 Qian Zhou1†
Qian Zhou1† Guanqiao Li2†
Guanqiao Li2† Yinshan Cui3Jingshu Xiang1Shu Zhu1
Yinshan Cui3Jingshu Xiang1Shu Zhu1 Shijun Li1Jingyu Huang1Yafang Wang1Ying Liu1Li Zhou1*
Shijun Li1Jingyu Huang1Yafang Wang1Ying Liu1Li Zhou1*- 1Guizhou Provincial Centre for Disease Control and Prevention, Guiyang, Guizhou, China
- 2College of Bioinformatics, Chongqing University of Post and Telecommunications, Chongqing, China
- 3Yunnan Pulis Biotechnology Co., Ltd., Kunming, Yunnan, China
Introduction: Bacillus cereus is a ubiquitous opportunistic human pathogen that causes food intoxications worldwide. However, the genomic characteristics and pathogenic mechanisms of B. cereus are still unclear.
Methods: Here, we isolated and purified nine strains of B. cereus (LY01-LY09) that caused vomiting, diarrhea and other symptoms from four foodborne outbreaks happened in Guizhou Province in southwest China from June to September 2021. After colony observation, Gram staining, microscopic examination and biochemical test, they were identified as B. cereus. The genomic characteristics, phylogenetic relationships and virulence factors of the isolated strains were analyzed at the genome level. Genome sequencing, comparative genomic analysis, secondary metabolite analysis and quantitative PCR were utilized to give a thorough exploration of the strains.
Results: We obtained the genome maps of LY01-LY09 and found that LY01-LY09 had a complex interspecific relationship with B. anthracis and B. thuringiensis. We also observed a contraction of gene families in LY01-LY09, and the contracted families were mainly associated with prophage, which contributed to the species diversity of B. cereus. The Hsp20 gene family underwent a rapid evolution in LY01-LY09, which facilitated the adaptation of the strains to adverse environmental conditions. Moreover, the LY01-LY09 strains exhibited a higher copy number in the non-ribosomal polypeptide synthetase (NRPS) genes and carried the complete cereulide synthetase (ces) gene cluster sequences. Considering that the NRPS system is a classical regulatory mechanism for emetic toxin synthesis, we hypothesized that LY01-LY09 could synthesize emetic toxins through the regulation of ces gene clusters by the NRPS system.
Discussion: These findings are important for further investigation into the evolutionary relationship between B. cereus and their related species, as well as the underlying mechanisms governing the synthesis and secretion of bacterial toxins.
1 Introduction
Foodborne pathogens frequently cause public health emergencies (Josephs-Spaulding et al., 2016; Antunes et al., 2020) and have become a critical concern in food safety in China (He and Shi, 2021). Pathogenic microorganisms can lead to both food poisoning and contamination. Microbial contamination poses a significant threat to food safety and the issue of foodborne illnesses resulting from such contamination is also highly critical (Tran et al., 2011; Pires et al., 2021). B. cereus is a frequently encountered foodborne pathogen, and B. cereus-related food poisoning primarily manifests as acute gastroenteritis characterized by symptoms such as vomiting, fatigue, nausea, and diarrhea (Kotiranta et al., 2000). B. cereus causes two distinct types of food poisoning based on symptoms: food intoxication (vomiting type) and food toxico-infection (diarrhea type). According to relevant statistics, the vomiting type accounts for 75.9% of B. cereus poisoning cases, while the diarrheal type 11.4%, thus making vomiting the predominant manifestation of B. cereus foodborne illness (Zhou et al., 2008). The diarrheal type is mainly attributed to the B. cereus production of enterotoxins, including hemolysin BL (HBL), non-hemolytic enterotoxin (NHE), and cytotoxin K (CytK) (Dietrich et al., 2021). In contrast, the vomiting type is caused by cereulide, an emetic toxin produced by B. cereus through a non-ribosomal polypeptide synthetase (NRPS), which is encoded by the cereulide synthetase (ces) gene cluster (Marxen et al., 2015; Carroll and Wiedmann, 2020). The ces gene cluster, about 24 kb long, comprises seven genes, and is typically situated on a megaplasmid of B. cereus (Ehling-Schulz et al., 2006). The B. cereus-caused vomiting type of food poisoning is often mild, although severe and even lethal conditions might happen in rare cases (Stenfors Arnesen et al., 2008).
Genome sequencing technology allows for a comprehensive analysis of the molecular biological characteristics of pathogenic bacteria at the genetic level, and is widely used in epidemiological investigations of foodborne disease outbreaks (Li et al., 2021). This technology provides a crucial foundation for molecular epidemiological investigations of foodborne disease outbreaks caused by B. cereus, enabling not only pathogen identification, but also the detection and classification of virulence and drug resistance genes, and further the analysis of pathogenicity (Balloux et al., 2018). A previous study detected hemolytic (hblA, hblC, and hblD) and non-hemolytic (nheA, nheB, and nheC) enterotoxin genes in two B. cereus strains isolated from indoor air, suggesting that airborne isolates may cause diarrhea rather than vomiting (Premkrishnan et al., 2021). Toxigenic heterogeneity was found to be related to toxin genes such as nheABC, hblCDAB, cytK2, entFM, and CesB in the genomes of B. cereus sensu lato (s.l.) isolated from ready-to-eat foods and powdered milk (Sánchez Chica et al., 2020). The characterization of the genomes of B. cereus and the identification of virulence-related genes are of great significance for determining the causes of foodborne poisoning. However, few studies have dealt with the genomic profiling of B. cereus isolated from food sources that induces symptoms such as vomiting and diarrhea.
In this study, we performed genome sequencing and comparative genetic analysis on nine strains of B. cereus (LY01-LY09) isolated from contaminated food linked to foodborne outbreaks in Guizhou Province, China to analyze the evolution of these strains at the genome level, explore their virulence factors, and evaluate their pathogenic potential. By analyzing the genomic information of LY01-LY09, our objective was to establish a fundamental basis for research on the pathogenic mechanisms of B. cereus, as well as for the development of antibiotics, disease prevention and control. This study is of great significance in comprehending the evolutionary traits and patterns of B. cereus, and may provide a rapid method for the detection of B. cereus strains.
2 Materials and methods
2.1 Outbreak investigation
From June to September 2021, four foodborne outbreaks had occurred in Ziyun County and Tongren City, Guizhou Province. Persons affected reported symptoms of gastroenteritis. Afterward, technicians from Guizhou Provincial Centre for Disease Control and Prevention carried out epidemiological and environmental investigations. Food samples were collected for laboratory test.
In the 23 cases, more than 86% experienced nausea or vomiting (Supplementary Table 1). Few reported abdominal pain, diarrhea, dizziness, fatigue, etc. In the three outbreaks happened in Ziyun County, all persons had eaten rice noodle (a popular local food) bought from the same vendors or stores. The Tongren outbreak occurred in a nursery, and all affected children had eaten meals served by the nursery kitchen (Supplementary Table 1). By laboratory test, B. cereus colonies were isolated from food samples. In addition, two out of four vomit samples from children of the Tongren outbreak yielded toxin-producing B. cereus. All those affected were managed symptomatically, except for six were hospitalized and given intravenous rehydration.
