
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 28 July 2023
Sec. Extreme Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1228266
 Samuel I. de Jong1
Samuel I. de Jong1 Dimitry Y. Sorokin1,2
Dimitry Y. Sorokin1,2 Mark C. M. van Loosdrecht1
Mark C. M. van Loosdrecht1 Martin Pabst1*
Martin Pabst1* Duncan G. G. McMillan1*
Duncan G. G. McMillan1*Proteomics has greatly advanced the understanding of the cellular biochemistry of microorganisms. The thermoalkaliphile Caldalkalibacillus thermarum TA2.A1 is an organism of interest for studies into how alkaliphiles adapt to their extreme lifestyles, as it can grow from pH 7.5 to pH 11. Within most classes of microbes, the membrane-bound electron transport chain (ETC) enables a great degree of adaptability and is a key part of metabolic adaptation. Knowing what membrane proteins are generally expressed is crucial as a benchmark for further studies. Unfortunately, membrane proteins are the category of proteins hardest to detect using conventional cellular proteomics protocols. In part, this is due to the hydrophobicity of membrane proteins as well as their general lower absolute abundance, which hinders detection. Here, we performed a combination of whole cell lysate proteomics and proteomics of membrane extracts solubilised with either SDS or FOS-choline-12 at various temperatures. The combined methods led to the detection of 158 membrane proteins containing at least a single transmembrane helix (TMH). Within this data set we revealed a full oxidative phosphorylation pathway as well as an alternative NADH dehydrogenase type II (Ndh-2) and a microaerophilic cytochrome oxidase ba3. We also observed C. thermarum TA2.A1 expressing transporters for ectoine and glycine betaine, compounds that are known osmolytes that may assist in maintaining a near neutral internal pH when the external pH is highly alkaline.
The biological membrane is where the cell meets its environment, forming the boundary between the cytoplasm and the outside world and also at the point where the cell senses environmental changes. They are composed of proteins, lipids, lipid-like molecules, and sugars. The way in which the cell senses these changes is through membrane-embedded and membrane-associated proteins (Boes et al., 2021). Proteins in the membrane have diverse functions, from the obvious bioenergetic processes, such as facilitating the active transport of useful solutes into the cell, to broader regulatory functions. The dual function is also common, such as for an enzyme found in the membrane-bound pathway for cellular respiration, an enzyme known as the F1Fo ATP synthase. On the one hand, its purpose is to harness both a proton or sodium gradient alongside membrane potential to regenerate ATP, a form of chemical cellular energy; on the other hand, it is also thought to regulate cytoplasmic pH (Krah et al., 2018).
The membranes of extremophiles are even more important as a physical barrier, as they have a much more protective role against the adverse environment outside the cell. For example, conditions including high concentrations or high temperatures, high salinity, extreme acidity, or alkalinity are common in extreme environments. Thermophiles adapt by producing more saturated lipids or by branching fatty acids, and sometimes lipids may have complex structures (Reizer et al., 1985; Russell and Fukunaga, 1990; Sinninghe Damsté et al., 2011), increasing their rigidity (Russell and Fukunaga, 1990). With alkaliphiles, the defining environmental pressure is a lack of environmental proton availability. This leads to a situation where there is a lower pH inside the cell than outside the cell – a so-called “inverted pH gradient.” In aerobic alkaliphiles, this is a mountainous challenge and a complete bioenergetics conundrum because cellular energy generation is coupled to this inverted proton gradient (Krulwich, 1995; Horikoshi, 2004; McMillan et al., 2007). When these two environmental pressures are combined in a given organism, the membrane is theoretically very sensitive to leakage, so it must prevent the loss of protons to the outside environment (Takami, 2000; Yumoto, 2002; Olsson et al., 2003; McMillan et al., 2007). Adaptations to minimize proton leakage are localized to the membrane (Koch, 1986; Krulwich, 1995). The architecture of the lipid bilayer is important for its protective properties, but for all other membrane processes, proteins are involved. Adaptations to cope with this environment are proposed to include a highly saturated and branched lipid composition (Siliakus et al., 2017), making solubilisation of such membranes to extract membrane proteins for downstream analysis a challenging process. This situation is a “nightmare scenario” in terms of a proteomics approach to understanding environmental adaptation at the cell membrane.
Proteomics of membrane proteins is challenging in general. The typical protocol for membrane protein detection and quantification relies on proteolytic digestion of soluble or solubilised proteins, followed by liquid-chromatography linked to mass-spectrometry (MS) detection. Subsequently, the processing of the data obtained is delineated using various database search tools. This yields a tremendous amount of information but invariably underrepresents the membrane proteins (Sievers, 2018). Most of the transmembrane proteins harbor a large number of hydrophobic TMHs. The hydrophobic nature of these proteins makes them insoluble in aqueous environments, a particular challenge in MS detection. Moreover, TMHs are protected from proteolytic digestion, a key requirement to generate peptide fragments for MS detection. With these proteins, only the short loops connecting such helices and soluble domains are accessible for proteolytic cleavage. To add to these challenges, membrane proteins suffer from low absolute expression levels because membrane “real estate” is severely limited (Fischer et al., 2006; Helbig et al., 2010). Solubilisation of membrane extracts is a potential approach for the extraction of integral membrane proteins, yet defining a universal protocol for that solubilisation is seemingly impossible (Schuck et al., 2003; Eshaghi et al., 2005). A key reason for the problems in building such a protocol is that every organism constructs its membrane to suit the conditions under which it grows, i.e., an organism will adapt the membrane composition to avoid compromising on membrane fluidity or polarity (Uda et al., 2001; Meador et al., 2014; Mendoza, 2014). In general, the literature on solubilising membranes for proteomics focuses on well-studied model organisms, such as Escherichia coli (Eshaghi et al., 2005). The field of membrane proteomics in extremophiles is poorly developed. For thermophiles, a few studies also focus on this subject. Two published works detail whole-cell studies in Clostridium thermocellum (Williams et al., 2007; Burton and Martin, 2012), and membrane solubilisation of the archaeon Sulfolobus solfataricus is summarized (Zaparty et al., 2009). For alkaliphiles, two studies have been reported. One investigates membrane solubilisation with urea in Alkalihalobacillus marmarensis (Altinisik Kaya et al., 2018), while the other focuses on a few other well-known yet ultimately ineffective detergents for Alkalimonas amylolytica (Wang et al., 2009). None of these studies rely on a comparative approach that assesses the contribution of each step to the protocol. Here, we attempted to address this shortcoming and advance the field of membrane proteomics for polyextremophiles.
In this study, we focus on a polyextremophile, the thermoalkaliphile Caldalkalibacillus thermarum TA2.A1. This microorganism is highly adapted to the harsh conditions that it lives in (pH 9.5 at 65°C) (Peddie et al., 1999) and has a high degree of variability in its ETC (de Jong et al., 2020). We reveal the membrane proteome by implementing different solubilisation techniques while considering the organism's physiological growth temperature. Specifically, we present an overview of the detected membrane proteome of C. thermarum TA2.A1, focusing particularly on its transporters and ETC. Our findings also resulted in a noteworthy increase in detectable proteins and peptides.
Caldalkalibacillus thermarum TA2.A1 was grown in 5-L flat-bottomed shake flasks at 65°C with a working volume of 1 L rich medium, the composition of which was described previously (McMillan et al., 2009). A smaller pre-inoculum was grown beforehand in a 500-ml round-bottomed shake flask with a 100-ml working volume. The larger shake flasks were inoculated at 1% from the pre-inoculum. The incubations lasted for ~18 h. Cells were harvested by centrifugation at 8,000 × g and were subsequently washed. The fraction appropriated to whole cell proteomics (±100 mg cell wet weight) was washed in ice-cold phosphate-buffered saline (PBS) and thereafter flash frozen with liquid nitrogen and stored at −80°C. The fraction used for membrane proteomics was washed in 50 mM Tris–HCl buffer, pH 8.0, with 10% (w/v) glycerol and 2 mM MgCl2, and thereafter also flash frozen and stored at −80°C.
