 Yang Deng1,2†
Yang Deng1,2† Yu-Qin Zhang
Yu-Qin Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol., 08 September 2023
Sec. Evolutionary and Genomic Microbiology
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1225746
This article is part of the Research TopicEnvironmental Remediation Strategies of New and Emerging Chemical ContaminantsView all 13 articles
Mycobacterium species exhibit high bioremediation potential for the degradation of polycyclic aromatic hydrocarbons (PAHs) that are significant environmental pollutants. In this study, three Gram-positive, rapidly growing strains (YC-RL4T, MB418T, and HX176T) were isolated from petroleum-contaminated soils and were classified as Mycobacterium within the family Mycobacteriaceae. Genomic average nucleotide identity (ANI; < 95%) and digital DNA–DNA hybridization (dDDH; < 70%) values relative to other Mycobacterium spp. indicated that the strains represented novel species. The morphological, physiological, and chemotaxonomic characteristics of the isolates also supported their affiliation with Mycobacterium and their delineation as novel species. The strains were identified as Mycobacterium adipatum sp. nov. (type strain YC-RL4T = CPCC 205684T = CGMCC 1.62027T), Mycobacterium deserti sp. nov. (type strain MB418T = CPCC 205710T = KCTC 49782T), and Mycobacterium hippophais sp. nov. (type strain HX176T = CPCC 205372T = KCTC 49413T). Genes encoding enzymes involved in PAH degradation and metal resistance were present in the genomes of all three strains. Specifically, genes encoding alpha subunits of aromatic ring-hydroxylating dioxygenases were encoded by the genomes. The genes were also identified as core genes in a pangenomic analysis of the three strains along with 70 phylogenetically related mycobacterial strains that were previously classified as Mycolicibacterium. Notably, strain YC-RL4T could not only utilize phthalates as their sole carbon source for growth, but also convert di-(2-ethylhexyl) phthalate into phthalic acid. These results indicated that strains YC-RL4T, MB418T, and HX176T were important resources with significant bioremediation potential in soils contaminated by PAHs and heavy metals.
• Polycyclic aromatic hydrocarbons (PAHs) are significant environmental pollutants caused by human industrial activities.
• The bioremediation of PAHs is a promising method of pollutant mitigation and Mycobacterium species have been previously reported to hold considerable bioremediation potential.
• In this study, three novel species of Mycobacterium were isolated from polluted soil samples, formally characterized, and evaluated for bioremediation potential.
• The three strains were found to exhibit considerable potential for bioremediation of PAHs due to their physiological and genomic characteristics.
• Thus, these newly described species represent an important resource for bioremediation efforts.
The Mycobacterium genus encompasses a large group of Gram-positive, rod-shaped, acid-fast bacteria of the phylum Actinomycetota, that was first proposed by Skerman et al. (1980). Early classification of Mycobacterium was based on growth rate, pigmentation, and clinical significance (Runyon, 1959). Indeed, the fundamental taxonomic division of the group was connected to growth rate, such that species were defined as either slow or rapid growers. Rapid growers exhibit visible growth from dilute inocula within 7 days, while slow growers require over 7 days to achieve visible growth. Subsequently, immunological methods, in addition to comparisons of cell wall components, homologous enzyme sequences, DNA–DNA homologies, plasmid profiles, and restriction endonuclease analyses were used to complement the quantitative growth studies and infer natural relationships among Mycobacterium strains. Stahl and Urbance conducted a thorough phylogenetic analysis of Mycobacterium in 1990 based on comparison of 16S rRNA sequences, revealing phylogenetic relationships that were consistent with previous classifications (Stahl and Urbance, 1990). Lévy-Frébault and Portaels further proposed minimal standards for describing Mycobacterium species in 1992 (Lévy-Frébault and Portaels, 1992). These standards included acid-alcohol staining fastness, DNA G + C content levels, and mycolic acid presence. The recommended minimal standards for describing a new slowly growing Mycobacterium species were consequently based on phenotypic and genomic analyses. Takewaki et al. (1994) revealed species-specific restriction site profiles within amplified dnaJ genes in 1994 that could differentiate most Mycobacterium species based on a combination of PCR and restriction fragment length polymorphism analyses. The relationships among mycobacterial species were also concomitantly evaluated using the sequences of 23S rRNA gene spacers (Stone et al., 1995) in addition to those of several housekeeping genes including hsp65 (Kim et al., 2005), gyrB (Kasai et al., 2000), rpoB (Tortoli, 2012), and gyrA (Guillemin et al., 1995). Gupta et al. (2018) proposed a revision of mycobacteria taxonomy in 2018 that redistributed the 150 species of Mycobacterium into five genera based on synapomorphies. The four newly proposed genera comprised the non-tuberculous mycobacteria and included Mycobacteroides, Mycolicibacter, Mycolicibacterium, and Mycolicibacillus. Non-tuberculous mycobacteria exhibit diverse genomic backgrounds and physiological characteristics, but produce remarkably similar disease manifestations within at-risk populations. Consequently, Tortoli et al. (2019) suggested not renaming clinically important organisms in 2019, including non-tuberculous mycobacteria, suggesting that the use of the previously established Mycobacterium genus exhibited the advantage of avoiding confusion in health care settings. The genus Mycobacterium currently comprises 195 species with validly published names.1 The primary ecological niche for some Mycobacterium is the diseased tissue of warm-blooded hosts. Most human infections are caused by either M. leprae or M. tuberculosis, although other Mycobacterium strains are opportunistic pathogens of humans, particularly in immunocompromised individuals (Hartmans et al., 2006). However, most Mycobacterium species are non-pathogenic to humans and are inhabitants of many natural environments, including freshwaters and soils (Falkinham, 2009). In addition, several strains of Mycobacterium have been isolated from soils contaminated with polycyclic aromatic hydrocarbons (PAHs) like gasoline and coal tar (Willumsen et al., 2001; Hormisch et al., 2004; Zhang et al., 2012).
PAHs have become a significant environmental concern due to their potential toxic, mutagenic, and carcinogenic properties (Zeng et al., 2004). Mycobacterium species are typical PAH-degrading bacteria and can degrade high molecular weight PAHs by introducing two oxygen atoms to PAH compounds through dioxygenase enzymes (Khan et al., 2001). M. vanbaalenii PYR-1 was first shown to degrade low molecular weight compounds like 4-ring pyrene (Pereira et al., 2020). The unique cell wall layer of mycobacteria, including the presence of mycolic acids, plays an important role in mycobacterial degradation of hydrophobic PAHs (Abbasnezhad et al., 2011). Indeed, pyrene-degrading mycobacteria can degrade diverse PAHs, including naphthalene, phenanthrene, and fluoranthene, and even five-ring compounds like benzo[α]pyrene via co-metabolic biodegradation, subsequently using these compounds as carbon sources (Kweon et al., 2011). Zeng et al. (2017) revealed that the PdoAB dioxygenase from Mycobacterium sp. NJS-P exerts versatile functions in oxidizing various PAHs, including up to five-ring compounds like benzo[α]pyrene. Further, pdoAB can be induced during bacterial growth on pyrene and phenanthrene, implicating its function in PAH degradation. Mycobacterium species in rhizosphere soils have also been shown to accelerate the degradation of organic contaminants like PAHs and enhance plant resistance to soil pollution (Dai et al., 2020). Moreover, Yang et al. (2021) recently observed that the pyrene-degrading bacterium, Mycobacterium sp. Pyr9 exhibited diverse plant growth-promoting characteristics and a high tolerance to harsh environments, suggesting a high potential for bioremediation application (Yang et al., 2021). Thus, pyrene-degrading Mycobacterium strains may contribute to improving the safety of agricultural products and human health in PAHs polluted environments.
Phthalates are widely used industrial compounds and intensively studied environmental pollutants that exhibit endocrine disrupting properties. Strain NK0301 was the first Mycobacterium strain observed to degrade phthalates. It could degrade di-(2-ethylhexyl) phthalate (DEHP) into 2-ethylhexanol and 1,2-benzenedicarboxylic acid (Nakamiya et al., 2005). Wright et al. (2020) subsequently observed that the marine phthalate-degrading strain Mycobacterium sp. DBP42 could degrade diverse phthalates by transforming them into phthalic acid (PA) via di-alkyl phthalate esters (DAPs), and was also able to utilize PA as a growth substrate via the protocatechuate branch of the β-ketoadipate pathway.
Notably, some rapidly growing mycobacterial strains that were previously classified as Mycolicibacterium are also phthalate-degrading bacteria. Fourteen molecular markers (4 CSIs and 10 CSPs) are unique to members of the former Mycolicibacterium genus, supporting its monophyly and genetic cohesiveness. These strains could be distinguished from other Mycobacteriaceae genera, in addition to other bacteria, based on the presence of conserved signature indels in genes encoding the LacI family transcriptional regulator, cyclase, CDP-diacylglycerol–glycerol-3-phosphate 3-phosphatidyltransferase, and CDP-diacylglycerol–serine O-phosphatidyltransferase (Gupta et al., 2018). A novel DEHP-degrading marine bacterium was identified in 2021 as Mycolicibacterium phocaicum RL-HY01 that was isolated from intertidal sediments polluted by municipal wastewaters. The strain could transform DEHP into PA by β-oxidation and de-esterification and was further utilized within the gentisate branch of the β-ketoadipate pathway (Ren et al., 2021). Strain YC-RL4T was previously isolated from petroleum-contaminated soil and belonged to the Mycobacterium genus. The strain could utilize phthalates as their sole carbon source for growth and could also transform DEHP into PA via MEHP (mono (2-ethylhexyl) phthalate), with PA further being utilized for growth via the benzoic acid (BA) degradation pathway (Ren et al., 2016). Currently, limited research could be referred on the degradation of polycyclic aromatic hydrocarbons (PAHs) in environmental pollution by strains of the genus Mycobacterium. Especially, the taxonomic status of these PAHs-degrading strains and the clear understanding of the functional genes and metabolic pathways involved in PAHs degradation are still unclear. In this study, strain YC-RL4T (= CPCC 205684T = CGMCC 1.62027T) and two other strains that were closely related to each other were characterized, and their functional genes, metabolic pathways and potential for degrading PAHs were evaluated. Furthermore, this study also identified that these strains with PAHs degradation potential also possessed the potential heavy metal resistance. This remarkable bioremediation potential held significant importance for their future applications in the field of bioremediation. Polyphasic taxonomic analyses revealed that the three strains were novel Mycobacterium species with considerable application potential for the bioremediation of PAH-degrading soils.
