 Camilo Gómez-Garzón
Camilo Gómez-Garzón Shelley M. Payne
Shelley M. Payne
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Microbiol. , 04 July 2023
Sec. Infectious Agents and Disease
Volume 14 - 2023 | https://doi.org/10.3389/fmicb.2023.1219359
Introduction: Feo is the most widespread and conserved system for ferrous iron uptake in bacteria, and it is important for virulence in several gastrointestinal pathogens. However, its mechanism remains poorly understood. Hitherto, most studies regarding the Feo system were focused on Gammaproteobacterial models, which possess three feo genes (feoA, B, and C) clustered in an operon. We found that the human pathogen Helicobacter pylori possesses a unique arrangement of the feo genes, in which only feoA and feoB are present and encoded in distant loci. In this study, we examined the functional significance of this arrangement.
Methods: Requirement and regulation of the individual H. pylori feo genes were assessed through in vivo assays and gene expression profiling. The evolutionary history of feo was inferred via phylogenetic reconstruction, and AlphaFold was used for predicting the FeoA-FeoB interaction.
Results and Discussion: Both feoA and feoB are required for Feo function, and feoB is likely subjected to tight regulation in response to iron and nickel by Fur and NikR, respectively. Also, we established that feoA is encoded in an operon that emerged in the common ancestor of most, but not all, helicobacters, and this resulted in feoA transcription being controlled by two independent promoters. The H. pylori Feo system offers a new model to understand ferrous iron transport in bacterial pathogens.
Iron acquisition is a major challenge for bacterial pathogens inside the host, and it is often a determining factor for infection and disease. Most of the iron in the host environment is tightly bound to proteins, hence not readily available for pathogens. Bacterial pathogens have evolved a diverse arsenal of systems for iron acquisition. This includes the secretion of siderophores, organic molecules that bind iron with high affinity. In response to infection and inflammation, the mammalian host may sequester iron and reduce its availability to the pathogen, a pathway known as nutritional immunity (Skaar, 2010; Barber and Elde, 2015).
Iron can exist in two forms, the oxidized state of iron, ferric iron (Fe3+), or the reduced state, ferrous iron (Fe2+). The more insoluble ferric form is commonly found in association with proteins and siderophores, while ferrous iron can exist as a free ion, especially under conditions of acidic pH and low oxygen tension, such as those present in the gastric tract (Mey et al., 2021). For this reason, the acquisition of free Fe2+ may be particularly relevant for bacterial gastric pathogens, and Feo constitutes the most widespread and conserved Fe2+ transporter in bacteria (Lau et al., 2016; Sestok et al., 2018).
Feo has been shown to contribute to virulence in numerous plant (Franza and Expert, 2013), animal, and human pathogens (Lau et al., 2016), including Xanthomonas oryzae (Pandey and Sonti, 2010), Salmonella enterica (Boyer et al., 2002), Legionella pneumophila (Robey and Cianciotto, 2002), and Helicobacter pylori (Velayudhan et al., 2000). Although this system is ubiquitous in bacteria, and important for virulence in some instances, its mechanism and the specific role of its components remain poorly understood. Most of the studies of the Feo system are based on Gammaproteobacteria [Pseudomonadota.] In these species, Feo is made up of three components: FeoA, FeoB, and FeoC, that have been shown to work as a polyprotein complex embedded in the inner membrane in the V. cholerae model (Figure 1A). FeoB is a large (~ 85 kDa) transmembrane protein with cytoplasmic N- and C-terminal domains; and its N-terminal domain (NFeoB) is an NTPase that shares homology with eukaryotic G-proteins, such as the human oncogene protein p21 Ras (Lau et al., 2016; Sestok et al., 2018). The catalytic activity of NFeoB is essential for the function of the transporter, and its nucleotide specificity varies among species; for example, NFeoB is a dual GTP/ATPase in V. cholerae and H. pylori, but solely a GTPase in E. coli (Shin et al., 2019, 2020). The other two components, FeoA and FeoC, are small (~8.5 kDa) cytoplasmic proteins with unknown functions though they are both required for iron uptake via Feo (Weaver et al., 2013).
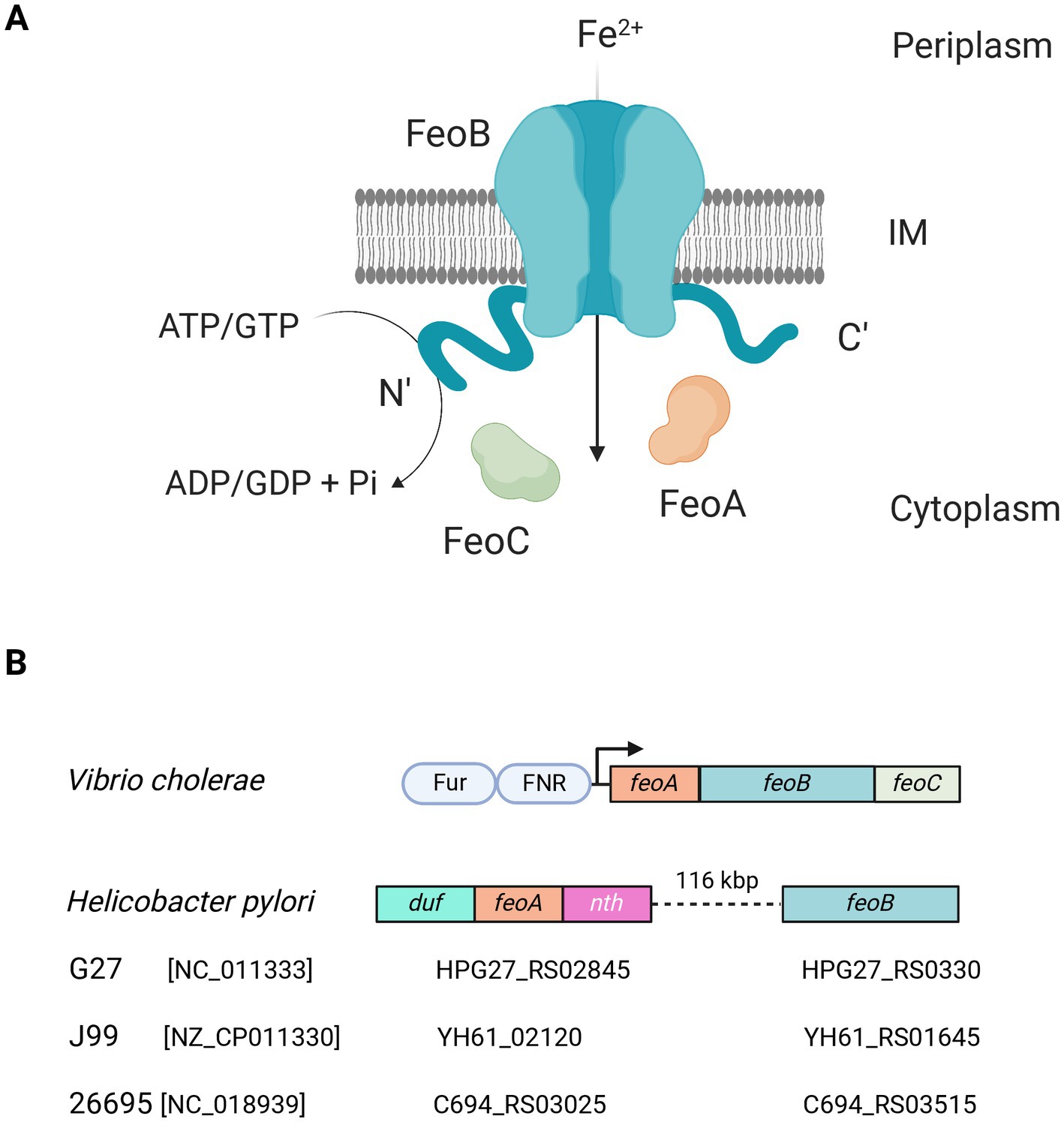
Figure 1. Current understanding of the Feo system. (A) Feo works as a large (>250 kDa) complex embedded in the inner membrane (IM), likely consisting of trimers of FeoABC units. The N- and C-terminal domains of FeoB remain in the cytoplasm, and the N-terminal domain has ATPase and GTPase activity, which is essential for Fe2+ uptake. This model is largely based on observations made in V. cholerae (Gómez-Garzón and Payne, 2020). (B) In V. cholerae—as well as in most of the Gammaproteobacteria group—the feo genes form an operon controlled by Fur and FNR (shown as pale blue ovals). Strikingly, in H. pylori the operon architecture is not conserved. Instead, feoA is located between duf and nth, and feoB is separated from feoA by about 116 kbp. Diagrams are not to scale. The locus tags of feoA and feoB for three representative H. pylori strains are shown below the diagram. The RefSeq accession number of each genome is shown in brackets. Figure created with BioRender.com.
In Gammaproteobacteria, the feoA, B, and C genes are encoded in an operon controlled by the Ferric Uptake Regulator (Fur) and the Fumarate and Nitrate Reduction regulatory protein (FNR), bacterial master regulators responsive to Fe2+ and O2, respectively. In previous studies of V. cholerae, we have determined that the Feo proteins likely work in a 1:1:1 stoichiometric ratio (Gómez-Garzón and Payne, 2020). We have also shown that FeoA is essential for the assembly of the complex, while FeoC, though not required for complex formation, is critical for function, as those complexes assembled in the absence of FeoC do not support iron uptake (Weaver et al., 2013; Stevenson et al., 2016).
Although research on the bacterial Feo system has primarily focused on Gammaproteobacteria species, alternative architectures of Feo have been identified in other groups (Sestok et al., 2018). For example, the commensal species Bacteroides fragilis has a single Feo protein containing a fusion of FeoA and FeoB homolog domains (Sestok et al., 2022). A common feature found when comparing the Feo system among species is that FeoA and FeoB orthologs (or their corresponding domains) are nearly universal, while FeoC is poorly conserved, being present in about 5% of bacterial proteomes, predominantly within the Gamma group (Gómez-Garzón et al., 2022). The specific role of FeoC as well as the functional significance of the different architectures of the Feo system are still to be determined.
By exploring these diverse architectures of Feo among bacteria, we found that H. pylori—an important human pathogen for its causal relationship with peptic ulcers and gastric cancer (Herrera and Parsonnet, 2009; Plummer et al., 2015; de Martel et al., 2020)—exhibits a unique arrangement of the feo genes. Feo is not an operon in this species, since feoA and feoB are separated by 116 kbp. feoB has canonical Fur-binding boxes in its putative promoter, while feoA is embedded between two genes in an operon-like arrangement with no evident Fur binding boxes (Figure 1B). Namely, feoA localizes between the nth gene (downstream), which encodes the endonuclease III, and an upstream gene annotated as a “hypothetical Domain of Unknown Function (DUF) 3,971-containing protein.” H. pylori is naturally competent and recombination events drive evolution of subpopulations within the host during infection (Blaser and Berg, 2001; Suerbaum and Josenhans, 2007; Linz et al., 2014; Kobayashi, 2016; Jackson et al., 2020). Consequently, H. pylori is characterized by having a highly plastic genome, with low synteny and the absence of several transcription factors commonly found in other species. For instance, H. pylori lacks FNR, and it is not rare that it lacks the operon structure of several systems, which is the case for feo. Thus, feoA was initially overlooked because it was not associated with feoB. In consequence, although FeoB likely contributes to virulence in H. pylori, whether feoA is also required for Fe2+ uptake remains unclear.
Characterizing the Feo system in H. pylori offers a new model in addition to that of Gammaproteobacteria to understand this major bacterial iron transporter. Equally important, this represents a model of Feo relevant for an important human pathogen. In this study, we conducted an initial characterization of the H. pylori Feo system. We determined the requirement of feoA; complex formation by FeoA and B; and transcriptional regulation of both genes, including the role of the transcriptional regulator Fur. Additionally, we modeled the evolutionary history of this feo architecture in the context of other helicobacters and the Campylobacterota [Epsilonproteobacteria] group.
All reagents and growth media were purchased from Sigma-Aldrich Chemical Company unless stated otherwise. E. coli and V. cholerae strains were routinely grown in Luria-Bertani (LB) broth (10 g/L tryptone, 5 g/L yeast extract, and 10 g/L NaCl in double-distilled water) or on LB agar (1.5% w/v bacteriological agar) at 37°C and 200 rpm for liquid media. These strains were preserved at −80°C in tryptic soy broth (TSB) with 20% glycerol.
Helicobacter pylori strains were grown and maintained following the protocols described in Whitmire and Merrell (2012): Freezer stocks were prepared in brain heart infusion (BHI) broth supplemented with 20% v/v glycerol and 10% v/v fetal bovine serum (FBS). For culturing in solid media, H. pylori was always grown in horse blood agar (HBA) plates containing 4% w/v Columbia agar base (BD Difco™), 5% v/v defibrinated horse blood (Remel™), 0.2% w/v β-cyclodextrin, and antibiotic supplementation (5 μg/mL trimethoprim, 8 μg/mL amphotericin B, 10 μg/mL vancomycin hydrochloride, 5 μg/mL cefsulodin sodium salt, and 0.33 μg/mL Polymyxin B Sulfate) at 37°C under microaerobic atmosphere. For liquid cultures, brucella broth (BB) supplemented with 10% v/v FBS and 10 μg/mL vancomycin was used. Cells were grown at 37°C with 100 rpm shaking under microaerobic atmosphere. In both cases, microaerobiosis was generated with CampyGen™ packets in a Oxoid™ AnaeroJar™ system (Thermo Fisher Scientific).
For strains harboring plasmids, antibiotics were used as follows: For E. coli, 50 μg/mL ampicillin, 8 μg/mL chloramphenicol, and 12.5 μg/mL tetracycline; for V. cholerae, 25 μg/mL ampicillin and 6.25 μg/mL tetracycline; and for H. pylori, 25 μg/mL kanamycin and 8 μg/mL chloramphenicol.
Bacterial strains and plasmids used in this study are listed in Supplementary Tables S1, S2, respectively.
