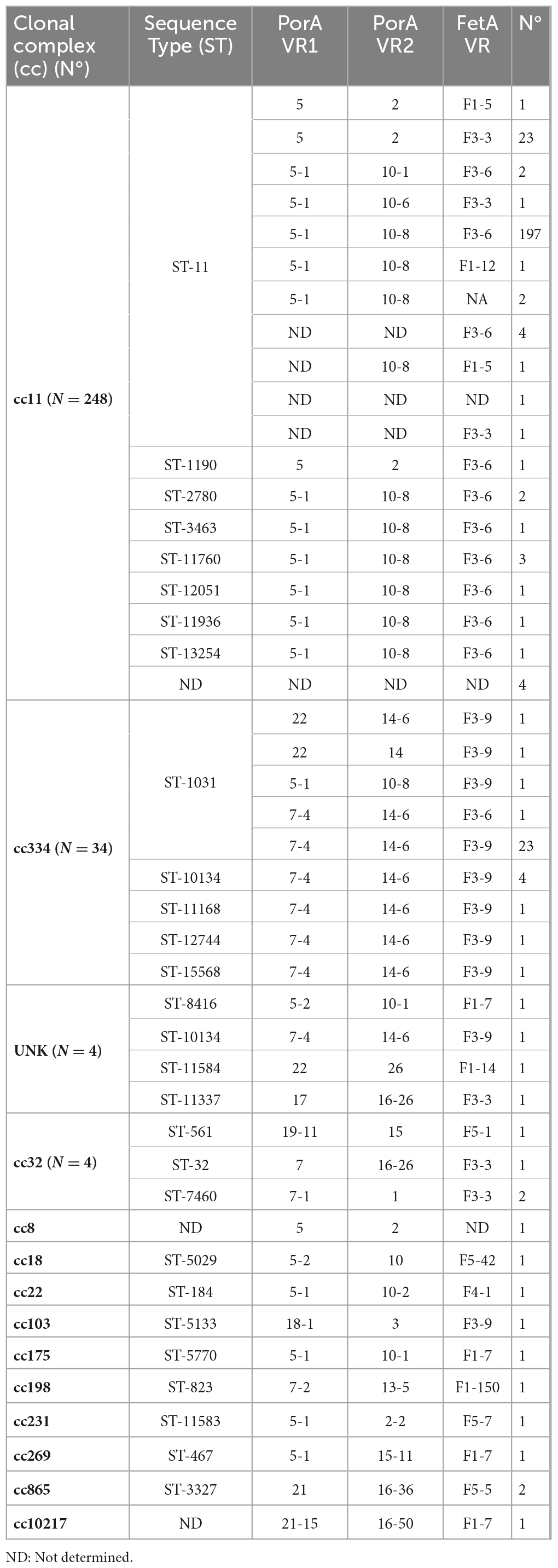
Table 2. Genotypic profile of invasive MenC, both from microbiological isolates and clinical specimens, collected in Italy from 2012 to 2020.
The finetype combination (PorA VR1, VR2: FetA VR) P1.5-1,10-8:F3-6 (N = 206), followed by P1.7-4,14-6: F3-9 (N = 30), P1.5,2: F3-3 (N = 23) was the most common.
Overall, 37 genotypes were found and C:P1.5-1,10-8:F3-6:ST-11(cc11) (N = 197), C:P1.5,2:F3-3:ST-11(cc11) (N = 23), and C:P1.7-4,14-6:F3-9:ST-1031(cc334) (N = 23) resulted the prevalent.
Comparative genomic analysis
Comparative genomic analysis was performed on 232 meningococcal isolates collected between 2012 and 2020 (Figure 1).
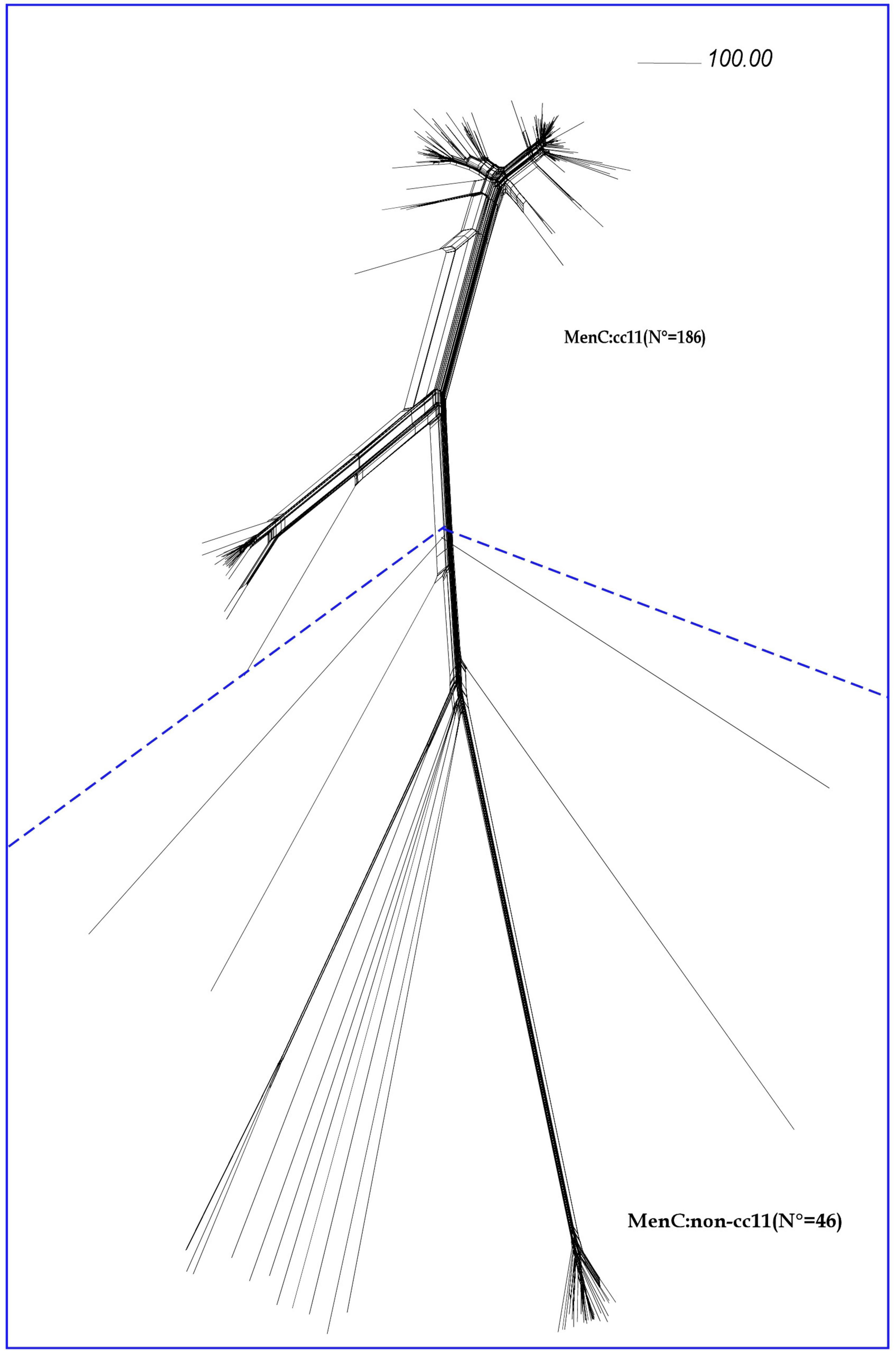