Strain YC-RL4T was isolated from a petroleum-contaminated soil sample collected from Heze City (35°16′ N, 115°28′ E), Shandong Province, China, as described by Ren et al. (2016). Strain MB418T was isolated from a gravel soil sample collected from the Gurbantunggut desert (45°22′ N, 88°20′ E) in Xinjiang. Strain HX176T was isolated from rhizosphere soil associated with a medicinal plant in Xinjiang (43°36′ N, 82°11′ E). About 2 g of each sample was suspended in 18 mL of 0.85% (w/v) NaCl solution. Then, 200 μL of 10−4 diluted soil suspensions were spread on humic acid agar medium, as previously described (Deng et al., 2022). After incubation for 2 weeks at 28°C, visible colonies were picked and streaked on peptone yeast glucose (PYG) medium containing peptone (3 g L−1), yeast extract (5 g L−1), glycerol (10 g L−1), betaine hydrochloride (1.25 g L−1), sodium pyruvate (1.25 g L−1), and agar (15 g L−1), with adjustment to pH 7.2. The cultures were again incubated at 28°C to obtain isolated colonies. Purified isolates were maintained in glycerol suspensions (20%, v/v) at −80°C.
The reference strain M. fluoranthenivorans JCM 14741T was obtained from the Japan Collection of Microorganisms (JCM), while strains M. frederiksbergense DSM 44346T and M. litorale DSM 45785T were acquired from the German Collection of Microorganisms and Cell Cultures (DSMZ).
Strain growth was evaluated on glucose–yeast extract–malt extract agar (GYM; DSMZ medium 65), Middlebrook 7H10 agar (MB7H10; Difco), nutrient agar (NA; Difco), and tryptic soy agar (TSA; Difco), with incubation at 28°C for 48–72 h. Cell motility was examined by microscopic observations and inoculation on semisolid GYM medium with 0.3% agar (w/v). The growth conditions of strains YC-RL4T, MB418T, and HX176T were evaluated over temperature, pH, and salt tolerance ranges. The temperature range of growth was determined in MB7H10 mediu (Difco) at 4, 15, 20, 22, 25, 28, 30, 32, 35, 37, 45, and 50°C. The growth range across pH was determined in GYM medium at pH 4.0–10.0, at intervals of 1.0 unit. NaCl tolerance was determined with cultivation at 28°C in modified GYM medium (pH 7.0, without Na+ and Cl−), and with NaCl supplemented at concentrations of 0–10.0% (w/v) at increasing increments of 1.0%. All growth condition assays were performed in triplicate.
Biochemical and physiological tests were conducted for strains YC-RL4T, MB418T, HX176T and other closely related strains. Oxidase activity was investigated using the API oxidase reagent (bioMérieux) according to the manufacturer’s instructions. Catalase activity was evaluated via production of bubbles after addition of a drop of 3% (v/v) hydrogen peroxide. Voges–Proskauer, H2S production, and starch hydrolysis tests were evaluated using a biochemical identification kit (Huankai Microbial) according to the manufacturers’ instructions. Additional biochemical characteristics including enzymatic activities, acid production from fermentation, and assimilation of carbon sources were evaluated using API ZYM, API 50CH, and Biolog GEN III MicroPlates (Biolog) according to the manufacturers’ instructions. API strip and Biolog results were recorded every 24 h after incubation at 28°C until all reactions had stabilized. The API and Biolog tests were performed in duplicate using consistent conditions.
Biomass for chemotaxonomic investigations of strains was obtained by cultivation in flasks on a rotary shaker (180 r.p.m.) using ISP2 broth as the medium and with incubation at 28°C for 3 days, except that cellular fatty acids and cell-wall mycolic acids extraction and analyses were conducted using cultures grown on Middlebrook 7H10 (MB7H10) medium. Cellular polar lipids were extracted, detected with two-dimensional TLC silica-gel 60 F254 thin-layer plates (10 × 10 cm, Merck), and analyzed, as previously described (Minnikin et al., 1984). Menaquinones were extracted and purified using reverse-phase HPLC, also as previously described (Minnikin et al., 1984). Cellular fatty acids were extracted, methylated, and analyzed using the Sherlock Microbial Identification System (MIDI) with the standard ACTIN1 database (version 6.0) according to the manufacturer’s instructions (Sasser, 2006). Diagnostic isomers of diaminopimelic acid in whole cell hydrolysates (6 N HCl, 120°C, 30 min) of the strains were subjected to thin-layer chromatography on cellulose plates (10 × 20 cm, Merck) using a previously described solvent system (Schleifer and Kandler, 1972). The cell-wall mycolic acids were determined following saponification, extraction and derivatization, and then separated using a gradient of methaand 2-propanol via high-performance liquid chromatography (HPLC) as recommended by the Sherlock Mycobacteria Identification System Operating Manual Version 1.0 (MIDI) (Butler and Guthertz, 2001; Zimenkov et al., 2015).
16S rRNA genes of strains were amplified using PCR and the universal bacterial primers 27F (5′-AGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-GGTTACCTTGTTACGACTT-3′). Purified PCR products were cloned into the vector pMD19-T (TaKaRa) and recombinant plasmids were transformed into Escherichia coli DH5α cells, followed by sequencing in Sangon Biotech (Shanghai, China). The 16S rRNA gene sequences of the isolates were compared with publicly available sequences in the EzBioCloud platform2 to determine the approximate taxonomic affiliations of the strains (Yoon et al., 2017a). Multiple sequence alignments and phylogenetic reconstructions of 16S rRNA genes were performed in MEGA (version 11) (Tamura et al., 2021). A phylogenetic tree was then inferred using neighbor-joining methods, and evolutionary distances were calculated using the Kimura 2-parameter substitution model (Shamsuzzaman et al., 2023). Maximum Parsimony and Maximum Likelihood phylogenetic methods were also used to evaluate the phylogenetic affiliations of the strains. The topologies of the resultant phylogenetic trees were evaluated using bootstrap analysis with 1,000 replicates (Deng et al., 2023).
Genome sequencing was conducted on an Illumina HiSeq 4,000 system platform at the BGI sequencing company (Shenzhen, China). To prepare sequencing libraries, genomic DNA was randomly sheared to construct three read libraries with lengths of 300 bp using a Bioruptor ultrasonicator (Diagenode, Denville, NJ, United States) and physico-chemical methods. The paired-end fragment libraries were then sequenced on the Illumina platform. Low quality reads (those with consecutive bases covered by fewer than five reads) were discarded. The sequenced reads were then assembled using the SOAPdenovo (version 1.05) assembly software program (Li et al., 2010). Estimated completeness and contamination values for the genomes were estimated using the CheckM pipeline (Parks et al., 2015). Digital DNA–DNA hybridization (dDDH) and average nucleotide identity (ANI) values between the strains and related strains were calculated using the Genome-to-Genome Distance Calculator (GGDC, version 3.0)3 (Auch et al., 2010) and with the ezbiocloud platform (Yoon et al., 2017b), respectively. Genome-based phylogeny of supermatrix approach from protein sequences of the bac120 gene set (a collection of 120 single-copy protein sequences prevalent in bacteria) was constructed by using EasyCGTree version 3.04 as described previously (Zhang et al., 2020). Evolutionary distances were calculated using the IQ-Tree software program (version 1.6.1) (Nguyen et al., 2015). The genome sequences of the strains of interest were downloaded from the NCBI genome database.5
Protein sequences encoded by the strains were predicted and annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP). Gene prediction was conducted for the genome assemblies using glimmer36 with Hidden Markov models. tRNA, rRNA, and sRNA identification was conducted with tRNAscan-SE (Chan et al., 2021), RNAmmer (Lagesen et al., 2007), and the Rfam database (Gardner et al., 2009), respectively. Tandem repeat annotation was conducted using the Tandem Repeat Finder7, with minisatellite and microsatellite DNAs identified based on the numbers and lengths of repeat units. Functional annotation was also conducted based on the best hits within BLAST analyses. In addition, general functional annotations were identified by comparisons against several databases included the Kyoto Encyclopedia of Genes and Genomes (KEGG), Clusters of Orthologous Groups (COG), Non-Redundant Protein (NR), Swiss-Prot, Gene Ontology (GO), TrEMBL, and EggNOG databases. Predictions of gene clusters involved in natural product formation were conducted using antiSMASH (Blin et al., 2019).
The bacterial pan-genome analysis (BPGA) pipeline (version 1.3) was used to assess the genomic diversity among Mycobacterium strains using default settings (Chaudhari et al., 2016). A total of 73 protein datasets were used for the pan-genome analysis, including for strains YC-RL4T, MB418T, HX176T, and 70 related mycobacterial strains that were previously classified as “Mycolicibacterium.” Orthologous gene/protein clusters (homologous families) were identified using the USEARCH clustering tool (Kim et al., 2014).
Cells of strains YC-RL4T, MB418T, and 205372T were Gram-positive, non-motile and rod-shaped. The three isolates grew well on GYM and MB7H10 agar, with weak growth on NA and TSA agar. Colonies of strains YC-RL4T and HX176T grown on GYM medium and incubated for 48 h were 1.0–2.5 mm in diameter, opaque, ivory, and convex with a smooth surface. Colonies of strain HX176T grown on GYM medium incubated for 48 h were 1.0–3.2 mm in diameter, dry, rough, and orange-colored with undulated/scalloped edges. All strains grew over the pH range of 6.0–7.0 (optimum: 7.0). Strain YC-RL4T grew in the presence of 0–5.0% (w/v) NaCl (optimum: 0–3.0%, w/v), and 10–37°C (optimum: 28°C), while strain MB418T grew at 0–5.0% (w/v) NaCl (optimum 0–3.0%, w/v), and 15–45°C (optimum: 28°C), and strain HX176T grew in the presence of 0–4.0% NaCl (w/v) (optimum: 0–2.0%), and at 15–28°C (optimum: 28°C). All strains exhibited positive catalase reactions, but negative results for oxidase, starch hydrolysis, Voges–Proskauer, and H2S production tests. All strains exhibited positive enzymatic activities for alkaline phosphatase, esterase (C4), leucine arylamidase, lipase (C14), naphthol-AS-BI-phosphohydrolase, and valine arylamidase, based on API ZYM strip tests. None of the strains could produce acid from fermentation of amidon, amygdalin, arbutin, D-arabinose, D-ardonitol, D-cellobiose, D-fucose, D-galactose, D-lactose, D-maltose, D-mannitol, D-melezitose, D-melibiose, D-raffinose, D-sorbitol, D-trehalose, D-turanose, dulcitol, erythritol, gentiobiose, glycogen, inulin, L-arabitol, L-fucose, L-rhamnose, L-sorbose, L-xylose, mehyl-α-D-glucopyranoside, mehyl-α-D-mannopyranoside, mehyl-β-D-xylopyranoside, N-acetylglucosamine, potassium 5-ketogluconate, salicin, or xylitol. The major differentiating features among strains YC-RL4T, MB418T, HX176T, and other species are shown in Table 1.