Primers used in this study are listed in Supplementary Table S3. The accession numbers for the H. pylori loci used for primer design are listed in Supplementary Table S4 PCRs were all done using high-fidelity Phusion Taq polymerase (New England BioLabs). Restriction and ligation reactions were carried out using NEB restriction enzymes and NEB T4 ligase, respectively. Plasmids were routinely purified using a QIAprep Spin Miniprep Kit by Qiagen. All constructs were initially cloned into E. coli TOP10 via CaCl2 heat-shock transformation. To confirm the sequence and directionality of the DNA constructs, the final products were submitted to Genewiz for Sanger DNA sequencing, and results were analyzed with the SnapGene v6.1.2 software.
For experiments in E. coli and V. cholerae purified genomic DNA from H. pylori 26695 (ATCC®) was used as a template to amplify HpfeoA with the primers HpFeoA-EcoRI-F and HpFeoA-EcoRI-R, and HpfeoB with the primers HpFeoB-EcoRI-F and HpFeoB-NotI-R. To generate pHpfeoA, the PCR product for HpfeoA was digested with EcoRI and cloned into the corresponding restriction site in pACYC184 in the same direction as the Pcamr promoter. Similarly, the PCR product for HpfeoB was digested with EcoRI and BamHI and cloned into the corresponding restriction sites in pWKS30 in the same direction as the Plac promoter to generate pHpfeoB.
The complementation vector for HpfeoA (pTMHpfeoA) in H. pylori G27 was constructed by substituting the gfp gene in pTM117 with HpfeoA from this strain. Briefly, gfp in pTM117 was removed by digestion with BamHI and PstI to then insert the HpfeoA coding sequence, which was previously amplified from H. pylori G27 genomic DNA—purified from overnight liquid cultures using a PureLink™ Genomic DNA Kit (Thermo Fisher Scientific)—with the primers HpFeoA-BamHI-F and HpFeoA-PstI-R, and treated with the same restriction enzymes.
Vibrio cholerae EPV6 was transformed with equimolar amounts of pACYC184 and pWKS30 (or derivatives) simultaneously by electroporation as previously described (Occhino et al., 1998). EPV6 cells carrying the Feo constructs under analysis were streaked onto different quadrants of LB agar plates, with and without 10 μM heme supplementation, and incubated at 37°C overnight. In these assays, functional Feo systems and empty vectors were used as positive and negative controls, respectively. To facilitate development of isolated colonies after 24 h, plates without heme were supplemented with 20 μM FeSO4 stabilized with 5 mM sodium ascorbate. For replication, all these assays were carried out in at least three separate plates under each condition; each plate was inoculated with individual colonies. Observable growth after incubation was considered a positive result.
feoA and feoB H. pylori isogenic deletion mutants were generated following the protocol described in Servetas et al. (2021). Specifically, each feo gene was disrupted via homologous recombination by transforming H. pylori G27 with splicing by overlap extension (SOE) PCR products containing the kanr marker with homologous flanking sequences for the feo gene. SOE PCR products for HpfeoA and HpfeoB were kindly donated by Dr. Nina Salama (Fred Hutch Cancer Center, Seattle, WA).
In short, 30–50 μL of concentrated H. pylori cell suspension prepared from 24 h liquid cultures are spotted onto prewarmed HBA plates and dried for 3–4 h at 37°C under microaerobiosis. Afterwards, 50–100 ng of the SOE PCR product are added on top of the spotted cells and incubated for 24 h at 37°C in microaerobic conditions. Then, cells are swabbed and resuspended in liquid media, and this suspension is plated onto HBA plates containing kanamycin. Plates are incubated at 37°C under microaerobiosis and continuously monitored for colony growth. Colonies that grew after 3–5 days were isolated and streaked on kanamycin-containing HBA plates. Successful recombination was confirmed by amplifying the feo loci with the primers Conf-HpfeoA-F and Conf-HpfeoA-R (for HpfeoA::kanr) and Conf-HpfeoB-F and Conf-HpfeoB-R (for HpfeoB::kanr) and sequenced to verify the insertion of the kanr marker.
For insertion of plasmids in H. pylori G27, the same transformation protocol described above was followed using a plasmid prep as the DNA source. Since all plasmid transformations in H. pylori were made with pTM117 derivatives, selection was done in HBA plates supplemented with kanamycin.
Nickel sensitivity of H. pylori feo mutants was assessed by growing them on HBA plates containing a gradient of Ni2+ concentration up to 250 μM. In order to expose the cells to the whole gradient, 50 μL of a concentrated cell suspension (prepared from a 24 h liquid culture) was spotted on the lowest-concentrated border of each plate and the plate was tilted to let the drop slip. Gradient plates were prepared by pouring 0 and 250 μM melted HBA separately on Petri dishes, as described in Weinberg (1959). Plain HBA plates (no Ni2+) and H. pylori WT were used as controls in this experiment. Likewise, a complementation strain for HpfeoA was also included to rule out polar effects of the HpfeoA deletion.
For expression in E. coli, transcriptional reporters (pGT- vectors as listed in Supplementary Table S2) were constructed by cloning the putative promoters under study upstream of the promoterless gfp gene in pGTXN3, between the BamHI and XmaI sites (for PfeoA and PfeoB) or XmaI and XbaI (for Pduf). Promoters were obtained by PCR from H. pylori G27 genomic DNA with the primers promHpFeoA-XmaI-F and promHpFeoA-BamHI-R for PfeoA, promHpFeoB-XmaI-F and promHpFeoB-BamHI-R for PfeoB, and prom-DUF-XmaI-F and prom-DUF-XbaI-R for Pduf. Equivalent transcriptional reporters compatible with H. pylori (pTM- vectors in Table X) were constructed in pTM117 with the same cloning strategy.
For gfp expression assays the protocol described in Carpenter et al. (2007) was followed with some modifications: H. pylori G27 and Δfur were grown for 48 h in liquid culture as described above Then 1.5 mL of each culture were washed twice with phosphate-buffered saline (PBS) and used in an SDS-PAGE gel to be immunoblotted with anti-GFP antibodies (JL-8 from Clonetech).
A C-terminal PstI restriction site was added in the HpfeoA CDS in pHpfeoA via QuikChange site directed mutagenesis (Xia et al., 2015) with the primers HpFeoA-PstC-F and HpFeoA-PstC-R (Supplementary Table S3).
Separately, a dsDNA probe carrying the epitope FLAG coding sequence with PstI sticky ends was generated by annealing the C-FLAG-PstI-Top and C-FLAG-PstI-Btm fragments. The annealed FLAG-coding probe was ligated with the PstI-treated pHpfeoA plasmid and transformed into E. coli TOP10. The final product of this ligation is referred to as pHpfeoAC-FLAG.
Protein samples from cell cultures were analyzed through 8–16% gradient SDS polyacrylamide gels prepared as described in Miller et al. (2016).
For immunoblot analysis, resolved proteins were tank-transferred from the polyacrylamide gel to an Immobilon®-P PVDF membrane (Merck Millipore). Then, GFP or FLAG-tagged proteins were detected using mouse monoclonal anti-GFP (JL-8 from Clonetech) or anti-FLAG monoclonal antibody (M2 from Sigma Aldrich), respectively; and visualized using horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Bio-Rad) followed by detection with Pierce™ ECL Western Blotting Substrate (Thermo Fisher Scientific). To ensure even levels of loading, total protein content in the immunoblotted samples was assessed by Coomassie staining using R-250 Brilliant Blue (Bio-Rad).
Iron depletion was induced with a rapid exposure to 2,2′-dipyridyl (dpp) adapted from the method described in Carpenter et al. (2007): H. pylori G27 and Δfur were grown overnight, cultures were divided in halves, and one half of each culture was exposed to 200 μM dpp for 1 h. After this incubation, RNA was isolated as described below.
Total RNA was isolated from ∼109 cells using RNA-Bee (Tel-Test, Inc.) per the manufacturer’s instructions. RNA was treated with TURBO™ DNase (Thermo Fisher Scientific) and then precipitated with cold ethanol and resuspended in diethyl pyrocarbonate (DEPC)-treated water (Thermo Fisher Scientific). Integrity of the isolated RNA was checked by electrophoresis in an agarose gel and the concentration, assessed with a NanoDrop® machine (Thermo Fisher Scientific). The RNA was retrotranscribed to cDNA using a SuperScript III system with random hexamers (Thermo Fisher Scientific). Additional reactions using water instead of retrotranscriptase were done and included in both PCR and qPCR runs as a negative control. cDNA was used as a template for PCR and qPCR amplifications.
For regular PCR amplification, a 1:2 cDNA dilution was used as a template. The primers used for obtaining the products shown below were: for A, RTq-junct-F and RTq-junct-R; for B, Duf-feoA-Locus-F and Duf-feoA-Locus-R; and for C, feoA-nth-F and feoA-nth-R. Amplifications were additionally performed on genomic DNA samples as a positive control.
qPCR was conducted with cDNA diluted in a 1:10 ratio with Power SYBR green (Thermo Fisher Scientific) in an Applied Biosystems ViiA 7 instrument with the following parameters: 50°C for 2 min and 95°C for 10 min; followed by 95°C for 15 s and 60°C for 1 min for 40 cycles, with the fluorescence recorded at 60°C. A melting curve was generated as follows: 90°C for 15 s, 60°C for 1 min, and then 95°C for 15 s with the fluorescence recorded every 0.05 s. Relative expression levels were calculated using the threshold cycle (ΔΔCT) method (Schmittgen and Livak, 2008). Each reaction produced only one melting curve, indicating that only one target had been amplified during the qPCR reaction. A single amplification product from these reactions was further verified by resoling the samples after the qPCR run through regular agarose electrophoresis.
All of these analyses used rpoD and gyrB as internal references. The primers used for qPCR were designed using the Primer3 algorithm (Untergasser et al., 2012) optimizing the parameters as recommended by the SYBR green manufacturer: RTq-HpfeoA-F and RTq-HpfeoA-R for HpfeoA, RTq-HpfeoB-F and RTq-HpfeoB-R for HpfeoB, for RTq-junct-F and RTq-junct-R the HpfeoA-B junction (junct), RTq-duf-F and RTq-duf-R for duf, RTq-pfr-F and RTq-pfr-R for pfr, RTq-rpoD-F and RTq-rpoD-R for rpoD, and RTq-gyrB-F and RTq-gyrB-R for gyrB.
For in vivo crosslinking of EPV6 cells, the protocol described in Stevenson et al. (2016) was followed with minor modifications: 50 mL of LB broth supplemented with the appropriate antibiotics were inoculated with overnight cultures in a 1:100 ratio. The culture was grown until mid-log phase (i.e., OD650 ≈ 0.6) at 37°C and 200 rpm. Cells were pelleted and washed twice with 25 mL of PBS. All centrifugations were done at 8,500 × g for 5 min. Cells were then treated with 25 mL of 0.6% v/v formaldehyde in PBS at room temperature for 6 min with gentle shaking. Then, the reaction was quenched by washing the cells with 10 mL 1.25 M glycine in PBS. Cells were washed with 25 mL PBS to remove the quenching solution, and the final cell pellet was frozen at −80°C until further use.
Cell pellets collected and preserved as described above were thawed on ice and resuspended in 5 mL of low-salt buffer, consisting of 100 mM sodium phosphate (pH 7.2), 10% v/v glycerol, 1 mM EDTA, 1 mM PMSF. Samples were sonicated to induce cell lysis, and cell debris was removed by centrifugation at 12,000 × g for 10 min. Total membrane pellets were separated from the cytoplasmic fractions at 50,000 rpm for 45 min using a TLA-100.3 rotor (Beckman Coulter). Total membrane pellets were washed twice with high-salt buffer (20 mM sodium phosphate (pH 7.2), 2 M KCl, 10% v/v glycerol, 1 mM EDTA, 1 mM PMSF) and once with 20 mM HEPES-NaOH buffer (pH 7.5) containing 1 mM PMSF. Both cytoplasmic and membrane fractions were preserved at −80°C for further processing.
Cytoplasmic and membrane fractions were first enriched in FLAG-tagged proteins via immunoprecipitation with the same anti-FLAG monoclonal antibody used for immunoblot analyses and SureBeadsTM Protein G Magnetic Beads (Bio-Rad) following the manufacturer’s instructions.
Immunoprecipitated samples were subsequently resolved via SDS-PAGE and stained with R-250 Brilliant Blue for no longer than 15 min. The desired bands were excised from the gel, and stored at 4°C in 500 μL of a 10% v/v methanol, 7.5% v/v acetic acid solution until processing.
Protein identification was provided by the UT Austin Center for Biomedical Research Support Biological Mass Spectrometry Facility (RRID: SCR_021728). Proteins were reduced with DTT and alkylated with iodoacetamide, then digested in-gel with trypsin and desalted with Millipore μ-C18 ZipTip pipette tips. Peptide samples were run by LC–MS/MS on a Thermo Ultimate 3000 RSLCnano UPLC in-line with an Orbitrap Fusion Tribrid mass spectrometer. The analytical column was a 75 μm × 25 cm Acclaim PepMap100 C18 column (Thermo Fisher Scientific). The data were collected with FT MS followed by data-dependent acquisition of ion trap MS/MS. Raw data were searched using Proteome Discoverer 2.5 via Sequest HT search engine using 10 ppm mass tolerance for the MS from the FT detector and 0.6 Da for MS/MS from the ion trap detector with fixed modification of carbamidomethylation of cysteine, and variable modifications of methionine oxidation, protein N-terminal acetylation, and protein N-terminal acetylation with Met loss. Validation with Proteome Software Scaffold 5 used a protein threshold of 99% confidence for 2 peptides at a peptide threshold of 1% FDR.
A 200 bp upstream of feoA, feoB, or duf CDS in representative genomes were used to construct the alignment for each feo architecture, i.e., operon or separate genes. These alignments were used as inputs for motif discovery and identification with the XSTREME algorithm (Grant and Bailey, 2021) through the MEME Suite (Bailey et al., 2015). CollecTF (Kiliç et al., 2016) was used as a database for annotating discovered motifs together with binding sequences for Fur and NikR reported in the literature (Delany et al., 2005; Arnold et al., 2012; Pich et al., 2012; Carpenter et al., 2013; Agriesti et al., 2014; Roncarati et al., 2016), which were introduced manually. All the other parameters were used with the values set by default.
