 Rui Liu
Rui Liu Mengfei Chen2
Mengfei Chen2 Zhenchuan Mao
Zhenchuan Mao Jianlong Zhao
Jianlong Zhao- 1College of Plant Protection, Gansu Agricultural University/Biocontrol Engineering Laboratory of Crop Diseases and Pests of Gansu Province, Lanzhou, Gansu, China
- 2State Key Laboratory of Vegetable Biobreeding, Institute of Vegetables and Flowers, Chinese Academy of Agricultural Sciences, Beijing, China
- 3School of Life Sciences, Sun Yat-Sen University, Guangzhou, China
Introduction: Root-knot nematode (RKN; Meloidogyne spp.) is one of the most infamous soilborne plant diseases, causing severe crop losses every year. Effector proteins secreted by RKNs play crucial roles during plant-nematode interaction. However, less is known about whether RKN effector proteins can impact the rhizosphere microbial environment.
Methods: In this study, we investigated the rhizosphere microbiome community of MiMIF-2 (a plant immunity-modulating effector) transgenic Arabidopsis thaliana with or without nematode infection using the Illumina high-throughput sequencing analysis.
Results and discussion: The results showed that the bacterial species richness index increased, while the fungi species richness index decreased in M. incognita-infected MiMIF-2 transgenic A. thaliana plants. The relative abundance of genera such as Clitopilus, Komagataeibacter, Lactobacillus, Prevotella, Moritella, Vibrio, Escherichia-Shigella, and Pseudomonas was reduced in MiMIF-2 transgenic A. thaliana plants compared to wild type, but was significantly increased after inoculation with M. incognita. The Cluster of Orthologous Genes (COG) function classification analysis revealed a decrease in the relative abundance of defense mechanisms, secondary metabolite biosynthesis, transport, and nematode infection catabolism-related functions in MiMIF-2 lines compared to the wild type. These differences may be the reason for the increased susceptibility of MiMIF-2 transgenic A. thaliana to nematode infection. Our results provide a new insight into RKN effector proteins and their association with the microbial community, host, and plant pathogens, which will lead to the exploration of new innovative ideas for future biological control of RKNs.
1. Introduction
Root-knot nematodes (RKNs), belonging to the genus Meloidogyne, are among the most destructive plant-parasite nematodes (Trudgill, 1997). RKNs have an exceptionally large host range, with more than 5,500 plant hosts, and cause economic losses worth more than 100 billion dollars every year (Abad et al., 2008; Elling, 2013). Among RKNs, Meloidogyne incognita is an obligate, biotrophic pathogen of many plant species and is believed to be the most harmful nematode species in traditional as well as protected agriculture (Jones et al., 2013). RKNs build remarkably sophisticated interactions with their host plants (Vieira and Gleason, 2019). During the interaction, the motile second-stage juveniles (J2s) of M. incognita enter the host root tip and migrate intercellularly to reach the host vascular cells to form hypertrophied feeding cells called giant cells (GCs) (Favery et al., 2016). In recent decades, most studies focus on elucidating the molecular mechanism of plant-nematode interactions. However, less is known about the functions of the rhizosphere microbiome during RKN parasitism.
It is widely recognized that the extent of microbial colonization of plants is a crucial factor affecting their health (Berendsen et al., 2012). There are intricate interactions in plants, microbes, and nematodes (Bais et al., 2006). In 2012, studies revealed the core endophytic bacterial microbiome in the model plant Arabidopsis thaliana, where the dominant phyla are Proteobacteria, Bacteroidetes, and Firmicutes (Bulgarelli et al., 2012; Lundberg et al., 2012). Tens of thousands of plant-associated bacterial strains have been isolated, and they modulate plant growth and development, thereby increasing the yield of crops (Hardoim et al., 2008). Any disruption to this “plant-soil-microorganism” interaction can play a significant role in the occurrence of diseases (Classen et al., 2015). The rhizosphere microbiome has different mechanisms to effectively control RKNs. Certain bacteria, such as Bacillus cereus (Yin et al., 2021), Pseudomonas aeruginosa (Siddiqui and Shaukat, 2003), and Brevundimonas diminuta (Zheng et al., 2008), can suppress nematode infection. Conversely, other bacteria such as Chitinophaga (Baquiran et al., 2013) and Pedobacter (Tian et al., 2011) can promote the growth of nematodes. Many fungi have also been reported to inhibit RKNs. Verticillium, Paecilomyces lilacinus (Wang et al., 2010), and Pochonia chlamydosporia (Galeano et al., 2003) are known to inhibit RKNs parasitism. By investigating the microbial communities affected by RKNs in tomato roots and analyzing their functional characteristics in the interactions between microbes, plants, and nematodes, studies have made an intriguing finding: the pathogenicity of nematodes led to a decrease in the prevalence of the prominent endophytic groups, specifically Streptomyces and Pseudomonas (Tian et al., 2015).
Effectors are small molecular compounds secreted by nematodes into host tissues to alter host physiology and assist in the infection process (Hogenhout et al., 2009). In the last 20 years, a large number of effectors of M. incognita have been reported, which are involved in promoting infection, migration, and parasitism of nematodes in plant roots (Mejias et al., 2019, 2022; Truong et al., 2021; Zhao et al., 2021). We previously reported that M. incognita secretes macrophage migration inhibitory factor-like proteins (MiMIFs) that suppress plant immunity and facilitate nematode parasitism. MiMIF-2 exhibited enzyme activities and has played a potential role in plant salicylic acid synthesis (Zhao et al., 2019, 2020). Despite the essential roles in modulating plant immune responses of nematode effectors, their relationship with rhizosphere microorganisms and hosts is largely unknown.
A previous report has shown that plant pathogens secrete effectors to manipulate the host microbiome (Snelders et al., 2018). Recently, a report also showed that the soil-borne fungal plant pathogen Verticillium dahlia secreted the effector Vdave1, which manipulated the host microbiome to facilitate colonization in tomato and cotton (Snelders et al., 2020). Despite several metagenomic studies conducted to explore the diversity of root microbial community upon RKN infection (Wu et al., 2022; Lu et al., 2023), the key factors that affect RKN development and the potential mechanisms of RKN inhibition have not been fully explored. In this study, we focused on the rhizosphere microbiome of MiMIF-2 ectopic expression in A. thaliana, with or without inoculation of M. incognita. The results showed that the effector-driven effect on the species of fungi and bacteria contributes to the plasticity of the host rhizosphere microbiome, which is critical for M. incognita parasitism.
2. Materials and methods
2.1. Plant growth conditions and nematode culture
MiMIF-2 transgenic A. thaliana lines were previously described (Zhao et al., 2019). Wild-type (WT) A. thaliana (Col-0) was used as a control. Surface-sterilized A. thaliana seeds were placed on Murashige and Skoog Medium (MS) (Sigma, St. Louis, MO, USA) and were incubated at 22°C with a 12-h photoperiod. Seedlings of 15 days old plants were transplanted into soil and were grown in the greenhouse at 22°C and 65% relative humidity with a 16-h light/8-h dark photoperiod.
Tomato plants (Solanum lycopersicum var. “Baiguo”) were used to reproduce M. incognita nematode culture. Infective pre-parasitic second-stage juveniles (pre-J2s) were collected after egg hatching (Zhao et al., 2019).