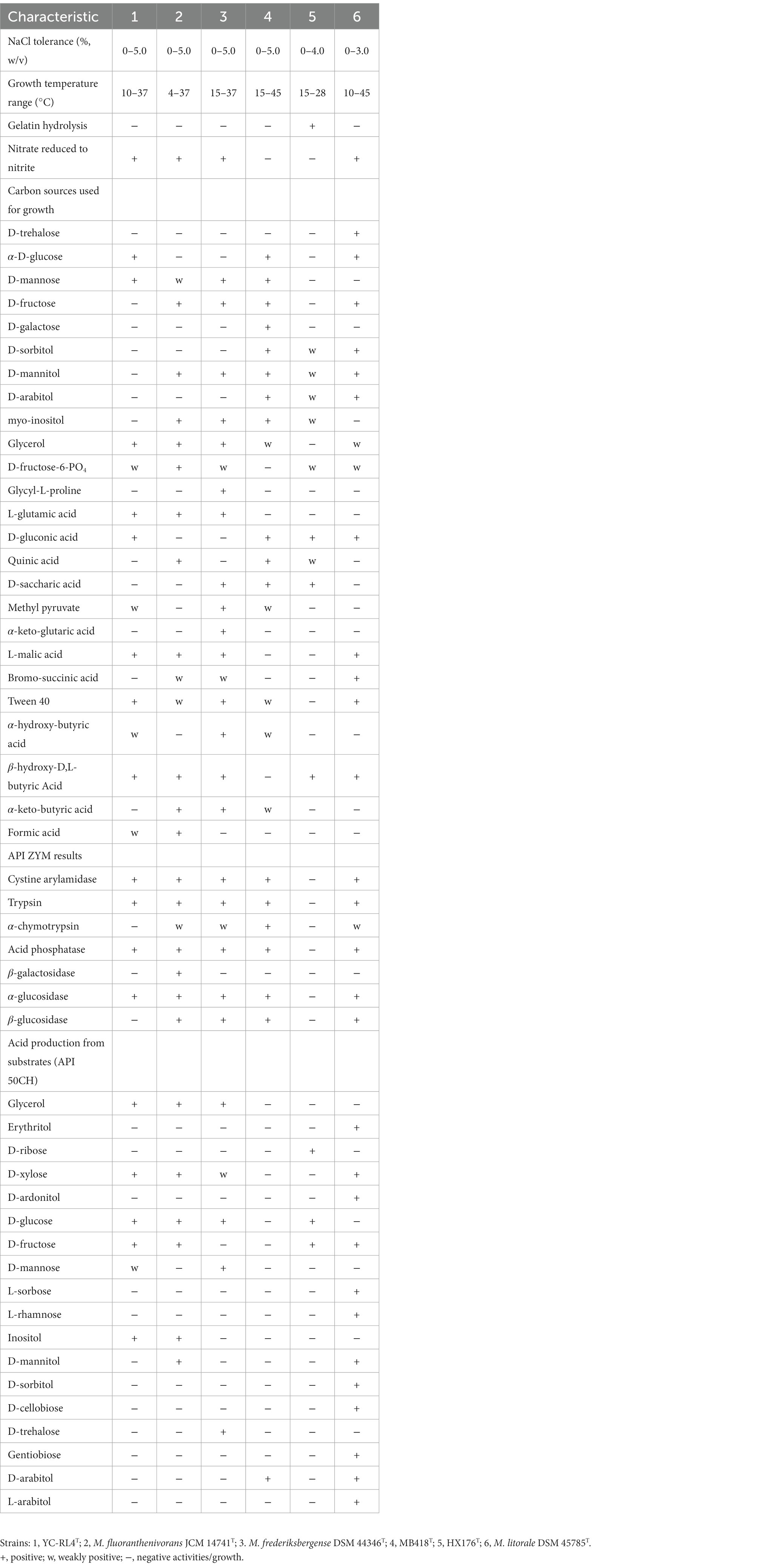
Table 1. Physiological characteristics of strains YC-RL4T, MB418T, HX176T, and closely related Mycobacterium type strains.
Strains YC-RL4T, MB418T, and HX176T exhibited chemotaxonomic properties consistent with their classification as Mycobacterium. The strains produced whole organism hydrolysates rich in meso-diaminopimelic acid (meso-A2pm), arabinose, galactose, glucose, mannose, and ribose. MK-9 (H2) was the predominant menaquinone and the polar lipid profile contained diposphatidylglycerol (DPG), phosphatidylethanolamine (PE), and phosphatidylinositol (PI) (Supplementary Figure S1). The three strains nevertheless exhibited differences in several chemotaxonomic characteristics relative to other Mycobacterium strains. The fatty acid profile of strain YC-RL4T comprised major levels of C16:0 (15.3%) and C17:1ω7c (15.3%), while the fatty acid profile of strain MB418T exhibited major levels of C17:1ω7c (47.7%) and sum in feature 9 (iso- C17:1ɷ9c/C16:0 10-methyl) (43.3%). Strain HX176T exhibited higher levels of C17:1ω7c (43.2%) and C19:1 trans 7 (30.9%), but lower C16:0 levels (8.8%), while also lacking sum in feature 9 (Supplementary Table S1). In addition, only the polar lipid profiles of strains MB418T and HX176T included glycolipids (GLs), contrasting with that of strain YC-RL4T that lacked GLs (Supplementary Figure S1). The HPLC profile of cell-wall mycolic acids from strain YC-RL4T, containing 60–70 carbon atoms of mycolic acids as the main peaks, was a little similar to that observed for M. fluoranthenivorans JCM 14741T and M. frederiksbergense DSM 44346T while quite different from that of strains MB418T, and HX176T. The HPLC pattern of cell-wall mycolic acids from strains MB418T and HX176T were more similar to that of M. litorale DSM 45785T than others, and the chromatogram of strain MB418T was distinct from that of strain HX176T (Supplementary Figure S2).
Nearly complete 16S rRNA gene sequences for strain YC-RL4T (1,527 bp, accession OQ096616), MB418T (1,509 bp, accession OP522341), and HX176T (1,508 bp, accession OQ096617) were obtained. BLAST searches of the 16S rRNA gene sequences against the GenBank database indicated that strains YC-RL4T, MB418T, and HX176T were closely related to Mycobacterium species of the family Mycobacteriaceae even though they were previously classified as the genus Mycolicibacterium. Strain YC-RL4T was most closely related to M. fluoranthenivorans JCM 14741T and M. frederiksbergense DSM 44346T, with 16S rRNA gene sequence similarities of 99.3 and 99.2%, respectively. The 16S rRNA gene of MB418T exhibited 98.5% nucleotide similarity to 16S rRNA genes of M. celeriflavum DSM 46765T and M. moriokaense DSM 44221T, but with 97.2% similarity to the 16S rRNA genes of strain YC-RL4T, and 96.2–98.3% similarity to 16S rRNA genes of other various Mycobacterium type strains. The 16S rRNA gene sequence of strain HX176T exhibited a nucleotide similarity of 98.3% to that of M. litorale DSM 45785T, 98.0% to that of strain YC-RL4T, and similarities of 95.8–97.9% to those from other Mycobacterium type strains (Supplementary Table S2). Phylogenetic analysis of the 16S rRNA gene sequences of the three isolates with other mycobacterial strains that were previously classified as “Mycolicibacterium” revealed that the strains belonged to the genus Mycobacterium. The 16S rRNA gene sequence of strain YC-RL4T formed a sub-clade with that of the type strain M. fluoranthenivorans JCM 14741T, which were together related to the 16S rRNA gene of M. frederiksbergense DSM 44346T, consistent with BLAST comparisons. The 16S rRNA gene of strain MB418T formed a well-supported sub-clade with the 16S rRNA gene of strain HX176T, which were together associated with that of M. litorale DSM 45785T (Figure 1). The whole-genome phylogenomic analysis recapitulated the 16S rRNA gene phylogenetic analysis, with the exception of the strain YC-RL4T genome being most closely related to that of M. frederiksbergense DSM 44346T (Supplementary Figure S3), contrasting with the 16S rRNA gene phylogenetic analysis.
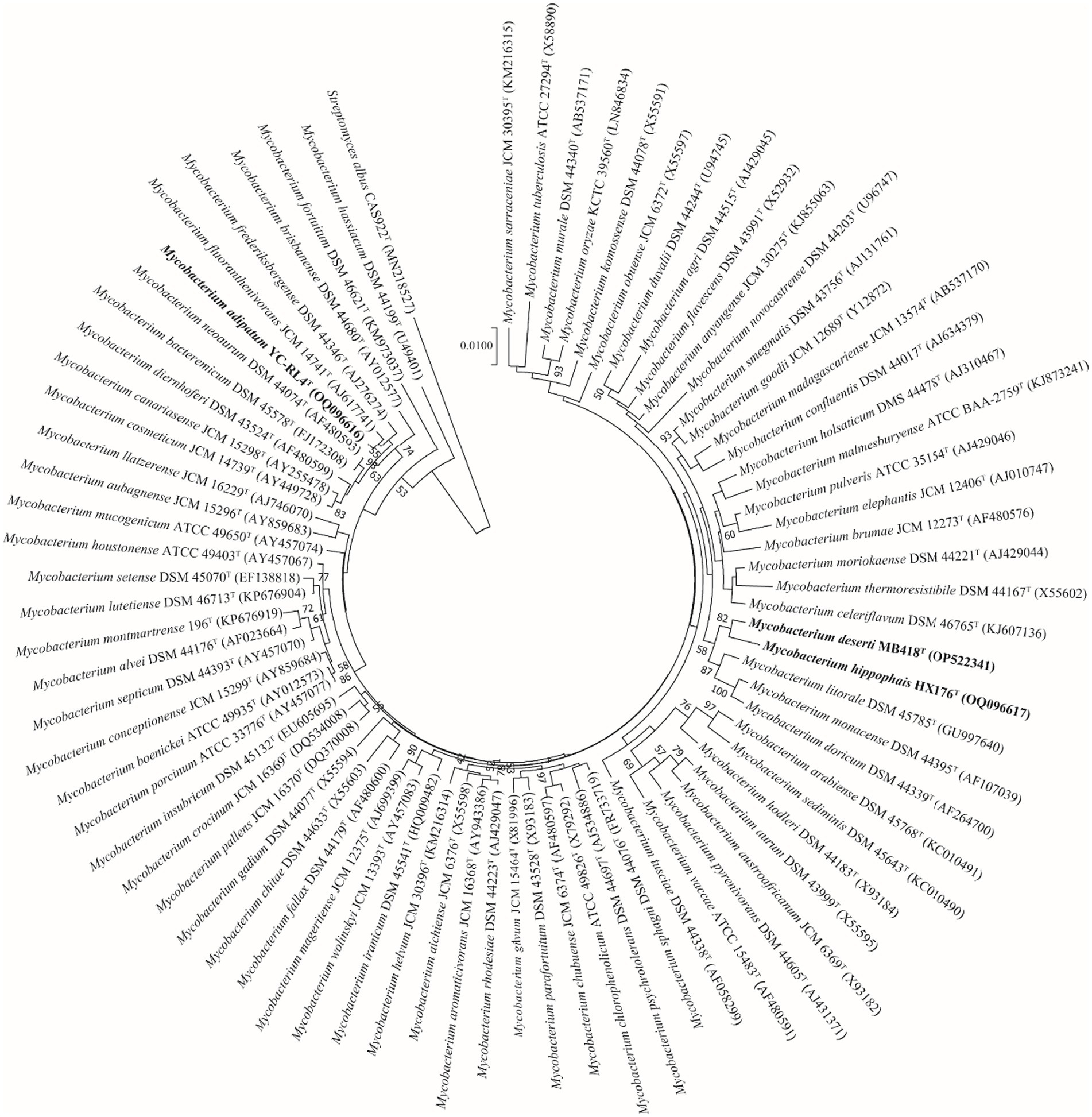
Figure 1. Neighbor-joining phylogenetic reconstruction of 16S rRNA gene sequences from strains YC-RL4T, MB418T, and HX176T along with those from other Mycobacterium. Bootstrap values above 50% are shown as percentages of 1,000 replicates. The 16S rRNA gene sequence of Streptomyces albus CAS922T (GenBank accession MN218527) was used as the outgroup. Scale bar indicates 0.01 nt substitutions per alignment site.
