
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 06 November 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1260422
 Deborah V. Hoyle1*†
Deborah V. Hoyle1*† Bryan A. Wee1†Kareen Macleod1
Bryan A. Wee1†Kareen Macleod1 Margo E. Chase-Topping1Andrew G. Bease1
Margo E. Chase-Topping1Andrew G. Bease1 Sue C. Tongue2David L. Gally1
Sue C. Tongue2David L. Gally1 Sabine Delannoy3
Sabine Delannoy3 Patrick Fach3Michael C. Pearce2
Patrick Fach3Michael C. Pearce2 George J. Gunn2Anne Holmes4‡Lesley Allison4‡
George J. Gunn2Anne Holmes4‡Lesley Allison4‡O26 is the commonest non-O157 Shiga toxin (stx)-producing Escherichia coli serogroup reported in human infections worldwide. Ruminants, particularly cattle, are the primary reservoir source for human infection. In this study, we compared the whole genomes and virulence profiles of O26:H11 strains (n = 99) isolated from Scottish cattle with strains from human infections (n = 96) held by the Scottish Escherichia coli O157/STEC Reference Laboratory, isolated between 2002 and 2020. Bovine strains were from two national cross-sectional cattle surveys conducted between 2002–2004 and 2014–2015. A maximum likelihood phylogeny was constructed from a core-genome alignment with the O26:H11 strain 11368 reference genome. Genomes were screened against a panel of 2,710 virulence genes using the Virulence Finder Database. All stx-positive bovine O26:H11 strains belonged to the ST21 lineage and were grouped into three main clades. Bovine and human source strains were interspersed, and the stx subtype was relatively clade-specific. Highly pathogenic stx2a-only ST21 strains were identified in two herds sampled in the second cattle survey and in human clinical infections from 2010 onwards. The closest pairwise distance was 9 single-nucleotide polymorphisms (SNPs) between Scottish bovine and human strains and 69 SNPs between the two cattle surveys. Bovine O26:H11 was compared to public EnteroBase ST29 complex genomes and found to have the greatest commonality with O26:H11 strains from the rest of the UK, followed by France, Italy, and Belgium. Virulence profiles of stx-positive bovine and human strains were similar but more conserved for the stx2a subtype. O26:H11 stx-negative ST29 (n = 17) and ST396 strains (n = 5) were isolated from 19 cattle herds; all were eae-positive, and 10 of these herds yielded strains positive for ehxA, espK, and Z2098, gene markers suggestive of enterohaemorrhagic potential. There was a significant association (p < 0.001) between nucleotide sequence percent identity and stx status for the bacteriophage insertion site genes yecE for stx2 and yehV for stx1. Acquired antimicrobial resistance genes were identified in silico in 12.1% of bovine and 17.7% of human O26:H11 strains, with sul2, tet, aph(3″), and aph(6″) being most common. This study describes the diversity among Scottish bovine O26:H11 strains and investigates their relationship to human STEC infections.
Shiga toxin-producing Escherichia coli (STEC) are a group of zoonotic pathogenic bacteria with a ruminant reservoir that cause gastrointestinal infections in humans (Kolenda et al., 2015; World Health Organization and Food and Agriculture Organization of the United Nations, 2018). Transmission to humans occurs via foodborne routes, as well as by direct contact with infected animals and through environmental contamination, particularly water (Kintz et al., 2017). Shiga toxin is the primary virulence factor responsible for severe pathology and is encoded by stx genes hosted on mobile lysogenic bacteriophage, which integrate into the bacterial genome at specific insertion sites (Bonanno et al., 2015). There are two main Shiga toxin proteins, namely, Stx1 and Stx2, encoded by different gene subtypes stx1a, 1c, 1d, 1e, and stx2a-o (Scheutz et al., 2012; Hughes et al., 2019; Gill et al., 2022), with the stx2a, 2c, and 2d subtypes associated with more serious disease (Friedrich et al., 2002; Persson et al., 2007). The majority of STEC are typically characterised by the presence of the locus of enterocyte effacement (LEE), which is required for the formation of attaching and effacing lesions in the intestine and encodes the intimin gene, eae, in addition to a number of other key virulence factors (Kaper et al., 2004). However, LEE is not essential for human pathogenicity, with some LEE-negative non-O157 STEC serotypes still capable of causing severe disease, mediated by other virulence determinants (Newton et al., 2009; Colello et al., 2019). The LEE is not specific to the STEC pathotype and is also found in the majority of enteropathogenic E. coli (EPEC), which cause non-haemorrhagic gastrointestinal illness in both animals and humans; EPEC are primarily distinguished from STEC by the absence of the stx gene (Denamur et al., 2021).
STEC cause a spectrum of clinical symptoms in humans, from uncomplicated diarrhoea to haemorrhagic enteritis, haemolytic uraemic syndrome (HUS), and, in exceptional cases, death. STEC serotypes that are responsible for the more severe, haemorrhagic disease presentations are further classified as enterohaemorrhagic (EHEC) (Nataro and Kaper, 1998). Globally, E. coli O157:H7 is the commonest STEC/ EHEC serotype and is often associated with large foodborne outbreaks of disease. However, a number of non-O157 STEC serotypes can also be classified as EHEC based on their disease and pathogenicity profile, with O26:H11 being the predominant non-O157 serotype of clinical relevance in human cases worldwide (Caprioli et al., 2005; Johnson et al., 2006). In Europe, O26:H11 frequently surpasses O157:H7 reported cases and is currently the leading serotype responsible for human STEC infection, including paediatric HUS (European Centre for Disease Prevention and Control, 2022).
The recent increase in the proportion of STEC clinical cases attributed to non-O157 serotypes may in part be due to improvements in diagnostic testing methods (Parsons et al., 2016). However, the emergence of two highly pathogenic, stx2-only positive O26:H11 clones, termed the new “European” and “French” clones, has also resulted in a true increase in O26:H11 incidence across Europe over the past decade (Bielaszewska et al., 2013; Zweifel et al., 2013; Delannoy et al., 2015). These clones have been particularly associated with disease outbreaks and hospitalisations in children, linked to the consumption of dairy products in France, Italy, and Romania (Severi et al., 2016; Jones et al., 2019; Loconsole et al., 2020).
Globally, O26:H11 strains can be grouped into two main multilocus sequence types (MLSTs), namely, ST21 and ST29 (Ogura et al., 2017). The majority of all stx-positive O26:H11 strains belong to ST21, which includes the predominant stx1-only strains as well as dual positive stx1 + stx2 strains and less common strains encoding stx2a only. ST29 comprises mostly stx-negative O26:H11 strains but also includes the newly emerging stx2a + “European” and stx2d + “French” clones. STEC harbour large virulence plasmids (pVF) that host genes for enterohaemolysin, ehxA, catalase-peroxidase, katP, serine protease, espP, and a type II effector protein, etpD (Fratamico et al., 2011; Bielaszewska et al., 2013). The main O26:H11 lineages are distinguished by the presence or absence of these pVF genes, with the ST21 lineage characterised by the ehxA+/katP+/espP+/etpD- gene profile. In contrast, the newly emerging and highly virulent ST29 European stx2+ clone bears a distinct ehxA+/katP-/espP-/etpD+ pVF gene profile (Bielaszewska et al., 2013; Ogura et al., 2017).
ST29 stx-negative O26:H11 strains that carry the eae gene are classed as EPEC; however, a subset of these strains has also been shown to carry the ehxA+/katP+/espP+/etpD- pVF gene profile typically seen in stx-positive ST21 strains, together with a range of additional virulence factors (Leomil et al., 2005). Such stx-negative strains have been termed “EHEC-like” because the acquisition of the stx gene through bacteriophage lysogeny could result in conversion to a highly virulent EHEC pathogenic strain profile (Bielaszewska et al., 2007; Bugarel et al., 2011). To distinguish between O26:H11 stx-negative EPEC and EHEC-like strains, an additional set of genetic markers has been proposed to assist in the identification of strains with EHEC potential (Bugarel et al., 2011; Delannoy et al., 2013a). These markers include the type III secretion system genes espK (Vlisidou et al., 2006), urease gene ureD (Steyert et al., 2011), and the open reading frame putative marker Z2098 (Delannoy et al., 2013b).
Scotland has a higher incidence of human STEC infections than the EU average and has reported an increased incidence of non-O157 serotypes isolated from clinical patients in recent years (Food Standards Scotland, 2020; Public Health Scotland, 2020). To assess the prevalence and distribution of STEC in Scottish cattle, two national cross-sectional surveys were conducted in 2002–2004 (Pearce et al., 2006, 2009) and 2014–2015 (Henry et al., 2017), from which a collection of bovine-sourced stx-positive and negative O26 E. coli strains were isolated (Pearce et al., 2006; Hoyle et al., 2021). The aim of this present study was to compare by whole-genome sequencing the O26:H11 strains isolated from bovine faecal samples collected through these two Scottish cattle surveys with clinical O26:H11 human strains isolated from patients and previously sequenced by the Scottish E. coli O157/STEC Reference Laboratory (SERL) (Food Standards Scotland, 2020). We also further examined how the Scottish bovine strains related to human-derived O26:H11 strains from across the wider UK (Dallman et al., 2021; Rodwell et al., 2023) and investigated their global O26:H11 phylogenetic context by comparison with the public collection of clonal complex 29 genomes deposited within EnteroBase (Zhou et al., 2020).
Ongoing analysis of strains from reservoir hosts such as cattle is essential for monitoring the microevolution and emergence of new pathogenic STEC and EHEC strains. These data inform on the risk and can assist in the public health management of this pathogen.
In total, 195 O26:H11 E. coli strains from Scottish cattle (n = 99) and the Scottish human strain collection (n = 96) were included in the analysis, together with 3 O177:H11 bovine strains that fell within the ST29 complex and a single bovine O103:H14 strain as an outgroup (Table 1; Supplementary Table 1).
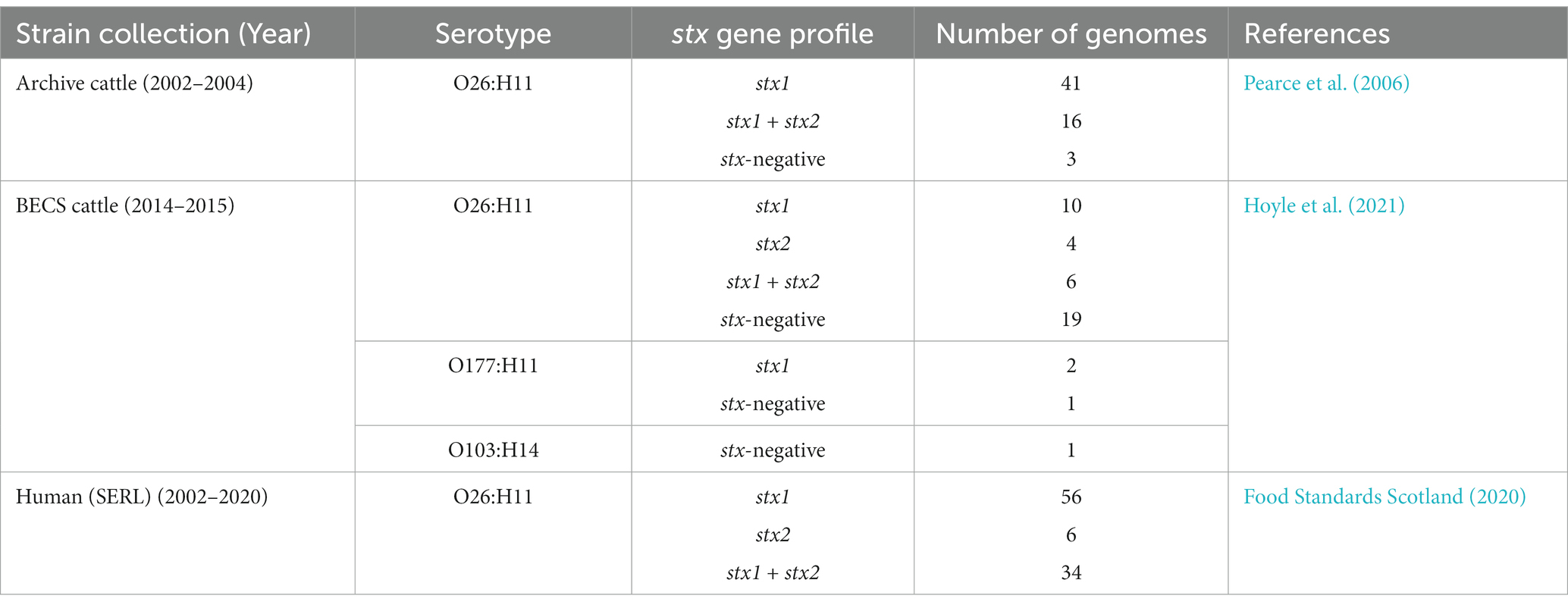
Table 1. Summary of the bovine and human bacterial strain genomes included in this study.
Bovine strains were originally isolated from cattle faecal pat samples that had been obtained during two cross-sectional surveys of Scottish cattle farms conducted between 2002–2004 (Pearce et al., 2006) and 2014–2015 (Henry et al., 2017; Hoyle et al., 2021), as previously described. In both surveys, the original faecal pat samples were collected by sampling discrete, dropped, faecal pats present on the ground of grazing land or the floor of pens.
The cattle strain collection comprised 60 isolates obtained from 35 herds, sampled in the 2002–2004 survey (Archive), and 43 isolates obtained from 29 herds in the 2014–2015 study (BECS). In the initial survey, Scotland was divided into six distinct geographical animal health district regions, as previously outlined (Pearce et al., 2006), and herds were therefore also grouped according to this geographic classification in the second survey.
Human clinical O26:H11 genomes were provided from a collection of genome sequences held by the SERL. Clinical O26:H11 human strains were originally isolated from faecal sample submissions received by the SERL between 2002 and 2020 that were PCR-positive for stx genes. A subset of the human genomes sequenced at the SERL was included in this study, selected as described below in phylogenetic analysis (2.3). Only a single representative genome from any outbreak-linked human strain was included in this comparative analysis.
