 Xueyuan Jiang1
Xueyuan Jiang1 Shaoshan Deng1,2
Shaoshan Deng1,2 Naisheng Lu1
Naisheng Lu1 Wen Yao2
Wen Yao2 Dong Xia1*
Dong Xia1* Weilong Tu1
Weilong Tu1 Hulong Lei1Peng Jia1
Hulong Lei1Peng Jia1 Yeqing Gan3
Yeqing Gan3- 1Shanghai Engineering Research Center of Breeding Pig, Livestock and Poultry Resources (Pig) Evaluation and Utilization Key Laboratory of Ministry of Agriculture and Rural Affairs, Institute of Animal Husbandry and Veterinary Science, Shanghai Academy of Agricultural Sciences, Shanghai, China
- 2College of Animal Science and Technology, Nanjing Agricultural University, Nanjing, China
- 3Meishan Pig Breeding Center of Jiading, Shanghai, China
Introduction: The gut microbiota closely relates to host health, whereas the relationship between gut microbiota and testosterone during the development of Meishan male pigs remains unclear. This study investigated the fecal microbiota composition and testosterone level during development in Meishan male pigs.
Methods: Fresh fecal samples of 20 healthy Meishan male pigs were individually collected at 10 and 22 weeks (wk) of age for testosterone content detection and bacteria pyrosequencing analysis. Anaerobic culture experiment of fecal bacteria in vitro was performed for bacteria pyrosequencing analysis.
Results: The fecal testosterone content increased significantly from 10 weeks (wk) to 22 wk of age (P < 0.05). Meanwhile, the boars at 22 wk had a lower abundance of phylum Bacteroidetes and Proteobacteria, and genus Alloprevotella, Prevotella_1, Prevotellaceae_NK3B31_group, and Streptococcus in the fecal microbiota composition (P < 0.05). but higher proportions of the phylum Actinobacteria, Firmicutes, Kiritimatiellaeota, and Tenericutes, and genus Clostridium_sensu_stricto_1, Muribaculaceae and Terrisporobacter than that at 10 wk (P < 0.05), and the Firmicutes to Bacteroidetes ratio was higher at 22 wk than 10 wk (P < 0.05). Moreover, the fecal testosterone level significantly correlated with the relative abundance of the phylum Actinobacteria, Firmicutes, and Tencuteseri, and genus Alloprevotella, Clostridium_sensu_stricto_1, Muribaculaceae, Prevotella_1 and Streptococcus. Furthermore, the in vitro experiments indicated that the abundance of the phylum Proteobacteria and genus Escherichia-Shigella reduced with the increase of supplemental testosterone level. In contrast, the proportion of Firmicutes phylum increased with additional testosterone levels.
Discussion: Testosterone could modulate the microflora structure. Meanwhile, the bacteria could degrade the testosterone in a dose testosterone-dependent manner. These results provide us with new insights into the relationship between the gut microbiome and testosterone and the contributions of the gut microbiome in physiological regulation in response to gonad development.
1 Introduction
It is well known that the gut microbiome plays a vital role in the health status of the host. Describing the complexity and changes of the intestinal microbiota may help understand its effects on overall animal health. Moreover, the gut microbiome is regulated by many factors, including environmental variables, host genetics, and even steroid hormones. For instance, cortisol increases the proliferation of Salmonella in primary porcine alveolar macrophages (Verbrugghe et al., 2011). Mudd et al. (2017) observed that the fecal Ruminococcus population negatively correlated to the serum cortisol level in piglets. Experiments on weaning piglets showed a significant correlation between elevated cortisol levels and pathogenic bacteria at weaning (Moeser et al., 2007; Tuchscherer et al., 2018). Estradiol and progesterone inhibited the growth of Helicobacter pylori in vitro culture (Hosoda et al., 2011). Progesterone could increase the relative abundance of the Bifidobacterium genus in the gut microbial composition (Nuriel-Ohayon et al., 2019). All these studies suggest that steroid hormones may influence the proliferation and metabolism of bacteria.
Testosterone is a steroid hormone secreted from the Leydig cells of the testes in males or the ovaries of females, and in small amounts by the adrenal glands. Testosterone plays an essential role in male sex maturation and spermatogenesis. Interestingly, sex differences in gut microbiota were reduced between emasculated male and female mice (Qin et al., 2010; Yurkovetskiy et al., 2013). Germ-free and SPF mice had differences in serum testosterone levels (Markle et al., 2013). Gonadectomy and hormone replacement significantly affected rodent intestinal microbiota composition (Org et al., 2016). These suggest that testosterone may interact with the gut microbiome.
The synthesis and secretion of testosterone in males vary with gonadal development. It has been reported that the plasma concentration of testosterone gradually increases during pubertal development and reaches peak level at sexual maturation in pigs (Allrich et al., 1983; Schwarzenberger et al., 1993). However, whether such developmental changes of testosterone affect intestinal microbial structure and function is yet to be being determined. Meishan pig is known as an early maturation breed, and the plasma testosterone level positively increases with age, even after sexual maturation (Kanematsu et al., 2006). Therefore, this study investigated the changes in the gut microbiome composition from 10 wk. to 22 wk. in Meishan male pigs, when the plasma testosterone concentration nearly increased twice (Kanematsu et al., 2006). Further, vitro experiments were conducted to verify the effect of testosterone on the gut microbiome. The discoveries will establish a foundation for understanding the interaction of testosterone and the gut microbiota in males.
2 Materials and methods
2.1 Animals and sampling
This experiment was approved by the Institutional Animal Care and Use Committee (IACUC) of the Shanghai Academy of Agricultural Sciences (Grant number: SAASPZ0520013). All animal care and experimental protocols followed the guidelines of the Laboratory Animal Guideline for Ethical Review of Animal Welfare (GB/T 35892-2018) set by the Standardization Administration of China.
A total of 20 healthy Meishan male pigs were investigated in this study, and all animals were given the same basal diet throughout the whole experiment. Fresh fecal samples were individually collected via rectal massage without contamination with the barn floor at 10 and 22 weeks (wk) of age, respectively. Each fecal sample was mixed homogeneously individually over ice and distributed for different analyses. The fecal sample for microbiome pyrosequencing analysis was filled in a tube with ethanol (1:3, v/v) and stored at −20°C until DNA extraction. The fecal sample for hormone analysis was stored at −20°C.
2.2 Determination of fecal testosterone concentration
Compared with blood sampling, fecal sample collection is non-invasive with less interference from acute stress (Sheriff et al., 2010). Fecal steroid hormones concentration can represent the systemic steroid hormones synthesis and secretion, especially for periodic investigation, and now it is commonly used and accepted as a means to study the changes of hormones in domestic and wild animals (Fujita et al., 2001; Sheriff et al., 2010; SankarGanesh et al., 2014). The fecal sample extraction and testosterone measurement followed the protocol described previously (Fujita et al., 2001). In brief, fecal samples were frozen-dried for 72 h, the sample weight before and after frozen-dried was recorded for the calculation of stool water content, and then 100 mg dried fecal powder was extracted in 3 mL of 80% aqueous methanol by vortex for 15 min. Following extraction, we centrifuged the suspension and recovered the supernatant liquid for testosterone measurement. The fecal testosterone was measured by using commercial enzyme-linked immunoassay (EIA) kits of testosterone (Cayman, Item No. 582701, inter-assay CV < 10.7%, intra-assay CV < 14.0%).
