 James G. Volmer
James G. Volmer Harley McRae
Harley McRae Mark Morrison
Mark Morrison
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Microbiol. , 01 September 2023
Sec. Biology of Archaea
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1268451
This article is part of the Research Topic The Methane Moment – Cross-Boundary Significance of Methanogens View all 20 articles
Methanogenic archaea (methanogens) represent a diverse group of microorganisms that inhabit various environmental and host-associated microbiomes. These organisms play an essential role in global carbon cycling given their ability to produce methane, a potent greenhouse gas, as a by-product of their energy production. Recent advances in culture-independent and -dependent studies have highlighted an increased prevalence of methanogens in the host-associated microbiome of diverse animal species. Moreover, there is increasing evidence that methanogens, and/or the methane they produce, may play a substantial role in human health and disease. This review addresses the expanding host-range and the emerging view of host-specific adaptations in methanogen biology and ecology, and the implications for host health and disease.
Methanogens are prokaryotic organisms that couple energy production and growth to the formation of methane. As such, all known methanogens are obligate methane producers, require anaerobic conditions for growth, and belong to the domain Archaea (Shalvarjian and Nayak, 2021). It is likely that methanogenesis was a dominant metabolic process around 3.5 billion years ago, with evidence suggesting it is one of the earliest mechanisms of metabolism (Lyu et al., 2018). Methanogens act as terminal electron acceptors in these anaerobic environments and are often found in habitats with a low abundance of other electron acceptors, such as sulfate (Lyu et al., 2018). Methanogens are found in a wide variety of anaerobic habitats, including wetlands, marine and freshwater sediments, soil, hot springs, landfills, rice paddy fields, and the digestive tracts of humans and other animals (Aschenbach et al., 2013; Buan, 2018). Although all currently known methanogens are obligate anaerobes, numerous species encode genes for oxygen resistance, suggesting they are capable of surviving short periods of oxygen exposure (Chibani et al., 2022). Historically, the microbial surveys of environmental microbiomes have largely accounted for methanogen diversity. However, with the intensification of high-throughput sequencing technologies applied to gut microbiome analyses, there has been an expansion in our awareness and understanding of the roles of host-associated Archaea, in particular methanogens.
Methanogens utilize a relatively narrow range of carbon sources, and the enzymes that catalyze their conversion to methane are often membrane-bound and coupled with ion (proton) translocation systems (Welte and Deppenmeier, 2011) that produce the electrochemical gradient for the synthesis of ATP (Pisa et al., 2007; Deppenmeier and Muller, 2008). As such, methanogenesis appears to be an obligatory step for energy production and growth of all methanogenic archaea, and the metabolic schema coordinating this process are currently separated into three broad groups: Hydrogenotrophic (Figure 1A), Methylotrophic (Figures 1B–D), and Acetoclastic (Figure 1E; Garcia et al., 2000) and are presented here in order of their current prevalence among host-associated lineages of methanogens. Hydrogenotrophic methanogenesis uses hydrogen to reduce carbon dioxide to methane, with some species additionally able to utilize formate. This form of methanogenesis is the most prevalent among characterized strains (and genomes), with the majority of Methanobacteriales, Methanomicrobiales, Methanococcales, Methanopyrales, and Methanocellales restricted to this pathway for energy production and growth. Methylotrophic methanogenesis involves the conversion of methanol, methylamines, and methylated thiols to methane. While most utilize a H2-dependent reduction of methylated compounds to methane (Fricke et al., 2006; Borrel et al., 2014; Sorokin et al., 2017) such as Methanosphaera stadtmanae DSMZ3091 and members of the order Methanomassiliicoccales; the existence of a small number of environmental isolates capable of H2-independent reduction of methanol to methane has long been known, and occurs in some host-associated strains (Hoedt et al., 2016). Acetoclastic methanogenesis, in which acetate is “split” during methanogenesis via the coordinated biochemistry of the acetyl-CoA decarbonyl (CdhCED) and carbon monoxide dehydrogenase (CdhAB) complex (Ferry, 1997; Welte and Deppenmeier, 2014) is currently the least common pathway encountered among characterised strains (and genomes) assigned to the order Methanosarcinales. This low abundance of host-associated acetolactic methanogens is counter to the high abundance observed in environmental samples, suggesting host physiology influences the overabundance of hydrogenotrophic methanogenesis observed in most animal gut microbiomes (Thauer et al., 2008).
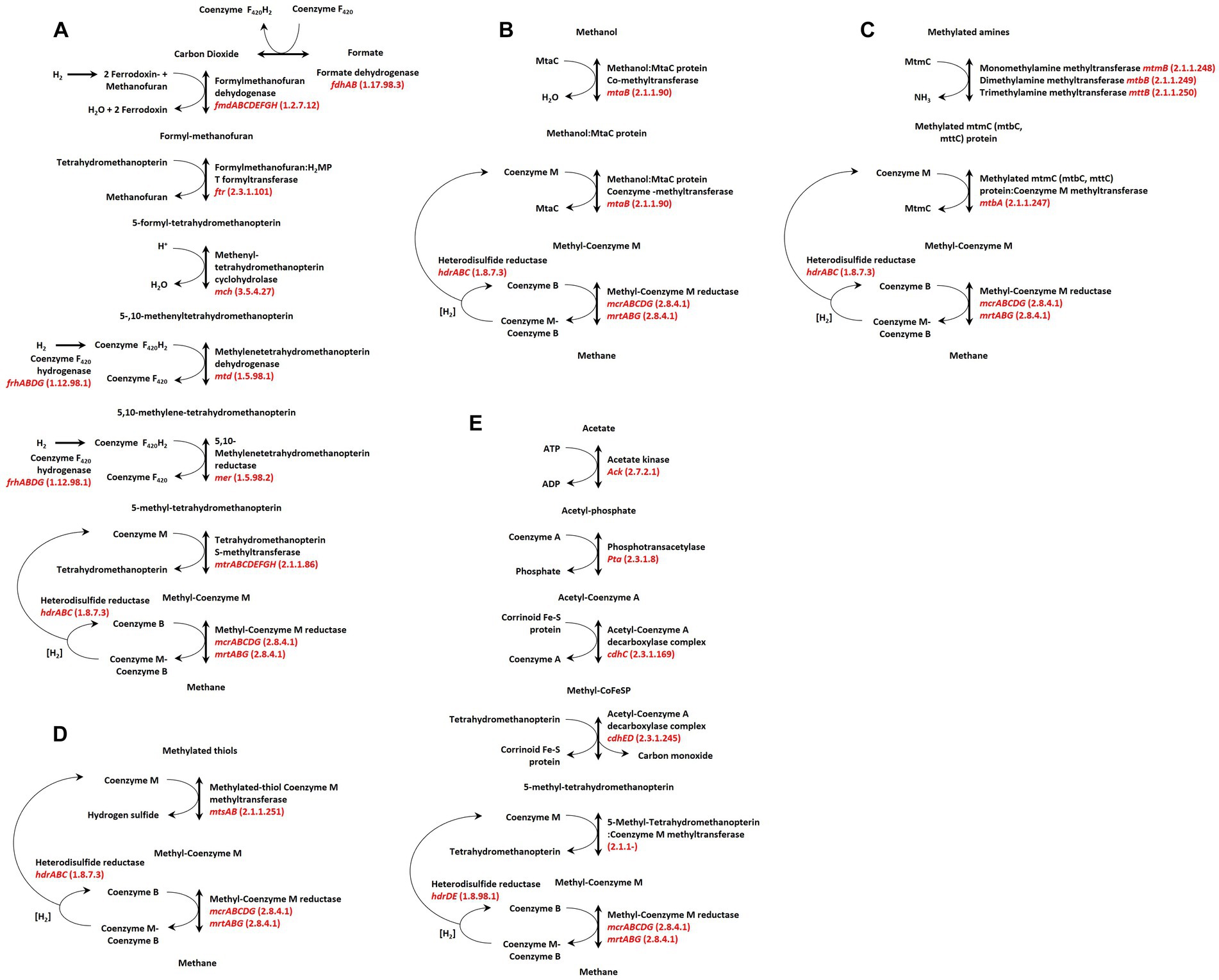
Figure 1. Common pathways of hydrogenotrophic, acetoclastic, and methylotrophic methanogenesis. (A) Acetoclastic pathway utilizing acetate, (B) hydrogenotrophic pathway utilizing carbon dioxide (or formate). (C–E) represent methylotrophic pathways for methylated thiols, methylated amines, methanol, respectively. Figure adapted from Gilmore et al. (2017), with additions based on available methanogenesis KEGG pathways (Kanehisa et al., 2016) and those described by Kurth et al. (2020).
Interestingly, short chain alcohols appear to play a role in the growth of at least some host-associated methanogens. For instance, Leahy et al. (2010) reported the synergistic effects of ethanol on the growth rate of Methanobrevibacter ruminantium during hydrogenotrophic growth. Additionally, a Methanosphaera spp. isolated from a Western Grey kangaroo (Macropus fulginosus) was shown to perform ethanol-dependent methanol reduction to methane, as well as the standard H2-dependent reduction observed in other Methanosphaera species (Hoedt et al., 2016). Similarly, a recent analysis of the bovine isolate Methanobrevibacter boviskoreani showed that ethanol, 1-propanol, and 1-butanol could serve as alternative electron donors for CO2 reduction to methane (Li et al., 2023). In contrast, while environmental isolates of Methanocorpusculum spp. are believed to utilize 2-propanol, butanol, and pentanol as a source of reducing power for CO2-dependent hydrogenotrophic methanogenesis, the recently isolated host-associated species Methanocorpusculum vombati and Methanocorpusculum petauri appear unable to utilize short chain alcohols in a similar manner (Volmer et al., 2023).
Methanogenesis is an important part of the Earth’s energy and carbon cycles, however excessive production of methane is undesirable, as it contributes to climate change. For this reason, there has been growing interest in methanogens, as methane is now known to be the second most important greenhouse gas following carbon dioxide (Lyu et al., 2018). Reducing emissions from agriculture is one area of particular importance, as livestock are the greatest contributors of anthropogenic methane, with enteric livestock methane emissions making up approximately 30% of all anthropogenically produced methane (Gilmore et al., 2017; Smith et al., 2021). Ruminant animals in particular are of significance, as they are farmed globally on larges scales and have been shown to emit more methane than some other animals, such as horses, macropodids and rabbits, even when scaled for size, feed intake, digesta retention time and gut capacity (Clauss et al., 2020). Methanogenesis also causes the loss of 6–10% of gross energy intake in ruminants and is therefore undesirable in terms of energy efficiency and feed costs (Pinares-Patiño et al., 2013). Ruminants, such as cattle, produce large amounts of methane due to the methanogenic archaea present in their gut microbiota, which consume hydrogen and other products of microbial fermentation. This process is known as interspecies hydrogen transfer and removes waste products that would otherwise limit the growth of microbial species required for fermentation in the gut (Bryant et al., 1967; Iannotti et al., 1973; Stams and Plugge, 2009). For these reasons, it is important that a deeper understanding of methanogens, especially those in high methane producing animals, is gained so that new techniques for reducing methane emissions can be developed.
Studies have shown that the amount of methane emitted by sheep is a heritable trait and that lower methane and higher methane emitting phenotypes exist (Pinares-Patiño et al., 2013; Johnson et al., 2022). Interestingly, one study found that the methanogen abundance in sheep deemed to be “high” and “low” methane emitters was similar, and increases in methane emission by certain animals appeared to be due to increases in expression of methanogenesis pathway genes, particularly those involved in hydrogenotrophic methanogenesis (Shi et al., 2014). This indicates that the composition of the gut methanogen community, rather than the total abundance of methanogens, contributed to differences in methane emissions. A similar study demonstrated that “high” and “low” methane phenotypes also exist in cattle and determined that Methanobrevibacter were more numerous in the “high” methane emitters, while the bacterial community in the “low” emitters included a higher abundance of Proteobacteria, in particular Succinivibrionaceae (Pope et al., 2011; Wallace et al., 2015). Furthermore, Martínez-Álvaro et al. (2020) found that high methane emitting ruminants were associated with a lower diversity of hydrogenotrophic methanogens, while low methane emitters were associated with an increase in methanogen species diversity across all three methanogenic pathways. Methanomethylophilus (a methylotrophic methanogen) was increased in lower methane emitting bovines, along with a decrease in Methanobrevibacter species. This study also highlighted that methane emissions in ruminants is affected by the complexity and diversity of the microbial community and the metabolism of these microbes. Together, these analyses suggests that both methanogens and other microorganisms play an important role in the different low and high methane emitting ‘ecotypes’. One such example is that rumen methanogen Methanobrevibacter ruminantium M1 is capable of binding to both protozoal and bacterial partners that produce hydrogen (Ng et al., 2016). Co-culture studies have also demonstrated inter-species H2 transfer from Ruminococcus flavefaciens, Ruminococcus albus, and Selenomonas ruminantium (Scheifinger et al., 1975; Latham and Wolin, 1977; Wolin et al., 1997). Bauchop and Mountfort (1981) showed that co-culture of a hydrogenotrophic methanogen with the fungi Neocallimastix frontalis resulted in a significant decrease in the concentrations of formate, ethanol and lactate. Methanogen have also been detected in cultures of ruminant fungi, such as Neocallimastix and Anaeromyces, though the exact fungi-methanogen interactions remain to be determined (Jin et al., 2011). Therefore, to fully understand methanogenesis, we need to learn more about these organisms and how they interact with other members of the gut microbiota.
There is evidence that the rumen microbiota is affected by the host, environment, diet and geographical location of the animal. Malik et al. (2021) investigated the effects of host species on the methanogen communities in ruminants by comparing the methane emissions and methanogen diversity of cattle and buffalo fed the same diet and kept in similar environments in the same geographical location. Cattle had higher overall methane emission levels compared to buffalo, but both species had a similar methane yield when calculated as grams of methane per dry matter intake (cattle on average had higher dry matter intake and higher body mass). There were some differences in the diversity of methanogen species present at very low abundances between the two host species, but overall, the rumen samples from cattle and buffalo had similar taxonomic profiles of their methanogens with the dominant methanogen genus being Methanobrevibacter for both host species. It is therefore likely that the methane yield may be more dependent on diet rather than host species for these types of hosts (Malik et al., 2021). However, cattle and buffalo are physiologically very similar and digest their food in the same way, so it may be expected that they would have similar archaeal profiles and methane yields when fed the same diet.
Various dietary interventions have been used to attempt to reduce methane emissions from ruminants. Poulsen et al. (2013) found that supplementing dairy cattle diets with rapeseed oil reduced their methane emissions. The reduction in methane emissions was found to be the result of an inhibitory effect on the abundance and activity of a novel methylotrophic Thermoplasmata group of methanogens, as the Methanobrevibacter and Methanosphaera species also present in the rumen samples were not decreased by the addition of rapeseed oil. However, as Methanobrevibacter species are often the most abundant in the rumen, this may not be an effective methane mitigation strategy. Tropical tree foliage supplementation also demonstrated a reduction in methanogens and enteric methane production (Alayón-Gamboa et al., 2023). Supplementing a diet of poor-quality roughage with sweet potato vine silage has also been shown to decrease methane emissions in female cattle (Ali et al., 2019). This study found that adding sweet potato vine silage to a low-quality roughage diet increased digestibility, decreased solid digesta retention time in the rumen and decreased production of methane per unit of digested dry matter. Decreased solid retention time has been linked to decreased methane emissions in other studies, likely due to the effects of food passage time on H2 concentrations, and therefore methanogen activity (Janssen, 2010). Further, plant secondary metabolites have also been demonstrated as a viable anti-methanogenic supplement ad libitum in sheep, though the predominant Methanobrevibacter populations remained consistent between the test and control groups (Malik et al., 2022). Denman et al. (2007) investigated the effects of supplementing cattle diets with the anti-methanogenic chemical bromochloromethane. They found that methane emissions were reduced by around 30% in cattle supplemented with bromochloromethane compared to cattle whose feed was not supplemented. Furthermore, through the use of clone libraries generated from DNA extracted from rumen samples, they found that there was a decreased abundance of the dominant Methanobrevibacter species and a more diverse population of other methanogen species in cows supplemented with bromochloromethane.
One of the most successful food additives for methane mitigation to date is bromoform containing seaweed, such as Asparagopsis species. These seaweeds have been shown to inhibit methanogenesis in ruminants by up to 98% and are effective at low concentrations (Glasson et al., 2022). Halogenated methane analogues, such as bromoform, are proposed to inhibit methanogenesis by competitively binding with key enzymes, such as methyl coenzyme M methyltransferase. Despite its effectiveness, there are some concerns about using bromoform as a feed additive, as it is potentially carcinogenic and has ozone-depleting properties. The effects of bromoform supplementation on animal health, as well as the potential for it to enter products for consumption such as meat and milk need to be considered. Most studies to date show no increased levels of bromoform in animal products and excrement after supplementation with Asparagopsis (Glasson et al., 2022). However, one study by Muizelaar et al. (2021) showed increased levels of bromoform in milk and urine from cattle in the early stages of Asparagopsis supplementation, but these levels were still below the recommended World Health Organization limit for bromoform. This study had to be terminated early, as many of the cows refused the food mix supplemented with seaweed and therefore had low food intakes. This may indicate that the supplementation of seaweed into the diet of cattle needs to be further optimized. Another potential risk of using bromoform containing seaweed as a supplement is the highly volatile nature of this compound. Bromoform can be converted into inorganic bromine in the atmosphere, which has ozone depleting properties (Glasson et al., 2022). For bromoform to be a viable methane mitigation strategy, the environmental impacts of Asparagopsis production on the atmosphere, as well as land use, storage and transportation need to be considered. As the cattle industry is so large in countries like Australia, it may not be feasible to grow enough Asparagopsis to supplement the feed of every ruminant raised for agricultural purposes.