2.2 Isolation, culture, and identification of bacterial strains
Food samples were approximately diluted, and the diluted solution was inoculated on Mannitol-Egg Yolk-Polymyxin (MYP) medium (Tryptone 10 g/L, Mannitol 10 g/L, Meat extract 1 g/L, NaCl 10 g/L and Phenol red 0.025 g/L) and Nutrient Agar (NA) medium. Number of colonies was enumerated after culturing at 37°C for 24 h (Anon, 1995). Five bacterial colonies from each plate were selected and inoculated on an NA inclined surface for pure culture at 37°C for 24 h. After being confirmed as Gram-positive, the morphology of the bacteria was observed under microscopic examination. Finally, the EasylD Biochemical Identification Kit for B. cereus (HuanKai microbial, China) was utilized to identify strains presenting in contaminated food.
2.3 Sequencing and assembling of B. cereus strain genomes
The genomic DNA of B. cereus strains was extracted using SDS, and total DNA was fragmented using the Covaris ultrasonic breaker (Covaris, East Sussex, UK). The library was constructed by the NEBNext®Ultra™ DNA Library Prep Kit (NEB, USA) in accordance with the manufacturer’s protocol. After a thorough quality inspection, the library was sequenced by Illumina NovaSeq 6000 platform (Illumina, San Diego, CA, USA) to obtain raw data, which were then filtered to acquire clean data. SOAP de novo (v2.04), SPAdes (v 3.15.5), and ABySS (v 2.0) assembly software were utilized for de novo assembly of high-quality sequencing data obtained from the B. cereus strains, while Contig Integrator for Sequence Assembly (CISA) software (Lin and Liao, 2013) was employed to obtain preliminary assembly results. The assembly output was optimized using GapClose (v1.12), which was employed to fill gaps and improve genome quality.
2.4 Genome annotation and collinearity analysis
GeneMarkS (Version 4.17) software was utilized to predict coding genes in the B. cereus strains, which were mapped to functional databases (E-value ≤ 1e–5) in Kyoto Encyclopedia of Genes and Genomes (KEGG),1 Transporter Classification Database (TCDB),2 pathogen–host interactions database (PHI),3 and carbohydrate active enzyme (CAZy)4 to obtain functional annotation of the protein-coding genes. In addition, repeated and tandem repeats were identified using RepeatMasker (Version open-4.0.5) and TRF (Tandem Repeats Finder, Version 4.07b), respectively. To search for homology, BLASTP software (Version 2.12.0+) was utilized to compare the protein-coding genes among the LY01-LY09 strains. The GENESPACE package of R language was employed for the visualization of genome collinearity.
2.5 Gene family clustering and phylogenetic tree construction
Furthermore, the complete genome sequences of 26 Bacillus strains were downloaded from the NCBI GenBank database, including four B. anthracis, two B. amyloliquefaciens, four B. thuringiensis, one B. pumilusstrain, ten B. cereus, two B. velezensis, and three B. subtilis. Strain names and accession numbers of the reference sequences are available in Figure 1. Altogether, 1269 single-copy orthologs were obtained from the whole-genome sequences of the 35 Bacillus strains (9 isolated and 26 downloaded) using OrthoFinder (v.2.5.4) (Emms and Kelly, 2019). Multiple alignments of amino acid sequences of orthologs were carried out by MUSCLE (v.3.8.1551) (Edgar, 2004). Conserved blocks from multiple alignments of protein sequences were obtained by Gblocks (Castresana, 2000). Maximum likelihood (ML) tree was constructed using RAxML v.8.2.12 software (Stamatakis, 2014) with 1000 bootstrap replicates by PROTGAMMAILGF model, which was built using ProtTest (v.3.4.2) software (Darriba et al., 2011). Tree visualization was completed by FigTree (v.1.4.4) software. The expansion and contraction analysis of gene family was performed by CAFE (v.4.2.1) software, afterward representative species on the branch were selected.
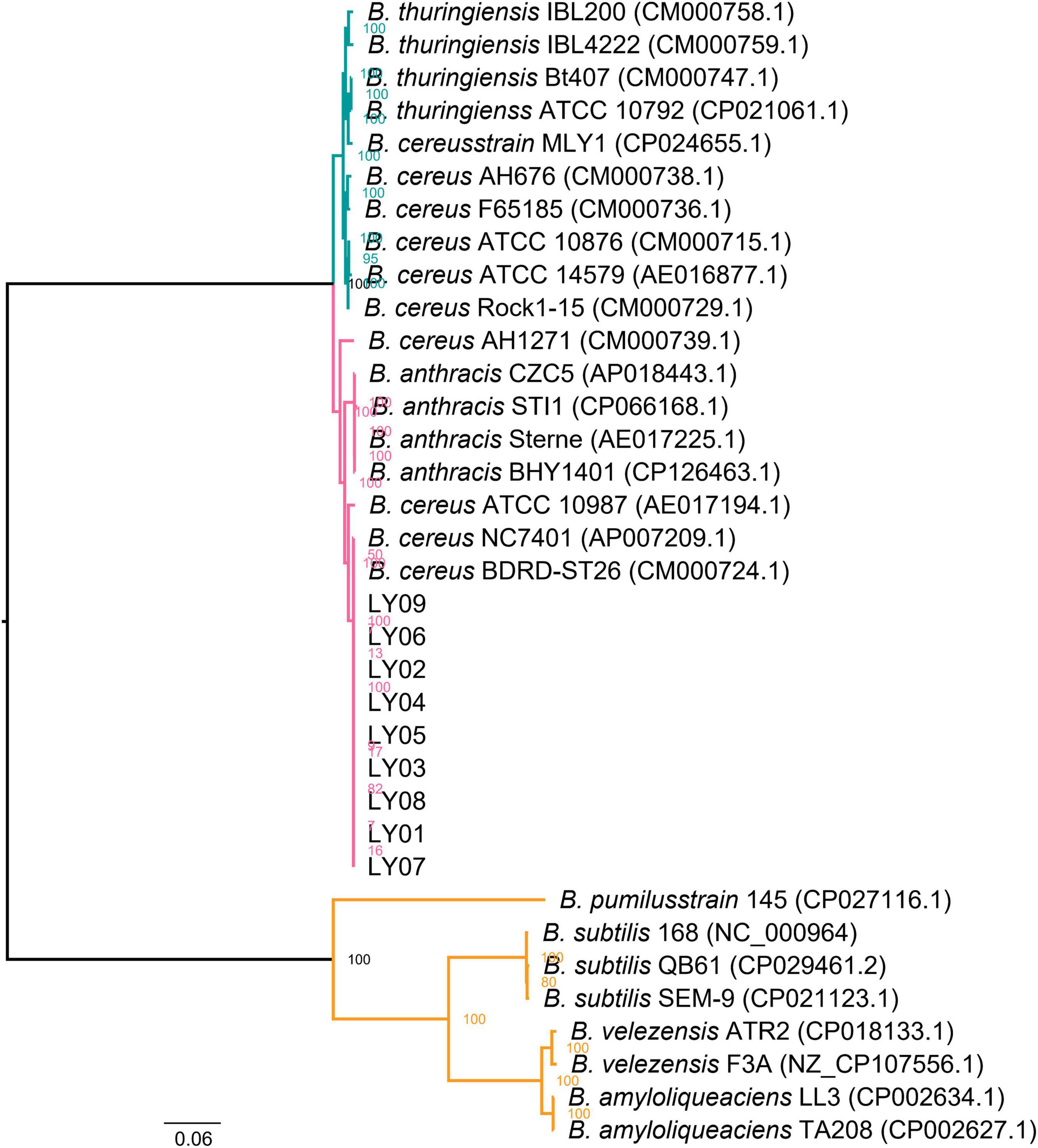
Figure 1. Phylogenetic analysis of the 9 Bacillus isolates and 26 Bacillus strains downloaded from databases based on whole genome sequence. Strain names and accessions of the downloaded strains are available in the figure. Bootstrap frequencies were obtained with 1,000 replicates.