Figure 1. Core genome analysis by cgMLST on MenC genomes. Neighbor-net phylogenetic network based on a comparison of 1605 core genome loci of genomes from invasive meningococcal isolates of serogroup C (MenC) belonging to cc11 (N = 186) and to different ccs (MenC:non-cc11), (N = 46). The scale bar represents a pairwise allelic difference of 100.
According to cgMLST analysis, all the MenC genomes belonging to cc11 (N = 186) clustered in lineage 11, separated from the MenC genomes belonging to others ccs (MenC:non-cc11) (N = 46).
As observed in Figure 2, lineage 11 split into two sub lineages: 11.1 and 11.2. The latter was prevalent during the entire study period.
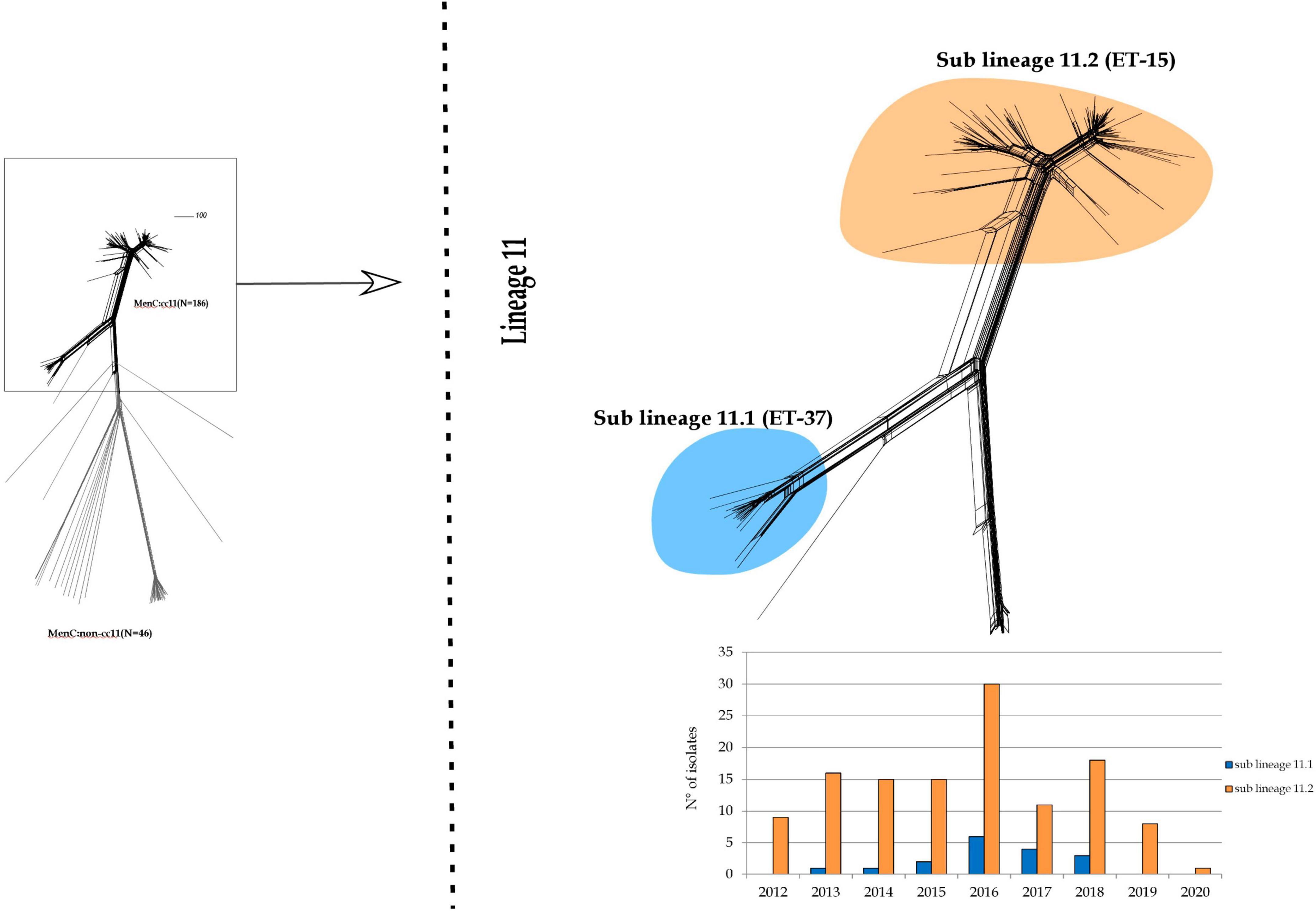
Figure 2. Core genome analysis by cgMLST on MenC:cc11. Invasive MenC belonged to cc11 clustering in a single main lineage (lineage 11) spitting into two sub lineages: 11.1 and 11.2.