Detailed methods used by the SERL for extraction, PCR, library preparation, sequencing, and analysis have been described elsewhere (Food Standards Scotland, 2020). In brief, genomic DNA was extracted either manually with the DNeasy Blood and Tissue Kit (Qiagen, Crawley, UK) or with the QIAsymphony using the QIA DSP DNA Mini Kit (Qiagen). Libraries were prepared using the Nextera XT kit and sequenced on the Illumina MiSeq, producing paired-end reads of 250 bp. Sequencing reads were processed in BioNumerics using the wgMLST and E. coli genotyping plug-in tools. The assembly was performed using SPAdes, and basic assembly metrics were calculated for quality assessment. Sequencing reads were also processed using the Scottish Microbiology Reference Laboratory Edinburgh Bioinformatics Pipeline (SMiRLWBP). Trimmomatic (Bolger et al., 2014) was used to remove bases with a Phred score < 30 from the trailing edge. KmerID (Chattaway et al., 2017) identified bacteria species, and the GeneFinder tool mapped reads to a panel of serotype and virulence genes using Bowtie2 (Langmead et al., 2009). Only in silico predictions of serotype and virulence that matched a gene determinant at >80% nucleotide identity and over >80% target gene length were accepted. MLST alleles of seven housekeeping genes (adk, fumC, gyrB, icd, mdh, purA, and recA) were determined using the Metric-Oriented Sequence Typer (MOST) (Tewolde et al., 2016). Shiga toxin gene subtyping was performed using a combined mapping and BLAST approach as previously described (Ashton et al., 2015).
The funding bodies approved and authorised informed consent documentation for farm survey participants to enable the collection of dropped faecal pat samples from participant land. Permission had been granted and consent obtained for the samples, strains, and data to be used for further research. All farm participants’ personal data were handled in accordance with the UK Data Protection Act (1998) and are now handled in accordance with the UK General Data Protection Regulation (GDPR 2018).
The de-identified bacterial genomes from human clinical samples were obtained from the National Health Service (NHS) Lothian following ethics approval from the biorepository bank in NHS Lothian covering the sequencing of samples (20/ES/0061).
DNA was extracted from the cattle-sourced bacterial strains using the DNeasy Blood and Tissue Kit (Qiagen, Crawley, UK). The quantity of DNA was measured using the Qubit Fluorimeter 3.0 (Thermo Fisher Scientific) with the dsDNA Assay HS Kit. The stx subtype for all bovine isolates was determined initially by PCR as previously described (Pearce et al., 2006; Hoyle et al., 2021), and library preparation, sequencing and analysis for serotype, MLST, and Shiga toxin subtyping were performed at the SERL, as described above.
A subset of the Scottish human strain genomes held by the SERL that differed by fewer than 50 cgMLST (core-genome MLST) alleles from bovine strains was determined using an ad-hoc cgMLST schema with chewBBACA v2.5.5 (Silva et al., 2018), using allele profiles downloaded from EnteroBase1 and made available for comparison with the bovine genomes. This subset of human strain genomes was provided as paired Illumina raw reads for full comparative analysis with all bovine strain genomes. Raw reads were quality filtered using bbduk (v38.45) with the settings ‘k = 19 mink = 11 hdist = 1 ktrim = r minoverlap = 12 qtrim = rl trimq = 20 minlength = 50’ (Bushnell, 2019). All genomes were assembled using SPAdes (v3.15.3) with the --careful option (Bankevich et al., 2012). Criteria for inclusion were 50–51% GC, a total assembly length of 4.5–6.5 Mbp, a duplication ratio below 1.021, and a minimum N50 of 40 Kbp. Genome alignment was performed using Parsnp v1.5.6 with reference genome AP010953.1 (O26:H11 strain 11368) (4.03 Mbp, 35,612 polymorphic sites) (Treangen et al., 2014). Recombinant SNPs were filtered using Gubbins with default settings (v3.0.0) (Croucher et al., 2015). A maximum likelihood tree (IQTree v2.1.2) was constructed from the filtered core-genome alignment of 198 sequences with the TVMe+ASC model and 1,000 bootstraps (Minh et al., 2020). Figures were generated using iTol (Letunic and Bork, 2021).
All genomes were screened against a panel of 2,710 virulence genes from the E. coli-specific virulence gene database (Date downloaded: 10 September 2021) using Abricate (v1.0.1), supplemented with 139 additional gene alleles of interest, run with default parameters (−-minid 80, −-mincov 80) (Supplementary Table 2)2 (Escherichia coli Virulence Factors, 2021; Seemann, 2022a). The percent identity was recorded, and the gene target was categorised as positive or negative based on whether the gene was detected using Abricate threshold parameters of minimum 80% coverage and 80% identity. Additional targets included the espK, ureD, and Z2098 genes, which were thought to be indicative of the potential pathogenicity of O26:H11 stx-negative strains following the acquisition of the stx gene (Bugarel et al., 2011; Delannoy et al., 2013a). Sequences for these genes were sourced from the O26:H11 reference strain 11368, GenBank accession number AP010953. Genomes were additionally screened using Abricate for the presence of four phage insertion site genes commonly associated with stx1 and stx2 bacteriophage insertion into the O26 E. coli serogroup: yecE and wrbA for stx2 and yehV and sbcB for stx1 (Bonanno et al., 2015).
To examine the potential for antimicrobial resistance, strain genomes were screened for acquired antimicrobial resistance genes (ARGs) using StarAMR (v0.5.1), and the ResFinder gene database (downloaded 7 September 2021) using default parameters (--pid-threshold 98, --percent-length-overlap 60) (Bharat et al., 2022; Florensa et al., 2022). Cattle strains positive for ARGs were submitted to the web-based Mobile Genetic Element Finder3 on 17–30 October 2022, to examine whether identified ARGs were associated with particular mobile genetic elements (Johansson et al., 2021).
The local arrangement of genes within the genome was examined for the single integron-bearing bovine strain and for the exploration of bacteriophage insertion site genes in bovine strains using Artemis 18.1.0 (Carver et al., 2012). Insertion site genes were identified using the navigator tool with primer sequences, according to Bonanno et al. (2015). Nucleotide and amino acid sequences were extracted as FASTA and verified in BLAST4 and UniProt5 (Camacho et al., 2009; The UniProt Consortium, 2021).
The potential carriage of any prophage sequences was assessed for all bovine stx-negative strains by submission to PHASTER6 in March/November 2022 (Arndt et al., 2016).
Publicly available genomes from the E. coli ST29 complex (n = 8,511; Supplementary Table 3) were downloaded from EnteroBase on 16 March 2022 (Zhou et al., 2020). The HeirCC HC-1100 cluster “2” was used to filter genome assemblies, and we further selected genomes for which “Country” metadata were available (n = 8,332, from 36 countries).
A core-genome alignment of the downloaded EnteroBase genomes in the E. coli ST29 complex, together with 198 of the genomes from this study, was generated using Snippy (v4.6.0) against the O26:H11 strain 11368 genome (AP010953.1), and the pairwise distances between genomes were calculated using Disty (v0.1.0) (Disty McMatrixface, 2021; Seemann, 2022b). A conservative threshold of 200 core SNPs was used to capture clusters of epidemiologically linked isolates (Dallman et al., 2015). Distances were compared within herds, between herds, and between cattle and humans within Scotland, as well as between cattle and non-UK country O26:H11 genomes.
Fisher–Freeman–Halton exact tests for comparing category proportions were performed in StatXact Version 11 (Cytel Inc., Cambridge, MA, US). Associations between binary virulence gene occurrence, stx profile, and host species for all stx-positive O26:H11 genomes were performed using non-metric multidimensional scaling (NMS), PC-ORD software version 7.04 (MJM Software Design, Gleneden Beach, OR, US). Strains that were stx-negative (n = 22) were excluded from the analysis since the almost complete separation in virulence profiles between ST396 and the two differing clusters of ST29 prevented the model from reaching a stable solution. Genes were excluded from the analysis where they were present in <4% or > 96% of the samples, identical to or highly correlated with other genes (Supplementary Table 4; Supplementary Figure 1). The final NMS was run with 30 genes and 173 strain genomes. NMS was used with a grower distance measure. The dimensionality of the dataset was determined by plotting an inverse measure of fit (“stress”) to the number of dimensions. Optimal dimensionality was based on the number of dimensions with the lowest stress. A three-dimensional solution was shown to be optimal. Several NMS runs were performed for each analysis to ensure that the solution was stable and represented a configuration with the best possible fit. On this basis, 500 iterations were used for each NMS run, using random starting coordinates.
All O26:H11 strains belonged to one of the three sequence types: ST21 (87.2%, 170/195), ST29 (8.7%, 17/195), and ST396 (2.6%, 5/195), or other single-locus variants of these STs (n = 3) (Figure 1). All stx-positive O26:H11 bovine strains belonged to ST21, as did the majority of the human strains included in this analysis, whilst all stx-negative O26:H11 bovine strains belonged to either the ST29 or ST396 lineages. Three bovine O177:H11 strains that were also typed as ST29 were included in the phylogeny: two stx1a-positive strains from a single herd, which also yielded a stx-negative O26:H11 strain, and one stx-negative strain from a herd yielding stx2a + stx1a-positive O26:H11 strains. This ST distribution of O26:H11 strains in Scotland is highly similar to that reported for human cases in England (Dallman et al., 2021) and broadly in line with the most recent phylogenetic analysis of worldwide O26:H11 genomes by Long et al. (2022), in which 84 and 16% of strains were identified as ST21 and ST29, respectively.
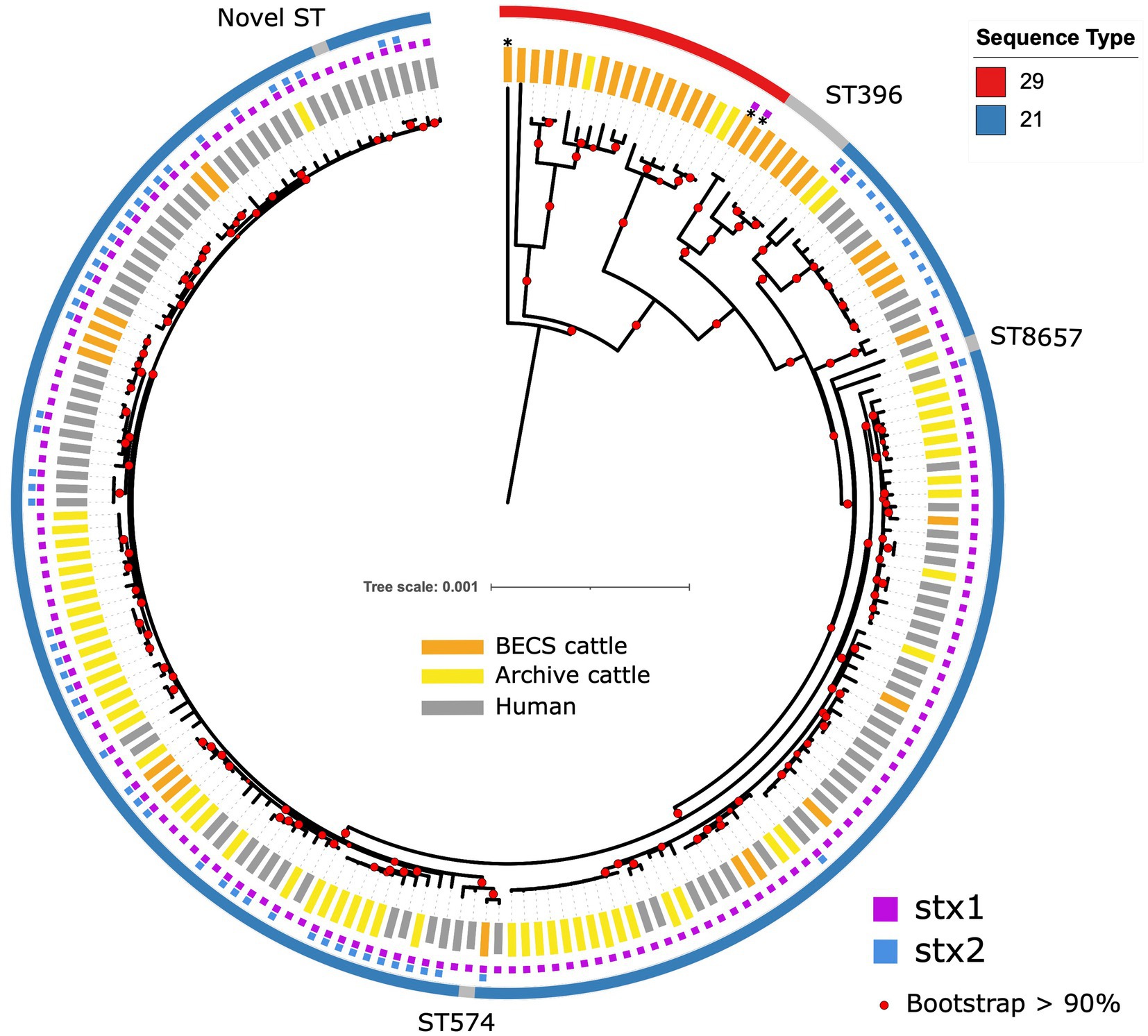
Figure 1. Maximum likelihood core-genome phylogenetic analysis of Scottish bovine and human O26:H11 strains (n = 195) and three O177:H11 strains (*) analysed in this study. The inner ring shows the presence of the stx1 and stx2 genes and the outer ring shows the MLST number. The tips are coloured by source and dataset [cattle – BECS (orange) or Archive (yellow); human clinical (grey)]. Red circles indicate branches with >90% bootstrap support. Tree scale is in substitutions per site.
Within the ST21 lineage, bovine and human stx-positive strains were interspersed within a relatively diverse phylogeny, comprising three main clades (Figure 1). The first clade contained stx2a-only positive O26:H11 cattle and human strains, which falls within the ST21C1a sub-lineage, as described by Ogura et al. (2017). The second clade comprised predominantly stx1a-positive strains only, with two exceptions in human clinical strains, where stx2a was also present. The third clade contained a combination of both stx1-only and dual stx2 + stx1-positive strains. Strain ST and stx subtypes are outlined in Supplementary Table 1.