Membrane preparation was performed as described earlier (McMillan et al., 2007), with modifications. Briefly, samples were thawed gradually by placing them on top of a layer of ice. Extraction started with a lysis step in 50 mM Tris-HCl buffer, pH 8.0, with 10% (w/v) glycerol, 2 mM MgCl2, 1 mM phenylmethylsulphonyl fluoride, and 1 mM dithiothreitol. Lysis was initiated by the addition of 1.2 mg ml−1 lysozyme and 0.2 mg DNaseI. Lysed cells were subsequently passed through a cell disruptor twice at 20,000 psi. As a trial, the number of passages through the cell disruptor was decreased to a single passage, but that yielded no discernible difference (Supplementary Figure 1). The resulting liquid fraction, discarding the denatured fraction, was centrifuged at 8,000 × g for 10 min. The resulting supernatant was ultracentrifuged at 180,000 × g for 45 min. The supernatant was discarded, and the pellet was resuspended in ~2 ml of 50 mM Tris-HCl buffer, pH 8.0, with 10% (w/v) glycerol and 2 mM MgCl2. When required, extracted membranes were flash frozen and stored at −80°C. C. thermarum TA2.A1 membranes were solubilised in 200 μM ammonium-bicarbonate (ABC) buffer with either 2% (w/v) n-tetradecylphosphocholine (FOS-choline-12, Anatrace) or 0.01% (w/v) sodium dodecyl sulfate (SDS) for 30 min at 50°C, 65°C, or 80°C, while gently shaking at 300 rpm. Extracted membrane samples were diluted to a final concentration of 4 mg ml−1 protein. After solubilisation, samples were frozen at −20°C until further preparation.
The following protocols were based on extensive work done before (den Ridder et al., 2022, 2023) to optimize the protocols. For whole cell proteomics, the pellet stored after washing with PBS was thawed, and 29 mg of the sample was used and transferred to a LoBind Eppendorf tube. The pellet was dissolved in 0.175 ml of 1 M triethylammonium bicarbonate buffer, and 0.15 g of glass beads (105–212 μm, Sigma Aldrich) were added. The sample was subjected to a vortexing regime that was repeated thrice; the sample was vortexed at maximum speed for 90 s and then placed on ice for 30 s. Afterward, the sample was incubated at 80°C for 3 min at 1,000 rpm and subsequently ultrasonicated for 10 min. The sonicated sample was centrifuged, and the resulting supernatant (250 μl) was carefully pipetted into a new LoBind Eppendorf tube. The proteins were then precipitated by adding 62.5 μl of 6.1 N tricholoacetic acid by vortexing and subsequent incubation at 4°C for 30 min. After centrifugation, the resulting pellet was washed with 200 μl of ice-cold acetone and then centrifuged again. The washed pellet was redissolved in 50 μl of 6 M urea. The sample was then subjected to the proteolytic digestion protocol as described below.
After thawing, in the case of the membrane extract, samples were transferred to a LoBind Eppendorf tube, vortexed, and an additional 50 μl of 200 mM ABC buffer was added. A total of 100 μl (≈100 μg protein) of that mix was used for reduction and alkylation. For the whole cell sample, proteolytic digestion occurred straight after lysate preparation. In this case, the preceding protocol yielded 30 μg of protein. For the following part, two volumes are given for every step; the lower volume corresponds to the whole cell sample and the higher volume to the membrane extracts. The reduction was performed by incubation with 8.5 or 30 μl of 10 mM dithiothreitol at 37°C for 60 min at 300 rpm. Alkylation was subsequently performed by adding 8.5 or 30 μl of 20 mM iodoacetamide and incubating at 20°C for 30 min in the dark. Samples were then further diluted with 285 or 400 μl ABC, and 5 or 20 μl of 0.1 μg μl−1 trypsin was added for digestion. Digestion occurred for ~18 h at 37°C and 300 rpm. After digestion, all samples were treated equally again. After digestion, peptides were extracted by solid-phase extraction in an Oasis HLB 96-well μElution Plate (Waters) according to manufacturer specifications. Peptides were eluted in two phases: (1) with 200 μl of 2% formic acid in 80% methanol and (2) with 200 μl of 1 mM ABC in 80% methanol. The peptides were then collected in an Integrated SpeedVacTM System (Thermo Scientific) at 50°C for 2 h. Samples were stored at −20°C until analysis.
Shotgun proteomic analysis was performed as described earlier (Kleikamp et al., 2021; Pabst et al., 2021). Speed vac dried samples were dissolved in 20 μl of 3% acetonitrile plus 0.1% trifluoroacetic acid. Protein concentrations were estimated by NanoDrop ND-1000 spectrophotometry (Thermo Scientific). An aliquot corresponding to ~0.5 μg of protein was analyzed using a one-dimensional shotgun proteomics approach using a nano-liquid chromatography system consisting of an EASY nano-LC 1200 (Acclaim PepMap RSLC RP C18 separation column, 50 × 150 mm, 2 μm, Cat. No. 164568) and a QE plus Orbitrap mass spectrometer (Thermo Fisher Scientific, Germany). The flow rate was maintained at 350 nl/min over a linear gradient from 5 to 25% solvent B over 88 min, then from 25 to 55% over 60 min, followed by back equilibration to starting conditions. Data were acquired from 5 to 240 min. Solvent A was H2O containing 0.1% formic acid, and solvent B consisted of 80% ACN in H2O and 0.1% FA. The Orbitrap was operated in data-dependent acquisition (DDA) mode, acquiring peptide signals from 385 to 1,250 m/z at 70 K resolution in full MS mode with a maximum ion injection time (IT) of 75 ms and an automatic gain control (AGC) target of 3E6. The top 10 precursors were selected for MS/MS analysis and subjected to fragmentation using higher-energy collisional dissociation (HCD). MS/MS scans were acquired at 17.5 K resolution with an AGC target of 2E5 and an IT of 75 ms, a 2 m/z isolation width, and normalized collision energy (NCE) of 28. Data were analyzed against the proteome database from C. thermarum [GenBank: CP082237.1, strain Caldalkalibacillus thermarum TA2.A1 complete genome, Tax ID: 559292 (de Jong et al., 2020)] using PEAKS Studio X (Bioinformatics Solutions Inc., Waterloo, Canada), allowing for 20 ppm parent ion and 0.02 m/z fragment ion mass error, three missed cleavages, carbamidomethylation and methionine oxidation, and N/Q deamidation as variable modifications. Peptide spectrum matches were filtered against 1% false discovery rates (FDR), and identifications with ≥2 unique peptides were considered significant. Results from PEAKS were exported to “proteins.csv,” listing the identified proteins.
Proteins from database searching files (Supplementary Table 1) were further processed using Python (version 3.9). Files were converted to “Pandas” dataframes and counted based on the following two criteria: “Area” > 0 and “Unique Peptides” ≥ 2. Venn diagrams were then visualized using the “Matplotlib” functions “venn2_unweighted” or “venn3_unweighted.” For the Venn diagrams concerning only membrane proteins, an additional criterion was added (TMH's ≥ 1). For the heat map, the raw absolute area was used directly, after filtering using Supplementary Table 2. The heat maps were visualized using the function ‘vizus.gene_exp_hmap' from the package ‘Bioinfokit'.