Another promising methane reduction strategy is the use of 3-Nitrooxypropanol (3-NOP). This molecule specifically targets and inactivates methyl-coenzyme M reductase (MCR), which is essential in catalyzing the final step of methanogenesis (Figure 1; Duin et al., 2016). In the last decade, numerous studies have shown the efficacy of 3-NOP in reducing enteric methane production, with average predicted reductions of 30% (Kim H. et al., 2020). In fact, some studies have reported the use of 3-NOP as a feed additive to reduce methane production by up to 82% (Vyas et al., 2016; McGinn et al., 2019). A recent analysis by Araújo et al. (2023) showed a reduction in methane emissions by ~49.3% (g/d) resulting in a reduction in gross energy intake loss by 42.5%. The methane mitigation potential of 3-NOP was also demonstrated with feedlot cattle fed a barley-based diet with canola oil and showed 65.5 to 87.6% reduction in emissions (Almeida et al., 2023). Several studies have also shown that 3-NOP poses no mutagenic or genotoxic potential and that Bovaer® 10, a food additive containing 3-NOP, was efficacious for methane reduction in dairy cows (Thiel et al., 2019; Bampidis et al., 2021). It was further shown that the supplement did not affect soil health and 3-NOP manure could be used as a nutrient source for forage crops (Owens et al., 2021). This positions 3-NOP as a promising feed additive for reducing methane emissions whist posing minimal impacts on the animal or surrounding environment.
Li et al. (2022) showed that altering the diet of cattle changed the microbial composition of the rumen and subsequently influenced methanogenesis. Their study showed that feeding a fiber-rich versus starch-rich diet resulted in two distinct microbiomes with differing carbohydrate degradation, H2 metabolism and methane production. The difference in the microbiome between the two diet types was associated with distinct substrate preferences and metabolic pathways of certain microbial species. The fiber-rich diet was found to increase the acetate to propionate ratio and selected for fibrolytic bacteria. The enrichment of fibrolytic bacteria can play a role in the adaptation to lignified diets, however it may also be associated with increased methane production and a decrease in energy conversion efficiency (Li et al., 2022). The starch-rich diet increased ruminal dissolved H2 levels, decreased CH4 production and enriched for amylolytic bacteria. The fiber-rich diet enriched for methanogenic hydrogenases from Methanobacteriota species, whereas the starch-rich diet enriched for hydrogenases from Firmicutes and Spirochaetota groups. These results show that modifying cattle diets to contain more starch-rich ingredients may help mitigate methane production. Interestingly, Li et al. (2022) also found that the marker gene for hydrogenotrophic acetogenesis was more abundant in cattle fed the fiber-rich diet. This is interesting because another method that has been proposed to reduce methane production is to enrich for acetogenic bacteria in the rumen, which compete for H2 and produce acetate rather than methane (Karekar et al., 2022). So far, acetogenic bacteria isolated from the rumen have not been able to outcompete methanogens in in vitro studies under normal circumstances, however there is evidence that increased H2 levels, may allow homo-acetogens to become more dominant hydrogen sinks in the gut environment (Karekar et al., 2022).
Defaunation (the removal of protozoa) has also been proposed as a potential method for reducing the methanogen population in the rumen and promoting acetogenesis. These protozoa can act as hosts for methanogens and protect them while providing a source of hydrogen. In vivo studies suggest that these protozoa are not essential to host animal health and defaunation has shown decreases in methane production of up to 49% in animals fed barley-based concentrates (Whitelaw et al., 1984). The direct feeding of reductive acetogens to ruminant livestock has so far shown only temporary success. In one study, methane emissions were reduced by up to 80% in rams supplemented with Peptostreptococcus productus (a reductive acetogen), before increasing to normal levels after less than a week (Nollet et al., 1998). This suggests that the acetogenic bacteria present in the rumen were not robust enough to take over and remain as the primary H2 sinks (Karekar et al., 2022). An increased understanding of how these microbes compete with methanogens in the rumen may provide insight into techniques that could enrich for acetogens and other hydrogenotrophic bacteria, so that they could outcompete methanogens and mitigate methane production from cattle and other ruminants.
To date there have been a number of studies investigating various methods to mitigate methane emissions from ruminant livestock, including the introduction of microorganisms into the rumen that would compete with methanogens for hydrogen (such as reductive acetogens), the elimination of protozoa in the rumen that form symbiotic relationships with methanogens, immunization of the host against methanogenic archaea, as well as dietary supplements and additives (Goopy, 2019). However, application of these techniques to alter the host microbiome or eradicate methanogens from the rumen have often shown limited success in vivo (Goopy, 2019) and no single methane mitigation technique has thus far been successfully implemented on a large scale in the agricultural industry. Methane production is a heritable trait (Pinares-Patiño et al., 2013), suggesting that it is possible to produce animals with a ‘low methane phenotype’ through selective breeding. However, this process would require a major shift in the agricultural industry that would be costly and time consuming, as identifying which animals are low methane emitters requires complex testing and it would take years of selective breeding to achieve the desired phenotype. Finally, vaccination against methanogens has repeatedly been suggested as a possible solution to enteric methane emissions (Baca-González et al., 2020), but this would require a vaccine that universally targets all methanogens, so that another methanogenic species does not expand to fill the niche. Clearly, the most likely way to produce such a vaccine requires better understanding of the genetics and molecular biology of host adaptation to identify those epitopes (targets) that generate a strong, multivalent and host-specific antibody response.
As well as ruminants, methanogens inhabit the gastrointestinal tract of Australian herbivores. The digestive anatomy of the rumen has some similarities, but differs to that of Australian marsupials, which can be classed as either foregut fermenters (Macropodidae) or hindgut fermenters (wombats, koalas, possums and gliders) (Hume, 1984). The gut microbiome also differs between ruminants and marsupials. However, the methanogens present in some macropodids have a similar taxonomic profile to those in ruminants, although present at substantially lower numbers (Evans et al., 2009). Despite the presence of methanogens in their digestive tract, macropodids have been shown to produce less methane than ruminants when fed the same diet (Madsen and Bertelsen, 2012). For this reason, investigating the diversity and metabolism of methanogens present in the digestive tracts of native Australian herbivores may provide insights into why these animals are ‘low’ methane emitters and provide approaches for the potential reduction of methane emissions in ruminants.
To date, there have been few studies aiming to characterize the methanogens present in Australian herbivores and determine why these animals are low methane emitters, and because of this, there are limited cultured isolates of marsupial associated methanogens. However, Evans (2011) isolated a Methanobrevibacter species (WBY1) from the forestomach digesta of a Tammar wallaby and obtained an enriched culture of a Thermoplasmatales (Methanomassiliicoccales) affiliated methanogen from the forestomach digesta of a Western Grey Kangaroo. WBY1 was found to grow only in the presence of CO2/H2 and the Methanomassiliicoccales-associated methanogen was found to utilize methylamines and H2 for methanogenesis, but an axenic culture was not achieved, which indicates that it may have relied on the bacteria present in the enrichment culture for specific metabolites. Subsequently, Hoedt et al. (2016) isolated a Methanosphaera species (sp. WGK6) from the digestive tract of a Western Grey Kangaroo that is capable of using ethanol and methanol, rather than H2 and methanol for methylotrophic methanogenesis. The proposed mechanism of this ethanol/methanol methanogenesis is a two-step oxidation of ethanol to acetate coupled with the reduction of methanol to methane. This mode of metabolism suggests that some Methanosphaera species have adapted to lower H2 environments and may be associated with lower methane emissions. Similar findings were recently confirmed in Methanobrevibacter, with Li et al. (2023) demonstrating that Methanobrevibacter boviskoreani, a rumen methanogen isolate, was capable of utilizing ethanol, 1-propanol, and 1-butanol as alternative electron donors in CO2-dependent methanogenesis. In a further study by Hoedt et al. (2018), a metagenomic analysis was used to compare Methanosphaera strains from different hosts and, interestingly, it was discovered that two genotypes exist. A larger (~2.9 Mbp) genotype was present in ruminant hosts and a smaller (~1.7 Mbp) genotype present in monogastric hosts, such as macropodids. The results of these findings demonstrate that the Methanosphaera genus is monophyletic and comprised of two genotypes, the larger of which is so far restricted to ruminant hosts. This demonstrates that Methanosphaera species have adapted to live in their specific host environments and their genome content reflects this. Recently, Volmer et al. (2023) successfully isolated two novel Methanocorpusculum species, M. petauri, and M. vombati from mahogany glider and wombat fecal samples, respectively. The Methanocorpusculum genomes were larger than those of Methanocorpusculum species found in environmental samples and showed a distinct phylogenetic separation from the environmental-associated genomes. The two novel Methanocorpusculum genomes encoded similar genes for methanogenesis, however, both were unable to utilize secondary alcohols in CO2-dependent methanogenesis, like the environmental isolate M. parvum.
There is evidence that macropodids may produce less methane than ruminants due to an increased presence of acetogens that consume more of the H2 produced in the kangaroo forestomach than in the rumen. Various studies, such as that by Ouwerkerk et al. (2009), have demonstrated the presence of acetogens in the macropodid forestomach. Godwin et al. (2014) used stable isotope probing to investigate the fate of H2 and CO2 in the kangaroo forestomach and the rumen. They performed in vitro fermentations using 13C labeled bicarbonate and CO2 with kangaroo forestomach and bovine rumen contents, in which the methane content in the headspace was measured at various intervals. They found that methane was detectable in the headspace of the rumen fermentation after only 3 h, whereas it took 7 days before a measurable quantity of methane was detected in the kangaroo fermentations. The methane produced in the kangaroo fermentations was also mostly unlabeled, which suggests that it did not originate from the CO2. This indicates that the methanogens present in the kangaroo forestomach may be less active than those in the rumen and also use a pathway other than hydrogenotrophic methanogenesis. Godwin et al. (2014) also found that kangaroo fermentations with 13C labelled bicarbonate produced highly labelled acetate, whereas the bovine fermentations produced only slightly labelled acetate. This demonstrates that reductive acetogenesis produces a larger amount of acetate in the kangaroo forestomach than the rumen. In addition to the in vitro fermentations, Godwin et al. (2014) used RNA stable isotope probing to identify bacterial species associated with CO2 and H2 metabolism in the kangaroo forestomach samples. They identified an OTU that is very close to the 16S sequence of Blautia coccoides, which is a known acetogen. Sequences with high similarity to this OTU were found in all the kangaroo samples, but only 40% of the rumen samples. One reason that acetogens are more dominant in the macropodid forestomach may be the shorter retention time of fiber when compared to the rumen (Karekar et al., 2022). Alternatively, Leng (2018) hypothesized that the morphology of the macropodid forestomach limits expansion and makes it more sensitive to high gas concentrations, which has led to selective pressure against methanogens through the excretion of immunogenic components by mucosal tissue in the cranial blind sac of the forestomach. Thus, leaving more opportunity for acetogens to fill the niche of H2 consumption without producing large amounts of gas. This hypothesis was based on the similarity of the cranial blind sac to other organ structures found in mammals, such as the appendix, which has been proposed to play a role in immune function and biofilm formation (Randal Bollinger et al., 2007). However, this theory proposed by Leng (2018) is yet to be backed up by experimental data. Overall, these results indicate that acetogenesis may play a larger role in CO2 and H2 metabolism in macropodids than in cattle, resulting in reduced methane emissions. However, the conditions that allow this less thermodynamically favorable pathway to operate in the macropodid forestomach are still not well understood. A better understanding of the methanogenic pathways utilized by methanogens in macropodids and their competition with other hydrogenotrophic bacteria may help to elucidate reduced methane production in ruminants.
Non-ruminant herbivores are also dependent on the recruitment and retention of microbes within different segments of their gastrointestinal tract in support of plant biomass conversion to nutrients. These adaptations to herbivory are outlined in detail by Mackie et al. (2000) but in brief detail, these animals may utilize either a sacciform, non-gastric region of the stomach (e.g., the macropodids, such as kangaroos and wallabies) or hindgut (caecum or colon) (White and Mackie, 1997). Many breeds of pigs including Duroc, Landrace, Yorkshire (Mao et al., 2011; Mi et al., 2019), and Erhualian (Zhu et al., 2011) also contain a substantial abundance of Methanobrevibacter. One study on Canadian pigs showed Methanoculleus spp. as additional major contributors to methane emissions through hydrogenotrophic methanogenesis (Barret et al., 2013). Colonic fermenters such as horses have shown an abundance of Methanobrevibacter (Lin and Miller, 1998; Fernandes et al., 2014) and Methanocorpusculum species (O'Donnell et al., 2013; Fernandes et al., 2014). Similarly, white and black rhinoceros are colonized by Methanobrevibacter and Methanocorpusculum, as well as Methanosphaera and Methanomassiliicoccales-related species (Luo et al., 2013; Gibson et al., 2019). In fact, Methanocorpusculum spp. was the most abundant methanogen in captive white rhinos at ~60% (Luo et al., 2013) and has also been found as the predominant taxon in Japanese thoroughbred horses and ponies (Lwin and Matsui, 2014). Smaller caecal fermenting animals also show methanogen colonization. Methanobrevibacter-related species have been isolated from rodents such as the feces of rats, with aging rats showing an increased abundance of total methanogens (Maczulak et al., 1989; Lin and Miller, 1998). Additionally, squirrels also contain Methanosphaera (Carey et al., 2013). The caecal contents of rabbits have also shown the presence of Methanosphaera and several Methanobrevibacter species (Kusar and Avgustin, 2010). Methanosphaera cuniculi was isolated in pure culture from the intestinal tract of a rabbit, providing genomic insights into the limited number of characterised Methanosphaera. The diet of the North American beaver consists entirely of woods, roots and aquatic plants, requiring a syntrophic relationship with fermentative bacteria to aid in digestion (Kohl et al., 2014). Interestingly, Methanosphaera, Methanobrevibacter, and Thermoplasmatales were detected from the caecum and feces, but Methanosphaera-associated OTUs accounted for more than 99% of archaeal reads (Kohl et al., 2014). Multiple sequences of Methanosphaera were detected across all samples, with a single OTU accounting for 85–90% of Methanosphaera sequences (Kohl et al., 2014). This shift toward Methanosphaera is likely driven by the production of methanol by bacterial fermentation of pectin derived from the plant-rich diet (Pieper et al., 1980; Revilla and González-SanJosé, 1998).
Figure 2 provides an overview of the prevailing evidence that the Domain Archaea, and the methanogens particularly are part of the human microbiome. That human methanogens are members of the human gut microbiota was first established principally by the efforts of Wolin and colleagues in the 1980s, including their isolation of the type strains of Methanobrevibacter smithii and Methanosphaera stadtmanae (Miller et al., 1982; Miller and Wolin, 1985), the latter of which prevailed for many years as the sole cultured member of the genus. More recently, the Methanomassiliicoccales (Methanomassiliicoccales and Methanomethylophilus; Figure 2) were confirmed to be present and capable of the utilization of methanol and other methylated compounds (Chaudhary et al., 2015). As such, the human-associated methanogens are similar to those isolated from other vertebrate animals in terms of their carbon utilization profile, but to date the metabolic capacity for methane formation and growth appears to be hydrogen-dependent.
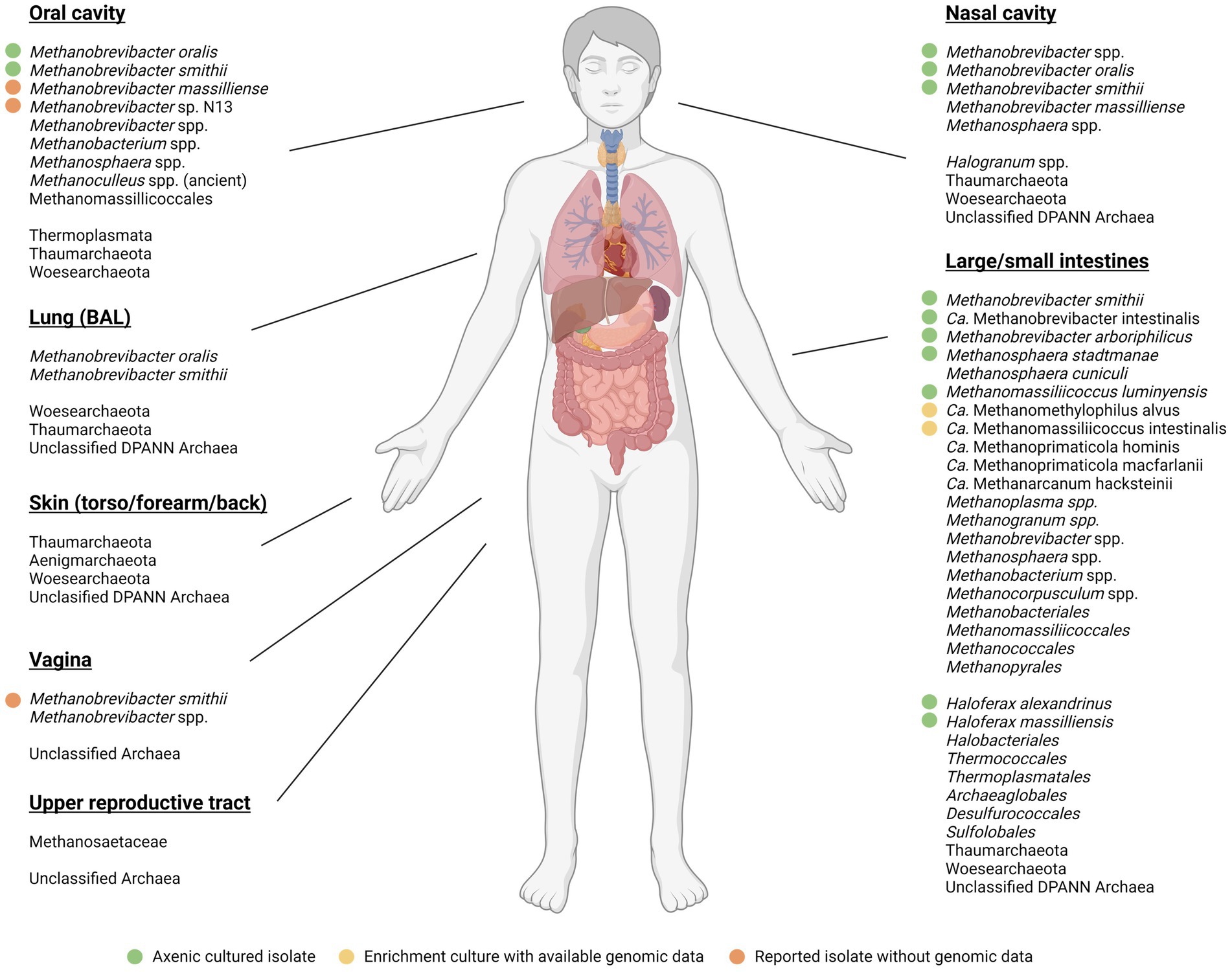
Figure 2. Methanogenic and other archaea detected across the human body. Samples included those from the oral cavity, nasal cavity, lungs (BAL), skin (torso/forearm/back), large/small intestine, vagina, and upper reproductive tract.  represents axenic isolates with available genomic data,
represents axenic isolates with available genomic data,  represents non-axenic enrichments with available genomic data,
represents non-axenic enrichments with available genomic data,  represents isolates with no available genomic data. No symbol represents identification through sequencing data alone. The Figure is adapted and augmented from Bang and Schmitz (2015) and Nkamga et al. (2017), respectively; and the anatomical diagram created with BioRender.com.
represents isolates with no available genomic data. No symbol represents identification through sequencing data alone. The Figure is adapted and augmented from Bang and Schmitz (2015) and Nkamga et al. (2017), respectively; and the anatomical diagram created with BioRender.com.