Amino acid sequences for RpoB and RpoC were concatenated when necessary and aligned using MUSCLE through MEGA X (Kumar et al., 2018) with all settings defined by default. The resulting alignments were used to construct the corresponding phylogenetic trees by the maximum likelihood method with the Jones-Taylor-Thornton model and the gamma distribution for evolutionary rates. Cutoff values were fixed at 95% for coverage, and trees were tested by bootstrapping with 300 replicates. Trees were plotted using iTOL v6 (Letunic and Bork, 2021).
The genomic context of each feoA gene was determined by manual inspection of the corresponding representative genome listed in Supplementary Table S4.
The HpFeoA-HpFeoB interaction was modeled using AlphaFold-Multimer (Evans et al., 2022) through the ChimeraX interface (Pettersen et al., 2021).
We examined the architecture and distribution of the feo genes in H. pylori. The separation of feoA from feoB as well as the association of feoA with the nth and duf genes are features conserved among H. pylori strains (Figure 1B). FeoB is required for ferrous iron transport in H. pylori (Velayudhan et al., 2000) and in some species, FeoB is sufficient for function and does not require accessory proteins like FeoA or FeoC (Lau et al., 2016; Sestok et al., 2018; Gómez-Garzón et al., 2022). To determine whether feoA is required for Feo function in H. pylori, we tested whether both genes are necessary to support iron uptake in the feo-null mutant strain V. cholerae EPV6. This strain harbors mutations in multiple iron transport systems, and it is therefore unable to grow in standard media such as LB agar, unless the medium is supplied with heme, for which it retains a functional transporter (Peng et al., 2016). Transforming EPV6 with a plasmid carrying a functional Feo system restores growth in non-supplemented media. We have extensively used EPV6 as a tool to unambiguously assess Feo function from diverse species. We cloned the H. pylori feoA and feoB genes (HpfeoA and HpfeoB) in separate, compatible vectors. We found that EPV6 is able to grow in non-supplemented medium only when both HpfeoA and HpfeoB are expressed; the presence of either HpfeoA or HpfeoB in the absence of the other gene did not support EPV6 growth (Figure 2). We obtained a similar result using E. coli H1771, an alternative indicator strain for iron transport (Hantke, 1987). In this strain, both HpfeoA and HpfeoB were required to alleviate iron starvation (Supplementary Figure S1). These results suggest that HpfeoA and HpfeoB are necessary and sufficient to assemble a functional iron transporter.
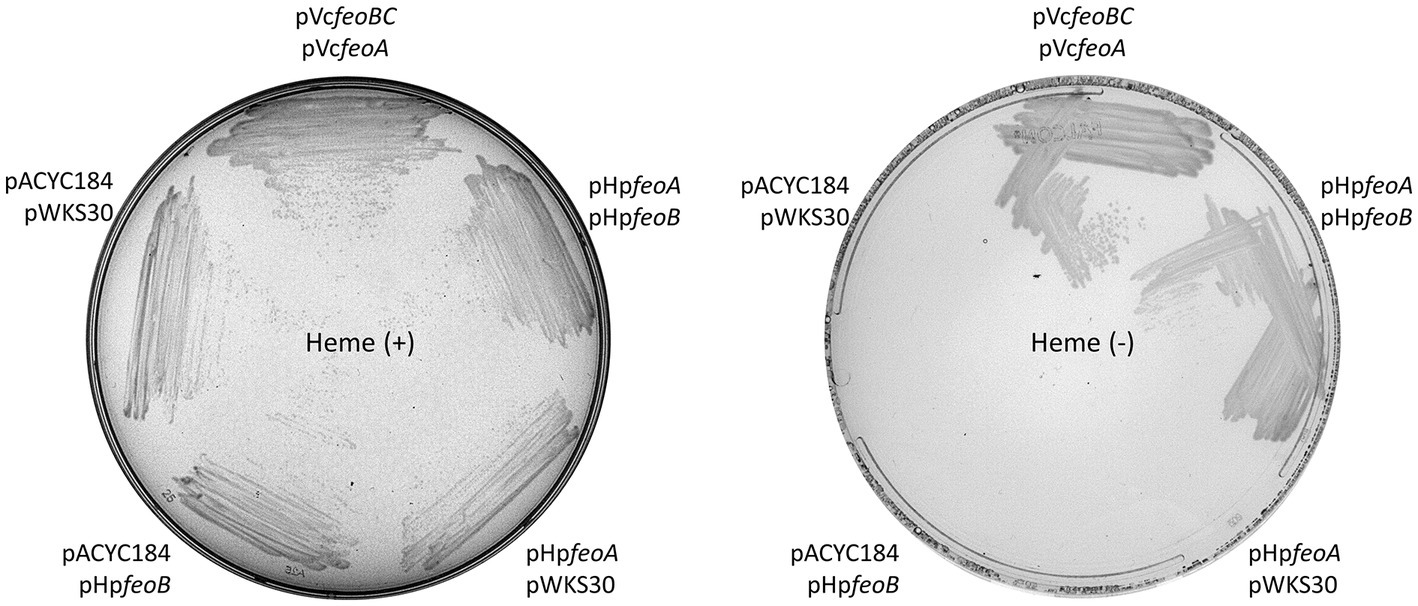
Figure 2. HpfeoA and HpfeoB are necessary and sufficient to support iron uptake. V. cholerae EPV6 requires heme supplementation to grow on LB agar in the absence of a functional iron transport system. EPV6 cells co-transformed with plasmids carrying HpfeoA and/or HpfeoB (pHpfeoA and pHpfeoB) were streaked on medium with (left panel) or without (right panel) heme. Expression of V. cholerae feo genes from the same backbones (pVcfeoA and pVcfeoBC) and the empty vectors in EPV6 served as positive and negative controls, respectively. The data are representative of multiple independent experiments with different transformants (biological replicates).
In order to directly assess the function of H. pylori feo genes in H. pylori G27, we constructed derivative ΔfeoA and ΔfeoB mutants, and determined their phenotypes. Deletion of feoB in H. pylori results in increased sensitivity to heavy metals, especially nickel (Ni2+) (Velayudhan et al., 2000). If both HpfeoA and HpfeoB participate in the same iron transport pathway, deletion of feoA should also lead to increased sensitivity to Ni2+. We tested this by comparing Ni2+ tolerance between ΔfeoA and ΔfeoB mutants of H. pylori G27. Consistent with this hypothesis, both deletion mutants exhibited increased sensitivity toward Ni2+ compared to the wild-type (WT) strain. Specifically, these mutants were unable to grow throughout the concentration gradient tested (up to 250 μM), while the WT strain did not show any inhibition in this range. In the absence of Ni2+, both deletion mutants grew similarly to the WT. In trans expression of feoA from a plasmid restored growth in media containing Ni2+ (Figure 3), demonstrating that the observed phenotype of ΔfeoA was not due to polar effects induced by the gene deletion.
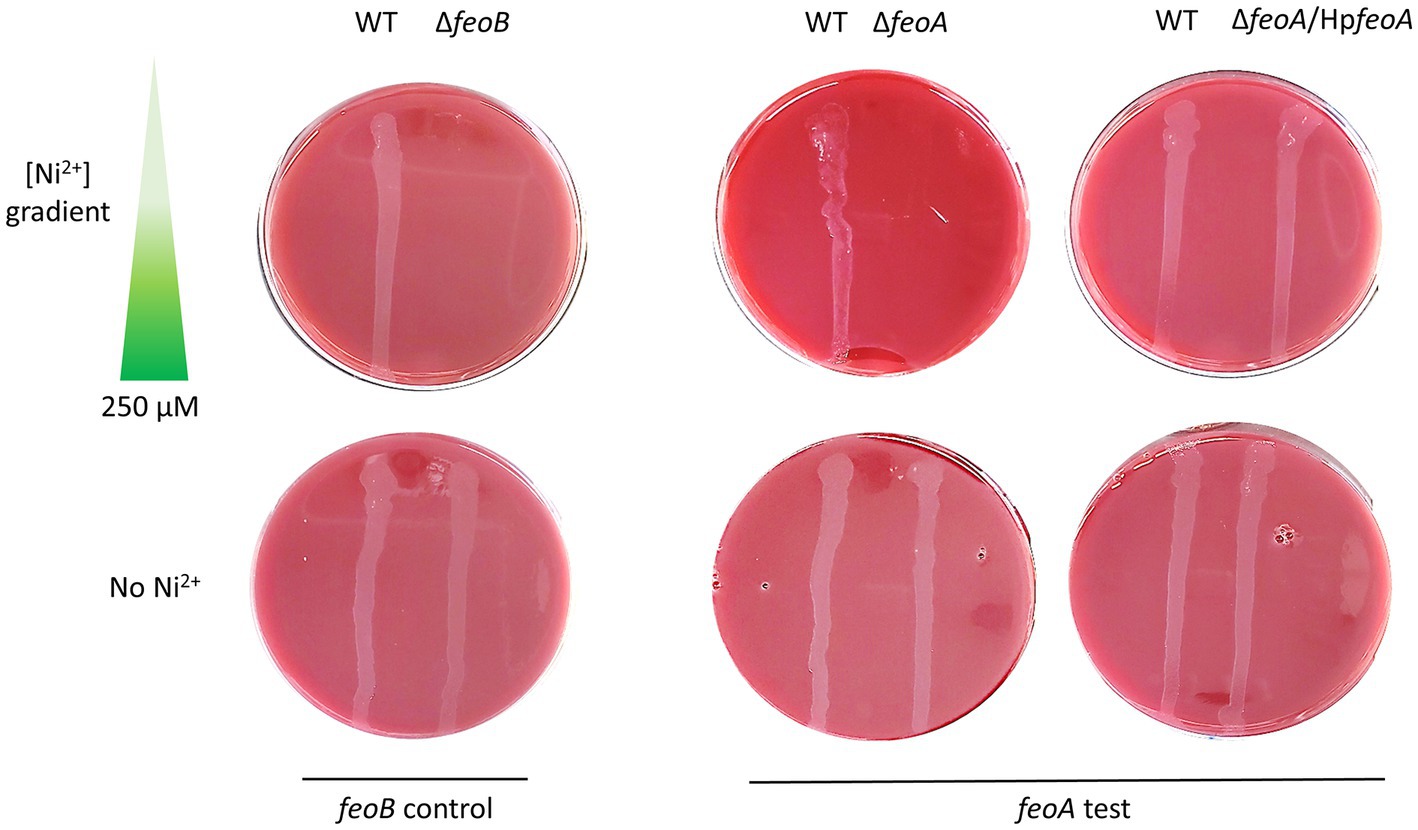
Figure 3. Deletion of feoA in H. pylori leads to increased sensitivity toward nickel. Similar to ΔfeoB (left panel), a ΔfeoA mutant (right panel) failed to grow on Ni2+ gradient HBA plates. Complementation of HpfeoA from a vector (pTMHpfeoA) restored the WT phenotype in the ΔfeoA mutant. All strains were able to grow on HBA plates without Ni2+ as shown in the bottom section of both panels. These plates correspond to a single representative experiment of multiple biological replicates.
Because feoA is an essential part of the H. pylori Feo system but is not linked to feoB as it is in most species, we wanted to determine how its genomic context affected expression. In H. pylori, feoA localizes between the endonuclease III-coding gene (nth) and an uncharacterized protein-coding gene (duf) as depicted in Figure 1B. The three genes in this cluster are oriented in the same direction and have no intergenic space between them, suggesting the three genes are in an operon. To explore this possibility, we first examined the transcription units in the H. pylori G27 genome as annotated in two publicly available repositories, BioCyc (ID number TU2BRX-234) (Karp et al., 2019) and ProOpDB (operon containing the locus tag HPSH112_04095, Supplementary Dataset S1; Taboada et al., 2012). These databases implement different, and independently developed, pipelines for operon inference, and they both predict the nth-feoA-duf cluster to be a single transcription unit.
To experimentally validate the existence of the nth-feoA-duf transcript, we used reverse-transcription PCR (RT-PCR) to amplify the junctions between these genes from mRNA of H. pylori (Figure 4). The RT-PCR tests yielded positive results for both gene junctions from the cDNA sample, confirming the existence of transcripts mapping across the junctions of these clustered genes. Notably, the existence of the nth-feoA-duf transcript is also supported by RNA-Seq data (Sharma et al., 2010). In sum, in silico and experimental evidence indicate that nth, feoA, and duf are co-transcribed in H. pylori G27, and likely comprise an operon.
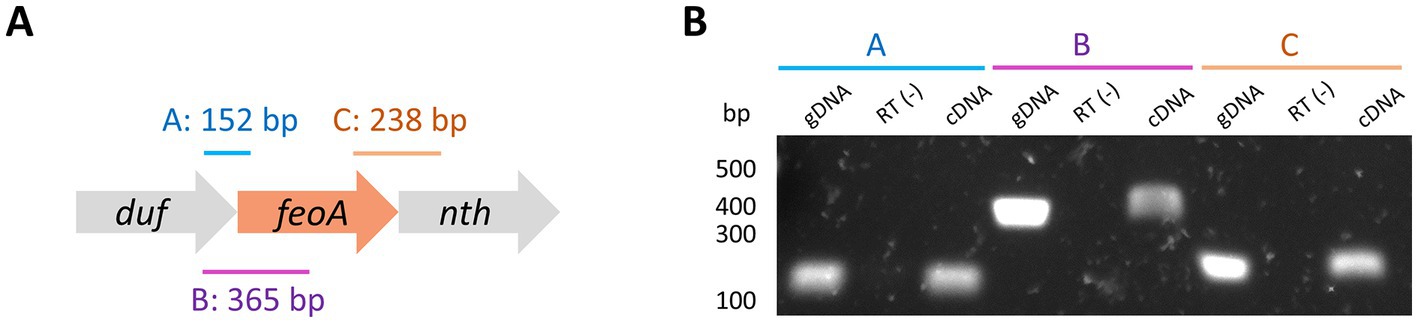
Figure 4. Deletion of the duf-feoA-nth transcription unit. (A) Three regions mapping the junctions between duf, feoA, and nth (labeled as A, B, and C) were amplified by PCR from cDNA produced from RNA of H. pylori G27. The expected size for each product is shown above the approximate location (scheme not to scale). (B) Results of PCR amplifications shown in A. gDNA refers to the positive controls for the amplification conditions using genomic DNA instead of cDNA. RT(−) corresponds to the negative controls for DNA contamination, where the reverse transcription was carried out in the absence of reverse transcriptase. Numbers on the left show the approximated size in bp as estimated from a DNA ladder.