We first performed a conventional whole-cell lysate shotgun proteomics experiment to detect the soluble protein fraction. The full genome of C. thermarum TA2.A1 (de Jong et al., 2020) contains 3,085 genes (NCBI; accession: CP082237), of which 1,244 (Figure 1A) were detected in this experiment (~40% of the genome). DTU's TMHMM tool was used to distinguish which of these proteins were likely integral membrane proteins, using a single TMH as the cut-off (Krogh et al., 2001). The C. thermarum TA2.A1 genome contains 754 proteins out of the total 3,085 that contain a single predicted TMH. It should be noted that proteins associated with the membrane (peripheral membrane proteins) but lacking a TMH are not “soluble proteins” and are also included in our analysis. Peripheral membrane proteins are membrane-associated via hydrophobic patches, lipid anchors, or lateral helices (Boes et al., 2021). However, these proteins cannot be identified from genetic information or be unequivocally identified or in a whole-cell proteome analysis without supporting biochemical data. Our analysis confirmed that only 124 of the 1,244 proteins detected contain a TMH. Considering that roughly a quarter of all proteins in the complete genome will be either integral or peripheral membrane proteins (Wallin and Von Heijne, 1998; Sawada et al., 2007), this reinforces the notion that the membrane proteome is generally underrepresented in conventional whole cell lysate proteomics experiments. Nevertheless, the whole-cell proteomics approach yields a membrane proteome with a considerable proportion of membrane proteins. For instance, the majority of the aerobic ETC proteins were detected. The integral membrane protein NADH dehydrogenase type I (Ndh-1) was detectable, as was the peripheral membrane protein NADH dehydrogenase type II (Ndh-2) (Godoy-Hernandez et al., 2019; Godoy-Hernandez and McMillan, 2021; Juergens et al., 2021). Other integral membrane proteins, such as succinate dehydrogenase (Sdh) and the cytochrome b6c1 complex (a hybrid complex III), were also detectable, as well as the cytochrome oxidase aa3 complex. The cytochrome oxidase aa3 complex is expected to be the primary terminal oxidase under fully aerobic conditions. However, we also detected a single subunit pertaining to the microaerophilic cytochrome oxidase ba3 complex. Surprisingly, the subunit detected contains 13 TMHs, while the soluble subunits were undetectable. However, caution should be taken with such a result because to confidently state that C. thermarum TA2.A1 expressed two terminal oxidases under the growth conditions we imposed, detecting the additional subunits provides greater clarity toward the notion of functionality. Finally, the ATP synthase was also detected, revealing that we can get very close to detecting a fully functional C. thermarum TA2.A1 pathway for oxidative phosphorylation from whole cell proteomics.
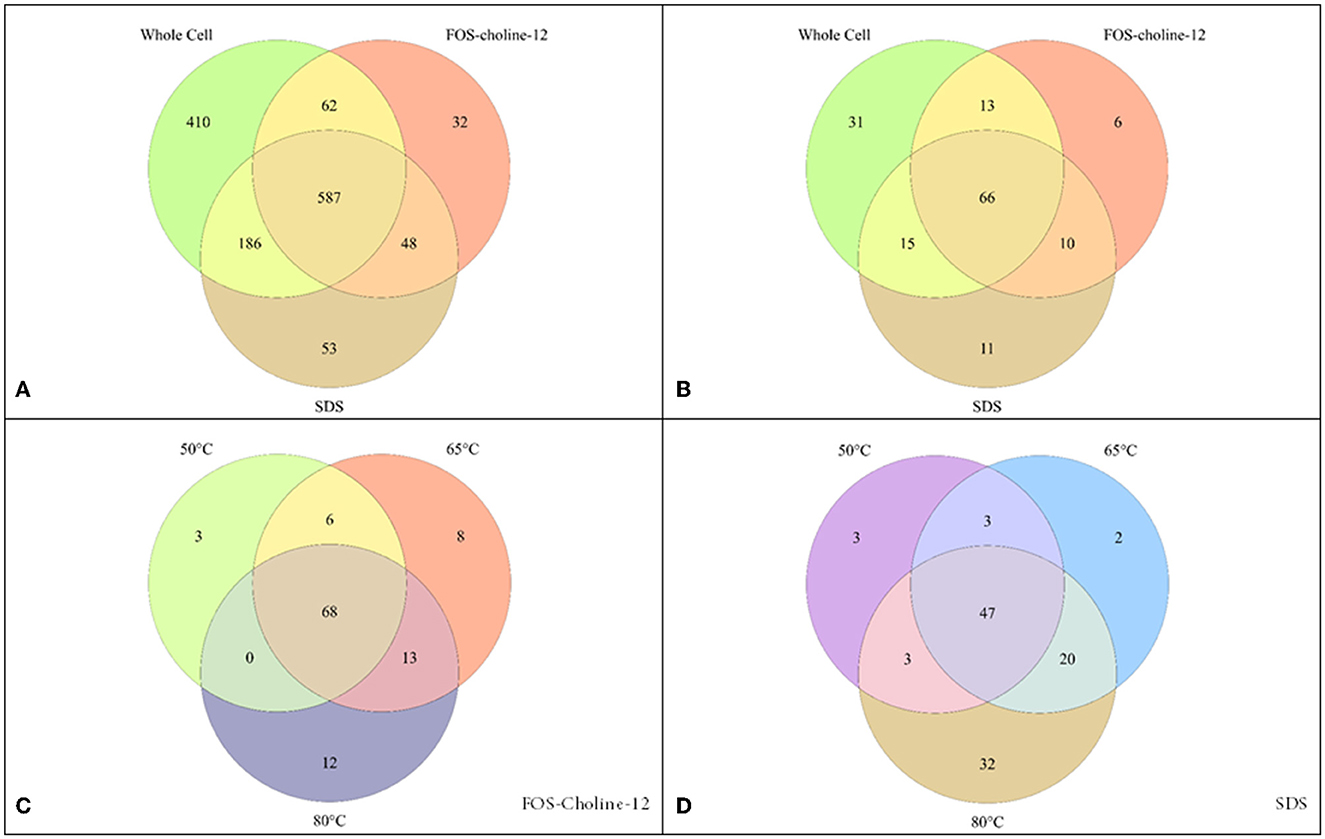
Figure 1. Detection of proteins by proteomics methods with different solubilisation methods. (A) covers the entire potential proteome, while (B–D) zoom in on solely the membrane proteins. (A, B) compare the detection using whole cell proteomics vs. that with solubilisation with FOS-choline-12 at 65°C and SDS at 80°C. (C, D) show the effect of solubilisation at different temperatures (50°C, 65°C, and 80°C) using either FOS-choline-12 (C) or SDS (D).
To better evaluate the possible expression of the cytochrome oxidase ba3, as well as many more functional proteins, increased detection is paramount. Unfortunately, membrane proteins are lesser in number in a cell, so a whole-cell proteome analysis including soluble proteins dwarfs the lower signals of membrane proteins (Tan et al., 2008). Isolating the membrane provides an initial step toward enrichment. Second, detergent solubilisation can singularly isolate proteins and is an easily accessible method to purify proteins for this reason (McMillan et al., 2007; Pullara et al., 2013; Mathieu et al., 2019). Proteins are in a crowded space in the biological membrane, and this crowding can prevent access by the proteases necessary for proteomics. Finally, as previously mentioned, for peripheral membrane proteins lacking biochemical description, we cannot rely on genetic information and cannot be unequivocally identified in whole-cell proteome analysis (Boes et al., 2021).
To obtain better coverage of the membrane proteome, a modified membrane extraction protocol was followed. While extracting the membrane fraction from the rest of the cell constituents added a very modest increase in protein hits, adding a membrane solubilisation step significantly increased the detection of membrane proteins (Figures 1A, B). The breadth of solubilisation methods available in the literature (Schuck et al., 2003; Eshaghi et al., 2005; Williams et al., 2007; Wang et al., 2009; Zaparty et al., 2009; Burton and Martin, 2012; Altinisik Kaya et al., 2018) leaves a plethora of options available, but it is well noted that there is no common optimal method. Given our knowledge of membrane proteins, we opted for the use of detergents. Most detergents are not compatible with mass spectrometry, thus limiting options. Fortunately, two extremely effective detergents for membrane protein solubilisation were feasible [FOS-choline-12 and sodium dodecyl sulfate (SDS)]. Additionally, the effect of the temperature (50°C, 65°C, and 80°C) used for solubilisation was assessed. For solubilisation with FOS-choline-12, using 65°C or 80°C yielded optimal results, while for SDS, 50°C was clearly optimal (Figures 1C, D). Additionally, cross-comparison between the two detergents showed that each detergent appears to solubilise the membrane protein fraction selectively (Figure 1B). Finally, we tested whether changing the digestion enzyme made a difference in protein detection. An in silico digestion (Supplementary Figure 2) showed potential improvement in the optimal peptide length range using both LysC and chymotrypsin as digestion enzymes. Contrary to expectations, neither increased the detection of membrane proteins significantly, having only a modest effect (Figures 2, 3). Combining all proteomics experiments allowed for the construction of an overview of all detected proteins containing a TMH, as well as zooming in on the ETC and known transporters.