2.2. Sample collection of rhizosphere soil
Homozygous T3 MiMIF-2 transgenic A. thaliana lines and WT were challenged with 200 pre-parasitic J2s of M. incognita, indicated by MiMIF-2+N and WT+N. WT and MiMIF-2 transgenic A. thaliana lines of uninoculated nematodes were used as the controls, indicated by WT and MiMIF-2. All the plants were grown in the laboratory of the Institute of Vegetable and Flower Research of the Chinese Academy of Agricultural Sciences, Beijing, China (116.19.32°E, 39.57.44°N). Rhizosphere soil samples around the A. thaliana roots were collected at 30 days post-inoculation (dpi) with nematodes. After excavating the roots of 30 Arabidopsis plants from the soil, the soil that was loosely attached to the roots was carefully removed. The soil that adhered to the Arabidopsis root system was brushed loose using a delicate brush. This soil was collected in sterile self-sealing bags and mixed thoroughly, and the amalgamated soil was divided into five equal portions. Each treatment was replicated five times with five seedlings per replication (Lundberg et al., 2012).
2.3. DNA extraction and PCR amplification
Microbial community genomic DNA was extracted using 0.5 g of soil per sample employing the E.Z.N.A.® soil DNA Kit (Omega Bio-tek, Norcross, GA, USA) as per the manufacturer's recommendations. The quality of the DNA extract was confirmed on 1% agarose gel, whereas the DNA concentration and purity were determined using the NanoDrop 2000 UV-vis spectrophotometer (Thermo Scientific, Wilmington, USA). Amplification of the hypervariable region, V3–V4 of the bacterial 16S rRNA gene, was performed using the primer pair 338F (5'-ACTCCTACGGGAGGCAGCAG-3') and 806R (5'-GGACTACHVGGGTWTCTAAT-3'), whereas the primer set SSU0817 forward (5′-TTAGCATGGAATAATRRAATAGGA-3′) and 1196 reverse (5′-TCT GGACCTGGTGAGTTTCC-3′) was used to amplify the V5–V7 region of the 18S rDNA gene through the ABI GeneAmp® 9700 PCR thermocycler (ABI, CA, USA).
The PCR method used for the amplification comprised of an initial denaturation at 95°C for 3 min, followed by 27 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and extension at 72°C for 45 s in each cycle with a final extension at 72°C for 10 min after which the reaction was maintained at 4°C. The PCR mixtures contained 5× TransStart FastPfu buffer 4 μl, 2.5 mM dNTPs 2 μl, forward primer (5 μM) 0.8 μl, reverse primer (5 μM) 0.8 μl, TransStart FastPfu DNA polymerase 0.4 μl, template DNA 10 ng, and ddH2O to make a final volume of 20 μl. PCR amplifications were performed in triplicate. The PCR products were electrophoresed using 2% agarose gel, and the band of interest was eluted using the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, USA) as per the recommendations of the manufacturer, and the eluted DNA was quantified using the Quantus™ Fluorometer (Promega, USA).
2.4. Illumina MiSeq sequencing
Purified amplicons pooled in equimolar volumes were paired-end sequenced (2 × 300) on an Illumina MiSeq platform (Illumina, San Diego, USA) following the standard protocols recommended by Majorbio Bio-Pharm Technology Co. Ltd. (Shanghai, China). The raw reads were deposited into the NCBI Sequence Read Archive (SRA) database (accession number: PRJNA954129).
2.5. Bioinformatics and statistical analyses
Splicing and filtering were performed on raw data to secure valid data and to ensure the accuracy and reliability of the results. Operational taxonomic units (OTUs) were clustered using 97% similarity. The Chao and Shannon indices were computed to figure out the alpha diversity and to determine the species richness in each soil sample. A principal coordinates analysis (PCoA) of each sample formulated on the Bray–Curtis distances was accomplished using the R package vegan (version 2.1). Linear discriminant analysis (LDA) effect size measurements (LEfSe) was performed to search for significantly different (P < 0.05) taxa between the two groups. Significantly different taxa were used to generate taxonomic cladograms, which illustrated the differences between sample classes on the website http://huttenhower.sph.harvard.edu/galaxy.
Graphical representations were created using GraphPad Prism 5 (GraphPad Software, Inc., La Jolla, CA, USA). The mean and standard error for each group of data were calculated using the ANOVA and Tukey's honestly significant difference test (P < 0.05) using SPSS 19.0 (SPSS Inc., Chicago, IL, USA).
3. Results
3.1. Sequencing and metagenome assembly
After Illumina paired-end sequence, quality evaluation, data filtering, and integration, a total of 933,427 bacterial 16S rRNA sequences (Supplementary Table S1) and 1,169,253 effective fungal 18S rDNA sequences (Supplementary Table S2) were generated from 20 samples. A total of 2,299 bacterial OTUs were assigned to 38 different phyla: WT contained 28, MiMIF-2 contained 32, WT+N contained 30, and MiMIF-2+N contained 32 (Table 1). A total of 492 fungal OTUs were assigned to 9 different phyla: WT contained 9, MiMIF-2 contained 9, WT+N contained 9, and MiMIF-2+N contained 8 (Table 2).

Table 1. General features of the high-throughput bacteria sequencing results in different samples.
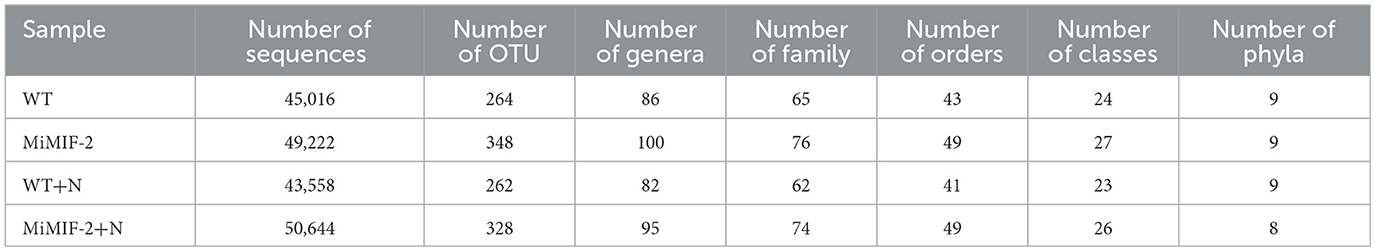
Table 2. General features of the high-throughput fungal sequencing results in different samples.
3.2. Composition and diversity of the rhizosphere microbiome with or without nematode infection
By comparing the α-diversities of the fungal species richness (Chao1) index, there was a significant difference between the before and after inoculation nematodes. However, the fungal species richness in MiMIF-2 was significantly reduced compared to WT, when inoculated with M. incognita (P < 0.05, Wilcoxon rank sum test; Figure 1A).