Sub lineage 11.1 grouped strains from sporadic IMD cases occurred between 2013 and 2018. Except for one, all MenC genomes belonging to sub lineage 11.1 were genotyped as C:P1.5,2:F3-3:ST-11(cc11) (Figure 3).
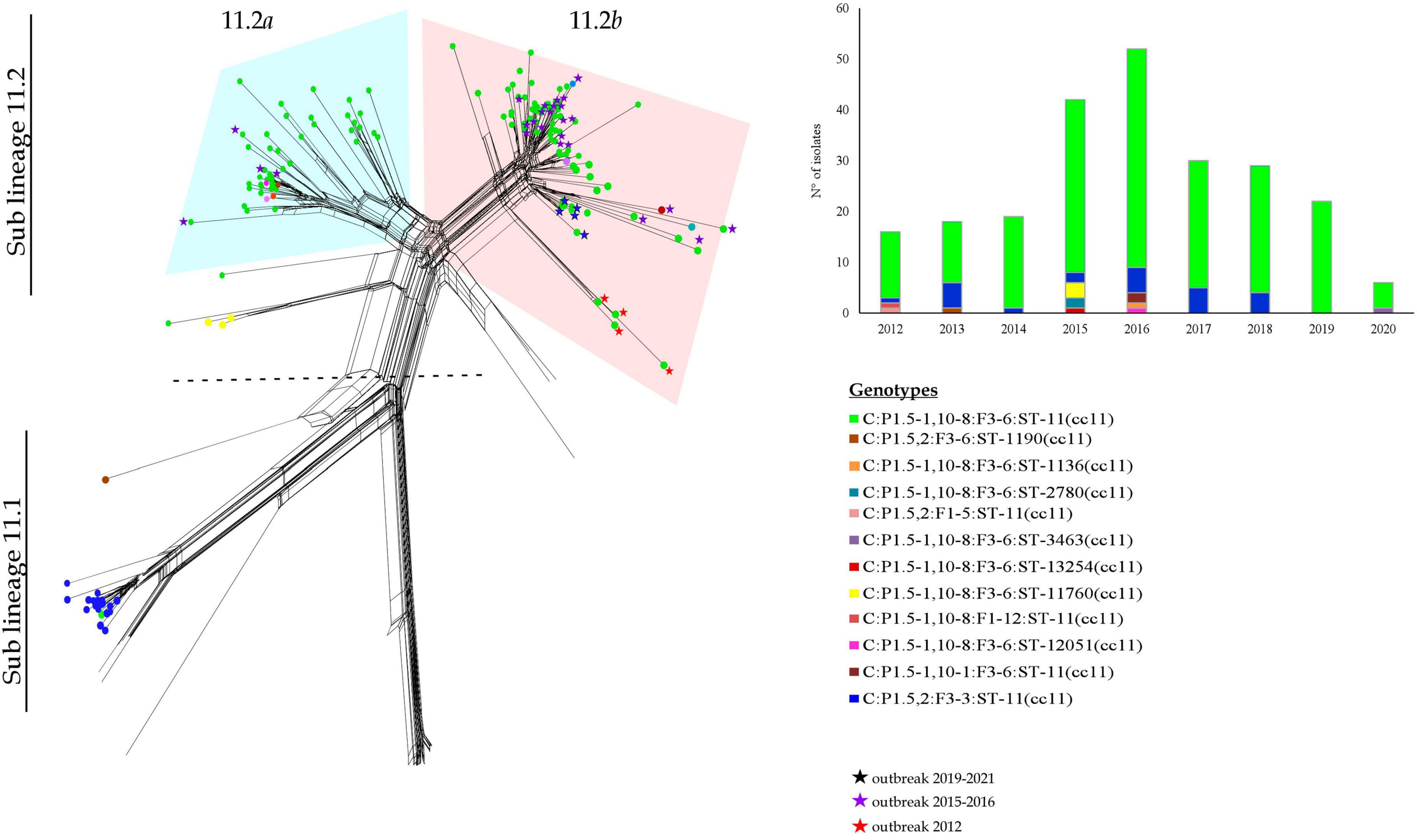
Figure 3. Core genome analysis by cgMLST on MenC:cc11 sub lineages 11.1 and 11.2 and temporal trend.