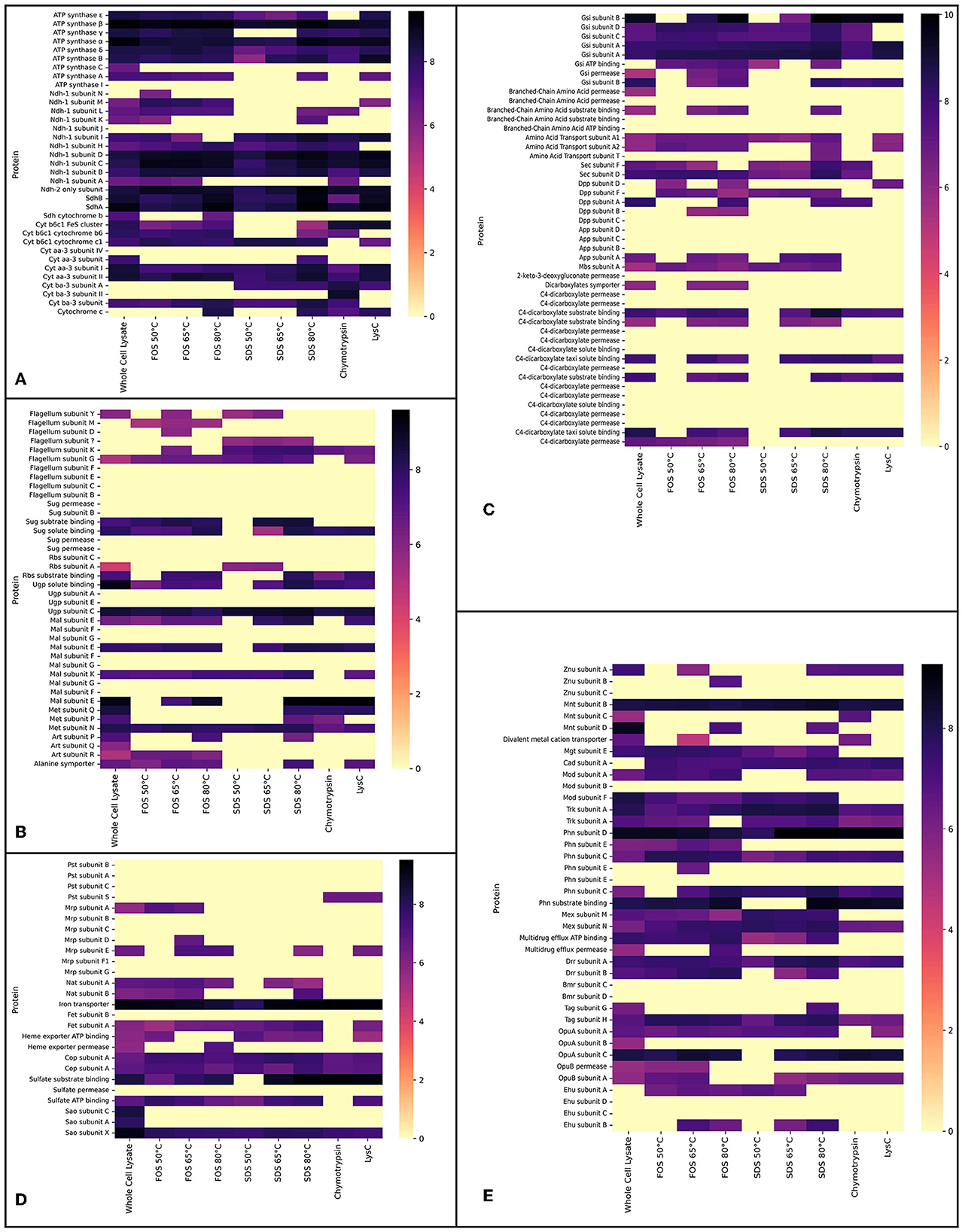
Figure 2. Proteomics data showing the logarithm (base 10) of absolute abundances detected in the whole cell lysate experiment and the experiments in which membrane extracts were solubilised with either FOS-choline-12 or SDS. For FOS-choline-12 and SDS, the temperature of solubilisation is given. Experiments with the alternative digestion enzymes, Chymotrypsin and LysC, are accrued together. (A) Heat map shows proteins of the ETC. (B) Heat map shows proteins of the flagellum and transporters of sugars amino acids. (C) Heat map shows transporters of primarily secondary metabolites; (A–C) together resemble heat maps corresponding to Figure 3. (D, E) Heat maps selectively show transporters of metals and other inorganic cations and anions, as in Figure 4. Heat maps show all subunits pertaining to a certain protein complex if at least a single subunit was detected in one of the conditions. Note that proteins absent here (but shown in both Figures 3, 4) were not detected by any method.
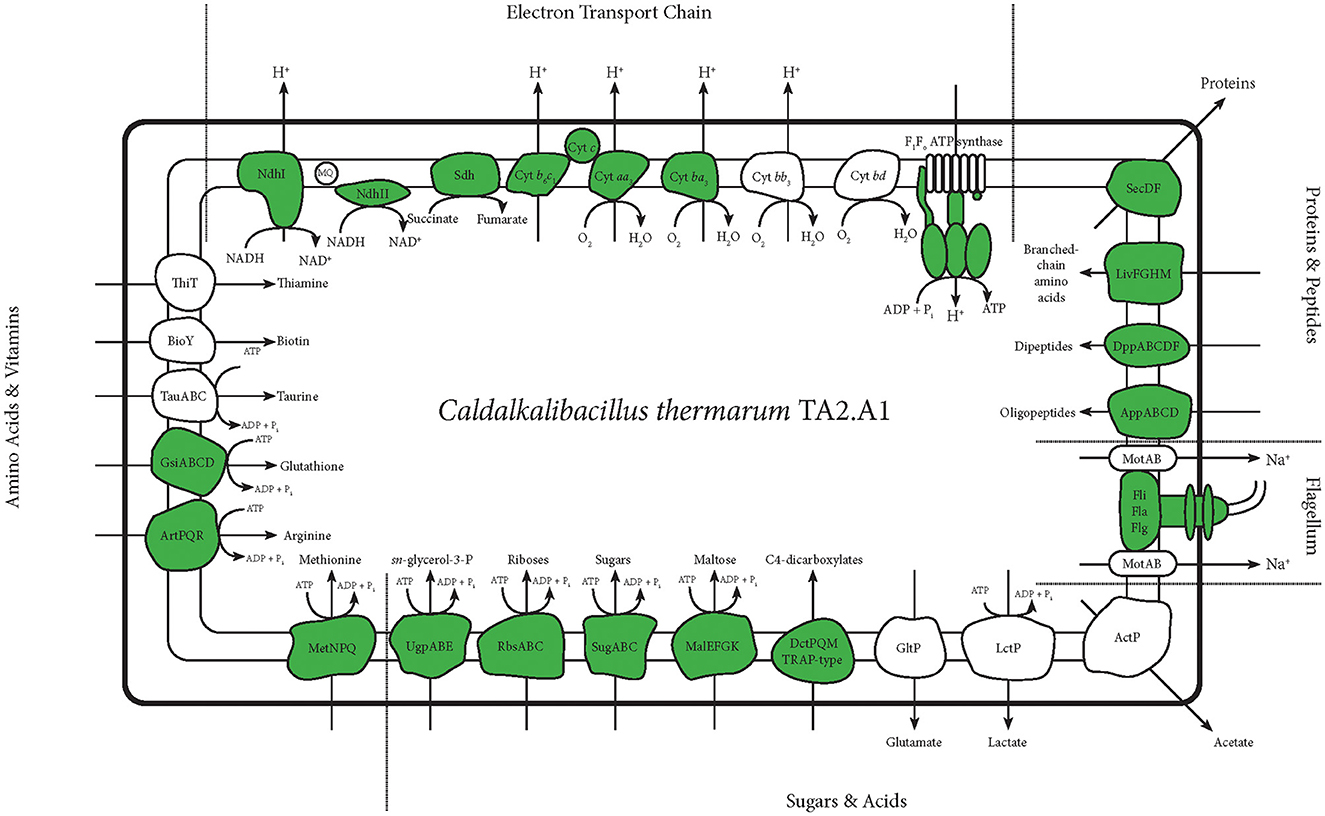
Figure 3. An overview of membrane proteins detected with any of the abovementioned protocols. This figure selectively shows all membrane proteins concerning the ETC and transporters concerning central carbon metabolism and anabolism. It also shows the flagellum. Detected proteins are depicted in green. For the ATP synthase and the flagellum, this figure is more detailed, and only the subunits detected are depicted in green.