Fur and FNR regulate the transcription of the feo operon in many species (Lau et al., 2016; Sestok et al., 2018). H. pylori lacks FNR, but encodes a Fur protein (Tomb et al., 1997). To examine the promoters controlling the expression of the feo genes in H. pylori, and a potential role of Fur in their regulation, we employed a gfp-based transcriptional reporter to test whether the sequences upstream of feoA and feoB have promoter activity (Figure 5A). Because feoA is the second gene of an operon, its transcription is likely controlled by the promoter of the first gene (i.e., duf). However, this does not rule out the possibility of feoA having its own, additional promoter. Thus, we included both the putative duf promoter and the sequence immediately upstream of feoA in our analyses. Similarly, we included the junction between both genes since this region should be present in the mRNA according to our prediction of the duf-feoA-nth transcription unit.
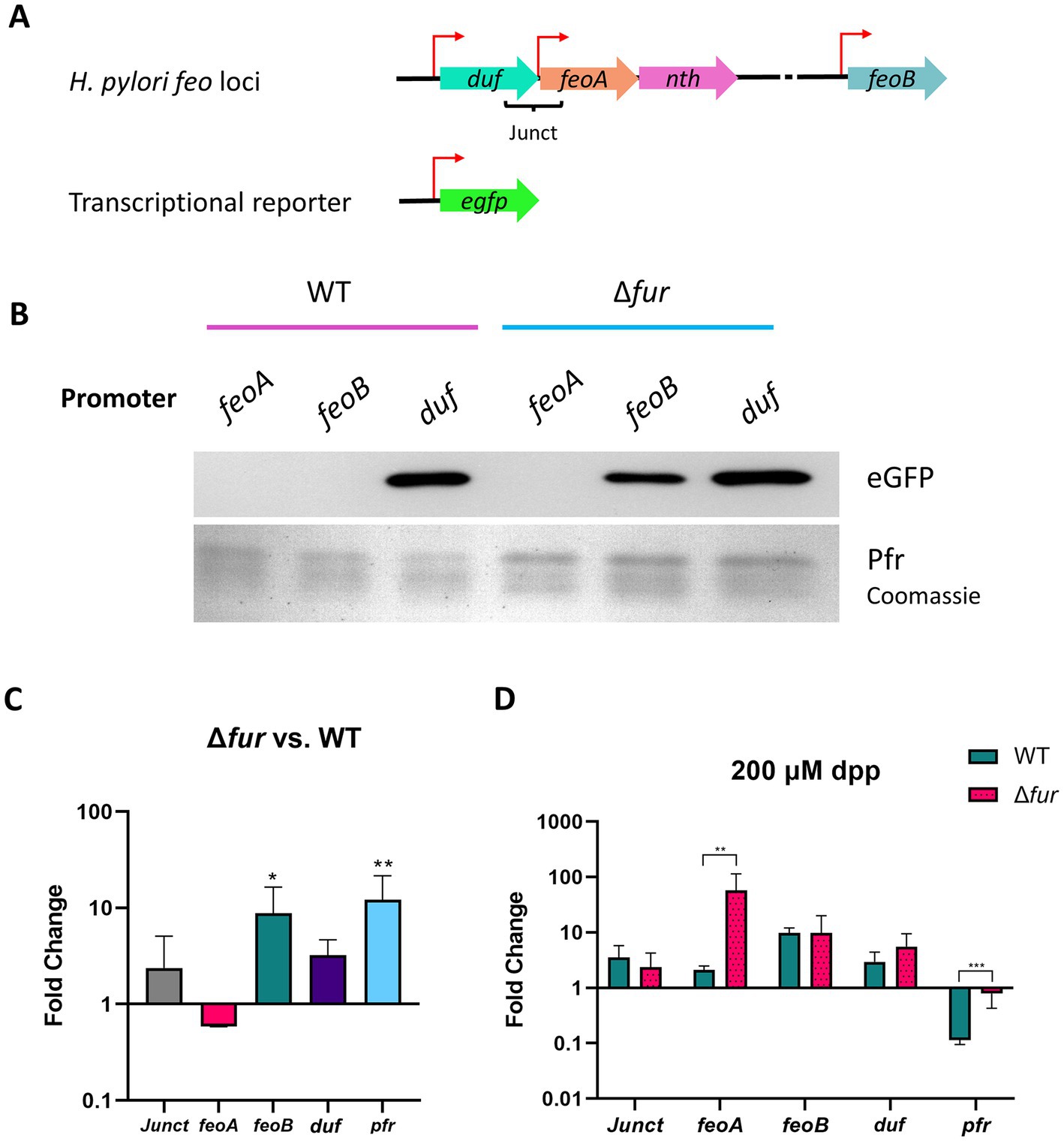
Figure 5. Activity of the putative feo promoters and gene transcription in H. pylori G27. (A) Schematic representation (not drawn to scale) of the transcriptional reporter used in these assays. Each putative promoter (depicted with red arrows) was amplified from H. pylori G27 genomic DNA and cloned upstream of a promoterless gfp gene in the pTM117 backbone. The junction between duf and feoA (Junct) is also shown. (B) Immunoblot analysis detecting GFP in H. pylori G27 strains, WT and Δfur, transformed with the promoter fusions shown in panel A. Ferritin (Pfr), shown in the bottom lane from a Coomassie-stained gel, was used as a control of a Fur-regulated gene (upregulated in the absence of Fur). Both gels were loaded with the same samples. (C) Relative fold changes in the expression levels of the HpfeoA-duf junction (Junct), HpfeoA, HpfeoB, duf, and pfr in the Δfur strain compared to the WT as determined by RT-qPCR. (D) Relative fold changes in gene expression in the Δfur and the WT strains upon iron depletion induced with 200 μM dpp. The p-values for C and D were determined by an unpaired, two-tailed Student’s t-test from the ΔCT values. Differences that were statistically significant are indicated (*p < 0.05, **p < 0.01, ***p < 0.001). The bars correspond to the relative means and standard deviations (error bars) from four biological replicates. Statistical analyses and bar graphs were generated with GraphPad Prism v9.5.0.
Helicobacter pylori G27 and a derivative Δfur strain were transformed with the plasmids bearing the gfp fusions. Detection of Pfr (ferritin) was included as a positive control for a Fur-regulated product in these assays (Bereswill et al., 2000). The activity of the duf promoter (Figure 5B) did not depend on the presence of Fur, as this construct yielded high levels of GFP in both strains as determined via immunoblot analysis. In contrast, GFP was synthesized from the feoB promoter only in the Δfur strain, indicating that this promoter is repressed by Fur in the WT background. Strikingly, GFP was synthesized in neither the WT nor the Δfur background from the construct carrying the feoA promoter. This suggests that the feoA promoter might depend on a regulatory network not directly linked to Fur, and the experimental conditions we used were not adequate to turn on its expression. Consistent with these results, GFP synthesis from the three promoters did not show any changes upon dpp addition in the Δfur strain (Supplementary Figure S2). Importantly, although the feoA promoter was not active under these conditions, transcripts encoding FeoA might still be present since the duf promoter is predicted to control the expression of the whole duf-feoA-nth unit. Thus, the duf promoter may work as the primary source of feoA transcription under certain conditions.
To further analyze how Fur affects the expression of the feo genes in H. pylori, we quantified the expression of the feo genes in both the WT and the Δfur strains via RT-qPCR (Figure 5C). Only the expression of feoB was significantly different between the WT and the Δfur backgrounds, with an average 10-fold increase in the absence of Fur. In contrast, changes in transcription levels for feoA, duf, and the junction between them were not significant. Taken together, the results of RT-qPCR assays agree with those obtained with the transcriptional reporter; feoB transcription is repressed by Fur in the WT strain, while duf and feoA do not appear to be directly regulated by Fur.
Because Fur is an iron-responsive transcription factor, we used RT-qPCR to assess how the expression of the feo genes in H. pylori responds to iron starvation, and whether such a response varies between the WT and the Δfur strains. We induced iron starvation with 2,2′-dipyridyl (dpp), a Fe2+ chelator routinely used to study Fur-mediated regulation in H. pylori (Merrell et al., 2003; Carpenter et al., 2007, 2009; Pich et al., 2012). Our results (Figure 5D) showed that pfr had the expected response for a Fur-regulated gene, insofar as dpp addition resulted in a significant downregulation of transcription in the WT strain, but this response was absent in the Δfur strain, indicating a direct Fur-dependent effect. Consistent with our previous results, changes of relative mRNA levels upon iron depletion did not vary significantly between the WT and the Δfur backgrounds for duf and its junction with feoA. Strikingly, we did not observe any significant change in the expression of feoB, as would have been expected for a Fur-regulated gene. This may suggest that our experimental conditions (i.e., 200 μM dpp over 2 h) were not enough to induce a measurable response in the feoB transcription levels; and, as discussed in detail below, the regulation of feoB by Fur is predicted to be more complex in H. pylori than in other species. Finally, feoA was upregulated in conditions of iron starvation but only in the Δfur strain; hence, this response might not be directly related to Fur, and, as anticipated from our immunoblot analysis, additional layers of regulation could be involved in the expression of this gene.
When transformed into a fur-null strain of E. coli, all three promoter-gfp fusions led to the synthesis of GFP (Supplementary Figure S3). Altogether, these results indicate that the sequences upstream of feoA, duf, and feoB are promoters of different strengths (reflected in the production of different GFP amounts under the same conditions); and transcription of HpfeoA relies on two promoters, that of the duf-feoA-nth operon; and that of HpfeoA, which might serve as a promoter for a feoA-nth suboperon.
To identify those transcription factors regulating the expression of the feo genes in H. pylori, including Fur, we searched for conserved binding motifs in the putative feo promoters using XSTREME (Grant and Bailey, 2021), a pipeline based on Hidden Markov Models (HMM). XSTREME identifies enriched motifs in aligned sequences, and compares them with those deposited in reference databases or provided by the user to find significant matches. We found that the H. pylori feoB promoter has conserved, overlapping binding boxes for the apo (dimeric, Fe2+-free) and holo (tetrameric, Fe2+-bound) forms of Fur (Figure 6 and Supplementary Figure S4), which are both active in H. pylori (Agriesti et al., 2014). Our analysis also identified binding sequences for the Ni2+-responsive transcription factor NikR. In addition, a previous study of the primary transcriptome of H. pylori identified at least two small anti-sense RNAs encoded within the feoB coding sequence (Sharma et al., 2010). Therefore, the expression of feoB in H. pylori is likely modulated by the presence of iron and nickel through a complex interplay of Fur and NikR and, potentially, small anti-sense RNAs.
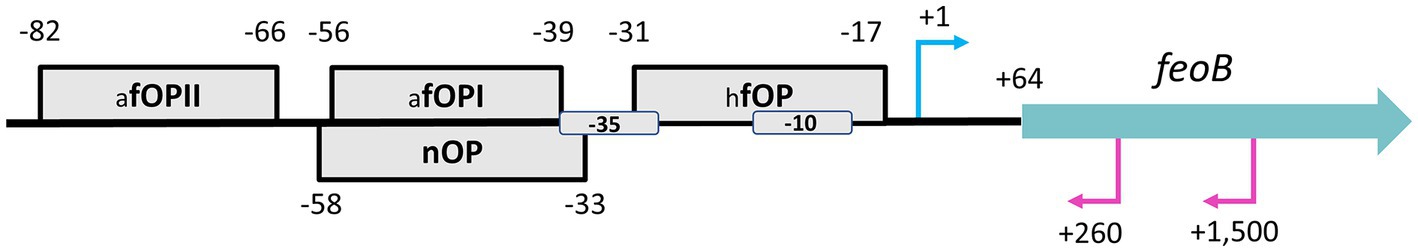
Figure 6. Schematic representation of the proposed HpfeoB promoter architecture. The gray boxes represent the identified operator sites for apo-Fur (afOP I and II), holo-Fur (hfOP), and NikR (nOP) with their relative positions indicated by the numbers around each box. All binding motifs were found in the same direction of HpfeoB, and their location in this scheme, either on the top or the bottom, is only for illustrative purposes. Proposed −10 and − 35 boxes are also shown. The transcription start site of HpfeoB (position +1) and the anti-sense RNAs (+260 and + 1,500) identified by Sharma et al. (2010) via RNA-Seq are depicted with the cyan (above the line) and magenta (below the line) arrows, respectively. The thick blue arrow represents the position and directionality of the HpfeoB coding sequence. This model is based on the H. pylori G27 genome and is not drawn to scale.
When examining the putative promoters of duf and feoA, we did not find any significant match for Fur or NikR binding sequences, nor for any other transcription factors. Thus, there is no evidence of direct Fur-dependent regulation on these genes. This supports our previous findings that there is not a single regulatory network controlling both feoA and feoB in H. pylori. In Figure 7, the first column on the right shows whether the feo operon architecture is conserved (filled in black) or not (white) in each species. Similarly, the second column shows the presence (filled black square) or absence (white) of the duf-feoA cluster. The red star indicates the node in which we hypothesize the initial operon split occurred, and the blue stars indicate the later feoA rearrangement events proposed in our model.