The genomes of strains YC-RL4T, MB418T, and HX176T were estimated to be 99.95, 100.00, and 100.00% complete, with estimated contamination of 0.62, 0.30, and 0.45%, respectively. The draft genome sizes of strains YC-RL4T, MB418T, and HX176T were 6.1, 5.6, and 5.9 Mbp, respectively, and were assembled from two contigs with an N50 length of 5,801,417 bp; 15 contigs with an N50 length of 710,882 bp; and 41 contigs with an N50 length of 302,245 bp, respectively (Supplementary Table S3). The genomic sequence for strain YC-RL4T encoded 5,881 total genes including 47 tRNA, 6 rRNA, 4 other ncRNA, and 83 pseudo genes. The genome of strain MB418T encoded 5,444 total genes including 47 tRNA, 3 rRNA, 3 other ncRNA, and 98 pseudo genes. The genome of strain HX176T encoded 5,692 genes including 46 tRNA, 4 rRNA, 3 other ncRNA, and 64 pseudo genes. The genomic G + C content of all three strains ranged between 66.5 and 69.3%. ANI values calculated between strains YC-RL4T, MB418T, HX176T, and other Mycobacterium species were all <86.9%, with corresponding dDDH values all <31.8% (Supplementary Table S4). The values were lower than the thresholds used to delineate bacterial species (i.e., ANI < 95–96% and dDDH <70%) (Kim et al., 2014), thereby indicating that the strains represented novel, uncharacterized species.
Polycyclic aromatic hydrocarbons ring-hydroxylating dioxygenases (PAH-RHDs) are critical for PAH degradation by aerobic bacteria, because they catalyze the initial oxidation of O2 atoms to form cisdihydrodiol and this step controls the PAH degradation rate (Kim et al., 2007; Chemerys et al., 2014). Among PAH-RHDs, aromatic ring-hydroxylating dioxygenases are particularly important in the aerobic bacterial degradation of aromatic compounds, because they catalyze the oxidation of the compounds during degradation. They typically catalyze the addition of two hydroxy groups to vicinal carbons, thereby disrupting aromaticity and yielding dihydrodiol compounds with cis, cis stereochemistry. The enzymes are biotechnologically important because they act as biocatalysts in the stereospecific synthesis of chiral synthons and the degradation of aromatic pollutants (Kahl and Hofer, 2003). Multiple genes encoding dioxygenases were identified in the genomes of strains YC-RL4T, MB418T, and HX176T. Specifically, four genes putatively encoding alpha subunits with homology to aromatic ring-hydroxylating dioxygenases were encoded by the genome of strain YC-RL4T, while genes putatively encoding alpha subunits of aromatic ring-hydroxylating dioxygenases were identified in the genomes of strains MB418T and HX176T (Supplementary Table S5).
Strains of the genus Mycobacterium could degrade PAHs to central intermediates via the o-phthalate and the β-ketoadipate pathway (Kim et al., 2007; Augelletti et al., 2020). The o-phthalate degradation pathway of Gram-positive bacteria involves oxygenation to form 3,4-dihydro-3,4-dihydroxyphthalate, dehydrogenation to 3,4-dihydroxyphthalate, and finally decarboxylation to generate protocatechuate (Bhattacharyya et al., 2023). Genes (phtAa) were identified that encoded homologous proteins involved in the degradation of phthalate in the genomes of strains YC-RL4T, MB418T, and HX176T (Supplementary Table S5). Thus, these three strains could likely degrade various types of phthalates, as previously suggested. In addition, genes of the β-ketoadipate pathway cluster (pcaC, pcaD, pcaG, and pcaH) were also identified in the genomes of strains YC-RL4T, MB418T, and HX176T (Supplementary Table S5), suggesting that anthracene degradation by these strains could proceed through the ortho-cleavage of protocatechuate (protocatechuic acid [PCA] 3,4-dioxygenase) (Habe et al., 2005). These protocatechuate catabolic genes were required for the complete degradation of PAHs to TCA cycle intermediates. Consequently, strains YC-RL4T, MB418T, and HX176T exhibited the potential for the bioremediation of PAHs and smaller ring aromatics.
Although heavy metals naturally exist in many environments (Masindi and Muedi, 2018), the ubiquitous distribution of heavy metals in natural environments is a consequence of global industrialization and urbanization that has led to negative impacts on human and environmental health. Microorganisms have adapted multiple resistance mechanisms to overcome the physiological stress from heavy metal exposure (Sherpa et al., 2020). Several genes were identified in the genomes of strains YC-RL4T, MB418T, and HX176T that encoded heavy metal resistance proteins. For example, the three strains exhibited the potential to mediate copper toxicity via copper-resistant genes (copC) identified in all of their genomes. Further, the copper-resistance gene copD was also identified in the genomes of strain MB418T and HX176T. Free form copper is highly toxic due to its ability to produce radicals during cycling between oxidized Cu(II) and reduced Cu(I) forms. Consequently, intracellular copper must remain complexed within a tightly controlled copper homeostatic system. Importantly, Cop proteins sequester excess copper in periplasms and outer membranes (Arnesano et al., 2003). Consequently, these proteins might confer copper resistance to strains YC-RL4T, MB418T, and HX176T by helping to sequester and accumulate copper in periplasms with copper binding proteins, thereby preventing toxic levels of copper from entering the cytoplasm.
In addition to the above, genes (merA and merB) that encoded mercury reductase enzymes were identified in the strain YC-RL4T genome that enable resistance to mercury stress. Mercury toxicity arises from the strong affinity of monomethyl-Hg and Hg2+ to sulfur atoms in cysteine residues and, hence, interference with protein structure and function. The mercury reductase encoded by merA and merB reduced the toxicity of Hg2+ to Hg0 (Møller et al., 2014). Specifically, organomercurial lyase (MerB) catalyzed the protonolysis of the carbon-mercury bond, resulting in the formation of ionic mercury and reduced hydrocarbon. The ionic mercury (Hg2+) was subsequently reduced to less reactive elemental mercury (Hg0) by mercuric reductase (MerA) (Di Lello et al., 2004). The presence of these genes involved in PAH-degradation and metal resistance genes in the genomes of these strains suggested that strains YC-RL4T, MB418T, and HX176T could be applied in bioremediation of soils contaminated with both PAHs and heavy metals.
The pan-genomes of strains comprise the entire gene repertoire for a given species across several populations, and their analysis can be informative for understanding the distribution of core, important genes shared by all strains of a species, in addition to strain-specific accessory genes that may be more dispensable (Reis and Cunha, 2021). A total of 401,362 protein-coding genes (Table 2) were encoded by the genomes of strains YC-RL4T, MB418T, HX176T, and 70 other related strains of the genus Mycobacterium, comprising 51,712 homologous families based on cluster analysis. A total of 734 core genes were shared by the 73 strains and comprise the core genome identified in this study. The core genes accounted for approximately 13.5% of the pan-genome for the 73 strains of the genus Mycobacterium. The identification of the “open” pangenome based on consideration of increasing numbers of strains was fit with a powerlaw regression function [f(X) = 5991.36×0.50], while the core genome identification fit an exponential regression [f(X) = 2838.85e−0.03X; Figure 2]. Thus, the relatively extensive open pangenome of the strains indicates that they have considerable repertoires of accessory genes, possibly due to extensive horizontal gene exchanges with various microbial species (Coyte et al., 2022). Aromatic ring-hydroxylating dioxygenase alpha subunit-encoding genes were identified in all of the 73 genomes, suggesting it was an important core gene shared by all strains and plays a critical role in PAH degradation by the strains. In addition, the copy numbers of these genes varied among different strains. The lowest number of copies was observed in the genome of M. brumae DSM 44177T, while the largest number of copies was observed in the genome of strain M. gadium DSM 44077T.