The first stx2a-only human strains isolated from clinical samples in Scotland were recorded in 2010 (Food Standards Scotland, 2020). We did not identify any stx2a-only positive bovine strains in the 2002–2004 cattle survey (n = 338 sampled herds); however, we isolated stx2a-only positive strains from two herds in the 2014–2015 survey (n = 110 sampled herds) that clustered with ST21 human stx2a-only strains (Table 1; Supplementary Table 1). This stx2a-only lineage has not been reported in Scottish cattle prior to this survey and is most likely explained by a relatively recent introduction into Scottish cattle after the estimated emergence of this clade in the mid-20th century (Ogura et al., 2017).
We did not identify any Scottish stx-positive bovine strains belonging to the newly emerging ST29, highly pathogenic stx2a-only new European clones (Bielaszewska et al., 2013; Karnisova et al., 2018) which have been isolated from clinical cases throughout Europe and at low levels in Japan (Ishijima et al., 2017). ST29 stx2a-only strains have been isolated from <3% of Scottish clinical O26:H11 infections (Food Standards Scotland, 2020). However, these strains were above the 50 cgMLST genetic distance threshold to Scottish bovine strains in our initial cgMLST-based screening, the limit for the selection of clinical strains for inclusion in the comparative phylogeny. Metadata reported for clinical submissions to the national reference laboratory in England indicated that ST29 stx2a-positive strains isolated from human cases reported within the UK were predominantly associated with travel abroad (Dallman et al., 2021). These data would suggest that if ST29 stx2a-only O26:H11 strains are present and circulating within the UK cattle population, this is not currently resulting in identified human infection.
We were interested in investigating the potential for O26:H11 pathogenicity in Scottish cattle regardless of stx status and therefore sequenced strains from each of the 19 herds yielding stx-negative isolates (n = 22). The majority of stx-negative strains belonged to ST29 (n = 17 strains), although four herds yielded minority strains (n = 5) in ST396, a single-locus variant of ST29 (Bielaszewska et al., 2013; Figure 1). The ST29 genomes were split into two distinct clades: a relatively conserved clade phylogenetically closer to the ST21 lineage and a more diverse clade at a greater distance. The separation of stx-negative ST29 into distinct lineages, with one sub-lineage phylogenetically closer to stx-positive ST21, is a broadly similar finding to that reported by Ogura et al. (2017). This also concurs with the population structure observed in ST29 stx-negative O26:H11 strains isolated from both the US and New Zealand cattle populations (Gonzalez-Escalona et al., 2016; Browne et al., 2018) and with the recent clade classification of stx-negative ST29 lineages by Long et al. (2022). Three Archive bovine strains that had been confirmed as stx-positive by PCR at the SERL prior to long-term cryostorage were found to be stx-negative on resuscitation, by both genome sequencing and repeat PCR. This may have been due to the spontaneous excision of the stx-encoding prophage.
Multiple O26:H11 isolates from individual herds were available for 10 Archive and 7 BECS herds, and 47 herds yielded a single strain per herd (Supplementary Table 1). Multiple strains within herds were compared to examine within-herd diversity. Overall, the closest observed relationship was within-herd/within-survey, with a minimum pairwise difference of 0 SNP and median of 3 (interquartile range, IQR = 5) (Figure 2; Supplementary Table 5), although strains within-herd from the BECS survey had closer SNP distances than those within-herd from the Archive survey. Ten stx1a genomes were available for a single Archive herd and gave a median pairwise difference of 4 SNPs and a maximum of 15 SNPs, clustered within a single node. These data suggest that within the study, O26:H11 isolates with the same stx profile spread clonally at the herd level, rather than supporting multiple lineage introductions across a herd cohort. A similar observation has been reported for O26:H11 within cattle herds in New Zealand (Browne et al., 2018). However, where the stx profile differed within a herd, the SNP difference was found to be 128 SNPs or more, reflecting the presence of different circulating lineages. Two herds yielded strains with the same stx subtype in both surveys; however, the genetic distance between these isolates was 69 SNPs or more. This also suggested the presence of distinct lineages, since this pairwise distance is higher than the expected variation from a single lineage over a 10-year period (Dallman et al., 2021).
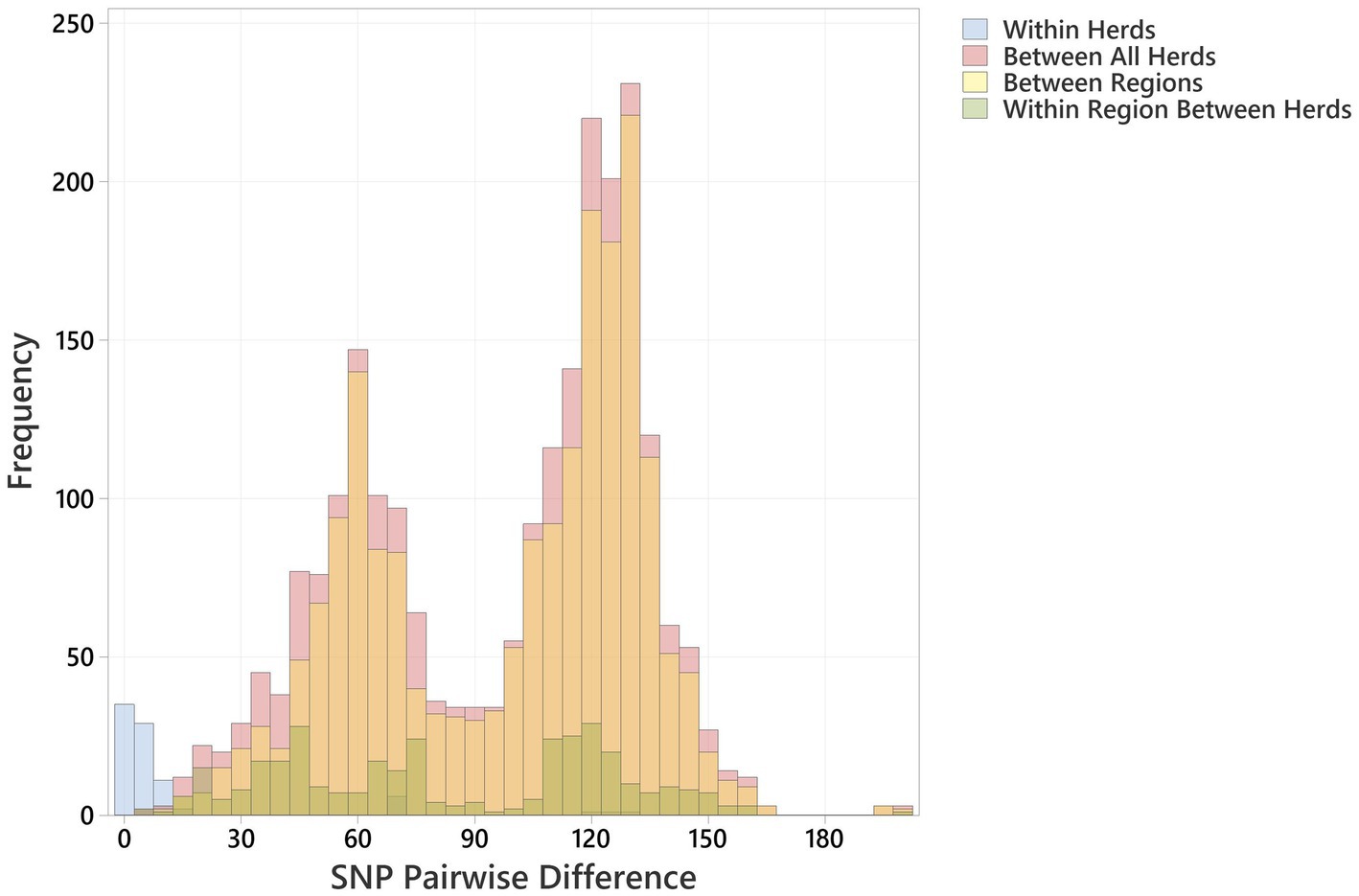
Figure 2. Histogram showing pairwise SNP differences <200 SNP, between bovine O26:H11 strains (n = 99), for the comparisons “Within Herd,” “Within Region” (excludes Within Herd), “Between Regions” and “Between All Herds.”
Regionally, across both cattle surveys, the median SNP difference between genomes from different herds within an animal health district was 76 (IQR 77), and between genomes across different animal health districts, the median SNP difference was 112 (IQR 63) (Figure 2; Supplementary Table 5). These data indicate that herds that were geographically closer showed closer genetic relationships between strains. This is contrary to observations on regional differences reported for New Zealand (Browne et al., 2018) and may be due to stock movements being more limited by geographical distance within Scotland, particularly for herds based on Scottish islands.
A total of 3,969 (47.6%) publicly available genomes within the HierCC:1100 (core-genome ST complex) clade 2 from EnteroBase were found to be within 200 core SNPs from Scottish bovine strain genomes (Supplementary Tables 3, 6). These genomes represented 24 countries; countries excluded that fell beyond the 200 SNP threshold were located in South America, Africa, or Asia, and yielded fewer than five genomes each, except for China, for which 11 genomes were available (Supplementary Table 6).
The distribution profiles for pairwise SNP differences between bovine strains, between human strains, and between cattle and humans within Scotland and across the rest of the UK were very similar (Figure 3). All showed a biphasic distribution, due to the presence of two major clusters of strains, with a similar median pairwise SNP difference in Scotland of cattle to cattle (between herds), cattle to human, and human to human of 110 (IQR 63), 114 (IQR 66), and 118 (IQR 70), respectively (Supplementary Table 5). The closest relationship between any bovine strain and a Scottish human strain was 9 SNPs between a BECS isolate (2014) and a human strain isolated in 2019. The closest relationship to any UK human strain was 7 SNPs between this same BECS strain and a non-Scottish UK human strain isolated from an individual with diarrhoea from the South of England in 2015 (Supplementary Table 6). For the latter example, the close relationship between the South England human strain and the Scottish cattle strain could be due to either the movement of cattle or of a bovine-contaminated food source from Scotland to South England or through human travel to and consequent infection within Scotland. This study does not allow us to draw any conclusions on directionality or source attribution; however, these data could indicate a common source reservoir for the majority of ST21C1b lineage strains found in cattle and humans within the UK.
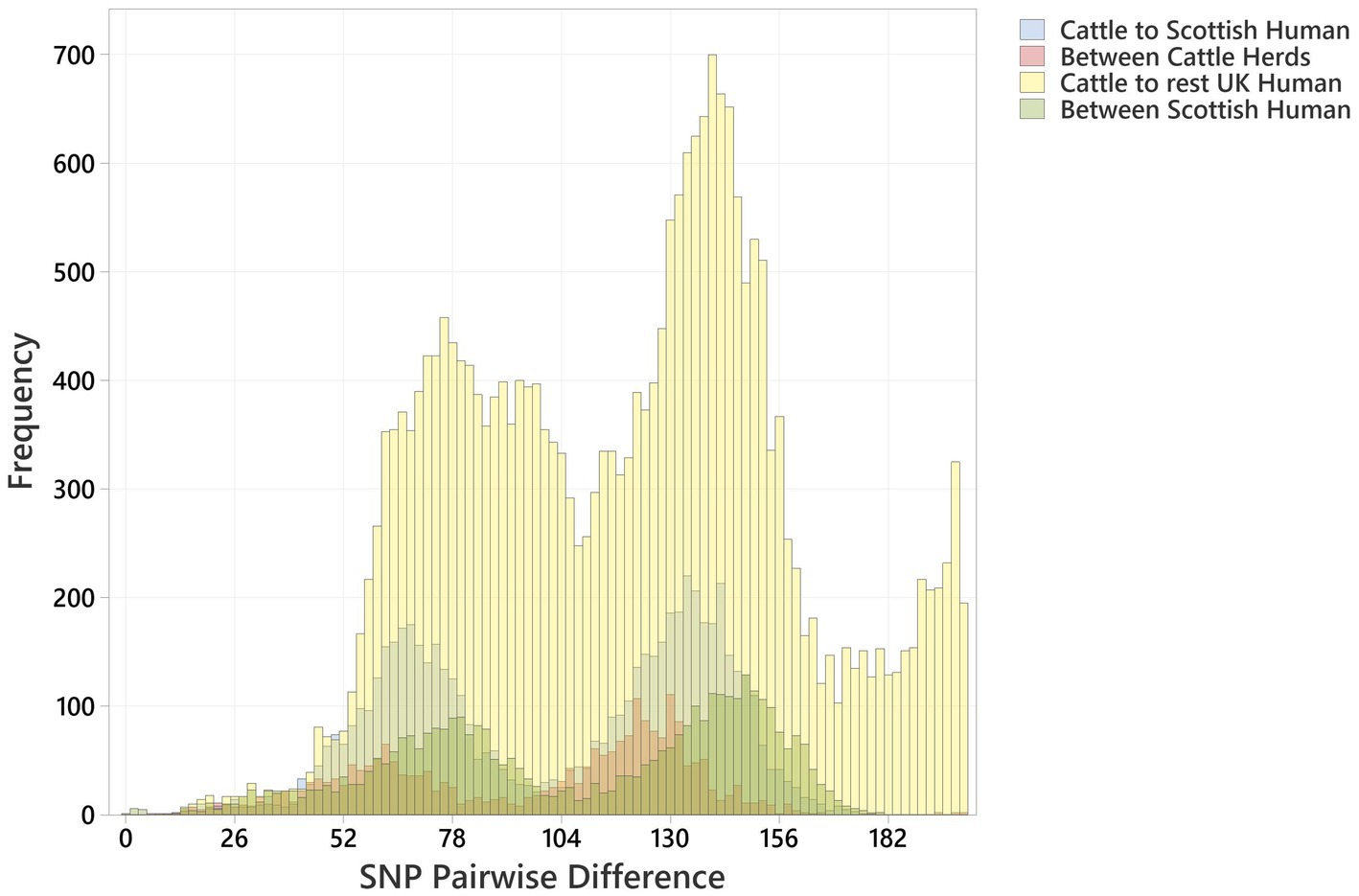
Figure 3. Histogram showing pairwise SNP differences between O26:H11 bovine strains (n = 99), Scottish human strains (n = 96), and rest of UK human strains (n = 1,217), displaying comparison with <200 SNP difference. Comparisons are given as “Cattle to Scottish Human,” “Between Scottish Cattle Herds,” “Cattle to rest UK Human,” and “Between Scottish Human.” Pairwise comparisons include bovine strains across both surveys.