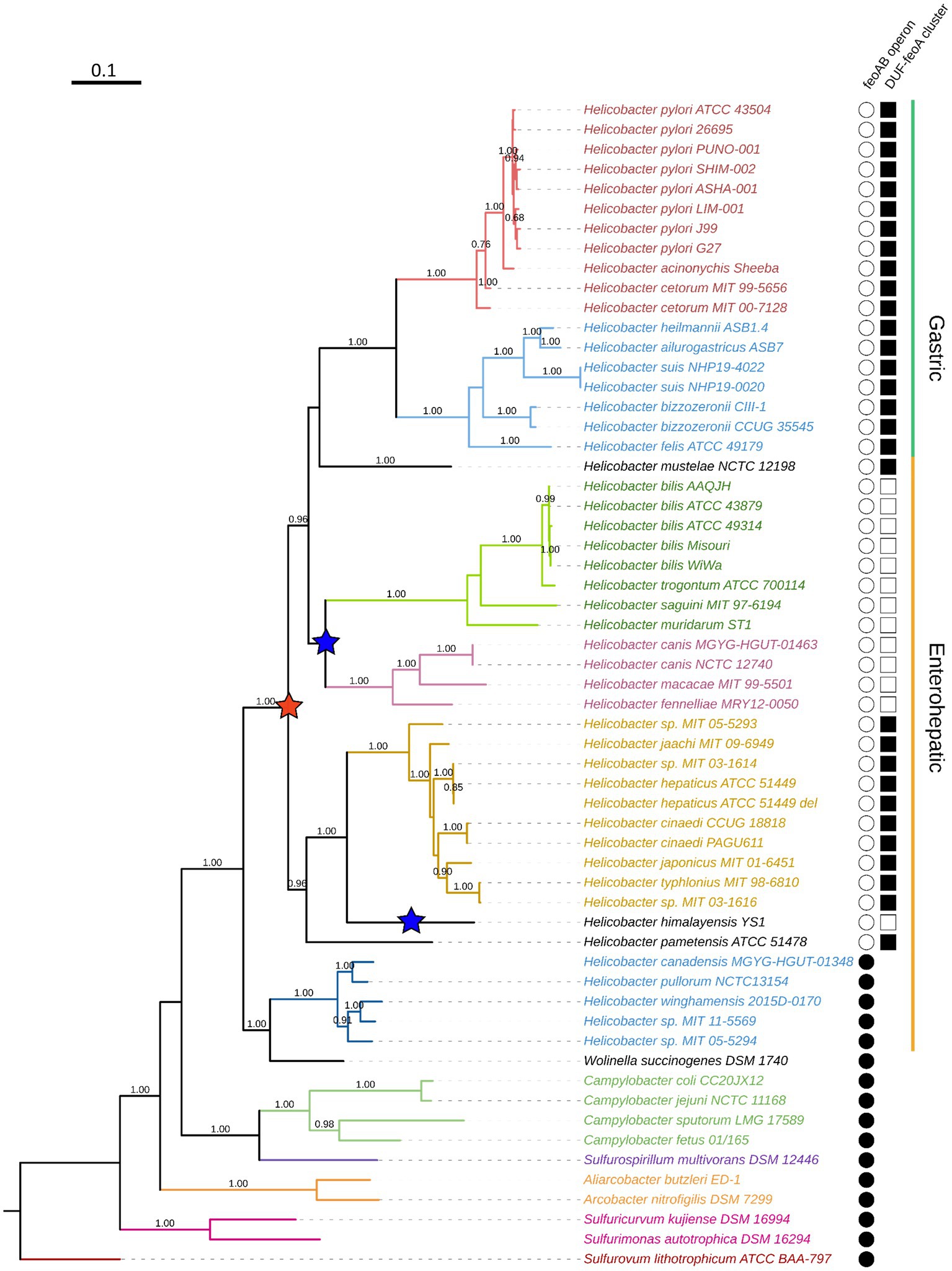
Figure 7. Phylogenetic reconstruction of the Campylobacterota group. This tree was constructed by the maximum likelihood method based on the alignment of concatenated RpoB-RpoC protein sequences. Bootstrap values were calculated with 300 replicates, and those values over 0.5 are shown on the corresponding nodes. Colors indicate clustering according to the currently proposed classification of the helicobacters. From top to bottom: H. pylori group (red), non-H. pylori gastric helicobacters (light blue); enterohepatic helicobacters groups A, B, C, and D (dark green, magenta, gold, and blue, respectively). The ones in black are not classified within these groups. Non-Helicobacter species are shown in the bottom and colored according to their genus affiliation: from Campylobacter spp. to Sulfurovum lithotrophicum (outgroup).
To determine how H. pylori evolved its unique architecture of the feo genes and whether this is a feature exclusive to this species, we carried out a phylogenetic reconstruction of the Campylobacterota [Epsilonproteobacteria] group to trace back the relative position of the feo loci. Our tree (Figure 7) was constructed based on the β and β’ unit of the RNA-polymerase (RpoBC), and recapitulated the current taxonomy of this group (Solnick and Vandamme, 2001; Dewhirst et al., 2005; Waite et al., 2017; Smet et al., 2018; Prada et al., 2022), insofar as the Helicobacter genus forms a monophyletic group together with Wolinella succinogenes. Within this group, the gastric helicobacters form a monophyletic group containing the clades of the H. pylori and the non-H. pylori helicobacters. The enterohepatic helicobacters formed a paraphyletic group made up of several discrete clades that other authors have proposed as genera with pending nomenclature, temporarily referred to as Helicobacter A, B, C, and D (Waite et al., 2017). In this text, we follow this temporary nomenclature as suggested by Waite and colleagues.
According to our phylogenetic tree, the split of the feo operon has a single evolutionary origin that dates to the common ancestor of most—but not all—the current helicobacters, excluding the Helicobacter D group. By contrast, our tree does not show a single node that differentiates those species with the duf-feoA cluster from those with other arrangements. Instead, the most parsimonious scenario given our tree is that in which the split of the ancestral feo operon correlated with the emergence of the duf-feoA cluster, but at least two independent additional rearrangements involving feoA took place later in the evolution of certain helicobacters: Namely, in H. himalayensis and the common ancestor of Helicobacter A and B. These latter groups are likely to comprise separate genera within the current Helicobacter classification, and are represented in our tree (Figure 7) by H. bilis, H. trogontum, H. saguini, and H. muridarum (Helicobacter A); and H. canis, H. macacae, and H. fennelliae (Helicobacter B).
The synteny of the feoA locus across the helicobacters (Figure 8) shows that both the feo operon and the association between the nth, duf, and mltG genes are conserved in the Helicobacter D group and W. succinogenes; while the duf-feoA-nth arrangement is prevalent in most of the helicobacters lacking the operon structure (i.e., all but the D group). This finding is consistent with the hypothesis of an ancestral feo operon that split upon the divergence of the ancestor shared by the gastric helicobacters and the groups A, B, and C. H. himalayensis as well as Helicobacter A and B do not share synteny of the feoA locus, suggesting that these groups underwent independent rearrangements of feoA after the primary split event of the ancestral operon. Since the species of group B conserves the order feoA, nth, cheX, fliN observed in other groups, we propose as the most parsimonious scenario is that group A evolved a second relocation of feoA, albeit alternative models are plausible.
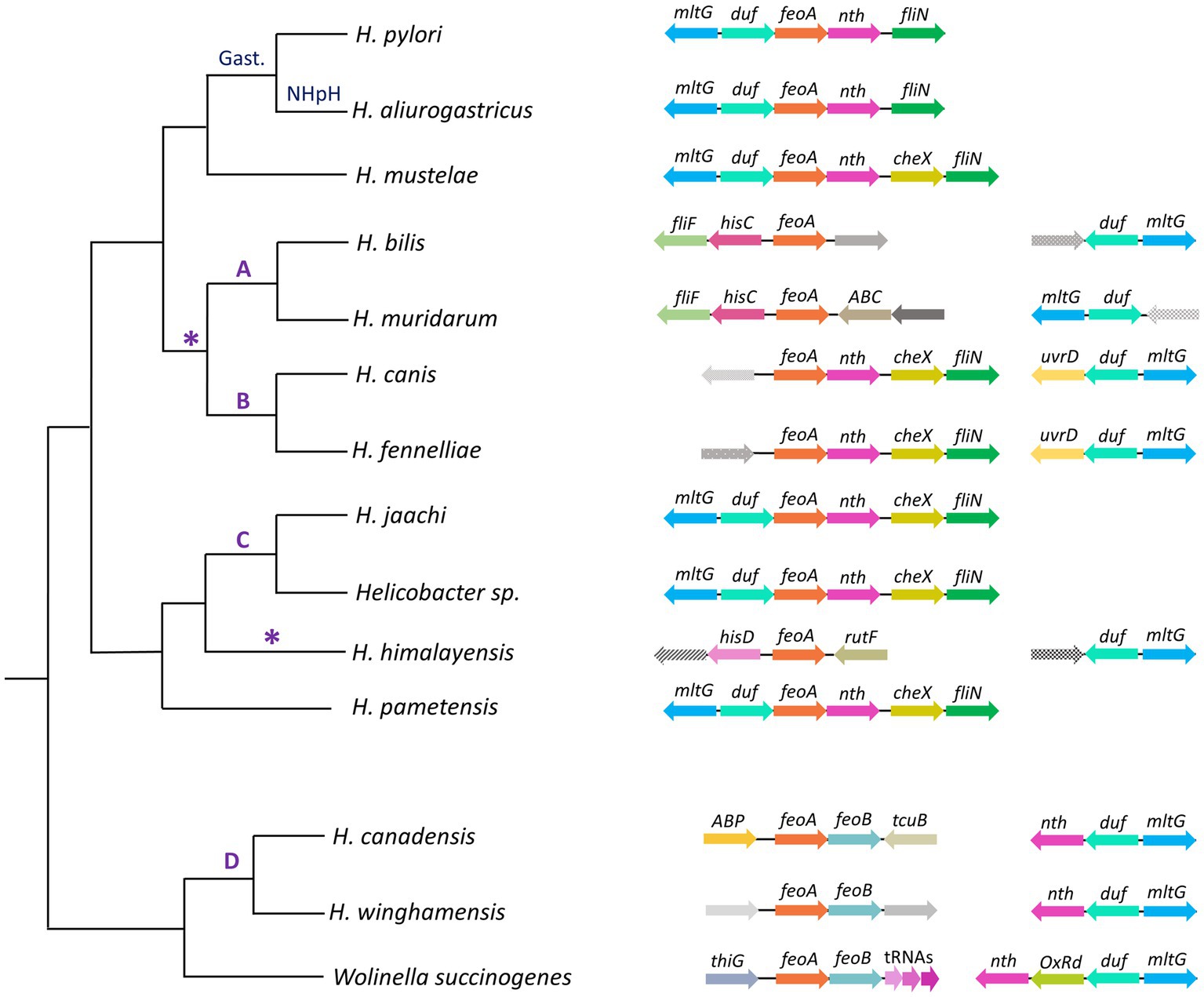
Figure 8. Genomic context of feoA loci among Helicobacter species. This cladogram shows the phylogenetic relationship as found in the organismal tree above for representative species of the gastric (Gast.) and non-H. pylori helicobacters (NHpH) as well as for the groups A, B, C, and D of the enterohepatic helicobacters (as indicated above the corresponding node). The AB node and the H. himalayensis branch, in which additional feoA rearrangements might have taken place, are indicated with an asterisk (*). The genomic context of the feoA and duf loci are shown after each species following the gene annotation deposited in GeneBank for each genome. Distances and gene sizes are not drawn to scale.
Considering the probable ancestral operon structure of feo as well as our promoter analyses showing that Fur and NikR likely regulate the expression of HpfeoB but not HpfeoA, we sought to determine whether the nucleotide sequences underlying this regulation existed in the ancestral operon and were conserved only in feoB upon operon excision. To investigate the feasibility of this hypothesis, we applied the same HMM-based approach described above to look for conserved transcription factor binding motifs in promoters of the feo operon in Helicobacter D and other Campylobacterota species. Indeed, this analysis showed that the putative promoter of the feo operon has a distribution of potential binding sequences for Fur and NikR similar to that of the feoB promoter (Supplementary Figure S5).
The current mechanistic model of the Feo system posits that FeoA, FeoB, and FeoC interact to assemble a large complex embedded in the inner membrane (Stevenson et al., 2016). We used a vector encoding a C-terminal FLAG-tagged version of H. pylori FeoA (HpFeoAC-FLAG) to test whether this protein forms a membrane complex in association with HpFeoB in EPV6. Expression in trans of HpfeoAC-FLAG and HpfeoB supports EPV6 growth in non-supplemented LB agar, the same as co-expression of the HpfeoB together with HpfeoA (without the epitope tag), indicating that the C-terminal FLAG tag in HpFeoA does not affect its function.
EPV6 cells transformed with plasmids harboring HpfeoAC-FLAG and HpfeoB were crosslinked in vivo with formaldehyde and then visualized via immunoblot analysis to identify complex formation. In addition to the HpFeoAC-FLAG monomer, there were two additional bands reactive to the anti-FLAG antibody that appeared upon crosslinking (Figure 9A). The approximate sizes of the bands are consistent with a HpFeoAC-FLAG dimer (20 kDa) and the large transmembrane Feo complex (250 kDa), as observed in the V. cholerae model (Stevenson et al., 2016).