Methanobrevibacter smithii typically represents the most dominant methanogen in the GI tract, with prevalence of nearly 95% (Dridi et al., 2009, 2011b; Dridi, 2012) and relative abundance of up to 10% in some studies (Eckburg et al., 2005). Methanosphaera stadtmanae are abundant and prevalent in ~30% of individuals (Dridi et al., 2012b). The Methanomassiliicoccales, represented by Methanomassiliicoccus luminyensis, are the least common, with a prevalence of 4–50% of individuals tested and increase in relative abundance with age (Dridi et al., 2012b; Vanderhaeghen et al., 2015). The use of culture-independent methods identified Candidatus Methanomethylophilus alvus (Borrel et al., 2012) and Candidatus Methanomassiliicoccus intestinalis (Borrel et al., 2013) as additional methanogens in the human gut. Methanogens have also been identified among the communities of other body sites, with Methanobrevibacter oralis and Methanomassiliicoccales spp. detected in the oral cavity (Li et al., 2009; Horz et al., 2012). Although phylogenetically similar, M. oralis and M. smithii show adaptations to their respective biological niches, with M. oralis isolates lacking the capacity to utilize formate (Ferrari et al., 1994) and M. smithii encoding for bile salt hydrolase (bsh) genes (Gaci et al., 2014). Other members of the Methanobrevibacter genus found in humans include M. arboriphilus (Khelaifia et al., 2014) and M. massiliense (Huynh et al., 2017), though there is currently little information about their respective prevalence or abundance. Interestingly, cultivation of oral methanogens from three individuals with severe periodontitis also identified a novel Methanobrevibacter species designated N13 (Huynh et al., 2015). Recently, Chibani et al. (2022) and colleagues, produced a comprehensive catalog of genomes recovered from human gut metagenomes. This analysis identified almost 100 archaeal strains (99% ANI), the majority of which were members of Methanobacteriales (87.15%), as would be expected. Interestingly, genomes were also recovered from Methanomicrobiales (0.26%) and Halobacteriales (0.17%), which are scarcely reported as a part of the human gut microbiome. Despite M. luminyensis originally isolated from a human fecal sample (Dridi et al., 2012a), no representatives of this species were recovered. In fact, Ca. Methanoprimaticola hominis (originally Methanomassiliicoccales Mx06 or UBA71), Ca. Methanomethylophilus alvus, and Ca. Methanomassiliicoccus intestinalis represented the most abundant species of Methanomassiliicoccales. Further, it was also identified that M. smithii represents two distinct species, M. smithii and Ca. M. intestinii, with distinct genes encoding molybdate transport and adhesin-like proteins, as well as additional uncharacterised processes.
Representatives of the Methanobacteriales, Methanomicrobiales, Methanococcales, Methanopyrales, and Methanosarcinales have also been detected in the human gut via shotgun metagenomics sequencing (Scanlan et al., 2008; Bang and Schmitz, 2015). Along with the abovementioned M. arboriphilus, methyl coenzyme M reductase A (mcrA) clone sequences closely related to Methanoculleus chikugoensis were found, along with oral representatives of Methanobacterium congolense and M. mazei (Nava et al., 2012; Nguyen-Hieu et al., 2013). A separate study on longitudinal GIT biopsies retrieved Methanobrevibacter sequences related to M. filiformis and M. woesei, along with the first identification of Methanobacterium sequences within the ileum (Koskinen et al., 2017). This study additionally showed Methanobacteriaceae present within nasal samples. Methanogenic archaea identified as M. smithii have been detected in vaginal samples of individuals suffering from bacterial vaginosis (Belay et al., 1990; Grine et al., 2019a,b). Further, a study into the reproductive tract showed Methanosaetaceae within cervical mucus and peritoneal fluid samples (Li et al., 2018).
Given the diversity of human-associated methanogens identified by culture-independent techniques, it is important to perform functional analyses of cultured isolates to validate culture-independent findings. Current human methanogen isolates have a substantial bias toward M. smithii, which may be expected given the high prevalence and abundance of this species (Figure 3). Despite Chibani et al. (2022) identifying at least 16 species of GIT-associated methanogens, the vast majority have no axenic cultured representative including Ca. Methanoprimaticola, one of the most prevalent genera in the human gut. This bias toward Methanobrevibacter species is consistent across other mammalian hosts, with isolates from bovine, ovine and termite samples dominated by the genus (Figure 3). To understand the role of methanogens in human health, it is important to cultivate representatives from diverse species. Further, ~50% of all current methanogen isolates are human M. smithii and, as such, methanogen cultivation should also focus on recovering a variety of species from diverse animal hosts to provide phylogenetic and functional analyses on the wider role of host-associated methanogens.
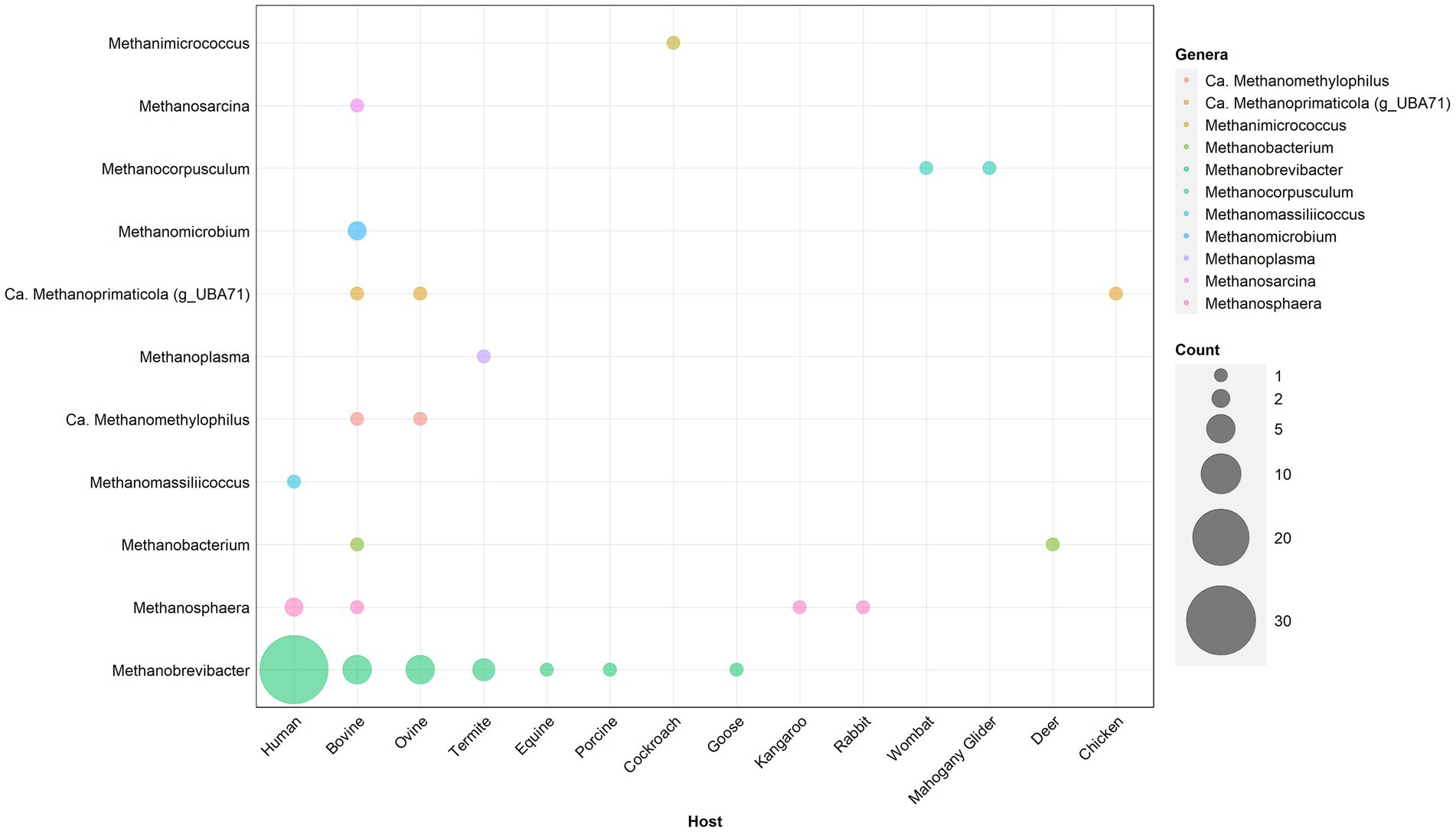
Figure 3. Taxonomic distribution of published methanogen isolates cultured from animal hosts. Only isolates which have been published with available whole genome sequencing have been included. The size of individual circles represents the number of isolates, with the color representing the respective animal from which the isolate was recovered. Isolates have been recovered from Methanobacteriales (Methanobrevibacter, Methanosphaera, and Methanobacterium), Methanomassiliicoccales (Methanomassiliicoccus, Methanomethylophilus, Methanoplasma, and Methanoprimaticola) (g_UBA71), Methanomicrobiales (Methanomicrobium and Methanocorpusculum), and Methanosarcinales (Methanimicrococcus and Methanosarcinales).
In summary then, and similar to other vertebrate hosts, methanogens (and the Domain Archaea) are a relatively small population of microbes that reside within the microbiomes resident at different sites throughout the human body. There has been a gradual but sustained increasing interest in Archaea, and specifically methanogens, and their relationship with human health and disease. In that context, Table 1 summarizes the associations between the relative and/or absolute abundance of methanogenic archaea with different non-communicable diseases and disorders. For the reasons outlined above, much of the interest has been directed toward digestive health and disease, and despite these associations, studies that dissect cause from consequence in these associations with the organic and/or functional diseases and disorders remain limited.
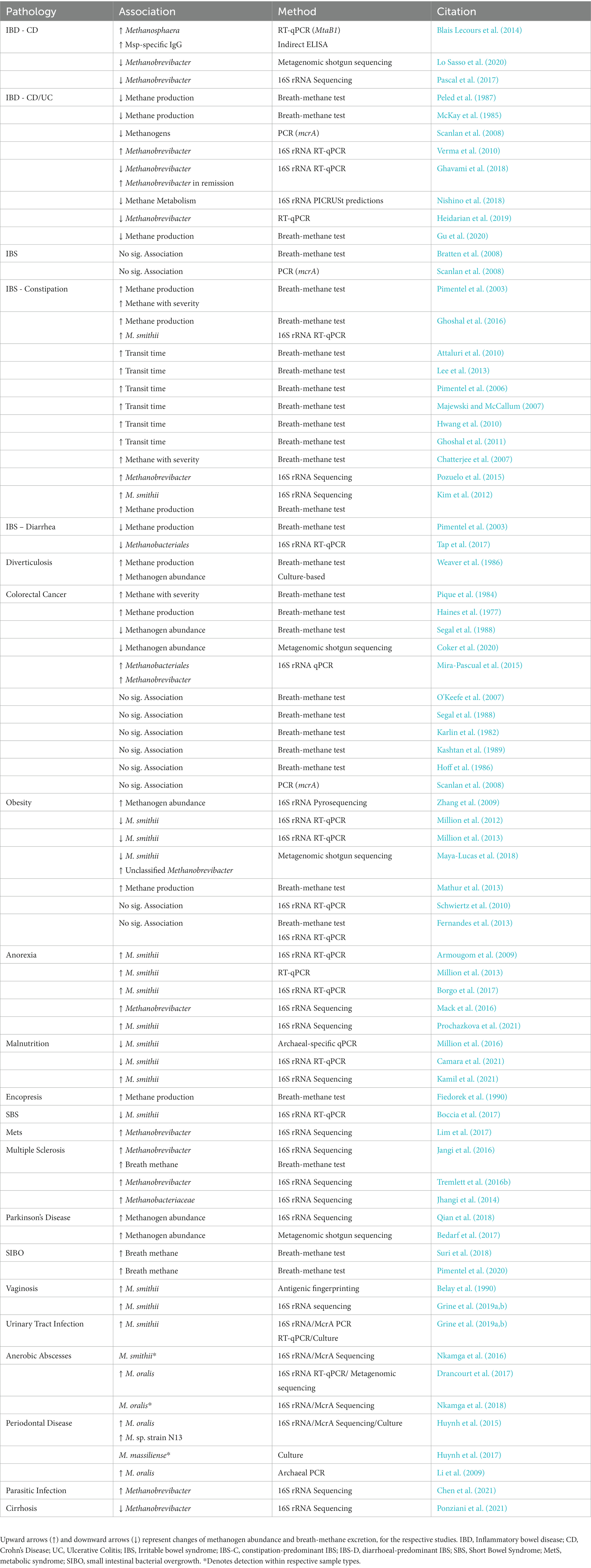
Table 1. Associations between methanogenic archaea and different diseases or disorders.
Methanogens occupy a key metabolic niche in anaerobic environments via their utilization of hydrogen gas, as well as the carbon dioxide and/or other simple carbon substrates produced by bacterial fermentation. This process is known as interspecies hydrogen (and carbon dioxide) transfer and serves to limit the build-up of hydrogen gas, which can inhibit bacterial fermentation and growth (Nakamura et al., 2010). The removal of these end products conserves the thermodynamic equilibrium of fermentation, maintaining ‘microbial homeostasis’ within the human GIT (Stams and Plugge, 2009; Sieber et al., 2012). For Methanosphaera, a source of methanol is necessary for growth, which can come in the form of free methanol (spirits, beer, wine), methyl esters of fatty acids (aspartame) and pectin (fruit and vegetable) (Toxicity, 2011). There is currently no evidence to suggest Methanosphaera can utilize pectin directly, so the degradation of pectin by pectinase-containing bacteria, such as Bacteroides spp., is necessary for methanol availability (Jensen and Canale-Parola, 1986; Dongowski et al., 2000). For Methanomassiliicoccales spp., methylated-amines produced from dietary carnitine, choline, trimethylamine N-oxide (TMAO) and phosphatidylcholine from meat, eggs, nuts, and fish can be utilized. These compounds are broken down by resident microbial communities, such as the conversion of TMAO to trimethylamine (TMA) by Enterobacteriaceae spp., to produce free methylated amines (Zeisel et al., 1983; Rebouche and Chenard, 1991; Spencer et al., 2011; Tang et al., 2013; Hoyles et al., 2018). Comparatively, there is little information on the role non-methanogenic archaea play in nutritional ecology of the gut. Given that halophilic archaeal sequences are frequently identified in high salt food products, it is reasonable to assume a portion of halophilic archaeal load may be directly associated with dietary intake (Kobayashi et al., 2000). However, some species of Halobacteriaceae are able to survive in salt concentrations similar to that of average salinity levels of healthy individuals (~140 mM sodium) (Fukushima et al., 2007). In addition, small pockets of concentrated luminal ions have also been identified within the colon, potentially acting as favorable micro-niches for these organisms (Naftalin and Pedley, 1995; Spring, 1998). Halophilic archaea have also demonstrated the ability to survive under anaerobic conditions, utilizing electron acceptors such as fumarate for the fermentation of compounds such as arginine (Oxley et al., 2010). Oxley et al. (2010) also noted the increase in luminal osmolality and organic solute concentration of IBD patients as a potential factor for the increase in halophilic archaea. Despite these linkages, the ecological and metabolic niche that halophilic archaea occupy within the GIT is currently inferential, however, with the isolation of human Haloferax spp. (Khelaifia and Raoult, 2016; Khelaifia et al., 2017), there is now an opportunity to better define the nutritional ecology of these organisms within the human gut. In fact, a recent study on the bacterial and archaeal composition of colorectal cancer patients showed an increased presence of the halophilic Natrinema sp. J7-2 and concurrent reduction in methanogens compared to control subjects (Coker et al., 2020). Additionally, the characterization of the archaeal community of South Korean individuals showed 42.47% archaeal positivity, with 95.54% of archaeal-positive fecal samples containing haloarchaea-associated sequences (Kim J. Y. et al., 2020). Although the average relative abundance of haloarchaea species was 9.63%, some individuals within the cohort displayed a haloarchaea-dominant archaeal community with up to 99.33% relative abundance (Kim J. Y. et al., 2020).