2.3 Anaerobic culture experiment of fecal bacteria in vitro
Fresh feces from 5 experimental castration boars were collected via rectal massage without contamination with the barn floor. In the laboratory, the feces were mixed evenly and sterilized with physiological saline at 1:9 (mass: volume). The diluted material was then homogenized and strained through a four-layer of cheesecloth. After centrifuging at 1,500 rpm for 15 min, the supernatant was taken for inoculation.
The medium for in vitro anaerobic culture was modified from that of Williams et al. (2005). The medium contained 4.0 g glucose, 1.0 g trypticase, 1.5 g Pipes, 0.6 g KCl, 0.6 g NaCl, 0.2 g CaCl2·2H2O, 0.5 g MgSO4·7H2O, 0.54 g NH4Cl, 1.0 mg resazurin solution, 4.1 g Na2CO3, 1.0 mg Haemin, 2.5 mg NiCl2·6H2O, 2.5 mg H3BO3, 2.5 mg Na2MoO4·2H2O, 0.5 mg SeO2, 0.5 mg CoCl2·6H2O, 0.314 mg NaVO3, 0.25 mg MnCl2·4H2O, 0.25 mg ZnCl2, 0.25 mg CuCl·2H2O, 0.2 mg FeSO4·7H2O, 68.5 μL acetic acid, 30 μL propionic, 18.4 μL butyric, 5.5 μL valeric, 5.5 μL 2-methyl-butyric, 4.7 μL iso-butyric, and 5.5 μL iso-valeric acids. The solution was filtered and sterilized by 0.22 μm.
The reducing agent contained 20.5 g Na2S·9H2O and 20.5 cysteines HCl dissolved in 1 L of boiled distilled water with nitrogen gas bubbling through it. This procedure was completed in a fume cupboard due to the danger of inhalation of toxic fumes. The vitamin/phosphate solution contained 0.0204 g biotin, 0.0205 g folic acid, 0.1640 g calcium D-pantothenate, 0.1640 g nicotinamide, 0.1640 g riboflavin, 0.1640 g thiamin HCl, 0.1640 g pyridoxine HCl, 0.0204 g para-aminobenzoic acid, 0.0205 g cyanocobalamin, dissolved in a 1 L of solution containing 54.7 g KH2PO4. This solution was then filter-sterilized into sterile bottles.
The anaerobic culture trial of fecal bacteria in vitro was designed with single factor variance, and the final concentrations of testosterone in the treatments were 200 μg/mL, 400 μg/mL, 800 μg/mL, and 1,200 μg/mL (The solvent for testosterone is DMSO). At the same time, the control group contained the same amount of DMSO as the treatments. Each treatment had six replicates, and each replicate had an 80 mL medium. After 10 min of carbon dioxide flushing, 1 mL reducing agent, 1 mL vitamin-phosphate solution, and 5 mL inoculant were added. After that, the culture was incubated in a shaker at 37°C and 180 rpm for 8 h.
2.4 Bacteria pyrosequencing analysis
The total DNA of the fecal microbial community was extracted from individual fecal samples and in vitro culture samples, as described previously (Jiang et al., 2020). Illumina Miseq PE250 sequencing was performed based on the bacterial 16S rRNA sequence V3-V4. The data were analyzed on the online platform of Majorbio Cloud Platform.1 The operational taxonomic units (OTU) picking with a 97% similarity cut-off was compiled with Qiime using default parameters. Taxonomic classification was performed based on the OTU database. The alpha-diversity indices, including Chao, Ace, Shannon, and Simpson, were calculated with the Mothur program2 and Rarefaction software. The community difference at 10 wk. and 22 wk. was evaluated using Principal Co-ordinates Analysis (PCoA) and Adonis test, and a significant difference was assigned at p < 0.05.
2.5 Statistical analysis
The statistical analyses were performed using SPSS 17.0 for Windows. Independent-Samples T-test was used to analyze the significant differences in different treatments. The results were presented as the mean ± standard error of the mean (SEM). Statistical significance was considered at p ≤ 0.05 for all analyses. Graph Pad Prism Version 5.01 software (Graph Pad Software, Inc., La Jolla, CA, United States) was used for mapping.
3 Results
3.1 Experiment in vivo
3.1.1 Testosterone concentration
As the male pigs grew from 10 wk. to 22 wk., the body weight increased significantly (p = 0.00, Figure 1A). Meanwhile, the fecal testosterone concentration of the male pigs increased significantly from 10 wk. to 22 wk. (p = 0.00, Figure 1B).

Figure 1. The developmental changes in male pigs. (A) The body weight; (B) the fecal testosterone content. Graph shows mean value ± SEM (N = 20). ** (p < 0.01) means significant difference between the males at the age of 10 weeks (10 wk) and 22 weeks (22 wk).
3.1.2 Fecal microbiota analysis
The 16S ribosomal RNA gene sequencing obtained 2,251,025 high-quality sequencing reads from 40 samples. Based on 97% species similarity, 1815 OTUs were obtained, which involved 476 different OTU classifications. The bacterial DNA pyrosequencing profile (Figure 2) observed that the Ace, Chao, and Simpson were higher at 22 wk. than at 10 wk. (p < 0.05). Principal Component Analysis (PCA)(a) and Principal Co-ordinates Analysis (PCoA) visually confirmed that the fecal microbial communities at 22 wk. distinctly separated from those at 10 wk. (Figure 3).

Figure 2. Fecal bacterial richness and alpha diversity index. (A) Shannon; (B) Ace; (C) Simpson; (D) Chao. *(p < 0.05) means significant difference between the males at the age of 10 weeks (10 wk) and 22 weeks (22 wk). N = 20.

Figure 3. Principal Component Analysis (PCA, A) and Principal Co-ordinates Analysis (PCoA, B) plots on OTU level. Adonis test showed sexual development time has a significant impact on the fecal microbial community. (A) Based on Bray-Curtis distance and R2 = 0.09, p = 0.00; (B) Based on weighted unifrac distance and R2 = 0.12, p = 0.00 at the OTU level. Red dots represent the male pigs at 10 weeks (10 wk), blue triangles represent the male pigs at 22 weeks (22). N = 20.
At the phylum level (Figure 4A), the majority proportions of sequences attributed to Bacteroidetes (>41.94%) and Firmicutes (>40.12%). Following the growth of the pigs from 10 wk. to 22 wk. (Figure 4B), there was a significant reduction of Bacteroidetes (p = 0.02) and Proteobacteria (p = 0.01) but a pronounced increase of Firmicutes (p = 0.02), Tenericutes (p = 0.01), Kiritimatiellaeota (p = 0.01) and Actinobacteria (p = 0.02). Moreover, the Firmicutes / Bacteroidetes ratio at 10 wk. was significantly lower than at 22 wk. (p = 0.02, Figure 4C).