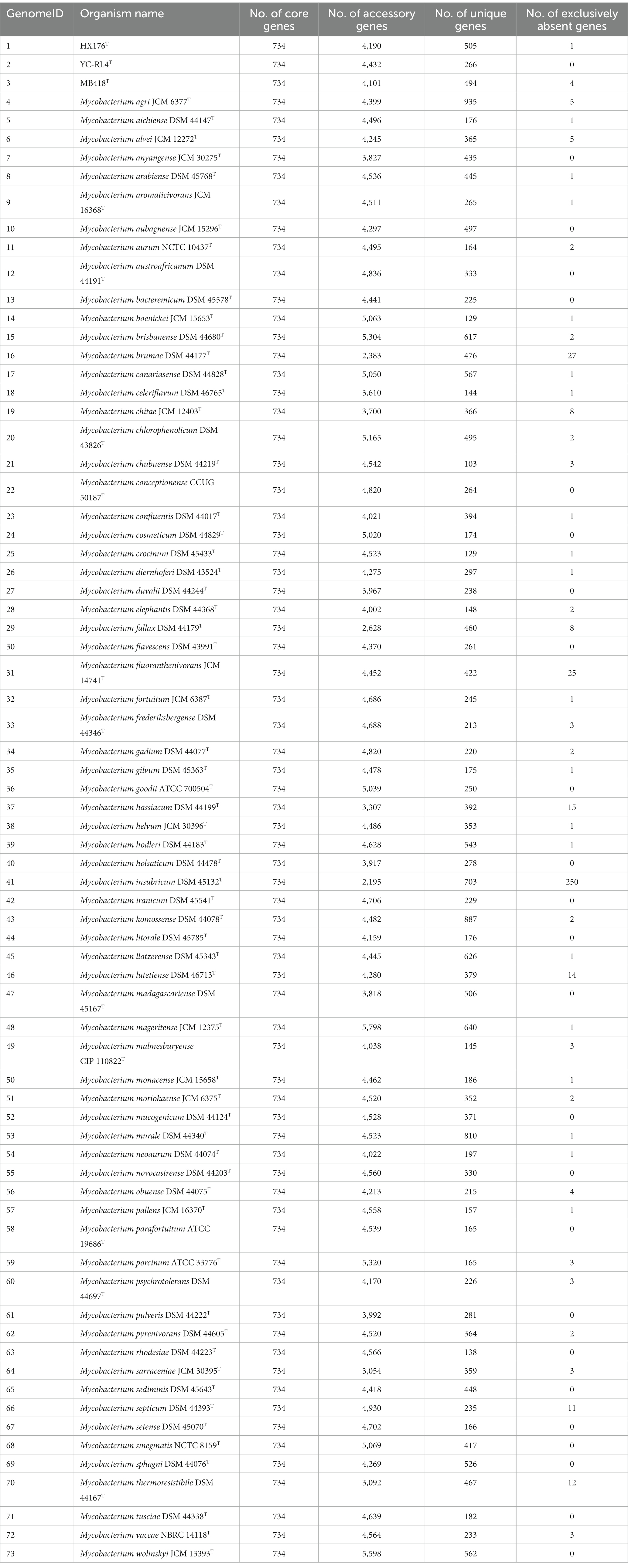
Table 2. Pan-genome information for Mycobacterium strains evaluated in this study, including strains YC-RL4T, MB418T, HX176T, and 70 others.
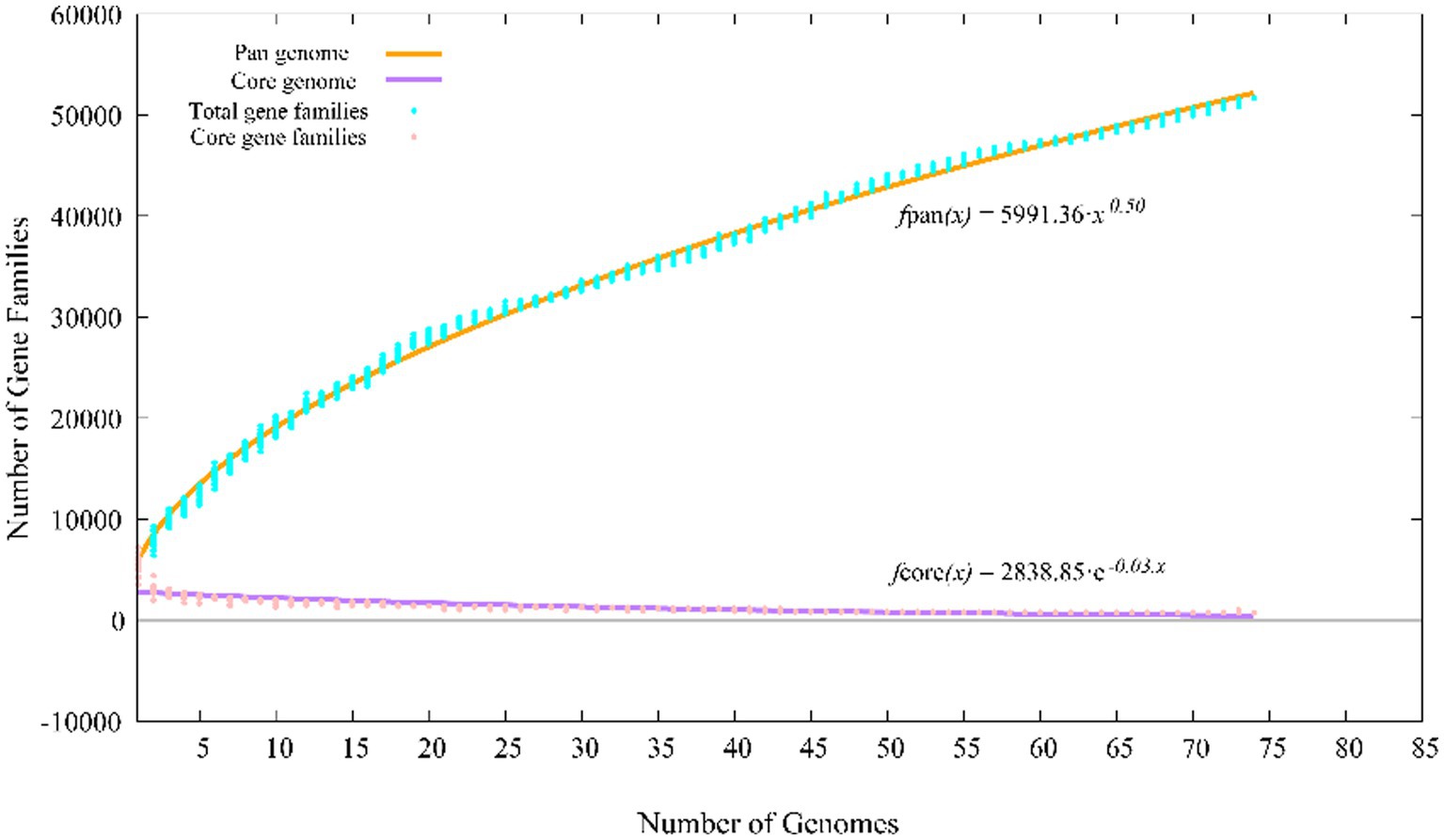
Figure 2. Pan-genome profiles of strains YC-RL4T, MB418T, HX176T and 70 related Mycobacterium strains based on protein clustering.
A functional classification of core, accessory, and unique genes was performed based on comparison to the COG database, revealing clear differences between core and accessory genes. Core genes were most associated with the COG categories of R (general function prediction only; 13.0% of genes), J (translation, ribosomal structure, and biogenesis; 12.4%), E (amino acid transport and metabolism; 9.1%) and I (lipid transport and metabolism; 8.2%) (Figure 3), while accessory and unique genes were most associated with the COG categories R (general function prediction only; 16.3 and 17.0%, respectively), K (transcription; 13.0 and 12.6%), and Q (secondary metabolite biosynthesis, transport, and catabolism; 8.8 and 9.3%). Thus, core genes were mostly involved in basic physiological functions, resulting in the overall prevailing phenotypes of the strains. In contrast, accessory genes related to tachytely evolution could increase the gene and functional diversity of their respective genomes. These accessory genes may be involved in metabolic pathways that critically ensure adaptations or functioning in variable ecological niches that differ among species (Vernikos et al., 2015).
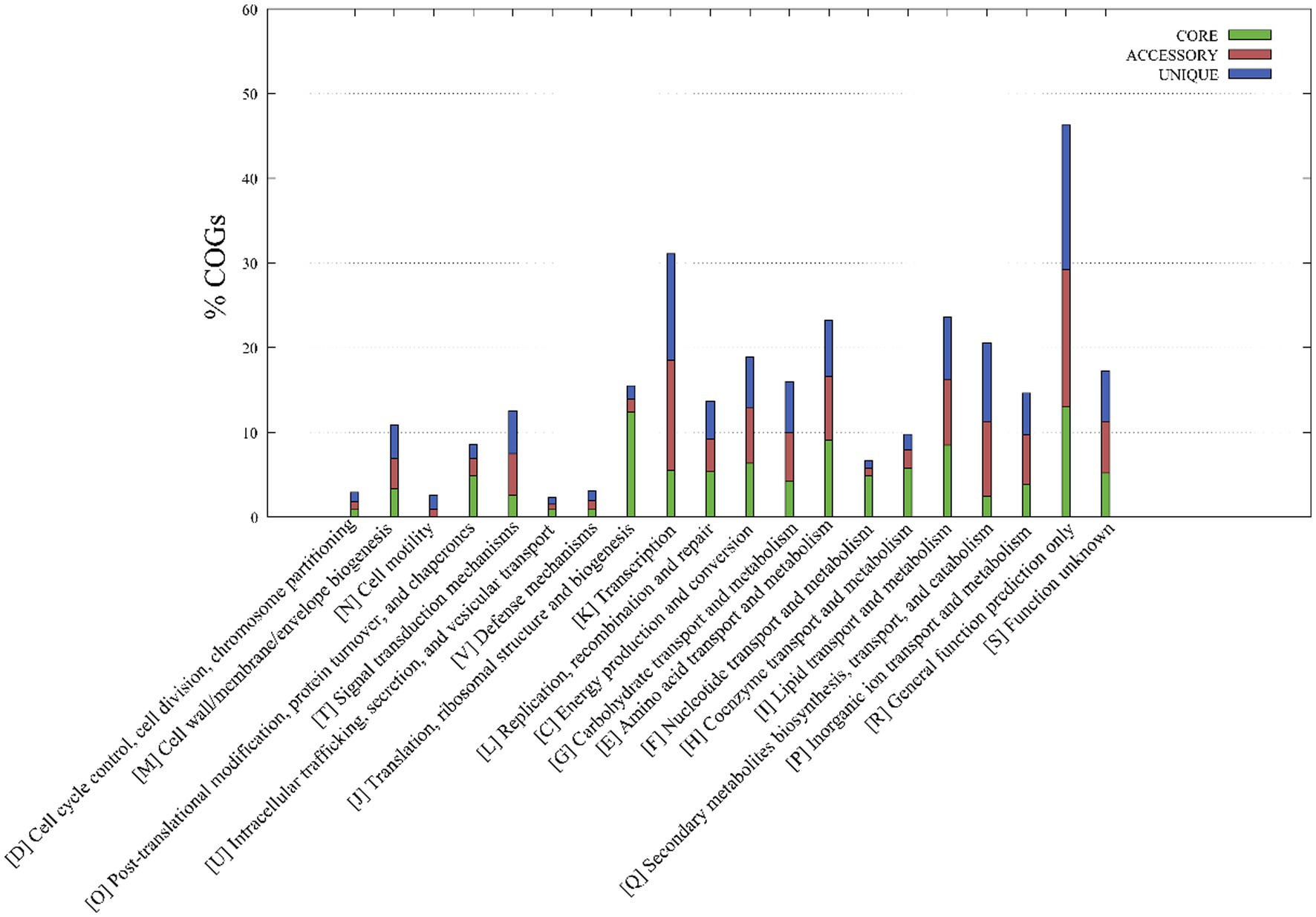
Figure 3. Metabolic pathways associated with the core, accessory, and unique genes of strains YC-RL4T, MB418T, HX176T, and 70 other related Mycobacterium strains. The metabolic annotations are based on clusters of orthologous genes (COG) database annotations.