Although there are currently no conclusive findings on the role of methanogenic archaea in human disease, there have been numerous associations made to intestinal-associated pathologies. Breath methane has historically been used to test for the presence of methanogens prior to the development of next-generation sequencing techniques. As summarized by de Lacy Costello et al. (2013), this technique involves the ingestion of a sugar, typically lactose, glucose or fructose, and analysis of alveolar methane over the subsequent 1–2 h period. An increase in the breath methane of constipation-predominant IBS (IBS-C) patients has been associated with increased severity and increased intestinal transit time (Pimentel et al., 2003; Chatterjee et al., 2007). Additionally, an increase in Methanobrevibacter, specifically M. smithii, has been associated with IBS-C by 16S rRNA sequencing (Pozuelo et al., 2015; Ghoshal et al., 2016). Conversely, individuals with diarrhoeal-predominant IBS show a reduction in both methane production and Methanobacteriales abundance (Pimentel et al., 2003; Tap et al., 2017). IBS broadly appears to have no significant association with breath methane or methanogen abundance, though failure to recognize and separate IBS-C/D patients may provide an explanation for these findings (Bratten et al., 2008; Scanlan et al., 2008).
Small intestine bacterial overgrowth (SIBO) is a symptom associated with IBD/IBS patients, in which there is a significant increase in small intestinal bacteria (Colombel et al., 2018). SIBO is relatively common in patients with UC, with ~30% presenting with the condition, compared to a lower prevalence observed in patients with CD (Sandborn, 2009; Lee et al., 2015). A recent study by Suri et al. (2018) showed delayed motility in SIBO to correlate with an increase in breath-methane levels. In a separate study on patients with IBD and SIBO, individuals categorized under IBS-C were more likely to be methane producers compared to IBS-D (58% compared to 28%) (Majewski and McCallum, 2007). Comparatively, individuals with IBS-D were more likely to be hydrogen producers. Given the implication of methanogens and methane in SIBO, recent recommendations by the American College of Gastroenterology include the terminology of intestinal methanogen overgrowth (IMO) to better represent the overgrowth of methanogens in the small intestine and colon (Pimentel et al., 2020). In fact, methane itself has been linked to a reduction in intestinal transit frequency. Jahng et al. (2012) used sections of guinea pig ileum submerged in a peristaltic bath to show an infusion of methane caused decreased peristaltic velocity and increased contraction amplitude, compared to increased peristalsis with hydrogen gas. Additionally, hydrogen was also shown to decrease transit time by 47% in the proximal colon (Jahng et al., 2012). This suggests a possible positive feedback loop between methanogen growth, methane production and increased retention times, caused by a neuromuscular transmitter-like effect of methane (Furnari et al., 2012; Triantafyllou et al., 2014).
Contradictory associations are observed in obese individuals, with an overall increase in the methanogen population but a shift away from M. smithii toward unclassified Methanobrevibacter, though other studies show no significant association (Zhang et al., 2009; Schwiertz et al., 2010; Million et al., 2012, 2013; Fernandes et al., 2013; Mathur et al., 2013; Maya-Lucas et al., 2018). Individuals with severe malnutrition show reduced M. smithii abundance, which may be explained by a lack of intestinal nutrients and thus bacterial fermentation (Million et al., 2016). Indeed, this was recently affirmed in patients with severe acute malnutrition, which showed M. smithii in only 4.2% of cases compared to 40.9% in control subjects (Camara et al., 2021). In contrast, individuals with anorexia show a significantly increase in M. smithii in multiple studies, as do individuals with metabolic syndrome, suggesting altered microbial communities could affect methanogen populations (Armougom et al., 2009; Mack et al., 2016; Borgo et al., 2017; Lim et al., 2017; Prochazkova et al., 2021).
While methanogens are historically characterized as commensal members of the gut microbiome, recent studies have provided evidence implicating methanogenic archaea in polymicrobial infections. For instance, 16S rRNA gene profiling studies have identified Methanobrevibacter, Methanobacterium, Methanosarcina, Methanosphaera, and Thermoplasmatales present in subgingival plaque (Belay et al., 1988; Kulik et al., 2001; Robichaux et al., 2003; Li et al., 2009; Horz et al., 2012). However, M. oralis is the only species to be significantly associated with periodontal disease, as summarised by Nguyen-Hieu et al. (2013). One of the most common treatment options for periodontitis is metronidazole and is one of the few widely used antibiotics with efficacy against methanogens such as M. oralis (Dridi et al., 2011a). Thus, the metronidazole-associated suppression of M. oralis may play a significant role in effective treatment (Nguyen-Hieu et al., 2013). Conversely, a separate study showed M. oralis to have no significant increase in prevalence for peri-implantitis, suggesting a potential for disease-specific associations in the oral microbiome (Belkacemi et al., 2018). Specific amplification of archaeal 16S rRNA and mcrA showed M. smithii sequences in chronic paravertebral abscess of a 41-year-old man (Nkamga et al., 2016). The group was also able to isolate M. oralis from a nasal sample of a patient suffering from chronic sinusitis. There is also growing evidence that methanogens may contribute to disease progression of brain abscesses. One recent study by Drancourt et al. (2017) showed a higher prevalence of archaeal species by PCR in brain abscess specimens compared with healthy controls. Additionally, metagenomics analysis identified M. oralis within multiple abscess samples, as well as several bacterial species, including Staphylococcus intermedius. Mice infected cerebrally with M. oralis, S. intermedius or both showed significantly increased mortality in all test cases compared to controls. Additionally, co-infection with M. oralis and S. intermedius showed an increased mortality rate compared to separate infections, suggesting a syntrophic relationship between the microbes. M. oralis was further observed in a community-acquired brain abscess of a 30-year-old woman along with Aggregatibacter actinomycetemcomitans, again suggesting a potential role for M. oralis in infections associated with anaerobic bacteria (Nkamga et al., 2018). These interesting finding could suggest that Methanobrevibacter species of the oral or gut microbiome are able migrate from their respective environments and, with bacterial partners, could contribute to the severity of infection. Screening for methanogenic archaea in other “unexpected” regions of the body could further elucidate their role in various disease states, such as tumor samples.
Using a murine model of archaeal airway exposure, Blais Lecours et al. (2011) showed that nasal administration of both M. smithii and M. stadtmanae biomass induced alveolar accumulation of granulocytes and macrophages, as well as thickening of the alveolar septa. While the effects from M. smithii challenge were relatively mild, there was a much stronger response toward M. stadtmanae, and in a separate study M. stadtmanae-induced pneumonitis in mice also caused a significant induction of B-cell-rich tertiary lymphoid tissues (Huppe et al., 2018). When the recruitment of B-cells was prevented by an agonist of sphingosine-1-phosphate receptor 1, a key regulator of lymphoid cells, M. stadtmanae-specific lung antibody titres were reduced along with airway leakage and neutrophilic inflammation (Huppe et al., 2018). In a murine model of airway inflammation, crude Methanosphaera and Methanobrevibacter extracts induced a TH17-dependent type IV hypersensitivity response (Bernatchez et al., 2017). Additionally, the Methanosphaera-specific immune response also presented with high titres of antigen-specific IgG1 and IgG2a, again showing the increased immunogenicity of Methanosphaera (Bernatchez et al., 2017). Collectively, these results suggest that human archaea, specifically M. stadtmanae, stimulate both arms of the immune system and induce a significant proinflammatory immune response. However, further work is needed to understand the archaea-induced inflammatory response in the progression and maintenance of gastrointestinal diseases such as IBD.
Bang et al. (2014) showed that M. smithii and M. stadtmanae were not recognized by Caco-2/BBe human epithelial cells, in terms of cytokine and antimicrobial peptide production, as was previously shown for intestinal commensal bacteria (Sansonetti, 2004). However, both Methanosphaera and Methanobrevibacter displayed a decreased growth rate and yield when exposed to a derivative of human cathelicidin, as well as a synthetic anti-lipoprotein peptide (Lpep) and porcine lysin NK-2, when supplemented in axenic culture (Bang et al., 2012). Similarly, M. luminyensis showed a high sensitivity to human cathelicidin, though it was significantly more resistant to Lpep and porcine lysin NK-2 (Bang et al., 2017). This mechanism was further explored by identifying specific Toll-like receptors (TLRs) for the recognition of methanogen-specific microbe-associated molecular patterns (MAMPs). Using human embryonic kidney (HEK) cells transfected with specific intracellular TLRs (3, 7, 8, and 9), Bang et al. (2014) showed no activation by RNA or DNA from heat-inactivated archaeal cell preparations. Similarly, no recognition was observed in TLR5 cells, which play an essential role in the recognition of flagella (Bang et al., 2014). This was not unexpected as there is no genetic evidence of flagellin-like genes within M. stadtmanae and only two predicted flagellin-like genes within the genome of M. smithii PS (Fricke et al., 2006; Samuel et al., 2007). TLR2, NOD1 and NOD2 were also tested for their role in the recognition of bacterial cell membrane components, such as lipid (TLR2) and murein (NOD1/NOD2) (Girardin et al., 2003a,b; Kataoka et al., 2006). Neither TLR2 nor NOD1/NOD2 cells displayed recognition of M. smithii and M. stadtmanae, suggesting the archaeal cell wall components are immunologically distinct from those of pathogenic bacterial species (Bang et al., 2014). However, contrary to these results, the stimulation of TLR knockout human monocyte BLaER1 cell lines showed not only M. stadtmanae itself but also preparations of M. stadtmanae RNA to elicit a TLR7- and TL8-specific immune recognition, with the latter showing a greater response (Vierbuchen et al., 2017). Additionally, the TLR8-specific response was able to induce the activation of the NLRP3 inflammasome (Vierbuchen et al., 2017). Thus, further work is warranted to better characterise the potential MAMPs of archaeal species and their associated TLR activation pathway. Despite this variation in response, both strains induced maturation of monocyte-derived dendritic cells (moDCs) through to up-regulation of CD197 and CD86 (Bang et al., 2014). Additionally, confocal and transmission electron microscopy (TEM) was used to show phagocytosis of the methanogens was required for activation of the moDCs (Bang et al., 2014). In a subsequent study, M. luminyensis showed a weak response in both moDCs and peripheral blood mononuclear cells (PBMCs), suggesting a lower immunogenic potential toward human immune cells (Bang et al., 2017).
With breath methane testing, individuals with IBD show reduced methane expulsion (McKay et al., 1985; Peled et al., 1987; Gu et al., 2020). Scanlan et al. (2008) further validated these results by PCR of the methanogenesis marker gene mcrA. This successfully showed a reduction in the abundance of methanogens in individuals with IBD, with UC patients showing a 24% reduction and patients with CD showing a 30% reduction (Scanlan et al., 2008). Subsequently, patients with CD showed a specific reduction of Methanobrevibacter, and a shift toward Methanosphaera, which may be responsible for the reduction in breath methane (Blais Lecours et al., 2014; Ghavami et al., 2018). Similarly, this was recently replicated in a population of Kazan IBD patients, which showed a significant reduction of Euryarchaeota, attributed to Methanobrevibacter, in patients with CD compared to those with UC (Lo Sasso et al., 2020). Methane metabolism has also been shown to be reduced in patients with IBD compared to control subjects (Nishino et al., 2018). Blais Lecours et al. (2014) specifically showed an increased prevalence of M. stadtmanae in patients with IBD compared to control subjects. Additionally, it was also shown that IBD patients produced a significant M. stadtmanae-specific IgG immune response compared to non-IBD healthy individuals and PBMCs produced a higher proinflammatory cytokine (TNFα) response when exposed to M. stadtmanae compared to M. smithii. Stimulation of moDCs also showed M. stadtmanae to elicit a significant proinflammatory cytokine response compared to M. smithii (Bang et al., 2014).
However, a study on patients with UC and CD from an Indian population showed a converse shift in methanogens, with an increase observed in Methanobrevibacter for both patient groups compared to controls (Verma et al., 2010). Individuals with short bowel syndrome (SBS) due to surgical intervention show a decrease in the abundance of M. smithii, possibly due to the physical restriction of extended retention times (Boccia et al., 2017). Conversely, individuals with diverticulosis showed an increase in Methanobrevibacter compared to standard IBD patients, potentially due to the diverticula creating micro-niches for the methanogens within the colon (Weaver et al., 1986).
Multiple studies show an increase of methanogens and methane production associated with colorectal cancer (CRC) and the stage of disease (Haines et al., 1977; Pique et al., 1984; Segal et al., 1988; Mira-Pascual et al., 2015). However, many studies also show no significant associations between the two, suggesting that the association between methanogens and CRC may involve complex factors that are currently not well understood (Karlin et al., 1982; Hoff et al., 1986; Kashtan et al., 1989; O'Keefe et al., 2007; Scanlan et al., 2008). Although, a recent study on CRC patients showed an enrichment in haloarchaea and a concurrent reduction in methanogens compared to control subjects (Coker et al., 2020).
Euryarchaeota have also been implicated in autoimmune diseases with potential links to the microbiome, such as an increase associated with shorter relapse time for pediatric multiple sclerosis patients (Tremlett et al., 2016a; Castillo-Alvarez et al., 2018) or a correlation to increased disease activity score in patients with rheumatoid arthritis (Picchianti-Diamanti et al., 2018). Recently, Wilmes et al. (2022) and colleagues showed that 2-hydroxypyridine, a metabolite associated with M. smithii and methane metabolism, was elevated in patients with Parkinson’s Disease. Further, this compound is a key molecule in disease pathogenesis and may indicate a causal role of M. smithii in Parkinson’s Disease. However, recent additional analyses by indicated that M. smithii was not the direct source of 2-hydroxypyridine and, as such, further validation is required to determine if M. smithii can produce this compound in vivo (Wilmes et al., 2023). Adults with asthma were also found to have a reduction in M. smithii compared to control subjects (Wang et al., 2018). Individuals with metabolic syndrome (MetS) were observed to have an increase in Methanobrevibacter compared to control subjects (Lim et al., 2017).
Despite the correlation of methanogens to various diseases, little information is available on whether methanogens are playing an active role or are simply responding to ecological changes. Some methanogens, namely the Methanomassiliicoccales, have been suggested as potential “archaebiotics” for their ability to metabolize TMA (Brugère et al., 2014). TMA is the precursor to uremic toxins TMA oxide (TMAO), which has been associated with cardiovascular diseases, and trimethylaminuria, which causes individuals to emit a pungent fishy odor (Ayesh et al., 1993; Wang et al., 2011; Koeth et al., 2013). Despite demonstrating that cultured isolates of Methanomassiliicoccales can metabolise TMA, there are no significant associations observed between these species and atherosclerotic cardiovascular disease (Jie et al., 2017). Similarly, no associations have been observed in patients with chronic kidney disease (CKD), which is also associated with increased levels of uraemic toxins, such as TMAO (Lau et al., 2018). As such, the diseased environment may be unfavorable for these methanogens and further work is required to access the potential of TMA metabolizing methanogens as “archaebiotics”.
Early research of host-associated methanogens was limited by numerous experimental constraints. One such example is the use of bacterial and archaea ‘universal’ primers, which are biased against the majority of archaeal lineages (Raymann et al., 2017). Pausan et al. (2019) demonstrated that a large number of archaeal 16S rRNA-targeting primer pairs demonstrate good coverage in silico but fail to detect Thaumarcheota, Woesearchaeota, and only captured a limited diversity of the Euryarchaeota. Additionally, host-associated archaea typically comprise a substantially smaller portion of DNA and contain fewer 16S rRNA gene copy numbers compared to the bacteria (Mahnert et al., 2018). Given their small relative abundance, it is also important to employ effective methods of cell lysis and DNA extraction as typical lysozyme-based lysis methods are ineffective at degrading the cell membranes of archaea (Bang and Schmitz, 2015). Typically, the lack of cultured isolates available for use as reference strains has also been a limitation, however, with the expansion large-scale metagenomic studies, archaeal metagenome-assembled genomes have substantially expanded the number of available representatives from the human gut (Chibani et al., 2022). Nevertheless, representative genomes of low abundance host-associated archaea and those of non-gut origin remain limited. To effectively characterize the host-associated archaeome, archaea-specific DNA extraction methods should be employed, as well as 16S rRNA primer pairs with wider archaeal specificity or metagenomic sequencing of a sufficient depth to detect low abundance archaeal populations. Further, targeted cultivation of novel archaeal lineages would improve reference databases used for culture-independent analyses.
Thomas et al. (2022) recently performed a comprehensive study of the abundance and diversity of archaea in the gastrointestinal tracts of 250 animal species (including mammals, birds and fish). They found that the absolute abundance and diversity of archaea in the gut of mammalian species is affected by host phylogeny, diet and intestinal tract physiology, whereas geographical location and host body mass had little effect. Using archaeal specific primers, they detected archaea at the Order- (10) and Family- (19) levels of taxonomy, from 175/250 (70%) of the candidate animal hosts sampled. In line with previous studies, they found the most abundant genera of methanogens to be Methanobrevibacter. Amplicons affiliated with Methanosphaera, along with Methanomethylophilaceae, Methanocorpusculum, and Methanomicrococcus species were commonly recovered from many samples. These dominant gut methanogens are rarely found in the environment but are closely related to some environmental lineages. This observation suggests that the species of methanogens that inhabit the gut have adapted and specialized to this environment (Thomas et al., 2022). Thomas et al. (2022) also discovered that their amplicons could be assigned to host-specific “clades,” especially for the genus Methanobrevibacter, suggesting host adaptation. This study also demonstrated that the abundance of methyl-reducing methanogens, such as Methanosphaera, was less affected by dietary fiber content than the hydrogenotrophic methanogens, probably because methanol is largely produced in the gut by the breakdown of pectin. Methyl-reducing methanogens made up around 40% of the total methanogen reads from the animal samples, which differs from environmental samples where this type of methanogenesis is much less common. In that context, the gut environment is likely favorable for this type of methanogenesis, due to the relatively large amounts of choline and pectin present in food and forages, which could have led to the transition from other types of methanogenesis to the methyl-reducing pathway in some gut associated groups, such as Methanosphaera spp. and the Methanomassiliicoccales. In summary, while Thomas et al. (2022) have proposed there may be several events of adaptation by methanogens to the gut environment, it is difficult to verify these postulates without some whole genome-based and/or biological research with cultured representatives of the major methanogen lineages from different animals hosts.