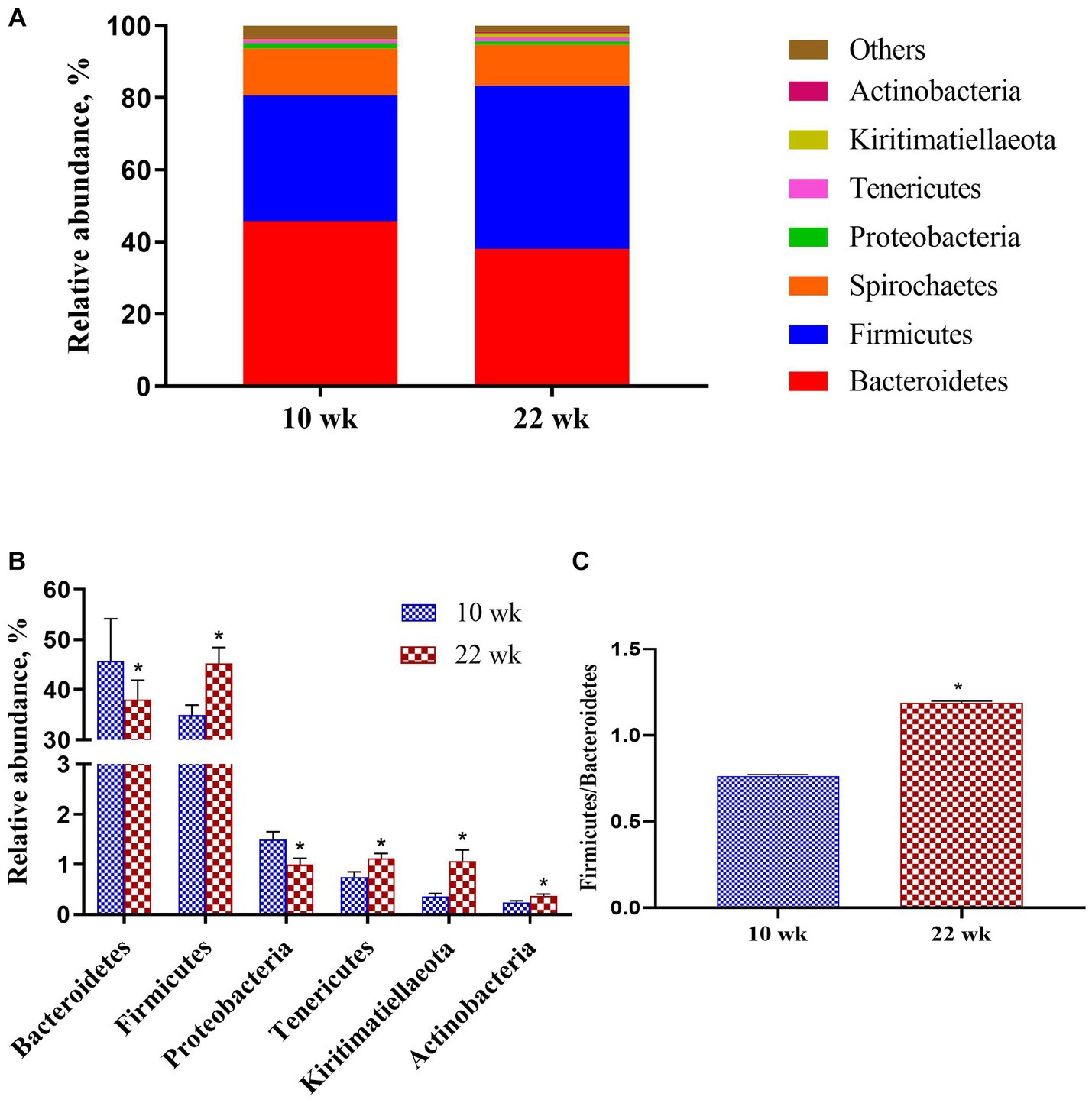
Figure 4. Taxonomic classification of the 16S rRNA gene sequences at the phylum level. (A) Fecal microbiota composition; (B) Composition difference; (C) the Firmicutes/Bacteroidetes ratio. Graph shows means value ± SEM (N = 20). * (p < 0.05) means significant difference between the males at the age of 10 weeks (10 wk) and 22 weeks (22 wk).
At the genus level (Figure 5), the proportions of Prevotellaceae_NK3B31_group (p = 0.00), Streptococcus (p = 0.00), Alloprevotella (p = 0.01), and Prevotella_1 (p = 0.00) of the fecal microbial composition were significantly lower at 22 wk. than that at 10 wk., while Clostridium_sensu_stricto_1 (p = 0.00), Muribaculaceae (p = 0.02) and Terrisporobacter (p = 0.03) were higher at 22 wk. than that at 10 wk.

Figure 5. The column chart identifying the significantly different taxa between the different age at the genus level. (A) Gut microbiota composition; (B) Composition difference. Graph shows means value ± SEM (N = 20). * (p < 0.05) means significant difference between the males at the age of 10 weeks (10 wk) and 22 weeks (22 wk).
Additionally, LEfSe analysis was performed to determine the abundance of specific microbial taxa between the male pigs at 10 wk. and 22 wk. (Figure 6). An LDA score (log10) greater than 3.5 was considered the threshold. The genera Prevotellaceae_NK3B31_group, Prevotella_1, Prevotellaceae, Streptococcus, Lachnospiraceae, Alloprevotella, Agathobacter, Sphaerochaeta, and Succinivibrio enriched at 10 wk., and the genera Ruminococcaceae_UCG-002, Turicibacter, Romboutsia, Muribaculaceae, Ruminococcaceae_NK4A214_group, Christensenellaceae_R-7_group and Lachnospiraceae_XPB1014_group improved at 22 wk.

Figure 6. Linear Discriminant Analysis Effect Size (LEfSe) of the most dominant bacteria in the fecal microbiota of the males at the age of 10 weeks (10 wk) (red) and 22 weeks (22 wk) (blue) at genus level (N = 20; all linear discriminant analysis [LDA] scores (log10) >3.5).
3.1.3 Correlation analysis between testosterone level and fecal differential microbe
Pearson’s correlation coefficient analysis (Figure 7) observed that the fecal testosterone level was significantly positively associated with the relative abundance of phylum Firmicutes (r = 0.56, p = 0.00), Tenericutes (r = 0.44, p = 0.00), and Actinobacteria (r = 0.72, p = 0.00), and genus Muribaculaceae (r = 0.52, p = 0.00), Clostridium_sensu_stricto_1 (r = 0.48, p = 0.00). However, the fecal testosterone level was significantly negatively associated with the relative abundance of genus Alloprevotella (r = −0.36, p = 0.02), Prevotella_1 (r = −0.43, p = 0.00), and Streptococcus (r = −0.47, p = 0.00).