All the genomes belonging to sub lineage 11.2 carried the fumC 640 G>A polymorphism characteristic of the ET-15 meningococci, and genotyped as C:P1.5-1,10-8:F3-6:ST-11(cc11) (Figure 3).
The sub lineage 11.2 can be distinct in two groups, here called 11.2a and 11.2b.
The 11.2a group included 51 genomes from isolates collected from 2012 to 2018. In particular, the majority of them belonged to the genotype C:P1.5-1,10-8:F3-6:ST-11(cc11), except for 2 MenC:cc11 with different finetypes—P1.5-1,10-8:F1-12 and P1.5-1,10-1:F3-6. Moreover, in this group clustered also 7 isolates non-ST-11, specifically ST-12051 (N = 1), ST-2780 (N = 2), ST-13254 (N = 1), ST-11760 (N = 3) (Figure 3).
Among the genomes in the 11.2a group, 47 genomes were identified in strains responsible of sporadic cases and 4 genomes from an outbreak occurred between 2015 and 2016.
The 11.2b group comprised 100 genomes from isolates collected from 2012 to 2020, and accounted the majority of meningococci circulating in Italy during the last years. Sixty of them were from sporadic IMD cases between 2012 and 2019, whereas 40 were from outbreaks occurred in Italy between the years 2012 and 2020 (Stefanelli et al., 2012, 2016b, 2019; Fazio et al., 2022). Genotype C:P1.5-1,10-8:F3-6:ST-11(cc11) was the predominant, 3 genomes showed different STs, ST-13254 (N = 1) and ST-2780 (N = 2). As shown in Figure 3, the majority of MenC of recent isolation, belonging to the sub lineage 11.2, was at the more distal region of the lineage.
As shown in Figure 4, (for isolates information see Supplementary Tables 1, 2), genetic relatedness was carried out between MenC:cc11 lineage 11 genomes from this study and a subset of genomes of MenC lineage 11 collected worldwide in the same time period available in the PubMLST database. Based on genomic comparison, all the genomes clustered according to the cc11 lineage structure as described by Lucidarme et al. (2015). In both sub lineages 11.1 and 11.2, Italian MenC were mostly intermixed with European isolates (Figure 4). Among the non-European isolates, the genome sequences from USA were more similar to the Italian MenC (Figure 4).
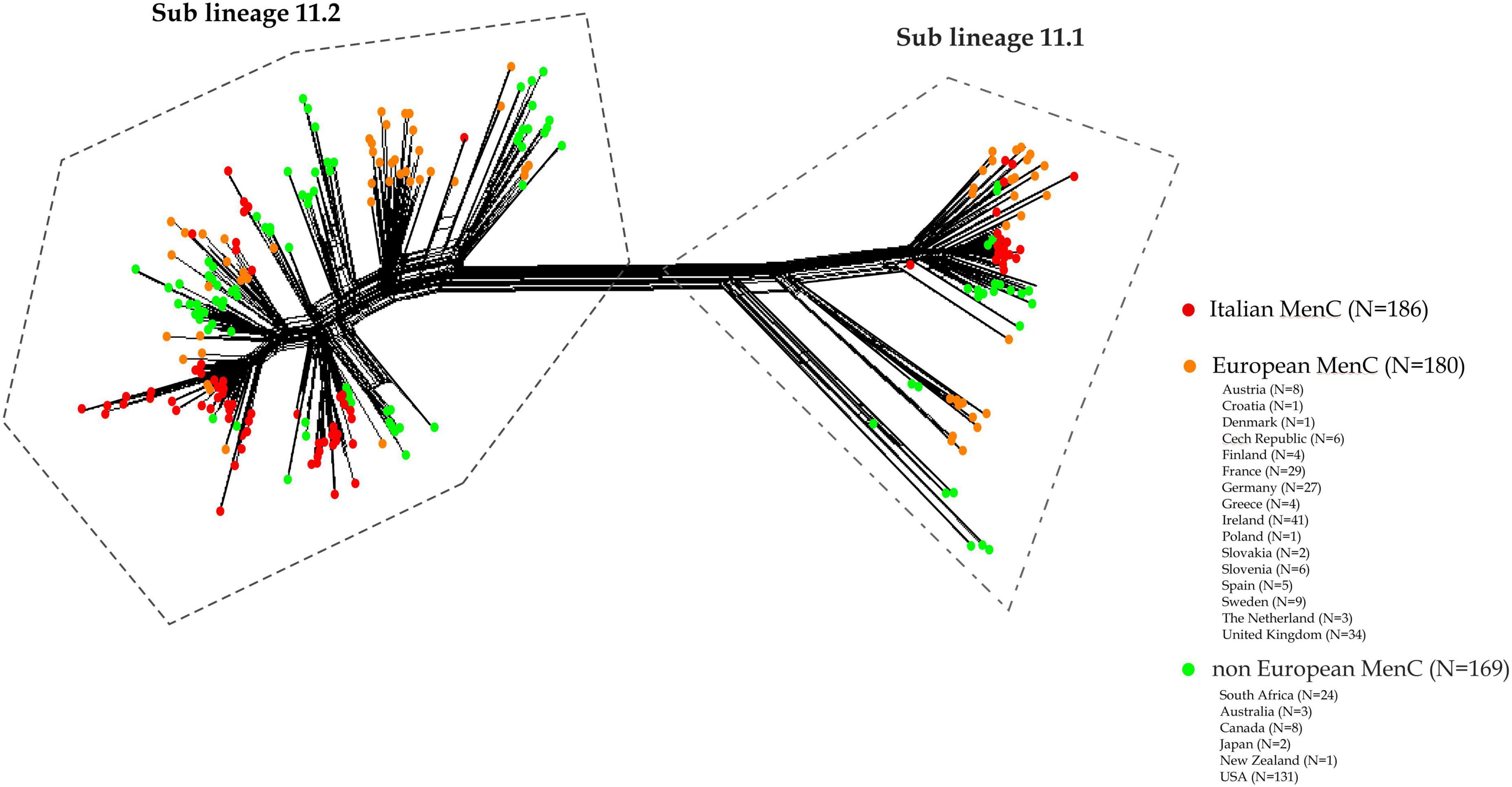
Figure 4. Core genome analysis by cgMLST on a subset of MenC:cc11 lineage 11 meningococcal genomes from Italy, European and non-European countries. Genetic relatedness of the Italian MenC:cc11 lineage 11 (N = 186) versus a subset of genomes collected worldwide during the same time period (N = 169, non-European; N = 180 European).