The main goal of our study was to identify as many membrane proteins as possible through the optimisation of membrane proteomics. In total, 158 membrane proteins were detected using the standard whole cell proteome method; although a full aerobic ETC was detected, not all subunits of each complex were found. Solubilisation enabled the detection of the other two remaining subunits pertaining to the microaerophilic cytochrome oxidase ba3 (Figure 2A). The aerobic and most efficient ETC C. thermarum TA2.A1 consists of Ndh-1, Sdh, cytochrome complex b6c1, cytochrome c, and cytochrome oxidase aa3. The expression of the F1Fo ATP synthase completes an aerobic oxidative phosphorylation pathway. All these complexes were detected (Figure 3).
Besides the expected aerobic ETC, somewhat surprisingly, ETC components that have been proposed to be more tuned toward a microaerophilic lifestyle, Ndh-2 and cytochrome oxidase ba3, were expressed under aerobic conditions. The ba3 complex proteins were among the few extra proteins detected after digestion with chymotrypsin or LysC instead of the standard trypsin digestion. The detection of Ndh-2 is in line with earlier research on this enzyme in C. thermarum TA2.A1, where it has been studied as a potential analog for drug targets (Heikal et al., 2014; Godoy-Hernandez et al., 2019; Godoy-Hernandez and McMillan, 2021). Finding the presence of cytochrome oxidase ba3 was more surprising, as it is only half as efficient as aa3 (Von Ballmoos et al., 2012). The expression could be due to the culturing conditions for this trial. A simple shake flask batch was used, which could have led to some oxygen limitation at the end of the batch phase, forcing C. thermarum TA2.A1 to adopt a more austere lifestyle. Such questions could be solved by culturing under more controlled conditions, such as a chemostat. We would expect that culturing at lower dissolved oxygen would also trigger the expression of the other two terminal oxidases, namely cytochromes bb3 and bd.
In terms of carbon import machinery, a range of different transporters was detected, pertaining to the SugABC, RbsABC, MalEFGK, DctPQM, and UgpABC complexes (Figures 2A, 3). Considering the use of sucrose as a carbon source, the detection of multiple subunits of SugABC was expected (Figure 2B). MalEFGK is annotated as a maltose importer but might transport sucrose as well; this dual specificity is described in Streptococcus mutans (Kilic et al., 2007). The detection of possibly two sucrose importers means this research has failed to highlight the genes encoding for the sucrose:sodium symporter that originally sparked the interest in C. thermarum TA2.A1 (Peddie et al., 1999, 2000). The detection of the TRAP-type C4-dicarboxylate importers DctPQM, RbsABC complex, and UgpABC complex (Figures 2A, 3) may be due to their scavenging functions rather than their favored presence under our culturing conditions. In line with other alkaliphiles, no defined medium for C. thermarum TA2.A1 exists, and it still requires the presence of tryptic peptone extract to grow, which could contain a substrate for all of these transporters. In agreement with this proposition, several amino acid importers, oligopeptide importers, and importers of related compounds, like glutathione, were also detected. Finally, the proteins translocase SecDF and the flagellum proteins were also detected.
Homeostasis of monovalent cations is crucial for any alkaliphile, and generally, this is coupled with proton transport in an antiport protein (Krulwich and Cheng, 1994). C. thermarum TA2.A1 has two types of sodium:proton antiporters; a few subunits of the multisubunit Mrp were detected under the conditions used. The active ATP powered NatABC was also expressed. C. thermarum TA2.A1 relies on the TrkAH system for potassium uptake. The low-affinity TrkAH system found in most bacteria has been highlighted as a strategy to combat saline stress (Kraegeloh et al., 2005; Guo et al., 2009). C. thermarum TA2.A1 is capable of growing up to 6% NaCl (Xue et al., 2006), perhaps in part due to its reliance on this potassium uptake system. Two proteins pertaining to the A subunit of the potassium:proton antiporter TrkAH were detected (Figures 2B, 4); the trkA genes encoding for the expression of these proteins are not part of a single operon or are they close to any of the other trkA or trkH genes. As an importer of inorganic phosphate, the subunit S of the Pts complex was detected only in the experiments using alternative digestion enzymes (Figures 2B, 4). For the import of trace metals, transporters for zinc, molybdate, magnesium, and manganese were detected (Figure 4).
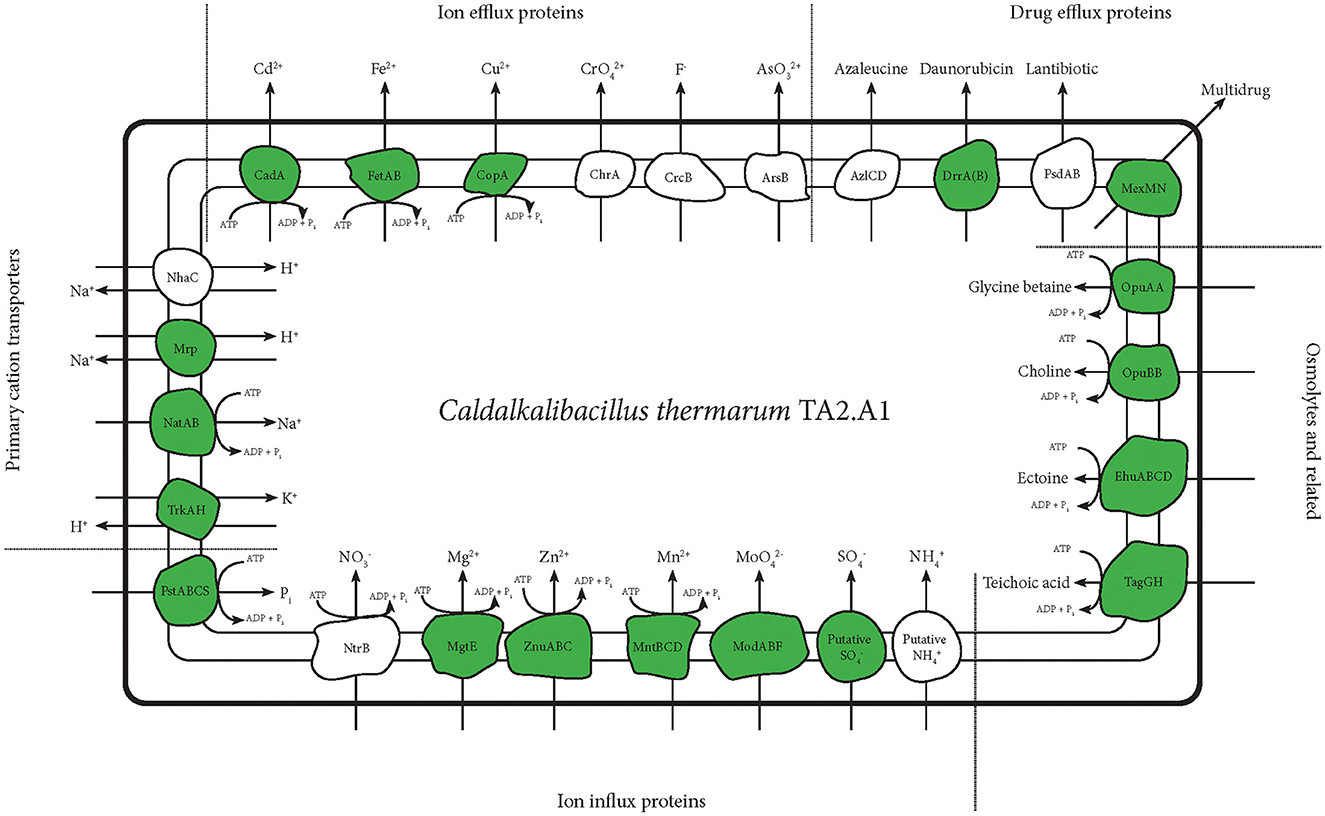
Figure 4. An overview of transporters detected with any of the abovementioned protocols. This figure selectively shows transporters of metals and inorganic cations and anions. Transporters related to drug efflux and osmolyte import are also shown. The teichoic acid importer is displayed, being regarded as an important protein for alkaliphiles (Aono and Uramoto, 1986; Aono et al., 1995, 1999), even though previous tests showed teichoic acid presence in C. thermarum TA2.A1 (Kleikamp et al., 2020). Detected proteins are depicted in green.