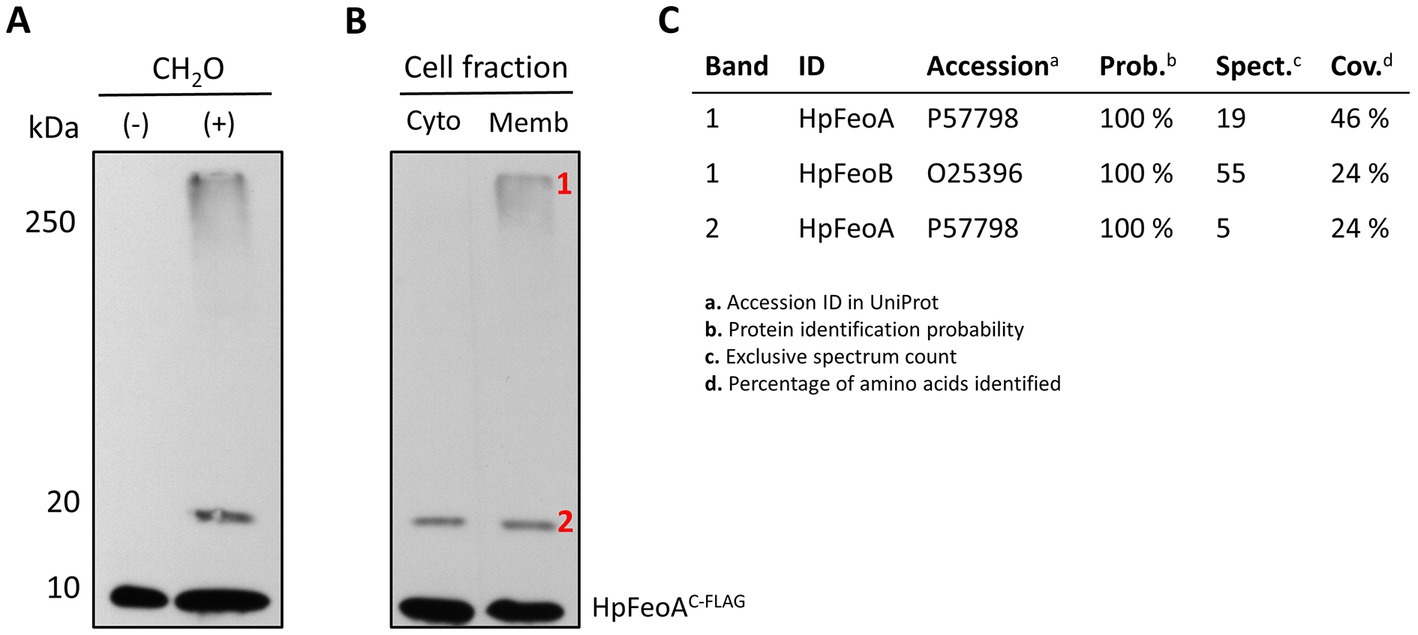
Figure 9. Formation of the transmembrane Feo complex by HpFeoA and HpFeoB in V. cholerae EPV6. (A) Immunoblot analysis of V. cholerae EPV6 co-transformed with plasmids carrying HpFeoAC-FLAG and HpFeoB. CH2O indicates whole cell pellets before (−) and after (+) formaldehyde crosslinking in vivo. (B) Samples obtained upon cell fractionation: cytoplasmic and membrane fractions (labeled as Cyto and Memb, respectively.) The numbers on the left indicate the estimated protein size in kDa. (C) Results for peptides matching HpFeoA and HpFeoB retrieved from mass spectroscopy analysis of those bands in the membrane fraction (labeled as 1 and 2) after immunoprecipitation with a monoclonal anti-FLAG antibody.
To determine whether the >250 kDa band corresponds to a transmembrane complex, we carried out cell fractionation via ultracentrifugation on the crosslinked sample. Immunoblot analysis of the cell fractions revealed that the large complex was indeed present only in the membrane fraction (Figure 9B). Similarly, to confirm the presence HpFeoAC-FLAG in both the 20 kDa and the >250 kDa bands, we enriched the crosslinked sample through immunoprecipitation using a monoclonal anti-FLAG antibody, excised the corresponding bands from the acrylamide gel, and analyzed them via mass spectrometry. Peptides mapping to HpFeoA were found in both samples, and peptides corresponding to HpFeoB were also identified in the >250 kDa band (Figure 9C). Together, these findings suggest that HpFeoA forms a dimer, and that HpFeoA and HpFeoB interact to form a complex in the cytoplasmic membrane similar to that observed in V. cholerae (Stevenson et al., 2016; Gómez-Garzón and Payne, 2020).
Attempts to validate these results with tagged HpFeoA or HpFeoB proteins in H. pylori were unsuccessful. The tagged proteins either were not detected or interfered with function.
Finally, we modeled the potential HpFeoA-HpFeoB interaction in silico through AlphaFold-Multimer (Evans et al., 2022), a recently released extension of the AlphaFold deep learning pipeline that allows to model protein–protein interactions from amino acid sequences. As shown in Figure 10A, our model predicts interacting residues in HpFeoB mapping to transmembrane, as well as the N-terminal, domains of the protein, which contrasts with the long-standing model in which FeoA interacts only with the N-terminal domain of FeoB. Our model also predicts that HpFeoB has a hinge close to the interface between the N-terminal and the transmembrane domains, about Ala208, which undergoes an important change upon HpFeoA binding. Namely, when compared the HpFeoA-bound form of HpFeoB to that of the protein by itself, our model shows that HpFeoA stabilizes a specific conformation of HpFeoB, by locking this hinge in place (Figure 10B and Supplementary Figure S6). This suggests that HpFeoA might trigger structural changes in HpFeoB, which could indicate a regulatory role. We identified those amino acids located in the interacting surfaces of both proteins (Figure 10C). These residues are likely to have a functional significance for the HpFeoA-HpFeoB interaction and thus they may be useful targets in further mutational analyses.
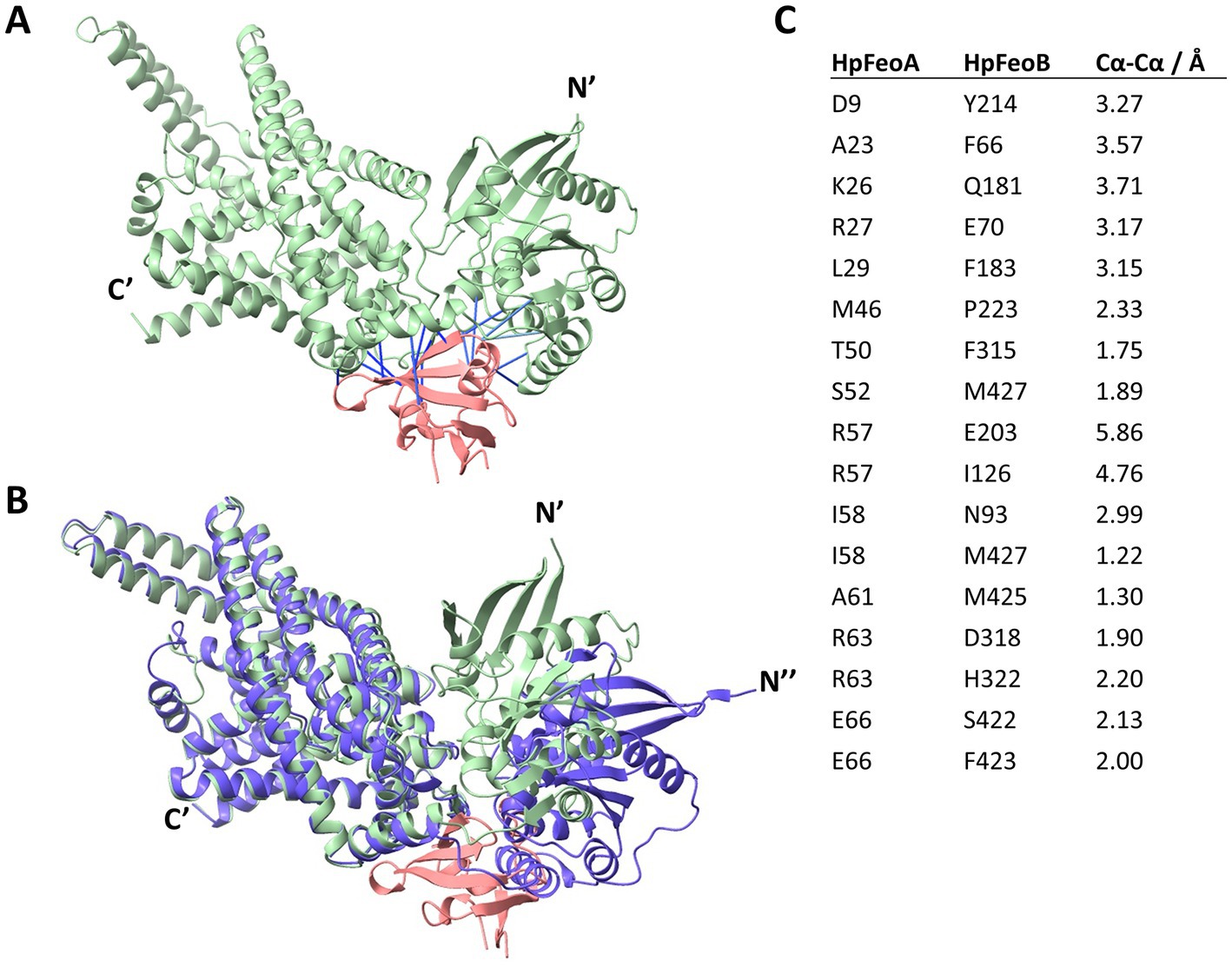
Figure 10. Structural model of the HpFeoA-HpFeoB interaction constructed with AlphaFold-Multimer. (A) 3D representation of the best model obtained for the interaction between HpFeoA (WP_000174130, in pink) and HpFeoB (WP_041201363, in green) by AlphaFold-Multimer. N′ and C′ correspond to the N- and C- termini of HpFeoB. The blue lines represent the predicted contacts between the two proteins, defined as residues at 3.00 Å or closer to each other. These contacts have a predicted aligned error value of 0. (B) Structural alignment of the HpFeoA-HpFeoB model shown in panel A and the 3D model for HpFeoB alone deposited in AlphaFold DB (ID: B5Z754_HELPG, shown in purple). N″ represent the N-terminus of the latter model as it does not align with that of the interaction model. (C) Predicted interacting peptides between HpFeoA and HpFeoB shown in (A), indicating the predicted distance between alpha carbon (Cα-Cα).
Helicobacter pylori is a widespread human commensal that colonizes the gastric mucosa. Chronic infection with H. pylori triggers an inflammatory response that, in some cases, may lead to the development of metaplasia and, ultimately, cancer. This bacterium is indeed considered the major risk factor for the development of peptic ulcers and gastric cancer worldwide (Herrera and Parsonnet, 2009; Plummer et al., 2015; de Martel et al., 2020). Therefore, H. pylori is an important human pathogen, and efforts to advance our understanding of this pathogen must be a priority.
By studying the evolutionary history of the bacterial ferrous iron transporter, Feo, we found the unique H. pylori gene architecture for this system. There are two feo genes in the H. pylori genome, feoA and feoB but, unlike in most species, they are not clustered in an operon, but separated by 116 kbp. This architecture is conserved among most helicobacters. We examined the functional implications of this arrangement by evaluating the requirement of HpfeoA and how HpfeoA and HpfeoB are regulated. We found that, although HpfeoA is encoded in an operon together with two other unrelated genes (nth and duf), it is needed for the function of the Feo transporter, demonstrating that H. pylori Feo is a two-component system encoded by distant loci.
The Feo system is widespread among bacteria, and although the feo genes are clustered in species other than Helicobacter, there is diversity in their number and organization (Lau et al., 2016; Sestok et al., 2018; Gómez-Garzón et al., 2022). For example, some species have two feoA genes, though whether both genes are needed or have different roles is yet to be determined. Also, it is common within the Firmicutes and Bacteroidetes that Feo is made up of a single protein containing a fusion of FeoA- and FeoB-like domains (Gómez-Garzón et al., 2022). A study in Rhodobacter capsulatus found that this bacterium has two feo gene clusters, but only one conserves iron transport activity, while the other one functions as a manganese transporter (Zappa and Bauer, 2013), showing that a duplication event may have led to the evolution of a homolog transporter. These studies suggest that gene rearrangements have shaped the evolution of Feo, and hence feo gene architecture may correlate with different mechanistic features. It was plausible, for example, that H. pylori dispensed with feoA, so that this gene has evolved separately and probably acquired a new function in the context of the operon it makes with nth and duf. However, our data ruled out this scenario, showing that both feoA and feoB are necessary and sufficient for iron transport, and deletion of either gene results in a similar phenotype (Figures 3, 4).
We propose that the split of the feo operon occurred early in the evolution of the Campylobacterota group, before the divergence of most Helicobacteriaceae genera, excluding only the ancestor of the species of the Helicobacter D group. We also hypothesize that this split resulted in the association of feoA and duf, but additional rearrangements of feoA took place later in the ancestors of the groups A and B (Figure 2). Our analyses indicate that the ancestral feo operon was likely regulated by Fur and NikR but only feoB conserved the ancestral promoter upon operon splitting, while feoA became dependent on the new operons it formed. Two questions emerge from this evolutionary scenario. First, how did HpfeoA acquire its own promoter? The current HpfeoA promoter may have retained key features of the ancestral promoter, but did not retain NikR/Fur regulation. Equally possible is that HpfeoA evolved a promoter de novo; it has been shown that random mutations can lead to the evolution of promoters (Yona et al., 2018). Second, has the split of the feo operon been positively selected in Helicobacter spp., i.e., how does the split of the feo operon affect the fitness of these species to thrive in the host environment? Since H. pylori is characterized by its highly plastic genome (Blaser and Berg, 2001; Suerbaum and Josenhans, 2007), and it is considered to comprise a panmictic population (Salaün et al., 1998), those factors preserving the split feo gene architecture in this group merit further study.
Little was known about HpfeoA and its role in iron uptake because the annotation of the first genome of H. pylori (Tomb et al., 1997) did not include this gene. This was likely due to its small size (230 bp) and location between two other unrelated genes, which made it difficult to be recognized by the annotation pipelines used at the time. In consequence, it has been largely assumed that H. pylori Feo relied solely on feoB. More recent genome annotations have identified feoA homologs throughout all the Campylobacterota group, and Müller et al., 2013 demonstrated through proteomics analysis that H. pylori 26,695 synthesizes the FeoA protein. Velayudhan and colleagues (Velayudhan et al., 2000) showed, using a mouse model, that feoB is a major contributor for virulence in H. pylori 4187E. Future studies will determine whether Feo is a major determinant for virulence in other clinically relevant H. pylori strains, and whether our findings regarding the requirement of feoA can be expanded to these models.