2.6 Virulence factor prediction of the B. cereus strains
Protein sequences of the nine B. cereus strains were subjected to BLAST with an E-value threshold ≤ 1e–5 against the virulence factor database (VFDB)5 (Liu et al., 2022). The resulting data were then integrated with corresponding functional annotations for each identified virulence factor and its target species, obtaining comprehensive annotation results for the virulence factors of the nine B. cereus strains. NRPS gene sequences were identified by BLAST and motif prediction were performed using the MEME website.6 The phylogenetic relationship of NRPS gene family was constructed based on IQ-TREE (v. 2.1.4-beta) according ML method.
2.7 Verification of virulence factors by q-PCR
The expressions of hblC, nheB and cesB of the nine B. cereus strains were detected by quantitative polymerase chain reaction (q-PCR). Specifically, primers and probes were designed based on B. cereus virulence factor genes (hblC, nheB, cesB). During PCR amplification, TaqMan probes were hydrolyzed by the action of Taq DNA polymerase to generate fluorescence signals. Cycle threshold (Ct) values ≤ 30 were regarded as positive. Amplification curves were obtained for the detection of B. cereus virulence factors genes.
2.8 Comparative genome analysis
The fastANI program (Jain et al., 2018) was used to calculate the average nucleotide identity (ANI) between the whole genome sequences of two B. cereus strains. In addition, MUMmer (v. 4.0.0beta2) (Kurtz et al., 2004) was used to analyze the single nucleotide polymorphism (SNP) and InDels among strains based on the whole genome sequence, with the genome of LY03 as a reference. CIRCOS (v.0.69-9) software (Krzywinski et al., 2009) was used to visualize SNP.
3 Results
3.1 Identification and characterization of isolates
A total of nine strains of bacteria were isolated and purified from food samples. The selective medium MYP was used to screen and identify the strains. White colonies surrounded by pink areas were observed on the MYP medium, while colonies formed rough white surface with irregular edges on the NA medium (Supplementary Figure 1A). This was consistent with the typical characteristics of B. cereus. The nine strains were stained with Gram’s stain and identified as Gram-positive (Supplementary Figure 1B). Finally, the strains were tested for starch hydrolysis, glucose fermentation, Voges-Proskauer (V-P) reaction, etc., using the identification kit. A diffuse growth out into the medium along and away from the stab was observed in the motility test, thus excluding the possibility of B. mycoides strains (Supplementary Table 2). Based on the aforementioned testing results, the strains isolated and purified in this study were identified as B. cereus according to the National Standard of the People’s Republic of China GB 4789.14 (2014).
3.2 Genome structure and linkage map of the LY01-LY09 strains
The nine strains of B. cereus had similar genome sizes, with all but the LY08 strain (5.68 Mb) possessing a genome size of 5.64 Mb. The GC content ranges from 35.23 to 35.25%, exhibiting a small variation as well. The lengths of their coding genes ranged from 4.75 Mb to 4.79 Mb, accounting for approximately 84.3–84.4% of their respective genome sizes (Supplementary Table 3). The proportions of repeat sequences (LINE, RC, SINE, and LTR) in each genome were similar among the strains (Supplementary Figure 2A). The number of transfer RNAs in each strain ranged from 92 to 93, with a length of 7,000–7,200 bp. Meanwhile, the number of small RNAs varied from 5 to 6, with lengths ranging from 400 to 550 nucleotides (Supplementary Figure 2B). CRISPR (clustered regularly interspaced short palindromic repeat sequences) elements counts ranged from 10 to 29, with lengths varying between 2,476 and 6,175 bp. Furthermore, the nine strains had similar numbers of CRISPR, genomic island (Gis) and prophage (Supplementary Figure 2C). Based on the KEGG database, the metabolic pathways of the LY01-LY09 cellular gene products and compounds were assigned to six categories: metabolism, genetic information processing, environmental information processing, cellular processes, organismal systems, and human diseases. At level B, a total of 41 metabolic pathways were involved, with a primary focus on amino acid metabolism, carbohydrate metabolism, membrane transport, caffeine and vitamin metabolism, as well as energy metabolism (Supplementary Figure 3A). Functional analysis of the transport proteins in the nine isolates was performed using the TCDB database. We found that the transport proteins were mainly primary active transporters and electrochemical potential-driven transporters (Supplementary Figure 3B). Based on the CAZy database, glycosyltransferase (GTs), Glycoside hydrolases (GHs), carbohydrate esterases (CEs) and auxiliary activities (AAs) were detected as the four enzyme classifications (Supplementary Figure 3C). In addition, we noticed that reduced virulence and unaffected pathogenicity were the phenotypes with the most matched genes (Supplementary Figure 3D).
3.3 Comparative genome analysis of the LY01-LY09 strains
Pairwise macrosynteny analysis revealed a high collinearity among the nine strains, with high conservation at the genome level, except for contig rearrangements on contig3 of LY02, and contig3 and contig6 of LY01 (Supplementary Figure 4). A further analysis detected a fission phenomenon on contig7 and contig9 only, compared with a high collinearity among the isolated strains and the reference B. cereus strain. The genomic characteristics of the nine strains of B. cereus were compared with each other, and the results showed that pairwise ANI values were greater than 99.98% (Supplementary Table 4). This may indicate that these nine strains were the same strain, which was consistent with the results of collinear analysis. In addition, the genomic sequences of the other eight strains were compared with LY03 to obtain mutation information (SNPs and InDels). We found few mutation sites between the genomes of the nine strains, indicating their low level of heterogeneity (Supplementary Figure 5).
3.4 Phylogenetic analysis based on whole genome sequencing
The evolutionary relationships between the nine isolated strains with other related Bacillus strains were analyzed using whole-genome ML phylogenetic tree (Figure 1). The LY01-LY09 strains showed 100 bootstrap frequency rooted at the branch. The LY01-LY09 strains exhibited a close genetic relationship with B. anthracis and B. thuringiensis, which formed sister groups with B. anthracis (Figure 1). In addition, B. thuringiensis, B. cereus and B. anthracis were in the same branch, the boundaries between some B. cereus and B. anthracis strains were not clear. The results showed that the genomes of the three Bacillus species were highly similar.