A study by De La Cuesta-Zuluaga et al. (2021) investigated the adaptations of Methanomassiliicoccales species, which are hydrogen dependent methylotrophs, to the mammalian gut and compared them to other Methanomassiliicoccales that inhabit non-host environments. This study confirmed the existence of two Methanomassiliicoccales clades, one enriched in the gut and one enriched in the environment (with some exceptions). They performed genomic comparisons between these two clades and found that genetic adaptations to the human gut included genome reduction and changes in abundance of genes involved in the shikimate pathway and bile resistance, which is consistent with gut adaptations shown by other methanogens. Gene clusters associated with eukaryote-like proteins and adhesion-like proteins (both are groups of membrane proteins involved in adhesion) were compared, and it was discovered that they were more likely to be found in the host-associated clade and members of the environmental clade enriched in host microbiomes. Adhesion factors are involved in syntrophic relationships with bacterial and eukaryotic organisms, and these results highlight that interaction with the host and other organisms in the microbiome may play an important role in gut adaptation (De La Cuesta-Zuluaga et al., 2021). The environmental clade members were found to have larger genomes and higher gene counts, and the results indicate that the genetic adaptations of Methanomassiliicoccales species differed based on their clade and not their habitat preference. This supports the hypothesis that adaptation to the gut environment occurred separately in each clade, as the host-associated species from each clade have developed different genomic adaptations to this environment.
Despite previous research into the methanogens that reside in the digestive tracts of animals, many host-associated methanogen species are still not well characterized and have not been cultured in isolation. The majority of studies involving gut-associated methanogens have focused on their abundance and ecology, rather than their functional and metabolic diversity. There has been some research into the adaptation of methanogens to the gut environment in general, but very few studies have looked into adaptations by methanogens to specific host animals and the functional diversity that accompanies these adaptations. Further research in this area could lead to a better understanding of their interactions with the host and the microbiome, as well as their roles in the gut environment.
Australia’s unique wildlife are becoming increasingly vulnerable due to anthropogenic factors, such as habitat destruction, agriculture and climate change, and a better understanding of their gut microbiota and nutritional ecology may help with conservation efforts in the future. The microbiome of native herbivores could provide biomarkers for vulnerable animals and give insights into their reactions to anthropogenic pressures. One study by Gibson et al. (2019) showed that, compared to their wild counterparts, captive rhinos had higher abundances of bacterial and archaeal taxa associated with agricultural animals, such as Methanobrevibacter, which dominate ruminant archaeal communities. Another study by Yan et al. (2022) demonstrated that Methanobrevibacter species were more prevalent in captive versus wild Bactrian Camels, and methanogens on the whole were more abundant in the captive animals. They hypothesize that the wild camels live in harsher environments and, as such, a decrease in methanogenesis in the gut allows for more efficient energy conversion of their food – a critical factor especially when food is scarce. These studies may also indicate that some of the gut microbial taxa found in wild animals were being overtaken by species that are more commonly found in domesticated animals. Similar shifts in gut methanogenic archaea in native Australian mammals may be used as markers to indicate potentially detrimental changes in nutritional and environmental conditions. For this to be effective, an understanding of the methanogens associated with these host animals in the wild and in captivity is required.
The unique diets of some marsupials, their geographical isolation, and the discovery of a non-canonical methanogenesis pathway in Methanosphaera sp. WGK6 indicates that it is possible that other, yet uncharacterised, marsupial-associated methanogens could use modified pathways. The discovery of such methanogens would help broaden our current understanding of methanogenesis, as well as possibly aiding our attempts to conserve Australian and global wildlife in the future.
It is evident that methanogenic archaea are prevalent members of host-associated microbiomes, with representatives found in diverse animal hosts from terrestrial and aquatic environments. Although high-throughput metagenomic sequencing has expanded our understanding of the prevalence and abundance of methanogens, there is a lack of cultured representative from the different host-associated species that would allow for functional analysis and validation of cultivation-independent analyses. To understand the role that methanogens play in the host-associated microbiomes of different animal species, it is imperative that diverse methanogen species are cultivated from a variety of animal hosts. More specifically, cultivation attempts should focus on the isolation of novel methylotrophic lineages, such as the Methanomethylophilaceae, as well as hydrogenotrophic methanogens other than Methanobrevibacter. In addition to characterizing these isolates individually, it is also important to perform in vitro and ex vivo transcriptomic and proteomic analyses on mixed communities of these methanogens and bacteria from the host animals, such as the methods described by Andersen et al. (2021). These “multi-omics” analyses will provide key information on the wider role of methanogens in the host-associated microbiome and identify strategies for mitigating methane emissions from Methanobrevibacter-dominated communities. Further, with the expansions of culture-independent data from the human microbiome, there remains an opportunity to use this data to complement analyses from other mammals and non-human isolates to improve our understanding of host-associated methanogens.
JV: Conceptualization, Writing – original draft, Writing – review & editing. HM: Writing – original draft, Writing – review & editing. MM: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The study was supported by an Australian Research Council Discovery Project (DP210103991) awarded to MM. JV was supported by the Research Training Program (RTP) scholarship provided by the Australian Federal Government and the Meat & Livestock Australia post-graduate technical assistance grant (Project code: B.STU.1909). HM is supported by a HDR scholarship from the University of Queensland. MM also acknowledges support of NHMRC CRE Digestive Health via GNT1170893.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alayón-Gamboa, J. A., Albores-Moreno, S., Jiménez-Ferrer, G., Alarcón-Zúñiga, B., Miranda-Romero, L. A., Pérez-Luna, E. J., et al. (2023). Tropical tree foliage supplementation in ruminants improves rumen fermentation and the bacterial profile and decreases methane production. Anim. Biotechnol., 1–13. doi: 10.1080/10495398.2023.2165935
Ali, A. I. M., Wassie, S. E., Korir, D., Merbold, L., Goopy, J. P., Butterbach-Bahl, K., et al. (2019). Supplementing tropical cattle for improved nutrient utilization and reduced enteric methane emissions. Animals 9. doi: 10.3390/ani9050210
Almeida, A. K., Cowley, F., McMeniman, J. P., Karagiannis, A., Walker, N., Tamassia, L. F. M., et al. (2023). Effect of 3-nitrooxypropanol on enteric methane emissions of feedlot cattle fed with a tempered barley-based diet with canola oil. J. Anim. Sci. 101. doi: 10.1093/jas/skad237
Andersen, T. O., Kunath, B. J., Hagen, L. H., Arntzen, M. Ø., and Pope, P. B. (2021). Rumen metaproteomics: closer to linking rumen microbial function to animal productivity traits. Methods 186, 42–51. doi: 10.1016/j.ymeth.2020.07.011
Araújo, T. L. R., Rabelo, C. H. S., Cardoso, A. S., Carvalho, V. V., Acedo, T. S., Tamassia, L. F. M., et al. (2023). Feeding 3-nitrooxypropanol reduces methane emissions by feedlot cattle on tropical conditions. J. Anim. Sci. 101. doi: 10.1093/jas/skad225
Armougom, F., Henry, M., Vialettes, B., Raccah, D., and Raoult, D. (2009). Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and methanogens in anorexic patients. PLoS One 4:e7125. doi: 10.1371/journal.pone.0007125
Aschenbach, K., Conrad, R., Reháková, K., Doležal, J., Janatková, K., and Angel, R. (2013). Methanogens at the top of the world: occurrence and potential activity of methanogens in newly deglaciated soils in high-altitude cold deserts in the Western Himalayas. Front. Microbiol. 4:e00359. doi: 10.3389/fmicb.2013.00359
Attaluri, A., Jackson, M., Valestin, J., and Rao, S. S. (2010). Methanogenic flora is associated with altered colonic transit but not stool characteristics in constipation without IBS. Am. J. Gastroenterol. 105, 1407–1411. doi: 10.1038/ajg.2009.655
Ayesh, R., Mitchell, S. C., Zhang, A., and Smith, R. L. (1993). The fish odour syndrome: biochemical, familial, and clinical aspects. BMJ 307, 655–657. doi: 10.1136/bmj.307.6905.655
Baca-González, V., Asensio-Calavia, P., González-Acosta, S., de la Lastra, J. M. P., and de la Nuez, A. M. (2020). Are vaccines the solution for methane emissions from ruminants? A systematic review. Vaccines (Basel) 8. doi: 10.3390/vaccines8030460
Bampidis, V., Azimonti, G., Bastos, M. L., Christensen, H., Dusemund, B., Fašmon Durjava, M., et al. (2021). Safety and efficacy of a feed additive consisting of 3-nitrooxypropanol (Bovaer(®) 10) for ruminants for milk production and reproduction (DSM nutritional products ltd). EFSA J. 19:e06905. doi: 10.2903/j.efsa.2021.6905
Bang, C., Schilhabel, A., Weidenbach, K., Kopp, A., Goldmann, T., Gutsmann, T., et al. (2012). Effects of antimicrobial peptides on methanogenic archaea. Antimicrob. Agents Chemother. 56, 4123–4130. doi: 10.1128/AAC.00661-12
Bang, C., and Schmitz, R. A. (2015). Archaea associated with human surfaces: not to be underestimated. FEMS Microbiol. Rev. 39, 631–648. doi: 10.1093/femsre/fuv010
Bang, C., Vierbuchen, T., Gutsmann, T., Heine, H., and Schmitz, R. A. (2017). Immunogenic properties of the human gut-associated archaeon Methanomassiliicoccus luminyensis and its susceptibility to antimicrobial peptides. PLoS One 12:e0185919. doi: 10.1371/journal.pone.0185919
Bang, C., Weidenbach, K., Gutsmann, T., Heine, H., and Schmitz, R. A. (2014). The intestinal archaea Methanosphaera stadtmanae and Methanobrevibacter smithii activate human dendritic cells. PLoS One 9:e99411. doi: 10.1371/journal.pone.0099411
Barret, M., Gagnon, N., Kalmokoff, M. L., Topp, E., Verastegui, Y., Brooks, S. P., et al. (2013). Identification of Methanoculleus spp. as active methanogens during anoxic incubations of swine manure storage tank samples. Appl. Environ. Microbiol. 79, 424–433. doi: 10.1128/AEM.02268-12
Bauchop, T., and Mountfort, D. O. (1981). Cellulose fermentation by a rumen anaerobic fungus in both the absence and the presence of rumen methanogens. Appl. Environ. Microbiol. 42, 1103–1110. doi: 10.1128/aem.42.6.1103-1110.1981
Bedarf, J. R., Hildebrand, F., Coelho, L. P., Sunagawa, S., Bahram, M., Goeser, F., et al. (2017). Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naïve Parkinson’s disease patients. Genome Med. 9:39. doi: 10.1186/s13073-017-0428-y
Belay, N., Johnson, R., Rajagopal, B. S., Conway de Macario, E., and Daniels, L. (1988). Methanogenic bacteria from human dental plaque. Appl. Environ. Microbiol. 54, 600–603. doi: 10.1128/aem.54.2.600-603.1988
Belay, N., Mukhopadhyay, B., Conway de Macario, E., Galask, R., and Daniels, L. (1990). Methanogenic bacteria in human vaginal samples. J. Clin. Microbiol. 28:1666. doi: 10.1128/jcm.28.7.1666-1668.1990
Belkacemi, S., Mazel, A., Tardivo, D., Tavitian, P., Stephan, G., Bianca, G., et al. (2018). Peri-implantitis-associated methanogens: a preliminary report. Sci. Rep. 8:9447. doi: 10.1038/s41598-018-27862-8
Bernatchez, E., Gold, M. J., Langlois, A., Blais-Lecours, P., Boucher, M., Duchaine, C., et al. (2017). Methanosphaera stadtmanae induces a type IV hypersensitivity response in a mouse model of airway inflammation. Physiol. Rep. 5. doi: 10.14814/phy2.13163
Blais Lecours, P., Duchaine, C., Taillefer, M., Tremblay, C., Veillette, M., Cormier, Y., et al. (2011). Immunogenic properties of archaeal species found in bioaerosols. PLoS One 6:e23326. doi: 10.1371/journal.pone.0023326
Blais Lecours, P., Marsolais, D., Cormier, Y., Berberi, M., Hache, C., Bourdages, R., et al. (2014). Increased prevalence of Methanosphaera stadtmanae in inflammatory bowel diseases: e87734. PLoS One 9. doi: 10.1371/journal.pone.0087734
Boccia, S., Torre, I., Santarpia, L., Iervolino, C., Del Piano, C., Puggina, A., et al. (2017). Intestinal microbiota in adult patients with short bowel syndrome: preliminary results from a pilot study. Clin. Nutr. 36, 1707–1709. doi: 10.1016/j.clnu.2016.09.028
Borgo, F., Riva, A., Benetti, A., Casiraghi, M. C., Bertelli, S., Garbossa, S., et al. (2017). Microbiota in anorexia nervosa: the triangle between bacterial species, metabolites and psychological tests. PLoS One 12:e0179739. doi: 10.1371/journal.pone.0179739
Borrel, G., Gaci, N., Peyret, P., O'Toole, P. W., Gribaldo, S., and Brugere, J. F. (2014). Unique characteristics of the pyrrolysine system in the 7th order of methanogens: implications for the evolution of a genetic code expansion cassette. Archaea 2014:374146. doi: 10.1155/2014/374146
Borrel, G., Harris, H. M., Parisot, N., Gaci, N., Tottey, W., Mihajlovski, A., et al. (2013). Genome sequence of "Candidatus Methanomassiliicoccus intestinalis" Issoire-Mx1, a third Thermoplasmatales-related methanogenic archaeon from human feces. Genome Announc. 1. doi: 10.1128/genomeA.00453-13
Borrel, G., Harris, H. M., Tottey, W., Mihajlovski, A., Parisot, N., Peyretaillade, E., et al. (2012). Genome sequence of "Candidatus Methanomethylophilus alvus" Mx1201, a methanogenic archaeon from the human gut belonging to a seventh order of methanogens. J. Bacteriol. 194, 6944–6945. doi: 10.1128/JB.01867-12
Bratten, J. R., Spanier, J., and Jones, M. P. (2008). Lactulose breath testing does not discriminate patients with irritable bowel syndrome from healthy controls. Am. J. Gastroenterol. 103, 958–963. doi: 10.1111/j.1572-0241.2008.01785.x
Brugère, J.-F., Borrel, G., Gaci, N., Tottey, W., O’Toole, P. W., and Malpuech-Brugère, C. (2014). Archaebiotics. Gut Microbes 5, 5–10. doi: 10.4161/gmic.26749
Bryant, M. P., Wolin, E. A., Wolin, M. J., and Wolfe, R. S. (1967). Methanobacillus omelianskii, a symbiotic association of two species of bacteria. Arch. Mikrobiol. 59, 20–31. doi: 10.1007/BF00406313
Buan, N. R. (2018). Methanogens: pushing the boundaries of biology. Emerg. Top. Life Sci. 2, 629–646. doi: 10.1042/ETLS20180031
Camara, A., Konate, S., Tidjani Alou, M., Kodio, A., Togo, A. H., Cortaredona, S., et al. (2021). Clinical evidence of the role of Methanobrevibacter smithii in severe acute malnutrition. Sci. Rep. 11:5426. doi: 10.1038/s41598-021-84641-8
Carey, H. V., Walters, W. A., and Knight, R. (2013). Seasonal restructuring of the ground squirrel gut microbiota over the annual hibernation cycle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 304, R33–R42. doi: 10.1152/ajpregu.00387.2012
Castillo-Alvarez, F., Perez-Matute, P., Oteo, J. A., and Marzo-Sola, M. E. (2018). The influence of interferon beta-1b on gut microbiota composition in patients with multiple sclerosis. Neurologia. doi: 10.1016/j.nrl.2018.04.006
Chatterjee, S., Park, S., Low, K., Kong, Y., and Pimentel, M. (2007). The degree of breath methane production in IBS correlates with the severity of constipation. Am. J. Gastroenterol. 102, 837–841. doi: 10.1111/j.1572-0241.2007.01072.x
Chaudhary, P. P., Gaci, N., Borrel, G., O'Toole, P. W., and Brugere, J. F. (2015). Molecular methods for studying methanogens of the human gastrointestinal tract: current status and future directions. Appl. Microbiol. Biotechnol. 99, 5801–5815. doi: 10.1007/s00253-015-6739-2
Chen, H., Mozzicafreddo, M., Pierella, E., Carletti, V., Piersanti, A., Ali, S. M., et al. (2021). Dissection of the gut microbiota in mothers and children with chronic Trichuris trichiura infection in Pemba Island Tanzania. Parasit Vectors 14:62. doi: 10.1186/s13071-021-04580-1
Chibani, C. M., Mahnert, A., Borrel, G., Almeida, A., Werner, A., Brugère, J. F., et al. (2022). A catalogue of 1,167 genomes from the human gut archaeome. Nat. Microbiol. 7, 48–61. doi: 10.1038/s41564-021-01020-9
Clauss, M., Dittmann, M. T., Vendl, C., Hagen, K. B., Frei, S., Ortmann, S., et al. (2020). Review: comparative methane production in mammalian herbivores. Animal 14, s113–s123. doi: 10.1017/S1751731119003161
Coker, O. O., Wu, W. K. K., Wong, S. H., Sung, J. J. Y., and Yu, J. (2020). Altered gut Archaea composition and interaction with Bacteria are associated with colorectal Cancer. Gastroenterology 159, 1459–1470.e1455. doi: 10.1053/j.gastro.2020.06.042
Colombel, J.-F., Shin, A., and Gibson, P. R. (2018). Functional gastrointestinal symptoms in patients with inflammatory bowel disease. Clin. Gastroenterol. Hepatol. doi: 10.1016/j.cgh.2018.08.001
De La Cuesta-Zuluaga, J., Spector, T. D., Youngblut, N. D., and Ley, R. E. (2021). Genomic insights into adaptations of trimethylamine-utilizing methanogens to diverse habitats including the human gut. mSystems 6. doi: 10.1128/mSystems.00939-20
de Lacy Costello, B. P., Ledochowski, M., and Ratcliffe, N. M. (2013). The importance of methane breath testing: a review. J. Breath Res. 7:024001. doi: 10.1088/1752-7155/7/2/024001
Denman, S. E., Tomkins, N. W., and McSweeney, C. S. (2007). Quantitation and diversity analysis of ruminal methanogenic populations in response to the antimethanogenic compound bromochloromethane. FEMS Microbiol. Ecol. 62, 313–322. doi: 10.1111/j.1574-6941.2007.00394.x
Deppenmeier, U., and Muller, V. (2008). Life close to the thermodynamic limit: how methanogenic archaea conserve energy. Results Probl. Cell Differ. 45, 123–152. doi: 10.1007/400_2006_026
Dongowski, G., Lorenz, A., and Anger, H. (2000). Degradation of pectins with different degrees of esterification by Bacteroides thetaiotaomicron isolated from human gut flora. Appl. Environ. Microbiol. 66, 1321–1327. doi: 10.1128/AEM.66.4.1321-1327.2000
Drancourt, M., Nkamga, V. D., Lakhe, N. A., Regis, J. M., Dufour, H., Fournier, P. E., et al. (2017). Evidence of archaeal methanogens in brain abscess. Clin. Infect. Dis. 65, 1–5. doi: 10.1093/cid/cix286
Dridi, B. (2012). Laboratory tools for detection of archaea in humans. Clin. Microbiol. Infect. 18, 825–833. doi: 10.1111/j.1469-0691.2012.03952.x
Dridi, B., Fardeau, M. L., Ollivier, B., Raoult, D., and Drancourt, M. (2011a). The antimicrobial resistance pattern of cultured human methanogens reflects the unique phylogenetic position of archaea. J. Antimicrob. Chemother. 66, 2038–2044. doi: 10.1093/jac/dkr251
Dridi, B., Fardeau, M. L., Ollivier, B., Raoult, D., and Drancourt, M. (2012a). Methanomassiliicoccus luminyensis gen. Nov., sp nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 62, 1902–1907. doi: 10.1099/ijs.0.033712-0
Dridi, B., Henry, M., El Khechine, A., Raoult, D., and Drancourt, M. (2009). High prevalence of Methanobrevibacter smithii and Methanosphaera stadtmanae detected in the human gut using an improved DNA detection protocol. PLoS One 4:e7063. doi: 10.1371/journal.pone.0007063
Dridi, B., Henry, M., Richet, H., Raoult, D., and Drancourt, M. (2012b). Age-related prevalence of Methanomassiliicoccus luminyensis in the human gut microbiome. APMIS 120, 773–777. doi: 10.1111/j.1600-0463.2012.02899.x
Dridi, B., Raoult, D., and Drancourt, M. (2011b). Archaea as emerging organisms in complex human microbiomes. Anaerobe 17, 56–63. doi: 10.1016/j.anaerobe.2011.03.001
Duin, E. C., Wagner, T., Shima, S., Prakash, D., Cronin, B., Yáñez-Ruiz, D. R., et al. (2016). Mode of action uncovered for the specific reduction of methane emissions from ruminants by the small molecule 3-nitrooxypropanol. Proc. Natl. Acad. Sci. U. S. A. 113, 6172–6177. doi: 10.1073/pnas.1600298113
Eckburg, P. B., Bik, E. M., Bernstein, C. N., Purdom, E., Dethlefsen, L., Sargent, M., et al. (2005). Diversity of the human intestinal microbial flora. Science 308, 1635–1638. doi: 10.1126/science.1110591
Evans, P. N. (2011). Culture- and molecular-based studies of the archaeal communities present in the forestomach microbiomes of the Tammar wallaby (Macropus eugenii) and Western Grey kangaroo (Macropus fuliginosus) St. Lucia: The University of Queensland].