The closest pairwise relationship observed between a Scottish bovine strain and an external country genome was 10 SNP between three bovine strains from Archive_24 and a Canadian strain (ESC_IB7316AA_AS), followed by 21 SNP between a 2019 French strain (ESC_FB8524AA_AS) and a BECS_5 strain, and 26 SNP between an Archive_14 strain and a 2018 isolate from the United States (ESC_RA4669AA_AS) (Figure 4; Supplementary Tables 3, 6). The strain source for all these closest genomes was designated as “Human” origin in the EnteroBase “Source Type” field. The 10 SNP difference between the Archive bovine strains and the human-sourced Canadian strain is unexpectedly close. The metadata associated with this Canadian strain indicate source as a human with gastroenteritis but do not provide information on the isolation date. The second-closest Canadian strain at 33 SNPs to a BECS bovine strain is attributed to the Canadian Food Inspection Agency. The nearest pairwise difference to a designated bovine source genome in the Canadian dataset was 134 SNPs in a Canadian bovine faecal sample collected in 2014.
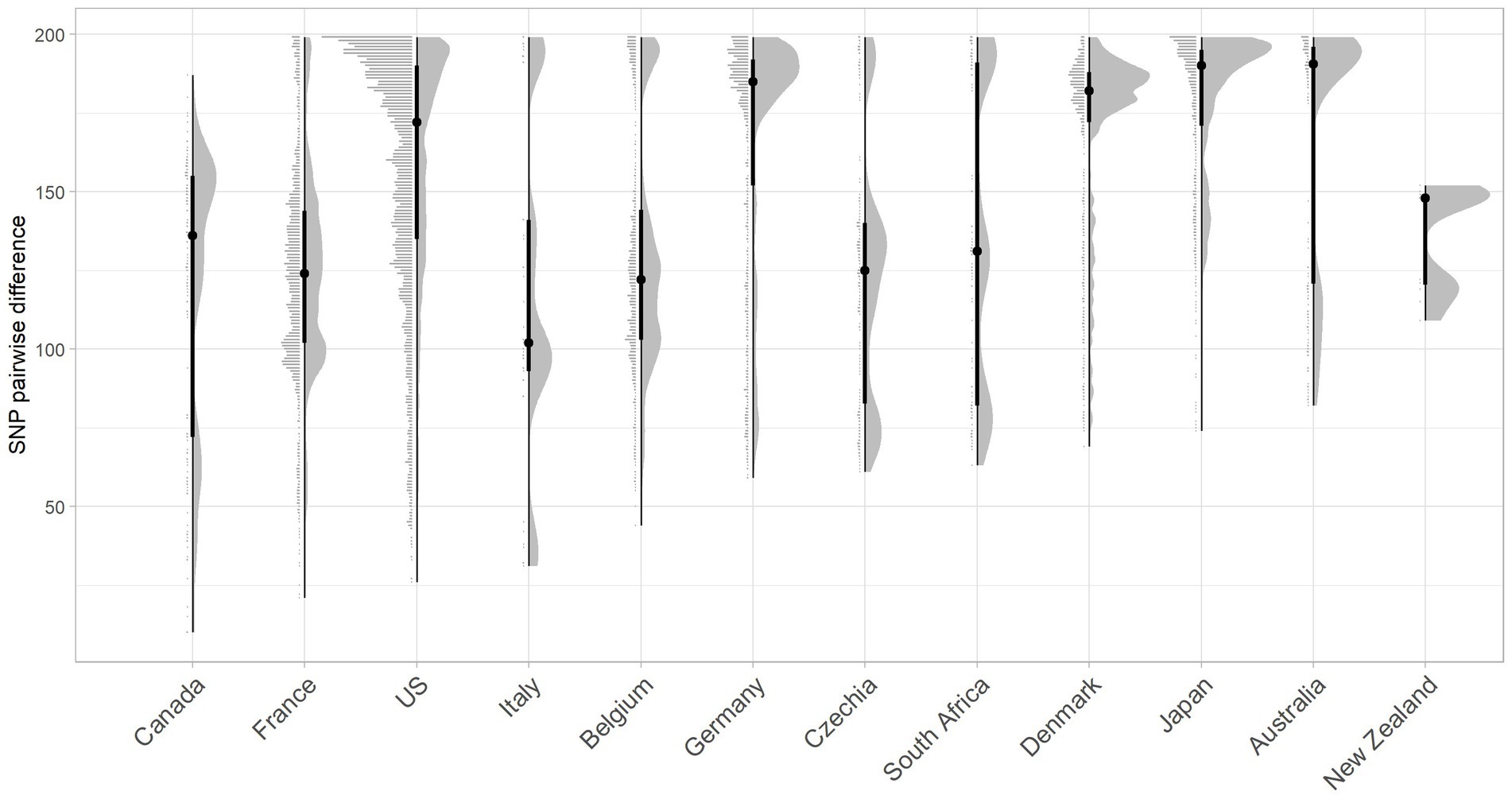
Figure 4. Raincloud plot for the pairwise SNP difference between bovine O26:H11 strain genomes (n = 99) to publicly available O26:H11 genomes from other countries present within the EnteroBase E. coli ST29 complex. Pairwise comparisons were displayed for bovine strains across both surveys to countries where five or more genomes match bovine strains <200 SNPs. Error bars illustrate the median and interquartile range.
Whilst two of the three non-UK strains that were closest matching to Scottish bovine strains were of North American origin, overall, considering the proportion of genomes matched within 200 SNPs, together with pairwise differences, Scottish cattle most closely matched strains from Europe than elsewhere, with the greatest commonality seen with France and Belgium (Figure 4; Supplementary Table 6). More than 60% of downloaded genomes from France, Italy, Belgium, and Germany were within 200 SNPs of our Scottish bovine strains, compared with less than 40% for North American strains. Japan was an unusual outlier, matching 80% of genomes within 200 SNPs of Scottish cattle. It has been previously noted that Japan imported relatively high levels of cattle from Western countries during the second half of the 20th century (Browne et al., 2019), which may account for the closer relationship of Scottish bovine strains to O26:H11 strains from Japan than to other Asian-Pacific countries. An analysis of source type by country was not performed due to a lack of available metadata within EnteroBase for the majority of the downloaded genomes.
Defining a core-genome alignment depends on the diversity and quality of the genomes included in the analysis. In this study, for the initial screen to identify Scottish human O26:H11 genomes that clustered with the bovine O26:H11 genomes, we used a reference-free cgMLST clustering approach, which would be less affected by sequence quality. For the subsequent in-depth analyses, we used the more conservative Parsnp whole-genome aligner to align the Scottish human and bovine genomes. For all comparisons of pairwise distances, we used the more robust short sequence mapping-based Snippy-Core-SNP approach, taken from the same core-genome alignment generated using Snippy-Core. The size of the alignment was approximately 276,190 SNPs. The pairwise distances calculated here are only meaningful when used to compare subsets of genomes that were included in the same core-genome alignment. This is because the core genome, by definition, is the collection of nucleotide positions that are conserved across all the genomes in the given alignment and can change according to the diversity of genomes being included.
All 195 O26:H11 genomes were screened against an E. coli-specific virulence gene database, together with selected additional gene targets (Escherichia coli Virulence Factors, 2021), and recorded as positive or negative according to the described Abricate threshold parameters (Supplementary Tables 4, 7). In total, a conserved set of 154 genes was identified as present in all genomes irrespective of stx status, including the key virulence factor genes eae, tir, cif, espA, and espB, encoded on the LEE pathogenicity island. Other common virulence genes present across all strains included fim D, fim F-H, gadX, iss, and lpfA, and the non-locus of enterocyte effacement effector (nle) genes nleB1, nleG7, nleG8, and nleH1. This observation concurs with the typical O26:H11 virulence profiles previously reported in bovine EPEC O26:H11 strains from the United States and in STEC O26:H11 worldwide (Gonzalez-Escalona et al., 2016; Long et al., 2022).
A total of 207 genes displayed differential occurrence, with distinct distributions noted according to stx status (Supplementary Table 7). All genomes were negative for the etpD gene, with 96.5% of all stx-positive strains (n = 167/173) showing the pVF profile typically observed in ST21 strains ehxA+/katP+/espP+/etpD- (Figure 5). Three stx-positive Archive bovine strains from different herds were ehxA-/katP-/espP-/etpD-, and one Archive bovine and one human strain were ehxA+/katP+/espP-/etpD-, with a further human strain having an ehxA+/katP-/espP-/etpD- pVF profile. The stx-negative ST396 strains bore an identical core virulence profile to the stx-positive strains, including the ehxA+/katP+/espP+/etpD- profile. A further 6 stx-negative Scottish cattle herds yielded 7 strains that were ehxA+/katP-/espP-/etpD-, whilst the remaining 9 stx-negative herds yielded 10 strains that were negative for all pVF genes. The espL and fimB genes showed similar distributions to ehxA across all stx profiles, excluding the stx-negative ST396 lineage, of which all but one strain was fimB-negative. All but one of the stx-negative strains, regardless of ST or pVF profile, also carried a distinct set of genes that were not detected in any stx-positive strains, including the genes aec17, aec18, aec22, aec23, hcp, and vgrG, which encode components of the type VI secretion system (Pukatzki et al., 2009), and the genes Z0263 and Z0265.
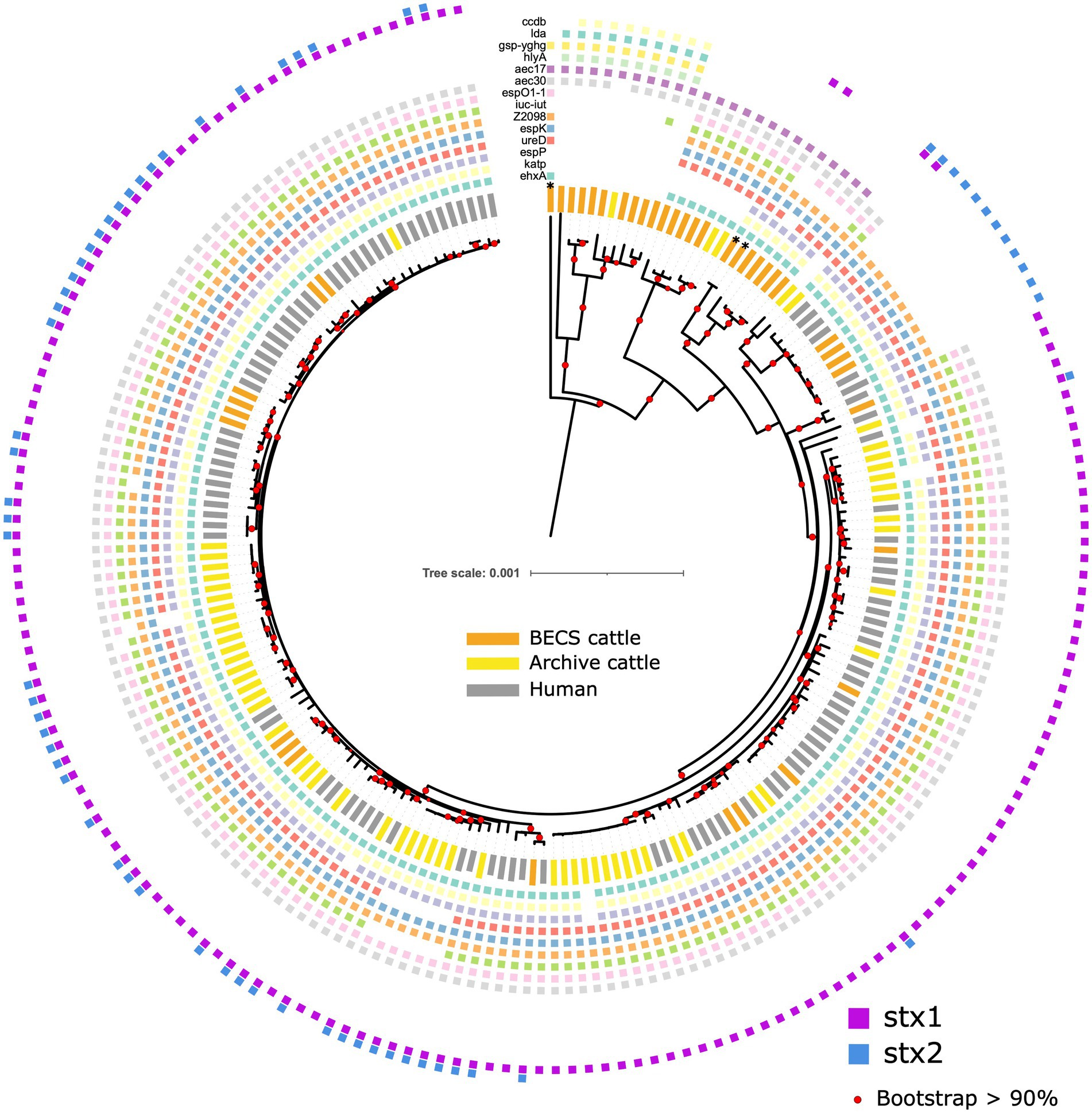
Figure 5. Maximum likelihood core-genome phylogenetic analysis of Scottish bovine and human O26:H11 strains (n = 195) and three O177:H11 strains (*), showing the presence of 14 selected key virulence genes or gene clusters. From inner to outer ring (ehxA, katP, espP, ureD, espK, Z2098, iuc-iut, espO1-1, aec30, aec17, hlyA, gsp-yghg, lda, and ccdb). Red circles indicate branches with >90% bootstrap support. Tree scale is in substitutions per site.
Other than stx subtype genes, the only gene specifically associated with stx-positive status was the iron regulatory protein 1 gene, irp1, detected in 98.9% (n = 171/173) of stx-positive strains, but not observed in stx-negative strains (Supplementary Table 7). In contrast, irp2 was detected in all strains, regardless of stx status. This is unusual, given that irp1 and irp2 are typically found together within a high-pathogenicity island. However, further analysis of a subset of these genomes using the Artemis genome browser did identify an irp1 variant allele in stx-negative strains, bearing a nine-base pair insertion sequence, which presumably reduced the alignment to below the set Abricate threshold parameters. The absence of irp1 in the two stx-positive strains was found to be due to a contig break within the gene. A further gene associated with stx-positivity was nleG5-1. This gene was found in 97.7% (n = 169/173) of stx-positive strains, but was detected in only 9.1% (n = 2/22) of stx-negative strains.