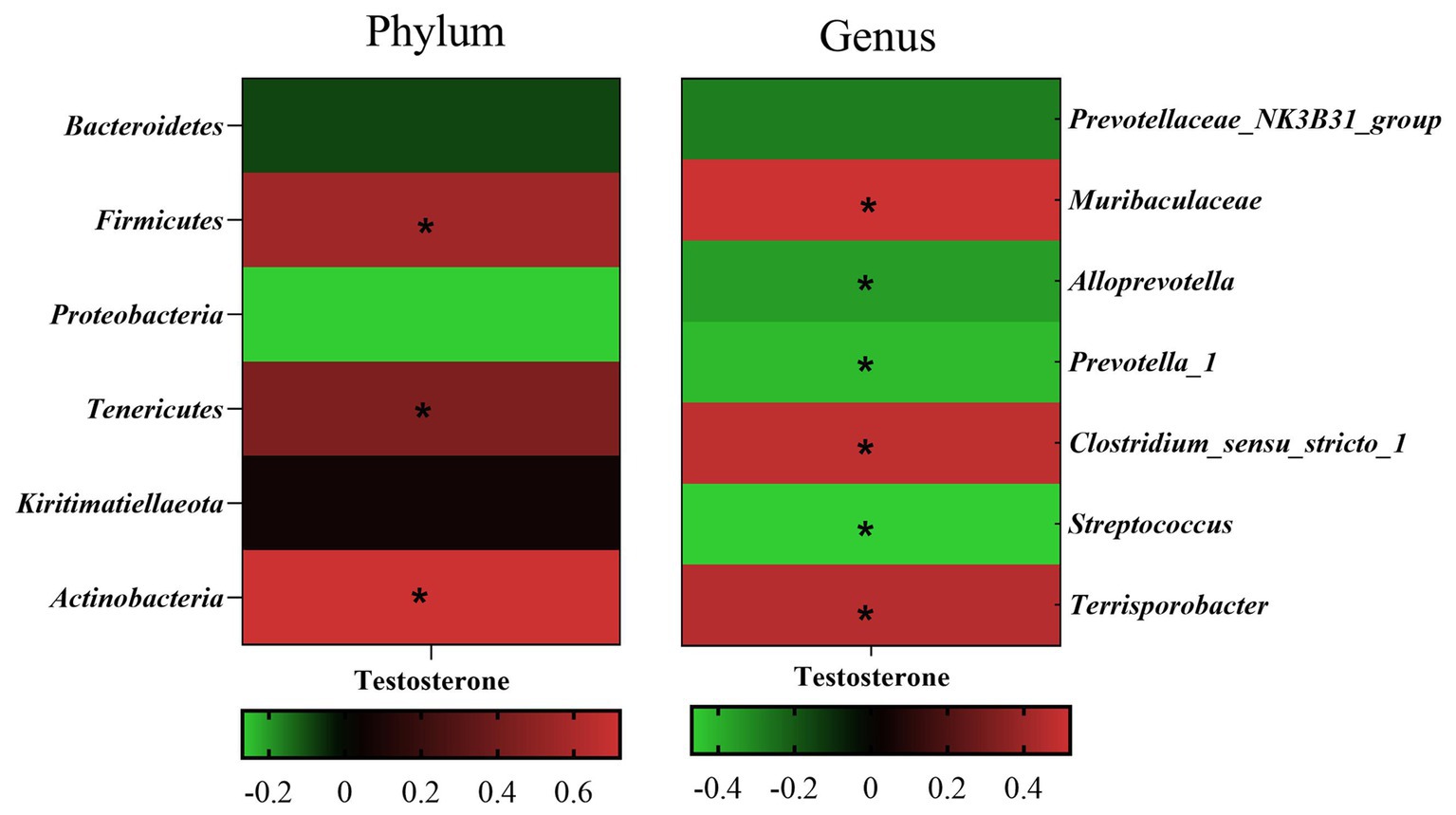
Figure 7. Pearson correlation analysis between fecal testosterone level and differential microbe at phylum level and genus level. The cells are colored based on the Pearson correlation coefficient between the testosterone level and differential microbe between different age, red represents positive correlation, green represents negative correlation, and * represents that the correlation was significant (p < 0.05).
3.2 Experiment in vitro
3.2.1 Depletion rate of testosterone in the microbial communities culture medium
The EIA assay determined the testosterone concentration in the culture solution and did not observe testosterone in the control group. However, the testosterone levels in the culture medium of the testosterone treatments (200 μg/mL, 400 μg/mL, 800 μg/mL, and 1,200 μg/mL) decreased significantly (p < 0.05) after 8 h culture, the depletion rate inclined with the increase of supplemental testosterone level (50.76 ± 2.61%, 66.20 ± 2.63%, 79.24 ± 1.14%, and 80.68 ± 1.01%, respectively, Figure 8).
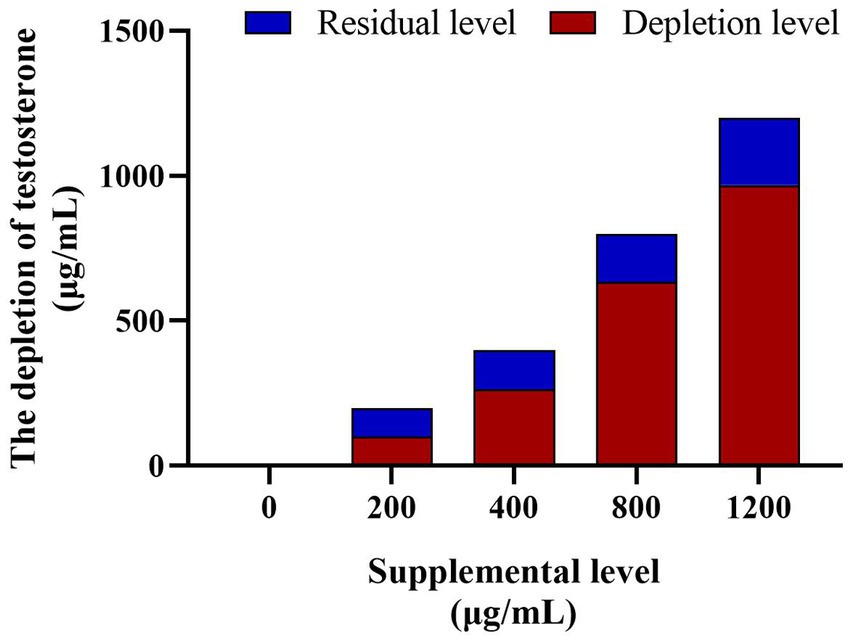
Figure 8. The testosterone depletion in culture medium among the different testosterone treatment group. The depletion rates of testosterone are 60.27, 66.20, 71.37%, and 78.23 in turn. N = 6.