Helicobacter pylori has closely coevolved with humans for more than 100,000 years (Moodley et al., 2012). The genome of this species exhibits an unusually high plasticity and has been largely shaped by horizontal gene transfer and recombination events (Garcia-Vallvé et al., 2002; Gressmann et al., 2005; Prada et al., 2022). It is not unexpected then that H. pylori lacks several operon arrangements and master regulators widely conserved among bacteria. Among the missing transcription factors in H. pylori is FNR, which, in Gammaproteobacteria, controls the expression of the feo operon in response to changes in oxygen tension. We found that the transcription of feoB in H. pylori is likely to be tightly regulated by Fur and NikR, hence H. pylori controls the expression of feoB depending on the availability of iron and nickel. Fur and NikR have been shown to form an intricate regulatory network in H. pylori (Delany et al., 2005; Danielli et al., 2010; Roncarati et al., 2016; Vannini et al., 2022). These transcription factors co-regulate genes essential for cell homeostasis, such as the exbB-exbD-tonB operon, which encodes a complex that provides energy to several ATP-driven transporters. In addition, Fur and NikR regulate the transcription of one another; namely, holo-NikR represses fur expression and holo-Fur represses NikR expression. Fur also impacts NikR expression as well as NikR-regulated genes and vice versa. The NikR and Fur regulons include important virulence factors and central metabolism genes (Vannini et al., 2022); for instance, NikR regulates the expression of the urease, essential for the colonization of the gastric mucosa; and Fur modulates the expression of cagA (involved in inflammatory response), pfr (ferritin), arsRS (master regulator), and amiE (amidase, critical in nitrogen metabolism), among others (Delany et al., 2005; Danielli et al., 2010; Roncarati et al., 2016).
Fur-mediated regulation is particularly complex in H. pylori. In addition to the interplay with NikR described above, both the holo (dimeric) and apo (monomeric) forms of Fur are active and bind different sequences in the DNA (Agriesti et al., 2014). Therefore, regulation by Fur is often the result of a competition between the two forms of the protein, on top of additional kinetic factors involving oligomerization as well as iron and DNA binding. This means that H. pylori Fur works like a commutator switch rather than like a simple ON/OFF switch. This feature, not reported in other species, likely evolved in H. pylori as a means to overcome the absence of other transcription factors (Agriesti et al., 2014). Based on sequence analysis, we anticipate that NikR and both forms of Fur are involved in the regulation of HpfeoB (Figure 7). In addition, a comprehensive RNA-Seq study on the primary transcriptome of this pathogen (Sharma et al., 2010) identified two small anti-sense RNAs encoded within the feoB coding sequences, which may form an additional layer of regulation.
We found that feoA transcripts are produced from two promoters (Figure 6), the promoter for duf—which also involves the nth gene and seems to be constitutively expressed—and an internal promoter. Interestingly, we found no evidence of Fur- or NikR-mediated regulation on feoA. Considering the stringent regulation to which feoB is subjected, we hypothesize that the regulation of the Feo system in H. pylori primarily relies on controlling feoB expression, while feoA transcripts may be readily available in the cell. Thus, feoB mRNA abundance might serve as a bottleneck for the assembly of the Feo transporter.
We used dpp-induced iron starvation to study regulation by Fur in H. pylori as reported in previous studies (Carpenter et al., 2007). These conditions led to significant repression of the pfr gene (Figure 6), indicating they serve to assess Fur-mediated response. However, dpp can bind metal cations other than iron, and Fur has been found responsive to these ions as well (Bereswill et al., 2000); thus, our results might reflect the effects of changes in iron availability along with other metals. The effects of copper on H. pylori gene regulation have been determined (Waidner et al., 2002), and neither the feoA nor the feoB operons were shown to be regulated by copper. Future studies should test additional metals, especially nickel, to determine whether any metals other than iron govern the expression of feoB in H. pylori.
While this study provides initial insight into Feo-mediated iron transport in H. pylori, those environmental conditions (i.e., changes in Fe2+ and Ni2+ concentration) necessary to induce a consistent response in HpfeoB and HpfeoA need further characterization. We did not find a specific set of experimental conditions in which the feoA promoter was up-regulated using our transcriptional reporter. Likewise, we did not observe a switch in feoB transcription in response to Fur in our RT-qPCR assay, though all our other analyses and the scientific literature show that Fur regulates this gene. We anticipate a high level of complexity in the regulation of these genes. Fur and NikR regulation on feoB and the ancestral feo operon are based on sequence analyses; hence, experimental validation, including evidence of physical protein-DNA interaction and effects of sequence variability (i.e., mismatches from the consensus sequence) on binding affinity, is needed to fully elucidate the role of these master regulators in modulating the expression of feoB.
In previous studies conducted in V. cholerae, we have found that FeoA, B, and C assemble a multimeric complex in the inner membrane (Stevenson et al., 2016), likely composed by trimers of FeoABC units, and that FeoB may form intermediate oligomers with FeoA or by itself before assembling the large complex. Consistent with these findings, some authors have observed that purified NFeoB or full-length FeoB forms trimers in vitro (Guilfoyle et al., 2009; Hagelueken et al., 2016; Seyedmohammad et al., 2016), although little is known about the relevance of these complexes in vivo. Here, we found that HpFeoA and HpFeoB form a membrane complex when expressed in V. cholerae, and that HpFeoA also formed dimers (Figure 9). However, a limitation of this study was that we were unable to assess Feo complex formation directly in H. pylori.
3D structural modeling of the HpFeoA-HpFeoB interaction using AlphaFold-Multimer (Evans et al., 2022) predicts an area of structural flexibility in HpFeoB that undergoes a structural shift when interacting with HpFeoA (Figure 10 and Supplementary Figure S6). This suggests that HpFeoA might interact with HpFeoB to trigger a regulatory response for either complex formation or iron uptake. Although it has been suggested that FeoA could regulate the NTPase activity of FeoB, FeoA does not affect the enzymatic activity of full-length FeoB (Lau et al., 2013; Gómez-Garzón and Payne, 2020). Therefore, we propose that structural changes in FeoB induced by FeoA may be involved in regulating pore opening or the assembly of the large complex. Our model yielded high confidence structures (Supplementary Figure S7); and thus, the putative interacting amino acids we identified should guide further studies seeking to discern the functional significance of this protein–protein interaction. Membrane-associated proteins, such as FeoB, present a major hurdle for both crystallographic analysis and in silico modeling; thus, mutational analysis is an important approach to fully characterize the interaction between HpFeoA and HpFeoB, especially in the absence of full-length crystal structures of FeoB. Also, there are some factors missing in the AlphaFold-Multimer model that may have an effect in vivo. For instance, the presence of an NTP molecule bound to HpFeoB may induce additional changes in this protein; oligomerization of HpFeoB as well as membrane localization may significantly change the way in which HpFeoB interacts with HpFeoA.
In summary, our studies identified the remarkable differences between the H. pylori Feo system and those of the Gammaproteobacteria group, which have largely been used as model organisms. Establishing the H. pylori Feo as a new model for Feo will represent a leap toward a more comprehensive understanding of this important bacterial transporter, especially in the context of bacterial pathogens.
The original contributions presented in the study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
CG-G and SP contributed to conception and design of the study. CG-G carried out the experimental procedures, statistical analyses, and wrote the first draft of the manuscript. SP contributed to the analysis of the data and experimental design. All authors contributed to manuscript revision, read, and approved the submitted version.
This work was funded by the National Institutes of Health (NIH) (grant R01 AI091957) to SP.
We thank Nina Salama (Fred Hutch Cancer Center, Seattle, WA) for donating the DNA used in the construction of the H. pylori mutants, and Scott Merrell (Uniformed Services University, Bethesda, MD) for donating the H. pylori G27 and Δfur strains and the pTM117 vector. Their guidance on H. pylori methods was invaluable for this work. We also thank Edward Marcotte (The University of Texas at Austin) for his insight into the use of AlphaFold-Multimer. We are grateful to Carolyn Fisher for her meticulous review, which gives this manuscript its final copacetic form. We thank Austin Pham for his work on the construction of the pHpfeoA vector.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1219359/full#supplementary-material
Agriesti, F., Roncarati, D., Musiani, F., Del Campo, C., Iurlaro, M., Sparla, F., et al. (2014). FeON-FeOFF: the Helicobacter pylori Fur regulator commutates iron-responsive transcription by discriminative readout of opposed DNA grooves. Nucleic Acids Res. 42, 3138–3151. doi: 10.1093/nar/gkt1258
Arnold, J., Mindiola, D., Agapie, T., Love, J., Dincă, M., Dauth, A., et al. (2012). Dissecting the role of DNA sequence in Helicobacter pylori NikR/DNA recognition. Dalton Trans. 41, 7946–7951. doi: 10.1039/C2DT30504F
Bailey, T. L., Johnson, J., Grant, C. E., and Noble, W. S. (2015). The MEME suite. Nucleic Acids Res. 43, W39–W49. doi: 10.1093/nar/gkv416
Barber, M. F., and Elde, N. C. (2015). Buried treasure: evolutionary perspectives on microbial Iron piracy. Trends Genet. 31, 627–636. doi: 10.1016/j.tig.2015.09.001
Bereswill, S., Greiner, S., Van Vliet, A. H. M., Waidner, B., Fassbinder, F., Schiltz, E., et al. (2000). Regulation of ferritin-mediated cytoplasmic iron storage by the ferric uptake regulator homolog (Fur) of Helicobacter pylori. J. Bacteriol. 182, 5948–5953. doi: 10.1128/JB.182.21.5948-5953.2000
Blaser, M. J., and Berg, D. E. (2001). Helicobacter pylori genetic diversity and risk of human disease. J. Clin. Investig. 107, 767–773. doi: 10.1172/JCI12672
Boyer, E., Bergevin, I., Malo, D., Gros, P., and Cellier, M. F. M. (2002). Acquisition of Mn(II) in addition to Fe(II) is required for full virulence of Salmonella enterica serovar typhimurium. Infect. Immun. 70, 6032–6042. doi: 10.1128/IAI.70.11.6032-6042.2002
Carpenter, B. M., Gancz, H., Gonzalez-Nieves, R. P., West, A. L., Whitmire, J. M., Michel, S. L. J., et al. (2009). A single nucleotide change affects Fur-dependent regulation of sodB in H. pylori. PLoS One 4:e5369. doi: 10.1371/journal.pone.0005369
Carpenter, B. M., Gilbreath, J. J., Pich, O. Q., McKelvey, A. M., Maynard, E. L., Li, Z. Z., et al. (2013). Identification and characterization of novel Helicobacter pylori apo-Fur-regulated target genes. J. Bacteriol. 195, 5526–5539. doi: 10.1128/JB.01026-13
Carpenter, B. M., McDaniel, T. K., Whitmire, J. M., Gancz, H., Guidotti, S., Censini, S., et al. (2007). Expanding the Helicobacter pylori genetic toolbox: modification of an endogenous plasmid for use as a transcriptional reporter and complementation vector. Appl. Environ. Microbiol. 73, 7506–7514. doi: 10.1128/AEM.01084-07
Danielli, A., Amore, G., and Scarlato, V. (2010). Built shallow to maintain homeostasis and persistent infection: insight into the transcriptional regulatory network of the gastric human pathogen Helicobacter pylori. PLoS Pathog. 6:e1000938. doi: 10.1371/journal.ppat.1000938
de Martel, C., Georges, D., Bray, F., Ferlay, J., and Clifford, G. M. (2020). Global burden of cancer attributable to infections in 2018: a worldwide incidence analysis. Lancet Glob. Health 8, e180–e190. doi: 10.1016/s2214-109x(19)30488-7
Delany, I., Ieva, R., Soragni, A., Hilleringmann, M., Rappuoli, R., and Scarlato, V. (2005). In vitro analysis of protein-operator interactions of the NikR and Fur metal-responsive regulators of coregulated genes in Helicobacter pylori. J. Bacteriol. 187, 7703–7715. doi: 10.1128/JB.187.22.7703-7715.2005
Dewhirst, F. E., Shen, Z., Scimeca, M. S., Stokes, L. N., Boumenna, T., Chen, T., et al. (2005). Discordant 16S and 23S rRNA gene phylogenies for the genus Helicobacter: implications for phylogenetic inference and systematics. J. Bacteriol. 187, 6106–6118. doi: 10.1128/JB.187.17.6106-6118.2005
Evans, R., O’Neill, M., Pritzel, A., Antropova, N., Senior, A., Green, T., et al. (2022). Protein complex prediction with AlphaFold-Multimer. bioRxiv :463034. doi: 10.1101/2021.10.04.463034
Franza, T., and Expert, D. (2013). Role of iron homeostasis in the virulence of phytopathogenic bacteria: an “à la carte” menu. Mol. Plant Pathol. 14, 429–438. doi: 10.1111/mpp.12007
Garcia-Vallvé, S., Janssen, P. J., and Ouzounis, C. A. (2002). Genetic variation between Helicobacter pylori strains: gene acquisition or loss? Trends Microbiol. 10, 445–447. doi: 10.1016/s0966-842x(02)02446-0
Gómez-Garzón, C., Barrick, J. E., and Payne, S. M. (2022). Disentangling the evolutionary history of Feo, the major ferrous Iron transport system in Bacteria. mBio 13:e03512–21. doi: 10.1128/mbio.03512-21
Gómez-Garzón, C., and Payne, S. M. (2020). Vibrio cholerae FeoB hydrolyzes ATP and GTP in vitro in the absence of stimulatory factors. Metallomics 12, 2065–2074. doi: 10.1039/d0mt00195c
Grant, C. E., and Bailey, T. L. (2021). XSTREME: comprehensive motif analysis of biological sequence datasets. bioRxiv :458722. doi: 10.1101/2021.09.02.458722
Gressmann, H., Linz, B., Ghai, R., Pleissner, K. P., Schlapbach, R., Yamaoka, Y., et al. (2005). Gain and loss of multiple genes during the evolution of Helicobacter pylori. PLoS Genet. 1:e43. doi: 10.1371/journal.pgen.0010043
Guilfoyle, A., Maher, M. J., Rapp, M., Clarke, R., Harrop, S., and Jormakka, M. (2009). Structural basis of GDP release and gating in G protein coupled Fe2+ transport. EMBO J. 28, 2677–2685. doi: 10.1038/emboj.2009.208
Hagelueken, G., Hoffmann, J., Schubert, E., Duthie, F. G., Florin, N., Konrad, L., et al. (2016). Studies on the X-ray and Solution structure of FeoB from Escherichia coli BL21. Biophys. J. 110, 2642–2650. doi: 10.1016/j.bpj.2016.05.018
Hantke, K. (1987). Ferrous iron transport mutants in Escherichia coli K12. FEMS Microbiol. Lett. 44, 53–57. doi: 10.1111/j.1574-6968.1987.tb02241.x
Herrera, V., and Parsonnet, J. (2009). Helicobacter pylori and gastric adenocarcinoma. Clin. Microbiol. Infect. 15, 971–976. doi: 10.1111/j.1469-0691.2009.03031.x
Jackson, L. K., Potter, B., Schneider, S., Fitzgibbon, M., Blair, K., Farah, H., et al. (2020). Helicobacter pylori diversification during chronic infection within a single host generates sub-populations with distinct phenotypes. PLoS Pathog. 16:e1008686. doi: 10.1371/journal.ppat.1008686
Karp, P. D., Billington, R., Caspi, R., Fulcher, C. A., Latendresse, M., Kothari, A., et al. (2019). The BioCyc collection of microbial genomes and metabolic pathways. Brief. Bioinformatics 20, 1085–1093. doi: 10.1093/bib/bbx085
Kiliç, S., Sagitova, D. M., Wolfish, S., Bely, B., Courtot, M., Ciufo, S., et al. (2016). From data repositories to submission portals: rethinking the role of domain-specific databases in CollecTF. Database 2016:baw055. doi: 10.1093/database/baw055
Kobayashi, I. (2016). “Genome evolution: Helicobacter pylori as an extreme model” in Helicobacter pylori research. eds. S. Backert and Y. Yamaoka (Tokyo: Springer), 217–232.