Other metal transporters, such as cadmium, copper and iron, were also detected. Notably, these were all described as exporters (Yoon and Silver, 1991; Rensing et al., 2000). The ferrous exporter FetAB was previously described as an exporter and implied as an important protein in avoiding problems with radical formation (Nicolaou et al., 2013). Several drug exporters were also detected, further shedding light on the possible mechanisms C. thermarum TA2.A1 might use to remain dominant in a given environment. Finally, several importers related to osmolytes were detected. The incorporation of transporters of glycine betaine and ectoine could be another method used by C. thermarum TA2.A1 to combat the challenges posed by the high pH at which it lives. Especially glycine betaine was abundantly available in the rich medium used; the strategy of utilizing compatible solutes should be perfectly viable.
This study is the first report on the proteome of C. thermarum TA2.A1, in which 1,398 proteins (45.3% of all potential proteins) were detected in at least one of the experiments. Of this, 158 contain at least a single TMH (11.3% of all identified proteins) and are therefore considered membrane proteins. The total coverage of the membrane proteome is 20.9%. C. thermarum TA2.A1 is an alkaliphile, and therefore this study focuses primarily on proteins identified in the ETC and on transporters. For an alkaliphile, homeostasis of protons, sodium, and potassium is crucial, and this study detected transporters for all of these compounds. Importers of various compounds related to central carbon metabolism and anabolism were also detected, as well as a complete ETC. Combined, this study gives a comprehensive overview of the membrane proteins required by an alkaliphile to survive and provides a valuable benchmark for further research on this organism. The authors specifically note its adaptability to a broad pH range as an area for further research. The conditions under which C. thermarum TA2.A1 employs its alternative terminal oxidases are also yet to be revealed. In that regard, performing solubilisation of membrane extracts as an addition to whole cell lysate proteomics will increase the chances of detecting these alternative terminal oxidases.
Here we report on a proteomic study where we investigated several sample preparation approaches to maximize the coverage for membrane proteins for C. thermarum TA2.A1. The whole cell proteomics experiment combined with proteomics of solubilised membrane extracts yielded 1,398 proteins (45.3% of the total proteome), of which 158 (11.3% of the discovered proteins; 20.9% of the membrane proteome) were membrane proteins. Interestingly, all different membrane solubilisation methods provided a unique set of membrane proteins. In terms of solubilisation of membrane proteins, we conclude that treatment with SDS at 50°C and the analysis of a sample treated with FOS-choline-12 at 65°C or 80°C proved most effective. The modest effect of alternative protease digestion, despite the promising in silico analysis, is a subtle but crucial warning that alternative complications, such as shielding of potential digestion sites, contain post-translational modifications such as glycosylations and lipidations.
The entire optimal ETC was detected using the combined methods for analysis, as well as two less efficient proteins – the Ndh-2 as an “alternative Complex I” (Biagini et al., 2006; Elguindy and Nakamaru-Ogiso, 2015) and the cytochrome oxidase ba3 as an alternative Complex IV. Additionally, we detected all proteins required for the transport of protons, sodium, and potassium, whose homeostasis is crucial to an alkaliphile. Finally, the expression of the glycine betaine and ectoine importers implies that C. thermarum TA2.A1 relies in part on osmolytes for its survival, something that has not been reported previously in the literature for this organism. Overall, this research has given a comprehensive overview of proteins that we expect to be expressed and shed light on some additional potentially interesting proteins. In short, this research provides a valuable benchmark for further research into alkaliphiles and C. thermarum TA2.A1 in particular.
Mass spectrometric raw data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository and are publicly available under the project code PXD042369.
SJ, DS, ML, MP, and DM conceptualized the study and wrote the manuscript. SJ performed biological work and extractions. MP performed proteomics experiments. All authors contributed to the article and approved the submitted version.
The study of SJ was funded by the SIAM Gravitation Grant from the Netherlands Organization for Scientific Research (024.002.002).
The authors would like to thank Martijn Wissink for his contribution to this project as part of his master's thesis. The authors would also like to thank Dita Heikens for her help with preparing samples for proteomics.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1228266/full#supplementary-material
Supplementary Figure 1. SDS-PAGE gel (Any kDTM Mini_PROTEAN® TGXTM, Bio-Rad) of extracted membranes. Ladder (Precision Plus ProteinTM Unstained Protein Standards, Bio-Rad) was loaded on either side of the samples; 1× pass indicates a sample of extracted membranes that only passed through the cell disruptor once, while 2× pass indicates a sample of extracted membranes that passed through the cell disruptor twice, confirming the regular protocol. Samples were diluted beforehand in MilliQ water to ensure 5 μg protein was loaded in the well. Samples were dyed with a mix of 2-mercaptoethanol and 4× Leammli Sample Buffer (Bio-Rad) according to the manufacturer's specifications. After running at 200 V for 45 min, the gel was submerged in SimplyBlueTM Safe Stain (Invitrogen) for 2 h and subsequently destained overnight in a 5% methanol and 7.5% acetic acid solution. This picture was taken afterwards.
Supplementary Figure 2. Violin plot showing an in silico digestion profile of the C. thermarum TA2.A1 membrane proteome using different digestion enzymes: Trypsin, LysC, chymotrypsin, and combining LysC with chymotrypsin. Sizes are relative to the number of peptides, distributed by peptide length over the y-axis. Horizontal lines indicate the range of peptide lengths (from 7 to 35 amino acids) considered ideal for detection by LC-MS.
Supplementary Table 1. Raw mass spectrometry data of all performed conditions. The absolute peak area and the number of unique peptides are shown for all proteins detected per condition. Some column headers are self-explanatory, as the name directly refers to a specific condition. For those that do not, the order is as follows: FOS, solubilised at 65°C (S1); FOS, solubilised at 65°C (S2); FOS, solubilised at 50°C (S3); FOS, solubilised at 65°C (S4); FOS, solubilised at 80°C (S5); SDS, solubilised at 50°C (S6); SDS, solubilised at 65°C (S7); and SDS, solubilised at 80°C (S8).
Supplementary Table 2. The locus tag, protein id, gene, and subunit identifier (Accession: CP082237) for proteins are shown in Figures 2–4. An arbitrary decision was made as to which complexes, aside from the ETC, should be included in Figures 2A, B, 3, 4 respectively.