3.5 Regulatory networks of the LY01-LY09 strains and identification of key genes
To understand genetic alterations of the LY01-LY09 strains and evolutionary forces driving these changes, we examined the interactions among the gene families that have expanded, contracted, and rapidly evolved (Supplementary Figure 6), and identified key genes in gene co-expression networks. The findings indicate that BA_0477, BA_3774, and BA_3775, which were closely associated with prophage (Supplementary Table 5), were the key genes within the contracted gene families of the LY01-LY09 strains (Supplementary Figure 7A). The genes belonging to the contracted gene families were primarily associated with phage terminase, phage portal protein, DNA binding, and extracellular region (Supplementary Figure 7B). Similarly, GO enrichment analysis were performed on the expanded gene families, which showed close relationships with DNA recombination, resolvase, N-terminal domain, Tn3 transposase DDE domain, recombinase conserved site and other related factors (Supplementary Figure 7C). To better understand species evolution, we investigated the complete genome sequences of B. anthracis, B. amyloliquefaciens, B. thuringiensis, B. velezensis, B. cereus and Clostridium botulinum, together with those of the nine Bacillus isolates. By doing so, we intended to explore the interactions among rapidly evolving gene families of the 15 strains. It was discovered that the key genes of these families were the same as those of the contracted gene families in the LY01-LY09 strains, and all of them were closely associated with prophage (Figure 2A; Supplementary Table 5). The Heat-shock protein 20 (Hsp20) family changed significantly in gene number among the rapidly evolving gene families. Structural analysis revealed that the Hsp20 families of LY09Scaffold63 and LY07LY09Scaffold61 were the most similar in terms of gene number and structure. We thus speculate that they performed similar functions as well (Figure 2B). The protein sequences of the Hsp20 families in LY01-LY09, B. thuringiensis, and B. velezensis exhibited significant variations due to point mutations and fragment insertions (Figure 2C).
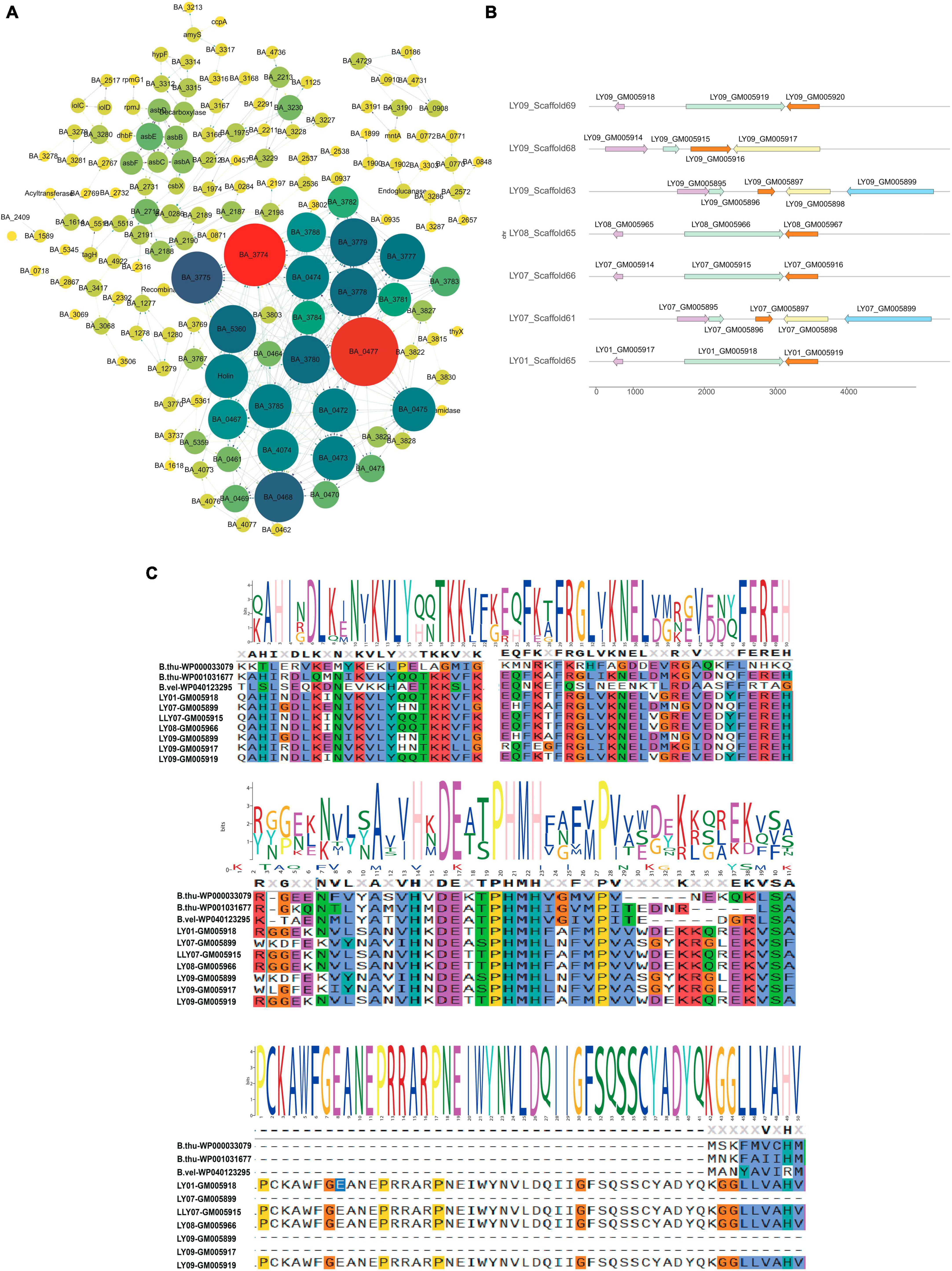
Figure 2. Functions of fast evolving genes and structure of heat-shock protein 20 (HSP20) B. anthracis, B. amyloliquefaciens, B. thuringiensis, B. velezensis, B. cereus and Clostridium botulinum downloaded from database and the nine Bacillus isolates. (A) Functions of fast-evolving genes in the 15 strains; (B) HSP20 gene structure of LY01-LY09; (C) Conserved motifs and loci of HSP20.
3.6 Virulence factors of the LY01-LY09 strains causing vomiting
We also observed a similar number of identical gene clusters across the nine B. cereus strains. Moreover, these gene clusters mainly focused on NRPS, leucine aminopeptidase (LAP), bacteriocin, and terpene (Figure 3A), among which the number of the NRPS genes was the highest. A further analysis of the NRPS genes revealed that Motif-1, Motif-2 and Motif-3 were the shared motifs among the genes (Figure 3B). In addition, the motif sequences were 2-3-1 and 3-2-1, indicating that the NRPS gene family in the LY01-LY09 strains was highly conservative. According to the maximum likelihood-based phylogenetic tree of NRPS proteins, all but one of the 27 NRPS proteins could be assigned to three groups (Figure 3C). Group 1 comprised of eight protein-coding genes, group 2 nine genes, and group 3 also nine genes.