Evans, P. N., Hinds, L. A., Sly, L. I., McSweeney, C. S., Morrison, M., and Wright, A.-D. G. (2009). Community composition and density of methanogens in the foregut of the Tammar wallaby (Macropus eugenii). Appl. Environ. Microbiol. 75, 2598–2602. doi: 10.1128/AEM.02436-08
Fernandes, K. A., Kittelmann, S., Rogers, C. W., Gee, E. K., Bolwell, C. F., Bermingham, E. N., et al. (2014). Faecal microbiota of forage-fed horses in New Zealand and the population dynamics of microbial communities following dietary change. PLoS One 9:e112846. doi: 10.1371/journal.pone.0112846
Fernandes, J., Wang, A., Su, W., Rozenbloom, S. R., Taibi, A., Comelli, E. M., et al. (2013). Age, dietary fiber, breath methane, and fecal short chain fatty acids are interrelated in Archaea-positive humans. J. Nutr. 143, 1269–1275. doi: 10.3945/jn.112.170894
Ferrari, A., Brusa, T., Rutili, A., Canzi, E., and Biavati, B. (1994). Isolation and characterization Ofmethanobrevibacter oralis sp. nov. Curr. Microbiol. 29, 7–12. doi: 10.1007/BF01570184
Ferry, J. G. (1997). Enzymology of the fermentation of acetate to methane by Methanosarcina thermophila. Biofactors 6, 25–35. doi: 10.1002/biof.5520060104
Fiedorek, S. C., Pumphrey, C. L., and Casteel, H. B. (1990). Breath methane production in children with constipation and encopresis. J. Pediatr. Gastroenterol. Nutr. 10, 473–477. doi: 10.1097/00005176-199005000-00010
Fricke, W. F., Seedorf, H., Henne, A., Kruer, M., Liesegang, H., Hedderich, R., et al. (2006). The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J. Bacteriol. 188, 642–658. doi: 10.1128/JB.188.2.642-658.2006
Fukushima, T., Usami, R., and Kamekura, M. (2007). A traditional Japanese-style salt field is a niche for haloarchaeal strains that can survive in 0.5% salt solution. Saline Syst. 3:2. doi: 10.1186/1746-1448-3-2
Furnari, M., Savarino, E., Bruzzone, L., Moscatelli, A., Gemignani, L., Giannini, E. G., et al. (2012). Reassessment of the role of methane production between irritable bowel syndrome and functional constipation. J. Gastrointestin. Liver Dis. 21, 157–163.
Gaci, N., Borrel, G., Tottey, W., O'Toole, P. W., and Brugere, J. F. (2014). Archaea and the human gut: new beginning of an old story. World J. Gastroenterol. 20, 16062–16078. doi: 10.3748/wjg.v20.i43.16062
Garcia, J. L., Patel, B. K., and Ollivier, B. (2000). Taxonomic, phylogenetic, and ecological diversity of methanogenic Archaea. Anaerobe 6, 205–226. doi: 10.1006/anae.2000.0345
Ghavami, S. B., Rostami, E., Sephay, A. A., Shahrokh, S., Balaii, H., Aghdaei, H. A., et al. (2018). Alterations of the human gut Methanobrevibacter smithii as a biomarker for inflammatory bowel diseases. Microb. Pathog. 117, 285–289. doi: 10.1016/j.micpath.2018.01.029
Ghoshal, U., Shukla, R., Srivastava, D., and Ghoshal, U. C. (2016). Irritable bowel syndrome, particularly the constipation-predominant form, involves an increase in Methanobrevibacter smithii, which is associated with higher methane production. Gut Liver 10, 932–938. doi: 10.5009/gnl15588
Ghoshal, U. C., Srivastava, D., Verma, A., and Misra, A. (2011). Slow transit constipation associated with excess methane production and its improvement following rifaximin therapy: a case report. J. Neurogastroenterol. Motil. 17, 185–188. doi: 10.5056/jnm.2011.17.2.185
Gibson, K. M., Nguyen, B. N., Neumann, L. M., Miller, M., Buss, P., Daniels, S., et al. (2019). Gut microbiome differences between wild and captive black rhinoceros – implications for rhino health. Sci. Rep. 9:7570. doi: 10.1038/s41598-019-43875-3
Gilmore, S. P., Henske, J. K., Sexton, J. A., Solomon, K. V., Seppala, S., Yoo, J. I., et al. (2017). Genomic analysis of methanogenic archaea reveals a shift towards energy conservation. BMC Genomics 18:639. doi: 10.1186/s12864-017-4036-4
Girardin, S. E., Boneca, I. G., Carneiro, L. A., Antignac, A., Jehanno, M., Viala, J., et al. (2003a). Nod1 detects a unique muropeptide from gram-negative bacterial peptidoglycan. Science 300, 1584–1587. doi: 10.1126/science.1084677
Girardin, S. E., Boneca, I. G., Viala, J., Chamaillard, M., Labigne, A., Thomas, G., et al. (2003b). Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem. 278, 8869–8872. doi: 10.1074/jbc.C200651200
Glasson, C. R. K., Kinley, R. D., de Nys, R., King, N., Adams, S. L., Packer, M. A., et al. (2022). Benefits and risks of including the bromoform containing seaweed Asparagopsis in feed for the reduction of methane production from ruminants. Algal Res. 64. doi: 10.1016/j.algal.2022.102673
Godwin, S., Kang, A., Gulino, L.-M., Manefield, M., Gutierrez-Zamora, M.-L., Kienzle, M., et al. (2014). Investigation of the microbial metabolism of carbon dioxide and hydrogen in the kangaroo foregut by stable isotope probing. ISME J. 8, 1855–1865. doi: 10.1038/ismej.2014.25
Goopy, J. P. (2019). Creating a low enteric methane emission ruminant: what is the evidence of success to the present and prospects for developing economies? Anim. Prod. Sci. 59, 1769–1776. doi: 10.1071/AN18457
Grine, G., Drouet, H., Fenollar, F., Bretelle, F., Raoult, D., and Drancourt, M. (2019a). Detection of Methanobrevibacter smithii in vaginal samples collected from women diagnosed with bacterial vaginosis. Eur. J. Clin. Microbiol. Infect. Dis. 38, 1643–1649. doi: 10.1007/s10096-019-03592-1
Grine, G., Lotte, R., Chirio, D., Chevalier, A., Raoult, D., Drancourt, M., et al. (2019b). Co-culture of Methanobrevibacter smithii with enterobacteria during urinary infection. EBioMedicine 43, 333–337. doi: 10.1016/j.ebiom.2019.04.037
Gu, P., Patel, D., Lakhoo, K., Ko, J., Liu, X., Chang, B., et al. (2020). Breath test gas patterns in inflammatory bowel disease with concomitant irritable bowel syndrome-like symptoms: a controlled large-scale database linkage analysis. Dig. Dis. Sci. 65, 2388–2396. doi: 10.1007/s10620-019-05967-y
Haines, A., Metz, G., Dilawari, J., Blendis, L., and Wiggins, H. (1977). Breath-methane in patients with cancer of the large bowel. Lancet 2, 481–483. doi: 10.1016/s0140-6736(77)91605-1
Heidarian, F., Alebouyeh, M., Shahrokh, S., Balaii, H., and Zali, M. R. (2019). Altered fecal bacterial composition correlates with disease activity in inflammatory bowel disease and the extent of IL8 induction. Curr. Res. Trans. Med. 67, 41–50. doi: 10.1016/j.retram.2019.01.002
Hoedt, E. C., Cuív, Ó., Evans, P. N., Smith, W. J. M., McSweeney, C. S., Denman, S. E., et al. (2016). Differences down-under: alcohol-fueled methanogenesis by archaea present in Australian macropodids. ISME J. 10, 2376–2388. doi: 10.1038/ismej.2016.41
Hoedt, E. C., Parks, D. H., Volmer, J. G., Rosewarne, C. P., Denman, S. E., McSweeney, C. S., et al. (2018). Culture- and metagenomics-enabled analyses of the Methanosphaera genus reveals their monophyletic origin and differentiation according to genome size. ISME J. 12, 2942–2953. doi: 10.1038/s41396-018-0225-7
Hoff, G., Bjorneklett, A., Moen, I. E., and Jenssen, E. (1986). Epidemiology of polyps in the rectum and sigmoid colon. Evaluation of breath methane and predisposition for colorectal neoplasia. Scand. J. Gastroenterol. 21, 193–198.
Horz, H. P., Seyfarth, I., and Conrads, G. (2012). McrA and 16S rRNA gene analysis suggests a novel lineage of Archaea phylogenetically affiliated with Thermoplasmatales in human subgingival plaque. Anaerobe 18, 373–377. doi: 10.1016/j.anaerobe.2012.04.006
Hoyles, L., Jiménez-Pranteda, M. L., Chilloux, J., Brial, F., Myridakis, A., Aranias, T., et al. (2018). Metabolic retroconversion of trimethylamine N-oxide and the gut microbiota. Microbiome 6:73. doi: 10.1186/s40168-018-0461-0
Hume, I. D. (1984). Microbial fermentation in herbivorous marsupials. Bioscience 34, 435–440. doi: 10.2307/1309633
Huppe, C. A., Blais Lecours, P., Lechasseur, A., Gendron, D. R., Lemay, A. M., Bissonnette, E. Y., et al. (2018). A sphingosine-1-phosphate receptor 1 agonist inhibits tertiary lymphoid tissue reactivation and hypersensitivity in the lung. Mucosal Immunol. 11, 112–119. doi: 10.1038/mi.2017.37
Huynh, H. T. T., Pignoly, M., Drancourt, M., and Aboudharam, G. (2017). A new methanogen “Methanobrevibacter massiliense” isolated in a case of severe periodontitis. BMC. Res. Notes 10:657. doi: 10.1186/s13104-017-2980-3
Huynh, H. T. T., Pignoly, M., Nkamga, V. D., Drancourt, M., and Aboudharam, G. (2015). The repertoire of Archaea cultivated from severe periodontitis. PLoS One 10:e0121565. doi: 10.1371/journal.pone.0121565
Hwang, L., Low, K., Khoshini, R., Melmed, G., Sahakian, A., Makhani, M., et al. (2010). Evaluating breath methane as a diagnostic test for constipation-predominant IBS. Dig. Dis. Sci. 55, 398–403. doi: 10.1007/s10620-009-0778-4
Iannotti, E. L., Kafkewitz, D., Wolin, M. J., and Bryant, M. P. (1973). Glucose fermentation products in Ruminococcus albus grown in continuous culture with Vibrio succinogenes: changes caused by interspecies transfer of H 2. J. Bacteriol. 114, 1231–1240. doi: 10.1128/jb.114.3.1231-1240.1973
Jahng, J., Jung, I. S., Choi, E. J., Conklin, J. L., and Park, H. (2012). The effects of methane and hydrogen gases produced by enteric bacteria on ileal motility and colonic transit time. Neurogastroenterol. Motil. 24:e192, 185–190. doi: 10.1111/j.1365-2982.2011.01819.x
Jangi, S., Gandhi, R., Cox, L. M., Li, N., von Glehn, F., Yan, R., et al. (2016). Alterations of the human gut microbiome in multiple sclerosis. Nat. Commun. 7:12015. doi: 10.1038/ncomms12015
Janssen, P. H. (2010). Influence of hydrogen on rumen methane formation and fermentation balances through microbial growth kinetics and fermentation thermodynamics. Anim. Feed Sci. Technol. 160, 1–22. doi: 10.1016/j.anifeedsci.2010.07.002
Jensen, N. S., and Canale-Parola, E. (1986). Bacteroides pectinophilus sp. nov. and Bacteroides galacturonicus sp. nov.: two pectinolytic bacteria from the human intestinal tract. Appl. Environ. Microbiol. 52, 880–887. doi: 10.1128/aem.52.4.880-887.1986
Jhangi, S., Gandhi, R., Glanz, B., Cook, S., Nejad, P., Ward, D., et al. (2014). Increased Archaea species and changes with therapy in gut microbiome of multiple sclerosis subjects. Neurology 82:S24.001
Jie, Z., Xia, H., Zhong, S.-L., Feng, Q., Li, S., Liang, S., et al. (2017). The gut microbiome in atherosclerotic cardiovascular disease. Nat. Commun. 8:845. doi: 10.1038/s41467-017-00900-1
Jin, W., Cheng, Y. F., Mao, S. Y., and Zhu, W. Y. (2011). Isolation of natural cultures of anaerobic fungi and indigenously associated methanogens from herbivores and their bioconversion of lignocellulosic materials to methane. Bioresour. Technol. 102, 7925–7931. doi: 10.1016/j.biortech.2011.06.026
Johnson, P. L., Hickey, S., Knowler, K., Wing, J., Bryson, B., Hall, M., et al. (2022). Genetic parameters for residual feed intake, methane emissions, and body composition in New Zealand maternal sheep. Front. Genet. 13:911639. doi: 10.3389/fgene.2022.911639
Kamil, R. Z., Murdiati, A., Juffrie, M., Nakayama, J., and Rahayu, E. S. (2021). Gut microbiota and short-chain fatty acid profile between Normal and moderate malnutrition children in Yogyakarta, Indonesia. Microorganisms 9. doi: 10.3390/microorganisms9010127
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Karekar, S., Stefanini, R., and Ahring, B. (2022). Homo-Acetogens: their metabolism and competitive relationship with Hydrogenotrophic methanogens. Microorganisms 10. doi: 10.3390/microorganisms10020397
Karlin, D. A., Jones, R. D., Stroehlein, J. R., Mastromarino, A. J., and Potter, G. D. (1982). Breath methane excretion in patients with unresected colorectal cancer. J. Natl. Cancer Inst. 69, 573–576.
Kashtan, H., Rabau, M., Peled, Y., Milstein, A., and Wiznitzer, T. (1989). Methane production in patients with colorectal carcinoma. Isr. J. Med. Sci. 25, 614–616.