3.2.2 Changes in bacterial diversity
The bacterial DNA pyrosequencing profile (Figure 9) observed that testosterone had no significant effect on richness estimators (Ace and Chao) of microbial communities of the in vitro culture. Nevertheless, the 400 μg/mL testosterone treatment achieved the most extensive Shannon index, higher than the 200 μg/mL testosterone and 1,200 μg/mL testosterone treatment (p < 0.05). Moreover, the Simpson of the 200 μg/mL testosterone treatment was significantly higher than other treatments (p < 0.05).
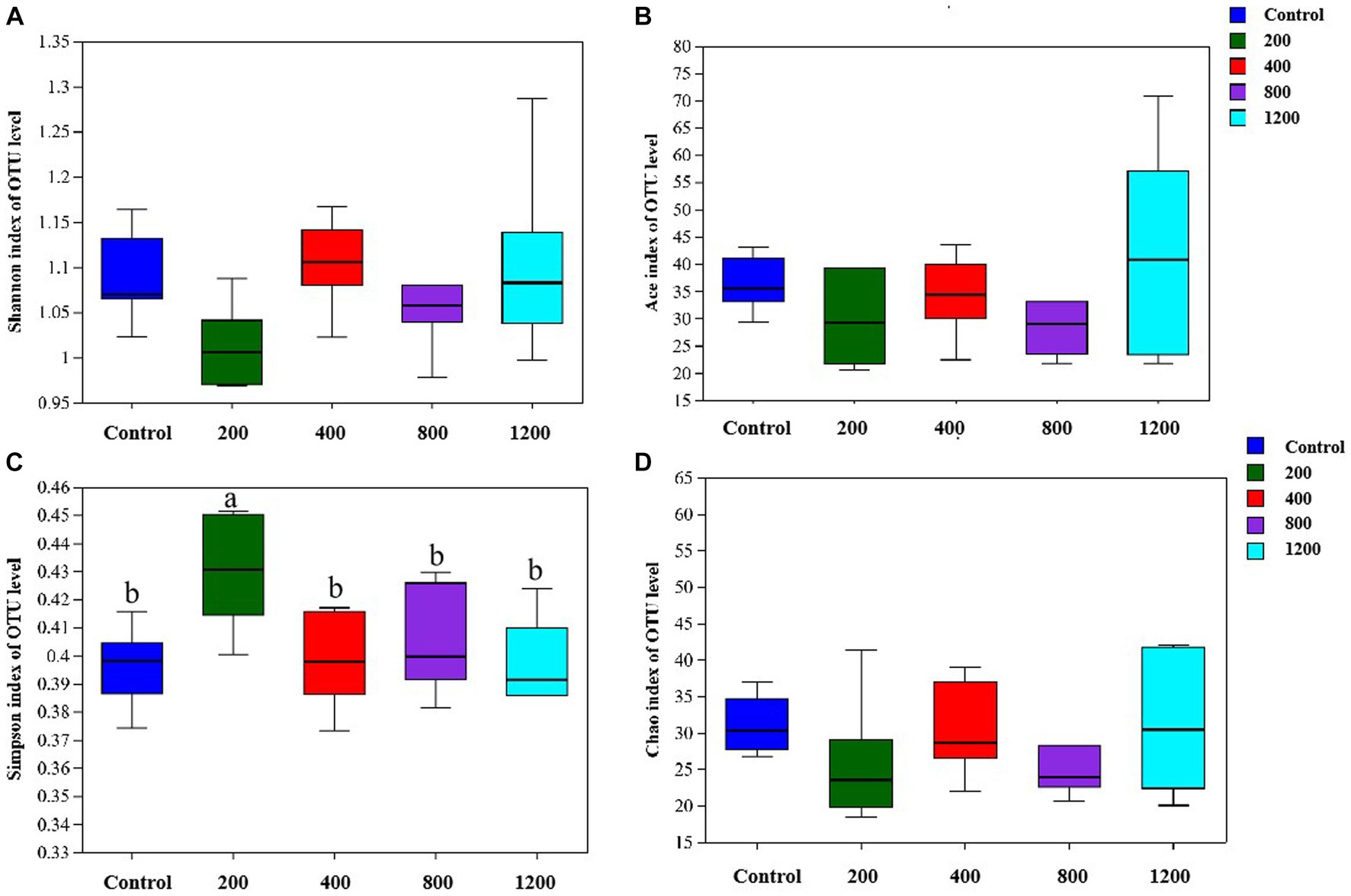
Figure 9. The richness and alpha diversity index of the culture microbial communities. (A) Shannon; (B) Ace; (C) Simpson; (D) Chao. Graph shows means value ± SEM (N = 6), and the bars with different letters indicate significant difference (p < 0.05).
Although there was no clear visual separation of groups within the testosterone treatments in Figure 10, the inter-group variation was higher than the inter-individual variation of the treatment. The PCA (Figure 10A) and PCoA (Figure 10B) visually confirmed the distinct separation of microbial communities at the OTU level (p = 0.00 and p = 0.03) among the five groups.

Figure 10. Principal Component Analysis (PCA, A) and Principal Co-ordinates Analysis (PCoA, B) plots on OTU level. Adonis test showed testosterone has a significant impact on the microbial community. (A) R2 = 0.37, p = 0.00; (B) R2 = 0.27, p = 0.03 at the OTU level. N = 6.
3.2.3 Microbiota analysis
The in vitro culture trial observed that Proteobacteria and Firmicutes were the dominant phylum (Figure 11A). The abundance of Proteobacteria in the 800 and 1,200 μg/mL testosterone treatments was significantly lower than in the 200 and 400 μg/mL testosterone treatments (p < 0.05, Figure 11B). The abundance of Firmicutes in the 800 and 1,200 μg/mL testosterone treatments was significantly higher than the 200 and 400 μg/mL testosterone (p < 0.05, Figure 11B).

Figure 11. Taxonomic classification of the 16S rRNA gene sequences at the phylum level for the testosterone treatment. (A) Gut microbiota composition; (B) Composition difference. Graph shows means value ± SEM (N = 6), and the bars with different letters indicate significant difference (p < 0.05).
At the genus level (Figure 12), with the increase in supplemental testosterone level, the proportion of Escherichia-Shigella decreased. In contrast, the abundance of Erysipelatoclostridium in the 800 μg/mL testosterone treatment was higher than the 200 and 400 μg/mL testosterone (p = 0.04 and p = 0.03).