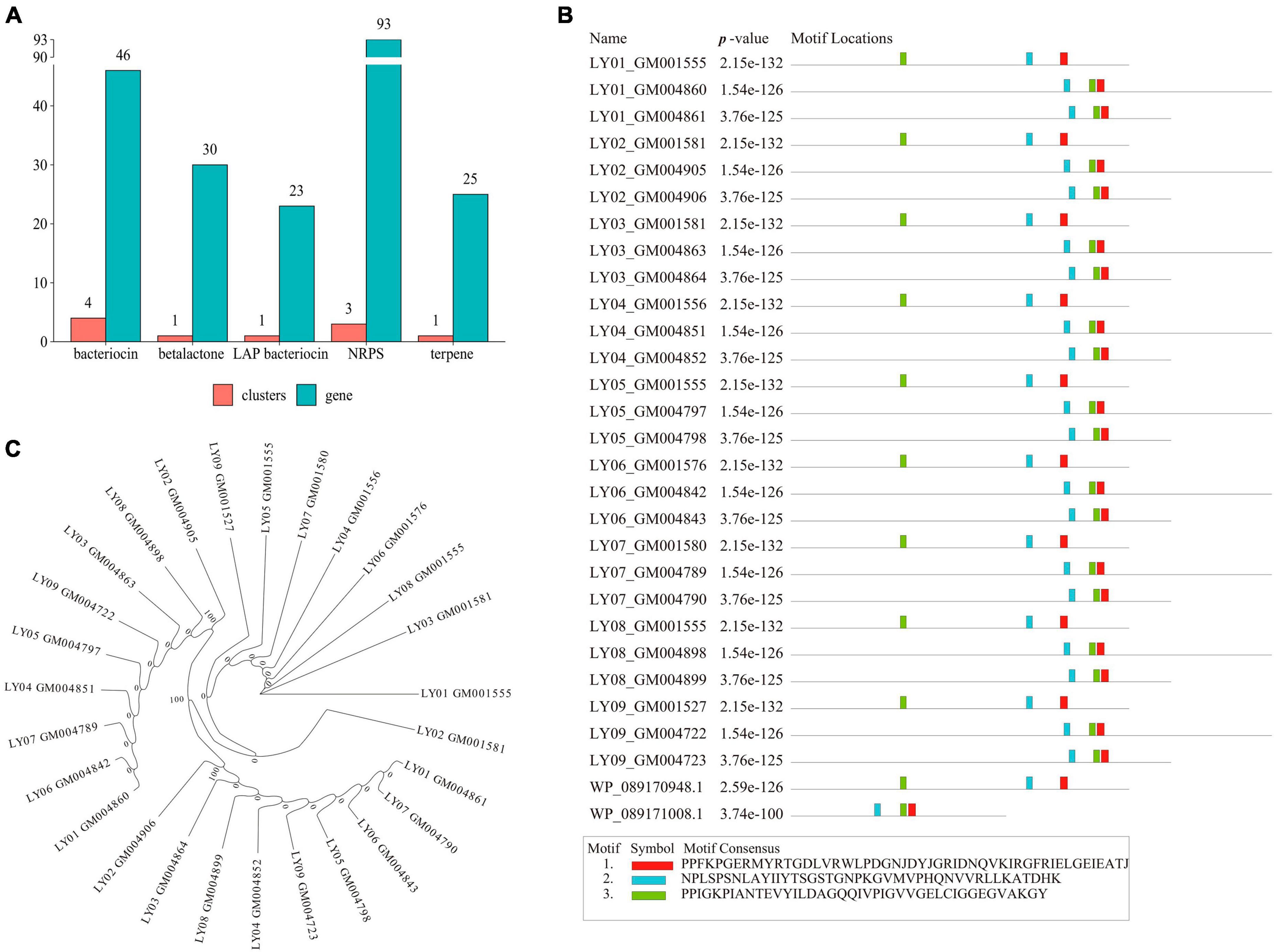
Figure 3. Gene clusters and NRPS gene analysis of B. cereus. (A) Gene cluster analysis of B. cereus; (B) Motif analysis of the NRPS genes; (C) Phylogenetic analysis of the NRPS genes.
The consistency of virulence factors among strains LY01-LY09 was further confirmed by analyzing the VFDB database (Table 1). The polysaccharide capsule involved the most virulence genes, totaling 16 in quantity. Capsular polysaccharide is a major virulence determinant for numerous bacteria, as it has the ability to evade host phagocytosis and immune responses, resulting in immunodeficiency and bacterial resistance against host defenses (Wang et al., 2023). Cereulide ranked second in virulence gene number (7 genes). It is an important toxin produced by pathogenic B. cereus, and is encoded by the ces gene cluster and synthesized by the regulation of RRPS (Kalbhenn et al., 2022). By q-PCR, hblC, nheB, and cesB were detected but only cesB was positive (Supplementary Table 6). In addition, the hemolysin BL gene (hblA, hblC, hblD) and non-hemolytic enterotoxin Nhe gene (nheC) associated with diarrhea toxins were discovered in the nine B. cereus strains (Table 1). The results showed that cesB had a high copy number, but the hblC gene was not detected in B. cereus.
Table 1. Analysis of virulence factors.
4 Discussion
According to annotation analysis of public databases, the metabolic pathways of the nine B. cereus isolates were mainly focused on amino acid metabolism, carbohydrate metabolism, membrane transport, caffeine, vitamin metabolism, and energy metabolism. Amino acid metabolism in organisms mainly includes the synthesis of proteins, peptides and other nitrogen-containing substances. Amino acids can also be broken down into α-keto acids, amines and carbon dioxide through deamination, transamination, combined deamination or decarboxylation. The synthesis of B. cereus toxins is catalyzed by multiple enzymes. Therefore, the metabolism of amino acids in B. cereus has potential functions in regulating the synthesis and expression of virulence factors (Tran et al., 2011; Yu et al., 2011). Carbohydrates function primarily as the energy source for organisms through glycolysis, tricarboxylic acid cycle, pentose phosphate pathway, gluconeogenesis, gluconeogenesis, etc. (Li et al., 2020). The functions of carbohydrates in LY01-LY09 fell into four categories according to the CAZy database, whose number of genes from high to low were glycosyltransferases (GTs), glycoside hydrolases (GHs), carbohydrate esterolytic enzymes (CEs), and oxidoreductase (AAs). GTs can catalyze the formation of glycosidic bonds between specific sugars and receptors. Their substrates encompass a range of biomolecules, including sugars, proteins, lipids, and other small molecules that are essential for the synthesis of biological compounds (Hsieh et al., 2014). The analysis of the metabolic pathways of B. cereus increases our understanding of the mechanism of synthesis and secretion of bacterial toxins.