Kataoka, H., Yasuda, M., Iyori, M., Kiura, K., Narita, M., Nakata, T., et al. (2006). Roles of N-linked glycans in the recognition of microbial lipopeptides and lipoproteins by TLR2. Cell. Microbiol. 8, 1199–1209. doi: 10.1111/j.1462-5822.2006.00702.x
Khelaifia, S., Caputo, A., Djossou, F., and Raoult, D. (2017). Draft genome sequence of a human-associated isolate of Haloferax alexandrinus strain arc-hr, an extremely halophilic archaea. New Microb. New Infect. 15, 44–45. doi: 10.1016/j.nmni.2016.11.012
Khelaifia, S., Garibal, M., Robert, C., Raoult, D., and Drancourt, M. (2014). Draft genome sequence of a human-associated isolate of Methanobrevibacter arboriphilicus, the lowest-G+C-content archaeon. Genome Announc. 2. doi: 10.1128/genomeA.01181-13
Khelaifia, S., and Raoult, D. (2016). Haloferax massiliensis sp. nov., the first human-associated halophilic archaea. New Microb. New Infect. 12, 96–98. doi: 10.1016/j.nmni.2016.05.007
Kim, G., Deepinder, F., Morales, W., Hwang, L., Weitsman, S., Chang, C., et al. (2012). Methanobrevibacter smithii is the predominant methanogen in patients with constipation-predominant IBS and methane on breath. Dig. Dis. Sci. 57, 3213–3218. doi: 10.1007/s10620-012-2197-1
Kim, H., Lee, H. G., Baek, Y.-C., Lee, S., and Seo, J. (2020). The effects of dietary supplementation with 3-nitrooxypropanol on enteric methane emissions, rumen fermentation, and production performance in ruminants: a meta-analysis. J. Anim. Sci. Technol. 62, 31–42. doi: 10.5187/jast.2020.62.1.31
Kim, J. Y., Whon, T. W., Lim, M. Y., Kim, Y. B., Kim, N., Kwon, M.-S., et al. (2020). The human gut archaeome: identification of diverse haloarchaea in Korean subjects. Microbiome 8:114. doi: 10.1186/s40168-020-00894-x
Kobayashi, T., Kimura, B., and Fujii, T. (2000). Haloanaerobium fermentans sp. nov., a strictly anaerobic, fermentative halophile isolated from fermented puffer fish ovaries. Int. J. Syst. Evol. Microbiol. 50, 1621–1627. doi: 10.1099/00207713-50-4-1621
Koeth, R. A., Wang, Z., Levison, B. S., Buffa, J. A., Org, E., Sheehy, B. T., et al. (2013). Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 19, 576–585. doi: 10.1038/nm.3145
Kohl, K. D., Miller, A. W., Marvin, J. E., Mackie, R., and Dearing, M. D. (2014). Herbivorous rodents (Neotoma spp.) harbour abundant and active foregut microbiota. Environ. Microbiol. 16, 2869–2878. doi: 10.1111/1462-2920.12376
Koskinen, K., Pausan, M. R., Perras, A. K., Beck, M., Bang, C., Mora, M., et al. (2017). First insights into the diverse human Archaeome: specific detection of Archaea in the gastrointestinal tract, lung, and nose and on skin. MBio 8. doi: 10.1128/mBio.00824-17
Kulik, E. M., Sandmeier, H., Hinni, K., and Meyer, J. (2001). Identification of archaeal rDNA from subgingival dental plaque by PCR amplification and sequence analysis. FEMS Microbiol. Lett. 196, 129–133. doi: 10.1111/j.1574-6968.2001.tb10553.x
Kurth, J. M., Op den Camp, H. J. M., and Welte, C. U. (2020). Several ways one goal—methanogenesis from unconventional substrates. Appl. Microbiol. Biotechnol. 104, 6839–6854. doi: 10.1007/s00253-020-10724-7
Kusar, D., and Avgustin, G. (2010). Molecular profiling and identification of methanogenic archaeal species from rabbit caecum. FEMS Microbiol. Ecol. 74, 623–630. doi: 10.1111/j.1574-6941.2010.00980.x
Latham, M. J., and Wolin, M. J. (1977). Fermentation of cellulose by Ruminococcus flavefaciens in the presence and absence of Methanobacterium ruminantium. Appl. Environ. Microbiol. 34, 297–301. doi: 10.1128/aem.34.3.297-301.1977
Lau, W. L., Savoj, J., Nakata, M. B., and Vaziri, N. D. (2018). Altered microbiome in chronic kidney disease: systemic effects of gut-derived uremic toxins. Clin. Sci. (Lond.) 132, 509–522. doi: 10.1042/CS20171107
Leahy, S. C., Kelly, W. J., Altermann, E., Ronimus, R. S., Yeoman, C. J., Pacheco, D. M., et al. (2010). The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS One 5:e8926. doi: 10.1371/journal.pone.0008926
Lee, J. M., Lee, K. M., Chung, Y. Y., Lee, Y. W., Kim, D. B., Sung, H. J., et al. (2015). Clinical significance of the glucose breath test in patients with inflammatory bowel disease. J. Gastroenterol. Hepatol. 30, 990–994. doi: 10.1111/jgh.12908
Lee, K. M., Paik, C. N., Chung, W. C., Yang, J. M., and Choi, M. G. (2013). Breath methane positivity is more common and higher in patients with objectively proven delayed transit constipation. Eur. J. Gastroenterol. Hepatol. 25, 726–732. doi: 10.1097/MEG.0b013e32835eb916
Leng, R. A. (2018). Unravelling methanogenesis in ruminants, horses and kangaroos: the links between gut anatomy, microbial biofilms and host immunity. Anim. Prod. Sci. 58, 1175–1191. doi: 10.1071/AN15710
Li, F., Chen, C., Wei, W., Wang, Z., Dai, J., Hao, L., et al. (2018). The metagenome of the female upper reproductive tract. Gigascience 7. doi: 10.1093/gigascience/giy107
Li, Y., Crouzet, L., Kelly, W. J., Reid, P., Leahy, S. C., and Attwood, G. T. (2023). Methanobrevibacter boviskoreani JH1(T) growth on alcohols allows development of a high throughput bioassay to detect methanogen inhibition. Curr. Res. Microb. Sci. 4:100189. doi: 10.1016/j.crmicr.2023.100189
Li, C. L., Liu, D. L., Jiang, Y. T., Zhou, Y. B., Zhang, M. Z., Jiang, W., et al. (2009). Prevalence and molecular diversity of Archaea in subgingival pockets of periodontitis patients. Oral Microbiol. Immunol. 24, 343–346. doi: 10.1111/j.1399-302X.2009.00514.x
Li, Q. S., Wang, R., Ma, Z. Y., Zhang, X. M., Jiao, J. Z., Zhang, Z. G., et al. (2022). Dietary selection of metabolically distinct microorganisms drives hydrogen metabolism in ruminants. ISME J. doi: 10.1038/s41396-022-01294-9
Lim, M. Y., You, H. J., Yoon, H. S., Kwon, B., Lee, J. Y., Lee, S., et al. (2017). The effect of heritability and host genetics on the gut microbiota and metabolic syndrome. Gut 66, 1031–1038. doi: 10.1136/gutjnl-2015-311326
Lin, C., and Miller, T. L. (1998). Phylogenetic analysis of Methanobrevibacter isolated from feces of humans and other animals. Arch. Microbiol. 169, 397–403. doi: 10.1007/s002030050589
Lo Sasso, G., Khachatryan, L., Kondylis, A., Battey, J. N. D., Sierro, N., Danilova, N. A., et al. (2020). Inflammatory bowel disease–associated changes in the gut: focus on Kazan patients. Inflamm. Bowel Dis. 27, 418–433. doi: 10.1093/ibd/izaa188
Luo, Y., Wright, A. D. G., Li, Y., Li, H., Yang, Q., Luo, L., et al. (2013). Diversity of methanogens in the hindgut of captive white rhinoceroses Ceratotherium simum. BMC Microbiol. 13:207. doi: 10.1186/1471-2180-13-207
Lwin, K. O., and Matsui, H. (2014). Comparative analysis of the methanogen diversity in horse and pony by using mcrA gene and archaeal 16s rRNA gene clone libraries. Archaea 2014:483574. doi: 10.1155/2014/483574
Lyu, Z., Shao, N., Akinyemi, T., and Whitman, W. B. (2018). Methanogenesis. Curr. Biol. 28, R727–R732. doi: 10.1016/j.cub.2018.05.021
Mack, I., Cuntz, U., Grämer, C., Niedermaier, S., Pohl, C., Schwiertz, A., et al. (2016). Weight gain in anorexia nervosa does not ameliorate the faecal microbiota, branched chain fatty acid profiles and gastrointestinal complaints. Sci. Rep. 6:26752. doi: 10.1038/srep26752
Mackie, R., Aminov, R., Gaskins, H., and White, B. (2000). Molecular microbial ecology in gut ecosystems. 8th International Symposium on Microbial Ecology, Halifax, Canada (1998, August 9–14)
Maczulak, A. E., Wolin, M. J., and Miller, T. L. (1989). Increase in colonic methanogens and total anaerobes in aging rats. Appl. Environ. Microbiol. 55, 2468–2473. doi: 10.1128/aem.55.10.2468-2473.1989
Madsen, J., and Bertelsen, M. F. (2012). Methane production by red-necked wallabies (Macropus rufogriseus). J. Anim. Sci. 90, 1364–1370. doi: 10.2527/jas.2011-4011
Mahnert, A., Blohs, M., Pausan, M.-R., and Moissl-Eichinger, C. (2018). The human archaeome: methodological pitfalls and knowledge gaps. Emerg. Top. Life Sci. 2, 469–482. doi: 10.1042/ETLS20180037
Majewski, M., and McCallum, R. W. (2007). Results of small intestinal bacterial overgrowth testing in irritable bowel syndrome patients: clinical profiles and effects of antibiotic trial. Adv. Med. Sci. 52, 139–142.
Malik, P. K., Trivedi, S., Kolte, A. P., Mohapatra, A., Bhatta, R., and Rahman, H. (2022). Effect of an anti-methanogenic supplement on enteric methane emission, fermentation, and whole rumen metagenome in sheep. Front. Microbiol. 13:1048288. doi: 10.3389/fmicb.2022.1048288
Malik, P. K., Trivedi, S., Mohapatra, A., Kolte, A. P., Sejian, V., Bhatta, R., et al. (2021). Comparison of enteric methane yield and diversity of ruminal methanogens in cattle and buffaloes fed on the same diet. PLoS One 16. doi: 10.1371/journal.pone.0256048
Mao, S. Y., Yang, C. F., and Zhu, W. Y. (2011). Phylogenetic analysis of methanogens in the pig feces. Curr. Microbiol. 62, 1386–1389. doi: 10.1007/s00284-011-9873-9
Martínez-Álvaro, M., Auffret, M. D., Stewart, R. D., Dewhurst, R. J., Duthie, C.-A., Rooke, J. A., et al. (2020). Identification of complex rumen microbiome interaction within diverse functional niches as mechanisms affecting the variation of methane emissions in bovine. Front. Microbiol. 11:e00659. doi: 10.3389/fmicb.2020.00659
Mathur, R., Amichai, M., Chua, K. S., Mirocha, J., Barlow, G. M., and Pimentel, M. (2013). Methane and hydrogen positivity on breath test is associated with greater body mass index and body fat. J. Clin. Endocrinol. Metab. 98, E698–E702. doi: 10.1210/jc.2012-3144
Maya-Lucas, O., Murugesan, S., Nirmalkar, K., Alcaraz, L. D., Hoyo-Vadillo, C., Pizano-Zarate, M. L., et al. (2018). The gut microbiome of Mexican children affected by obesity. Anaerobe. doi: 10.1016/j.anaerobe.2018.10.009
McGinn, S., Flesch, T., Beauchemin, K., Shreck, A., and Kindermann, M. (2019). Micrometeorological methods for measuring methane emission reduction at beef cattle feedlots: evaluation of 3-Nitrooxypropanol feed additive. J. Environ. Qual. 48, 1454–1461. doi: 10.2134/jeq2018.11.0412
McKay, L. F., Eastwood, M. A., and Brydon, W. G. (1985). Methane excretion in man--a study of breath, flatus, and faeces. Gut 26, 69–74. doi: 10.1136/gut.26.1.69
Mi, J., Peng, H., Wu, Y., Wang, Y., and Liao, X. (2019). Diversity and community of methanogens in the large intestine of finishing pigs. BMC Microbiol. 19:83. doi: 10.1186/s12866-019-1459-x
Miller, T. L., and Wolin, M. J. (1985). Methanosphaera stadtmaniae gen. Nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch. Microbiol. 141, 116–122. doi: 10.1007/BF00423270
Miller, T. L., Wolin, M. J., Conway de Macario, E., and Macario, A. J. (1982). Isolation of Methanobrevibacter smithii from human feces. Appl. Environ. Microbiol. 43, 227–232. doi: 10.1128/aem.43.1.227-232.1982
Million, M., Angelakis, E., Maraninchi, M., Henry, M., Giorgi, R., Valero, R., et al. (2013). Correlation between body mass index and gut concentrations of Lactobacillus reuteri, Bifidobacterium animalis, Methanobrevibacter smithii and Escherichia coli. Int. J. Obes. 37, 1460–1466. doi: 10.1038/ijo.2013.20
Million, M., Maraninchi, M., Henry, M., Armougom, F., Richet, H., Carrieri, P., et al. (2012). Obesity-associated gut microbiota is enriched in Lactobacillus reuteri and depleted in Bifidobacterium animalis and Methanobrevibacter smithii. Int. J. Obes. 36, 817–825. doi: 10.1038/ijo.2011.153
Million, M., Tidjani Alou, M., Khelaifia, S., Bachar, D., Lagier, J.-C., Dione, N., et al. (2016). Increased gut redox and depletion of anaerobic and methanogenic prokaryotes in severe acute malnutrition. Sci. Rep. 6:26051. doi: 10.1038/srep26051
Mira-Pascual, L., Cabrera-Rubio, R., Ocon, S., Costales, P., Parra, A., Suarez, A., et al. (2015). Microbial mucosal colonic shifts associated with the development of colorectal cancer reveal the presence of different bacterial and archaeal biomarkers. J. Gastroenterol. 50, 167–179. doi: 10.1007/s00535-014-0963-x
Muizelaar, W., Groot, M., van Duinkerken, G., Peters, R., and Dijkstra, J. (2021). Safety and transfer study: transfer of bromoform present in asparagopsis taxiformis to milk and urine of lactating dairy cows. Foods 10. doi: 10.3390/foods10030584
Naftalin, R., and Pedley, K. (1995). The sodium concentration of lateral intercellular spaces. J. Membr. Biol. 147, 105–107. doi: 10.1007/BF00235401
Nakamura, N., Lin, H. C., McSweeney, C. S., Mackie, R. I., and Gaskins, H. R. (2010). Mechanisms of microbial hydrogen disposal in the human colon and implications for health and disease. Annu. Rev. Food Sci. Technol. 1, 363–395. doi: 10.1146/annurev.food.102308.124101
Nava, G. M., Carbonero, F., Croix, J. A., Greenberg, E., and Gaskins, H. R. (2012). Abundance and diversity of mucosa-associated hydrogenotrophic microbes in the healthy human colon. ISME J. 6, 57–70. doi: 10.1038/ismej.2011.90
Ng, F., Kittelmann, S., Patchett, M. L., Attwood, G. T., Janssen, P. H., Rakonjac, J., et al. (2016). An adhesin from hydrogen-utilizing rumen methanogen Methanobrevibacter ruminantium M1 binds a broad range of hydrogen-producing microorganisms. Environ. Microbiol. 18, 3010–3021. doi: 10.1111/1462-2920.13155
Nguyen-Hieu, T., Khelaifia, S., Aboudharam, G., and Drancourt, M. (2013). Methanogenic archaea in subgingival sites: a review. APMIS 121, 467–477. doi: 10.1111/apm.12015
Nishino, K., Nishida, A., Inoue, R., Kawada, Y., Ohno, M., Sakai, S., et al. (2018). Analysis of endoscopic brush samples identified mucosa-associated dysbiosis in inflammatory bowel disease. J. Gastroenterol. 53, 95–106. doi: 10.1007/s00535-017-1384-4
Nkamga, V. D., Henrissat, B., and Drancourt, M. (2017). Archaea: essential inhabitants of the human digestive microbiota. Human Microb. J. 3, 1–8. doi: 10.1016/j.humic.2016.11.005
Nkamga, V. D., Lotte, R., Chirio, D., Lonjon, M., Roger, P. M., Drancourt, M., et al. (2018). Methanobrevibacter oralis detected along with Aggregatibacter actinomycetemcomitans in a series of community-acquired brain abscesses. Clin. Microbiol. Infect. 24, 207–208. doi: 10.1016/j.cmi.2017.08.021
Nkamga, V. D., Lotte, R., Roger, P. M., Drancourt, M., and Ruimy, R. (2016). Methanobrevibacter smithii and Bacteroides thetaiotaomicron cultivated from a chronic paravertebral muscle abscess. Clin. Microbiol. Infect. 22, 1008–1009. doi: 10.1016/j.cmi.2016.09.007
Nollet, L., Mbanzamihigo, L., Demeyer, D., and Verstraete, W. (1998). Effect of the addition of Peptostreptococcus productus ATCC 35244 on reductive acetogenesis in the ruminal ecosystem after inhibition of methanogenesis by cell-free supernatant of Lactobacillus plantarum 80. Anim. Feed Sci. Technol. 71, 49–66. doi: 10.1016/S0377-8401(97)00135-1
O'Donnell, M. M., Harris, H. M. B., Jeffery, I. B., Claesson, M. J., Younge, B., O' Toole, P. W., et al. (2013). The core faecal bacterial microbiome of Irish thoroughbred racehorses. Lett. Appl. Microbiol. 57, 492–501. doi: 10.1111/lam.12137
O'Keefe, S. J., Chung, D., Mahmoud, N., Sepulveda, A. R., Manafe, M., Arch, J., et al. (2007). Why do African Americans get more colon cancer than native Africans? J. Nutr. 137, 175s–182s. doi: 10.1093/jn/137.1.175S
Ouwerkerk, D., Maguire, A. J., McMillen, L., and Klieve, A. (2009). Hydrogen utilising bacteria from the forestomach of eastern grey (Macropus giganteus) and red (Macropus rufus) kangaroos. Anim. Prod. Sci. 49, 1043–1051. doi: 10.1071/EA08294
Owens, J., Hao, X., Thomas, B. W., Stoeckli, J., Soden, C., Acharya, S., et al. (2021). Effects of 3-nitrooxypropanol manure fertilizer on soil health and hydraulic properties. J. Environ. Qual. 50, 1452–1463. doi: 10.1002/jeq2.20276