Strains that were positive for stx2-only were distinguished from all other stx profiles, including negative strains, by the absence of the type VI secretory system gene aec30. However, other aec subtypes such as aec17-19, 22, and 23 were either absent in all stx strains, but detected in the majority of stx-negatives, or in the case of subtypes aec24-29, detected across all categories. Additional genes that were absent in stx2-positive strains, but observed in all other stx-positive strains and in up to 50% of negative strains included the espO1-1, iuc and iut genes (Supplementary Table 7).
A non-metric multidimensional scaling (NMS) ordination model was constructed to examine potential associations between binary virulence gene occurrence, stx profile, and host species for all stx-positive O26:H11 genomes (n = 173) (Figure 6; Supplementary Table 8A). A three-dimensional solution to the model was obtained, which explained 83.5% of the variation (axis 1 = 40.1%, axis 2 = 27.3%, and axis 3 = 16.1%). The graph was rotated to maximise the distance between cattle and human strains on axis 1 (Supplementary Figure 2). Axis 2 is explained by stx1 (Kendall’s tau, −4.28) and stx1 + stx2 (Kendall’s tau, 0.465) strains. Stx2 was located primarily on axis 3 (Kendall’s tau, 0.312) (Figure 6). A multi-response permutation procedure test (MRPP) found significant differences between human and bovine for stx1 (bovine versus human, p = 0.002) and stx1 + stx2 (bovine versus human, p = 0.002). There were no differences for stx2-only strains between bovine and humans (MRPP, p = 0.158). Using Kendall’s tau as an indicator, most genes were not highly correlated with the NMS axes, though a weak to moderate association was observed for axis 3 and the non-LEE effector genes nleC and nleG2-4 (Supplementary Table 8B). These data suggest that for our population, whilst some differences exist in virulence background between the differing stx subtypes and host source within the majority ST21 lineage, it was not possible to attribute this to specific genes. Virulence profiles within the stx2a-only clade were more conserved than for the stx1 and stx1 + stx2 strains, which supports the phylogenetic analysis and observation that these strains have appeared in both Scottish cattle and human strain populations only relatively recently.
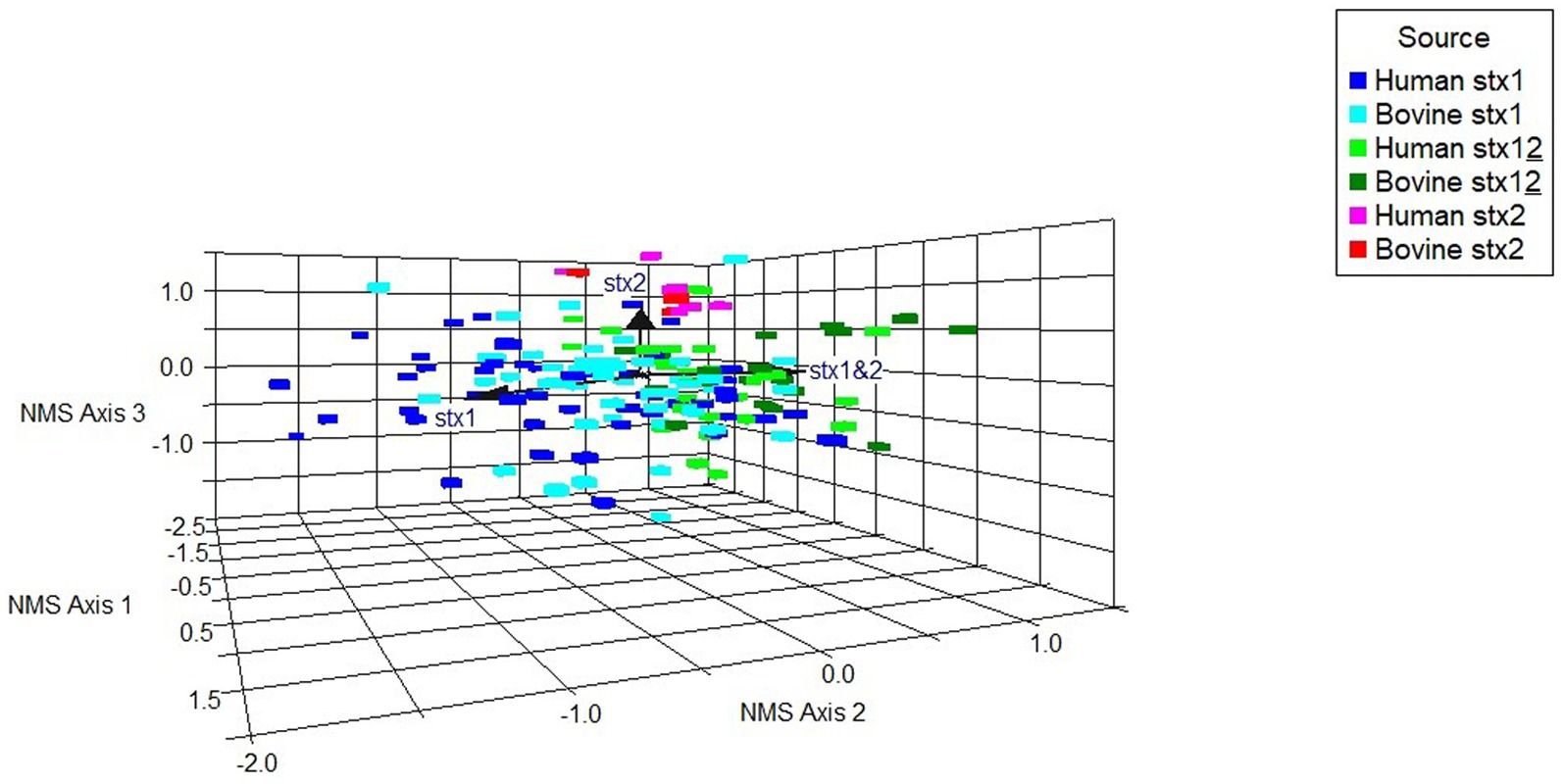
Figure 6. Non-metric multidimensional scaling ordination model to examine potential associations between a subset of differential gene occurrence for host source-stx profile combined (human, bovine; stx1: stx1, stx2: stx2, stx1 + stx2: stx12), for all stx-positive genomes (n = 173). Arrows indicate vector direction.
The 22 stx-negative strains from 19 herds were grouped into 2 core virulence profiles, “A” and “B,” across the ST29 and ST396 lineages, resulting in 3 distinct stx-negative populations overall (Figure 5; Supplementary Table 9).
The 12 “A” profile strains, which included all ST396 strains from four herds and ST29 strains from six herds, were located within the two clades phylogenetically closer to the stx-positive ST21 lineage. The majority of “A” strains carried ehxA, Z2201, and espO1-1, as well as the triplicate of genes ureD, espK, and Z2098, suggested as key markers for identifying E. coli with the potential for EHEC-type pathogenicity (Delannoy et al., 2013a). The presence of at least one of these three genes was always detected in all stx-positive bovine and human strains. ST396 is a less common ST variant of ST29, and all strains in this clade carried a greater complement of virulence genes, including the full ehxA+/katP+/espP+/etpD- pVF profile, as well as in all except one strain, the three espK, ureD, and Z2098 genes. ST396 stx2d-positive O26:H11 strains bearing the ehxA+/kapP+/espP+/etpD- profile have previously been reported in a minority of human HUS cases from Italy (Michelacci et al., 2022).
The virulence gene profile borne by these “A” profile stx-negative strains, together with the phylogenetic grouping, is consistent with an ST29C1 clade classification (Ogura et al., 2017; Long et al., 2022) and suggests that the strains are EHEC-like derivatives. The loss and acquisition of stx genes from O26 strains, both in vivo and in vitro, has been previously documented (Bielaszewska et al., 2007; Senthakumaran et al., 2018). Current diagnostic reliance on PCR testing for stx and eae genes only, may therefore potentially result in false-negative classification of EHEC strains that have lost the stx gene during laboratory isolation or within host. Our data concur with the proposal by Delannoy et al. (2013a) that additional genes, including ehxA, espK, ureD, and Z2098 should be included in diagnostic screening assays, and as shown here, are optimal gene markers for the identification of O26:H11 EHEC potential in livestock and animal products.
A further nine herds yielded 10 ST29 EPEC strains that were negative for ehxA and bore the virulence profile “B.” Distinguishing genes for this profile included b2972, the gsp cluster genes C-M, yghg, hlyA, lda(A-I), and ccdb (Figure 5; Supplementary Table 9). This virulence profile is consistent with the ST29C3 clade outlined by Long et al. (2022) and observed elsewhere (Leomil et al., 2005; Bugarel et al., 2011; Gonzalez-Escalona et al., 2016). Strains in this clade, whilst bearing a combination of virulence factors found across varying E. coli pathotypes (Kaper et al., 2004), do not appear to have the appropriate virulence background for EHEC pathogenicity following a potential recombination event with stx-bearing bacteriophage and are mostly represented by EPEC strains. The b2972 locus (pppA gene), yghG, and gsp(C-M) are located in a common gene cluster associated with the Type II secretion system found in both pathogenic and non-pathogenic E. coli strains (Tauschek et al., 2002; Yang et al., 2007). The pppA and yghG genes are also associated with the regulation of heat-labile (LT) toxin (Strozen et al., 2012; Lu et al., 2016; Wang et al., 2020). hlyA encodes α-haemolysin, an important cytotoxin found in uropathogenic E. coli (Ristow and Welch, 2016), whilst ccdb encodes a cytotoxin present in the toxin-antitoxin system targeting E. coli gyrase and is carried by the F plasmid (Bernard and Couturier, 1992). The lda genes, present in the locus of diffuse adherence, encode adhesins associated with atypical EPEC and have also been reported in an O26:H11 paediatric clinical strain (Scaletsky et al., 2005).
One herd (BECS_2) yielded both ST21 stx1a + stx2a-positive and ST396 stx-negative strains bearing very similar key virulence genes, including ehxA+/katP+/espP+/etpD-, though missing nleG-3, nleG2-4, nleG5-1, and fimB. This herd also yielded an ST29 O177:H11 stx-negative strain, which, whilst ehxA positive, was katP and espP negative. In contrast, a second herd (BECS_27) yielded both stx1a and stx-negative O26:H11 strains bearing the “B” virulence profile. A third herd (BECS_18) generated an ST29 O26:H11 group “A” stx-negative strain and an O177:H11 stx1a strain, both of similar virulence profiles, though locating to different clades within the phylogeny.
The stx gene is encoded by mobile bacteriophages, which integrates into the bacterial host genome at particular chromosomal insertion sites (Shaikh and Tarr, 2003; Rodríguez-Rubio et al., 2021). Insertion occurs within or adjacent to the host insertion site gene and typically causes disruption to the insertion site gene sequence. A number of integration sites have been identified for O26:H11 STEC, including the yecE, wrbA, yehV, and sbcB genes (Bonanno et al., 2015). In order to examine whether there was any evidence for the insertion of stx-phage at these sites across the collection of strains in this study, we compared the gene sequence identity obtained from the Abricate output with the presence and absence of stx and the stx subtype. We observed variability in nucleotide sequence percentage identity in the stx bacteriophage insertion site genes yecE and yehV, according to the stx status (Figure 7; Supplementary Table 10). yehV is one of the main insertion sites in O26:H11 for the stx1 bacteriophage and yecE for stx2 (Bonanno et al., 2015). For yecE, variation in nucleotide percentage identity was significantly associated with the presence of the stx2 gene (p < 0.001, test statistic 196, degrees of freedom 3), with 97% (64/66) of stx2-positive genomes showing a 90.6% identity and 98% (126/129) of stx1-only and stx-negative genomes showing 100% identity to yecE. In contrast, significant variation in percent identity for yehV appeared to be associated with stx1 status (p < 0.001, test statistic 90.8, degrees of freedom 6), with the majority of the ST21 stx1-positive genomes, as well as a subset of stx-negative ST29, showing 94.13% identity to yehV. There was limited variability in percent identity across the genomes for wrbA and sbcB: 99% of genomes showed 96.37% identity to wrbA (193/195) and 100% identity to sbcB (194/195).