Polyketide synthesis operons and others involved in non-ribosomal peptide synthesis (NRPS) are common in the genomes of Mycobacterium and are responsible producing cell wall-associated lipids, siderophores, and other biologically active molecules (Stinear et al., 2008). AntiSMASH analysis revealed the presence of 14–21 biosynthetic gene clusters (BGCs) in the genomes of strain YC-RL4T, MB418T, and HX176T that were especially enriched in genes encoding Type I polyketide synthases (PKSs) and non-ribosomal peptide synthases (NRPSs) (Supplementary Table S6). A BGC that was most similar to that encoding mycobactin was identified in all 73 genomes, consistent with the production of mycobactin being essential for Mycobacterium strains to access iron (McMahon et al., 2012). In addition, several putative BGCs exhibited low similarities to known clusters, indicating their potential for synthesizing putatively novel secondary metabolites.
In this study, three strains of the genus Mycobacterium (YC-RL4T, MB418T, and HX176T) were isolated from several petroleum-contaminated soil samples of China. The three strains exhibited highest similarity to Mycobacterium species of the family Mycobacteriaceae. The 16S rRNA gene sequence of strain YC-RL4T in comparison to other characterized Mycobacterium strains exhibited levels below the threshold for differentiating species (98.65%), while the 16S rRNA gene sequences of strains MB418Tand HX176T compared to other characterized Mycobacterium strains were above 98.65%. However, the dDDH values among the three strains and the other characterized Mycobacterium species were very below the threshold value (70%) used to delineate bacterial strains of the same species (46). Moreover, the ANI values among all three strains and other type species of Mycobacterium were much lower than the threshold for bacterial species delineation (95–96%) (Kim et al., 2014). Thus, strains YC-RL4T, MB418T, and HX176T likely represent three novel species of Mycobacterium. Chemotaxonomic, genomic, and phenotypic analyses of strains YC-RL4T, MB418T, and HX176T confirmed their identification as novel species. Lastly, the genomes of strains YC-RL4T, MB418T, and HX176T encoded proteins associated with PAH degradation and metal resistance, consistent with strain YC-RL4T previously being shown to degrade phthalates (Ren et al., 2016). Consequently, these three species may hold considerable application potential in the bioremediation of PAH and metal-contaminated soils.
(a.di.pa’tum. L. neut. adj. adipatum, fat, greasy)
Cells are Gram-positive, non-motile, and rod-shaped, producing opaque, ivory, and convex colonies on GYM and MB7H10 agar after cultivation at 48 h and 28°C. Cells are catalase-negative and oxidase-positive. Optimum growth was observed at 10–37°C, pH 6–7, and with NaCl concentrations ranging between 0 and 5%. Cells were negative for Voges–Proskauer, H2S production, and starch hydrolysis tests. Acetic acid, D-gluconic acid, D-mannose, glucuronamide, glycerol, L-glutamic acid, L-malic acid, propionic acid, tween 40, α-D-glucose, β-hydroxy-D, and L-butyric acid can be utilized as carbon sources. 3-methyl glucose, acetoacetic acid, bromo-succinic acid, citric acid, D-arabitol, D-aspartic acid, D-cellobiose, dextrin, D-fructose, D-fucose, D-galactose, D-galacturonic acid, D-glucose-6-PO4, D-glucuronic acid, D-lactic acid methyl ester, D-maltose, D-mannitol, D-melibiose, D-raffinose, D-saccharic acid, D-salicin, D-serine, D-sorbitol, D-trehalose, D-turanose, gelatin, gentiobiose, glycyl-L-proline, inosine, L-aspartic acid, L-fucose, L-galactonic acid lactone, L-histidine, L-pyroglutamic acid, L-rhamnose, L-serine, mucic acid, myo-inositol, N-acetyl neuraminic acid, N-acetyl-D-galactosamine, N-acetyl-D-glucosamine, N-acetyl-β-D-mannosamine, pectin, p-hydroxy-phenylacetic acid, quinic acid, stachyose, sucrose, α-D-lactose, α-keto-butyric acid, α-keto-glutaric acid, β-methyl-D-glucoside, and γ-amino-butryric acid are not used as carbon sources. D-fructose, D-glucose, D-xylose, esculin ferric citrate, glycerol, inositol, potassium 2-ketogluconate, and potassium gluconate can be assimilated and fermented to produce acid. Cells exhibit positive acid phosphatase, alkaline phosphatase, cystine arylamidase, esterase (C4), esterase lipase (C8), leucine arylamidase, lipase (C14), naphthol-AS-BI-phosphohydrolase, trypsin, valine arylamidase, and α-glucosidase activities via API ZYM strip assays. Whole-cell hydrolysates contain meso-diaminopimelic acid (meso-A2pm), arabinose, galactose, glucose, mannose, and ribose. Cellular polar lipid profiles comprise diposphatidylglycerol (DPG), phosphatidylethanolamine (PE) and phosphatidylinositol (PI). The primary menaquinone is MK-9 (H2). The major fatty acids were C16:0 (15.3%) and C17:1ω7c (15.3%). The mycolic acids primarily contain 60–70 carbon atoms. The type strain YC-RL4T (= CPCC 205684T = CGMCC 1.62027T) was isolated from a petroleum-contaminated soil sample from Shandong, China. The draft genome of the type strain is 6.1 Mbp in size and exhibits a genomic G + C content of 67.4%.
(de.ser’ti. L. gen. n. deserti, from a desert)
Cells are Gram-positive, non-motile, and non-spore forming. Ivory colonies develop on GYM and MB7H10 agar after incubation for 48 h at 28°C. Optimal growth was observed at 15–45°C, pH 6–7, and NaCl concentrations ranging from 0 to 5%. Cells were oxidase-positive and catalase-negative, but exhibited negative results for Voges–Proskauer, H2S production, and starch hydrolysis tests. Cells can utilize acetic acid, D-arabitol, D-fructose, D-galactose, D-gluconic Acid, D-mannitol, D-mannose, D-saccharic acid, D-sorbitol, myo-inositol, propionic acid, quinic acid, and α-D-glucose as sole carbon sources. API 50CH tests indicated that arbutin, D-lyxose, D-saccharose, D-tagatose, esculin ferric citrate, potassium 2-ketogluconate, and potassium gluconate are used as carbon sources. In contrast, amidon, amygdalin, D-arabinose, D-arabitol, D-ardonitol, D-cellobiose, D-fructose, D-fucose, D-galactose, D-glucose, D-lactose, D-maltose, D-mannitol, D-mannose, D-melezitose, D-melibiose, D-raffinose, D-ribose, D-sorbitol, D-trehalose, D-turanose, dulcitol, D-xylose, erythritol, gentiobiose, glycerol, glycogen, inositol, inulin, L-arabinose, L-arabitol, L-fucose, L-rhamnose, L-sorbose, L-xylose, mehyl-α-D-glucopyranoside, mehyl-α-D-mannopyranoside, Mehyl-β-D-xylopyranoside, N-acetylglucosamine, potassium 5-ketogluconate, salicin, and xylitol are not used as carbon sources. API ZYM analysis revealed the presence of positive acid phosphatase, alkaline phosphatase, cystine arylamidase, esterase (C4), leucine arylamidase, lipase (C14), naphthol-AS-BI-phosphohydrolase, trypsin, valine arylamidase, α-chymotrypsin, α-glucosidase, and β-glucosidase activities. Whole-cell hydrolysates contained meso-diaminopimelic acid (meso-A2pm), arabinose, galactose, glucose, mannose, and ribose. Cellular polar lipid profiles comprised diposphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylinositol (PI), and glycolipids (GL). The primary menaquinone was MK-9 (H2). The major fatty acids were C17:1ω7c (47.7%) and summed feature 9 (iso- C17:1ɷ9c/C16:0 10-methyl) (43.3%). The mycolic acids mainly contain 78–86 carbon atoms, with moderate amount of 76–84 carbon atoms, and minor of 62–70 carbon atoms. The type strain MB418T (= CPCC 205710T = KCTC 49782T) was isolated from a gravel soil sample from the Gurbantunggut desert, China. The draft genome of the type strain is 5.6 Mbp in length and exhibits a genomic G + C content of 66.5%.
(hip.po.pha’is. L. gen. n. hippophais, from Hippophae, a plant genus whose rhizosphere the type strain was isolated from)
Cells are Gram-positive, non-motile, and rod-shaped, producing yellow colonies. Cells were positive for oxidase activity, but negative for catalase, Voges–Proskauer, H2S production, and starch hydrolysis tests. Optimum growth was observed at 15–28°C, pH 6–7, and with NaCl concentrations ranging between 0 and 4%. Acetic acid, D-gluconic acid, D-saccharic Acid, propionic acid, and β-hydroxy-D,L-butyric acid can be used as substrates. API 50CH analysis revealed that D-fructose, D-glucose, D-ribose, esculin ferric citrate, potassium 2-ketogluconate, and potassium gluconate can be used as carbon substrates. Enzymatic activity analysis revealed the presence of alkaline phosphatase, esterase (C4), esterase lipase (C8), leucine arylamidase, lipase (C14), naphthol-AS-BI-phosphohydrolase, and valine arylamidase activities, but the lack of activity for acid phosphatase, cystine arylamidase, N-acetyl-β-glucosamimidase, trypsin, α-chymotrypsin, α-fucosidase, α-galactosidase, α-glucosidase, α-mannosidase, β-galactosidase, β-glucosidase, and β-glucuronidase. Whole-cell hydrolysates contained meso-diaminopimelic acid (meso-A2pm), arabinose, galactose, glucose, mannose, and ribose. Cellular polar lipid profiles comprised diposphatidylglycerol (DPG), phosphatidylethanolamine (PE), phosphatidylinositol (PI), and glycolipids (GL). The primary menaquinone was MK-9 (H2). The major fatty acids were C17:1ω7c (43.2%), C19:1 trans 7 (30.9%), and C16:0 (8.8%). The mycolic acids primarily contain 68–78 carbon atoms. The type strain HX176T (= CPCC 205372T = KCTC 49413T) was isolated from a rhizosphere soil sample of a medicinal plant in Xinjiang, China. The draft genome of the type strain is 5.9 Mbp in length and exhibits a genomic G + C content of 69.3%.