Oxley, A. P., Lanfranconi, M. P., Wurdemann, D., Ott, S., Schreiber, S., McGenity, T. J., et al. (2010). Halophilic archaea in the human intestinal mucosa. Environ. Microbiol. 12, 2398–2410. doi: 10.1111/j.1462-2920.2010.02212.x
Pascal, V., Pozuelo, M., Borruel, N., Casellas, F., Campos, D., Santiago, A., et al. (2017). A microbial signature for Crohn's disease. Gut 66, 813–822. doi: 10.1136/gutjnl-2016-313235
Pausan, M. R., Csorba, C., Singer, G., Till, H., Schöpf, V., Santigli, E., et al. (2019). Exploring the Archaeome: detection of archaeal signatures in the human body [original research]. Front. Microbiol. 10:e02796. doi: 10.3389/fmicb.2019.02796
Peled, Y., Weinberg, D., Hallak, A., and Gilat, T. (1987). Factors affecting methane production in humans. Gastrointestinal diseases and alterations of colonic flora. Dig. Dis. Sci. 32, 267–271. doi: 10.1007/BF01297052
Picchianti-Diamanti, A., Panebianco, C., Salemi, S., Sorgi, M. L., Di Rosa, R., Tropea, A., et al. (2018). Analysis of gut microbiota in rheumatoid arthritis patients: disease-related Dysbiosis and modifications induced by Etanercept. Int. J. Mol. Sci. 19. doi: 10.3390/ijms19102938
Pieper, H. J., Oplustil, E., and Barth, G. (1980). Reduction of methanol formation during alcoholic fermentation. Biotechnol. Lett. 2, 391–396. doi: 10.1007/BF00144243
Pimentel, M., Lin, H. C., Enayati, P., van den Burg, B., Lee, H. R., Chen, J. H., et al. (2006). Methane, a gas produced by enteric bacteria, slows intestinal transit and augments small intestinal contractile activity. Am. J. Physiol. Gastrointest. Liver Physiol. 290, G1089–G1095. doi: 10.1152/ajpgi.00574.2004
Pimentel, M., Mayer, A. G., Park, S., Chow, E. J., Hasan, A., and Kong, Y. (2003). Methane production during lactulose breath test is associated with gastrointestinal disease presentation. Dig. Dis. Sci. 48, 86–92. doi: 10.1023/A:1021738515885
Pimentel, M., Saad, R. J., Long, M. D., and Rao, S. S. C. (2020). ACG clinical guideline: small intestinal bacterial overgrowth. Off. J. Am. Coll. Gastroenterol. 115, 165–178. doi: 10.14309/ajg.0000000000000501
Pinares-Patiño, C. S., Hickey, S. M., Young, E. A., Dodds, K. G., MacLean, S., Molano, G., et al. (2013). Heritability estimates of methane emissions from sheep. Animal 7, 316–321. doi: 10.1017/S1751731113000864
Pique, J. M., Pallares, M., Cuso, E., Vilar-Bonet, J., and Gassull, M. A. (1984). Methane production and colon cancer. Gastroenterology 87, 601–605. doi: 10.1016/0016-5085(84)90532-8
Pisa, K. Y., Weidner, C., Maischak, H., Kavermann, H., and Muller, V. (2007). The coupling ion in the methanoarchaeal ATP synthases: H(+) vs. Na(+) in the a(1)a(o) ATP synthase from the archaeon Methanosarcina mazei Go1. FEMS Microbiol. Lett. 277, 56–63. doi: 10.1111/j.1574-6968.2007.00939.x
Ponziani, F. R., Picca, A., Marzetti, E., Calvani, R., Conta, G., Del Chierico, F., et al. (2021). Characterization of the gut-liver-muscle axis in cirrhotic patients with sarcopenia. Liver Int. 41, 1320–1334. doi: 10.1111/liv.14876
Pope, P. B., Smith, W., Denman, S. E., Tringe, S. G., Barry, K., Hugenholtz, P., et al. (2011). Isolation of Succinivibrionaceae implicated in Low methane emissions from Tammar wallabies. Science 333, 646–648. doi: 10.1126/science.1205760
Poulsen, M., Schwab, C., Borg Jensen, B., Engberg, R. M., Spang, A., Canibe, N., et al. (2013). Methylotrophic methanogenic Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat. Commun. 4. doi: 10.1038/ncomms2432
Pozuelo, M., Panda, S., Santiago, A., Mendez, S., Accarino, A., and Santos, J. (2015). Reduction of butyrate- and methane-producing microorganisms in patients with irritable bowel syndrome [article]. Sci. Rep. 5:12693. doi: 10.1038/srep12693
Prochazkova, P., Roubalova, R., Dvorak, J., Kreisinger, J., Hill, M., Tlaskalova-Hogenova, H., et al. (2021). The intestinal microbiota and metabolites in patients with anorexia nervosa. Gut Microb. 13, 1–25. doi: 10.1080/19490976.2021.1902771
Qian, Y., Yang, X., Xu, S., Wu, C., Song, Y., Qin, N., et al. (2018). Alteration of the fecal microbiota in Chinese patients with Parkinson's disease. Brain Behav. Immun. 70, 194–202. doi: 10.1016/j.bbi.2018.02.016
Randal Bollinger, R., Barbas, A. S., Bush, E. L., Lin, S. S., and Parker, W. (2007). Biofilms in the large bowel suggest an apparent function of the human vermiform appendix. J. Theor. Biol. 249, 826–831. doi: 10.1016/j.jtbi.2007.08.032
Raymann, K., Moeller, A. H., Goodman, A. L., and Ochman, H. (2017). Unexplored archaeal diversity in the great ape gut microbiome. mSphere 2. doi: 10.1128/mSphere.00026-17
Rebouche, C. J., and Chenard, C. A. (1991). Metabolic fate of dietary carnitine in human adults: identification and quantification of urinary and fecal metabolites. J. Nutr. 121, 539–546. doi: 10.1093/jn/121.4.539
Revilla, I., and González-SanJosé, M. L. (1998). Methanol release during fermentation of red grapes treated with pectolytic enzymes. Food Chem. 63, 307–312. doi: 10.1016/S0308-8146(98)00049-1
Robichaux, M., Howell, M., and Boopathy, R. (2003). Methanogenic activity in human periodontal pocket. Curr. Microbiol. 46, 53–58. doi: 10.1007/s00284-002-3807-5
Samuel, B. S., Hansen, E. E., Manchester, J. K., Coutinho, P. M., Henrissat, B., Fulton, R., et al. (2007). Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc. Natl. Acad. Sci. U. S. A. 104, 10643–10648. doi: 10.1073/pnas.0704189104
Sandborn, W. J. (2009). How to avoid treating irritable bowel syndrome with biologic therapy for inflammatory bowel disease. Dig. Dis. 27, 80–84. doi: 10.1159/000268125
Sansonetti, P. J. (2004). War and peace at mucosal surfaces. Nat. Rev. Immunol. 4, 953–964. doi: 10.1038/nri1499
Scanlan, P. D., Shanahan, F., and Marchesi, J. R. (2008). Human methanogen diversity and incidence in healthy and diseased colonic groups using mcrA gene analysis. BMC Microbiol. 8:79. doi: 10.1186/1471-2180-8-79
Scheifinger, C. C., Linehan, B., and Wolin, M. J. (1975). H2 production by Selenomonas ruminantium in the absence and presence of methanogenic bacteria. Appl. Microbiol. 29, 480–483. doi: 10.1128/am.29.4.480-483.1975
Schwiertz, A., Taras, D., Schafer, K., Beijer, S., Bos, N. A., Donus, C., et al. (2010). Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 18, 190–195. doi: 10.1038/oby.2009.167
Segal, I., Walker, A. R., Lord, S., and Cummings, J. H. (1988). Breath methane and large bowel cancer risk in contrasting African populations. Gut 29, 608–613. doi: 10.1136/gut.29.5.608
Shalvarjian, K. E., and Nayak, D. D. (2021). Transcriptional regulation of methanogenic metabolism in archaea. Curr. Opin. Microbiol. 60, 8–15. doi: 10.1016/j.mib.2021.01.005
Shi, W., Moon, C. D., Leahy, S. C., Kang, D., Froula, J., Kittelmann, S., et al. (2014). Methane yield phenotypes linked to differential gene expression in the sheep rumen microbiome. Genome Res. 24, 1517–1525. doi: 10.1101/gr.168245.113
Sieber, J. R., McInerney, M. J., and Gunsalus, R. P. (2012). Genomic insights into syntrophy: the paradigm for anaerobic metabolic cooperation. Annu. Rev. Microbiol. 66, 429–452. doi: 10.1146/annurev-micro-090110-102844
Smith, P., Reay, D., and Smith, J. (2021). Agricultural methane emissions and the potential for mitigation. Philos. Trans. R. Soc. London, Ser. A 379. doi: 10.1098/rsta.2020.0451
Sorokin, D. Y., Makarova, K. S., Abbas, B., Ferrer, M., Golyshin, P. N., Galinski, E. A., et al. (2017). Discovery of extremely halophilic, methyl-reducing euryarchaea provides insights into the evolutionary origin of methanogenesis. Nat. Microbiol. 2:17081. doi: 10.1038/nmicrobiol.2017.81
Spencer, M. D., Hamp, T. J., Reid, R. W., Fischer, L. M., Zeisel, S. H., and Fodor, A. A. (2011). Association between composition of the human gastrointestinal microbiome and development of fatty liver with choline deficiency. Gastroenterology 140, 976–986. doi: 10.1053/j.gastro.2010.11.049
Spring, K. R. (1998). Routes and mechanism of fluid transport by epithelia. Annu. Rev. Physiol. 60, 105–119. doi: 10.1146/annurev.physiol.60.1.105
Stams, A. J., and Plugge, C. M. (2009). Electron transfer in syntrophic communities of anaerobic bacteria and archaea. Nat. Rev. Microbiol. 7, 568–577. doi: 10.1038/nrmicro2166
Suri, J., Kataria, R., Malik, Z., Parkman, H. P., and Schey, R. (2018). Elevated methane levels in small intestinal bacterial overgrowth suggests delayed small bowel and colonic transit. Medicine (Baltimore) 97:e10554. doi: 10.1097/MD.0000000000010554
Tang, W. H., Wang, Z., Levison, B. S., Koeth, R. A., Britt, E. B., Fu, X., et al. (2013). Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 368, 1575–1584. doi: 10.1056/NEJMoa1109400
Tap, J., Derrien, M., Tornblom, H., Brazeilles, R., Cools-Portier, S., Dore, J., et al. (2017). Identification of an intestinal microbiota signature associated with severity of irritable bowel syndrome. Gastroenterology 152, 111–123.e118. doi: 10.1053/j.gastro.2016.09.049
Thauer, R. K., Kaster, A.-K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591. doi: 10.1038/nrmicro1931
Thiel, A., Schoenmakers, A. C. M., Verbaan, I. A. J., Chenal, E., Etheve, S., and Beilstein, P. (2019). 3-NOP: mutagenicity and genotoxicity assessment. Food Chem. Toxicol. 123, 566–573. doi: 10.1016/j.fct.2018.11.010
Thomas, C. M., Desmond-Le Quéméner, E., Gribaldo, S., and Borrel, G. (2022). Factors shaping the abundance and diversity of the gut archaeome across the animal kingdom. Nat. Commun. 13. doi: 10.1038/s41467-022-31038-4
Toxicity, C. O. (2011). CoT statement on the effects of chronic dietary exposure to methanol. Committee on toxicity of Chemicals in Food, consumer products and the environment Available at: https://cot.food.gov.uk/sites/default/files/cot/cotstatementmethanol201102revjuly.pdf
Tremlett, H., Fadrosh, D. W., Faruqi, A. A., Hart, J., Roalstad, S., Graves, J., et al. (2016a). Gut microbiota composition and relapse risk in pediatric MS: a pilot study. J. Neurol. Sci. 363, 153–157. doi: 10.1016/j.jns.2016.02.042
Tremlett, H., Fadrosh, D. W., Faruqi, A. A., Zhu, F., Hart, J., Roalstad, S., et al. (2016b). Gut microbiota in early pediatric multiple sclerosis: a case−control study. Eur. J. Neurol. 23, 1308–1321. doi: 10.1111/ene.13026
Triantafyllou, K., Chang, C., and Pimentel, M. (2014). Methanogens, methane and gastrointestinal motility. J. Neurogastroenterol. Motil. 20, 31–40. doi: 10.5056/jnm.2014.20.1.31
Vanderhaeghen, S., Lacroix, C., and Schwab, C. (2015). Methanogen communities in stools of humans of different age and health status and co-occurrence with bacteria. FEMS Microbiol. Lett. 362:fnv092. doi: 10.1093/femsle/fnv092
Verma, R., Verma, A. K., Ahuja, V., and Paul, J. (2010). Real-time analysis of mucosal flora in patients with inflammatory bowel disease in India. J. Clin. Microbiol. 48, 4279–4282. doi: 10.1128/JCM.01360-10
Vierbuchen, T., Bang, C., Rosigkeit, H., Schmitz, R. A., and Heine, H. (2017). The human-associated archaeon Methanosphaera stadtmanae is recognized through its RNA and induces TLR8-dependent NLRP3 Inflammasome activation. Front. Immunol. 8:1535. doi: 10.3389/fimmu.2017.01535
Volmer, J. G., Soo, R. M., Evans, P. N., Hoedt, E. C., Astorga Alsina, A. L., Woodcroft, B. J., et al. (2023). Isolation and characterisation of novel Methanocorpusculum species indicates the genus is ancestrally host-associated. BMC Biol. 21:59. doi: 10.1186/s12915-023-01524-2
Vyas, D., McGinn, S. M., Duval, S. M., Kindermann, M., and Beauchemin, K. A. (2016). Effects of sustained reduction of enteric methane emissions with dietary supplementation of 3-nitrooxypropanol on growth performance of growing and finishing beef cattle. J. Anim. Sci. 94, 2024–2034. doi: 10.2527/jas.2015-0268
Wallace, R. J., Rooke, J. A., McKain, N., Duthie, C.-A., Hyslop, J. J., Ross, D. W., et al. (2015). The rumen microbial metagenome associated with high methane production in cattle. BMC Genomics 16. doi: 10.1186/s12864-015-2032-0
Wang, Z., Klipfell, E., Bennett, B. J., Koeth, R., Levison, B. S., Dugar, B., et al. (2011). Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 472, 57–63. doi: 10.1038/nature09922
Wang, Q., Li, F., Liang, B., Liang, Y., Chen, S., Mo, X., et al. (2018). A metagenome-wide association study of gut microbiota in asthma in UK adults. BMC Microbiol. 18:114. doi: 10.1186/s12866-018-1257-x
Weaver, G. A., Krause, J. A., Miller, T. L., and Wolin, M. J. (1986). Incidence of methanogenic bacteria in a sigmoidoscopy population: an association of methanogenic bacteria and diverticulosis. Gut 27, 698–704. doi: 10.1136/gut.27.6.698
Welte, C., and Deppenmeier, U. (2011). “Chapter thirteen - proton translocation in methanogens” in Methods in enzymology. eds. A. C. Rosenzweig and S. W. Ragsdale, vol. 494 (New York: Academic Press), 257–280.
Welte, C., and Deppenmeier, U. (2014). Bioenergetics and anaerobic respiratory chains of aceticlastic methanogens. Biochim. Biophys. Acta 1837, 1130–1147. doi: 10.1016/j.bbabio.2013.12.002
White, B. A., and Mackie, R. I. (1997). Gastrointestinal microbiology. 1, Gastrointestinal ecosystems and fermentations.
Whitelaw, F. G., Eadie, J. M., Bruce, L. A., and Shand, W. J. (1984). Methane formation in faunated and ciliate-free cattle and its relationship with rumen volatile fatty acid proportions. Br. J. Nutr. 52, 261–275. doi: 10.1079/BJN19840094
Wilmes, P., Trezzi, J.-P., Aho, V., Jäger, C., Schade, S., Janzen, A., et al. (2023). Artefactual source of 2-hydroxypyridine. Res. Square. doi: 10.21203/rs.3.rs-1827631/v2
Wilmes, P., Trezzi, J. P., Aho, V., et al. (2022). An archaeal compound as a driver of Parkinson’s disease pathogenesis. PREPRINT (Version 1) available at Research Square.
Wolin, M. J., Miller, T. L., and Stewart, C. S. (1997). “Microbe-microbe interactions” in The rumen microbial ecosystem. eds. P. N. Hobson and C. S. Stewart (Heidelberg: Springer)
Yan, L., Tang, L., Zhou, Z., Lu, W., Wang, B., Sun, Z., et al. (2022). Metagenomics reveals contrasting energy utilization efficiencies of captive and wild camels (Camelus ferus). Integr. Zool. 17, 333–345. doi: 10.1111/1749-4877.12585
Zeisel, S. H., Wishnok, J. S., and Blusztajn, J. K. (1983). Formation of methylamines from ingested choline and lecithin. J. Pharmacol. Exp. Ther. 225, 320–324.
Zhang, H., DiBaise, J. K., Zuccolo, A., Kudrna, D., Braidotti, M., Yu, Y., et al. (2009). Human gut microbiota in obesity and after gastric bypass. Proc. Natl. Acad. Sci. U. S. A. 106, 2365–2370. doi: 10.1073/pnas.0812600106
Keywords: methanogen, methanogenic archaea, methane mitigation, archaea, host-associated archaea
Citation: Volmer JG, McRae H and Morrison M (2023) The evolving role of methanogenic archaea in mammalian microbiomes. Front. Microbiol. 14:1268451. doi: 10.3389/fmicb.2023.1268451
Edited by:
Amelia-Elena Rotaru, University of Southern Denmark, DenmarkReviewed by:
Alexander Mahnert, Medical University of Graz, AustriaCopyright © 2023 Volmer, McRae and Morrison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mark Morrison, bS5tb3JyaXNvbjFAdXEuZWR1LmF1
†This review is derived in part from the thesis submitted and approved as part of the PhD degree awarded to Dr. James Volmer, by the University of Queensland
‡Australian Gastrointestinal Research Alliance and National Health and Medical Research Council of Australia Centre of Research Excellence for Digestive Health
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.