By comparative genomics, we found more than 99.98% ANI between each pair of the nine B. cereus genomes. At the same time, the results of collinearity and SNP analyses also showed high similarity between strains. Therefore, we speculate these nine strains to be the same one, which caused the foodborne outbreaks. The results of phylogenetic and virulence factor analyses were consistent with the above conclusion. Interestingly, phylogenetic analysis revealed that the LY01-LY09 strains identified in this study was closely related to B. cereus and B. anthracis (Figure 1). B. cereus and B. anthracis, which are closely related, are members of the B. cereus s.l. group, as evidenced by the high collinearity and similarity in their chromosomes and proteins (Pilo and Frey, 2011). Therefore, it is difficult to discriminate B. cereus from B. anthracis, particularly for isolates exhibiting unusual biochemical or physiological characteristics. It is usually not enough when relying solely on genome-wide information to differentiate between the two species. Phenotype and plasmid content are also needed to take into consideration (Ehling-Schulz et al., 2019; Lin et al., 2022; Trunet et al., 2023). Phylogenetic analysis revealed that B. cereus and B. thuringiensis were clustered within the same clade, indicating a close evolutionary relationship. Previous research has indicated that B. cereus and B. thuringiensis are highly similar in genome. The only difference between the two strains is that B. thuringiensis can produce parasporal crystals in the outer spore membrane during spore formation, which is encoded by the insecticidal plasmids. The removal of this plasmid can result in the conversion of B. thuringiensis to B. cereus (Shu-sen et al., 2007; Venkateswaran et al., 2017). These results further confirm the high genetic similarity and close relationship between diarrheal toxin-producing B. cereus and B. thuringiensis.
By exploring the alterations in gene families across species, it was found that B. cereus, B. thuringiensis and B. anthracis have expanded significantly. Conversely, the gene families of the isolated B. cereus strains have contracted. We speculate that variations in the number of gene families may be responsible for the phenotypic differences of the three Bacillus species. This reflects genomic variations among different strains of the same species. To further investigate the relationship among the LY01-LY09 strains, we constructed an evolutionary tree based on single-copy genes. The analysis revealed that B. thuringiensis strains, B. anthracis strains, LY01 and LY09 formed a closely related clade, which is consistent with conclusions of previous studies. Previous research has shown that B. cereus s.l. can be classified into three separate clades. Most B. cereus can be classified into two clades: clade A, which is capable of producing emetic toxin, and clade B, which cannot. B. thuringiensis, an insect pathogen, mainly belongs to clade B, whereas B. anthracis was relatively conservative and restricted to clade A (Ehling-Schulz et al., 2019; Biggel et al., 2022; Carroll et al., 2022). As illustrated in Figure 1, the close clustering pattern between LY01-LY09 and B. anthracis suggests that LY01-LY09 may be pathogens that are harmful to humans as well. Previous studies have demonstrated that HSP20 exhibits chaperone activity, which can enhance cellular tolerance to damage and maintain normal cellular metabolism under various stresses, thus improving cell survival rates (Kranzler et al., 2016; Ling et al., 2022; Niu et al., 2022). LY07 and LY09 had more than one Hsp20 genes, some of which undergone point mutations and loss of long amino acids. Therefore, we speculated that the LY07 and LY09 strains may exhibit lower susceptibility to environmental stressors such as temperature, demonstrating greater adaptation to adverse conditions in comparison to the LY01 and LY08 strains.
Non-ribosomal polypeptide synthetase are the major systemic enzymes that regulate the synthesis of emetic toxins (Dommel et al., 2010). This study identified many NRPS genes in the LY01-LY09 strains, which may suggest that the LY01-LY09 strains can induce the vomiting-type food poisoning by synthesizing the emetic toxin cereulide regulated by NRPS. The NRPS biosynthetic machinery is a vast modular multi-enzyme complex that participates in the production of diverse natural products, including toxins, antibiotics, and surfactants. The emetic toxin cereulide is a typical product among these compounds (Bozhüyük et al., 2019). In a previous study, PCR was employed to explore the NRPS genes of an emetic strain of B. cereus, which confirmed that cereulide was produced via NRPS in B. cereus (Toh et al., 2004). NRPS plays a key role in the biosynthesis of emetic toxins (cereulide). The gene locus encoding Cereulide synthetase shows the typical structure of the NRPS genes clusters, which plays an important role in the synthetic expression of B. cereus cereulide (Magarvey et al., 2006; Makarasen et al., 2009). Meanwhile, we have identified seven genes (cesA, cesB, cesC, cesD, cesH, cesP, and cesT) within the emetic toxin synthetase gene cluster (ces gene cluster) in the LY01-LY09 strains. CesPTABCD can form a ces-operon in the megaplasmid pBCE4810, which is transcribed into a 23 kb polycistronic mRNA chain driven by the main promoter p1. Adjacent to CesPTABCD, cesH is transcribed as a single cistron by its own promoter PH and encodes a 31 kDa hydrolase (Ehling-Schulz et al., 2015; Lücking et al., 2015). CesT is involved in the synthesis of phosphopantetheinyl transferase encoded by cesP, which plays a crucial role in initiating non-ribosomal synthesis. In contrast, cesA and cesB are important in peptide assembly due to their high affinity for Nicotinamide adenine dinucleotide phosphate (NADPH) (Alonzo et al., 2015), while cesC and cesD are involved in encoding ATP-binding cassette (ABC) transporters (Dommel et al., 2010). The B. cereus strains isolated in this study carried a complete ces gene-cluster sequence and the diarrheal toxin nheC, as well as a substantial number of NRPS genes. We speculate that the isolated B. cereus regulated the ces gene cluster to synthesize emetic toxins mainly via the NRPS system, thereby inducing foodborne illness associated with vomiting (Ehling-Schulz et al., 2006).
5 Conclusion
In this study, nine strains of B. cereus (LY01-LY09) were successfully isolated from food samples of foodborne outbreaks. Their whole genome sequences were obtained by high-throughput sequencing. Based on this, the genetic relationship and gene family of LY01-LY09 and their related strains were studied via comparative genomics. The results showed that the nine strains belonged to B. cereus s.l and they were the same strain. Their genome has unique characteristics compared with B. thuringiensis and B. anthracis. According to the analysis of virulence factors of LY01-LY09, it was found that they all contained emetic toxin and NRPS gene clusters. Genomic characteristics of the strains were explained. These results provide insights for further study of the evolutionary relationship of B. cereus and the mechanism of toxin synthesis and secretion. This study provides a scientific basis for the prevention and rapid diagnosis of foodborne diseases at the genome level.
Data availability statement
The original contributions presented in this study are included in this article/Supplementary material, further inquiries can be directed to the corresponding author.
Author contributions
LZ designed the experiments and applied for fund. SZ, SL, JH, and YW went to the incidence sites to collect food samples and conducted the tests to identify the B. cereus strains. YL performed the q-PCR and prepared data. QZ, GL, YC, and JX have worked together to analyze data and wrote the manuscript, in which QZ drafted the Results and Discussion section. GL conducted comparative genome analysis. All authors participated in the rounds of manuscript revision, contributed to the manuscript, and approved its submission and publication.