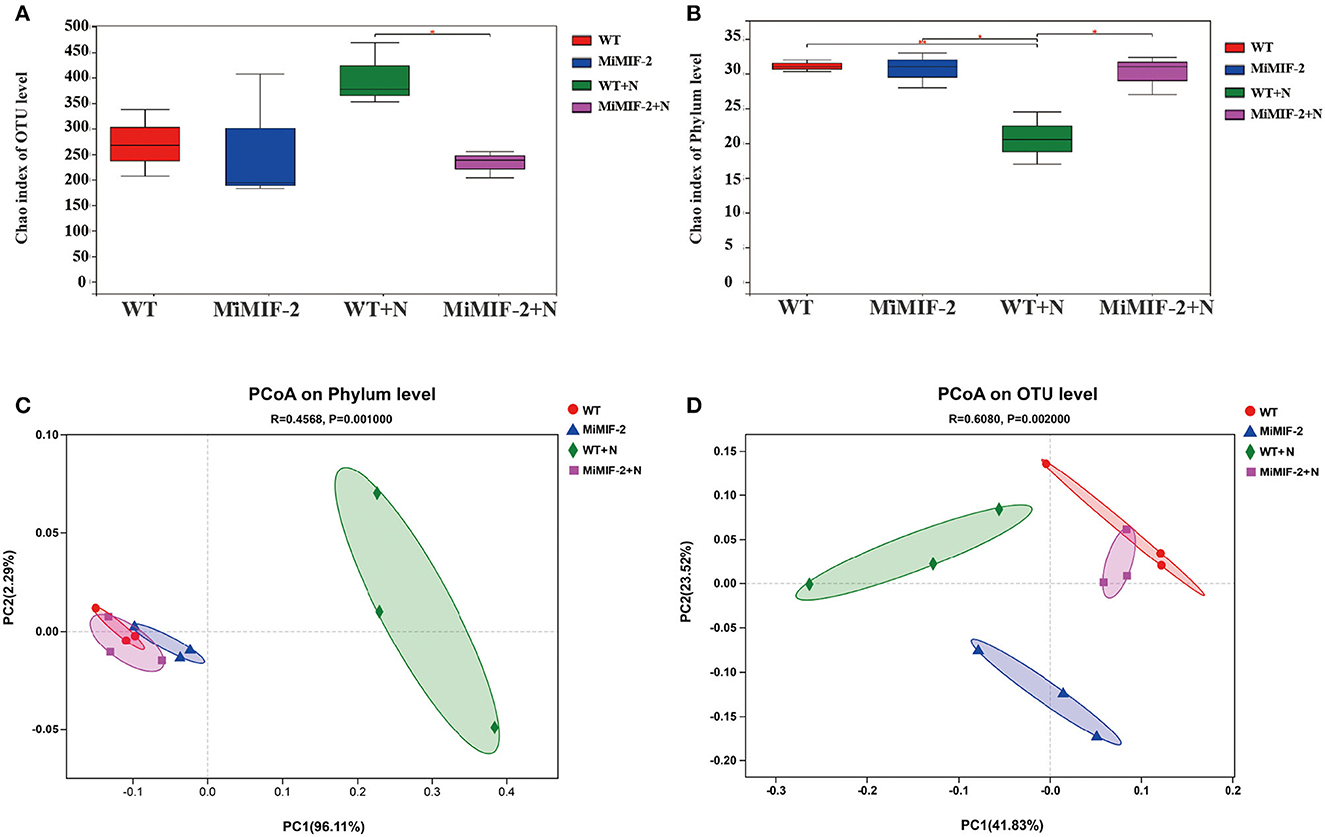
Figure 1. Diversity analysis: Alpha-diversity of the Arabidopsis rhizo-compartment microbiota in different samples. (A) fungal species richness (Chao1) index of OUT level; (B) bacterial species richness (Chao) index of phylum level (*indicates P < 0.05; **indicates P < 0.01; Wilcoxon rank sum test). PCoA of Bray–Curtis distances reveals that soil type is a major source of bacterial (C) and fungal (D) community variation in both the rhizosphere and roots.
In the case of bacterial species richness index, however, MiMIF-2 lines showed a significant increase compared to WT after inoculation with M. incognita. The bacterial species richness (Chao1) index was also reduced in wild-type Arabidopsis when compared with uninoculated WT (Figure 1B). Interestingly, the species richness index of WT inoculated with M. incognita was significantly lower than that of uninoculated samples (Figure 1B). The increase in bacterial species diversity might be the reason for the increased nematode susceptibility in MiMIF-2.
Statistical analysis of Bray–Curtis distances (β-diversity) was involved in the comparison of the composition of microbial communities. It was performed to determine the community treatment relationship between the samples. Concerning the phylum level, PCoA was performed on each sample, and the distribution of WT+N and other samples was significantly different for bacteria (Figure 1C). At the OTU level, PCoA of Bray–Curtis distances (β-diversity) revealed that WT, MiMIF-2, WT+N, and MiMIF-2+N microbiota in four types of soil exhibited a clear separation (Figure 1D).
OTUs of the four samples were assigned corresponding taxonomies based on the combined search results against the Greengenes and NCBI databases. The relative abundance of different phyla in the four samples was analyzed, of which Proteobacteria, Actinobacteria, and Firmicutes were the dominant bacterial phyla (Figure 2A). Ascomycota and Basidiomycota were the dominant fungal phyla in all soil samples, accounting for more than 90% of the total abundance in each sample (Figure 2B). After inoculation with nematodes, Firmicutes of WT+N were significantly reduced compared to uninoculated samples (Figure 2A). In community composition, Clitopilus, Candida, Meliniomyces, and Pseudeurotium were the dominant fungal genera in all soil samples (Figure 2C) whereas in the bacterium community composition, Komagataeibacter, Lactobacillus, Streptomyces, and Rhodanobacter were the dominant bacterial genera in all the rhizosphere bacteria. Lactobacillus of WT+N were significantly reduced compared to uninoculated samples. On the contrary, Streptomyces, Rhodanobacter, and Actinoallomurus of WT+N species increased compared with other samples (Figure 2D).
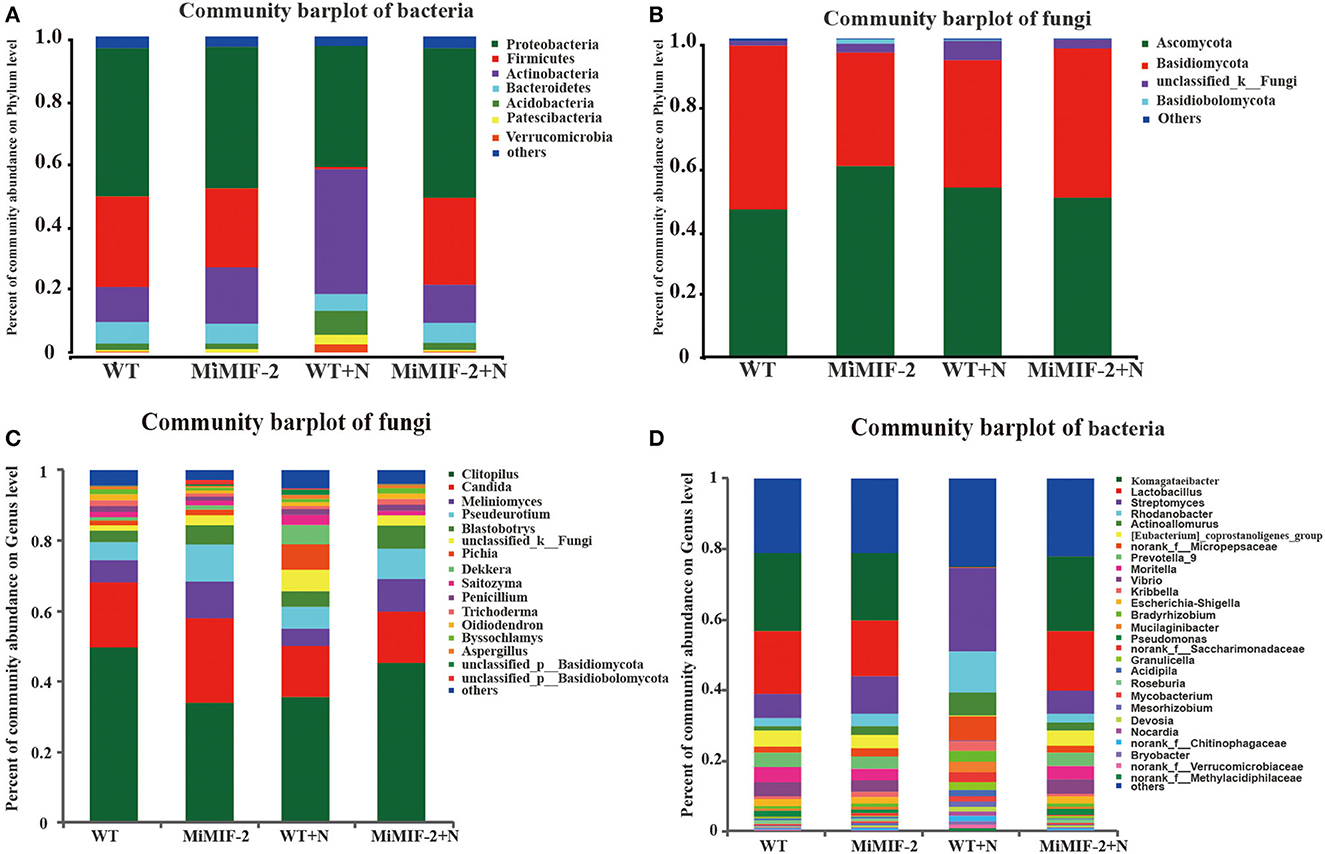
Figure 2. Community composition at the phylum and genus level of the rhizosphere soil samples of wild-type Arabidopsis thaliana (WT), MiMIF-2 transgenic A. thaliana lines (MiMIF-2), inoculated M. incognita MiMIF-2 transgenic A. thaliana lines (MiMIF-2+N) and WT (WT+N). Bacterial (A) and fungal (B) community composition at the phylum level. Fungal (C) and bacterial (D) community composition at the genus level.