Altinisik Kaya, F. E., Avci, F. G., Sayar, N. A., Kazan, D., Sayar, A. A., and Sariyar Akbulut, B. (2018). What are the multi-omics mechanisms for adaptation by microorganisms to high alkalinity? A transcriptomic and proteomic study of a bacillus strain with industrial potential. Omi. A J. Integr. Biol. 22, 717–732. doi: 10.1089/omi.2018.0127
Aono, R., Ito, M., Joblin, K. N., and Horikoshi, K. (1995). A high cell wall negative charge is necessary for the growth of the alkaliphile Bacillus lentus C-125 at elevated pH. Microbiology 141, 2955–2964. doi: 10.1099/13500872-141-11-2955
Aono, R., Ito, M., and Machida, T. (1999). Contribution of the cell wall component teichuronopeptide to pH homeostasis and alkaliphily in the alkaliphile Bacillus lentus C-125. J. Bacteriol. 181, 6600–6606. doi: 10.1128/jb.181.21.6600-6606.1999
Aono, R., and Uramoto, M. (1986). Presence of fucosamine in teichuronic acid of the alkalophilic Bacillus strain C-125. Biochem. J. 233, 291–294. doi: 10.1042/bj2330291
Biagini, G. A., Viriyavejakul, P., O'Neill, P. M., Bray, P. G., and Ward, S. A. (2006). Functional characterization and target validation of alternative complex I of Plasmodium falciparum mitochondria. Antimicrob. Agents Chemother. 50, 1841–1851. doi: 10.1128/AAC.50.5.1841-1851.2006
Boes, D. M., Godoy-Hernandez, A., and McMillan, D. G. G. (2021). Peripheral membrane proteins: Promising therapeutic targets across domains of life. Membranes 11, 346. doi: 10.3390/membranes11050346
Burton, E., and Martin, V. J. J. (2012). Proteomic analysis of clostridium thermocellum atcc 27405 reveals the upregulation of an alternative transhydrogenase-malate pathway and nitrogen assimilation in cells grown on cellulose. Can. J. Microbiol. 58, 1378–1388. doi: 10.1139/cjm-2012-0412
de Jong, S. I., van den Broek, M. A., Merkel, A. Y., de la Torre Cortes, P., Kalamorz, F., Cook, G. M., et al. (2020). Genomic analysis of Caldalkalibacillus thermarum TA2.A1 reveals aerobic alkaliphilic metabolism and evolutionary hallmarks linking alkaliphilic bacteria and plant life. Extremophiles 24, 923–935. doi: 10.1007/s00792-020-01205-w
den Ridder, M., Knibbe, E., van den Brandeler, W., Daran-Lapujade, P., and Pabst, M. (2022). A systematic evaluation of yeast sample preparation protocols for spectral identifications, proteome coverage and post-isolation modifications. J. Proteomics 261, 104576. doi: 10.1016/j.jprot.2022.104576
den Ridder, M., van den Brandeler, W., Altiner, M., Daran-Lapujade, P., and Pabst, M. (2023). Proteome dynamics during transition from exponential to stationary phase under aerobic and anaerobic conditions in yeast. Mol. Cell. Proteomics. 22, 552. doi: 10.1016/j.mcpro.2023.100552
Elguindy, M. M., and Nakamaru-Ogiso, E. (2015). Apoptosis-inducing factor (AIF) and its family member protein, AMID, are rotenone-sensitive NADH:ubiquinone oxidoreductases (NDH-2). J. Biol. Chem. 290, 20815–20826. doi: 10.1074/jbc.M115.641498
Eshaghi, S., Hedrén, M., Nasser, M. I. A., Hammarberg, T., Thornell, A., and Nordlund, P. (2005). An efficient strategy for high-throughput expression screening of recombinant integral membrane proteins. Protein Sci. 14, 676–683. doi: 10.1110/ps.041127005
Fischer, F., Wolters, D., Rögner, M., and Poetsch, A. (2006). Toward the complete membrane proteome: High coverage of integral membrane proteins through transmembrane peptide detection. Mol. Cell. Proteomics 5, 444–453. doi: 10.1074/mcp.M500234-MCP200
Godoy-Hernandez, A., and McMillan, D. G. G. (2021). The profound influence of lipid composition on the catalysis of the drug target nadh type ii oxidoreductase. Membranes. 11, 363. doi: 10.3390/membranes11050363
Godoy-Hernandez, A., Tate, D. J., and McMillan, D. G. G. (2019). Revealing the membrane-bound catalytic oxidation of NADH by the drug target type-II NADH dehydrogenase. Biochemistry 58, 4272–4275. doi: 10.1021/acs.biochem.9b00752
Guo, Y. H., Xue, Y. F., Liu, J., Wang, Q. H., and Ma, Y. H. (2009). Characterization and function analysis of a Halo-alkaline-adaptable Trk K+ uptake system in Alkalimonas amylolytica strain N10. Sci. China, Ser. C Life Sci. 52, 949–957. doi: 10.1007/s11427-009-0132-2
Heikal, A., Nakatani, Y., Dunn, E., Weimar, M. R., Day, C. L., Baker, E. N., et al. (2014). Structure of the bacterial type II NADH dehydrogenase: A monotopic membrane protein with an essential role in energy generation. Mol. Microbiol. 91, 950–964. doi: 10.1111/mmi.12507
Helbig, A. O., Heck, A. J. R., and Slijper, M. (2010). Exploring the membrane proteome-challenges and analytical strategies. J. Proteomics 73, 868–878. doi: 10.1016/j.jprot.2010.01.005
Juergens, H., Mielgo-Gómez, Á., Godoy-Hernández, A., ter Horst, J., Nijenhuis, J. M., McMillan, D. G. G., et al. (2021). Physiological relevance, localization and substrate specificity of the alternative (type II) mitochondrial NADH dehydrogenases of Ogataea parapolymorpha. Available online at: http://biorxiv.org/content/early/2021/04/28/2021.04.28.441406.abstract (accessed April 28, 2021).
Kilic, A. O., Honeyman, A. L., and Tao, L. (2007). Overlapping substrate speci city for sucrose and maltose of two binding protein-dependent sugar uptake systems in Streptococcus mutans. FEMS Microbiol. Lett. 266, 218–223. doi: 10.1111/j.1574-6968.2006.00522.x
Kleikamp, H. B. C., Lin, Y. M., McMillan, D. G. G., Geelhoed, J. S., Naus-Wiezer, S. N. H., Van Baarlen, P., et al. (2020). Tackling the chemical diversity of microbial nonulosonic acids - a universal large-scale survey approach. Chem. Sci. 11, 3074–3080. doi: 10.1039/c9sc06406k
Kleikamp, H. B. C., Pronk, M., Tugui, C., Guedes da Silva, L., Abbas, B., Lin, Y. M., et al. (2021). Database-independent de novo metaproteomics of complex microbial communities. Cell Syst. 12, 375–383. doi: 10.1016/j.cels.2021.04.003
Koch, A. L. (1986). The pH in the neighborhood of membranes generating a protonmotive force. J. Theor. Biol. 120, 73–84. doi: 10.1016/S0022-5193(86)80018-2
Kraegeloh, A., Amendt, B., and Kunte, H. J. (2005). Potassium transport in a halophilic member of the Bacteria domain: Identification and characterization of the K+ uptake systems TrkH and TrkI from Halomonas elongata DSM 2581T. J. Bacteriol. 187, 1036–1043. doi: 10.1128/JB.187.3.1036-1043.2005
Krah, A., Zarco-Zavala, M., and McMillan, D. G. G. (2018). Insights into the regulatory function of the 1 subunit from bacterial F-type ATP synthases: a comparison of structural, biochemical and biophysical data. Open Biol. 8, 1–11. doi: 10.1098/rsob.170275
Krogh, A., Larsson, B., Von Heijne, G., and Sonnhammer, E. L. L. (2001). Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Krulwich, T. A. (1995). Alkaliphiles: “basic” molecular problems of pH tolerance and bioenergetics. Mol. Microbiol. 15, 403–410.
Krulwich, T. A., Cheng, J., and Guffanti, A. A. (1994). The role of monovalent cation/proton antiporters in Na+-resistance and pH homeostasis in Bacillus: An alkaliphile versus a neutralophile. J. Exp. Biol. 196, 457–470.