Funding
This research was funded by the Guizhou Provincial Science and Technology Support Program [Qiankehe Support (2021) General 435]; the Guizhou Provincial Health Commission Science and Technology Fund (gzwkj 2023-492); and the Guizhou Province Infectious Disease Prevention and Control Talent Base Scientific Research Team Program (RCJD2105 and RCJD2102).
Conflict of interest
YS was employed by Yunnan Pulis Technology Co., Ltd.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1238799/full#supplementary-material
Footnotes
- ^ https://www.genome.jp/kegg
- ^ http://www.tcdb.org
- ^ www.phi-base.org
- ^ http://www.cazy.org/
- ^ http://www.mgc.ac.cn/VFs/
- ^ https://meme-suite.org/meme/
References
Alonzo, D., Magarvey, N., and Schmeing, T. (2015). Characterization of cereulide synthetase, a toxin-producing macromolecular machine. PLoS One 10:e0128569. doi: 10.1371/journal.pone.0128569
Anon (1995). Bacillus cereus in foods. Enumeration and Confirmation. Microbiological Methods. Rockville: AOAC International.
Antunes, P., Novais, C., and Peixe, L. (2020). Food-to-humans bacterial transmission. Microbiol Spectr. 8, 161–193. doi: 10.1128/microbiolspec.MTBP-0019-2016
Balloux, F., Brønstad Brynildsrud, O., van Dorp, L., Shaw, L., Chen, H., Harris, K., et al. (2018). From theory to practice: Translating whole-genome sequencing (WGS) into the clinic. Trends Microbiol. 26, 1035–1048. doi: 10.1016/j.tim.2018.08.004
Biggel, M., Jessberger, N., Kovac, J., and Johler, S. (2022). Recent paradigm shifts in the perception of the role of Bacillus thuringiensis in foodborne disease. Food Microbiol. 105:104025. doi: 10.1016/j.fm.2022.104025
Bozhüyük, K., Micklefield, J., and Wilkinson, B. (2019). Engineering enzymatic assembly lines to produce new antibiotics. Curr. Opin. Microbiol. 51, 88–96. doi: 10.1016/j.mib.2019.10.007
Carroll, L., Cheng, R., Wiedmann, M., and Kovac, J. (2022). Keeping up with the Bacillus cereus group: Taxonomy through the genomics era and beyond. Crit Rev Food Sci Nutr. 62, 7677–7702. doi: 10.1080/10408398.2021.1916735
Carroll, L., and Wiedmann, M. (2020). Cereulide synthetase acquisition and loss events within the evolutionary history of group III Bacillus cereus sensu lato facilitate the transition between emetic and diarrheal foodborne pathogens. mBio 11:e01263-20. doi: 10.1128/mBio.01263-20
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Darriba, D., Taboada, G., Doallo, R., and Posada, D. (2011). ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Dietrich, R., Jessberger, N., Ehling-Schulz, M., Märtlbauer, E., and Granum, P. (2021). The food poisoning toxins of Bacillus cereus. Toxins 13:98. doi: 10.3390/toxins13020098
Dommel, M., Frenzel, E., Strasser, B., Blöchinger, C., Scherer, S., and Ehling-Schulz, M. (2010). Identification of the main promoter directing cereulide biosynthesis in emetic Bacillus cereus and its application for real-time monitoring of ces gene expression in foods. Appl. Environ. Microbiol. 76, 1232–1240. doi: 10.1128/AEM.02317-09
Edgar, R. (2004). MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Ehling-Schulz, M., Frenzel, E., and Gohar, M. (2015). Food-bacteria interplay: Pathometabolism of emetic Bacillus cereus. Front. Microbiol. 6:704. doi: 10.3389/fmicb.2015.00704
Ehling-Schulz, M., Fricker, M., Grallert, H., Rieck, P., Wagner, M., and Scherer, S. (2006). Cereulide synthetase gene cluster from emetic Bacillus cereus: Structure and location on a mega virulence plasmid related to Bacillus anthracis toxin plasmid pXO1. BMC Microbiol. 6:20. doi: 10.1186/1471-2180-6-20
Ehling-Schulz, M., Lereclus, D., and Koehler, T. (2019). The Bacillus cereus group: Bacillus species with pathogenic potential. Microbiol. Spectr. 7.
Emms, D., and Kelly, S. (2019). OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 20:238. doi: 10.1186/s13059-019-1832-y
He, S., and Shi, X. (2021). Microbial food safety in China: Past, present, and future. Foodborne Pathog. Dis. 18, 510–518. doi: 10.1089/fpd.2021.0009
Hsieh, Y., Chiu, H., Huang, Y., Fun, H., Lu, C., Li, Y., et al. (2014). Purification, crystallization and preliminary X-ray crystallographic analysis of glycosyltransferase-1 from Bacillus cereus. Acta Crystallogr. F Struct. Biol. Commun. 70(Pt 9), 1228–1231. doi: 10.1107/S2053230X14014629
Jain, C., Rodriguez-R, L., Phillippy, A., Konstantinidis, K., and Aluru, S. (2018). High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat. Commun. 9:5114. doi: 10.1038/s41467-018-07641-9
Josephs-Spaulding, J., Beeler, E., and Singh, O. (2016). Human microbiome versus food-borne pathogens: Friend or foe. Appl. Microbiol. Biotechnol. 100, 4845–4863. doi: 10.1007/s00253-016-7523-7
Kalbhenn, E., Kranzler, M., Gacek-Matthews, A., Grass, G., Stark, T., Frenzel, E., et al. (2022). Impact of a novel pagr-like transcriptional regulator on cereulide toxin synthesis in emetic Bacillus cereus. Int. J. Mol. Sci. 23:11479. doi: 10.3390/ijms231911479
Kotiranta, A., Lounatmaa, K., and Haapasalo, M. (2000). Epidemiology and pathogenesis of Bacillus cereus infections. Microbes Infect. 2, 189–198. doi: 10.1016/s1286-4579(00)00269-0
Kranzler, M., Stollewerk, K., Rouzeau-Szynalski, K., Blayo, L., Sulyok, M., and Ehling-Schulz, M. (2016). Temperature exerts control of Bacillus cereus emetic toxin production on post-transcriptional levels. Front. Microbiol. 7:1640. doi: 10.3389/fmicb.2016.01640
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: An information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kurtz, S., Phillippy, A., Delcher, A., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Li, W., Cui, Q., Bai, L., Fu, P., Han, H., Liu, J., et al. (2021). Application of whole-genome sequencing in the national molecular tracing network for foodborne disease surveillance in China. Foodborne Pathog. Dis. 18, 538–546. doi: 10.1089/fpd.2020.2908
Li, Y., Zhao, M., Chen, W., Du, H., Xie, X., Wang, D., et al. (2020). Comparative transcriptomic analysis reveals that multiple hormone signal transduction and carbohydrate metabolic pathways are affected by Bacillus cereus in Nicotiana tabacum. Genomics 112, 4254–4267. doi: 10.1016/j.ygeno.2020.07.022