3.3. Predominant microbe species affected by M. incognita
The relative abundances of major fungal phyla and genus, including Basidiomycota, Clitopilus, and Candida, were different in MiMIF-2 transgenic A. thaliana (Figure 3A). Phylum Basidiomycota abundance in MiMIF-2 transgenic A. thaliana decreased compared with WT; however, the relative abundances increased after inoculation with M. incognita. Similar results were obtained during the analysis at the genus level. Clitopilus behaved similarly as Basidiomycota. Candida was abundant in MiMIF-2 transgenic A. thaliana; however, no differences were observed after inoculation with M. incognita (Figure 3B).
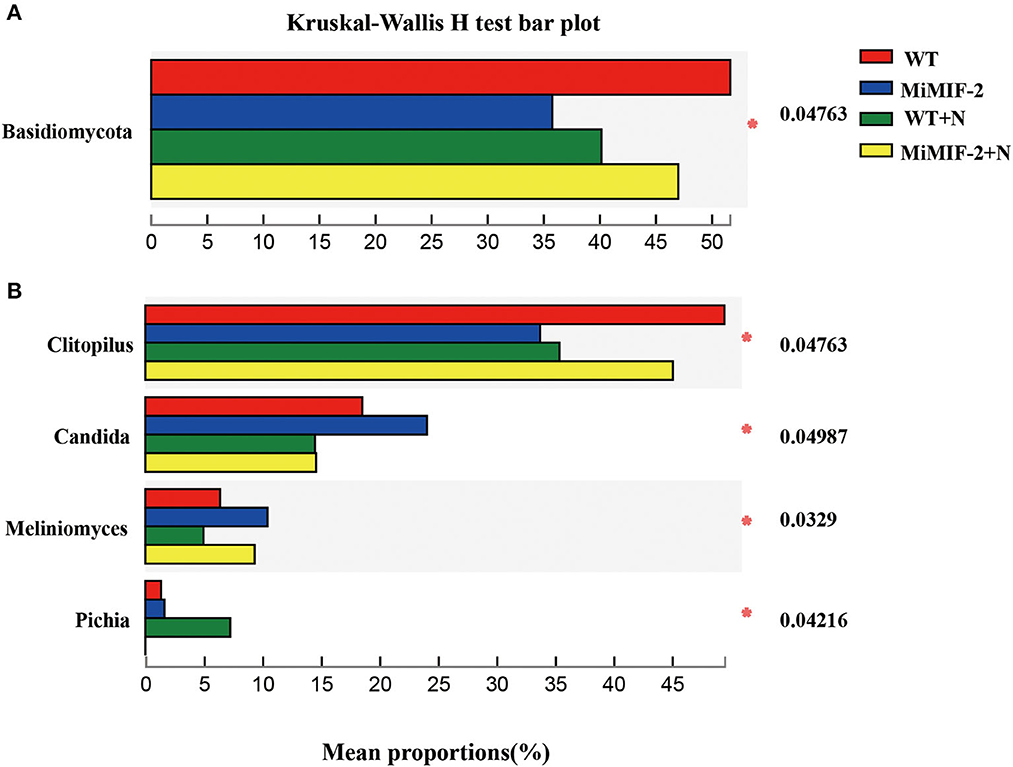
Figure 3. (A, B) The different fungal relative abundances of major phylum and genera among the different compartments. WT (red), MiMIF-2 (blue), WT+N (green), and MiMIF-2+N (yellow). The Kruskal–Wallis H-test was used to evaluate the significance of the difference between the indicated groups (n = 3, *indicates P < 0.05).
The analysis of bacterial phylum-level relative abundances revealed significant differences among Firmicutes, Actinobacteria, Acidobacteria, Epsilonbacteraeota, Spirochetes, and Chloroflexi. Firmicutes, Epsilonbacteraeota, Spirochetes, and Fusobacteria were significantly higher in MiMIF-2+N compared to those in WT+N. However, the richness of the phylum of WT was significantly reduced after inoculation with nematodes. Actinobacteria, Acidobacteria, and Chloroflexi were reduced in MiMIF-2+N compared to WT+N (Figure 4A). A similar phenomenon occurs at the genus-level relative abundances. More than 15 genera showed significant differences. For instance, Komagataeibacter, Lactobacillus, Prevotella, Moritella, Vibrio, Escherichia-Shigella, and Pseudomonas were reduced in MiMIF-2 lines compared to those in WT. However, these genera were significantly higher in MiMIF-2+N compared to those in WT+N (Figure 4B).