Discussion
Invasive meningococcal disease caused by MenC remains a public health concern due to the rapid onset and progression of the disease with fatal outcome and possible sequelae (Caugant and Brynildsrud, 2020). Italy ranks among low IMD incidence European countries of IMD (specifically 0.12 cases per 100 000 inhabitants in 2020) with a predominance of MenB, followed by MenC.6
The introduction of antimeningococcal C conjugated vaccine into the Italian NIP 2005–2007 contributed in the reduction of IMD due to this serogroup (Neri et al., 2015; Pezzotti et al., 2018) in the following years, mostly among children for whom the vaccination is targeted and recommended (Neri et al., 2015).
Although in Italy most of the MenC IMD cases occur sporadically, clusters and local outbreaks caused by hypervirulent C:P1.5-1,10-8,F3-6:ST-11(cc11) have been reported (Fazio et al., 2009; Stefanelli et al., 2012, 2016b). A large cluster of MenC:cc11 in Tuscany contributed to the increase of cases in the country (Stefanelli et al., 2016a,b) up to 44 and 43% of all the IMD cases occurred in 2015 and 2016, respectively (Pezzotti et al., 2018). In 2018, a local outbreak was identified in Sardinia due to a switched strain MenB:cc11 (Stefanelli et al., 2019). Between December 2019 and January 2020, an outbreak caused by MenC:cc11 occurred in a limited area in the Northern part of Italy (Fazio et al., 2022).
Outbreaks caused by MenC:cc11 have also been reported in the past in some European countries (De Schrijver and Maes, 2003; Ali et al., 2014; Taha et al., 2016) and the USA (Tzeng et al., 2017). In 2015, it was reported the first large-scale outbreak of MenC in Africa (Nnadi et al., 2017; Bozio et al., 2018), caused by the spread of a new MenC strain belonging to cc10217 (De Schrijver and Maes, 2003; Nnadi et al., 2017).
Although antimicrobial resistance in N. meningitidis does not currently represent an issue, resistant isolates were reported worldwide, mostly against antimicrobials used for prophylaxis (Acevedo et al., 2019). In agreement with that reported in others countries (Acevedo et al., 2019), in Italy meningococcal isolates resistant to penicillin, ciprofloxacin or rifampicin are rare, here representing the 2, 0.3 and 0.9%, respectively.
Molecular profiling of gyrA target gene for ciprofloxacin resistance showed mutations particularly at T91I amino acid position. Ciprofloxacin-resistance strains have also been described in other European countries, the USA, South America and in China (Acevedo et al., 2019). Resistance to rifampicin is correlated to the alterations in the rpoB gene. However, strains that harbor a modified rpoB gene are rare, identified in Europe and also in our country (Acevedo et al., 2019). Rifampicin resistant meningococci represent a great concern, especially in those countries where rifampicin is used as the first-line choice for chemoprophylaxis (Acevedo et al., 2019). PenA gene mutations and mosaicism represent the main molecular mechanisms associated with penicillin resistance (Taha et al., 2007).
Overall, in this study, 82% of MenC belonged to the genotype C:P1.5-1,10-8:F3-6:ST-11(cc11).
The core genome comparison of MenC, here analyzed, confirmed the phylogenetic structure of cc11 lineage 11, as described by Lucidarme et al. (2015), with two main sub-lineages: 11.1 and 11.2. Here, the majority of MenC belonged to the sub lineage 11.2, comprising genomes of strains causing both sporadic IMD cases or outbreaks.
Based on our previously study on MenC strains circulating in Italy (Lo Presti et al., 2019), using a Bayesian method to reconstruct the epidemiological dynamics, the C:P1.5-1,10-8:F3-6:ST-11(cc11) genotype segregated into two distinct clades, both apparently originating from UK, but in a different time periods. The study suggested a possible first introduction in our country between 2007 and 2011, where this variant was spreading among several European countries (Lo Presti et al., 2019). The second possible introduction could be reconducted between 2013 and 2014 (Lo Presti et al., 2019). It is likely that this strain has spread in Italy and remains stable as suggested by genomic and phylogenetic analysis, here reported.
In conclusion, this study provides a comprehensive characterization on antimicrobial susceptibility profiles and genetic characteristics of invasive meningococcal serogroup C collected over 20 years. Comparative genomic analysis on MenC isolates has provided valuable information on MenC population structure identifying the hypervirulent lineages and also on clusters and outbreaks occurred in our country. These results highlight the need to continue an active IMD surveillance to monitor the emergence and persistence of particular meningococci, such as those belonging to the cc11 lineage and related sub lineages and to compare them with those identified worldwide.