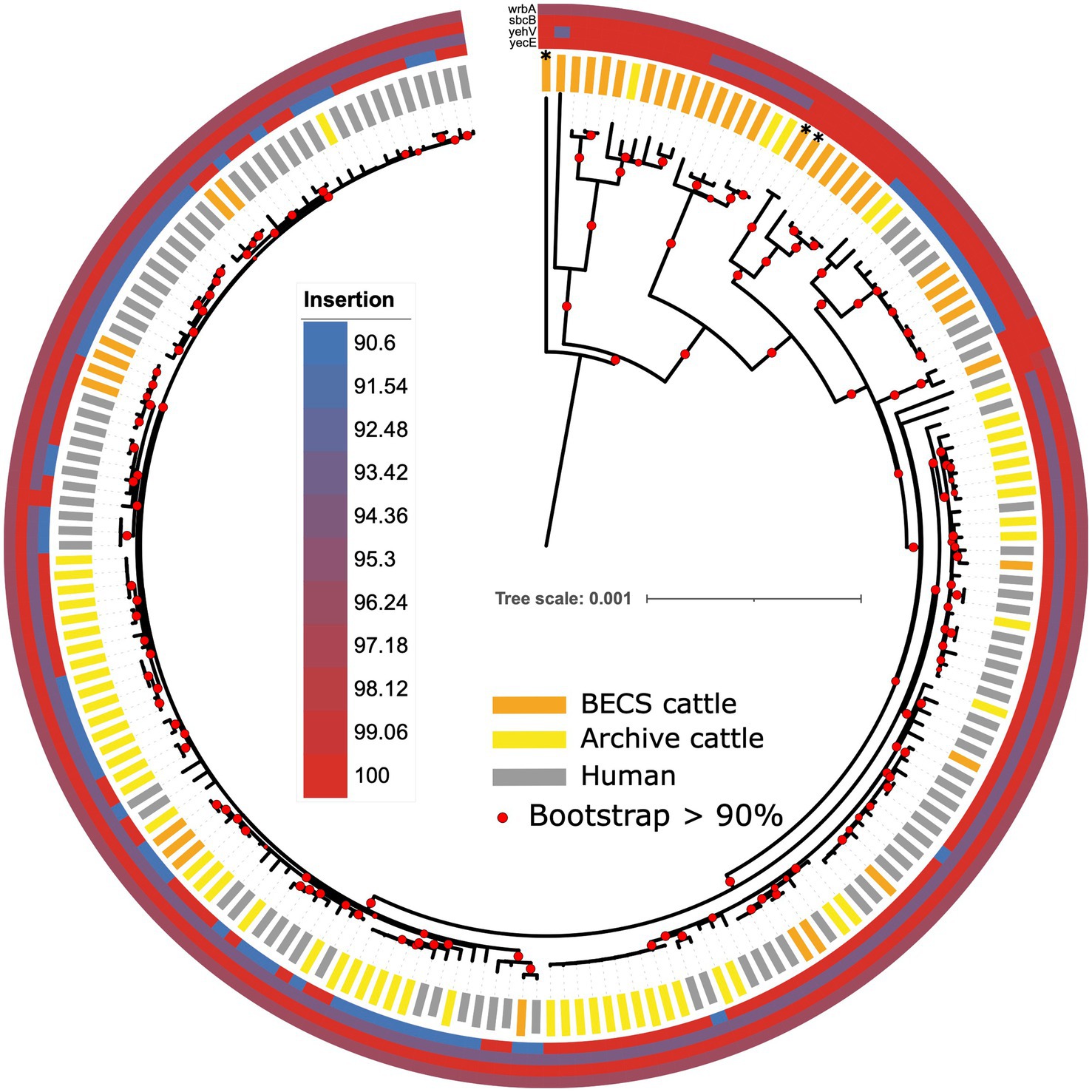
Figure 7. Maximum likelihood core-genome phylogenetic analysis of Scottish bovine and human O26:H11 strains (n = 195) and three O177:H11 strains (*) showing the sequence conservation of four known phage insertion sites (yecE, yehV, sbcB, and wrbA). Red circles indicate branches with >90% bootstrap support. Tree scale is in substitutions per site.
The individual insertion site gene size, sequence, and gene arrangements in the vicinity of the yecE and yehV genes were examined for a subset of bovine strains across the different stx categories using the Artemis genome viewer (Carver et al., 2012; Supplementary Table 1). In the stx-negative and stx1-positive genomes examined, yecE was 819 base pairs (bp) in size and was located within the consecutive gene sequences yecD, yecE, yecN, cmoA, and cmoB. In contrast, the four stx2-only positive bovine strains contained a truncated 111-bp fragment directly located next to the yecD gene, the latter immediately adjacent to a contig break. The truncated 111-bp fragment had 100% homology to bases 1–96 of the full-length yecE. A further 816 bp gene was located on an alternate contig at a different location within the genome, adjacent to the yecN, cmoA, and cmoB genes, in most cases flanked by an integrase gene. This 816 bp gene showed 0% homology to yecE between bases 1–70 and 99% homology between bases 71–819. All 13 stx1 + stx2 strains examined also showed a truncated 816 bp yecE gene adjacent to an integrase gene, and seven of these additionally bore the 111-bp fragment. This suggests the potential occurrence of an integration event at the yecE site, resulting in the disruption of this gene in the stx2-positive strains, which was not observed in the stx1-only and stx-negative strains.
We examined the yehV (mlrA), gene length, and arrangement in 6 stx1-positive, 13 stx1 + stx2, and 22 stx-negative cattle O26:H11 genomes. All stx1-positive, 11/13 stx1 + stx2 and the 7 “A” profile ST29 stx-negative genomes showed <100% homology to yehV and contained a 648 bp gene of 94.13% identity to yehV, flanked in all cases by yehW and the integrase IntQ_1 or IntQ_2 genes (Hall et al., 2021). The presence of the truncated yehV gene flanked by integrase intQ genes is highly suggestive of a phage insertion event in these 7 “A” profile ST29 stx-negative strains. In contrast, 9/10 stx-negative ST29 profile “B” strains with 100% homology to yehV, as well as the three O177:H11 strains, contained a full-length 732 bp yehV (mlrA) gene, flanked by yehW and the sensory histidine kinase gene ypdA-1. One “B” profile ST29 stx-negative strain contained two smaller gene fragments, and two stx1 + stx2 strains bore the full-length gene.
The ST396 “A” profile stx-negative strains carried a greater complement of virulence genes than the ST29 stx-negative strains. However, the yehV local gene arrangement in these ST396 strains did not show evidence of phage integration or interruption, with an intact 732 bp yehV gene. We examined the gene arrangement in these strains at another potential bacteriophage insertion site, the torS-T intergenic region (González-Escalona et al., 2019); however, this region was uninterrupted in all the ST396 strains.
All stx-negative cattle genomes were submitted to PHASTER for the identification of any stx-prophage regions; however, the results were inconclusive (Supplementary Table 9). Both intact and incomplete prophage regions with homology to stx-prophage as the first or second-listed most common phage were identified in the majority of the stx-negative strains. Due to the limitations of short-read genome assembly, long-read sequencing would be required to confirm the presence of any inserted prophage in stx-negative strains. However, given the distinct gene arrangement in the locality of yehV, together with the virulence background present in the “A” profile strains, it would seem probable that they had either the potential for acquiring stx or had previously been stx-positive and subsequently lost the stx gene from an integrated prophage.
Acquired antimicrobial resistance genes (ARGs) were identified in silico for 12.1% (12/99) of the O26:H11 bovine strains, 15.6% of the cattle herds (10/64) (Table 2). The commonest genes detected were sul2 and tet, found in six herds; aph(3″) and aph(6″) in five herds; and blaTEM and dfrA, each detected in four herds. Three-quarters of resistant strains (9/12) were positive for more than one ARG (median 4.5, range 1–5), with the commonest combination being aph(3″), aph(6″), together with one or more further ARGs. Seventeen human strains (17.7%) carried ARGs (Table 2), with the commonest being aph(3″) and aph(6″) in 13 strains, followed by sul2 and floR in eight strains. Multiple resistances were also common in human strains (median 4, range 1–9).
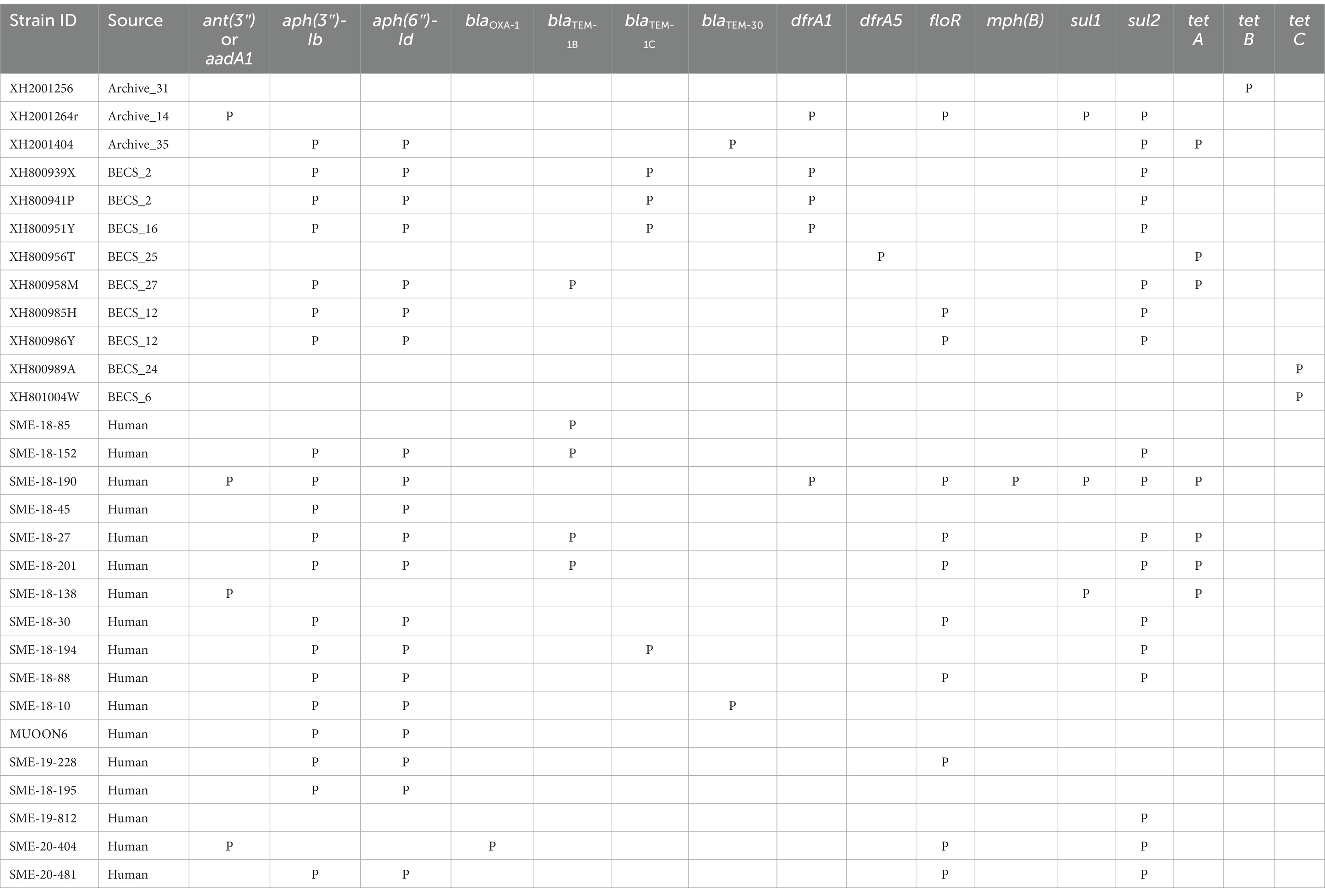
Table 2. Summary of antimicrobial resistance genes detected in the Scottish bovine and human O26:H11 genomes by STARAMR and ResFinder databases, where P indicates genome positive for respective genes.
Strains carrying resistance were screened for the presence of mobile genetic elements (Supplementary Table 11). One bovine Archive genome carried a clinical class 1 integron with a typical cassette arrangement of 5′-intI1, dfrA1, aadA1, qacEΔ1, and sul1-3′ with an additional two genes, floR and sul2, found in close proximity on the same contig. Two multiply-resistant human strains also carried clinical class 1 integrons. Across all genomes, blaTEM genes were found on transposon Tn2 in six cases, and in cattle, dfrA1 was associated with the composite transposon cn_4568_IS26 in two herds.
The resistome seen in the bovine isolates was very similar to that observed in the Scottish human dataset, with the commonest ARGs conferring resistance to streptomycin and spectinomycin aminoglycosides, sulphonamides, tetracyclines, and beta-lactam agents such as ampicillin, the latter class designated as critically important antimicrobials for human health by the World Health Organization (2019). The proportion of the bovine strains carrying ARGs is in line with that reported in a collection of O26:H11 strains from home (non-travel-associated) human STEC O26 cases from England and Wales isolated during 2015 (Day et al., 2017), but slightly lower than described in a more recent report on human case clonal complex 29 STEC isolates in England between 2014 and 2021 (Rodwell et al., 2023). Our results differ markedly from the very high AMR prevalence reported in a collection of O26:H11 strains from feedlot cattle in the United States (Gonzalez-Escalona et al., 2016); however, this likely reflects differences in the management systems and associated antimicrobial usage of the livestock systems between the two countries.
Overall, these data are in accordance with current antimicrobial usage observed within the bovine sector in the UK, with beta-lactams, tetracycline, and streptomycin being the most frequently prescribed antimicrobials in both beef and dairy cattle (Humphry et al., 2021; RUMA, 2021). Given ruminants are the primary reservoir source for human infection within the UK and antimicrobial therapy is generally not indicated in human STEC infection (Tarr and Freedman, 2022), this agreement in the resistance profile between the Scottish cattle and human genomes is not unexpected.
In conclusion, within the study herds, all stx-positive cattle O26:H11 strains fell within the ST21 lineage, and no ST29 stx-positive strains were identified. Bovine and clinical human strain genomes were relatively well interspersed, with the stx subtype generally clade-specific. Highly pathogenic stx2a-only ST21 was identified in two herds from the second cattle survey and in human strains from 2010 onwards. Where multiple strains were available from individual herds, we observed limited variability within the stx subtype, suggesting that the same stx subtype strains typically spread clonally at the farm level rather than supporting multiple lineage introductions across a cohort. Half of the stx-negative survey herds yielded O26:H11 strains with virulence profiles similar to those observed in stx-positive strains, including the genes ehxA, espK, and Z2098, which have been proposed as markers for “EHEC-like” potential. These data suggest that the reservoir of O26:H11 in Scottish cattle bearing a genomic background compatible with EHEC potential and therefore of public health concern may be greater than would be expected based on the detection of the STEC markers stx and eae alone.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ebi.ac.uk/ena, PRJEB57355; https://www.ncbi.nlm.nih.gov/, PRJNA419720.
The studies involving humans were approved by the biorepository bank for the National Health Service (NHS) Lothian, Scotland, covering the sequencing of samples and use of de-identified bacterial genomes from human clinical samples (20/ES/0061). The studies were conducted in accordance with the local legislation and institutional requirements. The human samples used in this study were acquired from a by- product of routine care or industry. Written informed consent for participation was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and institutional requirements. Ethical approval was not required for the studies involving animals in accordance with the local legislation and institutional requirements, because this study uses only strains from cattle faecal pat samples obtained from the BECS study doi: 10.1017/S0950268817002151 and from the national survey reported in doi: 10.1128/AEM.72.1.653-659.2006 conducted by Scotland’s Rural College (SRUC). Scotland’s Rural College did not require the study to be reviewed or approved by an ethics committee as per the local legislation where the studies were conducted, because samples were industry by-products (cattle faecal pats) collected from the ground of grazing land and the floor of pens, which involved no animal contact or intervention. The funding bodies approved and authorised informed consent documentation for farm survey participants, to enable collection of dropped faecal pat samples from participant land. Permission had been granted and written consent obtained from farm participants for the samples, strains and data to be used for further research. All farm participant personal data were handled in accordance with the UK Data Protection Act (1998) and are now handled in accordance with the UK General Data Protection Regulation (GDPR 2018).