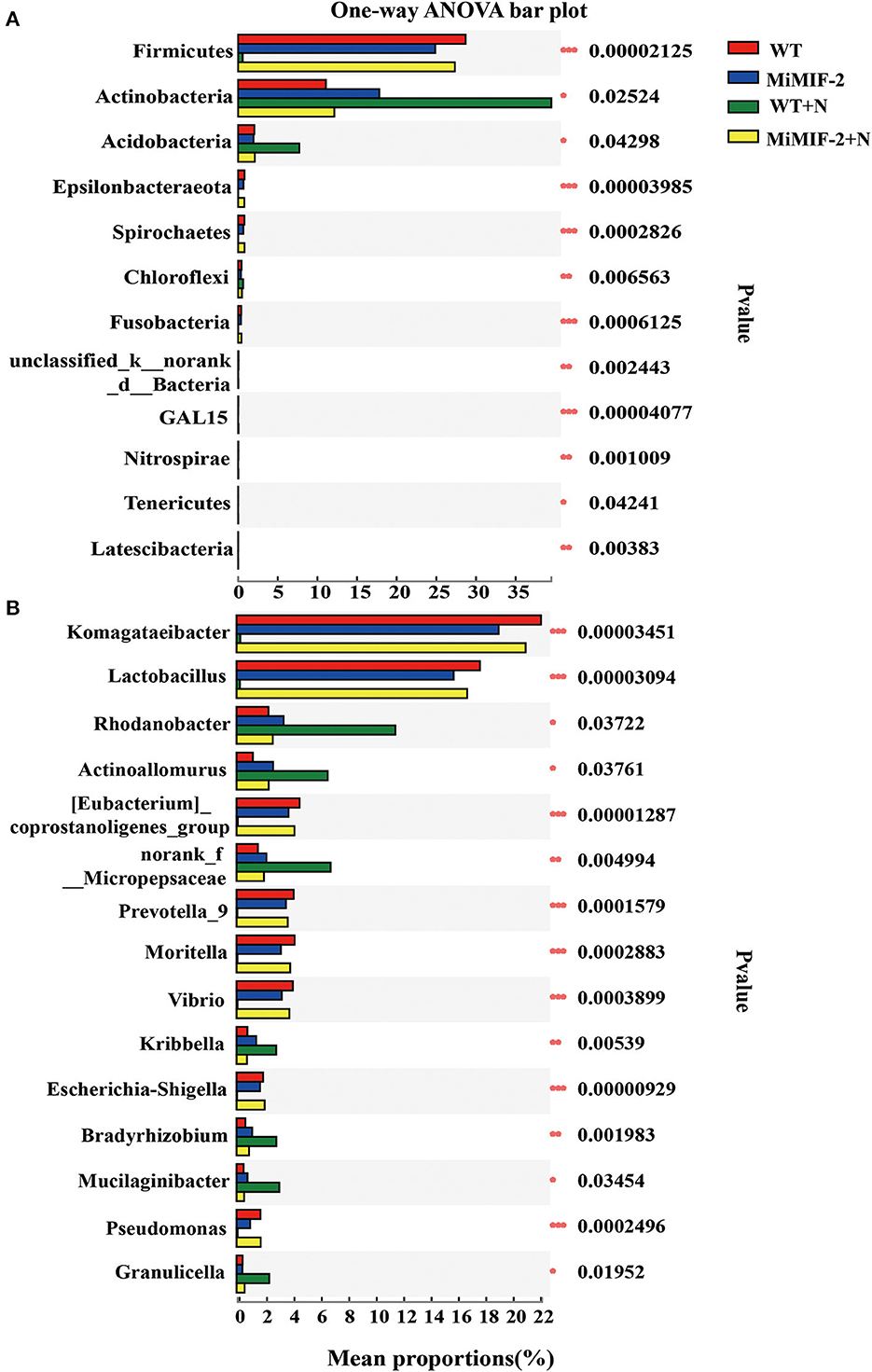
Figure 4. The different bacterial relative abundances of major phylum (A) and genera (B) among the different compartments. WT (red), MiMIF-2 (blue), WT+N (green), and MiMIF-2 + N (yellow). The Kruskal–Wallis H-test was used to evaluate the significance of the difference between the indicated groups (n = 3, *indicates P < 0.05; **indicates P < 0.01; ***indicates P < 0.001).
3.4. Microbial communities with statistically significant differences
The LEfSe was used to identify the dominant phylotypes responsible for the differences among the samples. The groups were shown in cladograms, and LDA scores of 4 (Supplementary Tables S3, S4) or greater were confirmed using LEfSe (Figure 5). In MiMIF-2+N, 11 groups of bacteria were significantly enriched, while no fungi were detected at a significant level. The bacteria with the highest LDA value in MiMIF-2+N was Firmicutes (logarithmic LDA score = 4.73). In WT+N, 4 groups of bacteria and 3 groups of fungi were significantly enriched. The bacterium and fungus with the highest LDA value in WT+N were Gammaproteobacteria (logarithmic LDA score = 4.57) and Pleurotaceae (logarithmic LDA score = 4.87), respectively. In WT, 5 groups of bacteria and 5 groups of fungi were detected at a significant level. The bacterium and fungus with the highest LDA value in WT were Proteobacteria (logarithmic LDA score = 4.98) and Entolomataceae (logarithmic LDA score = 4.96), respectively. In MiMIF-2, 4 groups of bacteria and 6 groups of fungi were detected at a significant level. The bacterium and fungus with the highest LDA value in MiMIF-2 were Actinobacteriota (logarithmic LDA score = 4.79) and Saccharomycetes (logarithmic LDA score = 4.84), respectively. Community structure and predominant taxa of different samples may account for differences in sensitivity to nematodes.
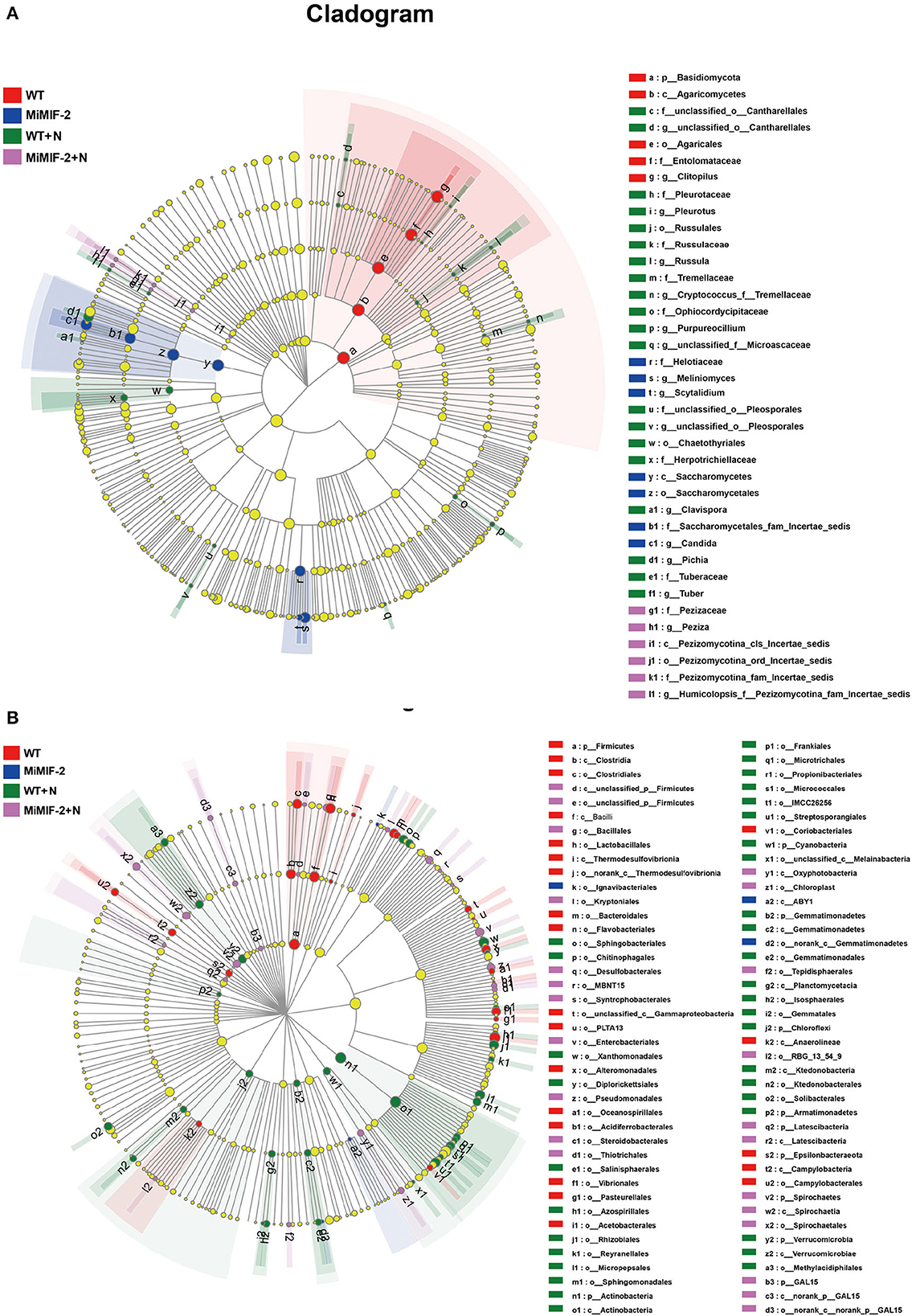
Figure 5. LEfSe cladogram of the aggregated groups of WT, MiMIF-2, WT+N, and MiMIF-2+N. A range of bacterium (A) and fungus (B) taxa from phylum to genus level was associated with WT (red), MiMIF-2 (blue), WT+N (green), and MiMIF-2+N (violet) (α = 0.05, LDA > 4.0, the size of circles is proportional to each taxon's mean relative abundance). The yellow circles represent the absence of significantly different taxa.