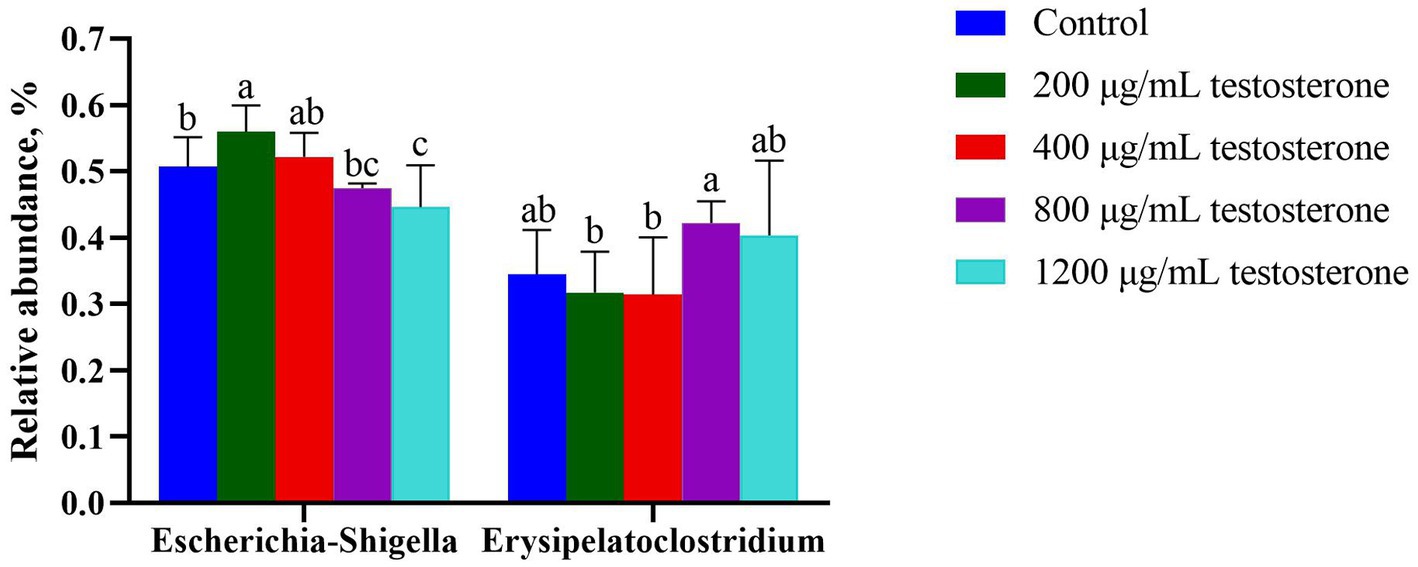
Figure 12. The column chart identifying the significantly different taxa among the testosterone treatment group at the genus level. Graph shows means value ± SEM (N = 6), and the bars with different letters indicate significant difference (p < 0.05).
4 Discussion
The present study revealed the relationship between testosterone and the composition of intestinal microorganisms during the gonad development of Meishan male pigs. It is well known that testosterone has a significant effect on the physiology and behavior of animals. Still, it has been more challenging to show that testosterone differences lead to differences in the gut microbiome of animals. In the present study, male Meishan pigs had higher fecal testosterone at 22 wk. of age than 10 wk. of age, which is consistent with previous studies that the plasma testosterone level in male Meishan pigs increased gradually from birth to 30 wk. of age and positively correlated with the bodyweight development (Kanematsu et al., 2006). Meanwhile, the fecal microbiome had higher Ace, Chao, and Simpson at 22 wk. of age than that at 10 wk., and the microbial communities at 22 wk. of age grouped differently from 10 wk. in the PCA and PCoA. Furthermore, in vitro trials confirmed that the different testosterone treatments (0, 200, 400, 800, 1,200 μg/mL) had distinct separation of microbial communities at the OTU level of PCoA. The present study via in vivo and in vitro trials showed that the intestinal microbial composition changed with testosterone levels.
As previously reported, Firmicutes and Bacteroidetes dominated the fecal microbiota at the phylum level (Xiao et al., 2017; Wang H. et al., 2019; Guo et al., 2020). The Firmicutes to Bacteroidetes ratio was relevant in signaling human gut microbiota status (Ley et al., 2006). It can be linked to overall changes in bacterial profiles at different stages of life (Mariat et al., 2009). The study of Mariat et al. (2009) also showed that the Firmicutes to Bacteroidetes ratio undergoes an increase from birth to adulthood. Consistently, the Firmicutes to Bacteroidetes ratio was higher at 22 wk. of male pigs, and the abundance of phylum Firmicutes positively correlated with testosterone content. Furthermore, the in vitro trial indicated that the mass of Firmicutes increased parallel to the increase in testosterone dosage. These suggest that the development of testosterone secretion may be attributed to changes in the fecal microbiota associated with advancing age.
The phylum Proteobacteria includes several gastrointestinal pathogens, such as diarrheagenic Escherichia coli (Cordonnier et al., 2016; Wang X. et al., 2019). Morgan et al. (2012) observed expansive Proteobacteria in intestinal inflammation, including irritable bowel syndrome and metabolic syndrome. Moreover, high abundances, or ‘blooms’ of Proteobacteria in the gastrointestinal tract, have been suggested as a microbial signature of dysbiosis in mammals (Shin et al., 2015). Numerous studies indicated that the abnormal expansion of Proteobacteria is a potential diagnostic microbial characteristic of gut microbiota imbalance and epithelial dysfunction (Litvak et al., 2017). Streptococcus is an early colonizer in the gut of pigs, and an increase in the abundance of Streptococcus may also initiate infection. Here, as male pigs developed from 10 wk. to 22 wk., the fecal testosterone level increased, while the proportion of Proteobacteria phylum and Streptococcus genera decreased. Moreover, the in vitro trial indicated that testosterone treatments inhibited Escherichia-Shigella growth. These suggest that testosterone may be advantageous for formatting a healthy gut microbial community in male pigs, combined with the developmental changes in the Firmicutes to Bacteroidetes ratio and fecal testosterone.
In addition, the present study observed that the abundance of the phylum Actinobacteria, Kiritimatiellaeota, and Tenericutes, and genera Alloprevotella, Clostridium_sensu_stricto_1, Muribaculaceae, Prevotellaceae_NK3B31_group, Prevotella_1, and Terrisporobacter differed from 10 wk. to 22 wk. The abundance of phylum Actinobacteria and Tenericutes, and genera Clostridium_sensu_stricto_1 and Muribaculaceae positively correlated with the fecal testosterone content, whereas genera Alloprevotella, Prevotella_1 negatively correlated with the fecal testosterone level. However, data about the changes in bacteria and testosterone were inconsistent. The phylum Actinobacteria is associated with synthesizing antibiotics, immunomodulatory compounds, and metabolites, which are essential for animal health. Niu et al. (2015) observed that the abundance of Clostridium and Tenericutes in the gut positively correlated with the apparent crude fiber digestibility in pigs. Muribaculaceae may be associated with carbohydrate degradation (Lagkouvardos et al., 2019). Zhang et al. (2022) observed that the proportion of Alloprevotella and Muribaculaceae increased in the gut of piglets under cold stress. Women with Polycystic Ovary Syndrome had a reduced relative abundance of the phylum Tenericutes in the fecal microbiome (Lindheim et al., 2017). A clinical study showed that Alloprevotella was enriched in obese participants (Insenser et al., 2018). Diethylhexylphthalate-exposed mice had a decrease in blood testosterone levels and a reduction of Firmicutes and Paraprevotella in the fecal microbiota, while an increase in Bacteroidetes and Prevotella (Tian et al., 2019). A previous study showed that Prevotella could produce succinate and acetate, which could improve the gut barrier and exhibit anti-inflammatory function, the abundance of Prevotella decreased from 30% of all bacteria to 4.0% as pigs aged (Xiao et al., 2017). These discrepant data suggest that the relationship between testosterone and gut microbiota may differ in the developmental stage and health status.