DH: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Project administration, Writing – original draft, Writing – review & editing. BW: Data curation, Formal analysis, Visualization, Writing – original draft, Writing – review & editing, Investigation. KM: Investigation, Writing – review & editing. MC-T: Formal analysis, Writing – review & editing. AB: Investigation, Writing – review & editing. ST: Resources, Writing – review & editing. DG: Conceptualization, Writing – review & editing. SD: Writing – review & editing. PF: Writing – review & editing. MP: Writing – review & editing. GG: Resources, Writing – review & editing. AH: Conceptualization, Data curation, Investigation, Resources, Writing – original draft, Writing – review & editing. LA: Conceptualization, Data curation, Investigation, Resources, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was funded by the Wellcome Trust through a personal fellowship to DH (105832/Z/14/Z). Additional resources to DH and part funding for BW were provided through an Institute Strategic Programme grant from the Biotechnology and Biological Sciences Research Council (BBS/E/D/20002173). BW was also part-funded by the Novo Nordisk Foundation Fund (Grant: NNF16OC0021856: Global Surveillance of Antimicrobial Resistance).
The authors thank Sharif Shaaban for assisting with the initial ChewBBACA comparison of clinical and cattle genomes and Stella Mazeri for assisting with R script provision for figure generation. The authors are grateful to the staff of the Scottish O157/STEC Reference Laboratory for laboratory assistance and to all the farmers who participated in the original Archive and BECS cattle surveys. For the purpose of open access, the author has applied a CC-BY public copyright licence to any author-accepted manuscript version arising from this submission.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1260422/full#supplementary-material
1. ^ https://enterobase.warwick.ac.uk/schemes/Escherichia.cgMLSTv1
2. ^ https://github.com/phac-nml/ecoli_vf
3. ^ https://cge.food.dtu.dk/services/MobileElementFinder/
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Ashton, P. M., Perry, N., Ellis, R., Petrovska, L., Wain, J., Grant, K. A., et al. (2015). Insight into Shiga toxin genes encoded by Escherichia coli O157 from whole genome sequencing. PeerJ 3:e739. doi: 10.7717/peerj.739
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bernard, P., and Couturier, M. (1992). Cell killing by the F plasmid CcdB protein involves poisoning of DNA-topoisomerase II complexes. J. Mol. Biol. 226, 735–745. doi: 10.1016/0022-2836(92)90629-X
Bharat, A., Petkau, A., Avery, B. P., Chen, J. C., Folster, J. P., Carson, C. A., et al. (2022). Correlation between phenotypic and in silico detection of antimicrobial resistance in Salmonella enterica in Canada using Staramr. Microorganisms 10:292. doi: 10.3390/microorganisms10020292
Bielaszewska, M., Mellmann, A., Bletz, S., Zhang, W., Köck, R., Kossow, A., et al. (2013). Enterohemorrhagic Escherichia coli O26:H11/H-: a new virulent clone emerges in Europe. Clin. Infect. Dis. 56, 1373–1381. doi: 10.1093/cid/cit055
Bielaszewska, M., Prager, R., Köck, R., Mellmann, A., Zhang, W., Tschäpe, H., et al. (2007). Shiga toxin gene loss and transfer in vitro and in vivo during enterohemorrhagic Escherichia coli O26 infection in humans. ASM J. CD 73, 3144–3150. doi: 10.1128/AEM.02937-06
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Bonanno, L., Loukiadis, E., Mariani-Kurkdjian, P., Oswald, E., Garnier, L., Michel, V., et al. (2015). Diversity of Shiga toxin-producing Escherichia coli (STEC) O26:H11 strains examined via stx subtypes and insertion sites of stx and espk bacteriophages. Appl. Environ. Microbiol. 81, 3712–3721. doi: 10.1128/AEM.00077-15
Browne, A. S., Biggs, P. J., Wilkinson, D. A., Cookson, A. L., Midwinter, A. C., Bloomfield, S. J., et al. (2019). Use of genomics to investigate historical importation of Shiga toxin-producing Escherichia coli serogroup O26 and nontoxigenic variants into New Zealand. Emerg. Infect. Dis. 25, 489–500. doi: 10.3201/eid2503.180899
Browne, A. S., Midwinter, A. C., Withers, H., Cookson, A. L., Biggs, P. J., Marshall, J. C., et al. (2018). Molecular epidemiology of Shiga toxin-producing Escherichia coli (STEC) on New Zealand dairy farms: application of a culture-independent assay and whole-genome sequencing. Appl. Environ. Microbiol. 84:84. doi: 10.1128/aem.00481-18
Bugarel, M., Beutin, L., Scheutz, F., Loukiadis, E., and Fach, P. (2011). Identification of genetic markers for differentiation of Shiga toxin-producing, enteropathogenic, and avirulent strains of Escherichia coli O26. Appl. Environ. Microbiol. 77, 2275–2281. doi: 10.1128/AEM.02832-10
Bushnell, B. (2019). BBMap. Available at: sourceforge.net/projects/bbmap/
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Caprioli, A., Morabito, S., Brugère, H., and Oswald, E. (2005). Enterohaemorrhagic Escherichia coli: emerging issues on virulence and modes of transmission. Vet. Res. 36, 289–311. doi: 10.1051/vetres:2005002
Carver, T., Harris, S. R., Berriman, M., Parkhill, J., and McQuillan, J. A. (2012). Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469. doi: 10.1093/bioinformatics/btr703
Chattaway, M. A., Schaefer, U., Tewolde, R., Dallman, T. J., and Jenkins, C. (2017). Identification of Escherichia coli and Shigella species from whole-genome sequences. J. Clin. Microbiol. 55, 616–623. doi: 10.1128/JCM.01790-16
Colello, R., Krüger, A., Velez, M. V., del Canto, F., Etcheverría, A. I., Vidal, R., et al. (2019). Identification and detection of iha subtypes in LEE-negative Shiga toxin-producing Escherichia coli (STEC) strains isolated from humans, cattle and food. Heliyon 5:e03015. doi: 10.1016/j.heliyon.2019.e03015
Croucher, N. J., Page, A. J., Connor, T. R., Delaney, A. J., Keane, J. A., Bentley, S. D., et al. (2015). Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res. 43:e15. doi: 10.1093/nar/gku1196
Dallman, T. J., Byrne, L., Ashton, P. M., Cowley, L. A., Perry, N. T., Adak, G., et al. (2015). Whole-genome sequencing for national surveillance of Shiga toxin-producing Escherichia coli O157. Clin. Infect. Dis. 61, 305–312. doi: 10.1093/cid/civ318
Dallman, T. J., Greig, D. R., Gharbia, S. E., and Jenkins, C. (2021). Phylogenetic context of Shiga toxin-producing Escherichia coli serotype O26:H11 in England. Microb. Genome 7:7. doi: 10.1099/mgen.0.000551
Day, M., Doumith, M., Jenkins, C., Dallman, T. J., Hopkins, K. L., Elson, R., et al. (2017). Antimicrobial resistance in Shiga toxin-producing Escherichia coli serogroups O157 and O26 isolated from human cases of diarrhoeal disease in England, 2015. J. Antimicrob. Chemother. 72, 145–152. doi: 10.1093/jac/dkw371
Delannoy, S., Beutin, L., and Fach, P. (2013a). Discrimination of enterohemorrhagic Escherichia coli (EHEC) from non-EHEC strains based on detection of various combinations of type III effector genes. J. Clin. Microbiol. 51, 3257–3262. doi: 10.1128/JCM.01471-13
Delannoy, S., Beutin, L., and Fach, P. (2013b). Towards a molecular definition of enterohemorrhagic Escherichia coli (EHEC): detection of genes located on O Island 57 as markers to distinguish EHEC from closely related enteropathogenic E. coli strains. J. Clin. Microbiol. 51, 1083–1088. doi: 10.1128/JCM.02864-12
Delannoy, S., Mariani-Kurkdjian, P., Bonacorsi, S., Liguori, S., and Fach, P. (2015). Characteristics of emerging human-pathogenic Escherichia coli O26:H11 strains isolated in France between 2010 and 2013 and carrying the stx2d gene only. J. Clin. Microbiol. 53, 486–492. doi: 10.1128/JCM.02290-14
Denamur, E., Clermont, O., Bonacorsi, S., and Gordon, D. (2021). The population genetics of pathogenic Escherichia coli. Nat. Rev. Microbiol. 19, 37–54. doi: 10.1038/s41579-020-0416-x
Disty McMatrixface (2021). Available at: https://github.com/c2-d2/disty (Accessed December 13, 2022).
Escherichia coli Virulence Factors (2021). Available at: https://github.com/phac-nml/ecoli_vf (Accessed December 13, 2022).
European Centre for Disease Prevention and Control (2022). Shiga toxin-producing Escherichia coli (STEC) infection – annual epidemiological report for 2020. Available at: https://www.ecdc.europa.eu/en/publications-data/shiga-toxin-producing-escherichia-coli-stec-infection-annual-epidemiological-0 (Accessed December 12, 2022).
Florensa, A. F., Kaas, R. S., Clausen, P. T. L. C., Aytan-Aktug, D., and Aarestrup, F. M. (2022). ResFinder – an open online resource for identification of antimicrobial resistance genes in next-generation sequencing data and prediction of phenotypes from genotypes. Microb. Genomics 8:748. doi: 10.1099/mgen.0.000748
Food Standards Scotland (2020). Whole genome sequence typing and analysis of non-O157 STEC. Available at: https://www.foodstandards.gov.scot/publications-and-research/publications/whole-genome-sequence-typing-and-analysis-of-non-o157-stec (Accessed May 14, 2021).
Fratamico, P. M., Yan, X., Caprioli, A., Esposito, G., Needleman, D. S., Pepe, T., et al. (2011). The complete DNA sequence and analysis of the virulence plasmid and of five additional plasmids carried by Shiga toxin-producing Escherichia coli O26:H11 strain H30. Int. J. Med. Microbiol. 301, 192–203. doi: 10.1016/j.ijmm.2010.09.002
Friedrich, A. W., Bielaszewska, M., Zhang, W.-L., Pulz, M., Kuczius, T., Ammon, A., et al. (2002). Escherichia coli harboring Shiga toxin 2 gene variants: frequency and association with clinical symptoms. J. Infect. Dis. 185, 74–84. doi: 10.1086/338115
Gill, A., Dussault, F., McMahon, T., Petronella, N., Wang, X., Cebelinski, E., et al. (2022). Characterization of atypical Shiga toxin gene sequences and description of Stx2j, a new subtype. J. Clin. Microbiol. 60:e0222921. doi: 10.1128/jcm.02229-21
González-Escalona, N., Allard, M. A., Brown, E. W., Sharma, S., and Hoffmann, M. (2019). Nanopore sequencing for fast determination of plasmids, phages, virulence markers, and antimicrobial resistance genes in Shiga toxin-producing Escherichia coli. PLoS One 14:e0220494. doi: 10.1371/journal.pone.0220494
Gonzalez-Escalona, N., Toro, M., Rump, L. V., Cao, G., Nagaraja, T. G., and Meng, J. (2016). Virulence gene profiles and clonal relationships of Escherichia coli O26:H11 isolates from feedlot cattle as determined by whole-genome sequencing. Appl. Environ. Microbiol. 82, 3900–3912. doi: 10.1128/AEM.00498-16
Hall, R. J., Whelan, F. J., Cummins, E. A., Connor, C., McNally, A., and McInerney, J. O. (2021). Gene-gene relationships in an Escherichia coli accessory genome are linked to function and mobility. Microb. Genomics 7:000650. doi: 10.1099/mgen.0.000650
Henry, M. K., Tongue, S. C., Evans, J., Webster, C., Mckendrick, I. J., Morgan, M., et al. (2017). British Escherichia coli O157 in cattle study (BECS): to determine the prevalence of E. coli O157 in herds with cattle destined for the food chain. Epidemiol. Infect. 145, 3168–3179. doi: 10.1017/S0950268817002151
Hoyle, D. V., Keith, M., Williamson, H., Macleod, K., Mathie, H., Handel, I., et al. (2021). Prevalence and epidemiology of non-O157 Escherichia coli serogroups O26, O103, O111, and O145 and Shiga toxin gene carriage in Scottish cattle, 2014–2015. Appl. Environ. Microbiol. 87:e03142-20. doi: 10.1128/AEM.03142-20
Hughes, A. C., Zhang, Y., Bai, X., Xiong, Y., Wang, Y., Yang, X., et al. (2019). Structural and functional characterization of Stx2k, a new subtype of Shiga toxin 2. Microorganisms 8:4. doi: 10.3390/microorganisms8010004
Humphry, R. W., Henry, M. K., Reeves, A., Correia-Gomes, C., Innocent, G. T., Smith, R., et al. (2021). Estimating antimicrobial usage based on sales to beef and dairy farms from UK veterinary practices. Vet. Rec. 189:e28. doi: 10.1002/vetr.28
Ishijima, N., Lee, K. I., Kuwahara, T., Nakayama-Imaohji, H., Yoneda, S., Iguchi, A., et al. (2017). Identification of a new virulent clade in enterohemorrhagic Escherichia coli O26:H11/H- sequence type 29. Sci. Rep. 7:43136. doi: 10.1038/srep43136
Johansson, M. H. K., Bortolaia, V., Tansirichaiya, S., Aarestrup, F. M., Roberts, A. P., and Petersen, T. N. (2021). Detection of mobile genetic elements associated with antibiotic resistance in Salmonella enterica using a newly developed web tool: MobileElementFinder. J. Antimicrob. Chemother. 76, 101–109. doi: 10.1093/jac/dkaa390
Johnson, K. E., Thorpe, C. M., and Sears, C. L. (2006). The emerging clinical importance of non-O157 Shiga toxin-producing Escherichia coli. Clin. Infect. Dis. 43, 1587–1595. doi: 10.1086/509573