Data availability statement
The data presented in this study are deposited in online repositories (https://pubmlst.org/bigsdb?db=pubmlst_neisseria_isolates&page=plugin&name=Export). Accession numbers can be found in the Supplementary material.
Author contributions
PV: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Software, Writing – original draft. CF: Formal analysis, Writing – review and editing. AN: Formal analysis, Writing – review and editing. LA: Writing – review and editing. ACa: Writing – review and editing. FL: Writing – review and editing. SF: Writing – review and editing. ACi: Writing – review and editing. AF: Writing – review and editing. PS: Conceptualization, Supervision, Validation, Visualization, Writing – review and editing.
Funding
The authors declare financial support was received for the research, authorship, and/or publication of this article. This work was partly funded by the Italian Ministry of Health-CCM Projects: “Sorveglianza delle malattie invasive da Neisseria meningitidis, Streptococcus pneumoniae ed Haemophilus influenzae,” 2007–2009; “Sorveglianza delle malattie invasive da Neisseria meningitidis, Streptococcus pneumoniae ed Haemophilus influenzae,” 2012–2013; “Sorveglianza delle malattie invasive da Neisseria meningitidis, Streptococcus pneumoniae ed Haemophilus influenzae,” 2014–2015; and “Sorveglianza delle malattie invasive da Neisseria meningitidis, Streptococcus pneumoniae ed Haemophilus influenzae,” 2015–2016.
Acknowledgments
We thank Laura Daprai, Microbiology Laboratory, Fondazione IRCCS Cà Granda Ospedale Maggiore Policlinico, Milan, Italy; Daniela Lombardi, SeREMI, ASL AL, Regional reference Centre for Infectious Disease Surveillance, Alessandria, Italy; Anna Maria Barbui and Alessandro Bondi, Microbiology and Laboratory, Molinette Hospital, Turin, Italy; Marina Busetti and Roberta Fais, Microbiology Unit, Trieste University Hospital (ASUGI), Trieste, Italy; Alessandro Camporese, Microbiology and Virology Department, Santa Maria degli Angeli Regional Hospital, Pordenone, Italy; Carlo Tascini, University of Udine and Azienda Sanitaria Universitaria Friuli Centrale, Udine, Italy; Caterina Vocale, Microbiology Unit, IRCCS Azienda Ospedaliero-Universitaria di Bologna; Francesca Crea and Erika Coppo, San Martino Hospital, Genova; Antonella Vulcano, Carla Fontana, Silvia D’Arezzo, Marina Selleri, Paola Scognamiglio, Francesco Vario, and Maria Concetta Fusco, National Institute for the Infectious Diseases Lazzaro Spallanzani IRCCS, Microbiology Laboratory and Infectious Diseases Biorepository and SERESMI, Rome, Italy; Gian Maria Rossolini and Anna Maria Bartolesi, Clinical Microbiology and Virology Unit, Florence Careggi University Hospital, Florence, Italy; Chiara Azzari and Maria Moriondo, Department of Health Sciences, University of Florence, Anna Meyer Children’s University Hospital, Florence, Italy; Patrizia Isola, UOC Medicina di Laboratorio Livorno, Azienda USL Toscana Nord Ovest, Livorno, Italy; Irene Galanti, San Donato Hospital, Arezzo, Italy; Roberto Degli Innocenti, Nuovo Ospedale Santo Stefano, Prato, Italy; Paolo Lanzafame, Marina Gaino, and Rosanna Predazzer, Department of Microbiology, Santa Chiara Regional Hospital, Trento, Italy; Patrizia Innocenti, Elisabetta Pagani, Richard Aschbacher, and Brigitte Ladinser, Laboratory of Microbiology and Virology, Comprensorio Sanitario di Bolzano, Italy; Paolo Castiglia, Department of Clinical and Experimental Medicine-Hygiene and Preventive Medicine Unit, University-AOU of Sassari, Hospital of Sassari, Italy; Novella Carannante, Cotugno Hospital, Naples, Italy; Antonella Mencacci, Microbiology and Clinical Microbiology Section, Department of Medicine and Surgery, University of Perugia, Santa Maria della Misericordia Hospital, Perugia, Italy; Francesca Orecchioni, Clinical Analysis Laboratory, Section of Microbiology, Ospedali Riuniti di Ancona, Italy; Maria Chironna, Department of Biomedical Science and Human Oncology, Aldo Moro University of Bari, Italy; Anna Giammanco and Teresa Fasciana, Department of Health Promotion, Maternal-Childhood, Internal Medicine of Excellence “G. D’Alessandro,” University of Palermo, 90127 Palermo, Italy; Paolo Fazii, S. Spirito Hospital, Pescara, Italy; Graziella Soldato, Department of Prevention, Epidemiology and Public Health, ASL Pescara, Italy; Maria Golato, Unit of Clinical Pathology, SS Annunziata University Hospital, ASL Lanciano-Vasto-Chieti, Chieti, Italy; Teresa Lopizzo and Letizia Camardese, Unit of Clinical Microbiology and Virology, San Carlo Hospital, Potenza, Italy; Cristina Giraldi, Unit of Microbiology and Virology, Annunziata Hospital, Cosenza, Italy; and Alessandro Bisbano, Epidemiology Unit ASP Crotone, Calabria, Italy. We also thank Paolo Fortunato D’Ancona, Roberta Urciuoli, and Stefano Boros (Department Infectious Diseases, Istituto Superiore di Sanità, Rome, Italy) for the collaboration in the National Surveillance System of Invasive Bacterial Diseases and for the data collection at Istituto Superiore di Sanità. This publication made use of the Neisseria MLST website (http://pubmlst.org/neisseria/), developed by Keith Jolley and sited at the University of Oxford, UK (Stefanelli et al., 2016a). The development of this site has been funded by the Wellcome Trust and European Union.