While steroid hormones model the gut microbiome composition and diversity, the bacteria may influence testosterone secretion and metabolism. Neuman et al. (2015) reported that transplantation of male microbiome to females caused an increase in the testosterone and enrichment of E. coli and Shigella-like bacteria in females. Clostridium can convert glucocorticoids to androgens, a group of male steroid hormones. Genera Prevotella can use estradiol and progesterone as alternative sources for growth (Kornman and Loesche, 1982). The Firmicutes, Proteobacteria, and Actinobacteria can metabolize and degrade steroid hormones (García-Gómez et al., 2013). Fecal bacteria, such as Bacillus, Brevibacterium, Streptomyces, and Pseudomonas, could degrade testosterone (Yang et al., 2010; Shih et al., 2017). Present data agreed with the above that the consumption rate of testosterone increased gradually with the treated testosterone dosage in the culture medium, further illustrating that the bacteria could degrade testosterone.
To conclude, the present study found a significant correlation between the change in testosterone content and microflora. The vitro trials showed that testosterone could modulate the microflora structure. Meanwhile, the bacteria could degrade the testosterone in a dose testosterone-dependent manner. Whether such interaction contributes to testosterone and gut microbiome development changes remains unclear and further studies are needed.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: Raw read sequences of the 16S rRNA gene from Meishan pig feces-associated microbiota in this study are publicly available in the NCBI SRA depository within BioProject PRJNA998586, with BioSample accession numbers SAMN36707203-SAMN36707246.
Ethics statement
The animal study was approved by Institutional Animal Care and Use Committee (IACUC) of Shanghai Academy of Agricultural Sciences Shanghai Academy of Agricultural Sciences. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
XJ: Conceptualization, Data curation, Formal Analysis, Project administration, Writing – original draft, Writing – review & editing. SD: Investigation, Methodology, Writing – review & editing. NL: Investigation, Validation, Writing – review & editing. WY: Formal Analysis, Writing – review & editing. DX: Supervision, Writing – review & editing. WT: Formal Analysis, Writing – review & editing. HL: Formal Analysis, Writing – review & editing. PJ: Investigation, Writing – review & editing. YG: Resources, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work was supported by Youth Program of National Natural Science Foundation of China (Grant No. 32100033 and Grant No. 32002177).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Footnotes
References
Allrich, R. D., Christenson, R. K., Ford, J. J., and Zimmerman, D. R. (1983). Pubertal development of the boar: age-related changes in testicular morphology and in vitro production of testosterone and estradiol-17β. Biol. Reprod. 28, 902–909. doi: 10.1095/biolreprod28.4.902
Cordonnier, C., Le Bihan, G., Emond-Rheault, J. G., Garrivier, A., Harel, J., and Jubelin, G. (2016). Vitamin B12 uptake by the gut commensal bacteria Bacteroides thetaiotaomicron limits the production of Shiga toxin by enterohemorrhagic Escherichia coli. Toxins 8, 11–19. doi: 10.3390/toxins8010014
Fujita, S., Mitsunaga, F., Sugiura, H., and Shimizu, K. (2001). Measurement of urinary and fecal steroid metabolites during the ovarian cycle in captive and wild Japanese macaques, Macaca fuscata. Am. J. Primatol. 53, 167–176. doi: 10.1002/ajp.3
García-Gómez, E., González-Pedrajo, B., and CamachoArroyo, I. (2013). Role of sex steroid hormones in bacterial-host interactions. Biomed. Res. Int. 2013:928290. doi: 10.1155/2013/928290
Guo, L., Wu, Y., and Wang, C. (2020). Gut microbiological disorders reduce semen utilization rate in Duroc boars. Front. Microbiol. 11:581926. doi: 10.3389/fmicb.2020.581926
Hosoda, K., Shimomura, H., Hayashi, S., Yokota, K., and Hirai, Y. (2011). Steroid hormones as bactericidal agents to Helicobacter pylori. FEMS Microbiol. Lett. 318, 68–75. doi: 10.1111/j.1574-6968.2011.02239.x
Insenser, M., Murri, M., Del Campo, R., Martínez-García, M. Á., Fernández-Durán, E., and Escobar-Morreale, H. F. (2018). Gut microbiota and the polycystic ovary syndrome: influence of sex, sex hormones, and obesity. J. Clin. Endocrinol. Metab. 103, 2552–2562. doi: 10.1210/jc.2017-02799
Jiang, X., Lu, N., Zhao, H., Yuan, H., Xia, D., and Lei, H. (2020). The microbiome-metabolome response in the colon of piglets under the status of weaning stress. Front. Microbiol. 11:2055. doi: 10.3389/fmicb.2020.02055
Kanematsu, N., Jin, W. Z., Watanabe, G., and Taya, K. (2006). Age-related changes of reproductive hormones in young Meishan boars. J. Reprod. Dev. 52, 651–656. doi: 10.1262/jrd.18013
Kornman, K. S., and Loesche, W. J. (1982). Effects of estradiol and progesterone on Bacteroides melaninogenicus and Bacteroides gingivalis. Infect. Immun. 35, 256–263. doi: 10.1128/iai.35.1.256-263.1982
Lagkouvardos, I., Lesker, T. R., Hitch, T. C. A., Gálvez, E. J. C., Smit, N., Neuhaus, K., et al. (2019). Sequence and cultivation study of Muribaculaceae reveals novel species, host preference, and functional potential of this yet undescribed family. Microbiome 7:28. doi: 10.1186/s40168-019-0637-2
Ley, R. E., Turnbaugh, P., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Lindheim, L., Bashir, M., Münzker, J., Trummer, C., Zachhuber, V., Leber, B., et al. (2017). Alterations in gut microbiome composition and barrier function are associated with reproductive and metabolic defects in women with polycystic ovary syndrome (PCOS): a pilot study. PLoS One 12:e0168390. doi: 10.1371/journal.pone.0168390
Litvak, Y., Byndloss, M. X., Tsolis, R. M., and Baumler, A. J. (2017). Dysbiotic proteobacteria expansion: a microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 39, 1–6. doi: 10.1016/j.mib.2017.07.003
Mariat, D., Firmesse, O., Levenez, F., and Guimarăes, V. (2009). The Firmicutes / Bacteroidetes ratio of the human microbiota changes with age. BMC Microbiol. 9:123. doi: 10.1186/1471-2180-9-123