Jones, G., Lefèvre, S., Donguy, M. P., Nisavanh, A., Terpant, G., Fougère, E., et al. (2019). Outbreak of Shiga toxin-producing Escherichia coli (STEC) O26 paediatric haemolytic uraemic syndrome (HUS) cases associated with the consumption of soft raw cow’s milk cheeses, France, March to May 2019. Euro Surveill. 24:1900305. doi: 10.2807/1560-7917.ES.2019.24.22.1900305
Kaper, J. B., Nataro, J. P., and Mobley, H. L. T. (2004). Pathogenic Escherichia coli. Nat. Rev. Microbiol. 2, 123–140. doi: 10.1038/nrmicro818
Karnisova, L., Marejkova, M., Hrbackova, H., Mellmann, A., Karch, H., Fruth, A., et al. (2018). Attack of the clones: whole genome-based characterization of two closely related enterohemorrhagic Escherichia coli O26 epidemic lineages. BMC Genomics 19:647. doi: 10.1186/s12864-018-5045-7
Kintz, E., Brainard, J., Hooper, L., and Hunter, P. (2017). Transmission pathways for sporadic Shiga-toxin producing E. coli infections: a systematic review and meta-analysis. Int. J. Hyg. Environ. Health 220, 57–67. doi: 10.1016/j.ijheh.2016.10.011
Kolenda, R., Burdukiewicz, M., and Schierack, P. (2015). A systematic review and meta-analysis of the epidemiology of pathogenic Escherichia coli of calves and the role of calves as reservoirs for human pathogenic E. coli. Front. Cell. Infect. Microbiol. 5:23. doi: 10.3389/fcimb.2015.00023
Langmead, B., Trapnell, C., Pop, M., and Salzberg, S. L. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. doi: 10.1186/gb-2009-10-3-r25
Leomil, L., Pestana de Castro, A. F., Krause, G., Schmidt, H., and Beutin, L. (2005). Characterization of two major groups of diarrheagenic Escherichia coli O26 strains which are globally spread in human patients and domestic animals of different species. FEMS Microbiol. Lett. 249, 335–342. doi: 10.1016/j.femsle.2005.06.030
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Loconsole, D., Giordano, M., Centrone, F., Accogli, M., Casulli, D., de Robertis, A. L., et al. (2020). Epidemiology of Shiga toxin-producing Escherichia coli infections in Southern Italy after implementation of symptom-based surveillance of bloody diarrhea in the pediatric population. Int. J. Environ. Res. Public Health 17:17. doi: 10.3390/ijerph17145137
Long, J., Geng, J., Xu, Y., Jin, Y., Yang, H., Xi, Y., et al. (2022). Large-scale phylogenetic analysis reveals a new genetic clade among Escherichia coli O26 strains. Microbiol Spectr 10, e02525–e02521. doi: 10.1128/spectrum.02525-21
Lu, X., Fu, E., Xie, Y., and Jin, F. (2016). Electron acceptors induce secretion of enterotoxigenic Escherichia coli heat-labile enterotoxin under anaerobic conditions through promotion of GspD assembly. Infect. Immun. 84, 2748–2757. doi: 10.1128/IAI.00358-16
Michelacci, V., Montalbano Di Filippo, M., Gigliucci, F., Arancia, S., Chiani, P., Minelli, F., et al. (2022). Population analysis of O26 Shiga toxin-producing Escherichia coli causing hemolytic uremic syndrome in Italy, 1989-2020, through whole genome sequencing. Front. Cell. Infect. Microbiol. 12:842508. doi: 10.3389/fcimb.2022.842508
Minh, B. Q., Schmidt, H. A., Chernomor, O., Schrempf, D., Woodhams, M. D., von Haeseler, A., et al. (2020). IQ-TREE 2: new models and efficient methods for phylogenetic inference in the genomic era. Mol. Biol. Evol. 37, 1530–1534. doi: 10.1093/molbev/msaa015
Nataro, J. P., and Kaper, J. B. (1998). Diarrheagenic Escherichia coli. Clin. Microbiol. Rev. 11, 142–201. doi: 10.1128/CMR.11.1.142
Newton, H. J., Sloan, J., Bulach, D. M., Seemann, T., Allison, C. C., Tauschek, M., et al. (2009). Shiga toxin–producing Escherichia coli strains negative for locus of enterocyte effacement. Emerg. Infect. Dis. 15, 372–380. doi: 10.3201/eid1502.080631
Ogura, Y., Gotoh, Y., Itoh, T., Sato, M. P., Seto, K., Yoshino, S., et al. (2017). Population structure of Escherichia coli O26:H11 with recent and repeated stx2 acquisition in multiple lineages. Microb. Genome 3:3. doi: 10.1099/mgen.0.000141
Parsons, B. D., Zelyas, N., Berenger, B. M., and Chui, L. (2016). Detection, characterization, and typing of Shiga toxin-producing Escherichia coli. Front. Microbiol. 7:478. doi: 10.3389/fmicb.2016.00478
Pearce, M. C., Chase-Topping, M. E., McKendrick, I. J., Mellor, D. J., Locking, M. E., Allison, L., et al. (2009). Temporal and spatial patterns of bovine Escherichia coli O157 prevalence and comparison of temporal changes in the patterns of phage types associated with bovine shedding and human E. coli O157 cases in Scotland between 1998-2000 and 2002-2004. BMC Microbiol. 9:276. doi: 10.1186/1471-2180-9-276
Pearce, M. C., Evans, J., McKendrick, I. J., Smith, A. W., Knight, H. I., Mellor, D. J., et al. (2006). Prevalence and virulence factors of Escherichia coli serogroups O26, O103, O111, and O145 shed by cattle in Scotland. ASM J. CD 72, 653–659. doi: 10.1128/AEM.72.1.653-659.2006
Persson, S., Olsen, K. E. P., Ethelberg, S., and Scheutz, F. (2007). Subtyping method for Escherichia coli Shiga toxin (Verocytotoxin) 2 variants and correlations to clinical manifestations. J. Clin. Microbiol. 45, 2020–2024. doi: 10.1128/JCM.02591-06
Public Health Scotland (2020). STEC in Scotland, 2019. Enhanced surveillance and reference laboratory data - STEC in Scotland: enhanced surveillance and reference laboratory data. Available at: https://publichealthscotland.scot/publications/stec-in-scotland-enhanced-surveillance-and-reference-laboratory-data/stec-in-scotland-2019-enhanced-surveillance-and-reference-laboratory-data/ (Accessed December 12, 2022).
Pukatzki, S., McAuley, S. B., and Miyata, S. T. (2009). The type VI secretion system: translocation of effectors and effector-domains. Curr. Opin. Microbiol. 12, 11–17. doi: 10.1016/j.mib.2008.11.010
Ristow, L. C., and Welch, R. A. (2016). Hemolysin of uropathogenic Escherichia coli: a cloak or a dagger? Biochim. Biophys. Acta BBA 1858, 538–545. doi: 10.1016/j.bbamem.2015.08.015
Rodríguez-Rubio, L., Haarmann, N., Schwidder, M., Muniesa, M., and Schmidt, H. (2021). Bacteriophages of Shiga toxin-producing Escherichia coli and their contribution to pathogenicity. Pathogens 10:404. doi: 10.3390/pathogens10040404
Rodwell, E. V., Simpson, A., Chan, Y.-W., Godbole, G., McCarthy, N. D., and Jenkins, C. (2023). The epidemiology of Shiga toxin-producing Escherichia coli O26:H11 (clonal complex 29) in England, 2014–2021. J. Infect. 86, 552–562. doi: 10.1016/j.jinf.2023.04.006
RUMA (2021). Antibiotic use in cattle: dairy trend data and usage by beef farm type 2015-2019. RUMA. Available at: https://ruma.org.uk/wp-content/uploads/2022/07/Antibiotic-Use-in-936Cattle-Report.pdf (Accessed July 11, 2023).
Scaletsky, I. C. A., Michalski, J., Torres, A. G., Dulguer, M. V., and Kaper, J. B. (2005). Identification and characterization of the locus for diffuse adherence, which encodes a novel afimbrial adhesin found in atypical enteropathogenic Escherichia coli. Infect. Immun. 73, 4753–4765. doi: 10.1128/IAI.73.8.4753-4765.2005
Scheutz, F., Teel, L. D., Beutin, L., Piérard, D., Buvens, G., Karch, H., et al. (2012). Multicenter evaluation of a sequence-based protocol for subtyping Shiga toxins and standardizing Stx nomenclature. J. Clin. Microbiol. 50, 2951–2963. doi: 10.1128/JCM.00860-12
Seemann, T. (2022a). ABRicate. Available at: https://github.com/tseemann/abricate (Accessed December 13, 2022).
Seemann, T. (2022b). Snippy. Available at: https://github.com/tseemann/snippy (Accessed December 13, 2022).
Senthakumaran, T., Brandal, L. T., Lindstedt, B. A., Jørgensen, S. B., Charnock, C., and Tunsjø, H. S. (2018). Implications of stx loss for clinical diagnostics of Shiga toxin-producing Escherichia coli. Eur. J. Clin. Microbiol. Infect. Dis. 37, 2361–2370. doi: 10.1007/s10096-018-3384-6
Severi, E., Vial, F., Peron, E., Mardh, O., Niskanen, T., and Takkinen, J. (2016). Community-wide outbreaks of haemolytic uraemic syndrome associated with Shiga toxin-producing Escherichia coli O26 in Italy and Romania: a new challenge for the European Union. Euro Surveill. 21:21. doi: 10.2807/1560-7917.ES.2016.21.49.30420
Shaikh, N., and Tarr, P. I. (2003). Escherichia coli O157:H7 Shiga toxin-encoding bacteriophages: integrations, excisions, truncations, and evolutionary implications. J. Bacteriol. 185, 3596–3605. doi: 10.1128/jb.185.12.3596-3605.2003
Silva, M., Machado, M. P., Silva, D. N., Rossi, M., Moran-Gilad, J., Santos, S., et al. (2018). chewBBACA: a complete suite for gene-by-gene schema creation and strain identification. MGen 4:e000166. doi: 10.1099/mgen.0.000166
Steyert, S. R., Rasko, D. A., and Kaper, J. B. (2011). Functional and phylogenetic analysis of ureD in Shiga toxin-producing Escherichia coli. J. Bacteriol. 193, 875–886. doi: 10.1128/JB.00922-10
Strozen, T. G., Li, G., and Howard, S. P. (2012). YghG (GspSβ) is a novel pilot protein required for localization of the GspSβ type II secretion system secretin of enterotoxigenic Escherichia coli. Infect. Immun. 80, 2608–2622. doi: 10.1128/IAI.06394-11
Tarr, P. I., and Freedman, S. B. (2022). Why antibiotics should not be used to treat Shiga toxin-producing Escherichia coli infections. Curr. Opin. Gastroenterol. 38, 30–38. doi: 10.1097/MOG.0000000000000798
Tauschek, M., Gorrell, R. J., Strugnell, R. A., and Robins-Browne, R. M. (2002). Identification of a protein secretory pathway for the secretion of heat-labile enterotoxin by an enterotoxigenic strain of Escherichia coli. Proc. Natl. Acad. Sci. 99, 7066–7071. doi: 10.1073/pnas.092152899
Tewolde, R., Dallman, T., Schaefer, U., Sheppard, C. L., Ashton, P., Pichon, B., et al. (2016). MOST: a modified MLST typing tool based on short read sequencing. PeerJ 4:e2308. doi: 10.7717/peerj.2308
The UniProt Consortium (2021). UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 49, D480–D489. doi: 10.1093/nar/gkaa1100
Treangen, T. J., Ondov, B. D., Koren, S., and Phillippy, A. M. (2014). The harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 15:524. doi: 10.1186/s13059-014-0524-x
Vlisidou, I., Marchés, O., Dziva, F., Mundy, R., Frankel, G., and Stevens, M. P. (2006). Identification and characterization of EspK, a type III secreted effector protein of enterohaemorrhagic Escherichia coli O157:H7. FEMS Microbiol. Lett. 263, 32–40. doi: 10.1111/j.1574-6968.2006.00410.x
Wang, H., Sanz Garcia, R., Cox, E., and Devriendt, B. (2020). Porcine enterotoxigenic Escherichia coli strains differ in their capacity to secrete enterotoxins through varying YghG levels. Appl. Environ. Microbiol. 86:e00523-20. doi: 10.1128/AEM.00523-20
World Health Organization (2019). Critically important antimicrobials for human medicine, 6th revision. Geneva: World Health Organization.
World Health Organization and Food and Agriculture Organization of the United Nations (2018). Shiga toxin-producing Escherichia coli (STEC) and food: attribution, characterization, and monitoring. Available at: https://apps.who.int/iris/handle/10665/272871 (Accessed December 12, 2022).
Yang, J., Baldi, D. L., Tauschek, M., Strugnell, R. A., and Robins-Browne, R. M. (2007). Transcriptional regulation of the yghJ-pppA-yghG-gspCDEFGHIJKLM cluster, encoding the type II secretion pathway in enterotoxigenic Escherichia coli. J. Bacteriol. 189, 142–150. doi: 10.1128/JB.01115-06
Zhou, Z., Alikhan, N.-F., Mohamed, K., Fan, Y., the Agama Study Group, and Achtman, M. (2020). The EnteroBase user’s guide, with case studies on Salmonella transmissions, Yersinia pestis phylogeny, and Escherichia core genomic diversity. Genome Res. 30, 138–152. doi: 10.1101/gr.251678.119
Keywords: STEC, O26:H11, cattle, phylogenetic, whole genome sequencing (WGS), virulence, epidemiology
Citation: Hoyle DV, Wee BA, Macleod K, Chase-Topping ME, Bease AG, Tongue SC, Gally DL, Delannoy S, Fach P, Pearce MC, Gunn GJ, Holmes A and Allison L (2023) Phylogenetic relationship and virulence composition of Escherichia coli O26:H11 cattle and human strain collections in Scotland; 2002–2020. Front. Microbiol. 14:1260422. doi: 10.3389/fmicb.2023.1260422
Edited by:
Ruiting Lan, University of New South Wales, AustraliaReviewed by:
Camilla Sekse, Norwegian Veterinary Institute (NVI), NorwayCopyright © 2023 Hoyle, Wee, Macleod, Chase-Topping, Bease, Tongue, Gally, Delannoy, Fach, Pearce, Gunn, Holmes and Allison. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Deborah V. Hoyle, ZGVib3JhaC5ob3lsZUBlZC5hYy51aw==
†These authors share first authorship
‡These authors share last authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.