Mathieu, K., Javed, W., Vallet, S., Lesterlin, C., Candusso, M. P., Ding, F., et al. (2019). Functionality of membrane proteins overexpressed and purified from E. coli is highly dependent upon the strain. Sci. Rep. 9, 1–15. doi: 10.1038/s41598-019-39382-0
McMillan, D. G. G., Keis, S., Berney, M., and Cook, G. M. (2009). Nonfermentative thermoalkaliphilic growth is restricted to alkaline environments. Appl. Environ. Microbiol. 75, 7649–7654. doi: 10.1128/AEM.01639-09
McMillan, D. G. G., Keis, S., Dimroth, P., and Cook, G. M. (2007). A specific adaptation in the a subunit of thermoalkaliphilic F1Fo-ATP synthase enables ATP synthesis at high pH but not at neutral pH values. J. Biol. Chem. 282, 17395–17404. doi: 10.1074/jbc.M611709200
Meador, T. B., Gagen, E. J., Loscar, M. E., Goldhammer, T., Yoshinaga, M. Y., Wendt, J., et al. (2014). Thermococcus kodakarensis modulates its polar membrane lipids and elemental composition according to growth stage and phosphate availability. Front. Microbiol. 5, 1–13. doi: 10.3389/fmicb.2014.00010
Mendoza, D. (2014). Temperature sensing by membranes. Annu. Rev. Microbiol. 68, 101–116. doi: 10.1146/annurev-micro-091313-103612
Nicolaou, S. A., Fast, A. G., Nakamaru-Ogiso, E., and Papoutsakis, E. T. (2013). Overexpression of fetA (ybbL) and fetB (ybbM), encoding an iron exporter, enhances resistance to oxidative stress in Escherichia coli. Appl. Environ. Microbiol. 79, 7210–7219. doi: 10.1128/AEM.02322-13
Olsson, K., Keis, S., Morgan, H. W., Dimroth, P., and Cook, G. M. (2003). Bioenergetic properties of the thermoalkaliphilic Bacillus sp. strain TA2.A1. J. Bacteriol. 185, 461–465. doi: 10.1128/JB.185.2.461
Pabst, M., Grouzdev, D. S., Lawson, C. E., Kleikamp, H. B. C., de Ram, C., Louwen, R., et al. (2021). A general approach to explore prokaryotic protein glycosylation reveals the unique surface layer modulation of an anammox bacterium. ISME J. 2, 1–12. doi: 10.1038/s41396-021-01073-y
Peddie, C. J., Cook, G. M., and Morgan, H. W. (1999). Sodium-dependent glutamate uptake by an alkaliphilic, thermophilic Bacillus strain, TA2.A1. J. Bacteriol. 181, 3172–3177.
Peddie, C. J., Cook, G. M., and Morgan, H. W. (2000). Sucrose transport by the alkaliphilic, thermophilic Bacillus sp. strain TA2.A1 is dependent on a sodium gradient. Extremophiles 4, 291–296. doi: 10.1007/s007920070016
Pullara, F., Guerrero-Santoro, J., Calero, M., Zhang, Q., Peng, Y., Spåhr, H., et al. (2013). A general path for large-scale solubilization of cellular proteins: From membrane receptors to multiprotein complexes. Protein Expr. Purif. 87, 111–119. doi: 10.1016/j.pep.2012.10.007
Reizer, J., Grossowicz, N., and Barenholz, Y. (1985). The effect of growth temperature on the thermotropic behavior of the membranes of a thermophilic Bacillus. Composition-structure-function relationships. BBA - Biomembr. 815, 268–280. doi: 10.1016/0005-2736(85)90297-4
Rensing, C., Fan, B., Sharma, R., Mitra, B., and Rosen, B. P. (2000). CopA: An Escherichia coli Cu(I)-translocating P-type ATPase. Proc Natl Acad Sci U.S.A 97, 652–656. doi: 10.1073/pnas.97.2.652
Russell, N. J., and Fukunaga, N. (1990). A comparison of thermal adaptation of membrane lipids in psychrophilic and thermophilic bacteria. FEMS Microbiol. Lett. 75, 171–182. doi: 10.1016/0378-1097(90)90530-4
Sawada, R., Ke, R., Tsuji, T., Sonoyama, M., and Mitaku, S. (2007). Ratio of membrane proteins in total proteomes of prokaryota. Biophysics 3, 37–45. doi: 10.2142/biophysics.3.37
Schuck, S., Honsho, M., Ekroos, K., Shevchenko, A., and Simons, K. (2003). Resistance of cell membranes to different detergents. Proc Natl Acad Sci U.S.A 100, 5795–5800. doi: 10.1073/pnas.0631579100
Sievers, S. (2018). Membrane proteomics in gram-positive bacteria: two complementary approaches to target the hydrophobic species of proteins. Microb. Proteomics 8, 21–33. doi: 10.1007/978-1-4939-8695-8_3
Siliakus, M. F., van der Oost, J., and Kengen, S. W. M. (2017). Adaptations of archaeal and bacterial membranes to variations in temperature, pH and pressure. Extremophiles 21, 651–670. doi: 10.1007/s00792-017-0939-x
Sinninghe Damsté, J. S., Rijpstra, W. I. C., Hopmans, E. C., Weijers, J. W. H., Foesel, B. U., Overmann, J., et al. (2011). 13,16-Dimethyl octacosanedioic acid (iso-Diabolic Acid), a common membrane-spanning lipid of Acidobacteria subdivisions 1 and 3. Appl. Environ. Microbiol. 77, 4147–4154. doi: 10.1128/AEM.00466-11
Takami, H. (2000). Complete genome sequence of the alkaliphilic bacterium Bacillus halodurans and genomic sequence comparison with Bacillus subtilis. Nucleic Acids Res. 28, 4317–4331. doi: 10.1093/nar/28.21.4317
Tan, S., Hwee, T. T., and Chung, M. C. M. (2008). Membrane proteins and membrane proteomics. Proteomics 8, 3924–3932. doi: 10.1002/pmic.200800597
Uda, I., Sugai, A., Itoh, Y. H., and Itoh, T. (2001). Variation in molecular species of polar lipids from Thermoplasma acidophilum depends on growth temperature. Lipids 36, 103–105. doi: 10.1007/s11745-001-0914-2
Von Ballmoos, C., Ädelroth, P., Gennis, R. B., and Brzezinski, P. (2012). Proton transfer in ba 3 cytochrome c oxidase from Thermus thermophilus. Biochim. Biophys. Acta - Bioenerg. 1817, 650–657. doi: 10.1016/j.bbabio.2011.11.015
Wallin, E., and Von Heijne, G. (1998). Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 7, 1029–1038. doi: 10.1002/pro.5560070420
Wang, Q., Han, H., Xue, Y., Qian, Z., Meng, B., Peng, F., et al. (2009). Exploring membrane and cytoplasm proteomic responses of Alkalimonas amylolytica N10 to different external pHs with combination strategy of de novo peptide sequencing. Proteomics 9, 1254–1273. doi: 10.1002/pmic.200800244
Williams, T. I., Combs, J. C., Lynn, B. C., and Strobel, H. J. (2007). Proteomic profile changes in membranes of ethanol-tolerant Clostridium thermocellum. Appl. Microbiol. Biotechnol. 74, 422–432. doi: 10.1007/s00253-006-0689-7
Xue, Y., Zhang, X., Zhou, C., Zhao, Y., Cowan, D. A., Heaphy, S., et al. (2006). Caldalkalibacillus thermarum gen. nov., sp. nov., a novel alkalithermophilic bacterium from a hot spring in China. Int. J. Syst. Evol. Microbiol. 56, 1217–1221. doi: 10.1099/ijs.0.64105-0
Yoon, K. P., and Silver, S. (1991). A second gene in the Staphylococcus aureus cadA cadmium resistance determinant of plasmid pI258. J. Bacteriol. 173, 7636–7642. doi: 10.1128/jb.173.23.7636-7642.1991
Yumoto, I. (2002). Bioenergetics of alkaliphilic Bacillus spp. J. Biosci. Bioeng. 93, 342–353. doi: 10.1016/S1389-1723(02)80066-4
Keywords: (Poly-) extremophile, thermophile, alkaliphile, membrane, proteomics, mass spectrometry, solubilisation
Citation: de Jong SI, Sorokin DY, van Loosdrecht MCM, Pabst M and McMillan DGG (2023) Membrane proteome of the thermoalkaliphile Caldalkalibacillus thermarum TA2.A1. Front. Microbiol. 14:1228266. doi: 10.3389/fmicb.2023.1228266
Received: 24 May 2023; Accepted: 06 July 2023;
Published: 28 July 2023.
Edited by:
Philippe M. Oger, UMR5240 Microbiologie, Adaptation et Pathogenie (MAP), FranceReviewed by:
Alfredo Cabrera-Orefice, Goethe University Frankfurt, GermanyCopyright © 2023 de Jong, Sorokin, van Loosdrecht, Pabst and McMillan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Duncan G. G. McMillan, ZC5nLmcubWNtaWxsYW5AVFVEZWxmdC5ubA==; Martin Pabst, bS5wYWJzdEBUVURlbGZ0Lm5s
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.