3.5. Functional annotations and functional guilds for the different rhizosphere microbiomes
The relative abundance of PICRUSt inferred function is shown in Figure 6A. All soil rhizosphere microorganisms consist mainly of energy production and conversion, amino acid transport and metabolism, carbohydrate transport and metabolism, cell wall/membrane/envelope biogenesis, and general function prediction. Whether or not the nematode is inoculated, the relative abundance of all the COG function classifications in WT was slightly larger than that in MiMIF-2 lines (Supplementary Table S5). For example, defense mechanisms, secondary metabolites biosynthesis, transport, and catabolism may be related to nematode infection.
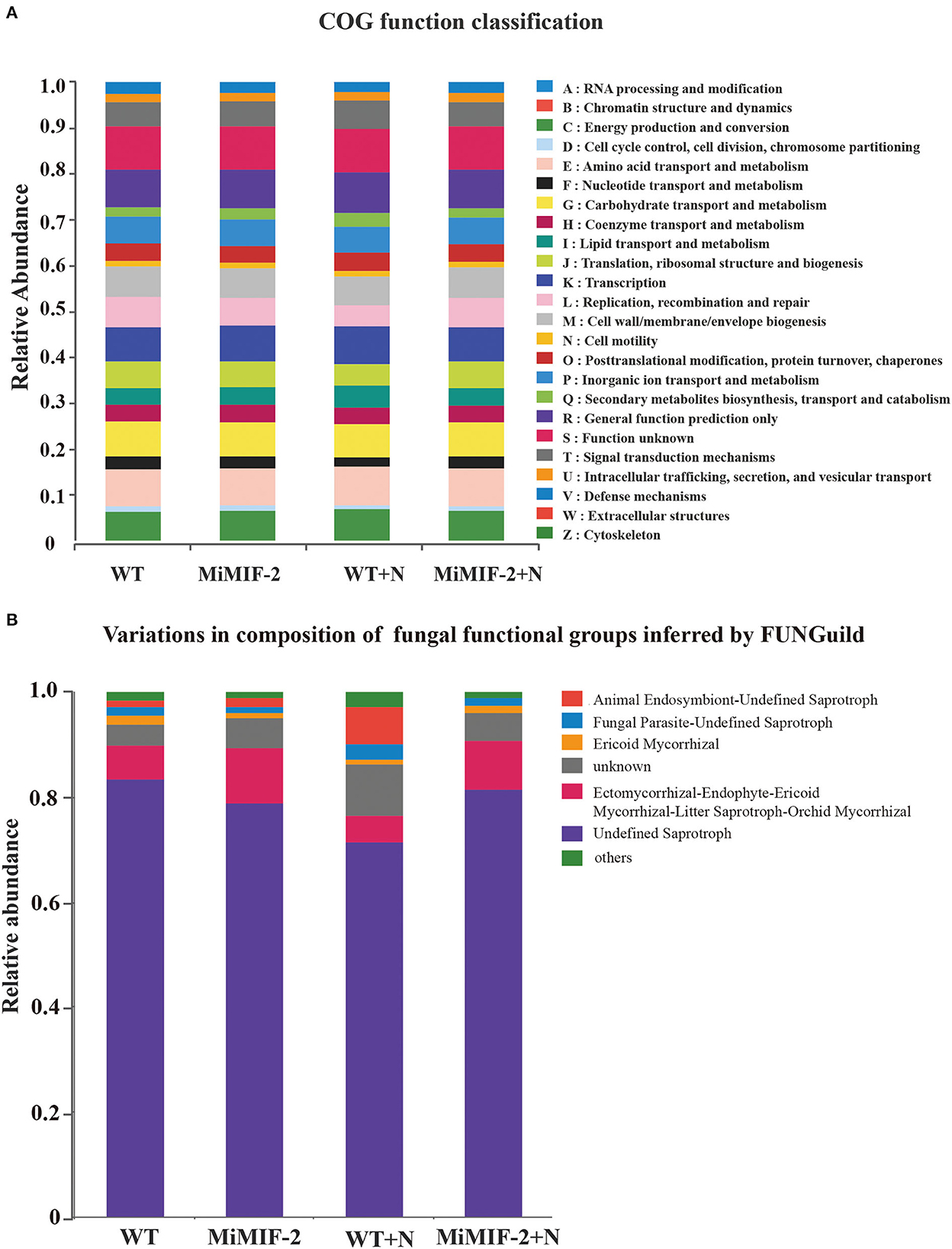
Figure 6. Relative abundance of the PICRUSt inferred function in different samples (A). Relative abundance of variations in the composition of fungal functional groups inferred by FUNGuild (B). The relative abundance is calculated by averaging the abundances of duplicate samples.
We used a manually curated set of designations based on FunGuild, a recently established fungal classification tool with rigorously defined and referenced trophic group assignments. The relative abundance of animal endosymbiont-undefined saprotroph was higher after inoculation with nematodes; however, it was almost none after transferring to the MiMIF-2 effector (Figure 6B).
4. Discussion
The rhizosphere microbes associated with plant roots are enormous, in the order of tens of thousands of species, where complex biological and ecological processes occur (Berendsen et al., 2012). It is universally accepted that the host's rhizosphere microbes play a significant role in its health and that hosts actively shape their microbiomes to prevent or suppress disease development (Pieterse et al., 2001; Lugtenberg and Kamilova, 2009). In previous studies, rhizosphere microbes play a key role in the relationship among the plant, soil, and pathogens (Trivedi et al., 2020). It has been well established that pathogens secrete effector molecules of various kinds during attempted host ingress to promote disease development, many of which target essential components of the host immune system. Our previous study showed that MiMIF-2 was an effector protein secreted from the cuticle of M. incognita, and it enters host cells during nematode parasitism; MiMIF-2 effector could suppress plant immunity by interacting with plant annexin proteins (Zhao et al., 2019). We provided evidence that salicylic acid (SA)-related marker genes and the content of SA were significantly reduced when MiMIF-2 transgenic Arabidopsis was challenged against pathogens (Zhao et al., 2020), roles of MiMIF-2 and annexins in root responses to soil microbes are expected. During the plant-microbiome interaction, beneficial microorganisms will improve plant resistance to pathogens and stresses. However, pathogens, such as RKNs, have evolved sophisticated means to interfere with plant immunities and recruit microbiome suits for themselves (Trivedi et al., 2020). Thus, we speculate that MiMIF-2 expression in planta will alter host immune responses and also manipulate the microbiome in favor of nematode parasitism. In recent years, similar reports have shown other pathogens manipulating the host immune system by secreting effectors to promote disease development, especially those of filamentous pathogens (Rovenich et al., 2014; Snelders et al., 2018). Moreover, researchers demonstrated that a fungal plant pathogen uses effector proteins to modulate microbiome compositions inside and outside the host (Snelders et al., 2020).