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lau, C. K. Y., Ishida, H., Liu, Z., and Vogel, H. J. (2013). Solution structure of Escherichia coli FeoA and its potential role in bacterial ferrous iron transport. J. Bacteriol. 195, 46–55. doi: 10.1128/JB.01121-12
Lau, C. K. Y., Krewulak, K. D., and Vogel, H. J. (2016). Bacterial ferrous iron transport: the Feo system. FEMS Micrbiol. Rev. 40, 273–298. doi: 10.1093/femsre/fuv049
Letunic, I., and Bork, P. (2021). Interactive tree of life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 49, W293–W296. doi: 10.1093/nar/gkab301
Linz, B., Windsor, H. M., McGraw, J. J., Hansen, L. M., Gajewski, J. P., Tomsho, L. P., et al. (2014). A mutation burst during the acute phase of Helicobacter pylori infection in humans and rhesus macaques. Nat. Commun. 5, 4165–4168. doi: 10.1038/ncomms5165
Merrell, D. S., Thompson, L. J., Kim, C. C., Mitchell, H., Tompkins, L. S., Lee, A., et al. (2003). Growth phase-dependent response of Helicobacter pylori to iron starvation. Infect. Immun. 71, 6510–6525. doi: 10.1128/IAI.71.11.6510-6525.2003
Mey, A. R., Gómez-Garzón, C., and Payne, S. M. (2021). Iron transport and metabolism in Escherichia, Shigella, and Salmonella. EcoSal Plus 9:eESP00342020. doi: 10.1128/ecosalplus.esp-0034-2020
Miller, A. J., Roman, B., and Norstrom, E. (2016). A method for easily customizable gradient gel electrophoresis. Anal. Biochem. 509, 12–14. doi: 10.1016/j.ab.2016.07.003
Moodley, Y., Linz, B., Bond, R. P., Nieuwoudt, M., Soodyall, H., Schlebusch, C. M., et al. (2012). Age of the association between Helicobacter pylori and man. PLoS Pathog. 8:e1002693. doi: 10.1371/journal.ppat.1002693
Müller, S. A., Findeiß, S., Pernitzsch, S. R., Wissenbach, D. K., Stadler, P. F., Hofacker, I. L., et al. (2013). Identification of new protein coding sequences and signal peptidase cleavage sites of Helicobacter pylori strain 26695 by proteogenomics. J. Proteome 86, 27–42. doi: 10.1016/j.jprot.2013.04.036
Occhino, D. A., Wyckoff, E. E., Henderson, D. P., Wrona, T. J., and Payne, S. M. (1998). Vibrio cholerae iron transport: haem transport genes are linked to one of two sets of tonB, exbB, exbD genes. Molec. Microbiol. 29, 1493–1507. doi: 10.1046/j.1365-2958.1998.01034.x
Pandey, A., and Sonti, R. V. (2010). Role of the FeoB protein and siderophore in promoting virulence of Xanthomonas oryzae pv. Oryzae on rice. J. Bacteriol. 192, 3187–3203. doi: 10.1128/JB.01558-09
Peng, E. D., Wyckoff, E. E., Mey, A. R., Fisher, C. R., and Payne, S. M. (2016). Nonredundant roles of iron acquisition systems in Vibrio cholerae. Infect. Immun. 84, 511–523. doi: 10.1128/IAI.01301-15
Pettersen, E. F., Goddard, T. D., Huang, C. C., Meng, E. C., Couch, G. S., Croll, T. I., et al. (2021). UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 30, 70–82. doi: 10.1002/pro.3943
Pich, O. Q., Carpenter, B. M., Gilbreath, J. J., and Merrell, D. S. (2012). Detailed analysis of Helicobacter pylori Fur-regulated promoters reveals a Fur box core sequence and novel Fur-regulated genes. Mol. Microbiol. 84, 921–941. doi: 10.1111/j.1365-2958.2012.08066.x
Plummer, M., Franceschi, S., Vignat, J., Forman, D., and De Martel, C. (2015). Global burden of gastric cancer attributable to Helicobacter pylori. Int. J. Cancer 136, 487–490. doi: 10.1002/ijc.28999
Prada, C. F., Casadiego, M. A., and Freire, C. C. M. (2022). Evolution of Helicobacter spp.: variability of virulence factors and their relationship to pathogenicity. PeerJ 10:e13120. doi: 10.7717/peerj.13120
Robey, M., and Cianciotto, N. P. (2002). Legionella pneumophila feoAB promotes ferrous iron uptake and intracellular infection. Infect. Immun. 70, 5659–5669. doi: 10.1128/IAI.70.10.5659-5669.2002
Roncarati, D., Pelliciari, S., Doniselli, N., Maggi, S., Vannini, A., Valzania, L., et al. (2016). Metal-responsive promoter DNA compaction by the ferric uptake regulator. Nat. Commun. 7, 1–13. doi: 10.1038/ncomms12593
Salaün, L., Audibert, C., Le Lay, G., Burucoa, C., Fauchère, J. L., and Picard, B. (1998). Panmictic structure of Helicobacter pylori demonstrated by the comparative study of six genetic markers. FEMS Microbiol. Lett. 161, 231–240. doi: 10.1111/j.1574-6968.1998.tb12953.x
Schmittgen, T. D., and Livak, K. J. (2008). Analyzing real-time PCR data by the comparative CT method. Nat. Protoc. 3, 1101–1108. doi: 10.1038/nprot.2008.73
Servetas, S. L., Whitmire, J. M., and Merrell, D. S. (2021). “Generation of isogenic mutant strains of Helicobacter pylori” in Helicobacter pylori: methods in molecular biology. ed. S. M. Smith (New York, NY: Humana), 107–122.
Sestok, A. E., Brown, J. B., Obi, J. O., Sullivan, S. M. O., Garcin, E. D., Deredge, D. J., et al. (2022). A fusion of the Bacteroides fragilis ferrous iron import proteins reveals a role for FeoA in stabilizing GTP-bound FeoB. J. Biol. Chem. 298:101808. doi: 10.1016/j.jbc.2022.101808
Sestok, A. E., Linkous, R. O., and Smith, A. T. (2018). Toward a mechanistic understanding of Feo-mediated ferrous iron uptake. Metallomics 10, 887–898. doi: 10.1039/c8mt00097b
Seyedmohammad, S., Fuentealba, N. A., Marriott, R. A. J., Goetze, T. A., Edwardson, J. M., Barrera, N. P., et al. (2016). Structural model of FeoB, the iron transporter from Pseudomonas aeruginosa, predicts a cysteine lined, GTP-gated pore. Biosc. Rep. 36:e00322. doi: 10.1042/bsr20160046
Sharma, C. M., Hoffmann, S., Darfeuille, F., Reignier, J., Findeiß, S., Sittka, A., et al. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature 464, 250–255. doi: 10.1038/nature08756
Shin, M., Mey, A. R., and Payne, S. M. (2019). Vibrio cholerae FeoB contains a dual nucleotide-specific NTPase domain essential for ferrous iron uptake. Proc. Natl. Acad. Sci. U. S. A. 116, 4599–4604. doi: 10.1073/pnas.1817964116
Shin, M., Park, J., Jin, Y., Kim, I. J., Payne, S. M., and Kim, K. H. (2020). Biochemical characterization of bacterial FeoBs: a perspective on nucleotide specificity. Arch. Biochem. Biophys. 685:108350. doi: 10.1016/j.abb.2020.108350
Skaar, E. P. (2010). The Battle for Iron between bacterial pathogens and their vertebrate hosts. PLoS Pathog. 6:e100094:e1000949. doi: 10.1371/journal.ppat.1000949
Smet, A., Yahara, K., Rossi, M., Tay, A., Backert, S., Armin, E., et al. (2018). Macroevolution of gastric Helicobacter species unveils interspecies admixture and time of divergence. ISME J. 12, 2518–2531. doi: 10.1038/s41396-018-0199-5
Solnick, J. V., and Vandamme, P. (2001). “Taxonomy of the Helicobacter genus” in Helicobacter pylori: physiology and genetics. eds. H. Mobley, G. Mendz, and S. Hazell (Washington, DC: ASM Press), 39–51.
Stevenson, B., Wyckoff, E. E., and Payne, S. M. (2016). Vibrio cholerae FeoA, FeoB, and FeoC interact to form a complex. J. Bacteriol. 198, 1160–1170. doi: 10.1128/JB.00930-15
Suerbaum, S., and Josenhans, C. (2007). Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 5, 441–452. doi: 10.1038/nrmicro1658
Taboada, B., Ciria, R., Martinez-Guerrero, C. E., and Merino, E. (2012). ProOpDB: prokaryotic operon DataBase. Nucleic Acids Res. 40, D627–D631. doi: 10.1093/nar/gkr1020
Tomb, J.-F., White, O., Kerlavage, A. R., Clayton, R. A., Sutton, G. G., Fleischmann, R. D., et al. (1997). The complete genome sequence of the gastric pathogen Helicobacter pylori. Nature 388, 539–547. doi: 10.1038/41483
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3—new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Vannini, A., Roncarati, D., D’Agostino, F., Antoniciello, F., and Scarlato, V. (2022). Insights into the orchestration of gene transcription regulators in Helicobacter pylori. Int. J. Mol. Sci. 23:13688. doi: 10.3390/IJMS232213688
Velayudhan, J., Hughes, N. J., McColm, A. A., Bagshaw, J., Clayton, C. L., Andrews, S. C., et al. (2000). Iron acquisition and virulence in Helicobacter pylori: a major role for FeoB, a high-affinity ferrous iron transporter. Mol. Microbiol. 37, 274–286. doi: 10.1046/j.1365-2958.2000.01987.x
Waidner, B., Melchers, K., Ivanov, I., Loferer, H., Bensch, K. W., Kist, M., et al. (2002). Identification by RNA profiling and mutational analysis of the novel copper resistance determinants CrdA (HP1326), CrdB (HP1327), and CzcB (HP1328) in Helicobacter pylori. J. Bacteriol. 184, 6700–6708. doi: 10.1128/jb.184.23.6700-6708.2002
Waite, D. W., Vanwonterghem, I., Rinke, C., Parks, D. H., Zhang, Y., Takai, K., et al. (2017). Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. Nov.). Front. Microbiol. 8:682. doi: 10.3389/fmicb.2017.00682
Weaver, E. A., Wyckoff, E. E., Mey, A. R., Morrison, R., and Payne, S. M. (2013). FeoA and FeoC are essential components of the Vibrio cholerae ferrous iron uptake system, and FeoC interacts with FeoB. J. Bacteriol. 195, 4826–4835. doi: 10.1128/JB.00738-13
Whitmire, J. M., and Merrell, D. S. (2012). “Successful culture techniques for Helicobacter species: general culture techniques for Helicobacter pylori” in Helicobacter species: methods in molecular biology. ed. J. Houghton (Totowa, NJ: Humana Press), 17–27.
Xia, Y., Chu, W., Qi, Q., and Xun, L. (2015). New insights into the QuikChangeTM process guide the use of Phusion DNA polymerase for site-directed mutagenesis. Nucleic Acids Res. 43:e12. doi: 10.1093/nar/gku1189
Yona, A. H., Alm, E. J., and Gore, J. (2018). Random sequences rapidly evolve into de novo promoters. Nat. Commun. 9, 1530–1510. doi: 10.1038/s41467-018-04026-w
Keywords: Vibrio cholerae, Fur, NikR, nickel, operon, Helicobacter pylori, Feo, iron transport
Citation: Gómez-Garzón C and Payne SM (2023) Divide and conquer: genetics, mechanism, and evolution of the ferrous iron transporter Feo in Helicobacter pylori. Front. Microbiol. 14:1219359. doi: 10.3389/fmicb.2023.1219359
Edited by:
Stefan Schild, University of Graz, AustriaReviewed by:
Christine Josenhans, Ludwig Maximilian University of Munich, GermanyCopyright © 2023 Gómez-Garzón and Payne. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shelley M. Payne, cGF5bmVAdXRleGFzLmVkdQ==
†Present address: Camilo Gómez-Garzón, Human Biology Division, Fred Hutchinson Cancer Center, Seattle, WA, United States
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.