Lin, S., and Liao, Y. C. (2013). CISA: Contig integrator for sequence assembly of bacterial genomes. PLoS One 8:e60843. doi: 10.1371/journal.pone.0060843
Lin, Y., Briandet, R., and Kovács, Á (2022). Bacillus cereus sensu lato biofilm formation and its ecological importance. Biofilm 4:100070. doi: 10.1016/j.bioflm.2022.100070
Ling, Y., Ling, Z., and Zhao, R. (2022). Construction of a heat-resistant strain of Lentinus edodes by fungal Hsp20 protein overexpression and genetic transformation. Front. Microbiol. 13:1009885. doi: 10.3389/fmicb.2022.1009885
Liu, B., Zheng, D., Zhou, S., Chen, L., and Yang, J. (2022). VFDB 2022: A general classification scheme for bacterial virulence factors. Nucleic Acids Res. 50, D912–D917. doi: 10.1093/nar/gkab1107
Lücking, G., Frenzel, E., Rütschle, A., Marxen, S., Stark, T., Hofmann, T., et al. (2015). Ces locus embedded proteins control the non-ribosomal synthesis of the cereulide toxin in emetic Bacillus cereus on multiple levels. Front. Microbiol. 6:1101. doi: 10.3389/fmicb.2015.01101
Magarvey, N., Ehling-Schulz, M., and Walsh, C. (2006). Characterization of the cereulide NRPS alpha-hydroxy acid specifying modules: Activation of alpha-keto acids and chiral reduction on the assembly line. J. Am. Chem. Soc. 128, 10698–10699. doi: 10.1021/ja0640187
Makarasen, A., Yoza, K., and Isobe, M. (2009). Higher structure of cereulide, an emetic toxin from Bacillus cereus, and special comparison with valinomycin, an antibiotic from Streptomyces fulvissimus. Chem. Asian J. 4, 688–698. doi: 10.1002/asia.200900011
Marxen, S., Stark, T., Rütschle, A., Lücking, G., Frenzel, E., Scherer, S., et al. (2015). Depsipeptide intermediates interrogate proposed biosynthesis of cereulide, the emetic toxin of Bacillus cereus. Sci. Rep. 5:10637. doi: 10.1038/srep10637
Niu, H., Yang, M., Qi, Y., Liu, Y., Wang, X., and Dong, Q. (2022). Heat shock in Cronobacter sakazakii induces direct protection and cross-protection against simulated gastric fluid stress. Food Microbiol. 103:103948. doi: 10.1016/j.fm.2021.103948
Pilo, P., and Frey, J. (2011). Bacillus anthracis: Molecular taxonomy, population genetics, phylogeny and patho-evolution. Infect. Genet. Evol. 11, 1218–1224. doi: 10.1016/j.meegid.2011.05.013
Pires, S., Desta, B., Mughini-Gras, L., Mmbaga, B., Fayemi, O., Salvador, E., et al. (2021). Burden of foodborne diseases: Think global, act local. Curr. Opin. Food Sci. 39, 152–159. doi: 10.1016/j.cofs.2021.01.006
Premkrishnan, B., Heinle, C., Uchida, A., Purbojati, R., Kushwaha, K., Putra, A., et al. (2021). The genomic characterisation and comparison of Bacillus cereus strains isolated from indoor air. Gut Pathog. 13:6. doi: 10.1186/s13099-021-00399-4
Sánchez Chica, J., Correa, M., Aceves-Diez, A., Rasschaert, G., Heyndrickx, M., and Castañeda-Sandoval, L. (2020). Genomic and toxigenic heterogeneity of bacillus cereus sensu lato isolated from ready-to-eat foods and powdered milk in day care centers in Colombia. Foodborne Pathog. Dis. 17, 340–347. doi: 10.1089/fpd.2019.2709
Shu-sen, S., Li-li, J., and Fang, W. (2007). Study on genetic relationship between Bacillus thuringiensis and Bacillus cereus. Chin. J. Public Health 23, 321–323.
Stamatakis, A. (2014). RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stenfors Arnesen, L., Fagerlund, A., and Granum, P. (2008). From soil to gut: Bacillus cereus and its food poisoning toxins. FEMS Microbiol. Rev. 32, 579–606. doi: 10.1111/j.1574-6976.2008.00112.x
Toh, M., Moffitt, M., Henrichsen, L., Raftery, M., Barrow, K., Cox, J., et al. (2004). Cereulide, the emetic toxin of Bacillus cereus, is putatively a product of nonribosomal peptide synthesis. J Appl Microbiol. 97, 992–1000. doi: 10.1111/j.1365-2672.2004.02381.x
Tran, S., Guillemet, E., Ngo-Camus, M., Clybouw, C., Puhar, A., Moris, A., et al. (2011). Haemolysin II is a Bacillus cereus virulence factor that induces apoptosis of macrophages. Cell Microbiol. 13, 92–108. doi: 10.1111/j.1462-5822.2010.01522.x
Trunet, C., Cauquil, A., Hymery, N., Koullen, L., Postollec, F., and Coroller, L. (2023). Are Bacillus thuringiensis strains like any other Bacillus cereus strains? Phenotypic-based tools to locate Bacillus thuringiensis in the diversity of the Bacillus cereus sensu lato group. Res. Microbiol. 174:104077. doi: 10.1016/j.resmic.2023.104077
Venkateswaran, K., Singh, N., Checinska Sielaff, A., Pope, R., Bergman, N., van Tongeren, S., et al. (2017). Non-toxin-producing Bacillus cereus strains belonging to the B. Anthracis clade isolated from the International Space Station. mSystems 2:e00021-17. doi: 10.1128/mSystems.00021-17
Wang, Y. J., Li, L., Yu, J., Hu, H. Y., Liu, Z. X., Jiang, W. J., et al. (2023). Imaging of Escherichia coli K5 and glycosaminoglycan precursors via targeted metabolic labeling of capsular polysaccharides in bacteria. Sci. Adv. 9:eade4770. doi: 10.1126/sciadv.ade4770
Yu, C., Wang, Y., Xu, C., He, J., Zhang, Q., and Yu, Z. (2011). [Analyze and compare metabolic pathways of Bacillus cereus group]. Yi Chuan 33, 1057–1066. doi: 10.3724/sp.j.1005.2011.01057
Keywords: Bacillus cereus, comparative genomic analysis, virulence factor, metabolic pathway, foodborne outbreak
Citation: Zhou Q, Li G, Cui Y, Xiang J, Zhu S, Li S, Huang J, Wang Y, Liu Y and Zhou L (2024) Genomic characterization of Bacillus cereus isolated from food poisoning cases revealed the mechanism of toxin production. Front. Microbiol. 14:1238799. doi: 10.3389/fmicb.2023.1238799
Received: 12 June 2023; Accepted: 12 December 2023;
Published: 12 January 2024.
Edited by:
Yujie Hu, China National Center for Food Safety Risk Assessment, ChinaReviewed by:
Pengfei Yu, Hubei University of Medicine, ChinaZixin Peng, China National Center for Food Safety Risk Assessment, China
Copyright © 2024 Zhou, Li, Cui, Xiang, Zhu, Li, Huang, Wang, Liu and Zhou. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Li Zhou, emhvdWxpMDgxMkAxMjYuY29t
†These authors have contributed equally to this work and share first authorship