The 16S rRNA gene sequences of strains YC-RL4T, MB418T, and HX176T were deposited in GenBank under the accession numbers OQ096616, OP522341 and OQ096617, respectively. The whole genome shotgun data for the genomes of strains YC-RL4T, MB418T, and HX176T were deposited in the DDBJ, ENA, and GenBank databases under the accession numbers CP015596, JAODWD000000000, and JAPZPY000000000, respectively.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary material.
YD, TM, JW, and JS carried out the experiments. YD, YY, and Y-QZ conceived the research, analyzed the data, and prepared the manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by CAMS Innovation Fund for Medical Sciences (CIFMS, 2021-I2M-1-055), National Natural Science Foundation of China (32170021 and 81960712), Beijing Natural Science Foundation (5212018), Key project at central government level-the ability establishment of sustainable use for valuable Chinese medicine resources (2060302), and the National Infrastructure of Microbial Resources (NIMR-2021-3).
The authors sincerely thank Aharon Oren (The Institute of Life Sciences, The Hebrew University of Jerusalem, Jerusalem, Israel) and Bernhard Schink (Department of Biology, University of Konstanz, Konstanz, Germany) for their guidance in nomenclature.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1225746/full#supplementary-material
DPG, diphosphatidylglycerol; PE, phosphatidylethanolamine; PI, phosphatidylinositol; ANI, average nucleotide identity; dDDH, digital DNA–DNA hybridization; PAHs, polycyclic aromatic hydrocarbons; PAH-RHDs, polycyclic aromatic hydrocarbons ring-hydroxylating dioxygenases.
1. ^https://lpsn.dsmz.de/genus/mycobacterium
2. ^http://www.ezbiocloud.net/identify
3. ^http://ggdc.dsmz.de/ggdc.php
4. ^https://github.com/zdf1987/EasyCGTree
5. ^https://www.ncbi.nlm.nih.gov/genome/
Abbasnezhad, H., Gray, M., and Foght, J. M. (2011). Influence of adhesion on aerobic biodegradation and bioremediation of liquid hydrocarbons. Appl. Microbiol. Biotechnol. 92, 653–675. doi: 10.1007/s00253-011-3589-4
Arnesano, F., Banci, L., Bertini, I., Mangani, S., and Thompsett, A. R. (2003). A redox switch in CopC: an intriguing copper trafficking protein that binds copper(I) and copper(II) at different sites. Proc. Natl. Acad. Sci. U. S. A. 100, 3814–3819. doi: 10.1073/pnas.0636904100
Auch, A. F., Von Jan, M., Klenk, H. P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Augelletti, F., Tremblay, J., Agathos, S. N., Jousset, A., and Stenuit, B. (2020). Draft whole-genome sequence of the anthracene-degrading strain Mycolicibacterium frederiksbergense LB501T, isolated from a polycyclic aromatic hydrocarbon-contaminated soil. Microbiol. Resour. Announc. 9:e00671-20. doi: 10.1128/mra.00671-20
Bhattacharyya, M., Dhar, R., Basu, S., Das, A., Reynolds, D. M., and Dutta, T. K. (2023). Molecular evaluation of the metabolism of estrogenic di(2-ethylhexyl) phthalate in Mycolicibacterium sp. Microb. Cell Factories 22:82. doi: 10.1186/s12934-023-02096-0
Blin, K., Shaw, S., Steinke, K., Villebro, R., Ziemert, N., Lee, S. Y., et al. (2019). antiSMASH 5.0: updates to the secondary metabolite genome mining pipeline. Nucleic Acids Res. 47, W81–W87. doi: 10.1093/nar/gkz310
Butler, W. R., and Guthertz, L. S. (2001). Mycolic acid analysis by high-performance liquid chromatography for identification of Mycobacterium species. Clin. Microbiol. Rev. 14, 704–726. doi: 10.1128/cmr.14.4.704-726.2001
Chan, P. P., Lin, B. Y., Mak, A. J., and Lowe, T. M. (2021). tRNAscan-SE 2.0: improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 49, 9077–9096. doi: 10.1093/nar/gkab688
Chaudhari, N. M., Gupta, V. K., and Dutta, C. (2016). BPGA- an ultra-fast pan-genome analysis pipeline. Sci. Rep. 6:24373. doi: 10.1038/srep24373
Chemerys, A., Pelletier, E., Cruaud, C., Martin, F., Violet, F., and Jouanneau, Y. (2014). Characterization of novel polycyclic aromatic hydrocarbon dioxygenases from the bacterial metagenomic DNA of a contaminated soil. Appl. Environ. Microbiol. 80, 6591–6600. doi: 10.1128/aem.01883-14
Coyte, K. Z., Stevenson, C., Knight, C. G., Harrison, E., Hall, J. P. J., and Brockhurst, M. A. (2022). Horizontal gene transfer and ecological interactions jointly control microbiome stability. PLoS Biol. 20:e3001847. doi: 10.1371/journal.pbio.3001847
Dai, Y., Liu, R., Zhou, Y., Li, N., Hou, L., Ma, Q., et al. (2020). Fire Phoenix facilitates phytoremediation of PAH-cd co-contaminated soil through promotion of beneficial rhizosphere bacterial communities. Environ. Int. 136:105421. doi: 10.1016/j.envint.2019.105421
Deng, Y., Han, X. F., Jiang, Z. M., Yu, L. Y., Li, Y., and Zhang, Y. Q. (2022). Characterization of three Stenotrophomonas strains isolated from different ecosystems and proposal of Stenotrophomonas mori spnov. and Stenotrophomonas lacuserhaii sp. nov. Front. Microbiol. 13:1056762. doi: 10.3389/fmicb.2022.1056762
Deng, Y., Jiang, Z. M., Han, X. F., Su, J., Yu, L. Y., Liu, W. H., et al. (2023). Pangenome analysis of the genus Herbiconiux and proposal of four new species associated with Chinese medicinal plants. Front. Microbiol. 14:1119226. doi: 10.3389/fmicb.2023.1119226
di Lello, P., Benison, G. C., Valafar, H., Pitts, K. E., Summers, A. O., Legault, P., et al. (2004). NMR structural studies reveal a novel protein fold for MerB, the organomercurial lyase involved in the bacterial mercury resistance system. Biochemistry 43, 8322–8332. doi: 10.1021/bi049669z
Falkinham, J. O. 3rd. (2009). Surrounded by mycobacteria: nontuberculous mycobacteria in the human environment. J. Appl. Microbiol. 107, 356–367. doi: 10.1111/j.1365-2672.2009.04161.x
Gardner, P. P., Daub, J., Tate, J. G., Nawrocki, E. P., Kolbe, D. L., Lindgreen, S., et al. (2009). Rfam: updates to the RNA families database. Nucleic Acids Res. 37, D136–D140. doi: 10.1093/nar/gkn766
Guillemin, I., Cambau, E., and Jarlier, V. (1995). Sequences of conserved region in the a subunit of DNA gyrase from nine species of the genus Mycobacterium: phylogenetic analysis and implication for intrinsic susceptibility to quinolones. Antimicrob. Agents Chemother. 39, 2145–2149. doi: 10.1128/aac.39.9.2145
Gupta, R. S., Lo, B., and Son, J. (2018). Phylogenomics and comparative genomic studies robustly support division of the genus Mycobacterium into an emended genus Mycobacterium and four novel genera. Front. Microbiol. 9:67. doi: 10.3389/fmicb.2018.00067
Habe, H., Chung, J. S., Ishida, A., Kasuga, K., Ide, K., Takemura, T., et al. (2005). The fluorene catabolic linear plasmid in Terrabacter sp. strain DBF63 carries the beta-ketoadipate pathway genes, pcaRHGBDCFIJ, also found in proteobacteria. Microbiology 151, 3713–3722. doi: 10.1099/mic.0.28215-0
Hartmans, S., De Bont, J. A., and Stackebrandt, E. J. T. P. (2006). “The genus Mycobacterium--nonmedical” in The prokaryotes. eds. M. Dworkin, S. Falkow, E. Rosenberg, K. H. Schleifer, and E. Stackebrandt (New York, NY: Springer)
Hormisch, D., Hormisch, D., Brost, I., Kohring, G. W., Giffhorn, F., Kroppenstedt, R. M., et al. (2004). Mycobacterium fluoranthenivorans sp. nov., a fluoranthene and aflatoxin B1 degrading bacterium from contaminated soil of a former coal gas plant. Syst. Appl. Microbiol. 27, 653–660. doi: 10.1078/0723202042369866
Kahl, S., and Hofer, B. (2003). A genetic system for the rapid isolation of aromatic-ring-hydroxylating dioxygenase activities. Microbiology 149, 1475–1481. doi: 10.1099/mic.0.25976-0
Kasai, H., Ezaki, T., and Harayama, S. (2000). Differentiation of phylogenetically related slowly growing mycobacteria by their gyrB sequences. J. Clin. Microbiol. 38, 301–308. doi: 10.1128/jcm.38.1.301-308.2000
Khan, A. A., Wang, R. F., Cao, W. W., Doerge, D. R., Wennerstrom, D., and Cerniglia, C. E. (2001). Molecular cloning, nucleotide sequence, and expression of genes encoding a polycyclic aromatic ring dioxygenase from Mycobacterium sp. strain PYR-1. Appl. Environ. Microbiol. 67, 3577–3585. doi: 10.1128/aem.67.8.3577-3585.2001
Kim, H., Kim, S. H., Shim, T. S., Kim, M. N., Bai, G. H., Park, Y. G., et al. (2005). Differentiation of Mycobacterium species by analysis of the heat-shock protein 65 gene (hsp65). Int. J. Syst. Evol. Microbiol. 55, 1649–1656. doi: 10.1099/ijs.0.63553-0
Kim, S. J., Kweon, O., Jones, R. C., Freeman, J. P., Edmondson, R. D., and Cerniglia, C. E. (2007). Complete and integrated pyrene degradation pathway in Mycobacterium vanbaalenii PYR-1 based on systems biology. J. Bacteriol. 189, 464–472. doi: 10.1128/jb.01310-06
Kim, M., Oh, H. S., Park, S. C., and Chun, J. (2014). Towards a taxonomic coherence between average nucleotide identity and 16S rRNA gene sequence similarity for species demarcation of prokaryotes. Int. J. Syst. Evol. Microbiol. 64, 346–351. doi: 10.1099/ijs.0.059774-0