Community analysis of tomato root-associated with healthy and nematode-infected tomatoes indicated that nematode pathogenesis led to a decrease in the abundance of the main endophytic bacteria Streptomycetaceae and Pseudomonadales (Tian et al., 2015). After M. incognita infected tomato, the main components of the root microorganisms were Proteobacteria, Actinobacteria, Bacteroidetes, and Firmicutes (Cao et al., 2015). Rhizosphere microbiome structures difference alters to tomato wilt resistance (Kwak et al., 2018). There was a significant relationship between RKN disease and rhizosphere microbial diversity (Lu et al., 2023). It has been reported that many Actinomyces have strong biocontrol potential in the egg mass and adult worms of nematodes (Sun et al., 2006). Streptomyces are important producers of antibiotics and toxic metabolites, and Streptomyces species have been found to control fungal pathogens and nematodes, which are considered promising biological control agents (Krechel et al., 2002). The analysis of rhizosphere microorganisms' diversity revealed that the presence of MiMIF-2 caused an increase in the species richness of Actinobacteria, Acidobacteria, Firmicutes, and Proteobacteria, ultimately leading to a promoted sensitivity to nematodes.
Pathogenic microorganisms can manipulate the host microbiome through the immune system of salicylic acid (Wu et al., 2022), reactive oxygen species (Song et al., 2021), PTI immune response (Topalovi et al., 2020), etc., which is conducive to parasitism. In this study, the relative abundance of the rhizosphere microorganisms in MiMIF-2 transgenic lines decreased compared with the wild type in secondary metabolism synthesis and defense response. Collectively, we speculate that the MiMIF-2 effector plays a role in manipulating the rhizosphere microbial to promote the parasitism of M. incognita through immune response changes of the host.
5. Conclusion
Taken together, our study showed that MiMIF-2 expression in plants has an effect on the aggregation of bacteria and fungi. Interestingly, there was an increase in bacterial species and a decrease in fungi species when MiMIF-2 transgenic A. thaliana plants were infected with RKNs. We observed a decrease in defense mechanisms and secondary metabolites in MiMIF-2 transgenic A. thaliana, which could explain their increased susceptibility to nematode infection. These findings shed new light on the role of plant-parasitic nematode effector proteins and their interaction with microbial communities, plants, and pathogens, providing new clues for the biological control of RKNs.
Data availability statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/genbank/, PRJNA954129.
Author contributions
RL, MC, and KH: performed the experiments. RL, BL, ZM, JZ, and HL: contributed to the data analysis, discussion, wrote, reviewed, and corrected the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by the Youth Innovation Program of the Chinese Academy of Agricultural Sciences (Y2022QC06), the National Natural Science Foundation of China (32001878, 32260654, and 32172366), the Beijing Natural Science Foundation (6222054), and the China Agriculture Research System (CARS-23).
Acknowledgments
The authors would like to thank Prof. Heng Jian (College of Plant Protection, China Agricultural University, China) for his guidance on the experiment design.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2023.1217863/full#supplementary-material
References
Abad, P., Gouzy, J., Aury, J., Castagnone-Sereno, P., Danchin, E. G. J., and Deleury, E.. (2008). Genome sequence of the metazoan plant-parasitic nematode Meloidogyne incognita. Nat. Biotechnol. 26, 909–915. doi: 10.1038/nbt.1482
Bais, H. P., Weir, T. L., Perry, L. G., Gilroy, S., and Vivanco, J. M. (2006). The role of root exudates in rhizosphere interactions with plants and other organisms. Annu. Rev. Plant Biol. 57, 233–266. doi: 10.1146/annurev.arplant.57.032905.105159
Baquiran, J. P., Thater, B., Sedky, S., Ley, D., Crowley, P. D., and Orwin, P. M. (2013). Culture-independent investigation of the microbiome associated with the nematode Acrobeloides maximus. PloS ONE. 8, e67425. doi: 10.1371/journal.pone.0067425
Berendsen, R. L., Pieterse, C. M. J., and Bakker, P. A. H. M. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bulgarelli, D., Rott, M., Schlaeppi, K., Ver Loren Van Themaat, E., Ahmadinejad, N., Assenza, F., et al. (2012). Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature. 488, 91–95. doi: 10.1038/nature11336
Cao, Y., Tian, B., Ji, X., Shang, S., Lu, C., and Zhang, K. (2015). Associated bacteria of different life stages of Meloidogyne incognita using pyrosequencing-based analysis. J. Basic Microbiol. 55, 950–960. doi: 10.1002/jobm.201400816
Classen, A. T., Sundqvist, M. K., Henning, J. A., Newman, G. S., Moore, J. A. M., Cregger, M. A., et al. (2015). Direct and indirect effects of climate change on soil microbial and soil microbial-plant interactions: what lies ahead? Ecosphere. 6, 1–21. doi: 10.1890/ES15-00217.1
Elling, A. A. (2013). Major emerging problems with minor Meloidogyne species. Phytopathology. 103, 1092–1102. doi: 10.1094/PHYTO-01-13-0019-RVW
Favery, B., Quentin, M., Jaubert-Possamai, S., and Abad, P. (2016). Gall-forming root-knot nematodes hijack key plant cellular functions to induce multinucleate and hypertrophied feeding cells. J. Insect Physiol. 84, 60–69. doi: 10.1016/j.jinsphys.2015.07.013
Galeano, M., Verdejo-Lucas, S., Sorribas, F. J., and Ornat, C. (2003). Evaluation of a native and introduced isolate of Pochonia chlamydosporia against Meloidogyne javanica. Biocontrol. Sci. Techn. 13, 707–714. doi: 10.1080/09583150310001606282
Hardoim, P. R., van Overbeek, L. S., and Elsas, J. D. V. (2008). Properties of bacterial endophytes and their proposed role in plant growth. Trends Microbiol. 16, 463–471. doi: 10.1016/j.tim.2008.07.008
Hogenhout, S. A., Van der Hoorn, R. A. L., Terauchi, R., and Kamoun, S. (2009). Emerging concepts in effector biology of plant-associated organisms. Mol. Plant Microbe. Interact. 22, 115–122. doi: 10.1094/MPMI-22-2-0115
Jones, J. T., Haegeman, A., Danchin, E. G. J., Gaur, H. S., Helder, J., Jones, M. G. K., et al. (2013). Top 10 plant-parasitic nematodes in molecular plant pathology. Mol. Plant Pathol. 14, 946–961. doi: 10.1111/mpp.12057
Krechel, A., Faupel, A., Hallmann, J., Ulrich, A., and Berg, G. (2002). Potato-associated bacteria and their antagonistic potential towards plant-pathogenic fungi and the plant-parasitic nematode Meloidogyne incognita (kofoid and white) chitwood. Can. J. Microbiol. 48, 772–786. doi: 10.1139/w02-071
Kwak, M., Kong, H. G., Choi, K., Kwon, S., Song, J. Y., Lee, J., et al. (2018). Rhizosphere microbiome structure alters to enable wilt resistance in tomato. Nat Biotechnol. 36, 1100–1109. doi: 10.1038/nbt.4232