Conflict of interest
PS received a financial grant from GlaxoSmithKline (GSK) for a scientific proposal unrelated to this study.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1272123/full#supplementary-material
Footnotes
- ^ https://www.salute.gov.it/portale/documentazione/p6_2_2_1.jsp?id=543&lingua=italiano
- ^ https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32018D0945
- ^ http://eucast.org/clinicalbreakpoints/
- ^ http://pubmlst.org/neisseria/
- ^ https://pubmlst.org/bigsdb?db=pubmlst_neisseria_isolates&page=plugin&name= Genome Comparator
- ^ https://www.iss.it/sn-mbi-rapporti-iss
References
Acevedo, R., Bai, X., Borrow, R., Caugant, D. A., Carlos, J., Ceyhan, M., et al. (2019). The global meningococcal initiative meeting on prevention of meningococcal disease worldwide: Epidemiology, surveillance, hypervirulent strains, antibiotic resistance and high-risk populations. Expert. Rev. Vaccines 18, 15–30. doi: 10.1080/14760584.2019.1557520
Ali, A., Jafri, R. Z., Messonnier, N., Tevi-Benissan, C., Durrheim, D., Eskola, J., et al. (2014). Global practices of meningococcal vaccine use and impact on invasive disease. Pathog. Glob. Health 108, 11–20. doi: 10.1179/2047773214Y.0000000126
Birtles, A., Hardy, K., Gray, S. J., Handford, S., Kaczmarski, E. B., Edwards-Jones, V., et al. (2005). Multilocus sequence typing of Neisseria meningitidis directly from clinical samples and application of the method to the investigation of meningococcal disease case clusters. J. Clin. Microbiol. 43, 6007–6014. doi: 10.1128/JCM.43.12.6007-6014.2005
Bozio, C. H., Vuong, J., Dokubo, E. K., Fallah, M. P., McNamara, L. A., Potts, C. C., et al. (2018). Outbreak of Neisseria meningitidis serogroup C outside the meningitis belt-Liberia, 2017: An epidemiological and laboratory investigation. Lancet Infect. Dis. 18, 1360–1367. doi: 10.1016/S1473-3099(18)30476-6
Caugant, D. A., and Brynildsrud, O. B. (2020). Neisseria meningitidis: using genomics to understand diversity, evolution and pathogenesis. Nat. Rev. Microbiol. 18, 84–96. doi: 10.1038/s41579-019-0282-6
De Schrijver, K., and Maes, I. (2003). An outbreak of serogroup C meningococcal disease in the province of Antwerp (Belgium) in 2001-2002. Eur. J. Epidemiol. 18, 1073–1077. doi: 10.1023/A:1026100321871
Dickinson, F. O., Pérez, A. E., and Cuevas, I. E. (2012). Meningococcal disease serogroup C. Risk Manag. Healthc. Policy 5, 1–15. doi: 10.2147/RMHP.S12711
Fazio, C., Daprai, L., Neri, A., Tirani, M., Vacca, P., Arghittu, M., et al. (2022). Reactive vaccination as control strategy for an outbreak of invasive meningococcal disease caused by Neisseria meningitidis C:P1.5-1,10-8:F3-6:ST-11(cc11), Bergamo province, Italy, December 2019 to January 2020. Euro. Surveill. 27, 2100919. doi: 10.2807/1560-7917.ES.2022.27.24.2100919
Fazio, C., Neri, A., Tonino, S., Carannante, A., Caporali, M. G., Salmaso, S., et al. (2009). Characterisation of Neisseria meningitidis C strains causing two clusters in the north of Italy in 2007 and 2008. Euro. Surveill. 14, 19179. doi: 10.2807/ese.14.16.19179-en
Hong, E., Thulin Hedberg, S., Abad, R., Fazio, C., Enríquez, R., Deghmane, A. E., et al. (2013). Target gene sequencing to define the susceptibility of Neisseria meningitidis to ciprofloxacin. Antimicrob. Agents Chemother. 57, 1961–1964. doi: 10.1128/AAC.02184-12
Lo Presti, A., Neri, A., Fazio, C., Vacca, P., Ambrosio, L., Grazian, C., et al. (2019). Reconstruction of dispersal patterns of hypervirulent meningococcal strains of serogroup C:cc11 by phylogenomic time trees. J. Clin. Microbiol. 58, e1351–e1319. doi: 10.1128/JCM.01351-19
Lucidarme, J., Hill, D. M., Bratcher, H. B., Gray, S. J., du Plessis, M., Tsang, R. S., et al. (2015). Genomic resolution of an aggressive, widespread, diverse and expanding meningococcal serogroup B. C and W lineage. J. Infect. 71, 544–552. doi: 10.1016/j.jinf.2015.07.007
Neri, A., Pezzotti, P., Fazio, C., Vacca, P., D’Ancona, F. P., Caporali, M. G., et al. (2015). Epidemiological and molecular characterization of invasive meningococcal disease in Italy, 2008/09-2012/13. PLoS One 10:e0139376. doi: 10.1371/journal.pone.0139376