Markle, J. G., Frank, D. N., Mortin-Toth, S., Robertson, C. E., Feazel, L. M., Rolle-Kampczyk, U., et al. (2013). Sex differences in the gut microbiome drive hormone-dependent regulation of autoimmunity. Science 339, 1084–1088. doi: 10.1126/science.1233521
Moeser, A. J., Ryan, K. A., Nighot, P. K., and Blikslager, A. T. (2007). Gastrointestinal dysfunction induced by early weaning is attenuated by delayed weaning and mast cell blockade in pigs. Am. J. Physiol Gastr. L. 293, G413–G421. doi: 10.1152/ajpgi.00304.2006
Morgan, X. C., Tickle, T. L., Sokol, H., Gevers, D., Devaney, K. L., Ward, D. V., et al. (2012). Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol. 13:R79. doi: 10.1186/gb-2012-13-9-r79
Mudd, A. T., Berding, K., Wang, M., Donovan, S. M., and Dilger, R. N. (2017). Serum cortisol mediates the relationship between fecal Ruminococcus and brain N-acetylaspartate in the young pig. Gut Microbes 8, 589–600. doi: 10.1080/19490976.2017.1353849
Neuman, H., Debelius, J. W., Knight, R., and Koren, O. (2015). Microbial endocrinology: the interplay between the microbiota and the endocrine system. FEMS Microbiol. Rev. 39, 509–521. doi: 10.1093/femsre/fuu010
Niu, Q., Li, P., Hao, S., Zhang, Y., and Huang, R. (2015). Dynamic distribution of the gut microbiota and the relationship with apparent crude fiber digestibility and growth stages in pigs. Sci. Rep. 5:9938. doi: 10.1038/srep09938
Nuriel-Ohayon, M., Neuman, H., Ziv, O., Belogolovski, A., Barsheshet, Y., Bloch, N., et al. (2019). Progesterone increases bifidobacterium relative abundance during late pregnancy. Cell Rep. 27, 730–736.e3. doi: 10.1016/j.celrep.2019.03.075
Org, E., Mehrabian, M., Parks, B. W., Shipkova, P., Liu, X., Drake, T., et al. (2016). Sex differences and hormonal effects on gut microbiota composition in mice. Gut Microbes 7, 313–322. doi: 10.1080/19490976.2016.1203502
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
SankarGanesh, D., Ramachandran, R., Muniasamy, S., Saravanakumar, V. R., Suriyakalaa, U., Kannan, S., et al. (2014). A correlation of fecal volatiles and steroid hormone profiles with behavioral expression during estrous cycle of goat, Capra hircus. Gen. Comp. Endocrinol. 206, 178–183. doi: 10.1016/j.ygcen.2014.07.028
Schwarzenberger, F., Toole, G. S., Christie, H. L., and Raes, J. I. (1993). Plasma levels of several androgens and estrogens from birth to puberty in male domestic pigs. Acta Endocrinol. 128, 173–177. doi: 10.1530/acta.0.1280173
Sheriff, M. J., Krebs, C. J., and Boonstra, R. (2010). Assessing stress in animal populations: do fecal and plasma glucocorticoids tell the same story? Gen. Comp. Endocrinol. 166, 614–619. doi: 10.1016/j.ygcen.2009.12.017
Shih, C. J., Chen, Y. L., Wang, C. H., Wei, T. S., Lin, I. T., Ismail, W. A., et al. (2017). Biochemical mechanisms and microorganisms involved in anaerobic testosterone metabolism in estuarine sediments. Front. Microbiol. 8:15. doi: 10.3389/fmicb.2017.01520
Shin, N.-R., Whon, T. W., and Bae, J.-W. (2015). Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 33, 496–503. doi: 10.1016/j.tibtech.2015.06.011
Tian, X., Yu, Z., Feng, P., Ye, Z., Li, R., Liu, J., et al. (2019). Lactobacillus plantarum TW1-1 alleviates diethylhexylphthalate-induced testicular damage in mice by modulating gut microbiota and decreasing inflammation. Front. Cell. Infect. Microbiol. 9:221. doi: 10.3389/fcimb.2019.00221
Tuchscherer, M., Puppe, B., Tuchscherer, A., and Kanitz, E. (2018). Psychosocial stress sensitizes neuroendocrine and inflammatory responses to Escherichia coli challenge in domestic piglets. Brain Behav. Immun. 68, 274–287. doi: 10.1016/j.bbi.2017.10.026
Verbrugghe, E., Boyen, F., Van Parys, A., Van Deun, K., Croubels, S., Thompson, A., et al. (2011). Stress induced Salmonella Typhimurium recrudescence in pigs coincides with cortisol induced increased intracellular proliferation in macrophages. Vet. Res. 42:118. doi: 10.1186/1297-9716-42-118
Wang, H., Shou, Y., Zhu, X., Xu, Y., Shi, L., Xiang, S., et al. (2019). Stability of vitamin B12 with the protection of whey proteins and their effects on the gut microbiome. Food Chem. 276, 298–306. doi: 10.1016/j.foodchem.2018.10.033
Wang, X., Tsai, T., Deng, F., Wei, X., Chai, J., Knapp, J., et al. (2019). Longitudinal investigation of the swine gut microbiome from birth to market reveals stage and growth performance associated bacteria. Microbiome 7:109. doi: 10.1186/s40168-019-0721-7
Williams, B. A., Bosch, M. W., Verstegen, M. H. B., and Tamminga, S. (2005). An in vitro batch culture method to assess potential fermentability of feed ingredients for monogastric diets. Anim. Feed. Sci. Tech. 123-124, 445–462. doi: 10.1016/j.anifeedsci.2005.04.031
Xiao, Y., Li, K., Xiang, Y., and Zhou, W. (2017). The fecal microbiota composition of boar Duroc, Yorkshire, landrace and Hampshire pigs. Asian Australas. J. Anim. Sci. 30, 1456–1463. doi: 10.5713/ajas.16.0746
Yang, Y. Y., Borch, T., Young, R. B., Goodridge, L. D., and Davis, J. G. (2010). Degradation kinetics of testosterone by manure-borne bacteria: influence of temperature, pH, glucose amendments, and dissolved oxygen. J. Environ. Qual. 39, 1153–1160. doi: 10.2134/jeq2009.0112
Yurkovetskiy, L., Burrows, M., Khan, A. A., and Graham, L., Volchkov, P., Becker, L., Antonopoulos, D., Umesaki, Y., and Chervonsky, A. V. (2013). Gender bias in auto-immunity is influenced by microbiota. Immunity 39: 400–412. doi: 10.1016/j.immuni.2013.08.013
Keywords: Meishan male pigs, testosterone, microbiome, development, interaction
Citation: Jiang X, Deng S, Lu N, Yao W, Xia D, Tu W, Lei H, Jia P and Gan Y (2023) Fecal microbial composition associated with testosterone in the development of Meishan male pigs. Front. Microbiol. 14:1257295. doi: 10.3389/fmicb.2023.1257295
Edited by:
Congying Chen, Jiangxi Agricultural University, ChinaReviewed by:
Yu Pi, Chinese Academy of Agricultural Sciences, ChinaZhigang Song, Shandong Agricultural University, China
Copyright © 2023 Jiang, Deng, Lu, Yao, Xia, Tu, Lei, Jia and Gan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dong Xia, eGlhZG9uZ0BzYWFzLnNoLmNu