Lu, P., Shi, H., Tao, J., Jin, J., Wang, S., Zheng, Q., et al. (2023). Metagenomic insights into the changes in the rhizosphere microbial community caused by the root-knot nematode Meloidogyne incognita in tobacco. Environ. Res. 216, 114848. doi: 10.1016/j.envres.2022.114848
Lugtenberg, B., and Kamilova, F. (2009). Plant-growth-promoting rhizobacteria. Annu. Rev. Microbiol. 63, 541–556. doi: 10.1146/annurev.micro.62.081307.162918
Lundberg, D. S., Lebeis, S. L., Paredes, S. H., Yourstone, S., Gehring, J., Malfatt, S., et al. (2012). Defining the core Arabidopsis thaliana root microbiome. Nature. 488, 86–90. doi: 10.1038/nature11237
Mejias, J., Chen, Y., Bazin, J., Truong, N., Mulet, K., Noureddine, Y., et al. (2022). Silencing the conserved small nuclear ribonucleoprotein SmD1 target gene alters susceptibility to root-knot nematodes in plants. Plant. Physiol. 189, 1741–1756. doi: 10.1093/plphys/kiac155
Mejias, J., Truong, N. M., Abad, P., Favery, B., and Quentin, M. (2019). Plant proteins and processes targeted by parasitic nematode effectors. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00970
Pieterse, C. M. J., Van Pelt, J. A., Van Wees, S. C. M., Ton, J., Léon-Kloosterziel, K. M., Keurentjes, J. J. B., et al. (2001). Rhizobacteria-mediated induced systemic resistance: triggering, signalling and expression. Eur. J. Plant Pathol. 107, 51–61. doi: 10.1023/A:1008747926678
Rovenich, H., Boshoven, J. C., and Thomma, B. P. (2014). Filamentous pathogen effector functions: of pathogens, hosts and microbiomes. Curr. Opin. Plant Biol. 20, 96–103. doi: 10.1016/j.pbi.2014.05.001
Siddiqui, I. A., and Shaukat, S. S. (2003). Effects of pseudomonas aeruginosa on the diversity of culturable microfungi and nematodes associated with tomato: impact on root-knot disease and plant growth. Soil Biol. Biochem. 35, 1359–1368. doi: 10.1016/S0038-0717(03)00215-3
Snelders, N. C., Kettles, G. J., Rudd, J. J., and Thomma, B. P. H. J. (2018). Plant pathogen effector proteins as manipulators of host microbiomes? Mol Plant Pathol. 19, 257–259. doi: 10.1111/mpp.12628
Snelders, N. C., Rovenich, H., Petti, G. C., Rocafort, M., van den Berg, G. C. M., Vorholt, J. A., et al. (2020). Microbiome manipulation by a soil-borne fungal plant pathogen using effector proteins. Nat. Plants. 6, 1365–1374. doi: 10.1038/s41477-020-00799-5
Song, Y., Wilson, A. J., Zhang, X., Thoms, D., Sohrabi, R., Song, S., et al. (2021). FERONIA restricts pseudomonas in the rhizosphere microbiome via regulation of reactive oxygen species. Nat. Plants. 7, 644–654. doi: 10.1038/s41477-021-00914-0
Sun, M., Gao, L., Shi, Y., Li, B., and Liu, X. (2006). Fungi and actinomycetes associated with Meloidogyne spp. Eggs and females in China and their biocontrol potential. J. Invertebr Pathol. 93, 22–28. doi: 10.1016/j.jip.2006.03.006
Tian, B., Cao, Y., and Zhang, K. (2015). Metagenomic insights into communities, functions of endophytes, and their associates with infection by root-knot nematode, Meloidogyne incognita, in tomato roots. Sci. Rep. 5, 87. doi: 10.1038/srep17087
Tian, X., Cheng, X., Mao, Z., Chen, G., Yang, J., Xie, B., et al. (2011). Composition of bacterial communities associated with a plant–parasitic nematode Bursaphelenchus mucronatus. Curr. Microbiol. 62, 117–125. doi: 10.1007/s00284-010-9681-7
Topalovi, O., Bredenbruch, S., Schleker, A. S. S., and Heuer, H. (2020). Microbes attaching to endoparasitic phytonematodes in soil trigger plant defense upon root penetration by the nematode. Front Plant Sci. 11. doi: 10.3389/fpls.2020.00138
Trivedi, P., Jan, E. L. S. G., and Singh, B. K. S. (2020). Plant-microbiome interactions: from community assembly to plant health. Nat. Rev. Microbiol. 18, 607–621. doi: 10.1038/s41579-020-0412-1
Trudgill, D. L. (1997). Parthenogenetic root-knot nematodes (Meloidogyne spp.); How can these biotrophic endoparasites have such an enormous host range. Plant Pathol. 46, 26–32. 1997.d01-201.x
Truong, N. M., Chen, Y., Mejias, J., Soul,é, S., Mulet, K., Jaubert-Possamai, S., et al. (2021). The meloidogyne incognita nuclear effector mieff1 interacts with arabidopsis cytosolic glyceraldehyde-3-phosphate dehydrogenases to promote parasitism. Front. Plant Sci. 12, 480. doi: 10.3389/fpls.2021.641480
Vieira, P., and Gleason, C. (2019). Plant-parasitic nematode effectors – insights into their diversity and new tools for their identification. Curr. Opin. Plant Biol. 50, 37–43. doi: 10.1016/j.pbi.2019.02.007
Wang, J., Wang, J., Liu, F., and Pan, C. (2010). Enhancing the virulence of Paecilomyces lilacinus against Meloidogyne incognita eggs by overexpression of a serine protease. Biotechnol. Lett. 32, 1159–1166. doi: 10.1007/s10529-010-0278-9
Wu, W., Wang, J., Wang, Z., Guo, L., Zhu, S., Zhu, Y., et al. (2022). Rhizosphere bacteria from Panax notoginseng against Meloidogyne hapla by rapid colonization and mediated resistance. Front Microbiol. 13, 82. doi: 10.3389/fmicb.2022.877082
Yin, N., Zhao, J., Liu, R., Li, Y., Ling, J., Yang, Y., et al. (2021). Biocontrol efficacy of Bacillus cereus strain Bc-cm103 against Meloidogyne incognita. Plant Dis. 105, 2061–2070. doi: 10.1094/PDIS-03-20-0648-RE
Zhao, J., Li, L., Liu, Q., Liu, P., Li, S., Yang, D., et al. (2019). A MIF-like effector suppresses plant immunity and facilitates nematode parasitism by interacting with plant annexins. J. Exp. Bot. 70, 5943–5958. doi: 10.1093/jxb/erz348
Zhao, J., Mao, Z., Sun, Q., Liu, Q., Jian, H., Xie, B., et al. (2020). MiMIF-2 effector of Meloidogyne incognita exhibited enzyme activities and potential roles in plant salicylic acid synthesis. Int. J. Mol. Sci. 21, 3507. doi: 10.3390/ijms21103507
Zhao, J., Sun, Q., Quentin, M., Ling, J., Abad, P., Zhang, X., et al. (2021). A Meloidogyne incognita C-type lectin effector targets plant catalases to promote parasitism. New Phytol. 232, 2124–2137. doi: 10.1111/nph.17690
Keywords: rhizosphere microbiome, Meloidogyne incognita, Arabidopsis thaliana, MiMIF-2 effector protein, parasitism
Citation: Liu R, Chen M, Liu B, Huang K, Mao Z, Li H and Zhao J (2023) A root-knot nematode effector manipulates the rhizosphere microbiome for establishing parasitism relationship with hosts. Front. Microbiol. 14:1217863. doi: 10.3389/fmicb.2023.1217863
Received: 06 May 2023; Accepted: 28 June 2023;
Published: 19 July 2023.
Edited by:
Gang Yu, Shanghai Jiao Tong University, ChinaReviewed by:
Gao-feng Wang, Huazhong Agricultural University, ChinaMusharaf Ahmad, University of Agriculture, Peshawar, Pakistan
Copyright © 2023 Liu, Chen, Liu, Huang, Mao, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huixia Li, bGloeEBnc2F1LmVkdS5jbg==; Jianlong Zhao, emhhb2ppYW5sb25nQGNhYXMuY24=