Nnadi, C., Oladejo, J., Yennan, S., Ogunleye, A., Agbai, C., Bakare, L., et al. (2017). Large outbreak of Neisseria meningitidis serogroup C – Nigeria, December 2016-2017. MMWR Morb. Mortal Wkly. Rep. 66, 1352–1356. doi: 10.15585/mmwr.mm6649a3
Pelton, S. I. (2016). The global evolution of meningococcal epidemiology following the introduction of meningococcal vaccines. J. Adolesc. Health 59, S3–S11. doi: 10.1016/j.jadohealth.2016.04.012
Pezzotti, P., Miglietta, A., Neri, A., Fazio, C., Vacca, P., Voller, F., et al. (2018). Meningococcal C conjugate vaccine effectiveness before and during an outbreak of invasive meningococcal disease due to Neisseria meningitidis serogroup C/cc11, Tuscany, Italy. Vaccine 36, 4222–4227. doi: 10.1016/j.vaccine.2018.06.002
Read, R. C. (2019). Neisseria meningitidis and meningococcal disease: recent discoveries and innovations. Curr. Opin. Infect. Dis. 32, 601–608. doi: 10.1097/QCO.0000000000000606
Stefanelli, P., Fazio, C., Neri, A., Ciammaruconi, A., Balocchini, E., Anselmo, A., et al. (2016a). Genome-based study of a spatio-temporal cluster of invasive meningococcal disease due to Neisseria meningitidis serogroup C, clonal complex 11. J. Infect. 73, 136–144. doi: 10.1016/j.jinf.2016.05.003
Stefanelli, P., Miglietta, A., Pezzotti, P., Fazio, C., Neri, A., Vacca, P., et al. (2016b). Increased incidence of invasive meningococcal disease of serogroup C/clonal complex 11, Tuscany, Italy, 2015 to 2016. Euro. Surveill. 21:30176. doi: 10.2807/1560-7917.ES.2016.21.12.30176
Stefanelli, P., Fazio, C., Neri, A., Isola, P., Sani, S., Marelli, P., et al. (2012). Cluster of invasive Neisseria meningitidis infections on a cruise ship, Italy, October 2012. Euro. Surveill. 17:20336. doi: 10.2807/ese.17.50.20336-en
Stefanelli, P., Fazio, C., Sofia, T., Neri, A., and Mastrantonio, P. (2009). Serogroup C meningococci in Italy in the era of conjugate MenC vaccination. BMC Infect. Dis. 9:135. doi: 10.1186/1471-2334-9-135
Stefanelli, P., Fazio, C., Vacca, P., Palmieri, A., Ambrosio, L., Neri, A., et al. (2019). An outbreak of severe invasive meningococcal disease due to a capsular switched Neisseria meningitidis hypervirulent strain B:cc11. Clin. Microbiol. Infect. 25, e1–e111. doi: 10.1016/j.cmi.2018.07.014
Taha, M. K., Claus, H., Lappann, M., Veyrier, F. J., Otto, A., Becher, D., et al. (2016). Evolutionary events associated with an outbreak of meningococcal disease in men who have sex with men. PLoS One 11:e0154047. doi: 10.1371/journal.pone.0154047
Taha, M. K., Hedberg, S. T., Szatanik, M., Hong, E., Ruckly, C., Abad, R., et al. (2010). Multicenter study for defining the breakpoint for rifampin resistance in Neisseria meningitidis by rpoB sequencing. Antimicrob. Agents Chemother. 54, 3651–3658. doi: 10.1128/AAC.00315-10
Taha, M. K., Vázquez, J. A., Hong, E., Bennett, D. E., Bertrand, S., Bukovski, S., et al. (2007). Target gene sequencing to characterize the penicillin G susceptibility of Neisseria meningitidis. Antimicrob. Agents Chemother. 51, 2784–2792. doi: 10.1128/AAC.00412-07
Tzeng, Y., Bazan, J. A., Turner, A. N., Wang, X., Retchless, A. C., Read, T. D., et al. (2017). Emergence of a new Neisseria meningitidis clonal complex 11 lineage 11.2 clade as an effective urogenital pathogen. Proc. Natl. Acad. Sci. U. S. A. 114, 4237–4242. doi: 10.1073/pnas.1620971114
Keywords: invasive meningococcal disease, Neisseria meningitidis, whole genome sequencing, serogroup C, antimicrobial susceptibility
Citation: Vacca P, Fazio C, Neri A, Ambrosio L, Carannante A, Lista F, Fillo S, Ciammaruconi A, Fortunato A and Stefanelli P (2024) Antimicrobial susceptibility profiles and genotyping of Neisseria meningitidis of serogroup C, Italy, 2000–2020. Front. Microbiol. 14:1272123. doi: 10.3389/fmicb.2023.1272123
Received: 03 August 2023; Accepted: 17 October 2023;
Published: 03 January 2024.
Edited by:
Ainsley Nicholson, Centers for Disease Control and Prevention (CDC), United StatesReviewed by:
Amy Victoria Jennison, Queensland Health, AustraliaHussein O. M. Al-Dahmoshi, University of Babylon, Iraq
Copyright © 2024 Vacca, Fazio, Neri, Ambrosio, Carannante, Lista, Fillo, Ciammaruconi, Fortunato and Stefanelli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Paola Stefanelli, cGFvbGEuc3RlZmFuZWxsaUBpc3MuaXQ=