Kweon, O., Kim, S. J., Holland, R. D., Chen, H., Kim, D. W., Gao, Y., et al. (2011). Polycyclic aromatic hydrocarbon metabolic network in Mycobacterium vanbaalenii PYR-1. J. Bacteriol. 193, 4326–4337. doi: 10.1128/jb.00215-11
Lagesen, K., Hallin, P., Rødland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Lévy-Frébault, V. V., and Portaels, F. (1992). Proposed minimal standards for the genus Mycobacterium and for description of new slowly growing Mycobacterium species. Int. J. Syst. Bacteriol. 42, 315–323. doi: 10.1099/00207713-42-2-315
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., et al. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Masindi, V., and Muedi, K. L. J. H. M. (2018). “Environmental contamination by heavy metals” in Heavy metals. eds. H. E. D. M. Saleh and R. F. Aglan (Chichester: IntechOpen)
Mcmahon, M. D., Rush, J. S., and Thomas, M. G. (2012). Analyses of MbtB, MbtE, and MbtF suggest revisions to the mycobactin biosynthesis pathway in Mycobacterium tuberculosis. J. Bacteriol. 194, 2809–2818. doi: 10.1128/jb.00088-12
Minnikin, D., O'donnell, A., Goodfellow, M., Alderson, G., Athalye, M., Schaal, A., et al. (1984). An integrated procedure for the extraction of bacterial isoprenoid quinones and polar lipids. J. Microbiol. Methods 2, 233–241. doi: 10.1016/0167-7012(84)90018-6
Møller, A. K., Barkay, T., Hansen, M. A., Norman, A., Hansen, L. H., Sørensen, S. J., et al. (2014). Mercuric reductase genes (merA) and mercury resistance plasmids in high Arctic snow, freshwater and sea-ice brine. FEMS Microbiol. Ecol. 87, 52–63. doi: 10.1111/1574-6941.12189
Nakamiya, K., Hashimoto, S., Ito, H., Edmonds, J. S., Yasuhara, A., and Morita, M. (2005). Microbial treatment of bis (2-ethylhexyl) phthalate in polyvinyl chloride with isolated bacteria. J. Biosci. Bioeng. 99, 115–119. doi: 10.1263/jbb.99.115
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pereira, A. C., Ramos, B., Reis, A. C., and Cunha, M. V. (2020). Non-tuberculous mycobacteria: molecular and physiological bases of virulence and adaptation to ecological niches. Microorganisms 8:1380. doi: 10.3390/microorganisms8091380
Reis, A. C., and Cunha, M. V. (2021). The open pan-genome architecture and virulence landscape of Mycobacterium bovis. Microb Genom 7:000664. doi: 10.1099/mgen.0.000664
Ren, L., Jia, Y., Ruth, N., Qiao, C., Wang, J., Zhao, B., et al. (2016). Biodegradation of phthalic acid esters by a newly isolated Mycobacterium sp. YC-RL4 and the bioprocess with environmental samples. Environ. Sci. Pollut. Res. Int. 23, 16609–16619. doi: 10.1007/s11356-016-6829-4
Ren, L., Wang, G., Huang, Y., Guo, J., Li, C., Jia, Y., et al. (2021). Phthalic acid esters degradation by a novel marine bacterial strain Mycolicibacterium phocaicum RL-HY01: characterization, metabolic pathway and bioaugmentation. Sci. Total Environ. 791:148303. doi: 10.1016/j.scitotenv.2021.148303
Runyon, E. H. (1959). Anonymous mycobacteria in pulmonary disease. Med. Clin. North Am. 43, 273–290. doi: 10.1016/s0025-7125(16)34193-1
Sasser, M (2006). Bacterial identification by gas chromatographic analysis of fatty acids methyl esters (GC-FAME). Newark, NY: Microbial ID Inc.
Schleifer, K. H., and Kandler, O. J. B. R. (1972). Peptidoglycan types of bacterial cell walls and their taxonomic implications. Bacteriol. Rev. 36, 407–477. doi: 10.1128/br.36.4.407-477.1972
Shamsuzzaman, M., Dahal, R. H., Kim, S., and Kim, J. (2023). Genome insight and probiotic potential of three novel species of the genus Corynebacterium. Front. Microbiol. 14:1225282. doi: 10.3389/fmicb.2023.1225282
Sherpa, M. T., Najar, I. N., Das, S., and Thakur, N. (2020). Distribution of antibiotic and metal resistance genes in two glaciers of North Sikkim, India. Ecotoxicol. Environ. Saf. 203:111037. doi: 10.1016/j.ecoenv.2020.111037
Skerman, V. B. D., Mcgowan, V., and Sneath, P. H. A. (1980). Approved lists of bacterial names. Med. J. Aust 30, 225–230. doi: 10.5694/j.1326-5377.1980.tb131800.x
Stahl, D. A., and Urbance, J. W. (1990). The division between fast- and slow-growing species corresponds to natural relationships among the mycobacteria. J. Bacteriol. 172, 116–124. doi: 10.1128/jb.172.1.116-124.1990
Stinear, T. P., Seemann, T., Harrison, P. F., Jenkin, G. A., Davies, J. K., Johnson, P. D., et al. (2008). Insights from the complete genome sequence of Mycobacterium marinum on the evolution of Mycobacterium tuberculosis. Genome Res. 18, 729–741. doi: 10.1101/gr.075069.107
Stone, B. B., Nietupski, R. M., Breton, G. L., and Weisburg, W. G. (1995). Comparison of Mycobacterium 23S rRNA sequences by high-temperature reverse transcription and PCR. Int. J. Syst. Bacteriol. 45, 811–819. doi: 10.1099/00207713-45-4-811
Takewaki, S. I., Okuzumi, K., Manabe, I., Tanimura, M., Miyamura, K., Nakahara, K. I., et al. (1994). Nucleotide sequence comparison of the mycobacterial dnaJ gene and PCR-restriction fragment length polymorphism analysis for identification of mycobacterial species. Int. J. Syst. Bacteriol. 44, 159–166. doi: 10.1099/00207713-44-1-159
Tamura, K., Stecher, G., and Kumar, S. (2021). MEGA11: molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38, 3022–3027. doi: 10.1093/molbev/msab120
Tortoli, E. (2012). Phylogeny of the genus Mycobacterium: many doubts, few certainties. Infect. Genet. Evol. 12, 827–831. doi: 10.1016/j.meegid.2011.05.025
Tortoli, E., Brown-Elliott, B. A., Chalmers, J. D., Cirillo, D. M., Daley, C. L., Emler, S., et al. (2019). Same meat, different gravy: ignore the new names of mycobacteria. Eur. Respir. J. 54:1900795. doi: 10.1183/13993003.00795-2019
Vernikos, G., Medini, D., Riley, D. R., and Tettelin, H. (2015). Ten years of pan-genome analyses. Curr. Opin. Microbiol. 23, 148–154. doi: 10.1016/j.mib.2014.11.016
Willumsen, P., Karlson, U., Stackebrandt, E., and Kroppenstedt, R. M. (2001). Mycobacterium frederiksbergense sp. nov., a novel polycyclic aromatic hydrocarbon-degrading Mycobacterium species. Int. J. Syst. Evol. Microbiol. 51, 1715–1722. doi: 10.1099/00207713-51-5-1715
Wright, R. J., Bosch, R., Gibson, M. I., and Christie-Oleza, J. A. (2020). Plasticizer degradation by marine bacterial isolates: a Proteogenomic and Metabolomic characterization. Environ. Sci. Technol. 54, 2244–2256. doi: 10.1021/acs.est.9b05228
Yang, J., Gu, Y., Chen, Z., Song, Y., Sun, F., Liu, J., et al. (2021). Colonization and performance of a pyrene-degrading bacterium Mycolicibacterium sp. Pyr9 on root surfaces of white clover. Chemosphere 263:127918. doi: 10.1016/j.chemosphere.2020.127918
Yoon, S. H., Ha, S. M., Kwon, S., Lim, J., Kim, Y., Seo, H., et al. (2017a). Introducing EzBioCloud: a taxonomically united database of 16S rRNA gene sequences and whole-genome assemblies. Int. J. Syst. Evol. Microbiol. 67, 1613–1617. doi: 10.1099/ijsem.0.001755
Yoon, S. H., Ha, S. M., Lim, J., Kwon, S., and Chun, J. (2017b). A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 110, 1281–1286. doi: 10.1007/s10482-017-0844-4
Zeng, K., Hwang, H. M., Dong, S., Shi, X., Wilson, K., Green, J., et al. (2004). Photochemical transformation and phototoxicity of 1-aminopyrene. Environ. Toxicol. Chem. 23, 1400–1407. doi: 10.1897/03-415
Zeng, J., Zhu, Q., Wu, Y., Chen, H., and Lin, X. (2017). Characterization of a polycyclic aromatic ring-hydroxylation dioxygenase from Mycobacterium sp. NJS-P. Chemosphere 185, 67–74. doi: 10.1016/j.chemosphere.2017.07.001
Zhang, D. F., Cui, X. W., Zhao, Z., Zhang, A. H., Huang, J. K., and Li, W. J. (2020). Sphingomonas hominis sp. nov., isolated from hair of a 21-year-old girl. Antonie Van Leeuwenhoek 113, 1523–1530. doi: 10.1007/s10482-020-01460-z
Zhang, Y., Zhang, J., Fang, C., Pang, H., and Fan, J. (2012). Mycobacterium litorale sp. nov., a rapidly growing mycobacterium from soil. Int. J. Syst. Evol. Microbiol. 62, 1204–1207. doi: 10.1099/ijs.0.033449-0
Keywords: Mycobacterium adipatum, Mycobacterium deserti, Mycobacterium hippophais, genome, bioremediation potential
Citation: Deng Y, Mou T, Wang J, Su J, Yan Y and Zhang Y-Q (2023) Characterization of three rapidly growing novel Mycobacterium species with significant polycyclic aromatic hydrocarbon bioremediation potential. Front. Microbiol. 14:1225746. doi: 10.3389/fmicb.2023.1225746
Edited by:
Vishal Tripathi, Graphic Era University, IndiaReviewed by:
Anukool Vaishnav, Agroscope, SwitzerlandCopyright © 2023 Deng, Mou, Wang, Su, Yan and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanchun Yan, eWFueWFuY2h1bkBjYWFzLmNu; Yu-Qin Zhang, eXpoYW5nQGltYi5wdW1jLmVkdS5jbg==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.